

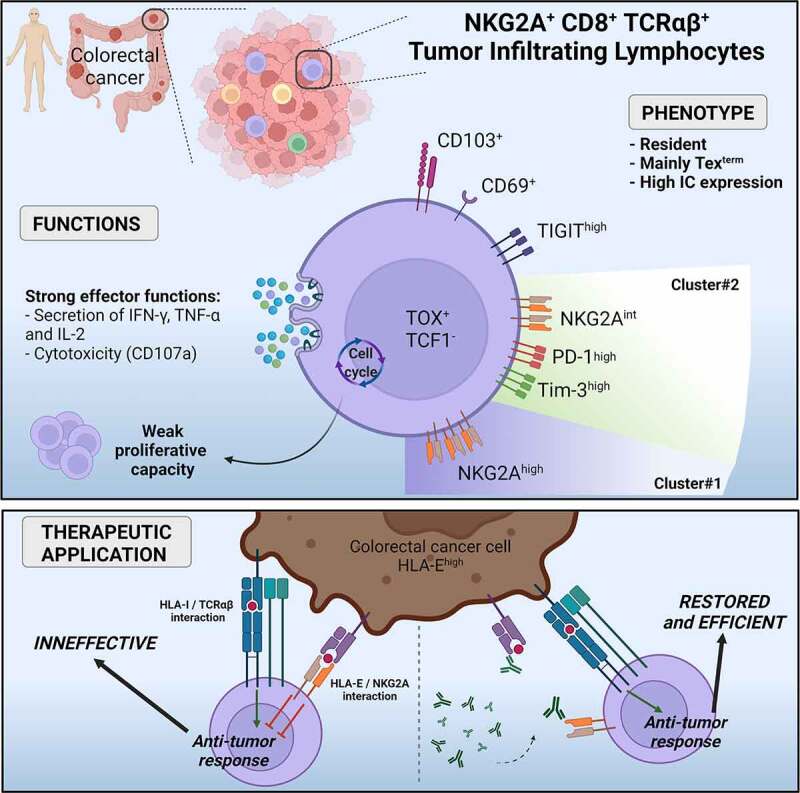
Abstract
Recently, the inhibitory CD94/NKG2A receptor has joined the group of immune checkpoints (ICs) and its expression has been documented in NK cells and CD8+ T lymphocytes in several cancers and some infectious diseases. In colorectal cancer (CRC), we previously reported that NKG2A+ tumor-infiltrating lymphocytes (TILs) are predominantly CD8+ αβ T cells and that CD94 overexpression and/or its ligand HLA-E were associated with a poor prognosis. This study aimed to thoroughly characterize the NKG2A+ CD8+ TIL subpopulation and document the impact of NKG2A on anti-tumor responses in CRC. Our findings highlight new features of this subpopulation: (i) enrichment in colorectal tumors compared to paired normal colonic mucosa, (ii) their character as tissue-resident T cells and their majority terminal exhaustion status, (iii) co-expression of other ICs delineating two subgroups differing mainly in the level of NKG2A expression and the presence of PD-1, (iv) high functional avidity despite reduced proliferative capacity and finally (v) inhibition of anti-tumor reactivity that is overcome by blocking NKG2A. From a clinical point of view, these results open a promising alternative for immunotherapies based on NKG2A blockade in CRC, which could be performed alone or in combination with other IC inhibitors, adoptive cell transfer or therapeutic vaccination.
KEYWORDS: NKG2A, CD8+ TILs, colorectal cancer, immune checkpoint inhibitor
Graphical Abstract
Introduction
Over the past 15 years, immunotherapies (ITs) have been developed and optimized to enhance anti-tumor immune responses in many cancer models, including T-cell transfer therapies, vaccines, and immune checkpoint inhibitors (ICis).1 Since 2011 and the first approval of ipilimumab (anti-cytotoxic T-lymphocyte antigen 4 (CTLA-4)) for the treatment of melanoma by U.S. FDA, six therapeutic mAbs targeting the programmed-cell death protein 1/programmed-cell death ligand 1 (PD-1/PD-L1) axis have been tested in many clinical trials2 in a large subset of tumor types including colorectal cancer.
Colorectal cancers are classified into two groups based on microsatellite status, distinguishing MSI (Microsatellite instable, 15%) tumors with a hyper-mutable phenotype leading to an increase in tumor infiltrating lymphocytes (TILs) and MSS (Microsatellite stable, 85%) tumors which have a low tumor mutational burden (TMB) and are considered as “cold tumors”.3 ICi responses in colorectal cancers are consistent with this classification since MSI-high (MSI-H)/deficient DNA mismatch repair (dMMR) metastatic colorectal cancers respond to anti-PD-1 mAb pembrolizumab,4,5 approved by FDA for the first-line treatment of these metastatic colorectal cancers in 2020.6 In addition, therapy combining the anti-PD-1 mAb nivolumab with ipilimumab has been shown to improve overall and progression-free survival compared with monotherapies in these cancers.7,8 These responses were related to a high density of TILs, associated with high TMB.5 Despite these reported successes, the majority of colorectal cancer patients still not benefit from effective treatments because MSI-H/dMMR metastatic colorectal cancers account for only 4 to 5% of all metastatic colorectal cancers.9 Many clinical trials are currently combining strategies to treat proficient DNA mismatch repair (pMMR/MSS) metastatic colorectal cancers and refractory dMMR/MSI metastatic colorectal cancers by first promoting an effective anti-tumor response with different approaches, and then optimizing it with ICis.6
New classifications have been proposed to characterize colorectal cancers more faithfully. A first one was proposed by J. Galon and colleagues based on the quantification of CD3+ and CD8+ cytotoxic lymphocytes at the tumor edge and core, from low (Immunoscore 0) to high (Immunoscore 4) immune cell density.10,11 In 2015, an international consortium proposed a new robust classification composed by four consensus molecular subtypes (CMS) based on their genomic signature, immune infiltrate and mutations.12 The CMS1 subtype is characterized by a hypermutated and MSI phenotype associated with strong immune activation and represents about 14% of colorectal cancers. The “Canonical” CMS2 subtype (37%) exhibits strong Wnt and MYC signaling activation and the “Metabolic” CMS3 subtype (13%) a metabolic dysregulation. In the last CMS4 subtype (23%), termed “Mesenchymal”, activation of TGF-β signaling, stromal invasion, and angiogenesis are observed. The remaining 13% of samples have mixed features and are unclassified, underlying the limitations of this classification, although it should improve the choice of the most appropriate IT for a majority of colorectal cancer patients.
In this context, it remains crucial to improve the global knowledge of all ICs expressed by TILs in colorectal cancer that may alter the anti-tumor immune response. Among these ICs, the Natural Killer group protein 2A (NKG2A) is now considered as a new one. Initially described by Carretero et al. in 1997, NKG2A forms, together with the CD94 chain, an inhibitory receptor, expressed by NK cells but also CD8+ T cells.13 The non-polymorphic and non-classical HLA class I molecule HLA-E, ubiquitously expressed by cells in normal tissues, is the unique ligand of CD94/NKG2A heterodimer, inducing inhibition of both NK-cell and T-cell responses.14 HLA-E overexpression has been reported in many solid tumors and we have recently shown its overexpression in 23% of colorectal tumors, preferentially in MSI compared to MSS colorectal tumors (45% versus 19%, respectively) associated with a worse prognosis.15,16 We also demonstrated that a high number of CD94+ intraepithelial TILs in close contact with tumor cells were independently associated with worse overall survival of colorectal cancer patients.16 In support of our study, two recent studies demonstrated that NKG2A blockade could enhance T cell- but also NK cell-mediated anti-tumor activity in preclinical models17,18 and in an in vitro model.19
Overall, targeting NKG2A in colorectal cancer could be a therapeutic opportunity for treatment optimization, due to the presence of NKG2A on CD8+ TILs and the overexpression of HLA-E by colorectal tumors. The current study aimed to decipher the NKG2A+ CD8+ TIL subpopulation in colorectal cancer, in terms of molecular and functional characteristics, in order to strengthen the relevance of its targeting for patients with metastatic colon cancer.
Materials and methods
Patients and specimens
Forty patients undergoing surgery for colorectal cancer without prior chemotherapy or radiotherapy, at the “Centre Hospitalier Universitaire de Nantes” or the “Nouvelles Cliniques Nantaises” (France), were included in this study. Pathological staging of all patients was assessed according to the 8th edition of the TNM (Tumor Node Metastasis) staging system for colorectal cancer published by the International Union against Cancer (UICC 8th edition). Histological subtyping was reviewed according to the 4th edition of World Health Organization classification of tumors of the digestive system. All tissues were processed according to the guidelines of the French Ethics Committee for research on human tissues. The institutional board of the University Hospital of Nantes approved this study. Our tissue biocollection was registered with the French Ministry for Higher Education and Research (DC-2014-2206) with approval from the ethic committee (CPP Ouest IV – Nantes). Our study was conducted in accordance with the Declaration of Helsinki. Each patient included in this study signed an informed consent form. Table 1 lists the clinicopathological features of the 35 colorectal cancer patients whose tumors were analyzed ex vivo by flow cytometry.
Table 1.
Clinicopathological features of colorectal cancer patients included in ex vivo multiparametric analysis
| All tumors (n = 35) | MSI tumors (n = 8, 22.9%) | MSS tumors (n = 27, 77.1%) | |
|---|---|---|---|
| Age | |||
| (mean (range)) | 68.8 (36–88) | 74.9 (42–88) | 67.0 (36–82) |
| Gender | |||
| Women (N (%)) | 15 (42.9) | 4 (50.0) | 11 (40.7) |
| Men (N (%)) | 20 (57.1) | 4 (50.0) | 16 (59.3) |
| Tumor location | |||
| Right (N (%)) | 15 (42.9) | 7 (85.7) | 8 (29.6) |
| Left (N (%)) | 15 (42.9) | 0 (0.0) | 15 (55.6) |
| Rectum (N (%)) | 5 (14.2) | 1 (14.3) | 4 (14.8) |
| Histological subtypes | |||
| Adenocarcinoma not otherwise specified (N (%)) | 29 (82.9) | 6 (75.0) | 23 (85.2) |
| Mucinous adenocarcinoma (N (%)) | 6 (17.1) | 2 (25.0) | 4 (14.8) |
| CMS classification | |||
| CMS1 (N (%)) | 5 (14.3) | 4 (50.0) | 1 (3.7) |
| CMS2 (N (%)) | 11 (31.3) | 0 (0.0) | 11 (40.7) |
| CMS3 (N (%)) | 5 (14.3) | 1 (12.5) | 4 (14.8) |
| CMS4 (N (%)) | 3 (8.6) | 0 (0.0) | 3 (11.1) |
| Unclassified (N (%)) | 3 (8.6) | 1 (12.5) | 2 (7.4) |
| NA (N (%)) | 8 (22.9) | 2 (25.0) | 6 (22.3) |
| UICC stage | |||
| I (N (%)) | 4 (11.4) | 0 (0.0) | 4 (14.8) |
| II (N (%)) | 17 (48.6) | 7 (85.7) | 10 (37.0) |
| III (N (%)) | 13 (37.1) | 0 (0.0) | 13 (48.1) |
| IV (N (%)) | 1 (2.9) | 1 (14.3) | 0 (0.0) |
Microsatellite instability and DNA mismatch repair status
Microsatellite instability or mismatch repair status was performed for diagnosis purposes by PCR or immunohistochemistry studies, respectively, as previously described.16 Microsatellite instability status was determined using pentaplex PCR with five markers: BAT-25, BAT-26, NR-21, NR-22, NR-24.20 Briefly, genomic DNA was extracted from 10 µm thick tissue sections of formalin-fixed, paraffin-embedded colorectal tumor tissues after manual macrodissection using the iPrepTM ChargeSwitch® Forensic kit (Invitrogen), and according to the manufacturer’s instructions. A colorectal tumor was considered as microsatellite instable if at least two of these five markers showed microsatellite instability.21 Mismatch repair status was assessed by immunohistochemistry using the following mAbs: MLH1 (Agilent Technologies, ES05, RRID:AB_2877720), MSH2 (Agilent Technologies, FE11, RRID:AB_2889974), MSH6 (Agilent Technologies, EP49, RRID:AB_2889975) and PMS2 (Agilent Technologies, EP51, RRID:AB_2889977).
Mechanical dissociation of colorectal tissues
Fresh samples (tumor and paired normal colonic mucosa) were recovered in MACS Medium tissue storage solution (Miltenyi Biotec) and dissected for flow cytometry analyses as previously described.16 Briefly, tissues were minced into small fragments at room temperature in RPMI 1640 medium (Gibco) and transferred to a GentleMACS C tube (Miltenyi Biotec) for non-enzymatic mechanical dissociation with a GentleMacs Dissociator (Miltenyi Biotec). After a filtration through a 40 µm cell strainer (Dutscher), the suspension was centrifuged and suspended in medium.
Ex vivo flow cytometry staining
1x106 cells from fresh or frozen (thawed with DNase I Solution (Stemcell)) dissociated tissues (tumor and paired normal colonic mucosa) were centrifuged and then incubated in tubes for each multiparametric panel in 100 µL of PBS with 2.5 µg of Human BD Fc Block (BD Biosciences, RRID:AB_2869554) to block the non-specific binding of fluorescent antibodies to cellular Fc-γ receptors. After 10 min at room temperature and centrifugation, cells were incubated in 150 µL of Brilliant Stain Buffer (BD Biosciences, RRID:AB_2869750) containing appropriate concentrations of specific or isotype control antibodies for 30 min at 4°C.
The first multiparametric panel was designed to determine the profile of NKG2A+ infiltrate and the anti-human mAbs used were: CD3-BUV395 (BD Biosciences, UCHT1, RRID:AB_2744387), T-cell receptor (TCR)αβ-BB700 (BD Biosciences, IP26, RRID:AB_2743389), TCRγδ-BV650 (BD Biosciences, B1, RRID:AB_2738628), Vα24JαQ TCR chain-BV711 (BD Biosciences, 6B11, RRID:AB_2738108), CD56-APC (BD Biosciences, B159, RRID:AB_398601), CD94-PE (BD Biosciences, HP-3D9, RRID:AB_396201) and NKG2A-FITC (Miltenyi Biotec, REA110, RRID:AB_2733623). The second multiparametric panel was designed to document the T-cell infiltrate and its expression of five ICs and the anti-human mAbs used were: CD3-BUV395 (BD Biosciences, UCHT1, RRID:AB_2744387), CD4-BUV496 (BD Biosciences, SK3, RRID:AB_2744422), CD8-APC (BioLegend, RPA-T8, RRID:AB_314132), PD-1-BV421 (BD Biosciences, EH12.1, RRID:AB_2739399), T-cell immunoglobulin, and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT)-PECy7 (BioLegend, A15153G, RRID:AB_2632929), T-cell immunoglobulin and mucin-domain 3 (Tim-3)-PE (BioLegend, F38-2E2, RRID:AB_2116576), lymphocyte-activation gene 3 (Lag3)-BV650 (BioLegend, 11C3C65, RRID:AB_2632951) and NKG2A-FITC (Miltenyi Biotec, REA110, RRID:AB_2733623). The third multiparametric panel was designed to identify the differentiation stage of NKG2A+ CD8+ αβ TILs and the anti-human mAbs used were: CD3-BUV496 (BD Biosciences, UCHT1, RRID:AB_2870222), CD8-BUV395 (BD Biosciences, RPA-T8, RRID:AB_2722501), TCRαβ-BV605 (BD Biosciences, IP26, RRID:AB_2742702), NKG2A-BV786 (BD Biosciences, 131411, RRID:AB_2872378), CD95-PECy7 (BD Biosciences, DX2, RRID:AB_10894384), CD45RO-BB515 (BD Biosciences, UCHL1, RRID:AB_2744408), CCR7-APC-R700 (BD Biosciences, 3D12, RRID:AB_2744304) and killer cell lectin-like receptor 1 (KLRG1)-APC (BioLegend, 14C2A07, RRID:AB_2571925). The fourth multiparametric panel was designed to investigate the exhaustion status of NKG2A+ CD8+ αβ TILs and the anti-human mAbs used were: CD3-BUV395 (BD Biosciences, UCHT1, RRID:AB_2744387), TCRαβ-BB700 (BD Biosciences, IP26, RRID:AB_2743389), CD4-BUV496 (BD Biosciences, SK3, RRID:AB_2744422), CD8-BUV805 (BD Biosciences, RPA-T8, RRID:AB_2873737), NKG2A-FITC (Miltenyi Biotec, REA110, RRID:AB_2733623), PD-1-BV421 (BD Biosciences, EH12.1, RRID:AB_2739399), Tim-3-BV786 (BD Biosciences, 7D3, RRID:AB_2741100), CD69-PECy7 (BD Biosciences, FN50, RRID:AB_1727509), CD103-BUV735 (BD Biosciences, Ber-ACT8, RRID:AB_2872913), TOX-AF647 (Cell Signaling Technology, E6G5O, RRID:AB_2904195) and TCF1-PE (Cell Signaling Technology, C63D9, RRID:AB_2798483). Isotypic controls were also performed for each multiparametric panel with antibodies from the same manufacturer. For each panel, Fixable Viability Stain 780 (BD Biosciences, RRID:AB_2869673) was used to exclude dead cells and staining was performed at the same time as the antibodies. After three washes in PBS 0.1% BSA, the stained cells were acquired in the viable cell gate on a BD LSRFortessa X-20 flow cytometer using BD FACSDiva software 8.0 (BD Biosciences) and analyzed with this software for the first three panels. For the last one, Foxp3/Transcription Factor Staining Buffer Set (eBioscience) was used according to the manufacturer’s recommendation for the staining with both mAbs TOX-AF647 and TCF1-PE. Stained cells were then acquired on a Symphony A5.2 flow cytometer using the 9.0.1 version of BD FACSDiva software. FlowJo software (v10.7) was also used to analyze results and generate tSNE plots on pre-gated viable NKG2A+ CD8+ CD3+ TILs (n = 9).
Colorectal cancer cell line
The HCT116 cell line (RRID: CVCL_0291) was derived from an MSI colonic adenocarcinoma and cultured in RPMI 1640 medium with 10% FBS (Eurobio), 2 mM GlutaMAX-I (Gibco), 100 U/mL penicillin (Gibco) and 0.1 mg/mL streptomycin (Gibco).
Generation of TILs
TILs from six colorectal cancer patients, three MSS and three MSI, were obtained after culture of tumor fragments (approximately 1mm3) during 3 weeks in RPMI 1640 medium supplemented with 8% human serum (intern production from healthy donors (“Etablissement Français du Sang”)), 2 mM GlutaMAX-I (Gibco), 100 U/mL penicillin, 0.1 mg/mL streptomycin, 1 µg/mL Amphotericin B (Sigma-Aldrich), 0.1 mg/mL gentamycin (Sigma-Aldrich) and 150 U/mL of human rIL-2 (Proleukin, Novartis), as previously reported.16 Fragments were removed at the end of the 3 weeks by filtration through a 40 µm cell stainer.
Sorting of NKG2A− and NKG2A+ CD8+ TIL subpopulations
First, CD8+ TILs were collected by magnetic cell sorting using the REAlease CD8 MicroBead kit by positive selection (Miltenyi Biotec) according to the manufacturer’s instructions. After cell sorting, CD8+ TILs were expanded by stimulation with Phytohemagglutinin-L (Sigma-Aldrich) and 150 U/mL human rIL-2 in the presence of irradiated allogeneic feeder cells (PBMC and B-EBV cells), as previously described.22 Second, for extensive phenotypic and functional characterizations, CD94/NKG2A negative and positive CD8+ TILs were sorted using anti-PE MicroBeads UltraPure Kit (Miltenyi Biotec). Briefly, CD8+ TILs were stained with PE-conjugated anti-CD94 mAb (BD Biosciences, HP-3D9, RRID:AB_396201) for 30 minutes at 4°C in PBS 0.1% BSA (Sigma-Aldrich) and then washed twice in PBS 2 mM EDTA 0.5% BSA. After centrifugation at 300 g for 10 min, cells were resuspended in anti-PE microBeads Ultrapure, incubated for 15 min at 4°C and washed again. Finally, magnetic cell sorting was performed on MACS LS columns with a MACS separator (Miltenyi Biotec). The purity of each subpopulation was checked before and after cell sorting and routinely by flow cytometry. Four days later, sorted cells were amplified in culture as described above.
T-cell clone generation
The T-cell clone (C178-A) from the NKG2A+ CD8+ TILs of patient C178 was obtained by cloning the polyclonal population in U-bottomed 96-well plates (Falcon) by limited dilution and amplification was induced as described above. This T-cell clone was selected based on its high expression of NKG2A and its ability to recognize the HCT116 colorectal cancer cell line.
Test for mycoplasma
All cells used (colorectal cancer cell line, TILs …) were checked weekly for mycoplasma contamination by performing the HEK-Blue Detection kit (InvivoGen) following the manufacturer’s instructions. Briefly, cell supernatants were deposited on mycoplasma sensor cells, inoculated in HEK-Blue Detection medium. These cells will eventually detect mycoplasma through their Toll-likereceptor 2, which induces a signaling cascade leading to the activation of the transcription factor NK-κB. The NF-κB-inductible secreted embryonic alkaline phosphatase was then detected by this simple colorimetric assay, with a blue/purple color.
T-cell activation by anti-CD3 mAb
NKG2A− and NKG2A+ subpopulations were stimulated in U-bottomed 96-well plate (NUNC) with 400 ng/mL of plate-bound anti-CD3 mAb (produced by OKT3, ATCC CRL-8001, RRID:CVCL_2665) for 5 hours in the presence of 10 µg/mL of Brefeldin A (Sigma-Aldrich). After fixation with PBS 4% paraformaldehyde (VWR) for 10 minutes, cells were washed in PBS 0.1% BSA 0.1% Saponin (Sigma-Aldrich), centrifuged then stained with anti-human TNF-α-FITC (BioLegend, Mab11, RRID:AB_315258) and IFN-γ-APC (BioLegend, B27, RRID:AB_315443) or IL-2-APC (BioLegend, MQ1-17H12, RRID:AB_315098) mAbs for 30 minutes in PBS 0.1% BSA 0.1% Saponin. For the CD107a degranulation assay, cells were stimulated for 3 hours without Brefeldin A in the presence of anti-human CD107a-AF647 mAb (BioLegend, H4A3, RRID:AB_1227506) added at the beginning of stimulation. Stained cells were acquired in the singlet-cell gate on a BD FACSCanto II flow cytometer using the same software as above.
T-cell proliferation assay
150,000 NKG2A− and NKG2A+ cells were activated for 5 days in U-bottomed 96-well plate (NUNC) with plate-bound anti-CD3 mAb (OKT3) at different concentrations ranging from 12.5 ng/mL to 200 ng/mL in the presence of 1 µM CellTrace CFSE (Invitrogen). CFSE staining of cells was performed according to the manufacturer’s recommendations. Cells were acquired in the singlet-cell gate on a BD FACSCanto II flow cytometer using the same software as above.
CD8+ TILs reactivity assay against colorectal cancer cell line
The HCT116 colorectal cancer cell line was pre-treated or not with human recombinant IFN-γ (Eurobio) for 48 hours at 500 U/mL. A 5-h coculture was performed in the presence of 10 µg/mL Brefeldin A at 37°C between NKG2A− or NKG2A+ CD8+ TIL C178 subpopulations and HCT116 cell line (Effector:Target ratio 1:2). Cells were then stained with anti-human CD8-PE mAb (BioLegend, RPA-T8, RRID:AB_314126) for 30 minutes in PBS 0.1% BSA. After fixation with PBS 4% paraformaldehyde for 10 minutes, cells were washed in PBS 0.1% BSA 0.1% Saponin, centrifuged then stained with anti-human IFN-γ-APC mAb for 30 minutes in PBS 0.1% BSA 0.1% Saponin. The stained cells were acquired in the singlet-cell gate on a BD FACSCanto II flow cytometer using the same software as above.
Agonist antibody assay
Anti-human CD94 mAb (R&D systems, 131412, RRID:AB_2234380) was plated in U-bottomed 96-well plate (NUNC) at 1 µg/mL after dilution in PBS for 1 h at 37°C. After two washing steps with PBS, 20,000 NGK2A− and NKG2A+ cells were added per well and simultaneously stimulated with soluble anti-CD3 mAb (OKT3, 500 ng/mL) overnight at 37°C. Supernatants were collected and IFN-γ secretion was determined by IFN-γ-specific ELISA (Invitrogen), according to the manufacturer’s recommendations.
Blocking antibody assay
To assess the impact of NKG2A/HLA-E interaction, C178-A T-cell clone was preincubated with 10 µg/mL of either purified anti-NKG2A (Beckman Coulter, Z199, RRID:AB_131495) or purified anti-CD94 (Beckman Coulter, HP3-B1, RRID:AB_131651) and HCT116 cell line with 10 µg/mL of purified anti-HLA-E (BioLegend, 3D12, RRID:AB_1659247) mAbs during 15 min at room temperature. The same conditions were performed for the isotypic controls (purified mouse IgG2b (Invitrogen), purified mouse IgG2a (R&D systems) and purified mouse IgG1κ (BioLegend)). After a 3-h coculture at 37°C of C178-A T-cell clone with the HCT116 cell line (ratio 1:2) in the presence of anti-human mAb CD107a-AF647, degranulation of the T-cell clone was assessed by flow cytometry. TNF-α production was detected by ELISA (Invitrogen), according to the manufacturer’s recommendations, after a 12-h coculture at 37°C.
RNA extraction
For transcriptomic comparison of NKG2A− et NKG2A+ cells, biologic duplicates of total RNA from resting and OKT3-activated (coated at 500 ng/mL, for 12 hours at 37°C) NKG2A− and NKG2A+ CD8 TILs (n = 4) were extracted using the RNeasy Mini kit (Qiagen) following manufacturer’s instructions. RNA quantification was determined with a NanodropND-1000 spectrophotometer (Thermo Fisher Scientific) and quality was assessed using the Agilent RNA 6000 Nano kit (Agilent Technologies) with a 2100 Bioanalyzer instrument (Agilent Technologies), according to the manufacturer’s instructions.
To determine the CMS classification of colorectal tumors (n = 27), tissue homogenization was performed with Fastprep-24 (MP Biomedicals), and RNA extraction, evaluation of total tumor RNA quantities and quality were performed as described above.
3’ RNA sequencing
The QIAseq UPX 3’ transcriptome kit (Qiagen) was performed with 2.5 ng of each RNA sample according to the manufacturer’s manual, with two independent libraries for each study. Briefly, a reverse transcription reaction was performed in a 96-well plate with anchored oligo-dT primers tagging each sample with a unique identifier (CellID) and each molecule with a unique molecular index (UMI). Following reverse transcription also incorporating a template switching, all individually tagged cDNAs were combined into two tubes for the two library constructions (NKG2A− and NKG2A+ cells and colorectal tumors). The amplified DNA was fragmented, end-repaired, and A-tailed within a single, controlled multienzyme reaction. Then, the prepared DNA fragments were ligated at their 5′ ends with a sequencing platform-specific adapter. A universal library amplification step introduced a unique sample index and ensured that DNA fragments containing the Cell ID and UMI were sufficiently amplified for NGS. Quality controls for the final libraries were determined using the Agilent DNA 7500 kit (Agilent Technologies) with the 2100 Bioanalyzer and using the Qubit dsDNA HS assay kit (Invitrogen) with the Qubit Fluorometer (Invitrogen), and quantification of the final libraries was performed using the KAPA Library Quantification kit (Roche). Libraries (1.2pM) were sequenced with the NextSeq 500 High Output V2 kit (150 cycles) (Illumina) using paired-end sequencing (R1:100 cycles, R2:27 cycles, IR:6cycles).
Transcriptomic analysis
Primary analysis was performed on each library on GeneGlobe (Qiagen, www.qiagen.com/GeneGlobe) and consisted of demultiplexing cells, aligning reads to the reference genome (GRCh38 (Human)) and counting UMIs. Reads less than 25 bp without a CellID or matching CellID, not mapped to the genome, or not annotated were dropped. The primary analysis led to two matrices containing the counts of genes in samples from both libraries, reflecting the abundance of mRNA in these samples. For differential expression between resting and stimulated NKG2A− and NKG2A+ cells, secondary analyses were then run with DEApp,23 an interactive web interface using three different methods: edgeR, limma-voom, and DESeq2. Volcano plots were generated by the VolcaNoseR web application.24 We decided to set a significance threshold of -log10(p-value) > 3 and a fold change threshold of log2(Fold Change) > 2. Genes with fewer than 10 counts in each sample were discarded as well as genes that did not have a homogeneous distribution of counts within groups. For CMS classification determination, the R package CMS classifier has been used.
Statistics
Statistical analyses were performed using GraphPad Prism 8 software or RStudio software (version 1.3.1093). All statistical details can be found in the figure legends. In all cases, a p-value of less than 0.05 was considered statistically significant (*), p < 0.01 (**), p < 0.001 (***) and p < 0.0001 (****) as highly significant. For non-significant results, (ns) was indicated.
Results
Colorectal tumor infiltrating CD8+ T cells overexpress the inhibitory NKG2A receptor irrespective of microsatellite status and CMS classification
Ex vivo phenotype analysis of fresh tumor tissues using multiparametric flow cytometry showed that the major cell-subset expressing the inhibitory NKG2A chain in colorectal tumors was the αβ TIL subset (Figure 1a). With a gating strategy on viable CD94+NKG2A+ lymphoid cells (Supplemental Figure 1a), we showed that 75.7% were CD3+TCRαβ+ cells, 21.4% were NK cells (CD3−CD56+) and only 2.9% were γδ TILs (medians of n = 20 colorectal tumor tissues). We then focused on CD8+ TILs, because CD94/NKG2A receptor is not expressed by CD4+ TILs16,17 (Supplemental Figure 1b). We observed a significantly higher frequency of CD8+ T cells expressing NKG2A in colorectal tumors compared with normal paired mucosa (means of 13.73% ± 7.68 vs. 10.08% ± 5.91, n = 29, p < 0.05) (Figure 1(b,c)), ranging from 1.1 to 40.9%. Of the 29 patients tested, only one had a particularly high frequency of NKG2A+ CD8+ cells within the normal mucosa (52.5%) compared with the tumor (18.9%), although it did not have any particular clinical features (Figure 1c).
Figure 1.
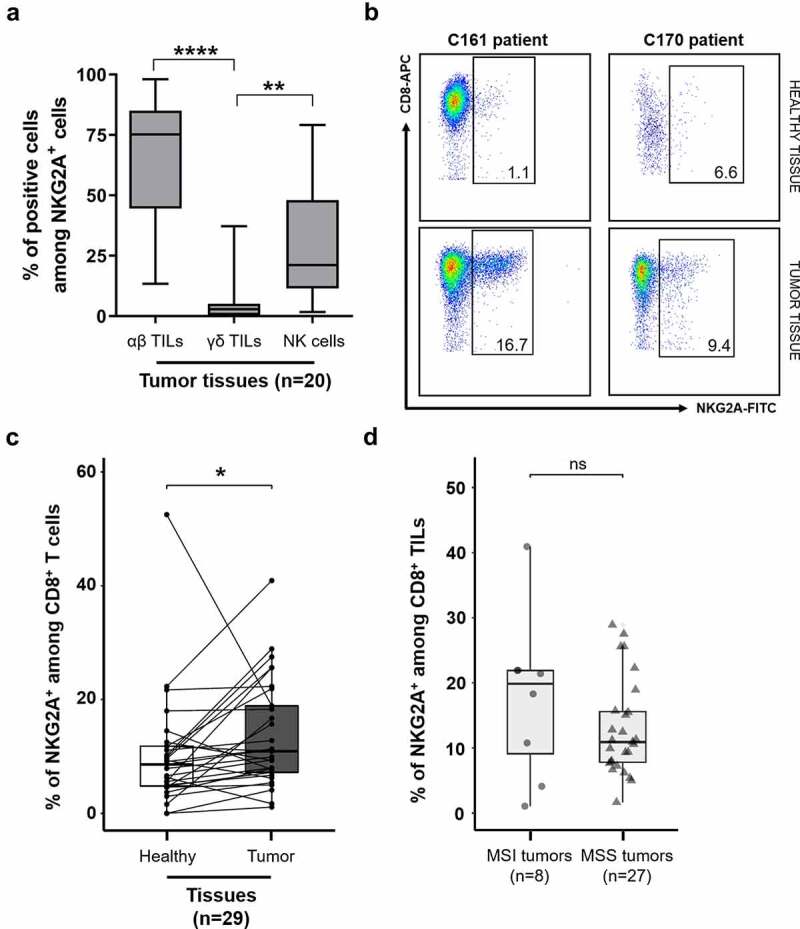
Ex vivo expression of NKG2A is highest on CD8 αβ T cells infiltrating colorectal tumors, regardless of MSS/MSI status. (a) Distribution of viable NKG2A+ lymphoid cells among αβ T cells (CD3+TCRαβ+ cells), γδ T cells (CD3+TCRγδ+ cells), and NK cells (CD3−CD56+) in 20 colorectal tumors; Friedman’s test, followed by Dunn’s multiple comparisons test. (b) Representative density plots of NKG2A expression on CD3+CD8+ T cells in healthy tissues (1.1 and 6.6%, top) and tumor tissues (16.7 and 9.4%, bottom), respectively from C161 and C170 patients. (c) Paired frequencies of NKG2A+ cells among CD8 T cells in paired tumor and normal tissues (n = 29); Wilcoxon paired t test. (d) Frequencies of NKG2A+ cells among CD8+ TILs according to MSS/MSI status (n = 35); Mann–Whitney test.
To identify a potential group of colorectal tumors containing a high amount of CD8+ TILs overexpressing NKG2A that may be more eligible for immunotherapies targeting this inhibitory receptor, we analyzed the frequencies of these cells according to MSS/MSI status or CMS classification. No correlation was found between the frequency of NKG2A+ CD8+ TILs and the MSS/MSI status of tumors, with heterogeneous frequencies among each subgroup (17.55% ± 9.16 for MSI tumors (n = 8) and 12.46% ± 5.84 for MSS tumors (n = 27)) (Figure 1d). CMS classification was determined for 27 tumors in the cohort with the R CMS classifier package12 and the proportions of each subgroup were similar to those described in the literature with 18.5% CMS1, 40.8% CMS2, 18.5% CMS3, 11.1% CMS4, and 11.1% unclassified tumors. Again, due to the substantial heterogeneity in the percentages of NKG2A+ CD8+ TILs within the CMS subgroups, no relationship has been revealed between this classification and their frequency (Supplemental Figure 2).
Similarly, we did not find any evidence of a relationship between the other clinicopathological features of the 35 patients (pTNM stage, age, sex, or tumor location), and the frequency of NKG2A+ CD8+ TILs (data not shown).
NKG2A+ CD8+ TILs are tissue-resident cells harboring terminally exhausted features
We first identified the cell differentiation stage of NKG2A+ CD8+ TILs by multiparametric flow cytometry analysis performed on freshly resected samples from nine colorectal tumors. Based on their high expression of CD45RO and CD95 and lack of CCR7 expression, we showed that NKG2A+ CD8+ TILs, as well as NKG2A− CD8+ TILs, possessed a T effector memory (TEM) phenotype with a frequency of 86% and 88.6%, respectively (Figure 2a and Supplemental Figure 3a).
Figure 2.
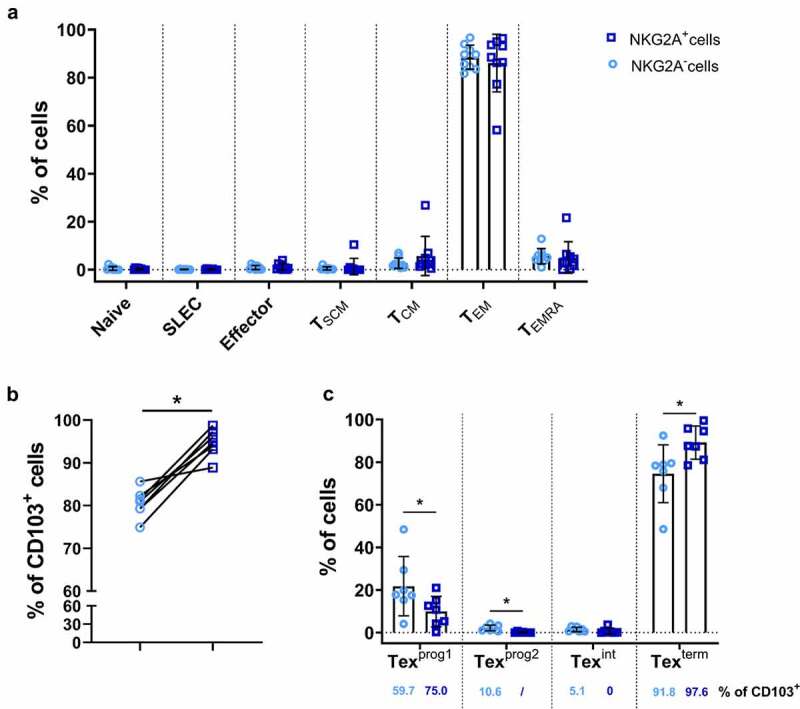
NKG2A+CD8+ TILs are tissue-resident cells harboring terminally exhausted features. (a) Proportion of NKG2A+ (dark blue squares) and NKG2A− (light blue circles) CD8+ TILs from 9 colorectal tumors for each T-cell differentiation stage, defined as follows: Naive (CCR7+CD45RO−CD95−), Short-lived effector cell (SLEC) (CCR7−KLRG1+CD45RO+CD95−), Effector (CCR7−KLRG1−CD45RO+CD95−), TSCM (CCR7+CD45RO−CD95+), TCM (CCR7+CD45RO+CD95+), TEM (CCR7−CD45RO+CD95+) and TEMRA (CCR7−CD45RO−CD95+). (b) Frequencies of CD103+ cells among NKG2A+ (dark blue squares) and NKG2A− (light blue circles) CD8+ TILs from 7 colorectal tumors; Wilcoxon paired t test. (c) Proportion of NKG2A+ (dark blue squares) and NKG2A− (light blue circles) CD8+ TILs from 7 colorectal tumors for each T-cell exhaustion stage, defined as follows: Texprog1 (TCF1+CD69+), Texprog2 (TCF1+CD69−), Texint (TCF1−CD69−) and Texterm (TCF1−CD69+); Wilcoxon paired t test. The percentage of CD103+ cells in the different subgroups is indicated below each histogram.
Based on CD103 expression by colorectal cancer-derived CD8+ TILs, we also showed that almost all NKG2A+ CD8+ TILs were tissue-resident, with a higher frequency than that observed with NKG2A− CD8+ TILs (95% versus 80%, Figure 2b). Finally, we investigated their exhaustion status based on the four exhausted CD8+ T-cell subsets recently described.25 We showed by multiparametric flow cytometry analysis performed on frozen resected samples from 7 colorectal tumors that NKG2A+ CD8+ TILs were in a large majority (average 90%) terminally exhausted (named Texterm) and that a minority corresponds to progenitor 1 exhausted (Texprog1) (Figure 2c and Supplemental Figure 3b). Both stages express TOX and CD69 but only Texprog1 are TCF1+. Furthermore, analysis of CD103 expression by the different stages of exhaustion confirmed that Texprog1 and Texterm are predominantly tissue-resident cells among NKG2A− CD8+ TILs (medians of 59.7% and 91.8%, respectively) and even more so among NKG2A+ CD8+ TILs (medians of 75.0% and 97.6%, respectively) (Figure 2c).
NKG2A+ CD8+ TILs are distributed in two subsets with differential expression levels of ICs
Because of their exhausted phenotype, we analyzed the expression of four other ICs, namely, PD-1, Tim-3, TIGIT, and Lag3 on NKG2A+ CD8+ TILs and compared it with that of NKG2A− CD8+ TILs (representative examples in Figure 3a). While the frequencies of cells expressing PD-1 (66.2% of NKG2A− cells vs. 69.3% of NKG2A+ cells), TIGIT (81.9% vs. 84.5%) and Lag3 (1.2% vs. 1.5%) (medians, n = 35) were similar in both subpopulations, the percentage of cells expressing Tim-3 was significantly higher among the NKG2A+ T-cell subset (26.5% vs. 49.7%, medians, n = 35, p = 0.00002) (Figure 3b). Moreover, interestingly, the expression levels of these ICs were significantly higher on NKG2A+ cells than of NKG2A− cells, especially those of PD-1 and TIGIT (RFI of 62.7 vs. 98.0 and RFI of 132.0 vs. 166.9, respectively) (Figure 3a). As shown in Figure 3c, NKG2A+ cells co-expressed a higher number of other ICs compared to NKG2A− cells (44% vs. 31% co-expressing more than 2 other ICs; 19% vs. 4% co-expressing the 4 ICs tested). To complete this analysis, we performed an unsupervised analysis of our multiparametric data on pre-grouped viable NKG2A+ CD8+ CD3+ TILs after concatenating tumor tissue samples tested (n = 9) using the FlowJo software. tSNE density plots and associated histograms, shown in Figure 3d, clearly identified the existence of two subgroups among this subpopulation differing in the expression level of some ICs. A first cluster (encircled in orange) representing 62.3% of NKG2A+ CD8+ TILs express moderate levels of NKG2A but high levels Tim-3, TIGIT and above all PD-1. The second cluster (encircled in blue), representing 37.6% of NKG2A+ CD8+ TILs, featured a very high expression level of NKG2A associated with high expression level of TIGIT but an absence of Tim-3 and PD-1 expression. Lag3 expression levels, low to moderate, were roughly the same in the two subgroups.
Figure 3.
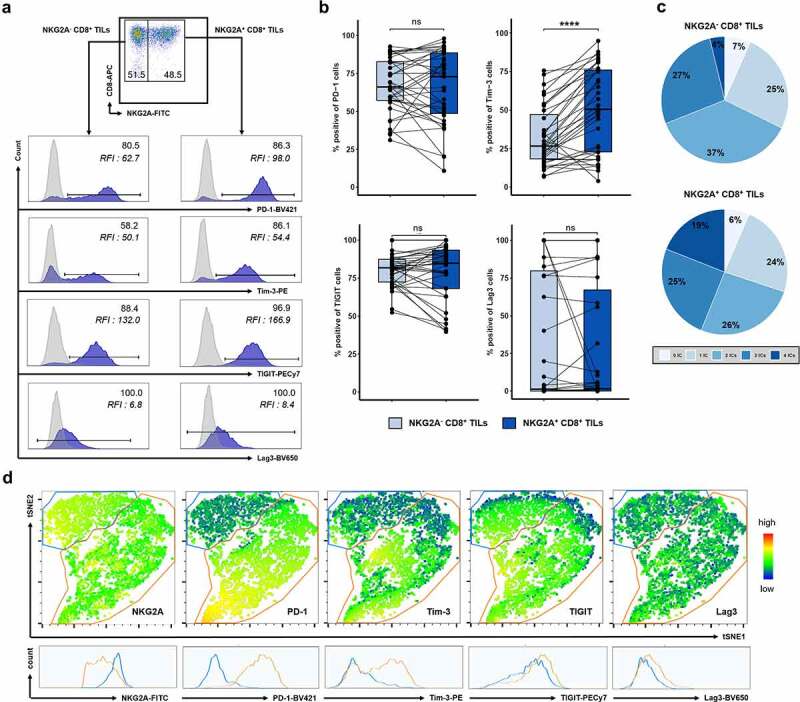
NKG2A+CD8+ TILs co-express other ICs at different levels highlighting two distinct subsets. (a) Representative histograms showing the expression of PD-1, Tim-3, TIGIT, or Lag3 on NKG2A− (left panels) and on NKG2A+ (right panels) CD8+ TILs from C169 patient. Results were expressed as positive cells and Relative Fluorescence Intensity (RFI) defined as the ratio of specific fluorescence (median fluorescence of cells incubated with specific Abs) over non-specific fluorescence (median fluorescence of cells incubated with isotypic controls). (b) Paired frequencies (n = 35) of NKG2A− (light blue) and NKG2A+ (dark blue) CD8+ TILs expressing PD-1, Tim-3, TIGIT, or Lag3; Wilcoxon paired t test. (c) Pie chart analysis of the co-expression of the 4 ICs on NKG2A− and NKG2A+ CD8+ TILs. (d) Density tSNE plots from FlowJo software on pre-gated viable CD3+ CD8+ NKG2A+ TILs after concatenation of all analyzed tissue samples (n = 9). The two clusters identified on the basis of the differential expression of NKG2A are encircled in blue (higher expression) and orange (lower expression). The expression level of the 5 ICs for each cluster is represented below by histograms.
NKG2A+ CD8+ TILs have greater functional capacity than NKG2A− CD8+ TILs, despite a reduced proliferative potential
To further characterize NKG2A+ CD8+ TILs, we generated in vitro polyclonal populations of TILs from tumor fragments of six colorectal cancer patients, 3 MSS (C10, C81, and C178) and 3 MSI (C65, C91, and C169). After sorting CD8+ T cells, we sorted NKG2A− and NKG2A+ cells for all six populations with at least 95% of purity, routinely verified during their amplifications by flow cytometry.
We performed transcriptomic analysis of the NKG2A− and NKG2A+ subpopulations of four CD8+ TIL pairs (C65, C81, C91, and C178). We first investigated the differential gene expression between these two subpopulations at rest (Figure 4a, EdgeR; Supplemental Figure 4a, limma-voom or Supplemental Figure 4b, DESeq2). The results showed that the NKG2A gene, KLRC1, was the only gene significantly upregulated in unstimulated NKG2A+ cells. Regarding the 44 genes identified as downregulated in NKG2A+ cells by at least one algorithm, 34% (15/44) were involved in cell proliferation such as CDC45, LIG1, or CLIC3 (Figure 4b), suggesting that NKG2A+ cells may have a reduced proliferative potential compared with NKG2A− cells. Other genes downregulated in NKG2A+ cells were related to diverse functions such as cell adhesion, metabolism, or intracellular trafficking.
Figure 4.
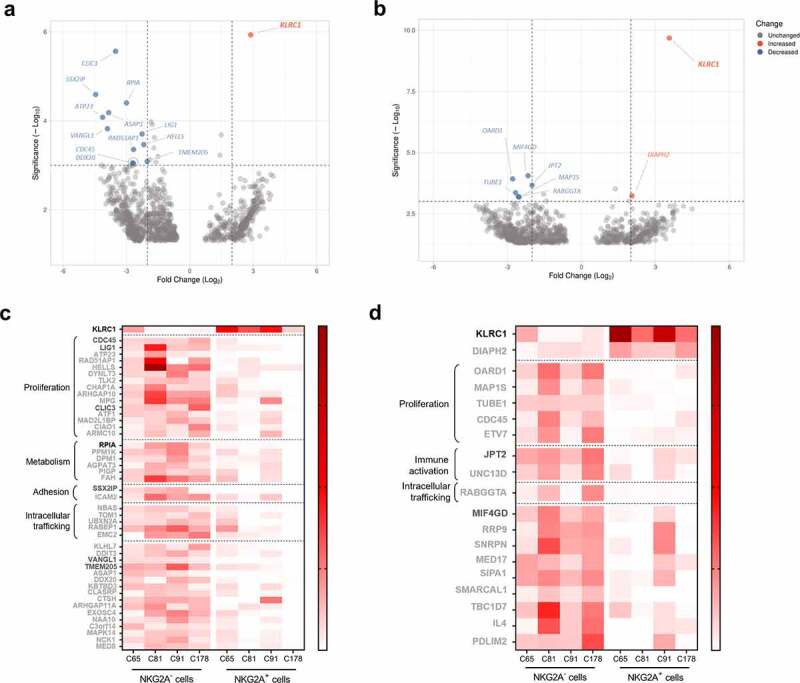
Comparative transcriptomic analysis of NKG2A+ and NKG2A− CD8+ TILs reveals downregulation of many genes involved in cell proliferation in NKG2A+ cells. Volcano plots of differentially expressed mRNAs between NKG2A− and NKG2A+ cells unstimulated (a) and activated (c) (n = 4 pairs), using the EdgeR algorithm. Heatmaps showing the expression scale of up- and down-regulated genes identified by one (light gray), two (dark gray) or three (black) algorithms (DEseq2, EdgeR and/or limma-voom) in NKG2A+ cells unstimulated (b) and activated (d) (n = 4 pairs). Each column represents the average of two technical replicates.
We next studied the differential gene expression between NKG2A− and NKG2A+ subpopulations after TCR activation. Confirming results obtained at rest, KLRC1 was upregulated in NKG2A+ cells (Figure 4c, Supplemental Figure 4(c,d)). Interestingly, NKG2A mRNA level was significantly higher upon TCR stimulation of NKG2A+ cells compared with unstimulated NKG2A+ cells, as illustrated by the increased significance (Figure 4(a,c)). The analysis performed with the EdgeR algorithm also revealed an upregulation of the DIAPH2 gene in NKG2A+ cells, this gene being involved in the regulation of endosome dynamics (Figure 4c). Among the 17 genes downregulated in NKG2A+ cells compared to the negative ones, we again identified some genes related to cell proliferation such as CDC45, MAP1S, or TUBE1 (Figure 4d; Supplemental Figure 4c).
To validate the results of the transcriptomic analysis, we assessed the proliferation of NKG2A− and NKG2A+ CD8+ TILs from four patients using CFSE assay after a 5-day stimulation with various concentrations of anti-CD3 mAb. A representative example of the flow cytometry histograms is shown in Figure 5a (TILs from C10 patient), and the results for the four pairs tested represented in Figure 5b. The proliferation of NKG2A+ cells was lower than that of matched negative cells regardless of OKT3 concentration. At the highest concentration of OKT3 (200 ng/mL), NKG2A+ CD8+ TILs from MSS tumors (C10, C81, and C178) have a reduced proliferative capacity of 38% ± 7 compared with NKG2A− cells and that of NKG2A+ CD8+ TILs from the MSI tumor (C65) by 6.2%.
Figure 5.
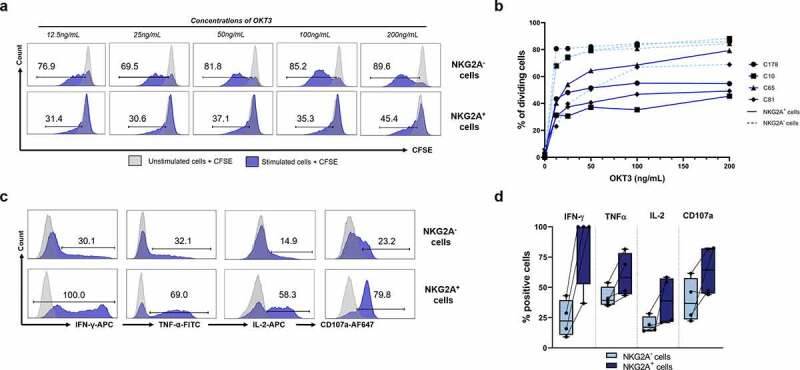
NKG2A+CD8+ TILs have better functional capacity than NKG2A−CD8+ TILs despite reduced proliferative potential. (a) Representative histograms of proliferation (CFSE assay) of NKG2A− (up) and NKG2A+ (down) CD8+ TILs from C10 patient after a 5-day culture in the presence of OKT3 mAb (from 0 to 200 ng/mL). (b) Proportion of proliferative NKG2A− (dashed light blue lines) and NKG2A+ (solid blue lines) CD8+ TILs in 4 patients, after a 5-day culture in the presence of OKT3 mAb (from 0 to 200 ng/mL). (c) Representative histograms of the percentage of NKG2A− (up) or NKG2A+ (down) CD8+ TILs from C81 patient producing IFN-γ, TNF-α, or IL-2 after a 5-hour stimulation (OKT3 mAb, 400 ng/mL) in the presence of brefeldin A (10 µg/mL) and expressing the surface degranulation marker CD107a after 3 hours in the presence of OKT3 mAb (400 ng/mL). (d) Percentage of NKG2A− (light blue) or NKG2A+ (dark blue) CD8+ TILs (n = 4) producing IFN-γ, TNF-α, IL-2 or CD107a in the same experimental conditions as (c).
We subsequently compared the functional capabilities of the NKG2A+ and NKG2A− subpopulations of four pairs of TILs (C65, C81, C169, and C178) in terms of major effector cytokine production (IFN-γ, TNF-α, and IL-2) and degranulation capability (CD107a detection) by flow cytometry. A representative example of the flow cytometry histograms is shown in Figure 5c (TILs from C81 patient), and the results for the four pairs tested represented in Figure 5d. After TCR activation (OKT3 mAb, 400 ng/mL), without NKG2A engagement (nor from any other IC), NKG2A+ CD8+ TILs displayed higher cytokine responses than NKG2A− CD8+ TILs with 84.2% vs. 24.2% of IFN-γ-producing cells, 60.2% vs. 41.8% of TNF-α-producing cells and 39.2% vs. 19.1% of IL-2-producing cells, respectively (means of n = 4 pairs). Similar results were observed for T-cell degranulation capacity, with an average of 63.8% of NKG2A+ degranulating cells versus 39.3% of NKG2A− cells (means of n = 4 pairs). Altogether, these results highlighted the improved baseline functional capacities of NKG2A+ CD8+ TILs compared to NKG2A− CD8+ TILs.
Targeting NKG2A restores optimal functions of NKG2A+ CD8+ TILs specific of colorectal tumor cells, especially in the presence of IFN-γ
Finally, we evaluated the functionality of the inhibitory receptor NKG2A and the impact of its engagement on the effector responses of NKG2A+ CD8+ TILs.
First, we assessed, by ELISA, the effect of an agonistic anti-CD94 mAb on IFN-γ secretion by NKG2A− and NKG2A+ subpopulations of CD8+ TILs, which were verified for purity and presented in Figure 6a (representative example of the TIL subpopulations from patient C178). IFN-γ production of activated NKG2A+ CD8+ TILs (mean of 141.2 ± 54.6 pg/mL) was reduced by 44.4 ± 14.4% after addition of an agonist anti-CD94 mAb (mean of 6 independent experiments in triplicate, p < 0.0001 compared to the isotypic control, Figure 6b). In contrast, the anti-CD94 mAb had no effect on the IFN-γ secretion of NKG2A− CD8+ TILs (mean of 111.4 ± 73.5 pg/mL) (p = 0.93 compared to the isotypic control condition, Figure 6b).
Figure 6.
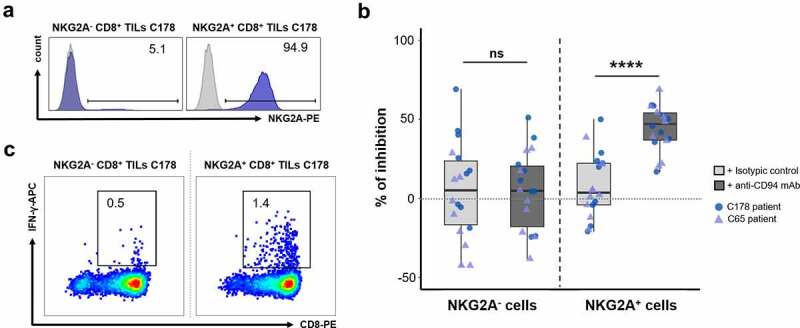
Inhibitory receptor CD94/NKG2A engagement induces impaired IFN-γ secretion in polyclonal NKG2A+CD8+ TILs. (a) Histograms showing the purity of the tested NKG2A− and NKG2A+ CD8+ TIL populations. (b) Percentages of inhibition of IFN-γ secretion by NKG2A− and NKG2A+ CD8+ TILs from C65 (purple triangles) and C178 (blue circles) patients in the presence of soluble OKT3 mAb (500 ng/mL) and coated isotypic control (light gray) or agonist anti-CD94 mAb (dark gray) (1 µg/mL). These percentages, calculated relative to the activated condition without antibody, were obtained from 3 independent experiments with 3 replicates (n = 9) for each patient; Wilcoxon paired t test. (c) Graphs showing the frequencies of reactive IFN-γ+ cells among NKG2A− and NKG2A+ CD8+ TIL populations after 5-hour coculture with the HCT116 cell line in the presence of brefeldin A (10 µg/mL).
As immunotherapy strategies consist in blocking the interaction between ICs and their ligands expressed by tumor cells, we further performed blocking experiments in a more physiological model of colorectal tumor-specific T-cell response. To this end, we first searched for the existence of NKG2A+ CD8+ TILs reactive toward colorectal tumor cell lines. We identified among polyclonal NKG2A+ CD8+ TILs from C178 patient, 1.4% of cells producing IFN-γ upon coculture with HCT116 cell line (Figure 6c). Among NKG2A− CD8+ TILs, only 0.5% of cells produced IFN-γ when cocultured with HCT116 cell line. Cloning of NKG2A+ CD8+ TILs by limiting dilution allowed us to obtain a NKG2A+ T-cell clone named C178-A, expressing the TCR Vβ14 chain (Figure 7a) and recognizing the HCT116 cell line in the shared HLA-A*02:01 context (personal data). Thus, as shown in Figure 7b, approximately 36% of cells expressed CD107a on their surface, a sign of degranulation, after coculture with the HCT116 cell line. Nevertheless, this degranulation capacity was threefold lower (10%) when the HCT116 cell line was pretreated with IFN-γ for 48 hours prior to coculture (Figure 7b). This inhibition was most likely due to the very significant increase in the expression of HLA-E molecules observed on IFN-γ-treated HCT116 cells (RFI of HLA-E labeling increasing from 11 to 45, Figure 7c), which would lead to a physiological enhancement of the NKG2A/HLA-E inhibitory pathway. Although IFN-γ treatment also increases PD-L1 expression by the HCT116 line, it will not affect the response of the clone as it does not express PD-1.
Figure 7.
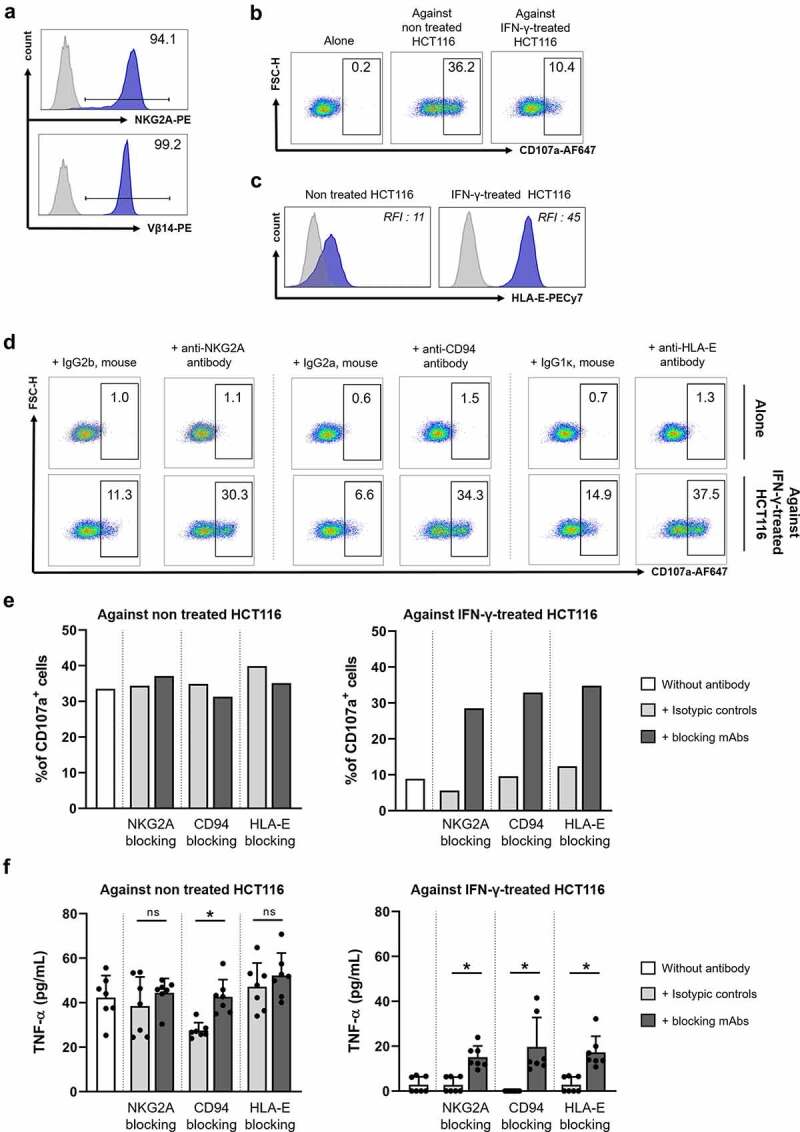
Blocking the CD94/NKG2A/HLA-E axis restores anti-tumor responses of a NKG2A+ T-cell clone. (a) Expression of NKG2A and Vβ14 by the C178-A T-cell clone. (b) Density plots showing the C178-A T-cell clone cytotoxic response (degranulation marker CD107a) when cultured alone (left) and upon coculture with HCT116 cell line without (middle) and after (right) IFN-γ treatment (500 U/mL, 48 hours). (c) HLA-E expression on HCT116 cell line before and after IFN-γ treatment (500 U/mL, 48 hours), with isotype control in light gray. (d) Density plots showing cytotoxic responses (degranulation marker CD107a) of C178-A T-cell clone alone (top) or against HCT116 cell line (bottom) pretreated with IFN-γ (500 U/mL, 48 hours) in the presence of a blocking mAb (anti-NKG2A, anti-CD94, or anti-HLA-E) or the corresponding isotypic control (10 µg/mL). (e) Frequencies of CD107a+ cells among C178-A T-cell clone or (f) concentrations of secreted TNF-α (pg/mL) by C178-A T-cell clone after stimulation with HCT116 cell line without (left) and after (right) IFN-γ treatment (500 U/mL, 48 h) in the presence of blocking mAb or isotypic control (10 µg/mL); Wilcoxon paired t test.
We then stimulated clone C178-A with the HCT116 cell line, pretreated or not with IFN-γ, in the presence or not of mAbs blocking each member of the NKG2A/HLA-E axis. As shown in Figure 7d, in the presence of anti-NKG2A, anti-CD94 or anti-HLA-E mAbs, the T-cell clone recovered its full degranulation capacity (30.3, 34.3, and 37.5% of CD107a+ cells, respectively), these mAbs having no effect on the unstimulated T-cell clone. In agreement with our hypothesis on the strengthening of this axis after IFN-γ treatment, the effect on T-cell clone’s degranulation of these three blocking mAbs was only observable during a coculture with IFN-γ-pretreated HCT116 cell line (Figure 7e). Finally, blocking the NKG2A/HLA-E axis also increased the production of TNF-α, from 2.828 to 15.12, 19.70, and 17.26 pg/mL, for anti-NKG2A, -CD94 and -HLA-E mAbs, respectively (means of 3 independent experiments, n = 7, p = 0.0156 for each mAb compared to the isotypic control conditions) (Figure 7f).
Discussion
Our study highlighted the enrichment in colorectal tumors of the NKG2A+ CD8+ TIL population and its high functional capacity despite a reduced proliferative potential. We generated from TILs of a colorectal cancer patient a NKG2A+ CD8+ T-cell clone specific to a colorectal cancer cell line which allowed us to formally demonstrate on the one hand, the inhibition of its anti-tumor reactivity in the presence of IFN-γ and on the other hand, the recovery of an effective anti-tumor response after blocking the NKG2A/HLA-E axis. In addition, we identified for the first time two subgroups of NKG2A+ CD8+ TILs based on the expression level of NKG2A and other ICs, which could help guide the choice of ICi treatments for colorectal cancer patients.
First, we showed that the frequency of CD8+ αβ T cells expressing NKG2A (average 14%) was significantly higher in colorectal tumors than in paired normal mucosa, a finding in line with our previous report on another cohort of patients.16 This enrichment in NKG2A+ CD8+ T cells in the tumor environment has also been reported in other solid tumors such as gastrointestinal cancers,19 head and neck squamous cell carcinoma (HNSCC)17 and lung cancer.26 While the work of Abd Hamid et al. showed that gastrointestinal cancer patients with advanced TNM stage have a significantly progressive increase in CD94/NKG2A+ CD8+ TIL population,19 our results did not reveal such differences, which may be due to the fact that our cohort includes a large majority of stages II and III tumors (30/35). In addition, the fact that the mean frequency of NKG2A+ CD8+ TILs did not differ according to the microsatellite status or CMS classification of our colorectal cancer patients is in favor of the potential involvement of NKG2A in the regulation of anti-tumor response in all subtypes of colorectal cancers.
When assessing the differentiation stage of intra-tumor NKG2A+ CD8+ T cells, we demonstrated that they were effector T cells, predominantly TEM cells, in accordance with previous studies associating them with an early effector or a late-stage effector memory phenotype, in HNSCC and gastrointestinal cancers, respectively.17,19 On the basis of CD103 expression, we also showed that almost all NKG2A+ CD8+ TILs were tissue-resident, with a higher frequency than that observed with NKG2A− CD8+ TILs (95% vs. 80%), in agreement with the results published in HNSCC.17 Moreover, according to the four exhausted CD8+ T-cell subsets recently described,25 we showed that more than 90% of NKG2A+ CD8+ TILs are Texterm (CD69+TCF1−), the remaining being Texprog1 (CD69+TCF1+). It is also interesting to note that these two subsets are the only ones corresponding to tissue-resident cells. Contrary to what was initially thought, recent data in the literature report anti-tumor reactivity of these exhausted CD8+ T cells and their increase in patients responding to anti-PD-1 therapy, suggesting a key role of these cells in the anti-tumor response.27,28
To complete this phenotype, we assessed the ex vivo expression of four additional ICs in the NKG2A+ and NKG2A− CD8+ subpopulations of TILs. We showed that the frequency of CD8+ TILs expressing PD-1 or TIGIT is similar in NKG2A− and NKG2A+ subpopulations, consistent with previous results in HNSCC patients.17 However, our results revealed an increased expression level of these ICs, as reported previously in lung cancer.19,26 Lag3 was poorly expressed, irrespective of NKG2A status. This low frequency of Lag3+ CD8+ TILs has also been reported in HNSCC although it is slightly higher in the NKG2A+ subset.17 Finally, we demonstrated a significantly higher frequency of Tim-3 positive cells among NKG2A+ CD8+ TILs compared to NKG2A− CD8+ TILs, explaining the overall increased frequency of NKG2A+ cells co-expressing four ICs. One of the key findings of our study was generated by unsupervised analysis of ex vivo multiparametric data of the co-expression of the four ICs by NKG2A+ CD8+ CD3+ TILs, leading to the identification of two clusters. A first majority cluster (about 62%) expresses NKG2A at a moderate level but at a high-level Tim-3, TIGIT, and above all PD-1. The second cluster (38%) is characterized by a high expression level of NKG2A and TIGIT but no expression of Tim-3 and PD-1. These results differ from those published in gastrointestinal cancers by Dong’s group, which showed exclusive co-expression of NKG2A and PD-1 by TILs.19 Thus, our results are the first to clearly discriminate the NKG2A+ CD8+ TIL population into two subpopulations based on the level of NKG2A expression: (i) one showing an exhaustive phenotype associated with moderate NKG2A expression and (ii) the other showing only TIGIT expression associated with high NKG2A expression. Interestingly, these two subgroups are in line with the scRNAseq data generated on CD8+ TILs from 12 colorectal cancer patients who identified two similar clusters, with only one expressing high levels of PDCD1 (PD-1) and HAVCR2 (Tim-3) mRNAs.29
Comparative transcriptomic analysis of four pairs of NKG2A+ and NKG2A− CD8+ TILs highlighted a reduced proliferative capacity of NKG2A+ cells as assessed by the downregulation of about 20 genes involved in cell proliferation, which we validated by a functional CFSE proliferation assay. Other downregulated genes revealed by transcriptomic analysis could negatively impact various functions of NKG2A+ cells such as cell adhesion, metabolism, or intracellular trafficking, which need further investigation. These results also showed an increase in KLRC1 gene expression after activation of NKG2A+ TILs. We confirmed in vitro that the NKG2A expression like that of PD-1 and Tim-3 (Supplemental Figure 5) was significantly upregulated following colorectal tumor infiltrating CD8+ T-cell activation. This increase in NKG2A expression could be explained by the upregulation of GATA-3 upon TCR signaling,30 which has been described as an important transcription factor regulating NKG2A gene expression.31 These findings strongly suggest that persistent activation of CD8+ T cells in colorectal tumors can promote their exhaustion by expressing numerous inhibitory receptors including NKG2A.
We then investigated the effector capabilities of NKG2A+ CD8+ TILs to assess their functional potential prior to the engagement of their NKG2A receptor. Our study highlighted that NKG2A+ CD8+ TILs displayed a higher functional capacity than NKG2A− CD8+ TILs with high production of IFN-γ and to a lesser extent TNF-α and IL-2 as well as T-cell degranulation. In accordance, Chen et al. reported a high level of IFN-γ produced by in vitro activation of NKG2A+ CD8+ T cells from the tissues of non-small cell lung carcinoma patients.26 Interestingly, melanoma-specific CD8+ TILs expressing PD-1 also possess higher functional avidity than PD-1− ones.32
Regarding the impact of NKG2A receptor engagement by its ligand, we first observed a halving of IFN-γ production by activated (OKT3) NKG2A+ CD8+ colorectal tumor-derived TILs after addition of an agonist anti-CD94 mAb. Abd Hamid et al. demonstrated that the treatment of antigen-specific NKG2A+ CTLs (derived from peripheral blood), with a combination of anti-CD94 and anti-NKG2A mAbs, enhanced their proliferation and tumor lysis upon stimulation with the HCT116 cell line pulsed with the corresponding peptide.19 To mimic a more pathophysiological context, we generated from a colorectal tumor TIL population a CD8+ T-cell clone strongly expressing NKG2A and specifically recognizing the colorectal tumor cell-line HCT116 in an autologous HLA-A*0201 context. We demonstrated that only blocking NKG2A (or its ligand) was sufficient to enhance both the cytokine and cytotoxic responses of this T-cell clone against HCT116 cell line pre-treated with IFN-γ, a treatment inducing high levels of surface HLA-E molecules. These results underline the role of IFN-γ in the tumor microenvironment in promoting the inhibition of the CD8 T-cell (and NK-cell) anti-tumor response via the NKG2A/HLA-E axis. Our findings in human colorectal cancer strengthen previous data on cancer mouse models treated with anti-NKG2A showing improved survival rates and tumor control as well as the synergy between a therapeutic cancer vaccine and NKG2A inhibition.17,18
All these results are of great interest considering the large number of human solid tumors including colon, lung, pancreas, stomach, liver, head, and neck tumor tissues, in which overexpression of the NKG2A receptor and/or its ligand HLA-E has been associated most often with a poor prognosis.15,17,18,33–39 In the same way, it has been shown in non-small-cell lung carcinoma, that CD8+ T-cell infiltrate strongly contributes to a better outcome when the tumor cells retain the expression of classical HLA class I and do not express HLA-E.39
From a therapeutic point of view, our study firstly supports the therapeutic use of monalizumab, a blocking anti-human NKG2A mAb, as new ICi in colorectal cancers.40,41 Combination therapies represent the next wave of clinical cancer treatment that enables to overcome the limitations associated with single-agent therapy. Furthermore, given that our results highlight the fact that all NKG2A+ CD8+ TILs, whatever the level of NKG2A expression, co-expressed TIGIT, the most appropriate therapeutic combination of ICis would be to combine anti-NKG2A and anti-TIGIT mAbs to treat colorectal cancer patients. This treatment could be complemented with anti-PD-1 therapy for colorectal cancer patients whose tumors have a high frequency of NKG2A+ TIGIT+ PD-1+ CD8+ TILs, corresponding to the intermediate NKG2A expression subgroup identified in this study. These therapies will have the advantage of not being dependent on current colorectal cancer classifications as our study does not demonstrate a difference in the frequency of NKG2A+ CD8+ TILs between different subclasses. This is particularly relevant for colon tumors displaying immunogenic profiles with both high immunoscore and IC expression, such as in MSI-H/dMMR tumors and probably a substantial proportion of MSS tumors.42–44 These strategies boosting effector T-cell functions could also be associated with chemotherapy, radiotherapy, anti-angiogenic therapy or MEK inhibitors.6 Finally, in a co-culture model of human colorectal tumor spheroids with immune cells, one study suggests the therapeutic potential of targeting NKG2A in combination with MHC class I Chain-related protein A and B (MICA/B).45
Finally, immunotherapies directed against NKG2A could be applied in pathologies other than cancer like some viral infections. Indeed, NKG2A has been described as a NK- and/or T-cell exhaustion checkpoint for several virus persistence such as in hepatitis C virus46 and poxvirus infections.47 Most recently, several studies have revealed functional exhaustion of antiviral CD8+ T lymphocytes in COVID-19 patients with the increased expression of NKG2A.48–50
In conclusion, our study confirms that NKG2A, as other IC expression by CD8+ TILs, associated with the immunoscore,51 should be weighed when considering patient selection for individual ICi protocol. In this regard, the work of Marisa et al. on three large independent cohorts of colorectal tumors suggests that prediction of colorectal cancer patient outcomes through evaluation of immune components in the tumor microenvironment will likely be improved by the integration of IC markers, the prognosis of patients being determined by the CTL/IC balance.52 Our results highlight the requirement to include the analysis of NKG2A (and TIGIT) inhibitory receptor expression in the selection of colorectal cancer patients for IC blocking immunotherapy that may induce an anti-tumor immune response and mediate tumor regression in situations of resistance to anti-PD-1 therapy.
Supplementary Material
Acknowledgments
The authors thank the Cytometry Facility “CytoCell” for expert technical assistance (Structure Fédérative de Recherche “Francois Bonamy”, Nantes, INSERM (UMS 016) CNRS (UMS 3556)).
Funding Statement
This work was supported by grants awarded by the Ligue contre le cancer Grand Ouest (Comités du Finistère, de Loire-Atlantique, Côtes d’Armor, Morbihan et Loir-et-Cher), DHU Oncogreffe, CHU Nantes (RC14-0416-1) and Cancéropôle Grand Ouest (Réseau Immunothérapie - Amgen RC16-0212-1). This work was performed in the context of the “LabEx IGO” program (ANR-11-LABX-0016-01);Ligue Contre le Cancer;
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Galon J, Angell HK, Bedognetti D, Marincola FM.. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39(1):11–16. doi: 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 2.de Miguel M, Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell. 2020;38(3):326–333. doi: 10.1016/j.ccell.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et al. Genomic correlates of immune-cell infiltrates in colorectal Carcinoma. Cell Rep. 2016;15(4):857–865. doi: 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. New Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marmorino F, Boccaccino A, Germani MM, Falcone A, Cremolini C. Immune checkpoint inhibitors in pMMR metastatic colorectal cancer: a tough challenge. Cancers. 2020;12(8):2317. doi: 10.3390/cancers12082317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz H-J, Morse MA, Desai J, Hill A, Axelson M, Moss RA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–1191. doi: 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Overman MJ, Lonardi S, Wong KYM, Lenz H-J, Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill A, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair–deficient/microsatellite instability–high metastatic colorectal cancer. JCO. 2018;36(8):773–779. doi: 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 9.Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. The Lancet. 2019;394(10207):1467–1480. doi: 10.1016/S0140-6736(19)32319-0. [DOI] [PubMed] [Google Scholar]
- 10.Galon JT, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, and Wind P. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 11.Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20(11):662–680. doi: 10.1038/s41568-020-0285-7. [DOI] [PubMed] [Google Scholar]
- 12.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carretero M, Cantoni C, Bellón T, Bottino C, Biassoni R, Rodríguez A, Pérez-Villar JJ, Moretta L, Moretta A, López-Botet M. The CD94 and NKG2-A C-type lectins covalently assemble to form a natural killer cell inhibitory receptor for HLA class I molecules. Eur J Immunol. 1997;27(2):563–567. doi: 10.1002/eji.1830270230. [DOI] [PubMed] [Google Scholar]
- 14.Braud VM, Allan DSJ, O’Callaghan CA, Söderström K, D’Andrea A, Ogg GS, Lazetic S, Young NT, Bell JI, Phillips JH, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391(6669):795–799. doi: 10.1038/35869. [DOI] [PubMed] [Google Scholar]
- 15.Bossard C, Bézieau S, Matysiak-Budnik T, Volteau C, Laboisse CL, Jotereau F, Mosnier J-F. HLA-E/β2 microglobulin overexpression in colorectal cancer is associated with recruitment of inhibitory immune cells and tumor progression. Int J Cancer. 2012;131(4):855–863. doi: 10.1002/ijc.26453. [DOI] [PubMed] [Google Scholar]
- 16.Eugène J, Jouand N, Ducoin K, Dansette D, Oger R, Deleine C, Leveque E, Meurette G, Podevin J, Matysiak T, et al. The inhibitory receptor CD94/NKG2A on CD8+ tumor-infiltrating lymphocytes in colorectal cancer: a promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Mod Pathol. 2020;33(3):468–482. doi: 10.1038/s41379-019-0322-9. [DOI] [PubMed] [Google Scholar]
- 17.van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marijt KA, Santegoets SJ, van Ham VJ, Ehsan I, Charoentong P, André P, et al. NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell. 2018;175(7):1744–1755.e15. doi: 10.1016/j.cell.2018.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, Bléry M, Bonnafous C, Gauthier L, Morel A, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175(7):1731–1743.e13. doi: 10.1016/j.cell.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abd Hamid M, Wang R, Yao X, Fan P, Li X, Chang X, Feng Y, Jones S, Maldonado-Perez D, Waugh C, et al. Enriched HLA-E and CD94/NKG2A interaction limits antitumor CD8+ tumor-infiltrating T lymphocyte responses. Cancer Immunol Res. 2019. Jun 18:canimm.0885.2018. doi: 10.1158/2326-6066.CIR-18-0885. [DOI] [PubMed] [Google Scholar]
- 20.Suraweera N, Duval A, Reperant M, Vaury C, Furlan D, Leroy K, Seruca R, Iacopetta B, Hamelin R. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology. 2002;123(6):1804–1811. doi: 10.1053/gast.2002.37070. [DOI] [PubMed] [Google Scholar]
- 21.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gervois N, Labarriere N, Le Guiner S, Pandolfino MC, Fonteneau JF, Guilloux Y, Diez E, Dreno B, Jotereau F. High avidity melanoma-reactive cytotoxic T lymphocytes are efficiently induced from peripheral blood lymphocytes on stimulation by peptide-pulsed melanoma cells. Clin Cancer Res. 2000;6:1459–1467. [PubMed] [Google Scholar]
- 23.Li Y, Andrade J. DEApp: an interactive web interface for differential expression analysis of next generation sequence data. Source Code Biol Med. 2017;12(1):2. doi: 10.1186/s13029-017-0063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goedhart J, Luijsterburg MS. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci Rep. 2020;10(1):20560. doi: 10.1038/s41598-020-76603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beltra JC, Manne S, Abdel-Hakeem M, Kurachi M, Giles J, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity. 2020;52(5):825–841. doi: 10.1016/j.immuni.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Xin Z, Huang L, Zhao L, Wang S, Cheng J, Wu P, Chai Y. CD8+ T cells form the predominant subset of NKG2A+ cells in human lung cancer. Front Immunol. 2020;10:3002. doi: 10.3389/fimmu.2019.03002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yost K, Satpathy A, Wells D, Qi Y, Wang C, Kageyama R, McNamara K, Granja J, Sarin K, Brown R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25(8):1251:1259. doi: 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang Y-Q, Wang Z-X, Zhong M, Shen L-J, Han X, Zou X, Liu X-Y, Deng Y-N, Yang Y, Chen G-H, et al. Investigating mechanisms of response or resistance to immune checkpoint inhibitors by analyzing cell-cell communications in tumors before and after programmed cell death-1 (PD-1) targeted therapy: an integrative analysis using single-cell RNA and Bulk-RNA sequencing data. OncoImmunology. 2021;10(1):1908010. doi: 10.1080/2162402X.2021.1908010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Zheng L, Zhang L, Hu X, Ren X, Zhang Z. Deep single-cell RNA sequencing data of individual T cells from treatment-naïve colorectal cancer patients. Sci Data. 2019;6(1):131. doi: 10.1038/s41597-019-0131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hernández-Hoyos G, Anderson M, Wang, C, Rothenberg E, and Alberola-Ila J . GATA-3 expression is controlled by TCR signals and regulates CD4/CD8 Differentiation. Immunity. 2003;19(1):83–94. doi: 10.1016/s1074-7613(03)00176-6. [DOI] [PubMed] [Google Scholar]
- 31.Marusina A, Kim DK, Lieto L, Borrego F, Coligan J. GATA-3 is an important transcription factor for regulating human NKG2A gene expression. J Immunol. 2005;174(4):2152–2159. doi: 10.4049/jimmunol.174.4.2152. [DOI] [PubMed] [Google Scholar]
- 32.Simon S, Vignard V, Florenceau L, Dreno B, Khammari A, Lang F, Labarriere N. PD-1 expression conditions T cell avidity within an antigen-specific repertoire. OncoImmunology. 2016;5(1):e1104448. doi: 10.1080/2162402X.2015.1104448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silva T, Crispim J, Miranda F, Hassumi M, de Mello J, Simões R, Souto F, Soares E, Donadi E, Soares C. Expression of the nonclassical HLA-G and HLA-E molecules in laryngeal lesions as biomarkers of tumor invasiveness. Histol Histopathol. 2011;26(26):1487–1497. doi: 10.14670/HH-26.1487. [DOI] [PubMed] [Google Scholar]
- 34.Gooden M, Lampen M, Jordanova ES, Leffers N, Trimbos JB, van der Burg SH, Nijman H, van Hall T. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8+ T lymphocytes. Proc Natl Acad Sci. 2011;108(26):10656–10661. doi: 10.1073/pnas.1100354108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Esch EMG, Tummers B, Baartmans V, Osse EM, ter Haar N, Trietsch MD, Hellebrekers BWJ, Holleboom CAG, Nagel HTC, Tan LT, et al. Alterations in classical and nonclassical HLA expression in recurrent and progressive HPV-induced usual vulvar intraepithelial neoplasia and implications for immunotherapy: HLA expression in recurrent and progressive uVIN. Int J Cancer. 2014;135(4):830–842. doi: 10.1002/ijc.28713. [DOI] [PubMed] [Google Scholar]
- 36.Zeestraten ECM, Reimers MS, Saadatmand S, Dekker J-WT, Liefers GJ, van den Elsen PJ, van de Velde CJH, Kuppen PJK. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br J Cancer. 2014;110(2):459–468. doi: 10.1038/bjc.2013.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andersson E, Poschke I, Villabona L, Carlson JW, Lundqvist A, Kiessling R, Seliger B, Masucci GV. Non-classical HLA-class I expression in serous ovarian carcinoma: correlation with the HLA-genotype, tumor infiltrating immune cells and prognosis. OncoImmunology. 2016;5(1):e1052213. doi: 10.1080/2162402X.2015.1052213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Z-Y, Y-g L, Wang L, Shi S-J, Yang F, Zheng G-X, Wen W-H, Yang A-G. Predictive value of HLA-G and HLA-E in the prognosis of colorectal cancer patients. Cell Immunol. 2015;293(1):10–16. doi: 10.1016/j.cellimm.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Yazdi MT, van Riet S, van Schadewijk A, Fiocco M, van Hall T, Taube C, Hiemstra PS, van der Burg SH. The positive prognostic effect of stromal CD8+ tumor-infiltrating T cells is restrained by the expression of HLA-E in non-small cell lung carcinoma. Oncotarget. 2016;7(3):3477–3488. doi: 10.18632/oncotarget.6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Hall T, André P, Horowitz A, Ruan DF, Borst L, Zerbib R, Narni-Mancinelli E, van der Burg SH, Vivier E. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J ImmunoTherapy Cancer. 2019;7(1). doi: 10.1186/s40425-019-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borst L, van der Burg SH, van Hall T. The NKG2A HLA-Eaxis as anovel checkpoint in the tumor microenvironment. Clin Cancer Res. 2020. May 14. clincanres.2095.2020. doi: 10.1158/1078-0432.CCR-19-2095. [DOI] [PubMed] [Google Scholar]
- 42.Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5(1):43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Housseau F, Llosa NJ. Immune checkpoint blockade in microsatellite instable colorectal cancers: back to the clinic. OncoImmunology. 2015;4(6):e1008858. doi: 10.1080/2162402X.2015.1008858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hermel D, Sigal D. The emerging role of checkpoint inhibition in microsatellite stable colorectal cancer. JPM. 2019;9(1):5. doi: 10.3390/jpm9010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Courau T, Bonnereau J, Chicoteau J, Bottois H, Remark R, Assante Miranda L, Toubert A, Blery M, Aparicio T, Allez M, et al. Cocultures of human colorectal tumor spheroids with immune cells reveal the therapeutic potential of MICA/B and NKG2A targeting for cancer treatment. J Immunotherapy Cancer. 2019;7(1):74. doi: 10.1186/s40425-019-0553-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang C, Wang X, Li S, Twelkmeyer T, Wang W, Zhang S, Wang S, Chen J, Jin X, Wu Y, et al. NKG2A is a NK cell exhaustion checkpoint for HCV persistence. Nat Commun. 2019;10(1):1507. doi: 10.1038/s41467-019-09212-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rapaport AS, Schriewer J, Gilfillan S, Hembrador E, Crump R, Plougastel BF, Wang Y, Le Friec G, Gao J, Cella M, et al. The inhibitory receptor NKG2A sustains virus-specific CD8+ T cells in response to a lethal poxvirus infection. Immunity. 2015;43(6):1112–1124. doi: 10.1016/j.immuni.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, Xu Y, Tian Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. 2020;17(5):533–535. doi: 10.1038/s41423-020-0402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yaqinuddin A, Kashir J. Innate immunity in COVID-19 patients mediated by NKG2A receptors, and potential treatment using Monalizumab, Cholroquine, and antiviral agents. MedHypotheses.2020;140:109777. doi: 10.1016/j.mehy.2020.109777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Antonioli L, Fornai M, Pellegrini C, Blandizzi C. NKG2A and COVID-19: another brick in the wall. Cell Mol Immunol. 2020;17(6):672–674. doi: 10.1038/s41423-020-0450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pagès F, Mlecnik B, Marliot F, Bindea G, F-s O, Bifulco C, Lugli A, Zlobec I, Rau TT, Berger MD, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. The Lancet. 2018;391(10135):2128–2139. doi: 10.1016/S0140-6736(18)30789-X. [DOI] [PubMed] [Google Scholar]
- 52.Marisa L, Svrcek M, Collura A, Becht E, Cervera P, Wanherdrick K, Buhard O, Goloudina A, Jonchère V, Selves J, et al. The balance between cytotoxic T-cell lymphocytes and immune checkpoint expression in the prognosis of colon tumors. JNCI. 2018;110(1):68–77. doi: 10.1093/jnci/djx136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.