

Abstract
Chimeric antigen receptor T cells (CAR-T cells) are engineered recombinant T cells, which were initially used to treat hematopoietic malignancies and are now widely used in the treatment of various diseases. Considering their intrinsic targeting efficiency, CAR-T cells show considerable potential in the treatment of autoimmune diseases. Furthermore, regulatory T cells (Treg), a subset of CD4 T cells exhibiting immunosuppressive functions, have attracted increasing attention regarding CAR-Treg cell production. In this review, we report on recent developments in preclinical and clinical studies on CAR-T cells in autoimmune diseases and provide an outlook on opportunities and challenges of CAR-T application in such diseases.
Keywords: Chimeric antigen receptor T cells, Cell immunotherapy, Autoimmune diseases
INTRODUCTION
Systemic autoimmune diseases are characterized by the production of autoantibodies and autoreactive T lymphocytes, which attack the body’s own organs (Figure 1). Autoimmune diseases suggest a breakdown of the body’s immune tolerance, but the causes are unknown (Rose, 2016; Wang et al., 2015). In terms of immune mechanisms, autoimmune diseases fall into two main categories: autoantibodies that destroy cells by binding to the cell surface and autoreactive cytotoxic T lymphocytes (CTLs) that recognize target cells through the formation of T cell receptor (TCR)-major histocompatibility complex class I (MHC-I) complexes (Wahren-Herlenius & Dörner, 2013). Clinical treatments for autoimmune diseases mainly rely on the use of immunosuppressants, which not only exhibit low specificity and potentially cause long-term infection but are generally unable to eliminate the source of the disease (Durcan et al., 2019; Schmidt, 2018; Wang et al., 2015). Fortunately, chimeric antigen receptor T (CAR-T) cells have emerged as a novel therapeutic strategy for autoimmune diseases.
Figure 1.

Mechanism of autoimmune diseases
Autoimmune diseases are characterized by an excessive host immune response, and are influenced by age, sex, region, genetics, infection, nutrition, environment, stress, and other factors (Wang et al., 2015). These diseases are also typically characterized by the presence of self-reactive B cells and T cells in the body (Wang et al., 2015). Tregs are natural immune regulatory cells that reduce the killing activity of T cells, establish immune surveillance, and prevent T cells from overreacting (Sakaguchi et al., 2008). Autoreactive CTLs recognize target cells by the binding of TCRs to a combination of MHC-I and autoantigen-derived peptides (Carreño et al., 2006). Subsequently, CTLs kill target cells by various mechanisms, leading to autoimmune disease (Halle et al., 2017). Autoantibodies can bind to cell surface markers, lyse cells, activate and block receptors, and form immune complexes with antibodies to deposit in blood vessels or tissues (Wang et al., 2015). Autoantibodies can mediate organ-specific or systemic damage through a variety of mechanisms, resulting in autoimmune disorders (Davidson & Diamond, 2001). CTL: Cytotoxic T lymphocyte; TCR: T cell receptor; FasL: Fas ligand; ADCC: Antibody-dependent cell-mediated cytotoxicity; Tregs: Regulatory T cells.
Chimeric antigen receptors (CARs) typically consist of a high-affinity antigen binding site on a monoclonal antibody (single chain fragment variant (scFv)) and a T cell-activating signal transduction domain, which allows them to recognize MHC-free antigens (Ramos & Dotti, 2011). Thus far, CARs have gone through four generations (Figure 2). First-generation CARs combined the scFV domains and CD4 (extracellular domain) with CD3ζ (intracellular domain), resulting in poor stability and clinical efficacy; second-generation CARs added additional co-stimulatory signaling domains, such as CD28 or 4-1BB, to CD3ζ, leading to enhanced in vivo expansion and persistence; and third- and fourth-generation CARs combined other co-stimulatory domains to exhibit different characteristics (June & Sadelain, 2018; Zhang et al., 2020).
Figure 2.
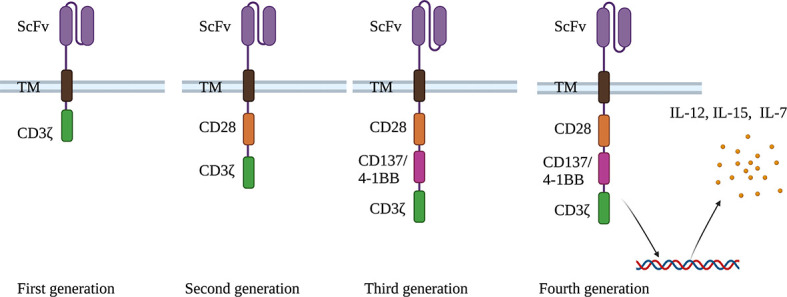
Four generations of CARs
First-generation CARs contain activation domain CDζ; Second-generation CARs contain an added co-stimulatory domain (CD28); Third-generation CARs contain another added co-stimulatory domain; and Fourth-generation CARs allow CAR-T cells to express certain cytokines, e.g., IL-12, IL-15, and IL-7, which significantly enhance T cell expansion. ScFv: Single chain antibody fragment; TM: Transmembrane domain; CD: Cluster of differentiation; IL: Interleukin.
Engineered CARs expressed in T cells or regulatory T (Treg) cells are called CAR-T and CAR-Treg cells, respectively. Methods for generating these kinds of T cells are similar. Briefly, the T cells or Treg cells are isolated from the blood of patients, then transduced with an overexpression vector encoding a synthetic receptor through viral or non-viral approaches (Zhang et al., 2020). Before injection into patients, CAR-Ts or CAR-Tregs need to be expanded to a specific number. To drive CAR-T cell expansion, the interleukin 2 (IL-2) cytokine and anti-CD3 antibodies are usually used by the cell processing centers (Makita et al., 2017). For Tregs, cell source is a major factor determining experimental success or failure. For example, Tregs directly induced by MHC peptides from the thymus (i.e., tTregs) are considered safer than peripheral Tregs (pTregs). Among tTregs, CD45RA+ cells are considered safer than CD45RO+ cells because CD45RO+ cells may contain pTregs or effector T cells, which can expand quickly in vitro, but also exhibit high cytotoxicity rather than immunosuppression (Rosado-Sánchez & Levings, 2020).
As a highly promising approach in cancer therapy, CAR-T cells play an important role in the treatment of hematological malignancies (Yu et al., 2019) and are now being investigated in solid tumors (Martinez & Moon, 2019) and autoimmune diseases. Previous studies have mainly focused on the therapeutic efficacy of CD8+ T cells (Maus et al., 2014). However, CD4+ T cells, another subset of T cells in peripheral blood, have gained attention in CAR engineering (Zhang et al., 2020). After binding to the target antigen in the body, CAR-T cells undergo activation and proliferation, and then become cytotoxic and finally kill the target cells (Sadelain et al., 2013). As a living drug, CAR-T cells show good targeting, persistence, and clinical activity, and persist in the peripheral blood of patients with B cell acute lymphoblastic leukemia (B-ALL), diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), and multiple myeloma (MM) for at least 3–6 months (Abramson, 2020; Dai et al., 2020; Finney et al., 2019; Majzner & Mackall, 2019; Raje et al., 2019; Turtle et al., 2016). Furthermore, 30%–40% of patients appear to achieve long-term remission, with few relapses occurring beyond 12 months, indicating a potential cure (Kersten et al., 2020). Although cytokine release syndrome (CRS) and neurological toxicity have been observed in patients with refractory B-cell lymphoma when treated with CD19-CAR-T cells (Acharya et al., 2019; Neelapu et al., 2017), clinical trials have also shown encouraging activity and disease relief (Abramson, 2020; Brudno et al., 2020; Raje et al., 2019; Schuster et al., 2017). Furthermore, such adverse reactions can be decreased through dosage reduction, steroid therapy, and antibody introduction (especially anti-IL-6R antibodies) (Acharya et al., 2019; Hartmann et al., 2017). Currently, five CAR-T cell therapies have been approved, including anti-CD19-CAR-T cells for lymphoma treatment (Abramson, 2020; Sermer & Brentjens, 2019) and anti-B cell maturation antigen (BCMA)-CAR-T cells for MM treatment (D’Agostino & Raje, 2020). Several phase I clinical trials of BCMA/CD19-CAR-T cells have been reported in refractory autoimmune diseases (Table 1), which may become a new therapeutic option. In addition, specifically designed CAR-T cells are showing better potency in treating autoimmune diseases than traditional ones, which we discuss in this review.
Table 1. Clinical trials of CAR-T or CAR-Treg cell treatment for autoimmune diseases.
| Specific autoantigen | Intervention | Disease | Phase | Status | Completion date | NCT ID |
| BCMA: B cell maturation antigen; CAR-T: Chimeric antigen receptor T cell; Dsg3: Desmosomal core glycoprotein-3. | ||||||
| BCMA
(CD269) |
Descartes-08 CAR-T | Myasthenia Gravis (MG) | I&II | Recruiting | 1 April 2022 | NCT04146051 |
| CT103A cell | Neuromyelitis optica spectrum disorder (NMOSD) | I | Recruiting | 31 December 2023 | NCT04561557 | |
| BCMA
& CD19 |
CD19/BCMA CAR T | Sjogren’s syndrome (SS) | I | Recruiting | 5 November 2024 | NCT05085431 |
| CD19/BCMA CAR T | Scleroderma | I | Recruiting | 8 October 2024 | NCT05085444 | |
| CD19 / BCMA CAR T | Immune Nephritis | I | Recruiting | 5 November 2024 | NCT05085418 | |
| CD19/BCMA CAR T | Systemic Lupus Erythematosus (SLE) | I | Recruiting | 10 September 2022 | NCT05030779 | |
| CD19 | Anti-CD19 CAR-T | Systemic Lupus Erythematosus (SLE) | I | Unknown | March 2018 | NCT03030976 |
| Anti-Dsg3 surface immunoglobulin | DSG3-CAAR T | Mucosal-dominant PV (mPV) | I | Recruiting | September 2026 | NCT04422912 |
CAR technology has now been extended to CAR-Treg therapy. Tregs are a subset of CD4+ T cells with immunosuppressive functions and include tTregs (directly induced by MHC peptides from the thymus), pTregs (stimulated by antigens in peripheral lymphoid tissue), and iTregs (induced in vitro). Tregs are characterized by the co-expression of CD25 and Foxp3. Foxp3 plays an important role in the function of Treg cells, and its mutation can lead to serious autoimmune diseases (Göschl et al., 2019). Treg cell-based therapy has been applied in several preclinical and clinical trials, such as graft versus-host disease (GvHD) in non-cytotoxic conditions and type I diabetes mellitus (T1D) to prolong islet survival time. However, traditional methods for amplifying Treg or gene-edited TCR-Treg cells are inefficient and MHC dependent. In addition, the number of cells required for infusion is very large. Moreover, non-specific immunosuppression may occur during injection. Therefore, CAR-Treg cells are gradually being developed to play a safer and more effective therapeutic role (Zhang et al., 2018). First, CAR-Treg cells can recognize target antigen on target cells through the CAR structure and inhibit effector T cell function. For example, Fas ligands (FasL) on Treg cells can bind to Fas on effector T cells to induce apoptosis. CTLA-4, another membrane protein, can competitively interact with antigen-presenting cells (APCs) to inhibit CD28-mediated T cell activation. On the other hand, IL-2 plays an important role in the proliferation and differentiation of effector T cells. However, CAR-Treg cells can competitively bind to IL-2 and inhibit effector T cell proliferation (Yamaguchi et al., 2011). CAR-Treg cells have been successfully applied in colitis, multiple sclerosis (MS), and T1D. Carcinoembryonic antigen (CEA) CAR-Tregs (SCA431, scFv, CD28, and CD3ζ) have been used in colitis treatment, with effective results (Blat et al., 2014). Myelin oligodendrocyte glycoprotein (MOG) CAR-Tregs (MOG scFv, CD28, and CD3ζ) have been used in MS treatment, showing effective control of inflammation (Fransson et al., 2012). CAR-Treg cells (insulin scFv, CD28, and CD3ζ) have also been used in T1D, showing successful treatment with no effects on recipient immunity (Tenspolde et al., 2019). Other potential clinical applications include treatment for GVHD, hemophilia A, and asthma. For example, HLA-A2 CAR-Tregs may be effective in preventing GvHD (MacDonald et al., 2016). Compared with polyclonal Tregs or TCR-Tregs, CAR-Tregs are MHC independent, with high target site specificity, low non-specific immunosuppression, and low sensitivity to IL-2 concentration.
In this article, we analyze the use of CAR-T and CAR-Treg cells in autoimmune diseases and discuss future prospects for these diseases (Table 2).
Table 2. Summary of therapeutic targets of CAR-T and CAR-Treg cells and established animal models for studies on autoimmune diseases.
| CAR | Disease | Target | Animal model | CAR reference |
| CAR-T: Chimeric antigen receptor T cell; CAR-Treg: Chimeric antigen receptor regulatory T cell; SLE: Systemic lupus erythematosus; T1D: Type 1 diabetes; MS: Multiple sclerosis; RA: Rheumatoid arthritis; PV: Pemphigus; TNP: 2,4,6-trinitrophenol; TPCR: Tripartite chimeric receptor; TNBS: 2,4,6-trinitrobenzenesulphonic acid; CEA: Carcinoembryonic antigen; DSS: Dextran sulfate sodium; AOM: Azoxymethane; MOG: Myelin oligodendrocyte glycoprotein; EAE: Encephalomyelitis; MBP: Myelin basic protein; CFA: Freund’s adjuvant; PT: Pertussis toxin; FITC: Fluorescein isothiocyanate; Dsg3: Desmosomal core glycoprotein-3; CAAR-T: Chimeric autoantibody receptor T cells. | ||||
| Anti-CD19 CAR-T | SLE | CD19 | MRL/lpr mice | Kansal et al., 2019;
Jin et al., 2021 |
| NZB×NZW F1 mice | Kansal et al., 2019 | |||
| Insulin/IGPR-reactive CAR-T | T1D | Insulin/IGPR-reactive TCR | NOD mice | Fishman et al., 2017 |
| mAb287-CAR-T | T1D | I-Ag7-B:9-23 (R3) antigen complex | NOD mice | Zhang et al., 2019 |
| Insulin-specific CAR-Treg | T1D | Insulin | NOD mice | Tenspolde et al., 2019 |
| TNP-TPCR Treg | Colitis | TNP | Hapten-mediated colitis mice | Elinav et al., 2009 |
| CEA-CAR-Treg | Colitis and colorectal cancer | CEA | DSS-induced colitis mice,
AOM and DSS-induced colorectal cancer mice |
Blat et al., 2014 |
| CARαMOG-FoxP3-Treg | MS | MOG | Chronic EAE mice | Fransson et al., 2012 |
| MBP-CAR Treg and MOG-CAR Treg | MS | MBP and MOG | Chronic EAE mice | De Paula Pohl et al., 2020 |
| Anti-FITC CAR-T | RA | Antigenic FITC-peptide | Collagen-induced arthritis (CIA) mice | Zhang et al., 2021 |
| Dsg3 CAAR-T | PV | Anti-Dsg3 surface immunoglobulin | Autoantigen-knockout mice
Passive transfer mice Skin graft mice |
Ellebrecht et al., 2016 |
SYSTEMIC LUPUS ERYTHEMATOSUS (SLE)
Introduction of disease
SLE is an autoimmune disease caused by the immune system mistakenly attacking healthy tissues in many parts of the body (Poole et al., 2009) and is characterized by red facial rashes (Tebbe & Orfanos, 1997). There is currently no cure for SLE, but conventional treatments include non-steroidal anti-inflammatory drugs, corticosteroids, immunosuppressants, hydroxychloroquine, and methotrexate (Murphy & Isenberg, 2013). As SLE significantly increases the risk of cardiovascular diseases, patients often die from SLE, and their life expectancy is usually short (Murphy & Isenberg, 2013).
SLE is caused by genetic susceptibility and environmental triggers (He & Sawalha, 2018; Yang et al., 2009). From the genetic perspective, multiple genes can affect the incidence rate of SLE. Studies have shown that HLA class I, class II, and class III genes are related to SLE, and genes such as IRF5, PTPN22, and STAT4 are also involved in disease progression (Yang et al., 2009). From an environmental perspective, lack of vitamin D as well as the use of certain drugs, such as procainamide, isoniazid, hydralazine, quinidine, and phenytoin, may induce lupus (He & Sawalha, 2018).
Application of CAR-T and CAR-Treg in SLE
Although CAR-T has been successfully applied in SLE animal models, CAR-Tregs remain relatively rare, but both show good prospects. For example, CAR-T therapy targeting CD19+ B cells in MRL-lpr mice can reverse diseased organ damage and prolong survival time with reduction in autoimmune antibody production (Kansal et al., 2019). In addition, after transfusing CD8+ T cells transduced with relevant A-MLV retrovirus, CD19 gene probes have proven that CAR-T can effectively treat mice with SLE, resulting in reduced CD19 gene expression in the spleen and extended lifespan in MRL-lpr mice. Similarly, researchers have used CAR-T in New Zealand black×New Zealand white (NZB×NZW) F1 mice by isolating, purifying, and modifying CD8+ T cells to eliminate potential disease enhancement of CD4+ T cells (Kansal et al., 2019). Specifically, CD19-targeted CAR-T plasmids are transduced into splenic CD8+ T cells through different viral transduction methods, then infused into mice with obvious SLE symptoms, resulting in CD19+ B cell hypoplasia, thus suggesting effective treatment (Figure 3) (Kansal et al., 2019). Jin et al. (2021) also infused MRL-lpr mice with the same anti-mouse CD19 CAR-T cells and tested different treatment strategies, e.g., CAR-T and monoclonal antibody therapies and different second-generation CAR-T cells, by adopting different co-stimulatory motifs (e.g., CD28 and 4-1BB) in the intracellular domain of CAR-T cells. For the former, they found that infusion of anti-CD19 CAR-T cells showed more sustained B cell depletion in MRL-lpr mice; for the latter, they found that CAR-T cells with 4-1BB showed better treatment effects. They also found that pretreating mice with low-dose total body irradiation (TBI) greatly improved the survival rate (Figure 3). Recently, CAR-T therapy targeting CD19 molecules on B cells was used for a female patient with severe SLE, who had failed to respond to various treatments (Mougiakakos et al., 2021). Results showed that 44 days after CAR-T cell infusion, autoimmune antibodies were undetectable and the disease condition was alleviated, with no obvious side effects (Figure 3). For CAR-Treg cells, the immune environment in SLE patients can significantly reduce the number and inhibitory function of Treg cells, and thus disrupt immune homeostasis (Kim et al., 2015). First, the large amount of IL-6 and interferon (IFN)-α secreted by dendritic cells (DCs) can hinder the inhibitory function of Treg cells, and IL-21 secreted by helper CD4+ T cells can also affect the survival and function of Treg cells (Guo et al., 2019). Second, the strong pro-inflammatory environment in SLE patients can also transform Treg cells to secrete IL-17 and other inflammatory cytokines. As CAR-Treg itself has monoclonal resistance, restoring the number of Treg cells and related inhibitory functions through CAR-Treg can effectively prevent autoimmune diseases (Haddadi et al., 2020). At the same time, SLE can induce many autoantibodies (~180), suggesting many autoantigens can be targeted, including nuclear antigens, cytoplasmic antigens, cell membrane antigens, phospholipid-related antigens, blood cells, endothelial cells, and nervous system antigens (Yaniv et al., 2015). Autologous Treg infusion can activate Tregs in inflamed skin, thus weakening the IFN-γ pathway and CD4+ effector cell infiltration (Dall’Era et al., 2019). Low-dose IL-2 treatment of 60 patients with active SLE was shown to be effective and restored tolerance (He et al., 2020). From these findings, CAR-Treg therapy for skin and kidney treatments could restore immune tolerance and reduce inflammation within the affected tissues.
Figure 3.
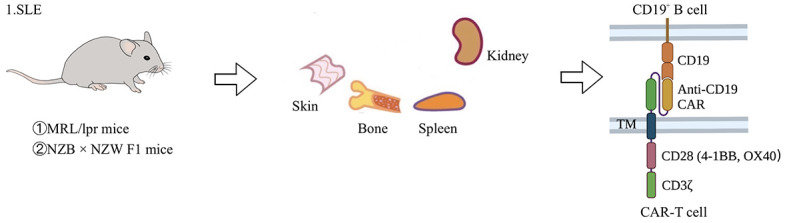
Application of CAR-T cells in animal model of SLE
SLE is an autoimmune disease caused by the immune system mistakenly attacking healthy tissues in many parts of the body. Currently, gene-edited T cells have been used in two mouse models: MRL/lpr mice and NZB×NZW F1 mice. MRL/lpr mice are produced by mating LG/J, AKR/J, C3H/HeDi, and C57BL/6J mouse strains, which are characterized by accumulation of double negative (CD4-CD8-) B220+ T cells (Murphy & Roths, 1978). NZB×NZW F1 mice are produced by crossing NZB and NZW mice (Helyer & Howie, 1963). In this animal model, anti-RNA-related antibodies are rarely produced and SLE pathogenesis is mainly due to suppression of Treg cells through its own MHC (Helyer & Howie, 1963). Treatment is to inhibit CD19+ B cells by CAR-T cells specifically targeting CD19, thereby curing SLE. SLE: Systemic lupus erythematosus; 4-1BB: Tumor necrosis factor receptor superfamily member 9; OX40: Tumor necrosis factor receptor superfamily member 4; lpr: Lymphoid proliferation gene; NZB: New Zealand black mouse; NZW: New Zealand white mouse; MHC: Major histocompatibility complex.
Currently, two phase I clinical studies are being carried out in China. One study at the recruitment stage (NCT05030779) aims to assess the safety and effectiveness of CD19/BCMA CAR-T cells in the treatment of refractory SLE. The second study (NCT03030976) aims to assess the safety and efficacy of second-generation CAR-T cells with an optimized hinge and transmembrane domain in the treatment of SLE patients, although the results are not yet known.
Of course, CAR-T treatment for SLE has several unique advantages and limitations. For its advantages, CAR-T can eliminate CD19+ B cells in a targeted manner, reduce the production of autoimmune antibodies, and show persistence in elimination functions. However, researchers have also identified a subgroup of B cells with lower IgM levels (IgMlo) in CAR-T-treated MRL-lpr mice that do not express CD19 targets (Kansal et al., 2019), with the CAR-T-treated MRL-lpr mice still retaining antibody-producing cells and showing incomplete disease treatment. In addition, control (TBI+phosphate-buffered saline) and TBI+CAR-T-treated mice show no significant differences in their concentrations of certain antibodies, such as serum anti-dsDNA and anti-nuclear antibodies (Jin et al., 2021). The non-reduction of these antibodies is a considerable issue in the treatment of SLE.
TYPE 1 DIABETES (T1D)
Introduction of disease
Also known as juvenile diabetes, T1D is characterized by patients producing little or no insulin from the β cells of the islets of Langerhans in the pancreas (Bluestone et al., 2010). Typical symptoms include frequent urination, thirst, increased hunger, and weight loss (Torpy et al., 2007), and potentially blurred vision, fatigue, and slow wound healing (Toni et al., 2017).
T1D is caused by the destruction of β cells (the only insulin-producing cells in the body) and subsequent progressive lack of insulin (Bluestone et al., 2010). In 70%–90% of cases, β cells are destroyed by one’s own immune system, while the specific reason for this is not fully understood (Katsarou et al., 2017). The remaining 10%–30% of patients show β cell destruction but no signs of autoimmune disease, and thus are diagnosed with idiopathic diabetes (Katsarou et al., 2017). At present, various theories have been espoused to explain the mechanisms of T1D, including genetic susceptibility with familial tendencies and environmental factors, e.g., maternal obesity, caesarean birth, and high dietary sugar intake (Norris et al., 2020).
Application of CAR-T and CAR-Treg in T1D
To date, CAR treatment for T1D has primarily focused on CAR-T, with some limited application of CAR-Treg. Notably, Treg cells can inhibit autoreactive T cells, which is essential for maintaining immune tolerance and homeostasis of the immune system, and even impacts the sex differences found in autoimmune diseases (Nie et al., 2015).
For CAR-T, researchers have redirected genetically modified T cells to pathogenic CD8+ T cells using MHC as an activation receptor (Fishman et al., 2017). Based on the conclusion that the signaling component of MHC-I molecules is the fusion product of b2 microglobulin (b2m) and CD3-ζ chain, studies have combined H-2Kd with the insulin B chain (amino acids 15–23 (InsB15-23)) and linked it to the N-terminus of the b2m/CD3ζ daughter chain. This design forms a specific MHC activation receptor, which specifically targets CD8+ T cells carrying TCRs with certain peptides and specifically inhibits insulin-responsive or pancreatic islet-specific glucose-6-phosphatase-related protein (IGRP)-responsive T cells. After using this CAR-T therapy, non-obese diabetic (NOD) mice are reported to show significant relief from insulitis and diabetes (Figure 4). Rather than targeting CD8+ T cells, Zhang et al. (2019) targeted the APC of CD4+ T cells in T1D NOD mice for CAR-T. This APC usually carries an I-Ag7-B:9-23 (R3) antigen complex, which consists of 9–23 residues of the insulin B chain (InsB9-23) bound to MHC class II molecules (I-Ag7), to stimulate the proliferation of CD4+ T cells. Thus, Zhang et al. (2019) established a monoclonal antibody (mAb287) against this I-Ag7-B:9-23 (R3) antigen complex for use as the extracellular domain of CAR-T, resulting in cytotoxic effects on APC and delayed T1D onset in NOD mice (Figure 4).
Figure 4.
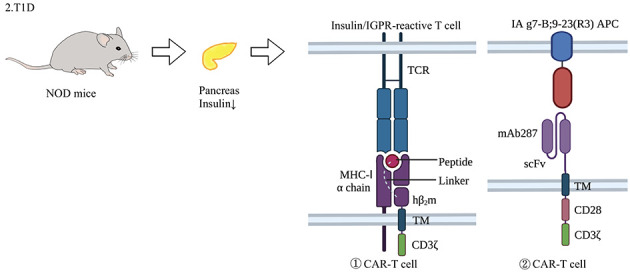
Application of CAR-T cells in animal model of type 1 diabetes
T1D is characterized by a progressive lack of insulin caused by the destruction of pancreatic β cells, leading to typical symptoms of diabetes. At present, the application of gene-edited T cells is limited to animal models such as NOD mice. NOD mice are established via inbreeding and selective breeding, resulting in pancreatic atrophy and fibrosis, decreased insulin secretion, and clinical symptoms resembling human diabetes (Makino et al., 1980). Various treatments have been established: e.g., construction of specific MHC activators by linking the H-2Kd and amino acid 15–23 (InsB15-23) complex of the insulin B chain peptide to the N-terminus of b2m/CD3ζ receptors, thereby targeting CD8+ T cells with specific peptides to inhibit insulin-responsive or IGRP-responsive T cells (Fishman et al., 2017); and establishing monoclonal antibodies (mAb287) against the I-Ag7-B:9-23 (R3) antigen complex for use as the extracellular domain of CAR-T to target CD4+T APCs (Zhang et al., 2019). TCR: T cell receptor; MHC-1α chain: Major histocompatibility complex-1α chain; mAb287: Monoclonal antibody287; scFv: Single chain Fv; NOD: Non-obese diabetic.
Before CAR-Treg was developed, cell therapy for T1D mainly focused on Treg cell re-infusion (Tang et al., 2004; Bluestone et al., 2015). However, T1D patients often present with a small number of CD4+CD25+ Tregs but a large expansion of autoreactive T cells. To solve this issue, researchers have long explored technologies for the rapid and massive proliferation of specific Tregs. For example, Tang et al. (2004) used a combination of anti-CD3, anti-CD28, and IL-2 antibodies to achieve 200 times purification of CD4+CD25+ Tregs in less than 2 weeks, with the amplified Tregs not only expressing classic cell surface antigens, but also inhibiting effector T cells in vitro and in vivo. They also infused related Treg cells into NOD mice, which showed effective reversal of the diabetic phenotype (Tang et al., 2004). In addition, Bluestone et al. (2015) reported on a phase I clinical trial that isolated and expanded Tregs from T1D patients and applied adoptive immunotherapy in 14 adult T1D patients. Their results showed that one year after infusion, Tregs remained at a quarter of the peak level and retained the FOXP3+CD4+CD25hiCD127lo phenotype, with no obvious clinical adverse reactions. Although the targeting of the above-mentioned Treg cells to self-reactive T cells is not yet clear, CAR-Treg therapy not only reduces the number of Treg cells required for an effective response, but also shows good specificity and low off-target effects. Tenspolde et al. (2019) first redirected T effector cells to Tregs through Foxp3 transduction, and then used CAR technology to direct CAR-Tregs to the pancreatic region of NOD mice. These transformed CAR-Tregs showed continuous inhibitory properties and pancreatic region specificity, with no apparent effects on overall immunity of the recipient.
However, CAR-Treg and CAR-T application also show limitations in current T1D treatment. For example, although Zhang et al. (2019) induced a delayed in T1D onset in NOD mice, treatment did not stop disease occurrence. In addition, Tenspolde et al. (2019) only found strong immunosuppressive function of CAR-Treg in vitro, not in live NOD mice. Therefore, it is necessary to discover further strategies to prolong and expand metastatic CAR-T cell activity. The stability of CAR-Treg cells is also worth attention as the transformation of CAR-Treg cells into effector T cells may further aggravate the disease. For example, research on mice has shown that, when exposed to inflammatory conditions, some Tregs will lose Foxp3 expression and undergo transformation, resulting in a pro-inflammatory phenotype (Zhou et al., 2009). In addition, disturbance in the balance between Treg and Th17 can induce immune disorders (Zhang et al., 2021b). Moreover, the separation and purification of CAR-T and CAR-Treg can be difficult as current clinical GMP reagents rarely reach 100% purity. Furthermore, our understanding of how CAR-T and CAR-Treg cells differentially proliferate and transport in the body remains incomplete. Therefore, CD8+ CAR-T cells that have not been isolated may expand and cause disease aggravation and possible adverse effects (Magnuson et al., 2015).
COLITIS
Introduction of disease
Colitis is a chronic inflammatory disease of the colon (Adams & Bornemann, 2013). The etiology of microscopic colitis is unknown, but is associated with autoimmune disorders, such as celiac disease, polyarthritis, and thyroid disorders (Langner et al., 2015). Exposure to medications, such as non-steroidal anti-inflammatory drugs, proton pump inhibitors, and selective serotonin reuptake inhibitors, is suspected to play a role in colitis, although their direct causal relationship has not been proven (Park et al., 2015).
Application of CAR-T and CAR-Treg in colitis
Application of 2,4,6-trinitrophenol (TNP)-tripartite chimeric receptor (TPCR) Tregs in treatment of colitis: Foxp3+ Tregs express the key transcription factor FOXP3, which plays a critical role in the maintenance of immune tolerance and homeostasis (Wang et al., 2021). Insufficient accumulation or impaired function of Treg cells in inflammatory sites is the main cause of a variety of autoimmune diseases: including autoimmune thyroiditis, gastritis, insulitis, arthritis, encephalomyelitis, and colitis (Maul et al., 2005). However, as direct Treg therapies may be inadequate for controlling autoimmunity and inflammation, strategies to induce or expand endogenous Treg cells in vivo may be a good option (Liang et al., 2021). In animal models, adoptive transfer of Treg cells can effectively prevent and improve autoimmune diseases caused by Treg cell abnormalities. As antigen-specific Treg cells are extremely rare, studies usually use adoptive transfer of a large number of non-specific Tregs or TCR-expressing Tregs to recognize specific MHC antigen peptide complexes isolated from transgenic mice (Chen et al., 2019). Using a transgenic method, Elinav et al. (2009) developed Treg cells expressing a chimeric receptor (CR) containing an antibody variable region specific for TNP, which fuses to the extracellular and transmembrane domains of CD28 co-stimulatory molecules and intracellular domain of stimulatory Fc-γ receptor chain (Figure 5). Transgenic mice expressing this receptor on all T cells are resistant to 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis (Elinav et al., 2008). For non-transgenic mice receiving a small amount of TPCR-expressing Treg cells, cells accumulate and activate in the inflamed colon. TNP-TPCR Tregs can cure acute experimental colitis via their antigen specificity, no MHC restriction, and co-stimulatory signal independence. In addition, TNBS has also been used to inhibit non-TNBS-induced colitis. Although, Tregs containing TNP-specific CR did not inhibit oxazolidone-induced colitis, the addition of trace amounts of TNBS cured oxazolidone-inflamed colons, thereby proving the “bystander effect” of TNBS (Elinav et al., 2008).
Figure 5.
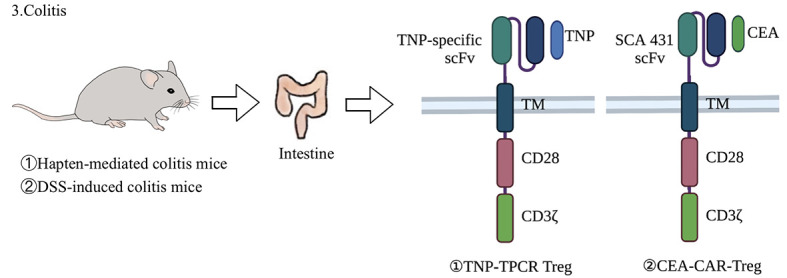
Application of CAR-Treg cells in animal model of colitis
Two CAR-Tregs have been applied in colitis. TNP-TPCR Tregs express a TNP-specific scFv, which is fused to the extracellular and transmembrane domain of CD28 co-stimulatory molecule and intracellular domain of stimulatory Fc-γ receptor chain. TNP-TPCR Tregs have been applied in hapten-mediated colitis mouse models, which are induced by TNBS or oxazolone (Elinav et al., 2008). CEA-CAR-Tregs express CEA-SCA431 CAR signaling domains fused with CD28-CD3ζ in CD4+CD25+ Treg cells. CEA-CAR-Tregs have been applied in dextran sulfate sodium (DSS)-induced colitis mouse models (Tanaka et al., 2003). TNP: 2,4,6-trinitrophenol; TPCR: Tripartite chimeric receptor; scFv: Single chain antibody fragment; CEA: Carcinoembryonic antigen.
To use TNP-TPCR Tregs in non-transgenic environments (such as in human diseases), Tregs must be transduced with a vector encoding CR, screened according to the expression level of TPCR, and finally amplified in vitro for clinical application (Elinav et al., 2009). Fontenot et al. (2003) transduced effector T cells with retrovirus or lentivirus encoding Foxp3 to partially obtain regulatory function. However, naturally occurring Tregs (nTregs) are highly resistant to retroviral and lentiviral vector transduction, which may hinder the pre-activation of Tregs, a necessary step for successful and effective retroviral transduction (Li et al., 2005) and thus clinical application. Li et al. (2005) described a pathway that can achieve efficient and reproducible retroviral transduction as well as selection and amplification of mouse nTregs, thus allowing rapid in vivo and in vitro amplification of antigen-specific nTregs that retain Foxp3 expression and inhibitory function. This enables TNP-TPCR Tregs to proliferate in an antigen-specific manner and accumulate in target organs for clinical treatment of colitis (Maldini et al., 2018).
TNP-TPCR Tregs have advantages over normal human Tregs. As mentioned above, their characteristic surface molecule (CR) is composed of an antibody variable region specific to TNP, extracellular and transmembrane domains of co-stimulatory molecule CD28, and intracellular domains of the stimulatory Fc-γ receptor chain. As such, TNP-TPCR Tregs exhibit antigen specificity, no MHC restriction, co-stimulatory signal independence, specific proliferation, good targeting, and rapid action. However, the resistance of nTregs to viral vector transduction is a challenge for TNP-TPCR Treg production, and a major problem for clinical application. In addition, adoptive transfer of engineered Treg cells contains some risks, e.g., the in vivo inflammatory environment may trigger the transformation of Treg cells into antigen-specific pathogenic effector T cells (McGovern et al., 2017). To solve this problem, techniques such as CRISPR can be used for gene editing. For example, Treg cells can be protected from inflammatory signals by editing cytokine receptors such as IL-6 and the tendency of Treg cells in autoimmune patients to produce inflammatory cytokines can be eliminated by removing IFN-γ or IL-17 genes from engineered Treg cells (Freen-van Heeren, 2021).
Application of CEA-CAR-Treg in treatment of colitis: Carcinoembryonic antigen (CEA)-CAR-Tregs are obtained by transducing specific CEA-SCA431 CAR signaling domains fused with CD28-CD3ζ in CD4+CD25+ Treg cells (Blat et al., 2014) (Figure 5), about 90% of which are Foxp3+. CEA-CAR-Tregs can effectively alleviate T cell metastatic colitis and inhibit the occurrence of colitis-related colorectal cancer induced by azoxymethane-dextran sodium sulfate (AOM-DSS) (Elinav et al., 2008). CEA-CAR-Tregs can reside and accumulate at sites that express CEAs, and mainly accumulate in the inflamed colon, with much lower concentrations found in the small intestine and other internal organs (Blat et al., 2014). They have many advantages, such as strong antigen specificity, no MHC restriction, specific proliferation, co-stimulatory signal independence, good targeting, and rapid action. As such, CEA-CAR-Tregs show great potential for improving ameliorated colitis and preventing colitis-associated colorectal cancer. However, CEA-CAR-Tregs have a short lifespan, accumulating and expanding in the colon for only about 7 days, then diminishing rapidly to undetectable levels at 9 days after injection (Zmievskaya et al., 2021). This sharp decline in signal intensity could be attributable to an immune response against potential epitopes on the CAR or against the luciferase reporter protein (Blat et al., 2014). Activation-induced cell death may also be responsible for the rather short longevity of CAR-Tregs in vivo. At present, their short lifespan has limited the clinical application of CEA-CAR-Tregs in the treatment of colitis, with no clinical trials yet reported.
MULTIPLE SCLEROSIS (MS)
Introduction of disease
MS is a chronic immune-mediated demyelinating disease of the central nervous system (Compston & Coles, 2008). Although its etiology remains elusive, environmental factors and susceptibility genes are involved in disease pathogenesis (Correale et al., 2017). MS is widely considered an autoimmune demyelinating disease, with autoimmune reactions by myelin-specific CD4+ T helper 1 (Th1) and Th17 cells (Sospedra & Martin, 2005). Results from immunological, genetic, and histopathological studies of MA patients further support the concept that autoimmunity plays a major role in the pathogenesis of MS (McFarland & Martin, 2007).
Application of CAR-T and CAR-Treg in MS
CAR treatments for MS have primarily focused on the transformation of CAR-Tregs. Fransson et al. (2012) used the lentiviral vector system to transduce CD4+ T cells to express the CAR-targeting myelin oligodendrocyte glycoprotein (MOG), which were then induced by mouse FoxP3 for differentiation into Tregs and CD28-CD3ζ signaling domains (Figure 6). They used CAR-Tregs to conduct both in vitro andin vivo experiments and found that CARαMOG-FoxP3-Tregs can continuously inhibit T cell proliferation in the presence of MOG+ cells and in the presence of activated macrophages. For the in vivo experiments, cells were administered to autoimmune encephalomyelitis (EAE) mice by intranasal (in) cell delivery. First, after 24 h of infusion of GFP and CARαMOG-FoxP3-co-expressing Tregs, immunohisto-chemical markers for myelination (myelin basic protein, MBP) and reactive astrogliosis (glial fibrillary acidic protein, GFAP) were recovered in treated mice compared to the controls, suggesting good targeting by the CAR-Tregs. Second, treatment of mice with an EAE score of 4 (hind limb paralysis) resulted in a reduction in the score in both the CD4+T cell and CAR-Treg groups. At day 7, only the CAR-Treg group showed continuous relief of symptoms and by day 25, the CAR-Treg group was asymptomatic. Symptom-free mice were rechallenged with a second EAE-inducing inoculum but remained healthy, demonstrating the sustained effect of engineered Tregs. Third, mice in the CAR-Treg group also showed decreased expression of IL-12 and IFN-γ mRNA in the brain, which may be due to the reduction in inflammation and inhibition of DC maturation (Fransson et al., 2012). In 2020, based on the bystander effect, researchers used both MBP-CAR Treg and MOG-CAR Treg therapies in an EAE model (Figure 6), and found that the combination of both significantly inhibited inflammation and myelin-specific T cell responses, thereby inhibiting EAE progress (De Paula Pohl et al., 2020).
Figure 6.
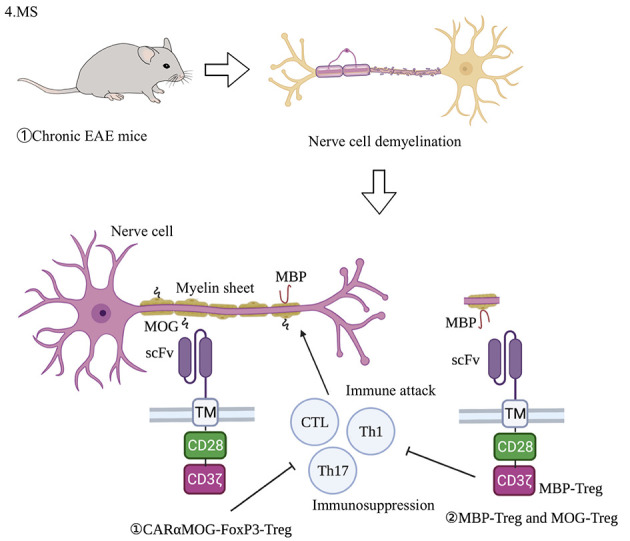
Application of CAR-Treg cells in animal model of MS
Chronic EAE mouse model is used in MS, induced by MOG35-55 and enhanced by Freund’s adjuvant (CFA) or pertussis toxin (PT) (Berard et al., 2010). CARαMOG-FoxP3-Tregs express MOG-targeting CAR, FoxP3, and CD28-CD3ζ signaling domains (Fransson et al., 2012). From in vitro experiments, CARαMOG-FoxP3-Tregs inhibit T cell proliferation in the presence of MOG+ cells. Based on the bystander effect, both MBP-CAR Treg and MOG-CAR Treg therapies have been used for EAE model treatment, with their combination significantly inhibiting inflammatory and myelin-specific T cell responses, and thereby EAE progress. EAE: Experimental autoimmune encephalomyelitis; MBP: Myelin basic protein; MOG: Myelin oligodendrocyte glycoprotein; scFv: Single chain antibody fragment; CTL: Cytotoxic T lymphocyte.
The advantages of CAR treatment for MS are obvious. First, intranasal delivery can facilitate the accumulation of CAR-Tregs in the correct place under a small dose, avoiding the risk of systemic exposure and entry into other vital organs. Second, CAR-Tregs show good targeting and durability. For example, if EAE is again induced by antigens, CAR-Tregs can still inhibit disease development. However, CAR-Treg therapy has some safety issues. First, because the nasal mucosa is highly vascularized, CAR-Tregs may enter blood circulation throughout the body. Second, after nasal injection, cells from the nasal mucosa may migrate to the brain and cerebrospinal fluid (CSF) through the lamina along the olfactory nerve pathway (Danielyan et al., 2009).
RHEUMATOID ARTHRITIS (RA)
Introduction of disease
RA is a common autoimmune disease characterized by an increase in rheumatoid factor (RF) and citrullinated protein antigen (ACPA) (Koivula et al., 2007), accompanied by joint swelling and systemic inflammation. In the early stage, autoantibodies and systemic inflammation are limited, but the adaptive response and tissue damage expand over time (van Venrooij et al., 2011).
Application of CAR-T and CAR-Treg in RA
For the treatment of RA, anti-fluorescein isothiocyanate (FITC) CAR-T has been applied to eliminate autoreactive B cells (Zhang et al., 2021a). Key steps include: determining autoantibody type in patients by enzyme-linked immunosorbent assay (ELISA), preparing FITC CAR-T and FITC-labeled autoantibody positive peptides; and eliminating autoreactive B cells by peptide targeting CAR-T. RA is characterized by the production of autoantibodies against citrulline antigens, so citrulline vimentin, citrulline type II collagen, citrulline fibrinogen and tenascin-C, and cyclic citrulline peptide-1 can be selected as autoantigens for targeting autoreactive B cells (Vossenaar et al., 2004). FITC can only be recognized by CAR-T and autoimmune B cells. Thus, anti-FITC CAR-T can be directed to various types of autoreactive B cells via recognition of FITC-labeled autoantigen peptides. In in vitro models, FITC CAR-T can recognize and kill hybridoma cells by producing antibodies against corresponding antigens, by binding with corresponding antigenic FITC-coupled peptides, and by recognizing and killing anti-COII antibody-producing B cells from collagen-induced arthritis (CIA) mice. FITC CAR-T also has cytotoxic effects on human RA autoimmune B cells in vitro (Figure 7) (Zhang et al., 2021a). Chimeric autoantibody receptor (CAAR) T cell (CAAR-T)-expressing citrullinated antigens have also been used to reduce the number of B cells resistant to citrullinated protein, while retaining protective B cells (Orvain et al., 2021). Anti-citrullinated vimentin CAR Tregs (CV CAR Tregs) also show potential in RA treatment but have not yet progressed beyond the concept stage (Orvain et al., 2021).
Figure 7.
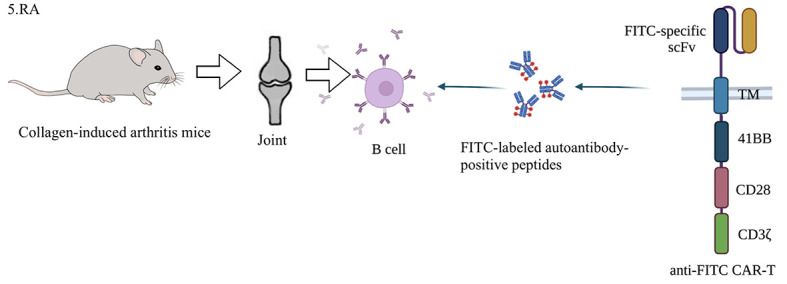
Application of CAR-T cells in animal model of RA
RA produces autoantibodies that cause joint swelling and systemic inflammation. Methods for eliminating autoimmune B cells by CAR-T are as follows: first, autoantibody type is determined by ELISA, and then anti-FITC CAR-T and FITC-labeled autoantibody-positive peptides are prepared to eliminate autoreactive B cells under peptide-mediated CAR-T toxicity in mice. In vitro experiments have shown that anti-FITC CAR-T cells recognize and kill anti-COII antibody-secreting B cells from CIA mice by binding with an antigenic FITC-conjugated peptide. CIA mice are RA mice induced by type II collagen and CFA. DBA/1 mice are commonly used (Kannan et al., 2005). RA: Rheumatoid arthritis; ELISA: Enzyme-linked immunosorbent assay; FITC: Fluorescein isothiocyanate; CIA: Collagen-induced arthritis; CFA: Complete Freund’s adjuvant.
Anti-FITC CAR-T is not prone to abrogation and shows few side effects. Studies have demonstrated that in the presence of specific antibodies, anti-FITC CAR-T cells preferentially kill target antibody-secreting cells rather than FCγR-expressing cells (such as macrophages expressing CD64) (Zhang et al., 2021a). Even if the amount of specific antibody is high, CAR-T cell cytotoxicity is still far less than that of antibody-secreting target cells, and this weak off-target effect can be reduced by blocking FCγR with irrelevant antibodies (Zhang et al., 2021a). This mechanism avoids the effects of FITC CAR-T on FCγR-expressing macrophages and natural killer (NK) cells. Avoiding excessive immune cell damage prevents excessive weakening of human immunity, thus reducing the risk of infection with other diseases or cancer development due to CAR-T treatment, while enhancing the specificity and sustainability of treatment. In addition, theoretically, anti-FITC CAR-T may not only directly eliminate high-level specific B cell receptor (BCR)-expressing autoantigen B cells, but also indirectly affect the division and differentiation of memory B cells to produce autoantibody-secreting plasma cells. The main limitation of this method is that its ability to eliminate autoreactive B cells has only been proven in vitro, and its toxic effects on autoimmune B cells and safety in vivo remain to be clarified. Second, the stability of autoantigens and FITC-coupling mediators needs to be improved. Stability could potentially be enhanced by optimizing the structure and increasing the molecular weight, e.g., adding antigenic peptides and immunogenic domains of antibodies (Rodgers et al., 2016).
PEMPHIGUS VULGARIS (PV)
Introduction of disease
PV is an autoimmune vesicular disease affecting skin and mucosa (Sanders & Nelson, 1965). PV is mediated by immunoglobulin G (IgG) autoantibodies against desmoglein (Dsg). Autoantibodies bind to keratinocyte adhesion proteins Dsg1 and Dsg3 (Amagai et al., 1991), which interferes with keratinocyte adhesion (Shimizu et al., 2002), resulting in spinous layer lysis and blister formation. Dsg3 plays a major role in PV (Amagai et al., 1991).
Application of CAR-T or CAR-Treg in pemphigus
In 2016, a recombinant TCR was developed by fusing the autoantigen (Dsg3) in the CAR structure with CD137-CD3ζ (Ellebrecht et al., 2016) (Figure 8), named a chimeric autoantibody receptor (CAAR) or B cell antibody targeting receptor (BAR) (Parvathaneni & Scott, 2018). Ellebrecht et al. (2016) used several mouse models and in vitro experiments to evaluate the activity of the different autoantibody fragments of CAAR-T, cytotoxic potencies in vivo andin vitro, and on-target affinity and off-target conditions in the presence of circulating soluble autoantibodies. Dsg3 is composed of five extracellular cadherin (EC) domains (Ohyama et al., 2012), with CAAR expressing EC1-4 showing the highest potential (Figure 8). This CAAR-T cell directly eliminates memory B cells with anti-Dsg3 surface Ig (sIg) and indirectly eliminates sIg-Dsg3-specific short-lived pathogenic antibodies producing plasma cells. In the presence of pathogenic IgG, Dsg3-CAAR-T is not be eliminated. CAAR-T cells exhibit mature specificity and only target pathogenic B cells and Dsg3 EC1-4 CAAR-T cells exhibit no off-target toxicity. Lee et al. (2020) also evaluated the cytotoxicity, pharmacological and toxicological effects, dose-response effects, and off-target effects of CAAR-T cells in a mouse model. They found that CAAR-T cells reduce anti-Dsg3 IgG B cells in mice by 83.1%–92.3%, without decreasing total pathogenic B cells, confirming the CAAR-T cells effectively target the pathogenic anti-Dsg3 B cell population in PV. In mouse models, Dsg3-CAAR-T does not follow a strict dose-response relationship, but shows a threshold-dose relationship, suggesting that a conservative fractional initial dose should be used in subsequent clinical studies (Ellebrecht et al., 2016). In addition, screening potential desmosome ligands of Dsg3 and membrane proteins that may be targeted by CAAR verified that Dsg3 EC1-4 CAAR-T exhibits little possibility of off-target toxicity, consistent with previous research (Ellebrecht et al., 2016).
Figure 8.
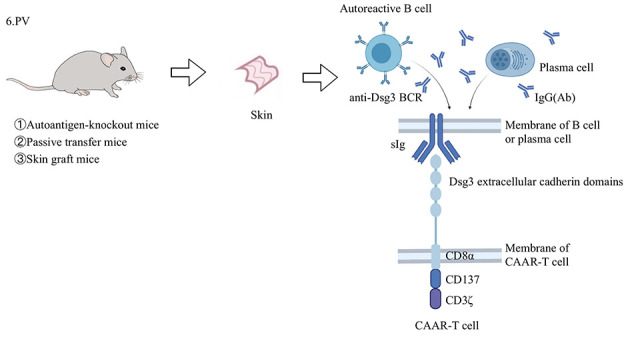
Application of CAAR-T cells in animal model of PV
PV develops in mucosa and skin, leading to a deadly autoimmune blistering disease. Three mouse models have been used in CAAR-T cell research. The “autoantigen-knockout mouse model” is a PV active model generated by transferring mDsg3-immunized splenocytes from Dsg3-/- mice to Rag2-/- immunodeficient mice (Amagai et al., 2000). The “passive transfer model” is established by injecting anti-mDsg3 IgG hybridoma cells into immunodeficient mice (Tsunoda et al., 2003). The “skin graft mouse model” is constructed by transplanting human skin grafts into immunodeficient mice and then transferring the hDsg3-secreting hybridoma cells of PV patients into mice (Ellebrecht et al., 2016). By fusing the EC domain (especially EC1-4) of Dsg3 into the CD137-CD3 ζ domain, CAAR-T cells can be directed to memory B cells and short-lived plasma cells, which express anti-Dsg3 sIg, thus curing the disease. PV: Pemphigus vulgaris; CAAR: Chimeric autoantibody receptor; Dsg3: Desmoglein3; mDsg3: Mouse desmoglein3; hDsg3: Human desmoglein3; EC: Extracellular cadherin; sIg: Surface immunoglobulin.
Overall, current preclinical research indicates that CAAR-T cells show high affinity and low toxicity. There are many obvious advantages of CAAR-T cells, including excellent targeting properties, limited toxicity and side effects, and effective continuous effects. CAAR-T cells can eliminate the autoreactive cloning of immune cells while retaining non-pathogenic B cells and patient immunity (Zmievskaya et al., 2021), which is significantly different from the B cell-targeted killing of CAR-T19 cells. In addition, the limited toxicity/side effects could be attributed to several reasons. First, current routine clinical treatment of PV includes corticosteroids (CS) and immunosuppressive agents (ISA) (Yanovsky et al., 2019). Treatment with high-dose corticosteroids and the anti-CD20 antibody rituximab takes a relatively long time to induce remission, which can cause long-term and systemic immunosuppression, potentially leading to repeated and serious infections (Schmidt et al., 2017; Siddiqi et al., 2018). Second, no off-target effects have been observed in CAAR-T cell experiments. In addition, because anti-Dsg3 B cells account for only a small part of total B cells in patients (Lee et al., 2020; Nishifuji et al., 2000), they are unlikely to cause CRS in CAR-T therapy (Ellebrecht et al., 2016). Moreover, more than 90% of PV patients relapse without repeated infusion of rituximab (anti-CD20 antibody) (Colliou et al., 2013), whereas CAAR-T cells are a “living” drug with the potential to expand and persist in vivo.
However, there are some concerns about CAAR-Ts: (1) Detection methods and mouse models are limited. Most of our understanding of the persistence and effects of CAAR-T (or CAR-T) cells comes from measuring their abundance in peripheral blood (Maldini et al., 2018), which cannot reflect all effects. Furthermore, hybridoma cells injected into mice are unlikely to have the same distribution pattern as patients’ autoreactive B cells (Galy, 2016; Maldini et al., 2018). (2) Similar to CAR-T cell therapy, CAAR-T is an individualized treatment plan, which is highly time-consuming and costly (MacLeod et al., 2017; Yanovsky et al., 2019). (3) At present, CAAR-T therapy is still at the preclinical test stage, so it is hard to predict the whole picture when used on patients. A phase I clinical trial on the maximum tolerated dose of Dsg3-CAAR-T in patients with mucosal dominant PV is currently in the recruitment stage (NCT04422912). However, in the PV mouse model, CAAR-T therapy represents a highly personalized treatment method, with great research value and potential.
PERSPECTIVES
Although CAR-T and CAR-Treg cells have a series of advantages, such as good targeting, good persistence, and small side effects, they still have shortcomings in terms of safety, effectiveness, persistence, and manufacturability. We will discuss the shortcomings of CAR-T and CAR-Treg from these four aspects and explore their future developments.
Regarding safety, there are several issues with CAR-T therapy, including cell response release syndrome (Breslin, 2007), neurotoxicity (Bonifant et al., 2016), and off-target identification attacks (Makita et al., 2017). At present, researchers have added relevant mechanisms to reduce the side effects of CAR-T cells and regulate their persistence and activity in human tumor-related fields. In the future, these mechanisms could be applied to the treatment of autoimmune diseases. The first mechanism is the activation of suicide genes. Herpes simplex virus-thymidine kinase (HSV-TK) and inducible caspase 9 (iCasp9) are two suicide genes that have been integrated into CAR-T cells (Bonini et al., 1997; Quintarelli et al., 2007; Zhang & Xu, 2017). The suicide system is activated via the administration of rimiducid, which controls the intensity of side effects by the induction of rapid CAR-T cell apoptosis (Bonini et al., 1997; Quintarelli et al., 2007; Zhang & Xu, 2017). The second mechanism is the construction of switches, such as the ON-switch system applied in tumors. In this system, engineered CARs are divided into two independent proteins. One protein usually contains an extracellular antigen-binding domain, while the other protein contains downstream signaling elements and co-stimulatory molecules (such as CD3ζ and 4-1BB) (Wu et al., 2015). Only through the activation of benign foreign molecules can the two proteins dimerize, allowing CAR-T cells to attack tumors but effectively controlling their abnormal activation (Wu et al., 2015). The third mechanism is the manufacture of bispecific linkers through the small molecule drug conjugate (SMDC) platform, which could potentially be applied for autoimmune diseases in the future (Gallo et al., 2021; Zhuang et al., 2019). These unique bispecific linkers are composed of FITC molecules and tumor homing molecules, which can accurately connect universal CAR-T cells and cancer cells, thereby causing local T cell activation (Gallo et al., 2021; Zhuang et al., 2019). In addition, adjusting the dose of adaptor molecules can precisely control CAR-T and other side effects (Gallo et al., 2021; Zhuang et al., 2019). CAR-T also has safety problems in administration. As mentioned above, intranasal delivery of CAR-T for the treatment of MS may be risky, and further experiments are needed to optimize dose and reduce risk (Danielyan et al., 2009).
CAR-T effectiveness is also limited by the lack of antigens (Schultz & Mackall, 2019). As mentioned previously, SLE MRL-lpr mice treated with CAR-T still retain IgMlo subgroups of B cells, and therefore CAR-T cannot completely cure the disease as antibody-producing cells still exist after treatment (Kansal et al., 2019). In addition, because of immune escape caused by antigen deletion, the long-term survival rate of CAR-T cells in leukemia is quite low (Schultz & Mackall, 2019). As such, bispecific targeting of CAR-T to circumvent the down-regulation of the CD19 antigen is considered a good direction for research (Schultz & Mackall, 2019). Another issue is the stability of the CAR structure after infusion, e.g., in vivo stability of FITC in the treatment of RA (Zhang et al., 2021a). Researchers have proposed the structural optimization of bifunctional antigen-specific targeting ligands, such as improvement in the coupled immunogenic domain containing antigen peptides and antibody fragments to increase the molecular weight of the medium (Zhang et al., 2021a).
Regarding persistence, CAR-T cannot guarantee its activity, stability, and continuous proliferation under inflammatory microenvironments in vivo. For example, Zhang et al. (2019) showed that CAR-T can induce a delay in T1D onset in NOD mice, but cannot stop the occurrence of T1D, which may be a problem for the expansion of CAR-T in vivo. To search for strategies to extend the metastatic activities of CAR-T cells, genome sequencing of CAR insertion sites in T cells has been established to better understand CAR-T cell function and persistence in vivo (Li et al., 2020). At the same time, modification of the CAR-T structure is also ongoing. Fourth-generation CARs have added factors (such as IL-2, IL-5, IL-12, and co-stimulatory ligands) to enhance the expansion, persistence, and anti-tumor activity of CAR-T cells (Chmielewski & Abken, 2015; Kueberuwa et al., 2018). Furthermore, CARs incorporating IL-2 receptor β-chain (Kagoya et al., 2018), telomerase reverse transcriptase co-transduction (Bai et al., 2015), or PI3K inhibitor treatment (Zheng et al., 2018) can improve in vivo persistence of CAR-T cells.
Concerning manufacturability, CAR-T therapy is an individualized treatment plan and complex cell manufacturing is required in a dedicated facility (Lyman et al., 2020). Furthermore, due to risks such as CRS, a high level of hospital care is required after the administration of CAR-T cells. Therefore, the price of treatment is usually high (Lyman et al., 2020). Thus, advances in CAR technology and market supervision should be strengthened to reduce the high costs of CAR-T therapy.
It is generally accepted that antigen-specific Tregs, including TCR-Tregs and CAR-Tregs, are superior to polyclonal Tregs in their suppression because they are antigen-specific and tend to migrate to a target organ harboring a specific antigen (Zheng et al., 2018). Moreover, CAR-Tregs hold advantages over TCR-Tregs as they are non-MHC-restricted and less dependent on IL-2 (Rosado-Sánchez & Levings, 2020). However, CAR-Tregs still exhibit some disadvantages and limitations.
Regarding CAR-Treg safety, the immunosuppressive phenotype of Tregs can change after losing Foxp3 expression under inflammatory microenvironments and engineered Treg cells may transform into antigen-specific pathogenic effector T cells that can aggravate disease symptoms (Juan et al., 2017). Several approaches to maintain the immune inhibitory phenotype of Tregs have been reported, e.g., treating Tregs with vitamin A derivative all-trans retinoic acid to sustain Treg stability and functionality (Zhou et al., 2010), administering a Treg-favoring microbiota to the gut (Atarashi et al., 2013), and inducing ectopic expression of the Foxp3 gene to stabilize the regulatory Treg phenotype (Beavis et al., 2011). It is well known that CAR-T cell treatment can cause side effects related to cytokine storm and neuronal cytotoxicity (Breslin, 2007), but whether CAR-Tregs also induce these adverse reactions remains to be determined. The tendency of Treg cells to produce inflammatory cytokines can be eliminated using CRISPR to remove IFN-γ or IL-17 genes (Freen-van Heeren, 2021). Off-target effects may also occur in CAR-Treg therapy. For example, when CAR-Tregs are given via nasal injection, cells from the nasal mucosa may migrate to the brain and cause damage (Danielyan et al., 2009), which could result in pan-immunosuppression, reduced immune responses against opportunistic infections, and possibly even cancer development (Mohseni et al., 2020). One way to control this may be to integrate a suicide gene into the genetically modified therapeutic cells before injecting them into the patient.
Regarding effectiveness, it is difficult to ensure that CAR-Treg cells are effective in all diseases. It is a necessary to characterize antibodies specific for self- or allo-antigens in order to construction efficient and specific CAR-Tregs. However, both the selection of CAR-targeted antigens and development of specific antibodies can be time-consuming and may be difficult in some disease models. If antigens are not chosen properly, CAR-Tregs may be ineffective, with low specificity and off-target effects.
Regarding CAR-Treg cell persistence, the exhaustion of CAR-Tregs can limit their efficacy in suppression. For example, Blat et al. (2014) reported that CEA-CAR-Tregs diminish rapidly in vivo, greatly limiting their clinical application in colitis. Because CD28 plays an important role in Treg development and expansion (Bour-Jordan & Bluestone, 2009), most CAR-Treg studies use second-generation CARs with a CD28 co-stimulatory domain to expand Tregs. However, several studies have indicated that substituting CD28 with CD137 are a co-stimulatory can ameliorate T cell exhaustion, thus improving CAR-T persistence (Long et al., 2015; Quintarelli et al., 2018). However, CD28 is critical for maintaining Treg homeostasis and plays a critical role in Treg proliferation, differentiation, and survival as well as IL-2 production and Foxp3 expression (Bour-Jordan & Bluestone, 2009). Thus, the inclusion of both CD28 and CD137 co-stimulatory domains may help maintain CAR-Tregs. More studies are needed to select the best co-stimulatory signals for optimizing CAR-Treg suppression.
The manufacture of CAR-Treg cells is complex and costly, which influences their translational success. Current CAR-Treg manufacturing presents several challenges in cell purification, yield, expansion, vector production/transduction, cryopreservation, and product release testing (Fritsche et al., 2020). For example, Elinav et al. (2009) experienced significant obstacles in vector transduction when producing TNP-TPCR Tregs. Several methods can be utilized to improve CAR-Treg manufacture. For example, the development of a multi-parameter flow cytometry panel with consistent marker profiles can simplify the isolation and sorting processes (Fritsche et al., 2020). Implementing self-dissolving or easily removable beads can simplify the activation process (Bernstroem et al., 2016). In CAR design and delivery, batch production using flexible module methods, such as UniCAR, not only improve safety and cost-benefit ratios (Koristka et al., 2018; Zhang et al., 2018) but also optimize non-viral platforms (transposon or CRISPR/Cas technologies) and improve safety and efficacy. Sleeping Beauty transposon systems hold great promise for reducing costs, eliminating the need for clinical-grade viral vector manufacture (Ramanayake et al., 2015), and lowering the risk of insertional oncogenesis (Gogol-Döring et al., 2016). Using CRISPR/Cas9 with non-viral delivery of template DNA to eliminate large genes and orthotopic placement of transgenic constructs opens new delivery opportunities (Schober et al., 2019). For CAR-Treg expansion, semi- or fully automated closed-system bioreactors with integrated gas transfer and fully automated process control could pave the way toward an automated and standardized ex vivo expansion procedure as well as improve safety and decrease the costs of manufacturing. Furthermore, implementation of allogeneic off-the-shelf products, elimination of endogenous TCRs and MHC molecules using genome-editing technologies, increased expression of NK cell inhibitory molecules, and application of iPSC-derived Treg technology may improve the safety of third generation CAR-Tregs at the manufacturing scale (Depil et al., 2020; MacDonald et al., 2019).
In conclusion, more studies are needed to improve the safety, effectiveness, persistence, and manufacturability of CAR-T and CAR-Treg cells. Despite their shortcomings, CAR-T and CAR-Treg cells show considerable potential for application in autoimmune diseases.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Z.W. and B.L. conceived the review. L.B. contributed to the introduction and pemphigus vulgaris section. H.W.C. contributed to the introduction and rheumatoid arthritis section. C.Q. contributed to the discussion and colitis and multiple sclerosis sections. X.C.B. contributed to the discussion and systemic lupus erythematosus, type 1 diabetes, and multiple sclerosis sections. X.C.B., L.B., H.W.C., and C.Q. contributed to the figures and tables. All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We thank the Shanghai Collaborative Innovation Center of Cellular Homeostasis Regulation and Human Diseases for providing constructive comments on our work and Dan Li, Jie-Qiong Chen, Qian-Ru Huang, Hao Cheng, and Xiao-Xia Wang for their valuable suggestions.
Funding Statement
This study was supported by the National Natural Science Foundation of China (81830051, 31961133011, 32130041); National Key R&D Program of China (2019YFA09006100); Shanghai Academic Research Leader (16XD1403800); Innovative Research Team of High-Level Local Universities in Shanghai; and Shanghai Collaborative Innovation Center of Cellular Homeostasis Regulation and Human Diseases
Contributor Information
Zeng Wang, Email: wz2573749689@126.com.
Bin Li, Email: binli@shsmu.edu.cn.
References
- 1.Abramson JS Anti-CD19 CAR T-cell therapy for B-cell non-hodgkin lymphoma. Transfusion Medicine Reviews. 2020;34(1):29–33. doi: 10.1016/j.tmrv.2019.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Acharya UH, Dhawale T, Yun S, Jacobson CA, Chavez JC, Ramos JD, et al Management of cytokine release syndrome and neurotoxicity in chimeric antigen receptor (CAR) T cell therapy. Expert Review of Hematology. 2019;12(3):195–205. doi: 10.1080/17474086.2019.1585238. [DOI] [PubMed] [Google Scholar]
- 3.Adams SM, Bornemann PH Ulcerative colitis. American Family Physician. 2013;87(10):699–705. [PubMed] [Google Scholar]
- 4.Amagai M, Klaus-Kovtun V, Stanley JR Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67(5):869–877. doi: 10.1016/0092-8674(91)90360-B. [DOI] [PubMed] [Google Scholar]
- 5.Amagai M, Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, Nishikawa T Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. The Journal of Clinical Investigation. 2000;105(5):625–631. doi: 10.1172/JCI8748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al Treg induction by a rationally selected mixture of clostridia strains from the human microbiota . Nature. 2013;500(7461):232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 7.Bai Y, Kan S, Zhou SX, Wang YT, Xu J, Cooke JP, et al Enhancement of the in vivo persistence and antitumor efficacy of CD19 chimeric antigen receptor t cells through the delivery of modified TERT mRNA . Cell Discovery. 2015;1:15040. doi: 10.1038/celldisc.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beavis PA, Gregory B, Green P, Cribbs AP, Kennedy A, Amjadi P, et al Resistance to regulatory T cell-mediated suppression in rheumatoid arthritis can be bypassed by ectopic foxp3 expression in pathogenic synovial T cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(40):16717–16722. doi: 10.1073/pnas.1112722108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berard JL, Wolak K, Fournier S, David S Characterization of relapsing-remitting and chronic forms of experimental autoimmune encephalomyelitis in C57BL/6 mice. Glia. 2010;58(4):434–445. doi: 10.1002/glia.20935. [DOI] [PubMed] [Google Scholar]
- 10.Bernstroem KE, Lieske N, Zhang H, Økern G, Aass H, Troseid A, et al 164 - optimized process for regulatory T cell activation and expansion using dynabeads™ Treg CD3/CD28 for clinical applications. Cytotherapy. 2016;18(6S):S96. [Google Scholar]
- 11.Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory t cells. Molecular Therapy. 2014;22(5):1018–1028. doi: 10.1038/mt.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Science Translational Medicine. 2015;7(315):315ra189. doi: 10.1126/scitranslmed.aad4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bluestone JA, Herold K, Eisenbarth G Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464(7293):1293–1300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ Toxicity and management in CAR T-cell therapy. Molecular Therapy Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276(5319):1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 16.Bour-Jordan H, Bluestone JA Regulating the regulators: co-stimulatory signals control the homeostasis and function of regulatory T cells. Immunological Reviews. 2009;229(1):41–66. doi: 10.1111/j.1600-065X.2009.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breslin S. 2007. Cytokine-release syndrome: overview and nursing implications. Clinical Journal of Oncology Nursing, 11 (1 Suppl): 37–42.
- 18.Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, et al Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Natur Medicine. 2020;26(2):270–280. doi: 10.1038/s41591-019-0737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carreño LJ, González PA, Kalergis AM Modulation of T cell function by TCR/pMHC binding kinetics. Immunobiology. 2006;211(1-2):47–64. doi: 10.1016/j.imbio.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Chen YH, Sun JH, Liu H, Yin G, Xie QB Immunotherapy deriving from CAR-T cell treatment in autoimmune diseases. Journal of Immunology Research. 2019;2019:5727516. doi: 10.1155/2019/5727516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chmielewski M, Abken H TRUCKs: the fourth generation of CARs. Expert Opinion on Biological Therapy. 2015;15(8):1145–1154. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 22.Colliou N, Picard D, Caillot F, Calbo S, Le Corre S, Lim A, et al Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Science Translational Medicine. 2013;5(175):175ra30. doi: 10.1126/scitranslmed.3005166. [DOI] [PubMed] [Google Scholar]
- 23.Compston A, Coles A Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 24.Correale J, Gaitán MI, Ysrraelit MC, Fiol MP Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain. 2017;140(3):527–546. doi: 10.1093/brain/aww258. [DOI] [PubMed] [Google Scholar]
- 25.D'Agostino M, Raje N Anti-BCMA CAR T-cell therapy in multiple myeloma: can we do better? Leukemia. 2020;34(1):21–34. doi: 10.1038/s41375-019-0669-4. [DOI] [PubMed] [Google Scholar]
- 26.Dai HR, Wu ZQ, Jia HJ, Tong C, Guo YL, Ti DD, et al Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. Journal of Hematology & Oncology. 2020;13(1):30. doi: 10.1186/s13045-020-00856-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dall'Era M, Pauli ML, Remedios K, Taravati K, Sandova PM, Putnam AL, et al Adoptive Treg cell therapy in a patient with systemic lupus erythematosus. Arthritis & Rheumatology. 2019;71(3):431–440. doi: 10.1002/art.40737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danielyan L, Schäfer R, von Ameln-Mayerhofer A, Buadze M, Geisler J, Klopfer T, et al Intranasal delivery of cells to the brain. European Journal of Cell Biology. 2009;88(6):315–324. doi: 10.1016/j.ejcb.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Davidson A, Diamond B Autoimmune diseases. The New England Journal of Medicine. 2001;345(5):340–350. doi: 10.1056/NEJM200108023450506. [DOI] [PubMed] [Google Scholar]
- 30.De Paula Pohl A, Schmidt A, Zhang AH, Maldonado T, Königs C, Scott DW Engineered regulatory T cells expressing myelin-specific chimeric antigen receptors suppress EAE progression. Cellular Immunology. 2020;358:104222. doi: 10.1016/j.cellimm.2020.104222. [DOI] [PubMed] [Google Scholar]
- 31.Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nature Reviews Drug Discovery. 2020;19(3):185–199. doi: 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 32.Durcan L, O'Dwyer T, Petri M Management strategies and future directions for systemic lupus erythematosus in adults. The Lancet. 2019;393(10188):2332–2343. doi: 10.1016/S0140-6736(19)30237-5. [DOI] [PubMed] [Google Scholar]
- 35.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao XM, Cho MJ, et al Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353(6295):179–184. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al CD19 CAR T cell product and disease attributes predict leukemia remission durability. The Journal of Clinical Investigation. 2019;129(5):2123–2132. doi: 10.1172/JCI125423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fishman S, Lewis MD, Siew LK, De Leenheer E, Kakabadse D, Davies J, et al Adoptive transfer of mRNA-transfected T cells redirected against diabetogenic CD8 T cells can prevent diabetes. Molecular Therapy. 2017;25(2):456–464. doi: 10.1016/j.ymthe.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fontenot JD, Gavin MA, Rudensky AY Foxp3 programs the development and function of CD4+CD25+ regulatory T cells . Nature Immunology. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 39.Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu BF, et al CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. Journal of Neuroinflammation. 2012;9:112. doi: 10.1186/1742-2094-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freen-van Heeren JJ Using CRISPR to enhance T cell effector function for therapeutic applications. Cytokine:X. 2021;3(1):100049. doi: 10.1016/j.cytox.2020.100049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fritsche E, Volk HD, Reinke P, Abou-El-Enein M Toward an optimized process for clinical manufacturing of CAR-Treg cell therapy. Trends in Biotechnology. 2020;38(10):1099–1112. doi: 10.1016/j.tibtech.2019.12.009. [DOI] [PubMed] [Google Scholar]
- 42.Gallo F, Korsak B, Müller C, Hechler T, Yanakieva D, Avrutina O, et al Enhancing the pharmacokinetics and antitumor activity of an α-amanitin-based small-molecule drug conjugate via conjugation with an Fc domain. Journal of Medicinal Chemistry. 2021;64(7):4117–4129. doi: 10.1021/acs.jmedchem.1c00003. [DOI] [PubMed] [Google Scholar]
- 43.Galy A Like angler fish, CAARs lure their prey. Molecular Therapy. 2016;24(8):1339–1341. doi: 10.1038/mt.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gogol-Döring A, Ammar I, Gupta S, Bunse M, Miskey C, Chen W, et al Genome-wide profiling reveals remarkable parallels between insertion site selection properties of the MLV retrovirus and the piggyBac transposon in primary human CD4+ T cells . Molecular Therapy. 2016;24(3):592–606. doi: 10.1038/mt.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Göschl L, Scheinecker C, Bonelli M Treg cells in autoimmunity: from identification to Treg-based therapies. Seminars in Immunopathology. 2019;41(3):301–314. doi: 10.1007/s00281-019-00741-8. [DOI] [PubMed] [Google Scholar]
- 46.Guo HF, Xun LR, Zhang RS, Hu FR, Luan J, Lao KJ, et al. 2019. Stability and inhibitory function of Treg cells under inflammatory conditions in vitro. Experimental and Therapeutic Medicine, 18 (4): 2443–2450.
- 47.Haddadi MH, Hajizadeh-Saffar E, Khosravi-Maharlooei M, Basiri M, Negahdari B, Baharvand H Autoimmunity as a target for chimeric immune receptor therapy: a new vision to therapeutic potential. Blood Reviews. 2020;41:100645. doi: 10.1016/j.blre.2019.100645. [DOI] [PubMed] [Google Scholar]
- 48.Halle S, Halle O, Förster R. 2017. Mechanisms and dynamics of t cell-mediated cytotoxicityin vivo. Trends in Immunology, 38 (6): 432–443.
- 49.Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Molecular Medicine. 2017;9(9):1183–1197. doi: 10.15252/emmm.201607485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He J, Zhang RJ, Shao M, Zhao XZ, Miao M, Chen JL, et al Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial. Annals of Rheumatic Diseases. 2020;79(1):141–149. doi: 10.1136/annrheumdis-2019-215396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He Y, Sawalha AH Drug-induced lupus erythematosus: an update on drugs and mechanisms. Current Opinion in Rheumatology. 2018;30(5):490–497. doi: 10.1097/BOR.0000000000000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Helyer BJ, Howie JB Renal disease associated with positive lupus erythematosus tests in a crossbred strain of mice. Nature. 1963;197:197. doi: 10.1038/197197a0. [DOI] [PubMed] [Google Scholar]
- 53.Jin XX, Xu Q, Pu CF, Zhu KX, Lu C, Jiang Y, et al Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cellular & Molecular Immunology. 2021;18(8):1896–1903. doi: 10.1038/s41423-020-0472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Juan M, ol Rego ME, Llobell A, Marzal B, Castell M, Boronat A Future of chimeric antigen receptors (CARs): could it drive solutions beyond cancer? Examples in autoimmune diseases. MOJ Immunology. 2017;5(3):00158. [Google Scholar]
- 55.June CH, Sadelain M Chimeric antigen receptor therapy. The New England Journal of Medicine. 2018;379(1):64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kagoya Y, Tanaka S, Guo TX, Anczurowski M, Wang CH, Saso K, et al A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nature Medicine. 2018;24(3):352–359. doi: 10.1038/nm.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kannan K, Ortmann RA, Kimpel D Animal models of rheumatoid arthritis and their relevance to human disease. Pathophysiology. 2005;12(3):167–181. doi: 10.1016/j.pathophys.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 58.Kansal R, Richardson N, Neeli I, Khawaja S, Chamberlain D, Ghani M, et al Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Science Translational Medicine. 2019;11(482):eaav1648. doi: 10.1126/scitranslmed.aav1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al Type 1 diabetes mellitus. Nature Reviews Disease Primers. 2017;3:17016. doi: 10.1038/nrdp.2017.16. [DOI] [PubMed] [Google Scholar]
- 60.Kersten MJ, Spanjaart AM, Thieblemont C CD19-directed CAR T-cell therapy in B-cell NHL. Current Opinion in Oncology. 2020;32(5):408–417. doi: 10.1097/CCO.0000000000000668. [DOI] [PubMed] [Google Scholar]
- 61.Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood. 2015;125(7):1107–1115. doi: 10.1182/blood-2014-04-566786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koivula MK, Heliövaara M, Ramberg J, Knekt P, Rissanen H, Palosuo T, et al Autoantibodies binding to citrullinated telopeptide of type II collagen and to cyclic citrullinated peptides predict synergistically the development of seropositive rheumatoid arthritis. Annals of the Rheumatic Diseases. 2007;66(11):1450–1455. doi: 10.1136/ard.2006.062919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koristka S, Kegler A, Bergmann R, Arndt C, Feldmann A, Albert S, et al Engrafting human regulatory t cells with a flexible modular chimeric antigen receptor technology. Journal of Autoimmunity. 2018;90:116–131. doi: 10.1016/j.jaut.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 64.Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Molecular Therapy Oncolytics. 2018;8:41–51. doi: 10.1016/j.omto.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langner C, Aust D, Ensari A, Villanacci V, Becheanu G, Miehlke S, et al Histology of microscopic colitis-review with a practical approach for pathologists. Histopathology. 2015;66(5):613–626. doi: 10.1111/his.12592. [DOI] [PubMed] [Google Scholar]
- 66.Lee J, Lundgren DK, Mao XM, Manfredo-Vieira S, Nunez-Cruz S, Williams EF, et al Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. The Journal of Clinical Investigation. 2020;130(12):6317–6324. doi: 10.1172/JCI138416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li D, Li N, Zhang YF, Fu HY, Feng MQ, Schneider D, et al Persistent polyfunctional chimeric antigen receptor T cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology. 2020;158(8):2250–2265.e20. doi: 10.1053/j.gastro.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li LQ, Godfrey WR, Porter SB, Ge Y, June CH, Blazar BR, et al CD4+CD25+ regulatory T-cell lines from human cord blood have functional and molecular properties of T-cell anergy . Blood. 2005;106(9):3068–3073. doi: 10.1182/blood-2005-04-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liang CL, Lu WH, Qiu FF, Li D, Liu HZ, Zheng F, et al Paeoniflorin ameliorates murine lupus nephritis by increasing CD4+FOXP3+ Treg cells via enhancing MTNFα-TNFR2 pathway . Biochemical Pharmacology. 2021;185:114434. doi: 10.1016/j.bcp.2021.114434. [DOI] [PubMed] [Google Scholar]
- 70.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature Medicine. 2015;21(6):581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lyman GH, Nguyen A, Snyder S, Gitlin M, Chung KC Economic evaluation of chimeric antigen receptor T-cell therapy by site of care among patients with relapsed or refractory large B-cell lymphoma. JAMA Network Open. 2020;3(4):e202072. doi: 10.1001/jamanetworkopen.2020.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. The Journal of Clinical Investigation. 2016;126(4):1413–1424. doi: 10.1172/JCI82771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.MacDonald KN, Piret JM, Levings MK Methods to manufacture regulatory T cells for cell therapy. Clinical and Experimental Immunology. 2019;197(1):52–63. doi: 10.1111/cei.13297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Molecular Therapy. 2017;25(4):949–961. doi: 10.1016/j.ymthe.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Magnuson AM, Thurber GM, Kohler RH, Weissleder R, Mathis D, Benoist C Population dynamics of islet-infiltrating cells in autoimmune diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(5):1511–1516. doi: 10.1073/pnas.1423769112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Majzner RG, Mackall CL Clinical lessons learned from the first leg of the CAR T cell journey. Nature Medicine. 2019;25(9):1341–1355. doi: 10.1038/s41591-019-0564-6. [DOI] [PubMed] [Google Scholar]
- 77.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29(1):1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 78.Makita S, Yoshimura K, Tobinai K Clinical development of anti-CD19 chimeric antigen receptor T-cell therapy for B-cell non-hodgkin lymphoma. Cancer Science. 2017;108(6):1109–1118. doi: 10.1111/cas.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maldini CR, Ellis GI, Riley JL CAR T cells for infection, autoimmunity and allotransplantation. Nature Reviews Immunology. 2018;18(10):605–616. doi: 10.1038/s41577-018-0042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martinez M, Moon EK CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Frontiers in Immunology. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maul J, Loddenkemper C, Mundt P, Berg E, Giese T, Stallmach A, et al Peripheral and intestinal regulatory CD4+CD25high T cells in inflammatory bowel disease . Gastroenterology. 2005;128(7):1868–1878. doi: 10.1053/j.gastro.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 82.Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao YB, June CH Adoptive immunotherapy for cancer or viruses. Annual Review of Immunology. 2014;32:189–225. doi: 10.1146/annurev-immunol-032713-120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McFarland HF, Martin R Multiple sclerosis: a complicated picture of autoimmunity. Nature Immunology. 2007;8(9):913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 84.McGovern JL, Wright GP, Stauss HJ Engineering specificity and function of therapeutic regulatory T cells. Frontiers in Immunology. 2017;8:1517. doi: 10.3389/fimmu.2017.01517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mohseni YR, Tung SL, Dudreuilh C, Lechler RI, Fruhwirth GO, Lombardi G The future of regulatory T cell therapy: promises and challenges of implementing CAR technology. Frontiers in Immunology. 2020;11:1608. doi: 10.3389/fimmu.2020.01608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mougiakakos D, Krönke G, Völkl S, Kretschmann S, Aigner M, Kharboutli S, et al CD19-targeted CAR T cells in refractory systemic lupus erythematosus. The New England Journal of Medicine. 2021;385(6):567–569. doi: 10.1056/NEJMc2107725. [DOI] [PubMed] [Google Scholar]
- 87.Murphy ED, Roths JB. 1978. A single gene model for massive lymphoproliferation with immune complex disease in new mouse strain MRL. In: Seno S, Takaku F, Irino S. Topics in Hematology. Amsterdam: Excerpta Medica, 69–72.
- 88.Murphy G, Isenberg D Effect of gender on clinical presentation in systemic lupus erythematosus. Rheumatology. 2013;52(12):2108–2115. doi: 10.1093/rheumatology/ket160. [DOI] [PubMed] [Google Scholar]
- 89.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. The New England Journal of Medicine. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nie J, Li YY, Zheng SG, Tsun A, Li B FOXP3+ Treg cells and gender bias in autoimmune diseases . Frontiers in Immunology. 2015;6:493. doi: 10.3389/fimmu.2015.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nishifuji K, Amagai M, Kuwana M, Iwasaki T, Nishikawa T Detection of antigen-specific B cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. Journal of Investigative Dermatology. 2000;114(1):88–94. doi: 10.1046/j.1523-1747.2000.00840.x. [DOI] [PubMed] [Google Scholar]
- 92.Norris JM, Johnson RK, Stene LC Type 1 diabetes-early life origins and changing epidemiology. The Lancet Diabetes & Endocrinology. 2020;8(3):226–238. doi: 10.1016/S2213-8587(19)30412-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ohyama B, Nishifuji K, Chan PT, Kawaguchi A, Yamashita T, Ishii N, et al Epitope spreading is rarely found in pemphigus vulgaris by large-scale longitudinal study using desmoglein 2-based swapped molecules. Journal of Investigative Dermatology. 2012;132(4):1158–1168. doi: 10.1038/jid.2011.448. [DOI] [PubMed] [Google Scholar]
- 94.Orvain C, Boulch M, Bousso P, Allanore Y, Avouac J Is there a place for chimeric antigen receptor-T cells in the treatment of chronic autoimmune rheumatic diseases. Arthritis & Rheumatology. 2021;73(11):1954–1965. doi: 10.1002/art.41812. [DOI] [PubMed] [Google Scholar]
- 95.Park T, Cave D, Marshall C Microscopic colitis: a review of etiology, treatment and refractory disease. World Journal of Gastroenterology. 2015;21(29):8804–8810. doi: 10.3748/wjg.v21.i29.8804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parvathaneni K, Scott DW Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Advances. 2018;2(18):2332–2340. doi: 10.1182/bloodadvances.2018018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Poole BD, Schneider RI, Guthridge JM, Velte CA, Reichlin M, Harley JB, et al Early targets of nuclear RNP humoral autoimmunity in human systemic lupus erythematosus. Arthritis & Rheumatology. 2009;60(3):848–859. doi: 10.1002/art.24306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Quintarelli C, Orlando D, Boffa I, Guercio M, Polito VA, Petretto A, et al Choice of co-stimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology. 2018;7(6):e1433518. doi: 10.1080/2162402X.2018.1433518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GMP, Pule M, Foster AE, et al Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. 2007;110(8):2793–2802. doi: 10.1182/blood-2007-02-072843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. The New England Journal of Medicine. 2019;380(18):1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ramanayake S, Bilmon I, Bishop D, Dubosq MC, Blyth E, Clancy L, et al Low-cost generation of good manufacturing practice-grade CD19-specific chimeric antigen receptor-expressing T cells using piggyBac gene transfer and patient-derived materials. Cytotherapy. 2015;17(9):1251–1267. doi: 10.1016/j.jcyt.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 102.Ramos CA, Dotti G Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opinion on Biological Therapy. 2011;11(7):855–873. doi: 10.1517/14712598.2011.573476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, et al Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(4):E459–E468. doi: 10.1073/pnas.1524155113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rosado-Sánchez I, Levings MK Building a CAR-Treg: going from the basic to the luxury model. Cellular Immunology. 2020;358:104220. doi: 10.1016/j.cellimm.2020.104220. [DOI] [PubMed] [Google Scholar]
- 105.Rose NR Prediction and prevention of autoimmune disease in the 21st century: a review and preview. American Journal of Epidemiology. 2016;183(5):403–406. doi: 10.1093/aje/kwv292. [DOI] [PubMed] [Google Scholar]
- 106.Sadelain M, Brentjens R, Rivière I The basic principles of chimeric antigen receptor design. Cancer Discovery. 2013;3(4):388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sakaguchi S, Yamaguchi T, Nomura T, Ono M Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 108.Sanders SL, Nelson CT Pemphigus and pemphigoid. Medical Clinics of North America. 1965;49(3):681–694. doi: 10.1016/S0025-7125(16)33314-4. [DOI] [PubMed] [Google Scholar]
- 109.Schmidt E, Spindler V, Eming R, Amagai M, Antonicelli F, Baines JF, et al Meeting report of the pathogenesis of pemphigus and pemphigoid meeting in Munich, September 2016. Journal of Investigative Dermatology. 2017;137(6):1199–1203. doi: 10.1016/j.jid.2017.01.028. [DOI] [PubMed] [Google Scholar]
- 110.Schmidt J Current classification and management of inflammatory myopathies. Journal of Neuromuscular Diseases. 2018;5(2):109–129. doi: 10.3233/JND-180308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schober K, Müller TR, Gökmen F, Grassmann S, Effenberger M, Poltorak M, et al Orthotopic replacement of T-cell receptor α- and β-chains with preservation of near-physiological T-cell function. Nature Biomedical Engineering. 2019;3(12):974–984. doi: 10.1038/s41551-019-0409-0. [DOI] [PubMed] [Google Scholar]
- 112.Schultz L, Mackall C Driving CAR T cell translation forward. Science Translational Medicine. 2019;11(481):eaaw2127. doi: 10.1126/scitranslmed.aaw2127. [DOI] [PubMed] [Google Scholar]
- 113.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al Chimeric antigen receptor T cells in refractory B-cell lymphomas. The New England Journal of Medicine. 2017;377(26):2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sermer D, Brentjens R. 2019. CAR T-cell therapy: full speed ahead. Hematological Oncology, 37 Suppl 1: 95–100.
- 115.Shimizu A, Ishiko A, Ota T, Tsunoda K, Koyasu S, Amagai M, et al Ultrastructural changes in mice actively producing antibodies to desmoglein 3 parallel those in patients with pemphigus vulgaris. Archives of Dermatological Research. 2002;294(7):318–323. doi: 10.1007/s00403-002-0341-z. [DOI] [PubMed] [Google Scholar]
- 116.Siddiqi HF, Staser KW, Nambudiri VE Research techniques made simple: CAR T-cell therapy. Journal of Investigative Dermatology. 2018;138(12):2501–2504.e1. doi: 10.1016/j.jid.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 117.Sospedra M, Martin R Immunology of multiple sclerosis. Annual Review of Immunology. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 118.Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Science. 2003;94(11):965–973. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tang QZ, Henriksen KJ, Bi MY, Finger EB, Szot G, Ye JQ, et al In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. Journal of Experimental Medicine. 2004;199(11):1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tebbe B, Orfanos CE Epidemiology and socioeconomic impact of skin disease in lupus erythematosus. Lupus. 1997;6(2):96–104. doi: 10.1177/096120339700600204. [DOI] [PubMed] [Google Scholar]
- 121.Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. Journal of Autoimmunity. 2019;103:102289. doi: 10.1016/j.jaut.2019.05.017. [DOI] [PubMed] [Google Scholar]
- 122.Toni G, Berioli MG, Cerquiglini L, Ceccarini G, Grohmann U, Principi N, et al Eating disorders and disordered eating symptoms in adolescents with type 1 diabetes. Nutrients. 2017;9(8):906. doi: 10.3390/nu9080906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Torpy JM, Lynm C, Glass RM JAMA patient page. Type 1 diabetes. JAMA. 2007;298(12):1472. doi: 10.1001/jama.298.12.1472. [DOI] [PubMed] [Google Scholar]
- 124.Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, et al Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. The Journal of Immunology. 2003;170(4):2170–2178. doi: 10.4049/jimmunol.170.4.2170. [DOI] [PubMed] [Google Scholar]
- 125.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al CD19 CAR-T cells of defined CD4+: CD8+ composition in adult B cell all patients . The Journal of Clinical Investigation. 2016;126(6):2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.van Venrooij WJ, van Beers JJBC, Pruijn GJM Anti-CCP antibodies: the past, the present and the future. Nature Reviews Rheumatology. 2011;7(7):391–398. doi: 10.1038/nrrheum.2011.76. [DOI] [PubMed] [Google Scholar]
- 127.Vossenaar ER, Després N, Lapointe E, van der Heijden A, Lora M, Senshu T, et al Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Research & Therapy. 2004;6(2):R142–R150. doi: 10.1186/ar1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wahren-Herlenius M, Dörner T Immunopathogenic mechanisms of systemic autoimmune disease. The Lancet. 2013;382(9894):819–831. doi: 10.1016/S0140-6736(13)60954-X. [DOI] [PubMed] [Google Scholar]
- 129.Wang LF, Wang FS, Gershwin ME Human autoimmune diseases: a comprehensive update. Journal of Internal Medicine. 2015;278(4):369–395. doi: 10.1111/joim.12395. [DOI] [PubMed] [Google Scholar]
- 130.Wang XX, Cheng H, Shen YG, Li B Metabolic choice tunes Foxp3+ regulatory T cell function. Advances in Experimental Medicine and Biology. 2021;1278:81–94. doi: 10.1007/978-981-15-6407-9_5. [DOI] [PubMed] [Google Scholar]
- 131.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350(6258):aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yamaguchi T, Wing JB, Sakaguchi S Two modes of immune suppression by Foxp3+ regulatory T cells under inflammatory or non-inflammatory conditions . Seminars in Immunology. 2011;23(6):424–430. doi: 10.1016/j.smim.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 133.Yang W, Ng P, Zhao M, Hirankarn N, Lau CS, Mok CC, et al Population differences in SLE susceptibility genes: STAT4 and BLK, but not PXK, are associated with systemic lupus erythematosus in Hong Kong Chinese . Genes & Immunity. 2009;10(3):219–226. doi: 10.1038/gene.2009.1. [DOI] [PubMed] [Google Scholar]
- 134.Yaniv G, Twig G, Shor DBA, Furer A, Sherer Y, Mozes O, et al A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmunity Reviews. 2015;14(1):75–79. doi: 10.1016/j.autrev.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 135.Yanovsky RL, McLeod M, Ahmed AR Treatment of pemphigus vulgaris: part 2 - emerging therapies. Expert Review of Clinical Immunology. 2019;15(10):1061–1071. doi: 10.1080/1744666X.2020.1672539. [DOI] [PubMed] [Google Scholar]
- 136.Yu JX, Hubbard-Lucey VM, Tang J The global pipeline of cell therapies for cancer. Nature Reviews Drug Discovery. 2019;18(11):821–822. doi: 10.1038/d41573-019-00090-z. [DOI] [PubMed] [Google Scholar]
- 137.Zhang B, Wang Y, Yuan YS, Sun JQ, Liu LL, Huang D, et al In vitro elimination of autoreactive B cells from rheumatoid arthritis patients by universal chimeric antigen receptor T cells. Annals of the Rheumatic Diseases. 2021a;80(2):176–184. doi: 10.1136/annrheumdis-2020-217844. [DOI] [PubMed] [Google Scholar]
- 138.Zhang EH, Xu HM A new insight in chimeric antigen receptor-engineered T cells for cancer immunotherapy. Journal of Hematology & Oncology. 2017;10(1):1. doi: 10.1186/s13045-016-0379-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang H, Zhao P, Huang H Engineering better chimeric antigen receptor T cells. Experimental Hematology & Oncology. 2020;9(1):34. doi: 10.1186/s40164-020-00190-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang L, Sosinowski T, Cox AR, Cepeda JR, Sekhar NS, Hartig SM, et al Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II: peptide complex modulate the progression of autoimmune diabetes. Journal of Autoimmunity. 2019;96:50–58. doi: 10.1016/j.jaut.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang QF, Lu WH, Liang CL, Chen YC, Liu HZ, Qiu FF, et al Chimeric antigen receptor (CAR) Treg: a promising approach to inducing immunological tolerance. Frontiers in Immunology. 2018;9:2359. doi: 10.3389/fimmu.2018.02359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang WQ, Liu X, Zhu YC, Liu XN, Gu YT, Dai XY, et al Transcriptional and posttranslational regulation of Th17/Treg balance in health and disease. European Journal of Immunology. 2021b;51(9):2137–2150. doi: 10.1002/eji.202048794. [DOI] [PubMed] [Google Scholar]
- 143.Zheng WT, O'Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, et al PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia. 2018;32(5):1157–1167. doi: 10.1038/s41375-017-0008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhou XY, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. 2009. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nature Immunology, 10 (9): 1000–1007.
- 145.Zhou XH, Kong N, Wang JL, Fan HM, Zou HJ, Horwitz D, et al Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu . The Journal of Immunology. 2010;185(5):2675–2679. doi: 10.4049/jimmunol.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhuang CL, Guan XH, Ma H, Cong H, Zhang WN, Miao ZY Small molecule-drug conjugates: a novel strategy for cancer-targeted treatment. European Journal of Medicinal Chemistry. 2019;163:883–895. doi: 10.1016/j.ejmech.2018.12.035. [DOI] [PubMed] [Google Scholar]
- 147.Zmievskaya E, Valiullina A, Ganeeva I, Petukhov A, Rizvanov A, Bulatov E Application of CAR-T cell therapy beyond oncology: autoimmune diseases and viral infections. Biomedicines. 2021;9(1):59. doi: 10.3390/biomedicines9010059. [DOI] [PMC free article] [PubMed] [Google Scholar]