

ABSTRACT
Air pollution might affect atherosclerosis through DNA methylation changes in cells crucial to atherosclerosis, such as monocytes. We conducted an epigenome-wide study of DNA methylation in CD14+ monocytes and long-term ambient air pollution exposure in adults participating in the Multi-Ethnic Study of Atherosclerosis (MESA). We also assessed the association between differentially methylated signals and cis-gene expression. Using spatiotemporal models, one-year average concentrations of outdoor fine particulate matter (PM2.5) and oxides of nitrogen (NOX) were estimated at participants’ homes. We assessed DNA methylation and gene expression using Illumina 450k and HumanHT-12 v4 Expression BeadChips, respectively (n = 1,207). We used bump hunting and site-specific approaches to identify differentially methylated signals (false discovery rate of 0.05) and used linear models to assess associations between differentially methylated signals and cis-gene expression. Four differentially methylated regions (DMRs) located on chromosomes 5, 6, 7, and 16 (within or near SDHAP3, ZFP57, HOXA5, and PRM1, respectively) were associated with PM2.5. The DMRs on chromosomes 5 and 6 also associated with NOX. The DMR on chromosome 5 had the smallest p-value for both PM2.5 (p = 1.4×10−6) and NOX (p = 7.7×10−6). Three differentially methylated CpGs were identified for PM2.5, and cg05926640 (near TOMM20) had the smallest p-value (p = 5.6×10−8). NOX significantly associated with cg11756214 within ZNF347 (p = 5.6×10−8). Several differentially methylated signals were also associated with cis-gene expression. The DMR located on chromosome 7 was associated with the expression of HOXA5, HOXA9, and HOXA10. The DMRs located on chromosomes 5 and 16 were associated with expression of MRPL36 and DEXI, respectively. The CpG cg05926640 was associated with expression of ARID4B, IRF2BP2, and TOMM20. We identified differential DNA methylation in monocytes associated with long-term air pollution exposure. Methylation signals associated with gene expression might help explain how air pollution contributes to cardiovascular disease.
KEYWORDS: Air pollution, fine particulate matter, oxides of nitrogen, DNA methylation, gene expression
INTRODUCTION
Air pollution exposure is a risk factor for cardiovascular disease (CVD), and several potential pathways have been proposed to explain associations, including progression of atherosclerosis [1,2], systemic inflammatory responses, and oxidative stress [3]. However, there is limited evidence concerning pathways that might mediate the effects of air pollution on CVD.
Growing literature suggests that environmental pollutants might influence epigenetic regulation of gene expression, such as that through DNA methylation [4]. Prior studies have shown associations between exposure to air pollutants and DNA methylation in mixed blood leukocytes [5–20]. In addition, studies have associated DNA methylation patterns with cardiovascular outcomes [5,21–24]. Moreover, a European study demonstrated that DNA methylation alterations of genes enriched in the ‘ROS/glutathione/cytotoxic granules’ and ‘cytokine signaling’ inflammatory pathways were associated with both air pollution and cardiovascular and cerebrovascular disease [10].
However, results from prior epigenome-wide studies of air pollution have been inconsistent and significant findings are seldom replicated. It is likely that the use of whole blood consisting of heterogeneous cell types and the identification of differentially methylated sites instead of regions contributed to the differences across studies. Prior studies measured DNA methylation in mixed leukocytes. As cell-specific epigenetic signatures have been reported [25], shifts in cell-type composition might confound results of these studies. In addition, previous studies typically conduct association analyses on the CpG level, which can have lower specificity than conducting association analyses on the region level [26]. DNA methylation measurements are continuous when aggregated over a large number of cells (e.g., proportion of cells methylated), are more densely distributed and spatially correlated across the genome, and are more vulnerable to measurement error [26]. Since DNA methylation of nearby CpG sites is strongly correlated, power can be increased by borrowing information from adjacent CpG sites to identify differentially methylated regions (DMRs) [26].
In the Multi-Ethnic Study of Atherosclerosis (MESA) cohort, we previously assessed the association between long-term ambient air pollution exposure and global DNA methylation (ALU and LINE-1 elements) and candidate-site DNA methylation in purified monocytes [8]. However, our prior study was limited by the site-by-site analysis used to identify differentially methylated sites and by the candidate-site approach that only interrogated <1% of the CpG sites in the DNA methylation array. In the current study, we expand on our previous work on global and candidate-site methylation to conduct an epigenome-wide study to identify DMRs associated with long-term exposure to ambient fine particulate matter (PM2.5) and oxides of nitrogen (NOX) in monocytes. Monocytes were selected because of their crucial role in atherosclerosis pathogenesis [27]. We utilized data from MESA, MESA Air, and the MESA Epigenomics and Transcriptomics Study. We also investigated potential biological relevance of air pollution-associated methylation signals by analysing their associations with the expression of nearby genes.[28]
METHODS
Study population
MESA is a prospective longitudinal study aimed to investigate early, or subclinical, atherosclerosis and began in July 2000, enrolling 6,814 adults aged 45–84 years who were free of CVD at baseline [29]. The population-based MESA cohort is diverse with 28% black, 12% Chinese-American, 22% Hispanic, and 38% white participants from six field centres (Baltimore, MD; Chicago, Illinois; Los Angeles, CA; New York, NY; St. Paul, MN, and Winston-Salem, NC). Participants received physical exams and questionnaires at baseline and during four follow-up visits over a 10-year period since baseline. MESA Air is an ancillary study that started in 2004 and recruited additional participants, measured supplementary outcomes, and assessed ambient air pollution exposures for PM2.5, NOX, and black carbon [30]. The MESA Epigenomics and Transcriptomics Study is another ancillary study that randomly selected over 1,200 participants at the fifth visit from Baltimore, New York, St. Paul, and Winston-Salem to generate genome-wide DNA methylation and gene expression data from CD14+ purified monocytes [31].
Our analytic sample included 1,207 participants with DNA methylation, gene expression, and air pollution data. There are no Chinese-American participants in our study because methylation and gene expression data were not obtained from participants in Los Angeles or Chicago, the predominant source of Chinese-American participants in MESA. The study protocol was approved by the Institutional Review Board at each site.
Air pollution assessment
MESA Air predicted likelihood-based 2-week average PM2.5 and NOX concentrations at participants’ residences from 1999 to 2012 using spatiotemporal models [32,33]. Air pollution monitoring data were obtained from Air Quality System monitors used by the Environmental Protection Agency, the Interagency Monitoring of Protected Visual Environments (IMPROVE) network, and MESA Air monitors located in MESA Air cities and participants’ homes [32,33]. Input into the spatiotemporal models included the air pollution monitoring data and other information such as geographic data (e.g., roadway density), meteorological data, and land use information. The spatiotemporal models performed well with cross-validation R2 >0.80 [33]. This method allows for the characterization of air pollution variability spatially and temporally, such as by seasons or other shorter time periods, in addition to the characterization of spatial and spatiotemporal correlation. For each participant, we computed the average ambient concentrations of PM2.5 and NOX at their home address over the 12 months prior to the blood draw used for DNA methylation and gene expression assessments.
DNA methylation and gene expression quantification
DNA methylation and gene expression were quantified from blood drawn at the fifth visit (April 2010-February 2012). Trained technicians purified monocytes from the blood for subsequent DNA and RNA extraction by following standardized protocols with comprehensive quality control measures. Monocyte purification was conducted at each site using anti-CD14-coated magnetic beads and AutoMACs automated magnetic separation unit (Miltenyi Biotec, Bergisch Gladbach, Germany). AllPrep DNA/RNA Mini Kit (Qiagen, Inc., Hilden, Germany) was used to extract DNA and RNA simultaneously from purified monocytes. Flow cytometry of 18 specimens showed that monocyte samples were consistently >90% pure.
We assessed the epigenome-wide methylation profile of purified monocytes using the Infinium HumanMethylation450 BeadChip (450k; Illumina, Inc. CA, USA), which measured methylation at >485,000 CpG sites. We used Illumina GenomeStudio software to summarize bead-level methylation data. For the statistical analysis, raw methylation calls were normalized and converted to M-values (log ratio of methylated to unmethylated intensities) [34]. To obtain genome-wide expression profile for monocytes, we used the Illumina HumanHT-12 v4 Expression BeadChip and Ilumina Bead Array Reader (Illumina, Inc. CA, USA) to assess expression of >48,000 transcripts [31].
To mitigate potential confounding by batch effects for both the DNA methylation and gene expression arrays, samples were randomly assigned to chips and positions. For quality control, we excluded probes designed for sequences on the X or Y chromosomes, probes with non-detectable methylation in over 90% of the samples (using a detection p-value cut-off of 0.05), and probes located within 10 bp of single nucleotide polymorphisms. 437,600 of the 480,000 CpG sites on the Infinium 450k remained after applying quality control exclusion. Additional details of data pre-processing using Bioconductor [35] packages including beadarray, neqc, limma, and lumi in R [36] and quality control methods have been previously described [31].
Other measures
Information on demographics, socioeconomic status, lifestyle factors, and body mass index were obtained through questionnaires (self- and interviewer-administered) and physical examination. Race/ethnicity, sex, and education were obtained via self-report at baseline. Food frequency questionnaire responses were used to estimate intake of methyl nutrients at the fifth visit. Nutrients were calculated using the Nutrition Data System for Research (NDS-R database; Nutrition Coordinating Center, Minneapolis, MN, USA) as previously described [37]. A physical activity survey adapted from the Cross-Cultural Activity Participation Study [38] was used to assess the frequency and time spent in various physical activities during a typical week in the previous month. Recent infection was defined as having a fever, cold or flu, urinary infection, bronchitis, sinus infection, pneumonia, tooth infection, or arthritis flare-up within the past two weeks. Since the monocyte samples were >90% pure, we calculated enrichment scores for neutrophils, B cells, T cells, and natural killer cells from a gene set enrichment analysis using the gene expression signature of each cell type obtained from previously defined lists to adjust for residual contamination from non-monocytes in the analysis [31].
Statistical analysis
Identification of differentially methylated regions
We adopted the bump hunting approach previously described [26] to the Infinium 450k array to search for associations at the region level rather than the individual site level. The bumphunter package in R was used to identify DMRs associated with long-term air pollution exposure [26]. This approach has been used by several recent epigenome-wide studies across diverse disease areas [39–43]. Consecutive probes on the 450k array were grouped into clusters where the maximum distance between two neighbouring probes was 300 bp and each cluster contained at least 5 probes. Of the 437,600 sites that passed our quality control criteria, 152,636 sites (35% of total) were successfully grouped into 18,293 clusters. For each probe within a cluster, we regressed the M-value on air pollution (PM2.5 or NOX) to estimate a coefficient for each CpG site, adjusting for age, sex, race/ethnicity (black, Hispanic, white), household income, education, neighbourhood socioeconomic status (a summary score based on principal factor analysis of United States census and American Community Survey measures at the census tract level), smoking (smoking status [never, former, current] and pack years), second-hand smoke, body mass index (continuous), recent infection, methyl nutrient intake (continuous folate, vitamin B12, vitamin B6, methionine, zinc), physical activity (continuous, MET-min/wk m-su), study site, and chip position. We also adjusted for residual sample contamination by non-monocytes by adjusting for enrichment scores for neutrophils, B cells, T cells, and natural killer cells. The ComBat function in the package SVA [44] was used to obtain methylation chip-adjusted M-values for use in regression analyses. These estimated coefficients were smoothed using locally estimated scatterplot smoothing, and candidate DMRs were identified as regions above or below the 97.5th percentile of the empirical distribution of the smoothed estimate. Statistical uncertainty for each candidate DMR was assessed by bootstrapping. We conducted 1,000 bootstrap samples, and each bootstrap sample generated null candidate regions. The empirical p-value for each observed candidate DMR was the percent of null regions that were as extreme (longer and higher average value) as the observed region. The false discovery rate (FDR) was controlled at 0.05 to account for multiple testing [45]. DMRs were plotted using the makeMethPlot function within the methylation package [46].
Site-specific analysis
Since 65% (284,964) of the 437,600 probes that passed quality control were not grouped into clusters for DMR analysis (based on criteria described above), we analysed them as individual CpG sites to maximize use of the Infinium 450k array. Linear models and robust empirical Bayes moderated t-statistics were used to assess the associations between PM2.5 or NOX and methylation chip-adjusted M-values at individual CpGs using the limma package from Bioconductor [47]. A separate linear model was fit for each CpG site, adjusting for the same set of covariates as above. The FDR was controlled at 0.05 to account for multiple comparisons [45].
Gene expression
For differentially methylated regions and sites, we used linear models to correlate their methylation with expression of genes within a 1 Mb genomic region centred on the CpG region or site in question, adjusting for age, sex, race/ethnicity, study site, residual cell contamination by non-monocytes (neutrophils, B cells, T cells, and natural killer cells), methylation chip position, and expression chip. For DMRs, the average M-value of all CpG sites within the DMR was used. To account for multiple comparisons, we used an FDR threshold of 0.05 [45].
Secondary analysis
Secondary analyses using an interaction term for air pollution and sex or air pollution and race/ethnicity were conducted to investigate if sex or race/ethnicity modifies the effect of air pollution on methylation.
Functional annotation analysis
We evaluated the sites of significant air pollution-associated signals for overlap with known and predicted potentially functional genomic regions. We used ChromHMM [48] to predict the chromatin states in monocytes in silico based on histone modifications in monocyte samples from the BLUEPRINT [49,50] (H3K27ac, H3K4me1, H3K4me3) and the Encyclopedia of DNA Elements (ENCODE) [51] (H3K36me3) projects. We also annotated the sites using DNase hypersensitive hotspot information from a sample of monocytes (Sample ID RO01746, data generated by the UW ENCODE group) and transcription factor binding sites detected in any cell type from ENCODE [51]. Data were accessed from the UCSC Genome Browser [52] and the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/).
RESULTS
Table 1 presents participant characteristics (n = 1,207). The mean age of the analytic sample was 70 years, and 52% were women. The sample was diverse, consisting of 47% white, 32% Hispanic, and 21% black participants. Over 90% of the participants were non-smokers, and 55% did not currently use alcohol. The participants were well-educated – over 65% of the participants received education beyond high school. Participants had a mean BMI of 29.7 kg/m2. The mean PM2.5 prediction was 10.7 µg/m3 (interquartile range [IQR] = 2.2 µg/m3), the mean NOX prediction was 28.7 ppb (IQR = 31.9 ppb).
Table 1.
Descriptive characteristics of 1,207 MESA participants
| Characteristic | n (%) or mean (SD) |
|---|---|
| or median [IQR] | |
| N | 1,207 |
| Age, years | |
| Mean (SD) | 69.6 (9.4) |
| Age category, years | |
| <65 | 421 (35%) |
| 65–74 | 378 (31%) |
| 75–84 | 331 (27%) |
| ≥85 | 77 (6%) |
| Sex | |
| Women | 623 (52%) |
| Men | 584 (48%) |
| Race/ethnicity | |
| White | 570 (47%) |
| Hispanic | 381 (32%) |
| Black | 256 (21%) |
| Site | |
| New York | 398 (33%) |
| Maryland | 301 (25%) |
| Minnesota | 457 (38%) |
| North Carolina | 51 (4%) |
| Body mass index, kg/m2* | |
| Mean (SD) | 29.7 (5.5) |
| Body mass index category (WHO categories), kg/m2* | |
| Normal or underweight (<25) | 237 (20%) |
| Grade 1 overweight (25–29.9) | 454 (38%) |
| Grade 2 overweight (30–39.9) | 458 (38%) |
| Grade 3 overweight (≥40.0) | 57 (5%) |
| Physical activity, MET-min/wk m-su)* | |
| Mean (SD) | 5,696.2 (7,205.6) |
| Smoking status* | |
| Never | 484 (40%) |
| Former | 604 (50%) |
| Current | 112 (9%) |
| Second-Hand smoke (hours per week)* | 3.7 (20.5) |
| Current alcohol use* | |
| No | 660 (55%) |
| Yes | 543 (45%) |
| Education* | |
| Less than high school | 176 (15%) |
| High school | 236 (20%) |
| Some college but no degree | 212 (18%) |
| Bachelor’s/Associate/Technical | 373 (31%) |
| Advanced degree | 208 (17%) |
| Income* | |
| <$24,999 | 297 (25%) |
| $25,000 – $49,999 | 371 (31%) |
| $50,000 – $99,999 | 333 (28%) |
| ≥$100,000 | 163 (14%) |
| Neighbourhood socioeconomic status score* | |
| Mean (SD) | −0.41 (1.1) |
| PM2.5, µg/m3 | |
| Mean (SD) | 10.7 (1.7) |
| Median [IQR] | 10.4 [2.2] |
| NOX, ppb | |
| Mean (SD) | 28.7 (18.5) |
| Median [IQR] | 19.1 [31.9] |
| SD, standard deviation; IQR; interquartile range; MET, metabolic equivalent of task; PM2.5, fine particulate matter; NOX, oxides of nitrogen; WHO, World Health Organization | |
| * Contains missing values | |
We identified four DMRs with methylation that was significantly (FDR<0.05) associated with PM2.5 (Table 2, Figure 1–4). The DMR with the smallest p-value (1.4×10−6) was located on chromosome 5 and overlaps the 5’ untranslated region (UTR) of SDHAP3 (succinate dehydrogenase complex, subunit A, flavoprotein pseudogene 3) and a predicted weak promoter (based on histone modifications in monocyte samples from the BLUEPRINT (H3K27ac, H3K4me1, H3K4me3) and ENCODE (H3K36me3) projects) (Table 2). On average, a 2.5 µg/m3 higher exposure to PM2.5 was associated with a 1.15-fold higher methylation at this DMR (Table 2, Figure 1). The second DMR was located 3,448 bases upstream of ZFP57 (ZFP57 zinc finger protein) on chromosome 6, and was on average 1.11-fold more methylated in those with a 2.5 µg/m3 higher exposure to PM2.5 (p = 2.1×10−5) (Table 2, Figure 2). This DMR also overlaps a predicted weak promoter. The last two DMRs were found in the exon of PRM1 (protamine 1) in chromosome 16 and the 5’ UTR region of HOXA5 (homeobox A5) in chromosome 7, and these two DMRs were associated with a 1.27-fold (p = 3.2×10−5) and 1.04-fold (p = 4.6×10−5) higher average methylation for a 2.5 µg/m3 higher exposure to PM2.5, respectively (Table 2, Figures 3 and 4). The DMR on chromosome 16 is predicted to overlap heterochromatin while the one on chromosome 7 is predicted to overlap both a weak promoter and heterochromatin. The DMR on chromosome 16 only spans one bp, but it belongs to a cluster of 5 CpGs. Although the identification of this DMR was based on borrowed information from other CpGs within the cluster, the DMR seems to be due only to the methylation status of one CpG, which might make this DMR less informative (Figure 3).
Table 2.
Differentially methylated regions for PM2.5 and NOX with FDR<0.05
| Chr | Start | End | Nearest Gene (hg19) |
Location Relative to Gene (hg19) |
CD14+ Chromatin State a | Average Fold Change in Methylation | P-value | FDR Cut-Off |
|---|---|---|---|---|---|---|---|---|
| PM2.5 | ||||||||
| 5 | 1,594,282 | 1,594,863 | SDHAP3 | Overlaps 5' UTR | Weak promoter | 1.15b | 1.4x10−6 | 1.3x10−5 |
| 6 | 29,648,379 | 29,649,024 | ZFP57 | Upstream | Weak promoter | 1.11b | 2.1x10−5 | 2.6x10−5 |
| 16 | 11,374,865 | 11,374,865 | PRM1 | Inside exon | Heterochromatin/low | 1.27b | 3.2x10−5 | 3.9x10−5 |
| 7 | 27,183,274 | 27,184,109 | HOXA5 | Overlaps 5' UTR | Weak promoter and Heterochromatin/low | 1.04b | 4.6x10−5 | 5.3x10−5 |
| NOX | ||||||||
| 5 | 1,594,282 | 1,595,048 | SDHAP3 | Overlaps 5' UTR | Weak promoter | 1.26c | 7.7x10−6 | 1.5x10−5 |
| 6 | 29,648,379 | 29,649,024 | ZFP57 | Upstream | Weak promoter | 1.25c | 9.8x10−6 | 3.0x10−5 |
PM2.5, fine particulate matter; NOX, oxides of nitrogen; FDR, false discovery rate; chr, chromosome; UTR, untranslated region.
aPrediction based on histone modifications in monocyte samples from the BLUEPRINT (H3K27ac, H3K4me1, H3K4me3) and ENCODE (H3K36me3) projects
bAverage fold change in methylation for every 2.5 µg/m3 higher exposure to PM2.5.
cAverage fold change in methylation for every 30 ppb higher exposure to NOX.
Figure 1.
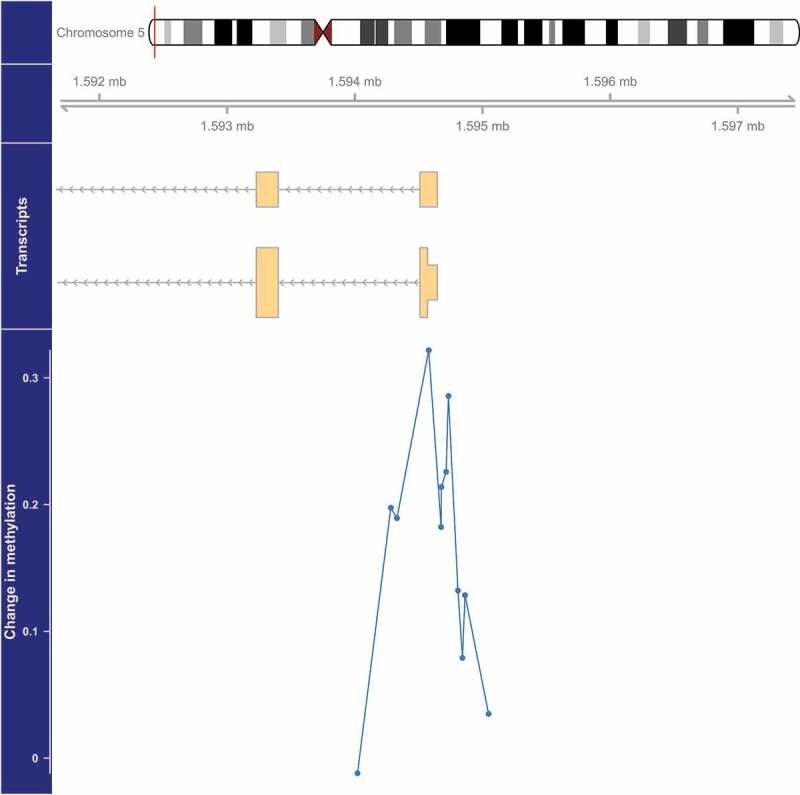
Genome-wide significant differentially methylated region associated with PM2.5 located at chromosome 5 (location: 1,594,282–1,594,863). The top portion of this plot is a karyogram of chromosome 5, with a red vertical bar indicating the location of interest. Below that is a graphic showing the exact chromosomal location in Mb, followed by a pictogram of the SDHAP3 gene (hg19). Tan boxes represent exons (vertically narrow portions are untranslated regions), and grey lines with arrows represent introns, as well as the direction of transcription (arrows pointing to the left indicate that this gene is transcribed from right to left, implying it is on the minus strand). The bottom section presents evidence for differential methylation of a DMR (blue line), based on a cluster of 12 CpGs (blue dots). The vertical axis for this section reflects the PM2.5 coefficient from the linear model. Plots were generated by using the function makeMethPlot from the package methylation.
Figure 2.
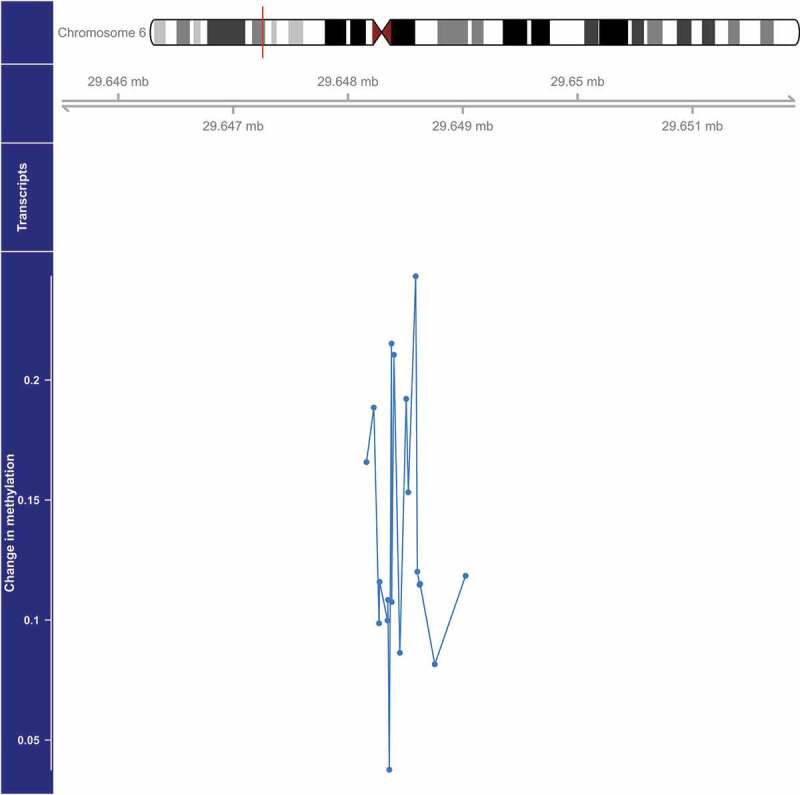
Genome-wide significant differentially methylated region associated with PM2.5 located at chromosome 6 (location: 29,648,379–29,649,024). The top portion of this plot is a karyogram of chromosome 6, with a red vertical bar indicating the location of interest. Below that is a graphic showing the exact chromosomal location in Mb (hg19). The bottom section presents evidence for differential methylation of a DMR (blue line), based on a cluster of 19 CpGs (blue dots). The vertical axis for this section reflects the PM2.5 coefficient from the linear model. Plots were generated by using the function makeMethPlot from the package methylation.
Figure 3.
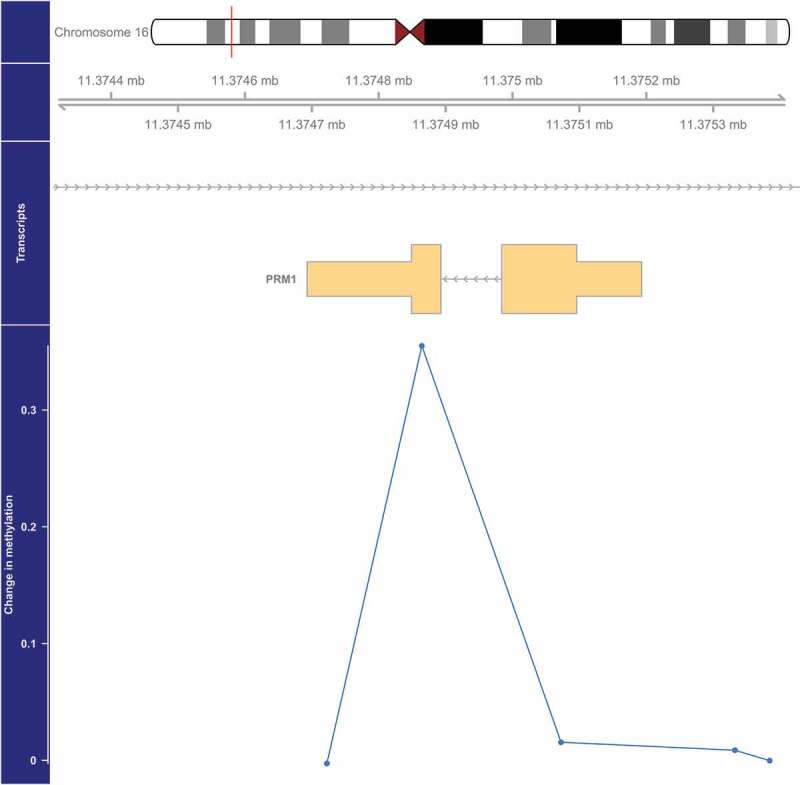
Genome-wide significant differentially methylated region associated with PM2.5 located at chromosome 16 (location: 11,374,865–11,374,865). The top portion of this plot is a karyogram of chromosome 16, with a red vertical bar indicating the location of interest. Below that is a graphic showing the exact chromosomal location in Mb, followed by a pictogram of the PRM1 gene (hg19). Tan boxes represent exons (vertically narrow portions are untranslated regions), and grey lines with arrows represent introns, as well as the direction of transcription (arrows pointing to the left indicate that this gene is transcribed from right to left, implying it is on the minus strand). The bottom section presents evidence for differential methylation of a DMR (blue line), based on a cluster of 5 CpGs (blue dots). The vertical axis for this section reflects the PM2.5 coefficient from the linear model. Plots were generated by using the function makeMethPlot from the package methylation.
Figure 4.
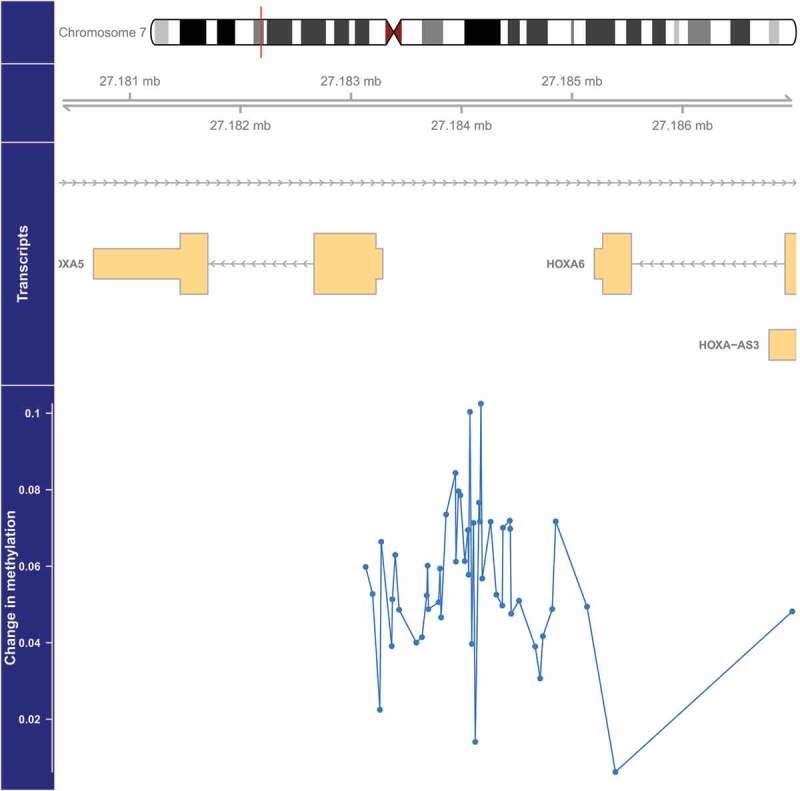
Genome-wide significant differentially methylated region associated with PM2.5 located at chromosome 7 (location: 27,183,274–27,184,109). The top portion of this plot is a karyogram of chromosome 7, with a red vertical bar indicating the location of interest. Below that is a graphic showing the exact chromosomal location in Mb, followed by pictograms of the HOXA5, HOXA6, and HOXA-AS3 genes (hg19). Tan boxes represent exons (vertically narrow portions are untranslated regions), and grey lines with arrows represent introns, as well as the direction of transcription (arrows pointing to the left indicate that this gene is transcribed from right to left, implying it is on the minus strand, and vice versa for arrows point to the right). The bottom section presents evidence for differential methylation of a DMR (blue line), based on a cluster of 47 CpGs (blue dots). The vertical axis for this section reflects the PM2.5 coefficient from the linear model. Plots were generated by using the function makeMethPlot from the package methylation.
For NOX, we identified two significant DMRs (FDR<0.05) that are the same as two of the DMRs identified for PM2.5, located on chromosomes 5 and 6 (Table 2; see Supplemental Material, Figures S1-S2). A 30 ppb higher exposure to NOX was associated with a 1.26-fold higher methylation in the DMR located on chromosome 5 (p = 7.7×10−6) and a 1.25-fold higher methylation in the DMR located on chromosome 6 (p = 9.8×10−6) (Table 2).
Of these four air pollution-associated DMRs, three were also associated with mRNA expression of nearby genes (Table 3). The DMR located on chromosome 7 was negatively associated with HOXA5 (β = −0.288; 95% CI: −0.332, −0.243; p = 1.5×10−34) and positively associated with HOXA9 (β = 0.168; 95% CI: 0.118, 0.218; p = 8.1×10−11) and HOXA10 (β = 0.079; 95% CI: 0.047, 0.112; p = 2.1×10−6) mRNA expression in monocytes. The DMR located in chromosome 16 was positively associated with DEXI (dexi homolog (mouse)) mRNA expression (β = 0.012; 95% CI: 0.004, 0.019; p = 2.8×10−3). The DMR located in chromosome 5 was positively associated with mRNA expression of MRPL36 (mitochondrial ribosomal protein L36) (β = 0.014; 95% CI: 0.005, 0.024; p = 3.7×10−3).
Table 3.
Significant (FDR<0.05) associations between methylation of PM2.5-associated DMRs and mRNA expression of nearby genes
| Chr | Start | End | Illumina Transcript | Gene (hg19) | β (95% CI) | P-value | FDR Cut-Off |
|---|---|---|---|---|---|---|---|
| 7 | 27,183,274 | 27,184,109 | ILMN_1753613 | HOXA5 | −0.288 (−0.332, −0.243) | 1.5x10−34 | 2.8x10−3 |
| 7 | 27,183,274 | 27,184,109 | ILMN_1739582 | HOXA9 | 0.168 (0.118, 0.218) | 8.1x10−11 | 5.6x10−3 |
| 7 | 27,183,274 | 27,184,109 | ILMN_1689336 | HOXA10 | 0.079 (0.047, 0.112) | 2.1x10−6 | 8.3x10−3 |
| 16 | 11,374,865 | 11,374,865 | ILMN_1738866 | DEXI | 0.012 (0.004, 0.019) | 2.8x10−3 | 1.1x10−2 |
| 5 | 1,594,282 | 1,594,863 | ILMN_1800197 | MRPL36 | 0.014 (0.005, 0.024) | 3.7x10−3 | 1.4x10−2 |
DMR, differentially methylated region; FDR, false discovery rate; CI, confidence interval.
Site-specific analyses identified three CpGs significantly associated with PM2.5 and one CpG associated with NOX at FDR<0.05 (Table 4). The PM2.5-associated CpG with the smallest p-value was cg05926640 (β = 0.049; 95% CI: 0.032, 0.067; p = 5.6×10−8), located on chromosome 1, 181,469 bases downstream of the TOMM20 (Translocase of Outer Mitochondrial Membrane 20) (Table 4). Cg05926640 is located within a DNase hypersensitive region (monocyte data from ENCODE), a transcription factor binding site (any cell type from UCSC Genome Browser), and a predicted strong enhancer region (Table 4). PM2.5 exposure was also positively associated with methylation of cg04310517, located on chromosome 16 (β = 0.052; 95% CI: 0.033, 0.072; p = 2.2×10−7) and cg09509909, located on chromosome 10 (β = 0.079; 95% CI: 0.049, 0.110; p = 4.6×10−7). NOX exposure was positively associated with methylation of cg11756214 on chromosome 19 (β = 0.078; 95% CI: 0.050, 0.106; p = 5.6×10−8). Additional details such as nearest gene annotation and chromatin state are listed in Table 4.
Table 4.
Differentially methylated sites for PM2.5 and NOX with FDR<0.05
| CpG | Chr | Nearest Gene | Distance |
Location Relative to Gene (hg19) |
CD14+ Chromatin State a | DNase HS CD14 + b | TFBS Any Cell c | β (95% CI) | P-value | FDR Cut-Off |
|---|---|---|---|---|---|---|---|---|---|---|
| PM2.5 | ||||||||||
| cg05926640 | 1 | TOMM20 | 181,469 | Downstream | Strong enhancer | Yes | Yes | 0.049 (0.032, 0.067) | 5.6x10−8 | 1.8x10−7 |
| cg04310517 | 16 | KREMEN2 | 3,954 | Upstream | Heterochromatin/low | No | No | 0.052 (0.033, 0.072) | 2.2x10−7 | 3.5x10−7 |
| cg09509909 | 10 | FGFBP3 | 3,489 | Upstream | Heterochromatin/low | Yes | Yes | 0.079 (0.049, 0.110) | 4.6x10−7 | 5.3x10−7 |
| NOX | ||||||||||
| cg11756214 | 19 | ZNF347 | 0 | 5' UTR | Weak promoter | Yes | Yes | 0.078 (0.050, 0.106) | 5.6x10−8 | 1.8x10−7 |
PM2.5, fine particulate matter; NOX, oxides of nitrogen; FDR, false discovery rate; chr, chromosome; DNAse HS, DNase hypersensitivity; TFBS, transcription factor binding site; ENCODE, The Encyclopedia of DNA Elements; CI, confidence interval.
aPrediction based on histone modifications in monocyte samples from the BLUEPRINT (H3K27ac, H3K4me1, H3K4me3) and ENCODE (H3K36me3) projects
bDNase hypersensitivity reported in a CD14+ monocyte sample (ENCODE).
cTFBS reported in any cell type available from the UCSC Genome Browser.
Of these air pollution-associated CpGs, only cg05926640 methylation was associated with cis-gene expression of genes within 1 Mb of the CpG site (Table 5). Cg05926640 methylation was negatively associated with mRNA expression of three ARID4B (AT-rich interaction domain 4B) transcripts and the coefficient for the transcript with the smallest p-value was −0.323 (95% CI: −0.453, −0.192; p = 1.3×10−6). It was also associated with IRF2BP2 (interferon regulatory factor 2 binding protein 2; β = −0.213; 95% CI: −0.318, −0.108; p = 7.3×10−5) and TOMM20 (β = 0.118; 95% CI: 0.044, 0.191; p = 1.7×10−3) mRNA expression in monocytes.
Table 5.
Significant (FDR<0.05) associations between cg05926640 methylation and mRNA expression of nearby genes
| CpG | Illumina Transcript | Gene (hg19) | β (95% CI) | P-value | FDR Cut-Off |
|---|---|---|---|---|---|
| cg05926640 | ILMN_2269564 | ARID4B | −0.323 (−0.453, −0.192) | 1.3x10−6 | 9.3x10−4 |
| cg05926640 | ILMN_2362982 | ARID4B | −0.227 (−0.333, −0.120) | 3.1x10−5 | 1.9x10−3 |
| cg05926640 | ILMN_1761334 | ARID4B | −0.274 (−0.406, −0.143) | 4.8x10−5 | 2.8x10−3 |
| cg05926640 | ILMN_1671005 | IRF2BP2 | −0.213 (−0.318, −0.108) | 7.3x10−5 | 3.7x10−3 |
| cg05926640 | ILMN_1679796 | TOMM20 | 0.118 (0.044, 0.191) | 1.7x10−3 | 4.6x10−3 |
FDR, false discovery rate; CI, confidence interval.
In secondary analyses, we did not find significant evidence of sex or race/ethnicity differences in the association of long-term air pollution exposure and DNA methylation in monocytes (interaction p >0.05).
DISCUSSION
We present results of an epigenome-wide analysis of DNA methylation and long-term PM2.5 and NOX exposure in monocytes from a multi-ethnic adult population. We identified four DMRs associated with PM2.5, two of which were also associated with NOX. Of these, three were associated with expression of nearby genes. Site-specific analyses identified three CpGs with methylation associated with PM2.5 and one CpG associated with NOX. DNA methylation of one of the PM2.5-associated CpGs was also associated with mRNA expression of nearby genes.
Our study builds upon the prior literature of air pollution and DNA methylation, and provides additional insights into the epigenome-wide associations specific to monocytes. Using methylomic and transcriptomic data from purified monocytes greatly improves the interpretability of our results especially with respect to signals that might be relevant to atherosclerosis. Although we have previously assessed the association between DNA methylation and PM2.5 and NOX, the prior candidate-site approach was limited by analysing <1% of the 450k array using a site-by-site analysis method [8]. Moreover, we adopted a bump hunting approach to identify DMRs to improve the sensitivity of our study. We also explored the functional relevance of significant DNA methylation signals using gene expression data. In addition, we utilized MESA Air’s sophisticated air pollution assessment, which provides participant-specific exposures that capture fine-scale spatial variability in air pollution. Finally, our results have greater generalizability than most previous studies due to the multi-ethnic nature of the MESA sample.
Three DMRs, located on chromosomes 7, 16, and 5 were associated with cis-expression of nearby genes. Methylation of the DMR located on chromosome 7 was associated with both PM2.5 exposure and expression of nearby homeobox cluster A (HOXA) genes including HOXA5, HOXA9, and HOXA10. This DMR is predicted to overlap a weak promoter region, providing evidence in favour of the hypothesis that this DMR affects gene expression. Two prior studies using whole blood associated long-term PM exposure with methylation of HOXA2 (p <1×10−3) and HOXA3 (p <1×10−5), both located within 40,000 bp of our HOXA5 DMR, although these associations did not reach statistical significance at the epigenome-wide level [9,53]. HOXA genes encode transcription factors crucial for patterning processes during vertebrate development [54] and haematopoietic differentiation [55]. Both HOXA9 and HOXA10 were hypomethylated in atherosclerotic aortas compared to normal aortas [56,57] and HOXA9 was overexpressed in atherosclerotic aortas [57]. Forced expression of HOXA10 in CD34+ progenitor cells increases the number of monocytes and reduces the number of B cells and natural killer cells produced [58]. In the carotid artery, disturbed flow, which regulates endothelial inflammation and monocyte-endothelial cell adhesion, induces hypermethylation of HOXA5 and downregulates its expression in endothelial cells [59]. In addition, HOX genes are differentially methylated in monocytes following lipopolysaccharide activation [60]. Taken together, these findings suggest that environmental exposures might induce differential methylation of HOXA genes, which might modulate haematopoietic differentiation and inflammation.
Both PM2.5 and NOX were positively associated with methylation of the DMR located on chromosome 5, which was positively associated with mRNA expression in MRPL36. MRPL36, a nuclear gene, encodes a member of the mitochondrial ribosomal proteins which are involved in mitochondrial protein synthesis [61]. The PM2.5-associated DMR located on chromosome 16 was associated with mRNA expression of DEXI, which encodes a transcript that is upregulated in emphysema tissue compared to normal tissue and might play a role in regulating inflammation [62].
The DMR located on chromosome 6 near ZFP57 was associated with both PM2.5 and NOX exposure in our study; however, it was not found to be associated with cis-gene expression. A study in a Swiss cohort reported that methylation of a DMR mapping to ZFP57 was associated with NO2 exposure [53].
Among CpG sites, cg05926640 (located 181,469 bp downstream of TOMM20) was most strongly associated with PM2.5 exposure. Methylation of this CpG might be directly related to alterations in gene expression because it is located within several potential functional features including a region of DNase hypersensitivity in monocytes, a transcription-factor binding site detected in many cell types including B cells, and a predicted strong enhancer region in monocytes. Cg05926640 methylation was negatively associated with mRNA expression of three genes, ARID4B, IRF2BP2, and TOMM20. ARID4B encodes a chromatin remodelling protein that might regulate genomic imprinting [63], is linked to various cancers [64,65], and is involved in spermatogenesis [66]. Interestingly, ARID4B has also been suggested to regulate haematopoiesis by controlling expression of HOX genes [65].
DNA methylation at cg05926640 was also positively associated with mRNA expression of its nearest gene TOMM20. TOMM20 encodes a subunit of the translocase of the outer mitochondrial membrane, which is the main import gate for most nuclear-encoded mitochondrial proteins [67]. As previously mentioned, the DMR located on chromosome 5 was positively associated with mRNA expression in MRPL36, which encodes a mitochondrial ribosomal protein. A study of Chinese adults reported that DMRs associated with long-term air pollution were significantly enriched in biological processes related to mitochondrial assembly [68]. Mitochondrial dysfunction can disrupt macrophage cholesterol homeostasis and contribute to inflammation and atherosclerosis [69]. Methylation of cg05926640 was also negatively associated with mRNA expression of nearby IRF2BP2. IRF2BP2 is a transcriptional corepressor that binds interferon regulatory factor 2, a negative regulator of many interferon-responsive genes [28]. In mice, IRF2BP2-deficient macrophages are inflammatory, have impaired cholesterol efflux, and worsened atherosclerosis [70]. Humans who are homozygous for a IRF2BP2 deletion polymorphism have lower IRF2BP2 protein levels in peripheral blood mononuclear cells and increased risk of coronary artery disease [70]. Taken together, this suggests that PM2.5 exposure might affect cholesterol homeostasis to promote inflammation and atherosclerosis.
We found cg11756214 within ZNF347 to be significantly associated with NOX. In a study of Korean adults, methylation of another CpG site located within ZNF347 (cg15050103) was statistically significantly associated with NO2 exposure [71]. Although cg11756214 was not associated with cis-gene expression in our study, we did not assess the potential associations with trans-gene expression.
Prior analysis in this cohort did not find long-term ambient air pollution to be associated with global DNA methylation as measured by methylation in Alu and LINE-1 repetitive elements [8]. In the prior study, PM2.5 significantly associated with methylation of the CpGs cg20455854, cg07855639, cg07598385, cg17360854, and cg23599683, which were previously associated with expression of nearby genes [8,31], but none of these CpGs reached statistical significance (FDR<0.05) in the current analysis.
Prior research in this cohort also demonstrated that methylation of cg05575921 was associated with smoking, carotid plaque score, and with mRNA expression of AHRR [72]. In addition, a study of non-smoking Taiwanese adults demonstrated that residing in areas with higher PM2.5 was associated with lower methylation of cg05575921 [73]. However, we did not find long-term exposure to ambient PM2.5 (β = −0.054; 95% CI: −0.116, 0.008) and NOX (β = −0.097; 95% CI: −0.218, 0.025) to be associated with methylation at cg05575921 (see Supplemental Material, Table S1). Possibly, we did not detect this association due to limited power. It is also possible that the AHRR pathway is more activated when exposures are high. The mean PM2.5 exposure in this study (10.7 μg/m3) was much lower than that of the Taiwanese study, where the mean PM2.5 exposures ranged from 27.3 to 39.8 μg/m3 for the four geographic areas.
Using mixed blood leukocytes from adults, a large number of studies have associated short- and intermediate-term air pollution exposure with DNA methylation [5,6,12–15,18,19,74] and long-term air pollution and DNA methylation [9–11,16,17,20,53,68,71,73,75–77]. Short-term air pollution exposure can trigger cardiovascular events, while long-term air pollution exposure can contribute to chronic cardiovascular processes such as atherosclerosis [1–3]. The majority of methylation sites or regions previously identified to be associated with long-term air pollution were not significantly associated with PM2.5 or NOX in our study. We might not have observed these associations due to fundamental differences between our studies, including the assessment of DNA methylation in monocytes versus mixed leukocytes and differences in study population. The prior studies adjusted for mixed blood cell type composition, most typically using the Houseman method [78], but the contributions of specific leukocytes, such as monocytes, cannot be deciphered. Moreover, our study included a racially/ethnically diverse cohort from the United States, whereas prior epigenome-wide studies, with few exceptions, generally included predominantly white cohorts. In addition, our DMR findings using the bump hunting method are not comparable to the site-specific methods used in most prior studies.
The cross-sectional nature of this study limits our ability to infer the causal effect of air pollution on changes in DNA methylation. We note, however, that our exposure estimates are for the one-year period preceding exposure. Since it is not plausible that DNA methylation causes air pollution, reverse causation is not a concern. Our analytic sample included slightly fewer black participants and slightly more Hispanic participants than overall participants from the four participating MESA field centres from the 5th examination, but we expect the impact of selection bias to be minimal in our analysis. Although we have adjusted for a wide range of potential confounders, there might be residual confounding or confounding by unmeasured factors or factors not included in the analysis. Our study was also limited by the small sample size. Finally, our analysis is a discovery effort, and future studies are needed to replicate our results and further elucidate the biological relevance of air pollution-associated methylation signals.
The bump hunting and site-specific analyses interrogated mutually exclusive sets of CpG sites. We used bump hunting where available, and sites on the Infinium 450k array that could not be grouped into clusters were not analysed using the bump hunting approach (See Methods for details about clustering); instead, non-clustered CpG sites were analysed using a site-specific approach. Since the bump hunting and site-specific analyses used separate sets of probes on the Infinium 450k array, we could not see if results based on one analysis were replicated by the other.
Using purified blood monocytes obtained from a multi-ethnic adult population, we identified differentially methylated regions and sites associated with long-term air pollution exposures. Some of the differentially methylated signals were also associated with expression of nearby genes. These genes are involved in inflammation and cholesterol homeostasis, which are relevant in atherosclerosis pathogenesis. Additional research in circulating monocytes is needed to elucidate whether DNA methylation mediates air pollution-associated diseases such as atherosclerosis and identify the functional relevance of the genomic regions identified via air pollution-associated methylation signals.
Supplementary Material
Acknowledgments
MESA and the MESA SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. This research was supported by contracts HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, 2R01 HL071759, and T32 HL007902 from the National Heart, Lung, and Blood Institute, and by grants UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420 from the National Center for Advancing Translational Sciences (NCATS). This publication was developed under STAR research assistance agreements, No. RD831697 (MESA Air), RD-83830001 (MESA Air Next Stage), and RD-83479601 awarded by the U.S. Environmental Protection Agency. It has not been formally reviewed by the EPA. The views expressed in this document are solely those of the authors and the EPA does not endorse any products or commercial services mentioned in this publication. This work was also supported by National Institute of Environmental Health Sciences (NIEHS) grants 1 F31 ES025475-01, P30 ES007033-01, P30 ES007033, and P50 ES015915-01. The MESA Epigenomics & Transcriptomics Study was funded by NIA grant 1R01HL101250-01 to Wake Forest University Health Sciences. The authors thank the other investigators, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI, USEPA, or NIEHS.
Funding Statement
This work was supported by the National Center for Advancing Translational Sciences [UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420]; National Heart, Lung, and Blood Institute [HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, 2R01 HL071759, and T32 HL007902]; National Institute of Environmental Health Sciences [1 F31 ES025475-01, P30 ES007033-01, P30 ES007033, and P50 ES015915-01]; National Institute on Aging [1R01HL101250-01]; U.S. Environmental Protection Agency [RD831697 (MESA Air), RD-83830001, and RD-83479601].
Disclosure statement
Gloria Chi completed this work while she was a student at the University of Washington. She is currently employed by Genentech, Inc.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Kaufman JD. Does air pollution accelerate progression of atherosclerosis?. J Am Coll Cardiol. 2010;56(22):1809–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kaufman JD, Adar SD, Barr RG, et al. Association between air pollution and coronary artery calcification within six metropolitan areas in the USA (the multi-ethnic study of atherosclerosis and air pollution): a longitudinal cohort study. Lancet. 2016;388(10045):696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brook RD, Rajagopalan S, Pope CA, et al. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American heart association. Circulation. 2010;121(21):2331–2378. [DOI] [PubMed] [Google Scholar]
- [4].Hou L, Zhang X, Wang D, et al. Environmental chemical exposures and human epigenetics. Int J Epidemiol. 2012;41(1):79–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Baccarelli A, Wright RO, Bollati V, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179(7):572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bind M-A, Lepeule J, Zanobetti A, et al. Air pollution and gene-specific methylation in the normative aging study. Epigenetics. 2014;9(3):448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bollati V, Baccarelli A, Hou L, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67(3):876–880. [DOI] [PubMed] [Google Scholar]
- [8].Chi GC, Liu Y, MacDonald JW, et al. Long-term outdoor air pollution and DNA methylation in circulating monocytes: results from the Multi-Ethnic Study of Atherosclerosis (MESA). Environ Health. 2016;15(1). DOI: 10.1186/s12940-016-0202-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].De F.c. Lichtenfels AJ, Van Der Plaat DA, De Jong K, et al. Long-term air pollution exposure, genome-wide DNA methylation and lung function in the lifelines cohort study. Environ Health Perspect. 2018;126(2):027004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fiorito G, Vlaanderen J, Polidoro S, et al. Oxidative stress and inflammation mediate the effect of air pollution on cardio- and cerebrovascular disease: a prospective study in nonsmokers: effect of Air Pollution on Cardio- and Cerebrovascular Disease. Environ Mol Mutagen. 2018;59:234–246. [DOI] [PubMed] [Google Scholar]
- [11].Gondalia R, Baldassari A, Holliday KM, et al. Methylome-wide association study provides evidence of particulate matter air pollution-associated DNA methylation. Environ Int. 2019;132:104723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li H, Chen R, Cai J, et al. Short-term exposure to fine particulate air pollution and genome-wide DNA methylation: a randomized, double-blind, crossover trial. Environ Int. 2018;120:130–136. [DOI] [PubMed] [Google Scholar]
- [13].Madrigano J, Baccarelli A, Mittleman MA, et al. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ Health Perspect. 2011;119(7):977–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mostafavi N, Vermeulen R, Ghantous A, et al. Acute changes in DNA methylation in relation to 24 h personal air pollution exposure measurements: a panel study in four European countries. Environ Int. 2018;120:11–21. [DOI] [PubMed] [Google Scholar]
- [15].Panni T, Mehta AJ, Schwartz JD, et al. A genome-wide analysis of DNA methylation and fine particulate matter air pollution in three study populations: KORA F3, KORA F4, and the normative aging study. Environ Health Perspect. 2016;124(7):983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Plusquin M, Guida F, Polidoro S, et al. DNA methylation and exposure to ambient air pollution in two prospective cohorts. Environ Int. 2017;108:127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sayols-Baixeras S, Fernández-Sanlés A, Prats-Uribe A, et al. Association between long-term air pollution exposure and DNA methylation: the REGICOR study. Environ Res. 2019;176:108550. [DOI] [PubMed] [Google Scholar]
- [18].Tarantini L, Bonzini M, Apostoli P, et al. Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environ Health Perspect. 2009;117(2):217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang C, Chen R, Shi M, et al. Possible mediation by methylation in acute inflammation following personal exposure to fine particulate air pollution. Am J Epidemiol. 2018;187(3):484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang C, O’Brien KM, Xu Z, et al. Long-term ambient fine particulate matter and DNA methylation in inflammation pathways: results from the sister study. Epigenetics. 2020a;15(5):524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Alexeeff SE, Baccarelli AA, Halonen J, et al. Association between blood pressure and DNA methylation of retrotransposons and pro-inflammatory genes. Int J Epidemiol. 2013;42(1):270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fernández-Sanlés A, Sayols-Baixeras S, Subirana I, et al. Association between DNA methylation and coronary heart disease or other atherosclerotic events: a systematic review. Atherosclerosis. 2017;263:325–333. [DOI] [PubMed] [Google Scholar]
- [23].Nakatochi M, Ichihara S, Yamamoto K, et al. Epigenome-wide association of myocardial infarction with DNA methylation sites at loci related to cardiovascular disease. Clin Epigenetics. 2017;9(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Turunen MP, Aavik E, Ylä-Herttuala S. Epigenetics and atherosclerosis. Biochim Biophys Acta BBA-Gen Subj. 2009;1790(9):886–891. [DOI] [PubMed] [Google Scholar]
- [25].Consortium RE, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jaffe AE, Murakami P, Lee H, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012;41(1):200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010;7(2):77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Childs KS. Identification of novel co-repressor molecules for interferon regulatory Factor-2. Nucleic Acids Res. 2003;31(12):3016–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bild DE, Bluemke DA, Burke GL, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol. 2002;156(9):871–881. [DOI] [PubMed] [Google Scholar]
- [30].Kaufman JD, Adar SD, Allen RW, et al. Prospective study of particulate air pollution exposures, subclinical atherosclerosis, and clinical cardiovascular disease: : the multi-ethnic study of atherosclerosis and air pollution (MESA Air). Am J Epidemiol. 2012;176(9):825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu Y, Ding J, Reynolds LM, et al. Methylomics of gene expression in human monocytes. Hum Mol Genet. 2013;22(24):5065–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cohen MA, Adar SD, Allen RW, et al. Approach to estimating participant pollutant exposures in the multi-ethnic study of atherosclerosis and air pollution (MESA air). Environ Sci Technol. 2009;43(13):4687–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Keller JP, Olives C, Kim S-Y, et al. A unified spatiotemporal modeling approach for predicting concentrations of multiple air pollutants in the multi-ethnic study of atherosclerosis and air pollution. Environ Health Perspect. 2015;123(4):301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587–2105–11–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].R Core Team . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- [37].Nettleton JA, Rock CL, Wang Y, et al. Associations between dietary macronutrient intake and plasma lipids demonstrate criterion performance of the Multi-Ethnic Study of Atherosclerosis (MESA) food-frequency questionnaire. Br J Nutr. 2009;102:1220–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ainsworth BE, Irwin ML, Addy CL, et al. Moderate physical activity patterns of minority women: the cross-cultural activity participation study. J Womens Health Gend Based Med. 1999;8(6):805–813. [DOI] [PubMed] [Google Scholar]
- [39].Chitrala KN, Hernandez DG, Nalls MA, et al. Race-specific alterations in DNA methylation among middle-aged African Americans and Whites with metabolic syndrome. Epigenetics. 2020;15(5):462–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gastoł J, Kapusta P, Polus A, et al. Epigenetic mechanism in search for the pathomechanism of diabetic neuropathy development in diabetes mellitus type 1 (T1DM). Endocrine. 2020;68(1):235–240. [DOI] [PubMed] [Google Scholar]
- [41].Mohandas N, Loke YJ, Mackenzie L, et al. Deciphering the role of epigenetics in self-limited epilepsy with centrotemporal spikes. Epilepsy Res. 2019;156:106163. [DOI] [PubMed] [Google Scholar]
- [42].Reimann B, Janssen BG, Alfano R, et al. The cord blood insulin and mitochondrial DNA content related methylome. Front Genet. 2019;10:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Serena C, Millan M, Ejarque M, et al. Adipose stem cells from patients with Crohn’s disease show a distinctive DNA methylation pattern. Clin Epigenetics. 2020;12(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Leek JT, Johnson WE, Parker HS, et al. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinforma Oxf Engl. 2012;28(6):882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Methodol. 1995; 289–300. [Google Scholar]
- [46].MacDonald JW 2015. jmacdon/methylation.
- [47].Smyth GK. 2005. Limma: linear models for microarray data. In: Bioinformatics and computational biology solutions using R and Bioconductor, Gentleman R, Carey V, Dudoit S, et al., editors. New York: Springer; p. 397–420. [Google Scholar]
- [48].Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9(3):215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Adams D, Altucci L, Antonarakis SE, et al. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol. 2012;30(3):224–226. [DOI] [PubMed] [Google Scholar]
- [50].Saeed S, Quintin J, Kerstens HHD, et al. Epigenetic programming during monocyte to macrophage differentiation and trained innate immunity. Science. 2014;345(6204):1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rosenbloom KR, Sloan CA, Malladi VS, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41(D1):D56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Karolchik D, Barber GP, Casper J, et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42(D1):D764–D770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Eze IC, Jeong A, Schaffner E, et al. Genome-wide DNA methylation in peripheral blood and long-term exposure to source-specific transportation noise and air pollution: the SAPALDIA study. Environ Health Perspect. 2020;128(6):67003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Krumlauf R. Hox genes in vertebrate development. Cell. 1994;78(2):191–201. [DOI] [PubMed] [Google Scholar]
- [55].Abramovich C, Humphries RK. Hox regulation of normal and leukemic hematopoietic stem cells. Curr Opin Hematol. 2005;12(3):210–216. [DOI] [PubMed] [Google Scholar]
- [56].Yamada Y, Horibe H, Oguri M, et al. Identification of novel hyper- or hypomethylated CpG sites and genes associated with atherosclerotic plaque using an epigenome-wide association study. Int J Mol Med. 2018;41(5):2724–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zaina S, Heyn H, Carmona FJ, et al. DNA methylation map of human atherosclerosis. Circ Cardiovasc Genet. 2014;7(5):692–700. [DOI] [PubMed] [Google Scholar]
- [58].Taghon T, Stolz F, Smedt MD, et al. HOX-A10 regulates hematopoietic lineage commitment: evidence for a monocyte-specific transcription factor. Blood. 2002;99(4):1197–1204. [DOI] [PubMed] [Google Scholar]
- [59].Dunn J, Simmons R, Thabet S, et al. The role of epigenetics in the endothelial cell shear stress response and atherosclerosis. Int J Biochem Cell Biol. 2015;67:167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Morgan R, Whiting K. Differential expression of HOX genes upon activation of leukocyte sub-populations. Int J Hematol. 2008;87(3):246–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kenmochi N, Suzuki T, Uechi T, et al. The human mitochondrial ribosomal protein genes: mapping of 54 genes to the chromosomes and implications for human disorders. Genomics. 2001;77(1–2):65–70. [DOI] [PubMed] [Google Scholar]
- [62].Edgar AJ, Birks EJ, Yacoub MH, et al. Cloning of dexamethasone-induced transcript. Am J Respir Cell Mol Biol. 2001;25(1):119–124. [DOI] [PubMed] [Google Scholar]
- [63].Wu M-Y, Tsai T-F, Beaud, et al. Deficiency of Rbbp1/Arid4a and Rbbp1l1/Arid4b alters epigenetic modifications and suppresses an imprinting defect in the PWS/AS domain. Genes Dev. 2006;20(20):2859–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Goldberger N, Walker RC, Kim CH, et al. Inherited variation in miR-290 expression suppresses breast cancer progression by targeting the metastasis susceptibility gene Arid4b. Cancer Res. 2013;73(8):2671–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wu M-Y, Eldin KW, Beaud, et al. Identification of chromatin remodeling genes Arid4a and Arid4b as leukemia suppressor genes. JNCI J Natl Cancer Inst. 2008;100(17):1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wu R-C, Zeng Y, Pan I-W, et al. Androgen receptor coactivator ARID4B is required for the function of sertoli cells in spermatogenesis. Mol Endocrinol. 2015;29(9):1334–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wurm CA, Neumann D, Lauterbach MA, et al. Nanoscale distribution of mitochondrial import receptor Tom20 is adjusted to cellular conditions and exhibits an inner-cellular gradient. Proc Natl Acad Sci U S A. 2011;108(33):13546–13551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wang M, Zhao J, Wang Y, et al. Genome-wide DNA methylation analysis reveals significant impact of long-term ambient air pollution exposure on biological functions related to mitochondria and immune response. Environ Pollut. 2020b;264:114707. [DOI] [PubMed] [Google Scholar]
- [69].Graham A. Mitochondrial regulation of macrophage cholesterol homeostasis. Free Radic Biol Med. 2015;89:982–992. [DOI] [PubMed] [Google Scholar]
- [70].Chen -H-H, Keyhanian K, Zhou X, et al. IRF2BP2 reduces macrophage inflammation and susceptibility to atherosclerosis. Circ Res. 2015;117(8):671–683. [DOI] [PubMed] [Google Scholar]
- [71].Lee MK, Xu C-J, Carnes MU, et al. Genome-wide DNA methylation and long-term ambient air pollution exposure in Korean adults. Clin Epigenetics. 2019;11(1). DOI: 10.1186/s13148-019-0635-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Reynolds LM, Wan M, Ding J, et al. DNA methylation of the aryl hydrocarbon receptor repressor associations with cigarette smoking and subclinical atherosclerosis. Circ Cardiovasc Genet CIRCGENETICS. 2015;115:001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Tantoh DM, Lee K-J, Nfor ON, et al. Methylation at cg05575921 of a smoking-related gene (AHRR) in non-smoking Taiwanese adults residing in areas with different PM2.5 concentrations. Clin Epigenetics. 2019a;11(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].De Nys S, Duca R-C, Nawrot T, et al. Temporal variability of global DNA methylation and hydroxymethylation in buccal cells of healthy adults: association with air pollution. Environ Int. 2018;111:301–308. [DOI] [PubMed] [Google Scholar]
- [75].Mostafavi N, Vlaanderen J, Portengen L, et al. Associations between genome-wide gene expression and ambient nitrogen oxides. Epidemiology. 2017;28(3):320–328. [DOI] [PubMed] [Google Scholar]
- [76].Tantoh DM, Wu M-F, Ho -C-C, et al. SOX2 promoter hypermethylation in non-smoking Taiwanese adults residing in air pollution areas. Clin Epigenetics. 2019b;11(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].White AJ, Kresovich JK, Keller JP, et al. Air pollution, particulate matter composition and methylation-based biologic age. Environ Int. 2019;132:105071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Houseman EA, Kelsey KT, Wiencke JK, et al. Cell-composition effects in the analysis of DNA methylation array data: a mathematical perspective. BMC Bioinformatics. 2015;16(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.