

Abstract
Mesenchymal stromal cells (MSCs) possess remarkable tumor tropism, making them ideal vehicles to deliver tumor-targeted therapeutic agents; however, their value in clinical medicine has yet to be realized. A barrier to clinical utilization is that only a small fraction of infused MSCs ultimately localize to the tumor. In an effort to overcome this obstacle, we sought to enhance MSC trafficking by focusing on the factors that govern MSC arrival within the tumor microenvironment. Our findings show that MSC chemoattraction is only present in select tumors, including osteosarcoma, and that the chemotactic potency among similar tumors varies substantially. Using an osteosarcoma xenograft model, we show that human MSCs traffic to the tumor within several hours of infusion. After arrival, MSCs are observed to localize in clusters near blood vessels and MSC-associated bioluminescence signal intensity is increased, suggesting that the seeded cells expand after engraftment. However, our studies reveal that a significant portion of MSCs are eliminated en route by splenic macrophage phagocytosis, effectively limiting the number of cells available for tumor engraftment. To increase MSC survival, we transiently depleted macrophages with liposomal clodronate, which resulted in increased tumor localization without substantial reduction in tumor-associated macrophages. Our data suggest that transient macrophage depletion will significantly increase the number of MSCs in the spleen and thus improve MSC localization within a tumor, theoretically increasing the effective dose of an anti-cancer agent. This strategy may subsequently improve the clinical efficacy of MSCs as vehicles for the tumor-directed delivery of therapeutic agents.
Keywords: cancer cell therapy, lentiviral transduction, mesenchymal stromal cells (MSCs), phagocytosis, splenic macrophage, stem cell transplantation, tumor homing
Introduction
Human mesenchymal stromal cells (MSCs), with their intrinsic ability to suppress inflammation and support healing and tissue repair [1], have emerged as a novel therapeutic approach for a variety of conditions, including tissue injury, inflammation, skin wounds and cancer [2–6]. With the capacity to be genetically modified with high efficiency using both viral and non-viral vectors, MSCs have the potential to be a valuable therapeutic tool for the delivery of soluble proteins. Additionally, MSCs display tumor tropism, rendering them, in principle, the ideal vehicle for tumor-targeted delivery of therapeutic agents [7,8]. Indeed, this concept of tumor-directed therapy is currently being studied in clinical trials [3,9–11].
The regulation of MSC migration, engraftment and survival within a tumor microenvironment, however, is not well defined [12]. Limitations to the study and utilization of MSCs for effective therapy include the low rate of MSC engraftment within the target site (0.1–1% of infused cells) [13–15] and the rapid rate of cell death after systemic injection, with the majority of cells dying within minutes to hours [16–18]. While a pulmonary first pass effect is widely accepted as underlying the initial clearance of MSCs [19], the rapid disappearance of cells after injection suggests that the host innate immune system may, at least partially, contribute to the elimination of intravenously infused MSCs [16,20].
In an effort to increase MSC localization in tumors, we sought to better understand the interaction between the host innate immune system and MSCs after intravenous infusion using a murine xenograft cancer model. We identified a striking difference in the tumor-directed migration and survival of intravenously infused MSCs in pan-immune deficient mice compared with nude mice, which lack only T cells. Moreover, transient depletion of macrophages with liposomal clodronate, which affects splenic but not pulmonary macrophages [21], can overcome this barrier and lead to significantly more MSCs localizing in the tumor. Together, our data suggest that splenic macrophages substantially contribute to rapid MSC clearance, and clinically feasible transient depletion fosters MSC survival and migration to tumors. This strategy could enhance the efficiency of MSC-delivered therapeutic agents.
Methods
Isolation and expansion of human MSCs
MSCs were isolated from the residual bone marrow obtained from collection bags of healthy donors undergoing marrow harvest for clinical indications. The protocol was classified as exempt from oversight by the institutional review board of Nationwide Children’s Hospital. Mononuclear cells were isolated using Ficoll-Paque Plus (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) gradient centrifugation, then placed in culture with maintenance medium (Dulbecco’s Modified Eagle’s Medium; Mediatech, Inc, Manassas, VA, USA) with L-glutamine supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. After 3 days, culture medium was replaced, effectively removing all non-adherent cells. Remaining cells were then trypsinized with 0.25% trypsin once ~80% confluence was reached and passaged at a density of 5000 cells/cm2. MSCs were identified using flow cytometric analysis by anti-CD90, anti- CD73, anti-CD105, anti-CD34, anti-CD45 and anti-CD3 monoclonal antibodies. MSC differentiation into adipocytes and osteoblasts in vitro was assessed by Oil Red O staining and Alizarin Red S staining, respectively, as previously described [22].
Tumor cell lines
MDA-MB-231 and U87MG were obtained from American Type Culture Collection (Manassas, VA, USA). SKNAS, CHP-134, CHLA-20, CHLA-136, ES-7, ES-8, ES-5838 and A673 were a gift from Dr Timothy Cripe. Renilla luciferase-expressing OS17 (rLUC OS17), OHS, OS25 and OS143B were a gift from Dr Ryan Roberts. CHLA-255 was a gift from Dr Shahab Asgharzadeh. CHLA-255, SKNAS, CHP-134, CHLA-20, CHLA-136, MDA-MB-231 and U87MG cells were expanded in Dulbecco’s Modified Eagle’s Medium (Mediatech, Inc) supplemented with L-glutamine, 1% penicillin/streptomycin and 10% FBS. OS17, OHS, OS25, OS143B, ES-7, ES-8, ES-5838 and A673 cells were expanded in Roswell Park Memorial Institute medium (Mediatech, Inc) supplemented with L-glutamine, 1% penicillin/streptomycin and 10% FBS.
In vitro migration assay
Conditioned medium (CM) was generated by incubating confluent cell cultures (~107 cells in 75 cm2) with the appropriate medium supplemented with 1% FBS and 1% penicillin/streptomycin at 37°C overnight. The medium was clarified by centrifugation (1000 g × 5 min), and the supernatant was then passed through a 0.45-μm pore filter. As a control, non-CM was generated in the same way as previously noted, except no cells were added to the plates. MSC migration was assessed using 24-well plates with 8-μm transwell inserts. Next, 2 × 104 MSCs in 100 μL of appropriate medium with 1% FBS were placed in the upper chamber, and 600 μL of CM and non-CM were placed in the lower chamber and incubated for 16 h at 37°C. Cell migration was quantified using the CytoSelect cell migration kit (Cell Biolabs, Inc, San Diego, CA, USA) according to the manufacturer’s instructions.
Human MSC transduction
Human MSCs were transduced with bicistronic lentiviral vector encoding firefly luciferase (ffLuc) and ZsGreen fluorescent protein. MSCs (10 000 cells/cm2) were incubated with vector at a multiplicity of infection of 10 in medium containing 100 μg/mL protamine sulfate for 24 h. The medium was replaced, and the cells were maintained in culture for 3 days. The cells were then transduced again for 24 h. Transduction efficiency was assessed by flow cytometry and immunofluorescence microscopy measuring ZsGreen fluorescence.
Animals and cell administration
Female 6- to 8-week-old athymic nude (nu/nu) mice were purchased from Envigo (Indianapolis, IN, USA), and NOD-scid IL2Rgam-manull (NSG, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice and Rag1 KO (B6.129S7-Rag1tm1Mom/J) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and used in accordance with institutional guidelines and approved protocols at The Research Institute at Nationwide Children’s Hospital. Tumors were created in nude, NSG and Rag-KO mice by subcutaneous injection of 5 × 106rLuc-expressing OS17 cells suspended in 100 μL phosphate-buffered saline and 50 μL Matrigel (Mediatech, Inc). Tumor size and body weight were measured twice weekly. Tumor volume was determined by caliper measurement using the formula volume = (width^2 × length)/2. Once the tumor volume reached 200–250 mm3, transduced MSCs (0.5 × 106 ffLuc MSCs/mouse) were intravenously injected.
In vivo depletion of natural killer cells and macrophages
Anti-asialo GM1 rabbit (Wako Chemicals USA, Inc, Richmond, VA, USA) antibody was injected intraperitoneally twice weekly for natural killer (NK) cell depletion in OS17 solid tumor-bearing nude mice. Rabbit serum was used in control mice. For macrophage depletion, OS17 solid tumor-bearing nude mice were injected with liposomal clodronate (Clodrosome; Encapsula NanoSciences LLC, Brentwood, TN, USA) intraperitoneally 6 days prior to MSC injection and then every 3 days to maintain depletion. Cell depletion confirmed by flow cytometric analysis of splenocytes for NK (CD49d+) or macrophage (F4/80+, CD11b+) content.
Bioluminescence imaging
Bioluminescence imaging (BLI) was performed using the Xenogen in vivoimaging system (PerkinElmer Inc, Hopkinton, MA, USA). Images were acquired at 1- to 3-min intervals until the peak signal was observed. Bioluminescence color images were analyzed using Living Image 2.11 software (PerkinElmer Inc). For quantification, a region of interest was manually applied to lung, spleen and tumor regions. The percentage of MSCs that migrated to tumor was calculated by taking the signal from the lungs at 4 h after injection to represent 100% of injected cells. Mice were injected with 150 mg/kg D-luciferin firefly potassium salt substrate (PerkinElmer Inc) intraperitoneally to track biodistribution of ffLuc MSCs. To monitor rLuc OS17 tumor, each mouse was intravenously injected with 15 μg coelenterazine substrate (PerkinElmer Inc).
Flow cytometry
Flow cytometric analyses used BD LSR II (BD Biosciences, San Jose, CA, USA) with commercially available antibodies. Results were analyzed using FlowJo software (Tree Star, Inc, Ashland, OR, USA).
Phagocytosis of MSCs
MSCs were labeled immediately prior to injection with CellVue Claret (Sigma-Aldrich, St Louis, MO, USA) per the manufacturer’s instructions. After MSC injection, freshly isolated spleen cells were analyzed by flow cytometry. Claret+F4/80+CD11b+ macrophages indicated macrophage engulfment of Claret-labeled MSCs.
Confocal microscopy
MSCs were labeled with DiI membrane stain (Life Technologies, Waltham, MA, USA) according to the manufacturer’s instructions. DiI-labeled MSCs were administered intravenously 4 h before animals were killed. Spleens were harvested and isolated single-cell suspension stained with F4/80 antibody. Cells were resuspended in phosphate-buffered saline, loaded to a cytospin cuvette (Shandon EZ Single Cytofunnel; Thermo Fisher Scientific, Waltham, MA, USA) and spun at 1000 rpm for 5 min. Cytospin slides were then dried and fixed with 4% paraformaldehyde, and nuclear staining was applied with 4’,6-diamidino-2-phenylindole for 10 min at room temperature. Coverslips were mounted on slides and imaged using a confocal microscope with ×63 objective (Zeiss LSM 710; Carl Zeiss Inc, Thornwood, NY, USA).
Histological analysis
Mice were killed according to approved protocol, and tissue samples were excised. Tissue samples were fixed in 10% buffered formalin and embedded in paraffin. Next, 5-μm thick sections were cut from paraffin-embedded blocks, deparaffinized with xylene and rehydrated with graded ethanols. Heated antigen retrieval was applied in citrate buffer by pressure cooker for 20 min. Endogenous peroxidase was quenched in 3% hydrogen peroxide for 10 min. Non-specific binding was blocked by incubation in 5% secondary antibody host serum for 10 min. Sections were incubated at 4°C for 16 h with monoclonal rabbit anti-human CD90 antibody 1:100 (clone EPR3132; Abcam, Cambridge, MA, USA) and then incubated with goat anti-rabbit biotinylated antibody 1:200 (Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. Immunodetection and color development were performed using the VECTASTAIN Elite ABC system and diaminobenzidine substrates (Vector Laboratories). Counterstaining was performed with Harris Hematoxylin (Sigma-Aldrich). The slides were dehydrated in graded ethanol and xylene.
Statistical analysis
Numerical data were expressed as mean ± standard deviation. Statistical analyses were performed using Prism 7 (GraphPad Software, La Jolla, CA, USA) or SAS 9.3 (SAS Institute, Cary, NC, USA). P < 0.05 was taken to be statistically significant.
Results
Characterization and lentiviral transduction of human MSCs
Human MSCs were transduced with a bicistronic lentiviral vector expressing ffLuc and ZsGreen under the control of viral long terminal repeats. Flow cytometric analysis of ex vivo-expanded ffLuc MSCs demonstrated uniform expression of CD90, CD73 and CD105 while lacking expression of hematopoietic markers CD45, CD34, CD3, CD14, CD19 and HLA-II (Figure 1A). The adherent, spindle-shaped cells underwent differentiation to osteocytes and adipocytes in vitro (Figure 1B). These data show that MSCs are not affected by lentiviral transduction or ffLuc and ZsGreen expression. This characterization is consistent with the defining criteria of the International Society for Cellular Therapy for MSCs [23].
Figure 1.
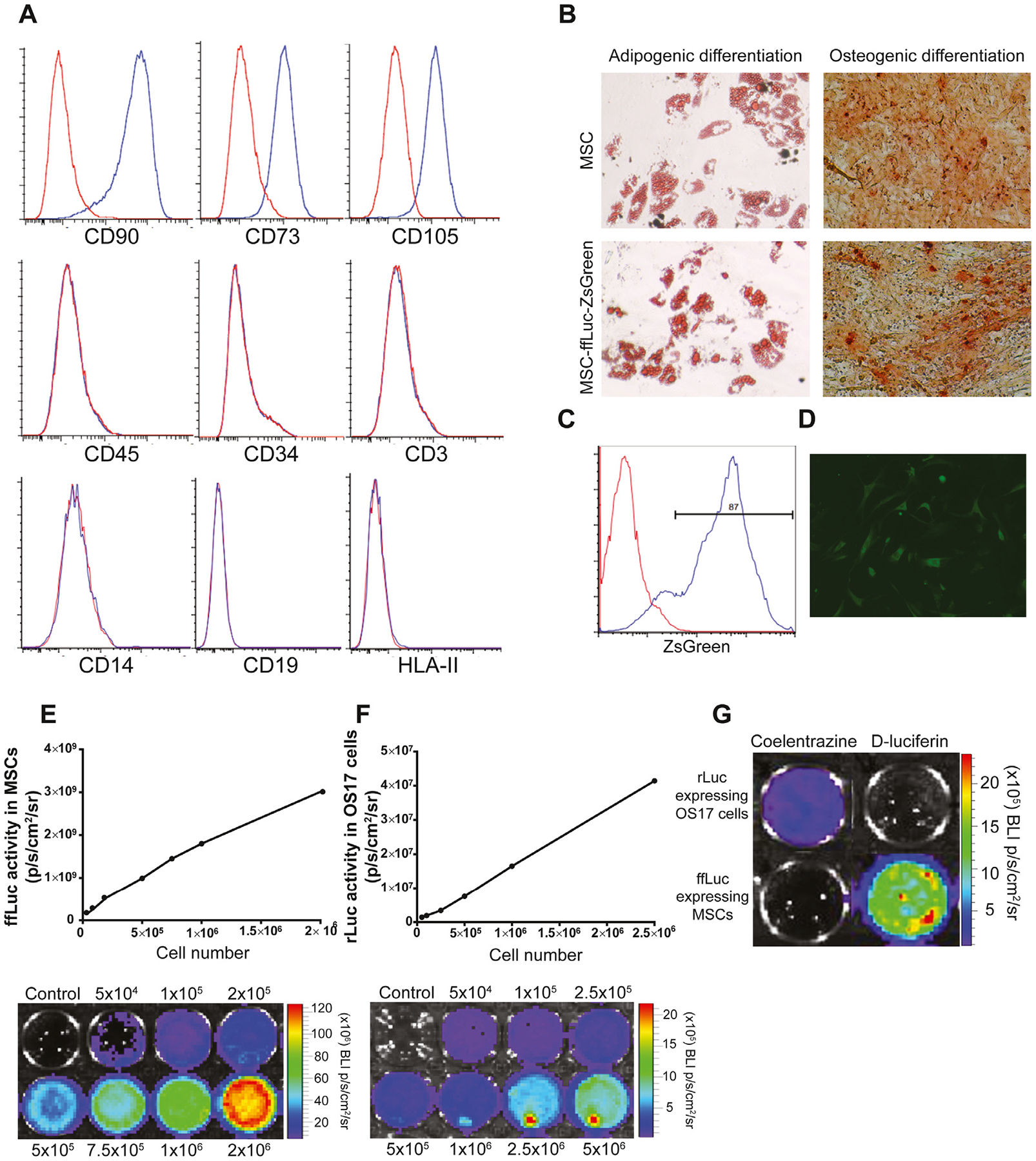
Characterization of human MSCs labeled with ffLuc and ZsGreen. (A) Flow cytometric analysis showed that ffLuc MSCs were positive for CD90, CD73 and CD105 and negative for hematopoietic cell markers CD45, CD34, CD3, CD14, CD19 and HLA-II. (B) The ffLuc MSCs differentiated into adipocytes (Oil Red O staining) and osteocytes (Alizarin Red S staining) in vitro (magnification ×20). (C) MSCs were transfected with ffLuc and ZsGreen reporter gene by lentiviral transduction. Transduction efficiency was analyzed by flow cytometry. (D) The ffLuc MSCs uniformly expressed ZsGreen within the cytosol. (E, F) In vitro analysis showed a linear correlation between the cell number and bioluminescence imaging signal of ffLuc MSCs and rLuc OS17, respectively. (G) The rLuc-expressing OS17 cells and ffLuc-expressing MSCs displayed specific reactivity with coelentrazine and D-luciferin, respectively.
Transduction was measured by expression of ZsGreen using fluorescence-activated cell sorting analysis (Figure 1C) and immunofluorescence microscopy (Figure 1D), which resulted in ~87% efficiency. The BLI signal intensity correlated with the number of transduced cells for ffLuc MSCs and rLuc OS17, respectively (Figure 1E,F). The ffLuc bioluminescent activity was detected when ffLuc MSCs were treated with D-luciferin, and rLuc was detected when OS17 tumor cells were treated with coelentrazine. There was no measurable cross-reactivity between 2 luciferase reporters (Figure 1G).
In vitro human MSC migration
We first determined the chemoattraction potency of each cell line for MSCs in vitro using a transwell assay of MSC migration toward the corresponding CM (Figure 2). Interestingly, while the transwell assay unambiguously identified cell lines that vigorously attract MSCs, the chemotactic potential was not uniform among cell lines derived from the same tumor type. For example, CHP-134 was the only neuroblastoma cell line of the 6 cell lines (CHLA-255, SKNAS, CHP-134, CHLA-15, CHLA-20, CHLA-136) assayed to attract MSCs. Three osteosarcoma cell lines (OS17, OHS, OS25) proved highly chemoattractive, whereas OS143B osteosarcoma cells did not show any activity. CM from the osteosarcoma cell line OS17 demonstrated the most robust chemotaxis and was therefore selected for this study. We also tested MSC migration toward CM taken from ES-7, ES-8, ES-5838, A673 Ewing’s sarcoma, MDA-MB-231 breast cancer and U87MG glioblastoma cell cultures. Transwell assays demonstrated a significant increase in MSC migration toward CM from A673 and U87MG cells compared with the control non-CM.
Figure 2.
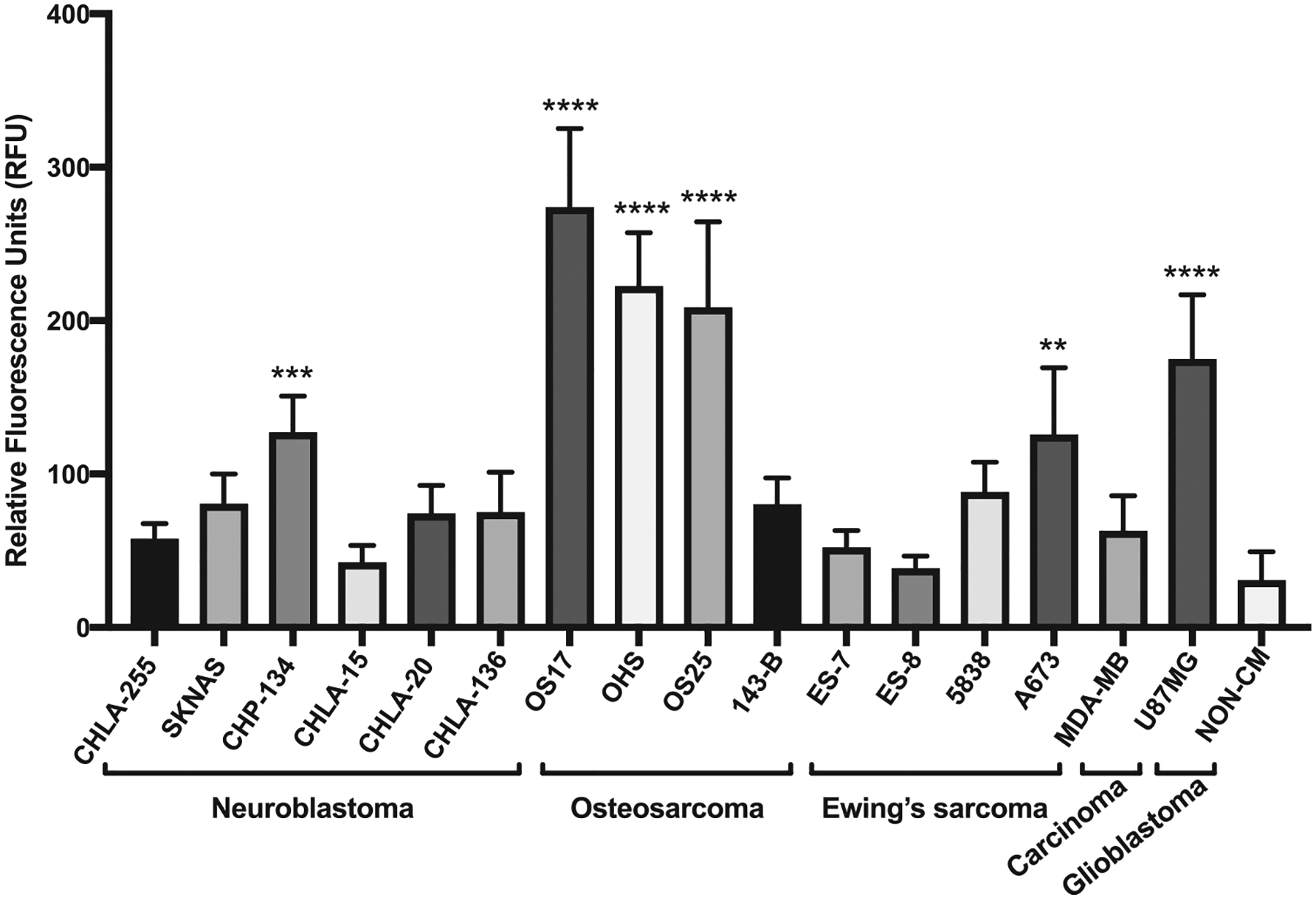
MSC migration assays toward various cancer cell CMs. The ffLuc MSCs significantly migrated toward CM from CHP-134, OS17, OHS, OS-25, A673 and U87MG cell lines in transwell migration assay. Representative of 2 independent experiments performed in triplicate. **P< 0.01, ***P< 0.001, ****P< 0.0001. RFU, relative fluorescence unit.
In vivo MSC migration in a nude mouse model
Next, rLuc OS17 cells were injected into the flank of nude mice, establishing a subcutaneous tumor. When a sufficient cohort of mice exhibited tumors of 200–250 mm3, these animals were used for our studies. Hence, all mice were intravenously infused with ffLuc MSCs in parallel on the same day, both animals with tumors of 200–250 mm3 and healthy controls. Two hours after infusion, the BLI signal intensity was detected predominantly in the lungs, as expected. However, the ffLuc MSC signal quickly dissipated and was undetectable in the lungs 3 days post-injection (Figure 3A).The bioluminescence emitted from MSCs was detected quantitatively (yet below the level of visualization relative to the lung signal) in the tumor as early as 2 h after injection (Figure 3B). Over the first 3 days, the signal intensity declined 40% (P < 0.0001), suggesting MSCs that arrived in the tumor subsequently migrated away or engrafted but did not survive in the tumor microenvironment (Figure 3B). From 3 to 14 days after injection, the signal increased (P < 0.0001), consistent with ongoing localization of MSCs in the tumor or intratumoral expansion of engrafted cells. The MSC signal was stable from day 14 to day 17 (P = 0.903) (Figure 3B).
Figure 3.
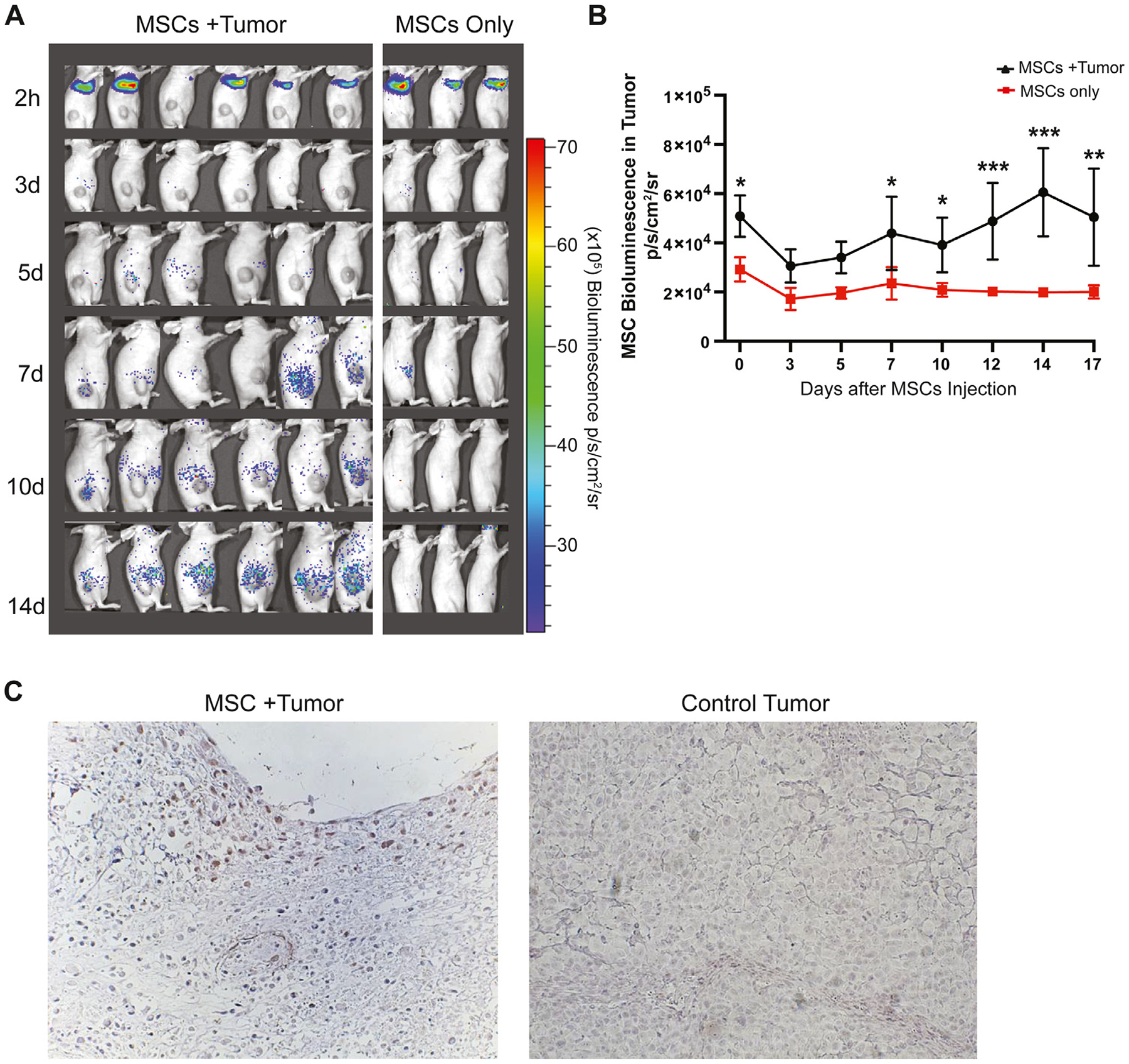
Biodistribution of ffLuc MSCs in OS17 tumor-bearing nude mice. (A) MSCs were injected intravenously and imaged 1 day, 3 days, 5 days, 7 days, 10 days, 14 days and 17 days after injection. Initial localization of MSCs in lung area and eventual localization in tumor area were demonstrated. (B) Bioluminescence signal intensity was quantified over the tumor regions. (C) Immunohistochemistry was performed with MSC marker anti-human CD90 antibody and Hematoxylin on tumor sections to detect MSCs. Representative images of tumor regions were taken 17 days after MSC injection of MSC- and PBS-injected mice (magnification ×20). Data were analyzed using a linear mixed-effects model and post hoc pairwise comparisons. Tukey-adjusted Pvalues were used to determine significance. *P< 0.05, **P< 0.01, ****P< 0.0001. PBS, phosphate-buffered saline.
Immunohistochemical staining of a histologic section of a tumor obtained 17 days after infusion revealed MSCs proximal to small blood vessels (Figure 3C), without individual MSCs dispersed in the tumor tissue. This observation suggests that after MSCs traffic to the tumor and cross the endothelium, the cells do not migrate throughout the tumor but remain near their point of tumor entry. Absence of MSC marker expression on tumor tissue was confirmed by immunofluorescence staining for CD90 and ZsGreen in ganglioside antigen GD2-expressing OS17 osteosarcoma tumor section obtained from the xenograft nude mouse model (see supplementary Figure 1).
MSC migration in nude versus NSG mouse models
The apparent clearance of MSCs led us to the hypothesis that MSC elimination by the host innate immune system may underlie the limited survival, migration and engraftment of MSCs we observed in our nude murine tumor models. To evaluate this question, we analyzed MSC migration in NSG mice with absent immune function compared with our nude mice, which lack only T cells.
Although the persistence of MSCs in the lungs was conspicuously different between the 2 murine models, the profile of MSC tumor migration and intratumoral expansion was comparable, albeit with greater magnitude of MSC localization in the NSG mice (Figure 4A). In both models, a similarly intense signal was observed over the lungs 2 h after injection. The bioluminescence signal over the lungs in NSG mice remained constant at 2 days, whereas the signal in nude mice dissipated visually and quantitatively. The NSG lung signal persisted over the course of observation.
Figure 4.
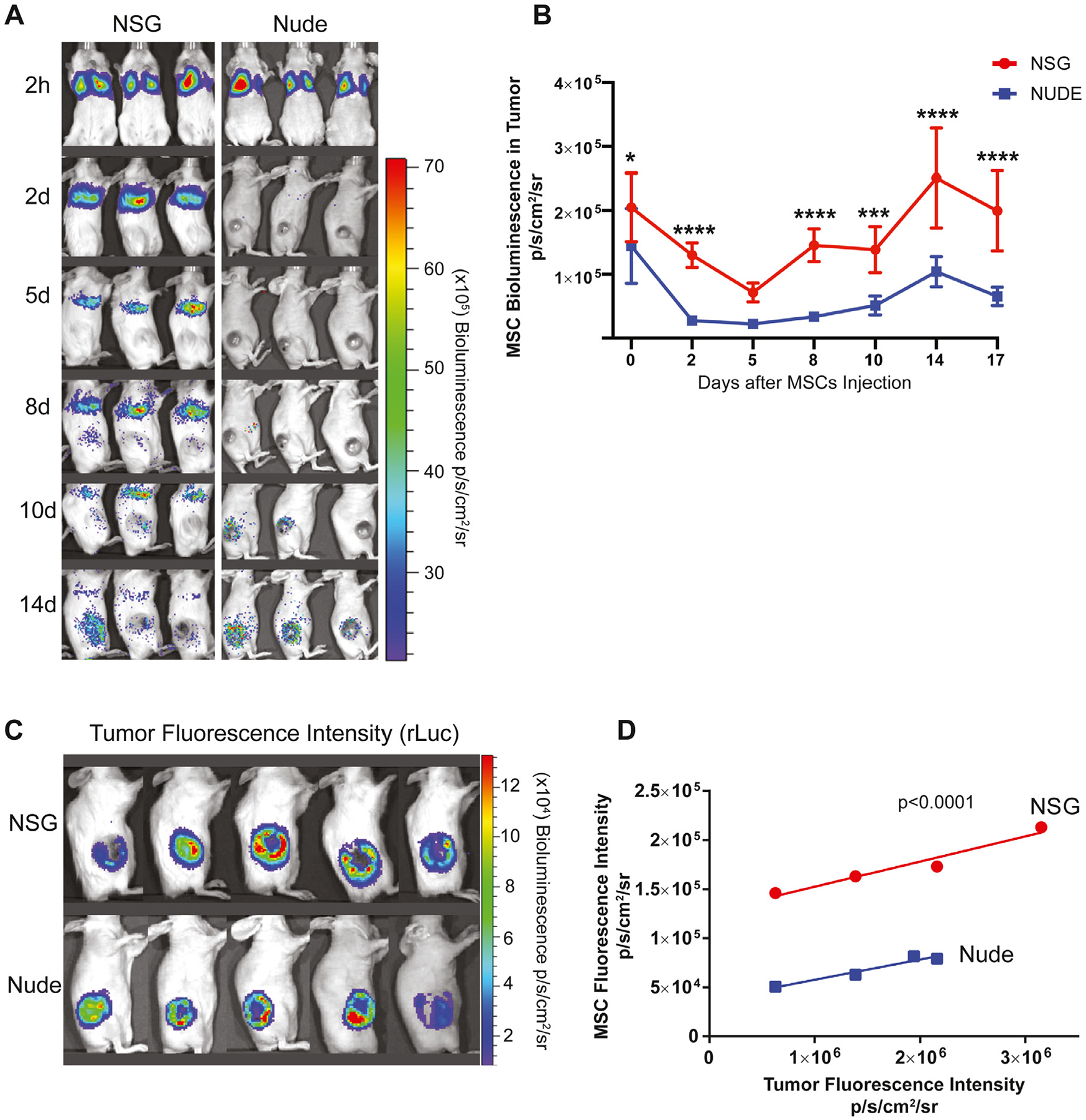
Biodistribution of MSCs in nude and NSG tumor models. (A) The ffLuc MSCs were injected intravenously into nude and NSG mice and imaged on day 1, day 2, day 5, day 8, day 10 and day 14 post-injection using Xenogen IVIS. (B) Bioluminescence signal was quantified in the tumors of nude and NSG groups. (C) Tumor sizes were measured with bioluminescence imaging using coelentrazine substrate in rLuc OS17 tumor-bearing nude and NSG animals. (D) Correlation between tumor sizes and MSC migration was quantified in both animal models. Data were analyzed using a linear mixed-effects model and post hoc pairwise comparisons. Tukey-adjusted Pvalues were used to determine significance. ***P< 0.001, ****P< 0.0001. IVIS, in vivo imaging system.
In both models, a strong signal was detected quantitatively over the tumor at 2 h (Figure 4B). In NSG mice, the signal decreased gradually, reaching the nadir on day 6 (P < 0.0001 relative to 2 h), at which time the signal was no different than that seen from the nude mice (P = 0.666). The signal then intensified to a maximum on day 14 (P < 0.0001 compared with day 2). At the peak signal intensity in both mouse models, the bioluminescence signal from NSG mice was stronger than the signal from the nude mice (P < 0.0001), suggesting a larger population of intratumoral MSCs resided in the tumors of NSG mice.
We found a positive correlation between tumor volume, determined by renilla luminescence, and MSC localization, determined by luciferase luminescence, in both nude and NSG mice. Interestingly, although the correlation was equivalent in the 2 models, there was a 3.5-fold greater ffLuc MSC signal per tumor volume in NSG compared with nude mice (P < 0.0001) (Figure 4C,D).
Our data show that MSCs have longer survival within the lungs and increased migration to the tumor in NSG mice compared with nude mice. Thus, an active effect of the immune system, not a passive trapping in the capillary beds, seems to underlie MSC clearance. The principal difference between these strains is the presence of fully competent NK cells, macrophages and B lymphocytes in the nude mice, rendering these 3 lineages the most likely candidates to mediate early MSC clearance.
Effect of NK cells
To assess the role of NK cells, we injected anti-asialo GM1 antibody (Wako Chemicals USA, Inc), which selectively depletes NK cells [24], into OS17 tumor-bearing nude mice. Flow cytometric analysis of splenocytes demonstrated 63% reduction in NK cells (P < 0.001) (Figure 5A). Importantly, the macrophage population was not affected (Figure 5B). NK-depleted and control mice were then infused with ffLuc MSCs. NK depletion, however, did not result in greater bioluminescence signal from the tumor, indicating there was no enhanced ffLuc MSC localization (Figure 5C). These data suggest that NK cells do not significantly contribute to the early clearance of intravenously infused MSCs.
Figure 5.
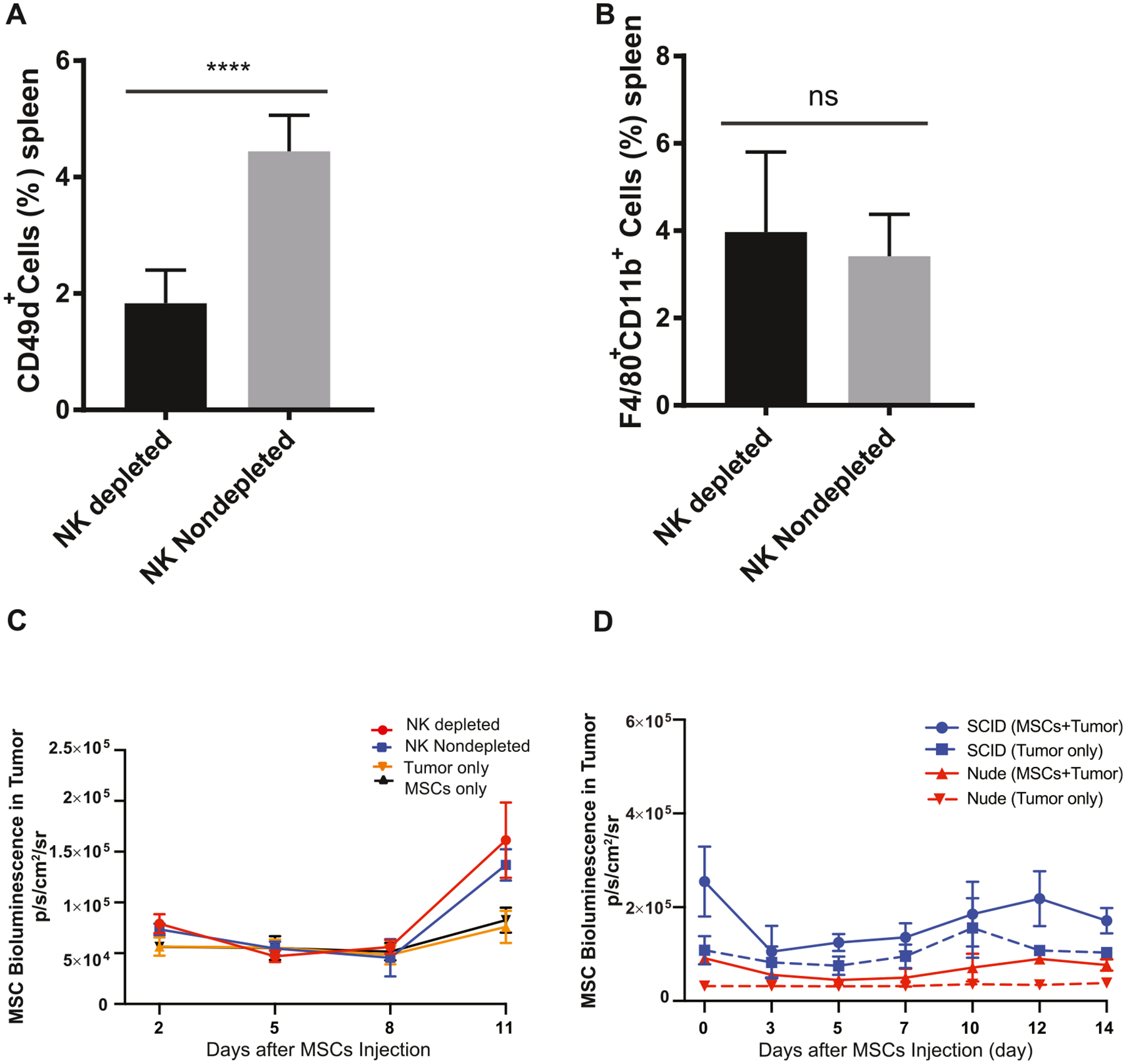
Effects of NK cells and B lymphocytes on MSC engraftment. (A) NK cells were depleted with ASGM1 antibody in spleen. (B) NK cell depletion did not have any impact on splenic macrophage levels. (C) MSC migration was analyzed on day 1, day 2, day 5, day 8 and day 11 by bioluminescence imaging and compared with NK cell non-depleted and control groups. Bioluminescence signal was quantified and analyzed in the tumor regions. (D) MSC migration was analyzed in SCID and nude mouse models. On day 1, day 3, day 5, day 7, day 10, day 12 and day 14 after MSC injection, animals were imaged using Xenogen IVIS, and MSC signal was analyzed in tumor regions, comparing each experimental cohort with their own strain-specific controls. Data were analyzed using a linear mixed-effects model and post hoc pairwise comparisons. Tukey-adjusted Pvalues were used to determine significance. *P< 0.05, **P< 0.01, ****P< 0.0001. ASGM1, anti-asialo GM1; IVIS, in vivo imaging system.
Effect of B cells
To examine whether B lymphocytes mediate MSC clearance, we generated a subcutaneous OS17 tumor model in severe combined immunodeficient (SCID) mice that lack T cells, similar to nude mice, but also mature B cells. Due to variations among murine strains, SCID and nude mice generate incomparable bioluminescence signals; therefore, we compared each experimental cohort with their own, strain-specific controls. As shown in Figure 5D, ffLuc MSCs persisted and migrated equally in SCID and nude hosts, with tumor signal intensities exceeding their controls by 2.02-fold and 2.61-fold, respectively. These findings suggest that B lymphocytes are not the effector cells mediating the killing of MSCs.
Effect of macrophages
To determine the impact of host macrophages on persistence of intravenously infused MSCs, we depleted macrophages in OS17 tumor-bearing nude mice with intraperitoneal liposomal clodronate. As is recognized for liposomal clodronate [25], the splenic macrophages were effectively depleted (71% reduction, P > 0.0001) (Figure 6A,B) but rapidly recovered after the liposomal clodronate course (see supplementary Figure 2A). By contrast, liposomal clodronate did not measurably affect pulmonary macrophages (Figure 6C). The intratumoral macrophages were modestly reduced but returned to baseline within 7 days (Figure 6D; also see supplementary Figure 2B). Intraperitoneal injection of liposomal clodronate did not affect the level of monocytes in peripheral blood (data not shown), as previously reported [26]. Next, ffLuc MSCs were intravenously infused into macrophage-depleted and non-depleted control mice. At 3 h post-infusion, the bioluminescence signal over the tumor was 11.4-fold greater in macrophage-depleted mice compared with controls (Figure 6E). The bioluminescence signal of the spleen was a striking 49.8-fold greater in macrophage-depleted mice 5 h post-infusion, whereas the signal intensity was 3.16-fold greater than controls at 48 h, which corresponds to the time course of macrophage repopulation in the spleen (Figure 6F). On day 11, the population of tumor-localized MSCs was 8.6-fold greater in macrophage-depleted mice compared with controls (P < 0.0001) (Figure 6G). That liposomal clodronate treatment depleted splenic but not pulmonary macrophages and the MSC bioluminescence signal persisted in the macrophage-depleted spleen until it was repopulated with macrophages suggests that splenic macrophages primarily mediate the rapid clearance of MSCs after intravenous infusion.
Figure 6.
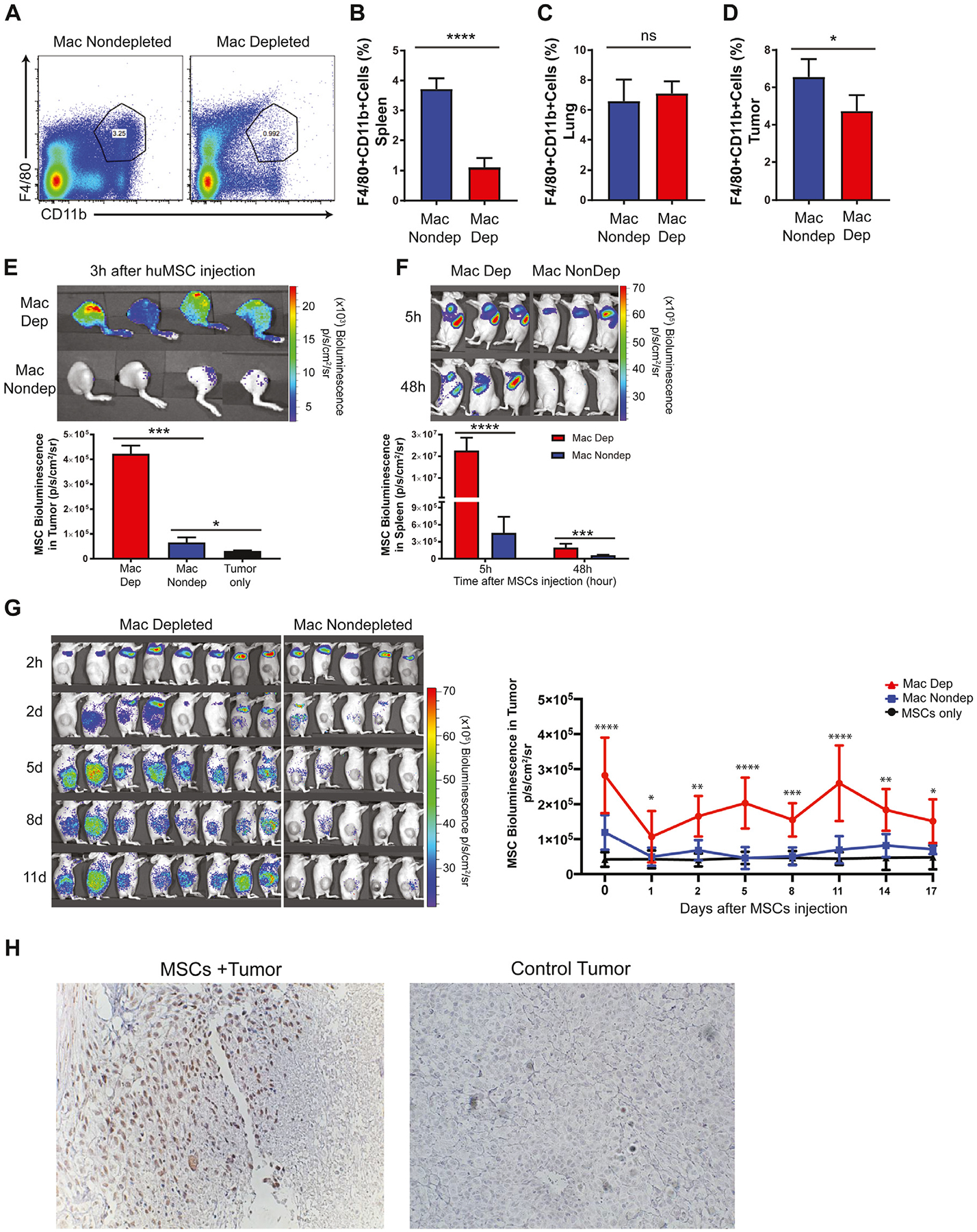
Effects of macrophage depletion on MSC engraftment and survival. (A, B) Flow cytometric analysis of macrophage depletion in spleen with liposomal clodronate. Percentage of F4/80+CD11b+ macrophages after liposomal clodronate administration in lung (C) and tumor (D) tissues. (E) After macrophage depletion, ffLuc MSCs were injected intravenously; 3 h after MSC injection, MSC migration to the tumor area was analyzed by bioluminescence imaging. (F) MSC localization in the spleen area was compared with the non-depleted group 5 h and 48 h after MSC injection. (G) Initial localization of MSCs in lung area and eventual localization in tumor area were imaged on day 1, day 2, day 5, day 8, day 11 and day 14 post-injection in macrophage-depleted and non-depleted control mice. Bioluminescence signal was quantified and analyzed in the tumor regions of animal groups. (H) On day 16, MSC engraftment in tumor regions of macrophage-depleted nude mice was confirmed by immunohistochemistry. Tumor sections harvested from macrophage-depleted nude mice exposed to ffLuc MSCs or negative control (PBS) were stained with human anti-CD90 antibody to show MSC incorporation within tumor sections. Data were analyzed using a linear mixed-effects model and post hoc pairwise comparisons. Tukey-adjusted P values were used to determine significance. *P < 0.05, ***P < 0.001, ****P < 0.0001. mac, macrophage; PBS, phosphate-buffered saline.
Immunohistochemical staining of a tumor section obtained from a macrophage-depleted mouse 16 days after infusion revealed MSCs prominently clustered around small blood vessels (Figure 6H). Similar to non-depleted animals (Figure 3C), we were unable to identify individual MSCs scattered within the tumor or numerous small foci of MSCs.
Macrophage phagocytosis of MSCs
MSC survival and migration were increased in our macrophage depletion model, suggesting macrophages mediate early clearance of intravenously infused cells. To determine whether macrophages directly eliminate MSCs by phagocytosis, we infused ffLuc MSCs labeled with Claret, a fluorescent membrane dye, into OS17 tumor-bearing nude mice. At 3 h and 24 h, single-cell suspensions prepared from freshly harvested spleens were assessed by flow cytometry. Flow cytometric analysis of the 3-h sample showed 0.908% ± 0.195% (n = 4) of F4/80+CD11b+ macrophages were positive for Claret fluorescence. At 24 h, 7.078% ± 0.988% (n = 4) of F4/80+ CD11b+ macrophages demonstrated Claret fluorescence (Figure 7A).
Figure 7.
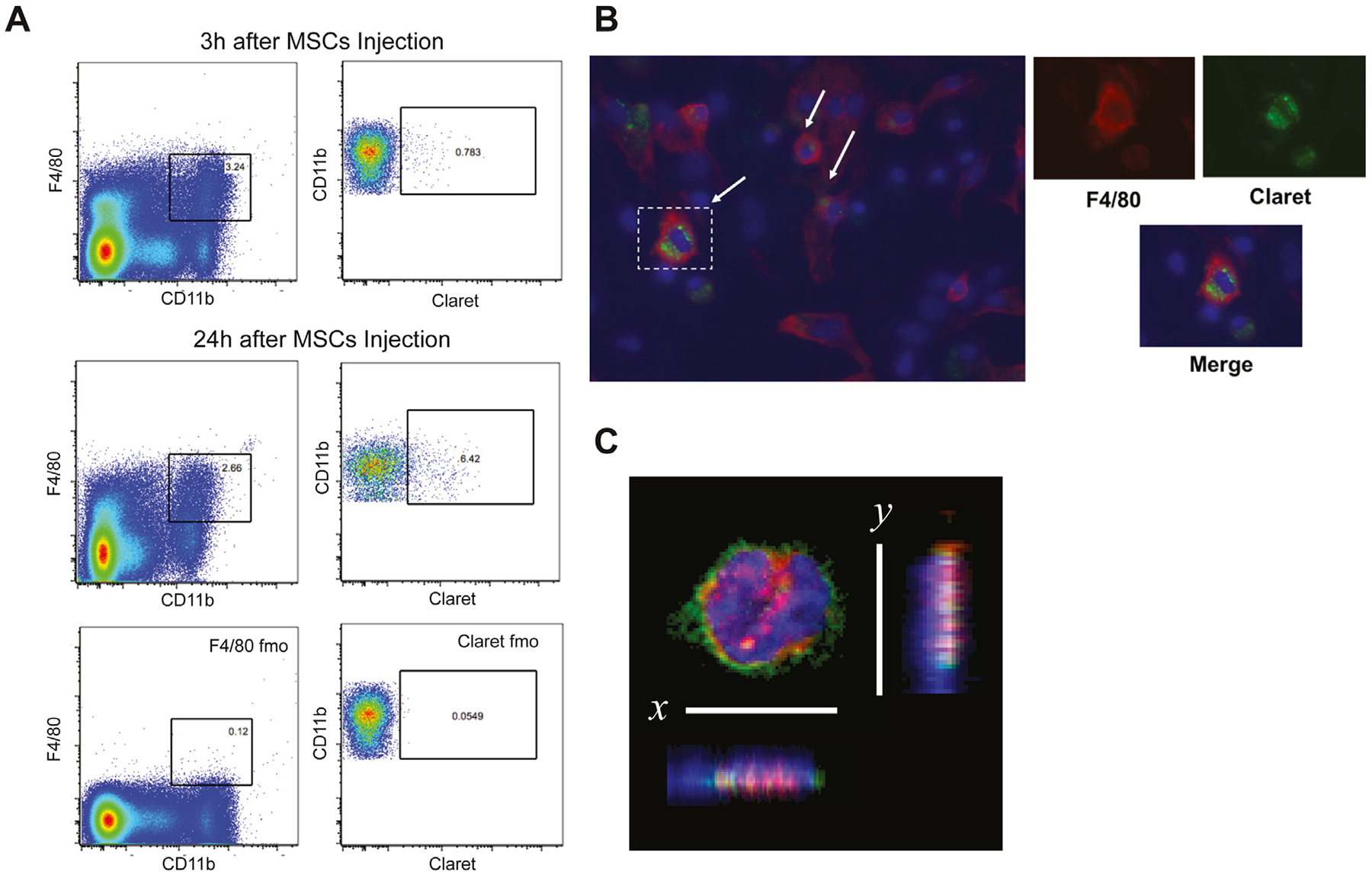
Analysis of spleen macrophages after MSC injection. (A) After intravenous injection of Claret-labeled MSCs, spleen cells were isolated and labeled with F4/80 and CD11b antibodies. Representative example of flow cytometric analysis at 3 h and 24 h in macrophage spleen cell population. After gating on macrophages (F4/80+CD11b+), Claret+ cells were identified according to the expression of Claret on this macrophage population. (B) Following isolation of spleen cells, cells were cultured for 3 days and stained with F4/80 antibody. Claret staining was found inside the macrophages. Smaller panels represent enlarged views of dotted rectangles. DAPI (blue) stained nucleus and anti-F4/80 antibody (red) stained macrophages. (C) MSCs were labeled with DiI membrane stain (red) and administered intravenously 4 h before animals were killed. Spleen cell suspension was stained with F4/80 antibody (green) and DAPI (blue), cytospinned and imaged with confocal microscopy. The image was digitally rotated along the x- or y-axis to demonstrate that the DiI signal (engulfed MSCs) was within the macrophage (magnification ×63). DAPI, 4’,6-diamidino-2-phenylindole; fmo, fluorescence minus one.
To pursue this notion, a sample of the single-cell suspension prepared from spleens obtained 3 h post-infusion was placed into a chamber slide cell culture. The medium was replaced the following day to remove nonadherent cells. After 3 days of culture, fluorescence microscopy demonstrated Claret-positive cells within the cytoplasm of F4/80+ macrophages (Figure 7B).
Finally, MSCs were labeled with DiI and injected into OS17 tumor-bearing nude mice. A single-cell suspension of splenic macrophages isolated 4 h after DiI-labeled MSC injection was stained with F4/80 and 4’,6-diamidino-2-phenylindole (DAPI). Suspension cells were centrifuged on glass slides using cytospin and imaged on confocal microscopy. Dil-positive cells within the cytoplasm of F4/80+ macrophages were demonstrated by confocal microscopy in a single optical section viewed in the x-y plane (Figure 7C). The image was digitally rotated along the x- or y-axis to demonstrate that the DiI signal (engulfed MSCs) was within the macrophage. These observations are consistent with our assertion that splenic macrophages phagocytose intravenously infused MSCs.
Discussion
After ex vivo-expanded MSCs are intravenously infused, <1% typically arrive in a solid tumor [13–15]. Contrary to the widely accepted notion that trapping in the pulmonary capillary beds is the principal basis for removal from the circulation [19], our data show that macrophages play an active role in clearance of infused MSCs. Moreover, although pulmonary macrophages phagocytose MSCs [27], we showed that splenic macrophages also phagocytose MSCs (Figure 7), substantially contributing to this first pass effect. Although there are few clinically feasible approaches for overcoming pulmonary clearance, the significance of our finding is that liposomal clodronate, and possibly other bisphosphonates, transiently depletes splenic macrophages, moderating clearance of infused MSCs. The end result is significantly more MSCs localizing in a solid tumor, which may, when applied clinically, be the underpinning of a more effective tumor-delivered therapy using MSCs as the vehicle.
Crosstalk between MSCs and macrophages has gained increasing attention in several research arenas. These mutual interactions have been attributed to cell-to-cell contacts and cytokine-mediated effects [28–30]. MSC-derived prostaglandin E2 promotes polarization of the macrophage M1 to M2 phenotype [31]. Similarly, Luz-Crawford et al. [32] showed that MSC-secreted IL-1R antagonist induces polarization of macrophages toward the M2 phenotype and inhibits B-cell differentiation via decreased tumor necrosis factor alpha production and increased IL-10 production. These regulatory functions of MSCs on macrophages have been demonstrated in various inflammatory conditions, including myocardial infarction, acute kidney and lung injury, atherosclerosis and inflammatory bowel disease [33–39]. Effects of MSCs on macrophages are not limited to soluble factors. The cytokine independent pathway is also involved in the MSC-induced polarization of macrophages. Braza et al. [27] showed, in an asthma model, that MSCs convert macrophages to an M2 phenotype after being phagocytosed by lung macrophages. Likewise, de Witte et al. [40] demonstrated that intravenously injected MSCs are rapidly phagocytosed by macrophages, which in turn results in polarization of macrophages to an anti-inflammatory phenotype. Moreover, these MSC-primed macrophages are capable of inducing Foxp3+ T regulatory cell formation. The mechanisms of macrophage phagocytosis of MSCs need to be further investigated. Whether apoptotic MSCs initiate phagocytosis or living MSCs are engulfed by phagoptosis remains unknown, and the two are not mutually exclusive. Galleu et al. [41] demonstrated that infused MSCs undergo perforin-induced apoptosis by recipient cytotoxic cells (CD8+ T cells and CD56+ NK cells). Subsequently, recipient phagocytes engulf apoptotic MSCs and induce immunosuppression by releasing indoleamine-2,3-dioxygenase. In another recent study, Gavin et al. [42] showed that complement components opsonize MSCs in plasma, which in turn induce MSC phagocytosis by classical and intermediate macrophages through the alternative complement pathway. This mechanism is consistent with our findings and might explain rapid clearance of MSCs (2–24 h) when administered systemically. In our study, we demonstrated that injected Claret- and DiI-labeled MSCs were phagocytosed by splenic macrophages within 3–24 h following intravenous injection. After initial entrapment in the lungs, MSCs moved systemically to the spleen, another major accumulation location [17]. However, transient depletion of splenic macrophages facilitated MSC survival and was correlated with greater MSC localization in the tumor area. MSCs have been reported to display various molecular mechanisms that either suppress tumor growth [43] or promote cancer progression by immune modulation of innate immune cells. MSCs can actively affect functions of macrophages by changing their polarization from the pro-inflammatory M1 to the IL-10-secreting immunosuppressive M2 phenotype [30,31,44]. However, exogenous MSCs as biological vehicles are a valuable treatment tool when engineered with immune-modulating genes [45,46]. We previously showed that direct injection of lentivirally transduced interferon gamma-expressing MSCs into the tumor activated the innate immune system by polarizing M2 tumor-associated macrophages (TAMs) to M1 macrophages, resulting in decreased tumor growth and increased survival in a neuroblastoma xenograft model [47]. Moreover, in our model, MSCs did not promote tumor growth, and the presence of MSCs did not impede the polarizing effect of interferon gamma [47]. An ostensible short-coming of transient systemic macrophage depletion for facilitating engineered MSC localization in a tumor is that TAMs may also be depleted from the tumor microenvironment. In our osteosarcoma model, we show that tumor-associated macrophages are reduced by only 20%, and the population recovers within 7 days (see supplementary Figure 2B), the time course observed for tumor-localized MSCs to begin expansion. Thus, our data predict that transient macrophage depletion will not remove the therapeutic benefits of TAM-targeted MSC therapy.
Whether tumor-localized MSCs expand within the tumor remains an open question [48]. This aspect of MSC/tumor biology is important, as MSC expansion within the tumor suggests that more cells are effectively available for anti-tumor activity than originally traffic to the tumor. Additionally, developing methods to increase intratumoral MSC expansion is a potential strategy to increase the number of engineered MSCs residing within a tumor available to deliver therapeutic agents. Using bioluminescence as an index of the relative size of the intratumoral MSC population, we found that MSCs expand beginning 3 days after infusion. Our histologic observations suggest the increase is due to in situ proliferation of MSCs that had previously trafficked to the spleen, in contrast to slow, ongoing localization; however, generating unambiguous evidence of proliferation is challenging on the background of a highly proliferating tumor.
MSCs have been reported to stimulate an innate immune response in vivo and in vitro [49,50]. Moreover, macrophage-mediated rejection of non-self (e.g., third-party) MSCs is primarily via phagocytosis [51]. Both observations are consistent with our findings. In addition to macrophages, NK cells are important constituents of innate immunity and can mediate rejection of non-self MSCs [52,53]. NK cells can recognize and lyse culture-expanded MSCs in vitro [54]. In our study, however, we did not find a deleterious effect of NK cells on MSC tumor trafficking. Thus, preserving NK cells while depleting macrophages will not only most effectively facilitate tumor localization of engineered MSCs but will also afford patients a level of innate immune protection during the time of transient macrophage depletion.
The significant limitation to our study is that our xenograft model, which developed by injecting human osteosarcoma cell line OS17 and human MSCs into B6 nude mice, by necessity, lacks host T cells. In addition to recognition of pathogens, T cells reject alloantigen-bearing cells. Although allogeneic MSCs are thought to stimulate a T-cell response [20,55,56], the primary immune response requires presentation of alloantigens by dendritic cells or other professional antigen-presenting cells and approximately 7 days to emerge. Given that human MSCs seem to localize to tumors relatively quickly, a primary T-cell response abrogating the delivery of engineered human MSCs to the tumor seems unlikely. In addition, liposomal clodronate depletes not only macrophages but also dendritic cells, which may limit the development of an alloimmune response, allowing repeated infusions of cells, if desired. Obviously, autologous MSCs could be used to obviate a concern for T-cell-mediated rejection, but such a strategy could complicate the clinical translation of our observations. Moreover, autologous cells would not likely overcome macrophage clearance of engineered MSCs, as this innate response is HLA-independent and is more likely a response to the non-physiologic setting of intravenously infused MSCs.
The next phase of this work is to address the role of T cells in fostering or hampering the delivery of engineered MSCs to the tumor microenvironment. In addition, we will strategically select anti-tumor agents for expression in MSCs in an effort to exploit the biological characteristics of MSC localization within a tumor as well as the specific Achilles’ heel of a designated solid tumor.
Conclusions
Our findings establish splenic macrophages as one of the principal mediators of the first pass effect that limits MSC localization to solid tumors and, possibly, other tissues in different applications. Our data suggest that a brief course of liposomal clodronate can transiently deplete splenic macrophages, leading to substantially greater MSC localization in a tumor. This strategy for delivering high levels of anti-cancer agents directly to solid tumors, obviating systemic toxicity, will be explored.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the technical assistance provided by the Flow Cytometry Core and Animal Research Center at Nationwide Children’s Hospital.
Funding
This work was supported by the CancerFree Kids Foundation, the Families for a Cure Foundation, The Research Institute at Nationwide Children’s Hospital and the Aflac Cancer & Blood Disorders Center.
Footnotes
Declaration of Competing Interest
The authors have no commercial, proprietary, or financial interest in the products or companies described in this article.
Supplementary materials
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.jcyt.2020.04.102.
References
- [1].D’Souza N, Rossignoli F, Golinelli G, et al. Mesenchymal stem/stromal cells as a delivery platform in cell and gene therapies. BMC medicine 2015;13:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999;284:143–7. [DOI] [PubMed] [Google Scholar]
- [3].Spaeth E, Klopp A, Dembinski J, et al. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene therapy 2008;15:730–8. [DOI] [PubMed] [Google Scholar]
- [4].Hofstetter CP, Schwarz EJ, Hess D, et al. Marrow stromal cells form guiding strands in the injured spinal cord and promote recovery. Proceedings of the National Academy of Sciences of the United States of America 2002;99:2199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer research 2005;65:3307–18. [DOI] [PubMed] [Google Scholar]
- [6].Studeny M, Marini FC, Dembinski JL, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. Journal of the National Cancer Institute 2004;96:1593–603. [DOI] [PubMed] [Google Scholar]
- [7].Wang S, Matuskova M, Bohovic R, et al. Genetically engineered mesenchymal stromal cells producing TNFα have tumour suppressing effect on human melanoma xenograft. J Gene Med 2015;17:54–67. [DOI] [PubMed] [Google Scholar]
- [8].Shah K Mesenchymal stem cells engineered for cancer therapy. Advanced drug delivery reviews 2012;64:739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dembinski JL, Wilson SM, Spaeth EL, et al. Tumor stroma engraftment of gene-modified mesenchymal stem cells as anti-tumor therapy against ovarian cancer. Cytotherapy 2013;15:20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Xu F, Shi J, Yu B, et al. Chemokines mediate mesenchymal stem cell migration toward gliomas in vitro. Oncol Rep 2010;23:1561–7. [DOI] [PubMed] [Google Scholar]
- [11].Studeny M, Marini FC, Champlin RE, et al. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer research 2002;62:3603–8. [PubMed] [Google Scholar]
- [12].Nitzsche F, Muller C, Lukomska B, et al. Concise Review: MSC Adhesion Cascade—Insights into Homing and Transendothelial Migration. Stem Cells 2017;35:1446–60. [DOI] [PubMed] [Google Scholar]
- [13].Waterman RS, Tomchuck SL, Henkle SL, et al. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an immunosuppressive MSC2 phenotype. PLoS One 2010;5:e10088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gao J, Dennis JE, Muzic RF, et al. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs 2001;169:12–20. [DOI] [PubMed] [Google Scholar]
- [15].Prockop DJ. Repair of tissues by adult stem/progenitor cells (MSCs): controversies, myths, and changing paradigms. Molecular therapy: the journal of the American Society of Gene Therapy 2009;17:939–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ankrum JA, Ong JF, Karp JM. Mesenchymal stem cells: immune evasive, not immune privileged. Nature biotechnology 2014;32:252–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009;27:2614–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Toma C, Wagner WR, Bowry S, et al. Fate of culture-expanded mesenchymal stem cells in the microvasculature: in vivo observations of cell kinetics. Circ Res 2009;104:398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fischer UM, Harting MT, Jimenez F, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem cells and development 2009;18:683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zangi L, Margalit R, Reich-Zeliger S, et al. Direct imaging of immune rejection and memory induction by allogeneic mesenchymal stromal cells. Stem Cells 2009;27:2865–74. [DOI] [PubMed] [Google Scholar]
- [21].Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. Journal of immunological methods 1994;174:83–93. [DOI] [PubMed] [Google Scholar]
- [22].Otsuru S, Gordon PL, Shimono K, et al. Transplanted bone marrow mononuclear cells and MSCs impart clinical benefit to children with osteogenesis imperfecta through different mechanisms. Blood 2012;120:1933–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006;8:315–7. [DOI] [PubMed] [Google Scholar]
- [24].Carroll JL, Nielsen LL, Pruett SB, et al. The role of natural killer cells in adenovirus-mediated p53 gene therapy. Molecular cancer therapeutics 2001;1:49–60. [PubMed] [Google Scholar]
- [25].Aichele P, Zinke J, Grode L, et al. Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. J Immunol 2003;171:1148–55. [DOI] [PubMed] [Google Scholar]
- [26].Fink K, Ng C, Nkenfou C, et al. Depletion of macrophages in mice results in higher dengue virus titers and highlights the role of macrophages for virus control. European journal of immunology 2009;39:2809–21. [DOI] [PubMed] [Google Scholar]
- [27].Braza F, Dirou S, Forest V, et al. Mesenchymal Stem Cells Induce Suppressive Macrophages Through Phagocytosis in a Mouse Model of Asthma. Stem Cells 2016;34:1836–45. [DOI] [PubMed] [Google Scholar]
- [28].Weiss ARR, Dahlke MH. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Frontiers in immunology 2019;10:1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mao F, Kang JJ, Cai X, et al. Crosstalk between mesenchymal stem cells and macrophages in inflammatory bowel disease and associated colorectal cancer. Contemporary oncology 2017;21:91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Carty F, Mahon BP, English K. The influence of macrophages on mesenchymal stromal cell therapy: passive or aggressive agents? Clinical and experimental immunology 2017;188:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chiossone L, Conte R, Spaggiari GM, et al. Mesenchymal Stromal Cells Induce Peculiar Alternatively Activated Macrophages Capable of Dampening Both Innate and Adaptive Immune Responses. Stem Cells 2016;34:1909–21. [DOI] [PubMed] [Google Scholar]
- [32].Luz-Crawford P, Djouad F, Toupet K, et al. Mesenchymal Stem Cell-Derived Interleukin 1 Receptor Antagonist Promotes Macrophage Polarization and Inhibits B Cell Differentiation. Stem Cells 2016;34:483–92. [DOI] [PubMed] [Google Scholar]
- [33].Dayan V, Yannarelli G, Billia F, et al. Mesenchymal stromal cells mediate a switch to alternatively activated monocytes/macrophages after acute myocardial infarction. Basic research in cardiology 2011;106:1299–310. [DOI] [PubMed] [Google Scholar]
- [34].Shen B, Liu J, Zhang F, et al. CCR2 Positive Exosome Released by Mesenchymal Stem Cells Suppresses Macrophage Functions and Alleviates Ischemia/Reperfusion-Induced Renal Injury. Stem cells international 2016;2016:1240301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ortiz LA, Dutreil M, Fattman C, et al. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proceedings of the National Academy of Sciences of the United States of America 2007;104:11002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ko JH, Lee HJ, Jeong HJ, et al. Mesenchymal stem/stromal cells precondition lung monocytes/macrophages to produce tolerance against allo- and autoimmunity in the eye. Proceedings of the National Academy of Sciences of the United States of America 2016;113:158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Li Q, Sun W, Wang X, et al. Skin-Derived Mesenchymal Stem Cells Alleviate Atherosclerosis via Modulating Macrophage Function. Stem cells translational medicine 2015;4:1294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang C, Chen J, Sun L, et al. TGF-beta signaling-dependent alleviation of dextran sulfate sodium-induced colitis by mesenchymal stem cell transplantation. Molecular biology reports 2014;41:4977–83. [DOI] [PubMed] [Google Scholar]
- [39].Simovic Markovic B, Nikolic A, Gazdic M, et al. Pharmacological Inhibition of Gal-3 in Mesenchymal Stem Cells Enhances Their Capacity to Promote Alternative Activation of Macrophages in Dextran Sulphate Sodium-Induced Colitis. Stem cells international 2016;2016:2640746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].de Witte SFH, Luk F, Sierra Parraga JM, et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells 2018;36:602–15. [DOI] [PubMed] [Google Scholar]
- [41].Galleu A, Riffo-Vasquez Y, Trento C, et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci Transl Med 2017;9: eaam7828. [DOI] [PubMed] [Google Scholar]
- [42].Gavin C, Meinke S, Heldring N, et al. The Complement System Is Essential for the Phagocytosis of Mesenchymal Stromal Cells by Monocytes. Frontiers in immunology 2019;10:2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Khakoo AY, Pati S, Anderson SA, et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. The Journal of experimental medicine 2006;203:1235–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nemeth K, Leelahavanichkul A, Yuen PS, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nature medicine 2009;15:42–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rivera-Cruz CM, Shearer JJ, Figueiredo Neto M, et al. The Immunomodulatory Effects of Mesenchymal Stem Cell Polarization within the Tumor Microenvironment Niche. Stem cells international 2017;2017:4015039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Almeida-Porada G, Atala AJ, Porada CD. Therapeutic Mesenchymal Stromal Cells for Immunotherapy and for Gene and Drug Delivery. Molecular therapy Methods & clinical development 2020;16:204–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Relation T, Yi T, Guess AJ, et al. Intratumoral Delivery of Interferongamma-Secreting Mesenchymal Stromal Cells Repolarizes Tumor-Associated Macrophages and Suppresses Neuroblastoma Proliferation In Vivo. Stem Cells 2018;36:915–24. [DOI] [PubMed] [Google Scholar]
- [48].Yagi H, Kitagawa Y. The role of mesenchymal stem cells in cancer development. Frontiers in genetics 2013;4:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li J, Ezzelarab MB, Cooper DK. Do mesenchymal stem cells function across species barriers? Relevance for xenotransplantation. Xenotransplantation 2012;19: 273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nature reviews Immunology 2008;8:726–36. [DOI] [PubMed] [Google Scholar]
- [51].Liu W, Xiao X, Demirci G, et al. Innate NK cells and macrophages recognize and reject allogeneic nonself in vivo via different mechanisms. J Immunol 2012;188:2703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Spaggiari GM, Capobianco A, Becchetti S, et al. Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood 2006;107:1484–90. [DOI] [PubMed] [Google Scholar]
- [53].Leibacher J, Henschler R. Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem cell research & therapy 2016;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Crop MJ, Korevaar SS, de Kuiper R, et al. Human mesenchymal stem cells are susceptible to lysis by CD8(+) T cells and NK cells. Cell transplantation 2011;20:1547–59. [DOI] [PubMed] [Google Scholar]
- [55].Nauta AJ, Westerhuis G, Kruisselbrink AB, et al. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood 2006;108:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Eliopoulos N, Stagg J, Lejeune L, et al. Allogeneic marrow stromal cells are immune rejected by MHC class I- and class II-mismatched recipient mice. Blood 2005;106:4057–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.