

Abstract
Organisms produce high levels of reactive oxygen species (ROS) to kill pathogens or act as signaling molecules to induce immune responses; however, excessive ROS can result in cell death. To maintain ROS balance and cell survival, mitophagy selectively eliminates damaged mitochondria via mitophagy receptors in vertebrates. In marine invertebrates, however, mitophagy and its functions remain largely unknown. In the current study, Vibrio splendidus infection damaged mitochondrial morphology in coelomocytes and reduced mitochondrial membrane potential (ΔΨm) and mitophagosome formation. The colocalization of mitochondria and lysosomes further confirmed that lipopolysaccharide (LPS) treatment increased mitophagy flux. To explore the regulatory mechanism of mitophagy, we cloned Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), a common mitophagy receptor, from sea cucumber Apostichopus japonicus (AjBNIP3) and confirmed that AjBNIP3 was significantly induced and accumulated in mitochondria after V. splendidus infection and LPS exposure. At the mitochondrial membrane, AjBNIP3 interacts with microtubule-associated protein 1 light chain 3 (LC3) on phagophore membranes to mediate mitophagy. After AjBNIP3 interference, mitophagy flux decreased significantly. Furthermore, AjBNIP3-mediated mitophagy was activated by ROS following the addition of exogenous hydrogen peroxide (H2O2), ROS scavengers, and ROS inhibitors. Finally, inhibition of BNIP3-mediated mitophagy by AjBNIP3 small interfering RNA (siRNA) or high concentrations of lactate increased apoptosis and decreased coelomocyte survival. These findings highlight the essential role of AjBNIP3 in damaged mitochondrial degradation during mitophagy. This mitophagy activity is required for coelomocyte survival in A. japonicus against V. splendidus infection.
Keywords: Apostichopus japonicus, Mitophagy, Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3, Reactive oxygen species, Microtubule-associated protein 1 light chain 3
INTRODUCTION
Invertebrate species depend on a series of innate immune responses that mediate physiological and pathological processes to eliminate pathogen invasion. Reactive oxygen species (ROS) are primarily produced as a byproduct of respiratory burst in innate immune cells and are critical for resisting pathogen invasion and promoting bactericidal capacity (Deng et al., 2009; Liu et al., 2010). Recently, mitochondrial ROS (mtROS) have also been shown to be important contributors to bactericidal capacity and immune signaling in innate immune cells (West et al., 2011). Typically, low levels of ROS are controlled by an efficient system of endogenous antioxidant scavengers. However, redox homeostasis in host cells is disrupted under excessive generation of ROS, which are not detoxified by cellular antioxidants (Fleury et al., 2002). Excessive ROS oxidize mitochondrial DNA, proteins, and lipids, resulting in strand breaks and base modifications, which lead to various pathologies, including cancer, neurological diseases, and cell death (Bjelland & Seeberg, 2003). Massive accumulation of ROS in the mitochondria can also trigger the release of additional ROS, further increasing oxidative stress and cell death (Suzuki et al., 2001). Moreover, the release of ROS from one mitochondrion can trigger release from its neighbors, resulting in a positive-feedback loop (Zorov et al., 2000). In contrast, mtROS production and mitochondrial DNA (mtDNA) damage can induce NLRP3 activation and subsequent NLRP3 inflammasome assembly (Zhou et al., 2011). Furthermore, mtROS regulate lipopolysaccharide (LPS)-mediated production of different cytokines, including proIL-1b (Mills & O’Neill, 2016; Tannahill et al., 2013). However, the functions of excessive mtROS and balance in invertebrates remain largely unknown.
Mitophagy, a term originally proposed by Lemasters (2005), is a cell survival mechanism responsible for clearing damaged mitochondria by forming double-membrane autophagosomes (Tolkovsky, 2009). Damaged mitochondria can generate excessive ROS, impair mitochondrial metabolic functions, and trigger apoptosis (Youle & Bliek, 2012). However, ROS can induce mitophagy, which, in turn, reduces ROS levels. Defective removal of damaged mitochondria can lead to ROS accumulation in the cell, resulting in hyperactivation of inflammatory signaling pathways, chronic systemic inflammation, and development of inflammatory diseases (Guo et al., 2014; Wu et al., 2016). Currently, the mechanisms of mitophagy can be classified into Parkin-dependent and Parkin-independent mitophagy (Youle & Narendra, 2011), with the latter mediated by mitophagy receptors. Mitophagy receptors are proteins localized to the outer mitochondrial membrane. Different types of mitophagy receptors have been identified in mammalian cells (Liu et al., 2014). A common feature of mitophagy receptors is that they harbor an LC3-interacting region (LIR) that interacts with microtubule-associated protein 1 light chain 3 (LC3), thus promoting the sequestration of mitochondria into the isolation membrane or phagophore (Wei et al., 2015).
Although other mitophagy proteins exist, Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), a vital mitophagy receptor, maintains cellular survival during hypoxia and nutritional deficiency (Wende et al., 2016). Accumulating evidence indicates that BNIP3-dependent mitophagy is required to limit ROS production and promote cell survival in cardiomyocytes, tumor cells, and hepatocytes (Merjaneh et al., 2017; Zhou et al., 2018a, 2018b). BNIP3-mediated mitophagy plays a neuroprotective role in neurodegenerative diseases such as Parkinson’s disease (Awan et al., 2014). BNIP3 was first discovered as an atypical member of the BH3-only proteins that interacted with adenovirus E1B-19K (Chen et al., 1997). The BH3 domain determines the biological roles and functions of BNIP3. Kubli et al. (2008) discovered that BNIP3 contains a single conserved cysteine residue in the N-terminus and a transmembrane domain (TMD, amino acid (aa) 164–184) in the C-terminal, with the latter being essential for BNIP3 localization to the outer mitochondrial membrane. At the outer mitochondrial membrane, BNIP3 interacts with LC3 to promote the sequestration and degradation of dysfunctional mitochondria (Yurkova et al., 2008). In addition, BNIP3 regulates mitophagy through other mechanisms. For example, BNIP3 inhibits PINK1 proteolytic processing to promote PINK1/Parkin-mediated mitophagy (Zhang et al., 2016). However, the roles and mechanisms of BNIP3 in invertebrates have not yet been studied.
The sea cucumber (Apostichopus japonicus) is a commercially important invertebrate species in Chinese aquaculture and uniquely depends on innate immunity to defend against invasive pathogens (Ma et al., 2006). Mitophagy, a cytoprotective negative feedback mechanism, can selectively eliminate damaged mitochondria and excessive ROS (Lu et al., 2013). In our previous research, we found that coelomocytes in A. japonicus generate a considerable ROS to promote inflammation and eradicate invading pathogens (Sun et al., 2020). However, whether mitophagy can be activated to eliminate damaged mitochondria and excessive ROS in A. japonicus remains poorly understood. Additionally, although BNIP3 has gained much attention in mitophagy (Ney, 2015; Roperto et al., 2019), it has not yet been determined in sea cucumbers. In the current study, we investigated the mechanism underlying BNIP3 mitigation of mitochondrial damage during pathogen invasion in A. japonicus. We first observed morphological changes in mitochondria and the occurrence of mitophagy after Vibrio splendidus infection. We then detected mitochondrial membrane potential (ΔΨm) and mitophagy flux of coelomocytes after LPS challenge at the cellular level. Subsequently, we investigated the significance of BNIP3-mediated mitophagy in pathogen-induced immune responses using in vitro and in vivo models to clarify the mechanism. Further investigation of the regulatory mechanisms of BNIP3 in mitophagy may reveal a novel negative feedback loop between BNIP3 expression and mitochondrial ROS generation and facilitate the development of novel therapeutic platforms for combating ROS-mediated inflammatory diseases.
MATERIALS AND METHODS
Ethics statement
The sea cucumbers and ICR mice used in the present study were commercially cultured animals. All experiments were conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All study protocols were approved by the Animal Ethics Committee of Ningbo University (Permit No.: 10168).
Experimental animals and V. splendidus challenge
Healthy sea cucumbers (weight: 125±15 g) were obtained from the Dalian Pacific Aquaculture Company (Dalian, China). All animals were acclimatized in 30 L of aerated natural seawater for three days (temperature of 16 °C and salinity of 28±1). For the immune-challenge experiment, 90 individuals were randomly distributed into six tanks. One tank served as the control, and the other five tanks were treated with live V. splendidus, the major pathogen of skin ulceration syndrome (SUS), at a final concentration of 1×107 CFU/mL. Coelomic fluids were randomly collected from three individuals (pooled as one sample), with five replicates performed in the control and challenge groups at 0, 6, 24, 48, and 72 h, respectively. The coelomocytes were filtered using a 200 mesh cell cribble to remove large tissue debris, then mixed with anticoagulant solution (0.02 mol/L EGTA, 0.48 mol/L NaCl, 0.019 mol/L KCl, and 0.068 mol/L Tri-HCl, pH 7.6) at a 1:1 (v:v) ratio and centrifuged at 800 g and 16 ℃ for 10 min. All harvested cells were washed twice with isotonic buffer (0.001 mol/L EGTA, 0.53 mol/L NaCl, and 0.01 mol/L Tri-HCl, pH 7.6) and ground into powder in liquid nitrogen using a mortar and pestle for time-course expression analysis at the mRNA and protein level. Finally, all samples were stored at −80 ℃ for RNA extraction.
LPS-exposed primary coelomocytes
Primary coelomocytes were isolated and cultured in vitro as described in our previous study (Zhang et al., 2014). Briefly, coelomic fluids from healthy sea cucumbers were filtered, mixed with anticoagulant solution, and centrifuged at 800 g for 10 min at 16 ℃ to collect coelomocytes. The harvested cells were washed twice with isotonic buffer and resuspended in Leibovitz’s L-15 cell culture medium (Invitrogen, USA) containing penicillin (100 U/mL) and streptomycin sulfate (100 mg/mL). The L-15 cell culture medium (500 µL, containing 1×106 cells) was dispensed into 24-well culture microplates and incubated at 16 °C. After incubation for 12 h, the cells were stimulated with 10 µg/mL LPS (Sigma, USA) purified from Escherichia coli (055: B5) for 1, 3, 6, 12, and 24 h. Cells untreated with LPS served as the controls.
Detection of mitochondrial structure and mitophagosome by transmission electron microscopy (TEM)
To confirm activation of mitophagy during pathogen infection, the sea cucumbers were exposed to V. splendidus at a final concentration of 1×107 CFU/mL for 48 h. Changes in mitochondrial morphology and mitophagosome appearance were then analyzed via TEM. Briefly, the coelomocytes were harvested by centrifugation at 800 g for 5 min at 16 °C, fixed in 2.5% glutaraldehyde overnight at 4 °C, and post-fixed for 1 h at room temperature with 1% osmium tetroxide. The fixed cells were washed with 0.1 mol/L phosphate-buffered saline (PBS, pH 7.4), dehydrated with ascending concentrations of ethanol (30% for 15 min, 50% for 15 min, 70% for 15 min, 90% for 15 min) and anhydrous acetone for 15 min three times, embedded in an epoxy resin (EMbed-812; Electron Microscopy Sciences, USA), and sliced with an ultramicrotome. Ultra-thin sections were collected on 300 mesh copper grids coated with formvar, then stained with uranyl acetate and lead citrate. The mitochondrial structure and mitophagosomes were observed using an electron microscope (H7650, Hitachi, Japan).
Measurement of ΔΨm
Under certain conditions, mitochondria lose their membrane potential (which reflects mitochondrial function) before being encapsulated in autophagosomes (Elmore et al., 2001). Here, ΔΨm analysis of coelomocytes was performed using JC-1 fluorescence dye (Beyotime, China). L-15 cell culture medium (500 µL containing 1×106 coelomocytes) was seeded into 24-well plates and treated with LPS for 0, 1, 3, 6, 12, and 24 h. According to the manufacturer’s instructions, the cells treated with LPS were incubated with 10 mg/mL JC-1 stain (lipophilic cationic probe 5,5’,6,6’-tetrachloro-l,l’,3,3’-tetraethylbenzimidazolcarbocyanine iodide, Beyotime, China) in the dark for 20 min at 37 °C, washed with 1×JC-1 staining buffer, and analyzed using flow cytometry (Becton Dickinson Biosciences, USA). The mitochondrial depolarizing agent carbonyl cyanide 3-chlorophenylhydrazone (CCCP) was used as a positive control (data not shown). The JC-1 monomers and polymers were visualized as green and red fluorescence, respectively. The red/green JC-1 fluorescence ratio was used as a sensitive measure of ΔΨm (Métivier et al., 1998).
Colocalization of mitochondria and lysosomes
Primary coelomocytes seeded in 24-well plates were treated with LPS for 0, 1, 3, 6, 12, and 24 h, and then collected for later use. Lyso-Tracker Red (Beyotime, China) and Mito-Tracker Green (Beyotime, China) were added to the cell culture medium at ratios of 1:13 333–1:20 000 and 1:5 000–1:50 000 to obtain final mixed working solution concentrations of 50–75 nmol/L and 20–200 nmol/L, respectively. The original coelomocyte medium was replaced with a mixed working solution, and cells were loaded with Mito-Tracker Green and Lyso-Tracker Red for 30–120 min according to the manufacturer’s protocols. The staining mixed solution was then removed, fresh cell culture was added, and the cells were observed under a laser confocal microscope (Zeiss LSM510, Germany). Green mitochondria located with red lysosomes formed orange fluorescent spots. The number of orange spots was then measured to quantify mitophagy.
Cloning and analysis of AjBNIP3
Based on the corresponding expressed sequence tags (ESTs) in our transcription data (Zhang et al., 2014), primers AjBNIP3-F (5'-ATGACGAGCACACCACGTAG-3') and AjBNIP3-R (5'-TCACATGCTGGATTTGGGC-3') were used to amplify the complete open reading frame (ORF) of AjBNIP3 using a PrimeScript RT Reagent Kit with gDNA Eraser (Takara, Japan). Amplified polymerase chain reaction (PCR) products were purified, ligated into the pMD19-T simple vector (Takara, Japan), and transformed into competent E. coli DH5α cells (Takara, Japan). Positive recombinant clones were verified by PCR screening and sequenced by Sangon Biotech (Shanghai, China). The cDNA sequence of AjBNIP3 was analyzed using the BLAST algorithm from the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov/blast), and the deduced aa sequences were analyzed using the Expert Protein Analysis System (http://www.expasy.org/). The molecular mass and theoretical isoelectric point (pI) of AjBNIP3 were calculated using the ProtParam tool (http://www.expasy.ch/tools/protparam.html). Domains in these aa sequences were detected using the Simple Modular Architecture Research Tool (SMART) (http://www.smart.emblheidelbergde/). Multiple alignment analysis of each protein was performed using DNAMAN software. A phylogenetic tree was constructed using the neighbor-joining method based on the distinct types of BNIP3 in MAGE v5.1 with 1 000 bootstrap replicates.
Real-time quantitative expression of AjBNIP3
Accumulating evidence suggests that the BNIP3 pathway is critical for autophagic degradation of damaged mitochondria (Wende et al., 2016). To investigate the involvement of AjBNIP3 in V. splendidus and LPS-induced mitophagy, we measured the mRNA expression levels of AjBNIP3 in response to V. splendidus infection and LPS exposure in vivo and in vitro. Coelomocytes from five samples in each group were collected at 0, 6, 24, 48, and 72 h, and primary coelomocytes were collected at 0, 1, 3, 6, 12, and 24 h in triplicate. Total RNA was extracted using TRIzol reagent (Takara, Japan) according to the manufacturer’s protocols, and cDNA was prepared using a PrimeScript RT Reagent Kit with gDNA Eraser (Takara, Japan). Amplification was conducted in a 20 μL reaction volume containing 8 μL of 1:50 diluted cDNA, 0.8 μL of each primer (F: 5'-TTAACAGCCGTCTGATCGTC-3' and R: 5'-CCAAAGTGAGGGAGGGAGA-3'), 10 μL of SYBR Green, and 0.4 μL of ROX (Takara, Japan). The PCR conditions were as follows: 95 °C for 5 min, then 40 cycles at 95 °C for 15 s, 60 °C for 20 s, and 72 °C for 20 s, followed by melting curve analysis. The baseline was automatically set by the software to maintain consistency. Each reaction was performed in triplicate, and expression levels were normalized to the expression of the Ajβ-actin gene (F: 5'-CCATTCAACCCTAAAGCCAACA-3' and R: 5'-ACACACCGTCTCCTGAGTCCAT-3'), which was used as an internal housekeeping control. The 2-ΔΔCT method was used to analyze the relative expression levels of candidate genes (Livak & Schmittgen, 2001), and the obtained value denoted the n-fold difference relative to the calibrator.
Polyclonal antibody preparation of AjBNIP3 and western blot analysis
The cDNA fragment encoding the predicted mature peptide of AjBNIP3 was amplified using specific primers AjBNIP3-F and AjBNIP3-R. The purified PCR products were inserted into the pET-28a expression vector with BamH I and Not I, then transformed intoE. coli BL (DE3) cells. After induction with isopropyl thiogalactoside (1 mmol/L) and purification by His-TrapTM Ni-agarose, the His-tagged recombinant AjBNIP3 (rAjBNIP3) protein was obtained. In addition, male ICR mice (20 g; 6–8 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (China) and housed for one week in sterile individually ventilated cages under a 12 h light/dark cycle at 22±2 °C. The mice were allowed to feed once every two days and were provided with distilled drinking water freely. For polyclonal antibody preparation, 100 μg of purified rAjBNIP3 protein was injected into a 4-week-old ICR mouse with an equal volume of complete Freund’s adjuvant. One week later, all mice were intramuscularly injected twice with 75 μg of rAjBNIP3 purified protein with an equal volume of incomplete Freund’s adjuvant at one-week intervals. After the last injection, antiserum was harvested from the eyes and stored at −80 °C until further western blot analysis. The mice were deprived of water and food the day before blood collection, then finally euthanized via cervical dislocation. In the western blot experiments, the collected coelomocytes were homogenized in 1×RIPA buffer supplemented with protease inhibitors (PMSF, 100 mmol/L, Beyotime, China). Protein concentration was determined using a bicinchoninic acid (BCA) assay (Sigma-Aldrich, USA). Protein lysates were separated using 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.22 μmol/L polyvinylidene difluoride membranes (Millipore Sigma, USA). The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline with Tween 20 for 2 h and incubated with anti-AjBNIP3 from mouse serum (1:200 dilution) overnight at 4 °C. Secondary horseradish peroxidase (HRP)-conjugated antibodies (1:10 000 dilution, Sangon Biotech, China) were used to treat membranes for 2 h at room temperature, followed by incubation with an ECL reagent (Bio-Rad, USA) for 3 min at room temperature. Ajβ-actin was used as an internal control.
Colocalization of AjBNIP3 and mitochondria
To confirm the regulatory relationship between AjBNIP3 and mitophagy, we studied the colocalization relationship between AjBNIP3 and mitochondria. Briefly, coelomocytes cultured in 6-well plates were treated with LPS for 24 h at 16 °C. The treated cells were then stained with Mito-Tracker Red CMXRos (200 nmol/L) for 30 min, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 5% bovine serum albumin (BSA). The cells were incubated with anti-AjBNIP3 from mouse serum (1:200 dilution) at 4 °C overnight. After washing three times with PBS, the cells were incubated with secondary antibody Alexa Fluor 488 conjugated goat anti-mouse IgG (1:500 dilution, Beyotime, China) for 1.5 h at room temperature. After staining with 4’,6-diamidino-2-phenylindole (DAPI) for 5 min, the cells were visualized using a laser confocal microscope.
Effects of exogenous hydrogen peroxide (H2O2) on AjBNIP3 mRNA and protein expression and mitophagy
To show the effects of excessive ROS on BNIP3-mediated mitophagy, coelomocytes were seeded in 6-well plates and treated with gradually increasing concentrations of H2O2 (0, 0.002, 0.02, 0.2, 2, and 20 μmol/L) for 12 h, which can induce mitochondrial stress and mtROS release (Zmijewski et al., 2010). Some cells were then collected for mRNA and protein expression analysis of AjBNIP3, and others were used to detect mitophagy flux by colocalizing mitochondria and lysosomes, as described above.
Effects of ROS inhibitors and scavengers on expression of AjBNIP3 and mitophagy
Several sources of ROS exist in cells, including NADPH oxidase (NOX) and mitochondria (Adam-Vizi & Chinopoulos, 2006). To further confirm that excessive ROS can activate BNIP3-mediated mitophagy, coelomocytes were seeded in 6-well plates and treated with ROS inhibitors (apocynin to inhibit ROS generation from NOX (10 mmol/L) and 5-hydroxydecanoate (5-HD, 100 μmol/L) to inhibit ROS production from mitochondria) and ROS scavenger (N-acetyl-l-cysteine (NAC), 1 mmol/L) for 12 h. To determine intracellular ROS and mtROS levels after LPS treatment, the coelomocytes were treated with peroxide-sensitive fluorescent probe 27-dichlorofluorescin diacetate (DCFH-DA, Sigma, USA) and MitoSOX Red mitochondrial superoxide indicator (Yeasen, China), then washed with PBS and observed using laser scanning confocal microscopy. Subsequently, some cells were collected for mRNA and protein expression analysis of AjBNIP3, and other cells were used to detect mitophagy flux by colocalizing mitochondria and lysosomes, as described above.
Co-immunoprecipitation of AjBNIP3 and AjLC3
In mammalian cells, LC3 is a critical component of mitophagosomes and BNIP3 is a mitophagy receptor (Yurkova et al., 2008). As a mitochondrial outer membrane protein, BNIP3 can interact with LC3 to recruit autophagosomes and then degrade identified targets (Zhou et al., 2018a). To further explore the interaction between AjBNIP3 and AjLC3, healthy sea cucumbers were treated with V. splendidus at a final concentration of 1×107 CFU/mL for 48 h, after which the coelomocytes were harvested for lysis. An equal amount of protein lysate was incubated with mouse anti-AjBNIP3 antibody overnight at 4 °C, followed by incubation with 40 µL of protein A/G agarose beads (Beyotime, China) for 3 h at 4 °C. The immune complexes were subjected to western blot analysis using rabbit anti-LC3 antibody (1:1 000 dilution; Abcam). Total protein lysates were also subjected to western blot analyses with anti-AjBNIP3 and anti-LC3 antibodies.
Microscale thermophoresis (MST) analysis of AjBNIP3 and AjLC3
To confirm whether AjBNIP3 interacts with AjLC3, MST was used for the first time to study the binding kinetics of recombinant His-AjBNIP3 to His-AjLC3. Fluorescent-labelled His-AjBNIP3 (10 μmol/L) was dissolved in a buffer containing 50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 10 mmol/L MgCl2, and 0.05% (v/v) Tween-20. A range of His-AjLC3 concentrations in the assay buffer (50 mmol/L Tris-HCl pH 7.8, 150 mmol/L NaCl, 10 mmol/L MgCl2, 0.05% Tween-20) were incubated with fluorescent-labelled protein (1:1, v/v) for 10 min at room temperature. A label-free instrument (NanoTemper Technologies GMBH, Germany) was used to detect changes in the size, charge, and conformation of the His-AjBNIP3 and His-AjLC3 proteins induced by binding. The His-AjLC3 and fluorescent-labelled His-AjBNIP3 mixture was loaded into the Monolith NT (NanoTemper, Germany). Label-free standard capillaries were measured with 20% LED power and 80% MST power. Finally, the curve was parameterized, and the dissociation constant (Kd) was calculated using the Kd fit function in Nano Temper Analysis v1.5.41.
Colocalization of AjBNIP3 and AjLC3 in coelomocytes
Coelomocytes were seeded in 6-well plates and treated with 10 μg/mL LPS for 12 h. Treated cells were washed with PBS, followed by fixation with 4% paraformaldehyde for 20 min and permeabilization with 0.1% Triton X-100. After blocking with 5% BSA for 60 min at room temperature, cells were incubated with primary antibodies (mouse anti-AjBNIP3, 1:200; rabbit anti-LC3, 1:500) overnight at 4 °C, followed by secondary antibodies (Alexa Fluor 488 conjugated goat anti-mouse IgG; Cy3 conjugated goat anti-rabbit IgG) for 1.5 h at room temperature. Nuclear staining was performed using DAPI (Beyotime, China) for 5 min. Images were obtained using a laser confocal microscope.
Colocalization of AjBNIP3 and AjLC3 in HeLa and HEK 293T cells
The ORFs of AjBNIP3 and AjLC3 were ligated into the pCMV-N-mCherry vector (Beyotime, China) and pIRES2-EGFP vector (Beyotime, China) to construct the pCMV-N-mCherry-AjBNIP3 and pIRES2-EGFP-AjLC3 plasmids, respectively. Thereafter, HeLa cells and HEK 293T cells in 6-well plates at 70%–80% confluence were co-transfected with the recombinant plasmids using Lipofectamine 6000 (Beyotime, China) according to the manufacturer’s instructions. At 24 and 36 h post-transfection, nuclei of the HeLa and HEK 293T cells were stained with DAPI and the colocalization of Cherry-AjBNIP3 and GFP-AjLC3 was observed with a confocal microscope.
AjBNIP3 interference
The small interfering RNA (siRNA) targeting AjBNIP3 (F: 5'-CCAACAUGGAGAAGCUAUUTT-3'; R: 5'-AAUAGCUUCUCCAUGUUGGTT-3') and negative control (F: 5'-UUCUCCGAACGUGUCACGUTT-3'; R: 5'-ACGUGACACGUUCGGAGAATT-3') were purchased from GenePharma Company (China), and transfected with siRNA using Lipofectamine 6000 (Beyotime, China) according to the manufacturer’s recommended conditions. The siRNA transfection mix was added to the cells for 24 and 48 h. Total RNA was extracted from cell culture lysates using TRIzol reagent (Invitrogen, USA) following the manufacturer’s instructions, and total protein was homogenized using 1×RIPA buffer supplemented with protease inhibitor to detect mRNA and protein expression levels of AjBNIP3. After interfering with AjBNIP3, cells were stimulated with LPS for 12 h, with mitophagy (using mitochondria-lysosome colocalization and AjBNIP3-AjLC3 protein colocalization), ROS levels, and coelomocyte apoptosis and survival then determined.
Detection of coelomocyte apoptosis and survival
Mitophagy is a basic cellular function that allows organisms to remove damaged mitochondria and excessive ROS, thereby reducing apoptosis and promoting cell survival (Tolkovsky, 2009; Youle & Bliek, 2012). Here, an Annexin V-FITC/PI Apoptosis Assay Kit (Beyotime, China) was used to analyze coelomocyte apoptosis according to the manufacturer’s instructions. The cells transfected with siRNA and subsequently exposed to LPS for 12 h were incubated with Annexin V/FITC, followed by PI staining and flow cytometry analysis (Becton Dickinson Biosciences, USA). The MTT assay was conducted. In brief, cells were diluted to a concentration of 1×104 cells/mL, and a 100 μL cell suspension was then transferred to a 96-well microplate well and incubated at 16 °C for 12 h. After treatment with AjBNIP3 siRNA and subsequent LPS, absorbance was measured at 570 nm using a microplate reader. The assay was repeated three times. To verify the effect of BNIP3-mediated mitophagy on the survival rate of sea cucumber coelomocytes, we treated the cells with different concentrations of lactate (5, 10, and 15 mmol/L) and challenged them with LPS for 24 h, then used the MTT and apoptosis kits to detect cellular viability.
Statistical analysis
All data in the different experimental groups are expressed as mean±standard error of the mean (SEM). Data shown in this research were obtained from at least three independent experiments. One-way analysis of variance (ANOVA) was applied to discern significant differences between the control and experimental groups for relative mRNA expression levels. Significant differences were determined at *P<0.05 and**P<0.01.
RESULTS
Effects of V. splendidus infection and LPS exposure on mitochondrial damage and mitophagy in coelomocytes
As shown in Figure 1A, TEM observations indicated that the mitochondria showed a normal cylindrical appearance with well-preserved cristae in the control group, but showed signs of damage, including irregular swelling and decreased cristae, in the V. splendidus infection group. Simultaneously, compared with the control, we observed typical double-membrane and lamellar mitophagosomes in the perinuclear area in the coelomocytes with damaged mitochondria (Figure 1B). Furthermore, at the cellular level, JC-1 staining showed that LPS exposure induced disruption of ΔΨm in coelomocytes (Figure 1C). Compared with the control, LPS treatment for 3, 6, and 12 h (P<0.05) dramatically decreased the ΔΨm of coelomocytes. In addition, the colocalization of mitochondria and lysosomes in the coelomocytes revealed that LPS exposure increased the number of mitophagosomes. As shown inFigure 1D, less overlap between the mitochondria and lysosomes was observed before LPS treatment. However, LPS stimulation increased the colocalization of mitochondria and lysosomes, as indicated by orange fluorescence, suggesting the formation of mitophagosomes. The mitophagosomes in the LPS-exposed group displayed significant induction from 6 h.
Figure 1.

Vibrio splendidus infection and LPS challenge induced mitochondrial damage and mitophagy in coelomocytes
A: Representative TEM images of morphology of damaged mitochondria (arrowhead) in coelomocytes of A. japonicus infected by V. splendidus for 48 h. B: Representative TEM images of autophagosomes engulfing mitochondria (arrowhead) in coelomocytes of A. japonicus infected by V. splendidus for 48 h. C: ΔΨm of coelomocytes after LPS treatment for 0, 1, 3, 6, 12, and 24 h determined by JC-1 staining and flow cytometry. *: P<0.05;**: P<0.01,n=3. D: Immunofluorescence colocalization analysis of Lyso-Tracker Red with Mito-Tracker Green in coelomocytes after LPS exposure for 0, 1, 3, 6, 12, and 24 h, with DAPI (blue) staining to identify nuclei. M: Mitochondria.
Effects of AjBNIP3 on mitophagy induced by V. splendidus infection and LPS exposure in coelomocytes
We detected the mRNA and corresponding protein levels of AjBNIP3 in response to V. splendidus infection and LPS exposure in vivo and in vitro. Results showed that V. splendidus and LPS challenge increased the AjBNIP3 mRNA and protein expression levels in a time-dependent manner, as demonstrated by quantitative PCR and western blot analysis (Figure 2A). Briefly, AjBNIP3 transcription increased 2.03-fold (P<0.05) and 3.30-fold (P<0.01) compared with the control group after bacterial challenge for 24 and 48 h, respectively, and peaked at 72 h (3.49-fold,P<0.01). Similarly, up-regulation ofAjBNIP3 expression was also detected at each time point in the LPS-challenged primary cultured cells. Peak expression of AjBNIP3 was detected at 24 h with a 3.29-fold (P<0.01) increase compared with that of the control group. Moreover, immunofluorescence staining showed that LPS treatment increased the protein expression level of AjBNIP3 and colocalization of AjBNIP3 with mitochondria (Figure 2B), suggesting an accumulation of AjBNIP3 in mitochondria and increase in mitophagy. To confirm the necessity of AjBNIP3 for mitophagy, we transfected coelomocytes with AjBNIP3 siRNA and detected interference efficiency. As shown in Figure 2C, AjBNIP3 interference significantly down-regulated the AjBNIP3 mRNA level by 43% (P<0.01), and the protein level was similarly reduced. Under these conditions,AjBNIP3 interference markedly inhibited LPS-induced mitophagy flux in coelomocytes, as indicated by the decrease in mitochondria-lysosome colocalization (Figure 2D).
Figure 2.

Effects of AjBNIP3 on mitophagy induced by V. splendidus infection and LPS challenge in coelomocytes
A: Quantification and immunoblots of AjBNIP3 in response toV. splendidus infection and LPS challenge in vivo and in vitro. *: P<0.05;**: P<0.01,n=5. B: Immunofluorescence colocalization analysis of AjBNIP3 in coelomocytes after LPS treatment for 0, 6, and 12 h with Mito-Tracker Red CMXRos, with DAPI (blue) staining to identify nuclei. C: Quantification and immunoblots of AjBNIP3 in coelomocytes after 24 and 48 h of transfection with or without AjBNIP3 siRNA. *: P<0.05;**: P<0.01,n=3. D: Immunofluorescence colocalization analysis of Lyso-Tracker Red with Mito-Tracker Green in coelomocytes after AjBNIP3 interference and subsequent LPS exposure for 12 h, with DAPI (blue) staining to identify nuclei.
Activation of AjBNIP3-mediated mitophagy by ROS
We previously found that ROS expression is up-regulated under pathogenic stress (Sun et al., 2020), and found that AjBNIP3 and mitophagy show the same expression trend under the same conditions in the current study. Here, we found that H2O2 induced AjBNIP3 mRNA and protein expression levels in coelomocytes in a dose-dependent manner (Figure 3A). Notably, coelomocytes treated with 2 and 20 μmol/L H2O2 showed a significant increase (1.66-fold, P<0.01 and 1.61-fold,P<0.01) in AjBNIP3 mRNA and protein expression compared to coelomocytes treated with 0.002, 0.02, and 0.2 μmol/L H2O2. Moreover, to visualize the levels of AjBNIP3-mediated mitophagy in vitro, we performed immunofluorescence on different concentrations of H2O2-treated cells and found an increase in mitochondria- and lysosome-positive puncta per cell in the high-level H2O2-treated group (Figure 3B). Additionally, LPS-stimulated coelomocytes were treated with ROS inhibitors (apocynin and 5-HD) and scavenger NAC to confirm the key role of ROS in AjBNIP3-dependent mitophagy activation. Results indicated that intracellular ROS levels decreased markedly following 5-HD and NAC treatment (Figure 3C). Similarly, there was a significant decrease in mtROS levels after 5-HD and NAC treatment (Figure 3D). Importantly, 5-HD and NAC treatment significantly inhibited AjBNIP3 mRNA and protein expression (Figure 3E) as well as mitophagy flux (Figure 3F).
Figure 3.

Activation of AjBNIP3-mediated mitophagy by ROS
A: Quantification and immunoblots of AjBNIP3 in coelomocytes after treatment with different concentrations of H2O2 for 12 h. *: P<0.05;**: P<0.01,n=3. B: Immunofluorescence colocalization analysis of Lyso-Tracker Red with Mito-Tracker Green in coelomocytes after treatment with different concentrations of H2O2 for 12 h, with DAPI (blue) staining to identify nuclei. C: Intracellular ROS levels determined by DCFH-DA after treatment with ROS inhibitors and scavenger for 12 h. *: P<0.05;**: P<0.01,n=3. D: Mitochondrial ROS levels determined by MitoSOX staining after treatment with ROS inhibitors and scavenger for 12 h. E: Quantification and immunoblots of AjBNIP3 in coelomocytes after treatment with ROS inhibitors and scavenger for 12 h. *: P<0.05;**: P<0.01,n=3. F: Immunofluorescence colocalization analysis of Lyso-Tracker Red with Mito-Tracker Green in coelomocytes after treatment with ROS inhibitors and scavenger for 12 h, with DAPI (blue) staining to identify nuclei.
Initiation of mitophagy by AjBNIP3 and AjLC3 interactions
According to the above results, AjBNIP3 can specifically activate mitophagy in coelomocytes, but it is unclear how it targets mitochondria to the autophagosomes. Therefore, we cloned AjBNIP3 (GenBank accession No.: OL770272) and analyzed the aa sequence of AjBNIP3 (Figure 4A). The complete ORF of AjBNIP3 was 570 bp, which encoded 189 residues, and AjBNIP3 molecular mass was predicted to be 20.41 kDa. We discovered that AjBNIP3 contained a LIR (WVEL), BH3 domain (GLRNTEWIW) in the N-terminal, and transmembrane domain (VLRMVVPSLILTNLITLGLGIVI) in the C-terminal, which were highly conserved (Supplementary Figures S1, S2). SMART analysis also indicated that AjBNIP3, which contains a BNIP3 domain, belongs to the BH3-only family (Figure 4B). Next, we investigated whether mitochondria-targeted AjBNIP3 interacts with LC3. We first predicted the combination model of AjBNIP3 and AjLC3 (Figure 4C). Co-immunoprecipitation experiments also demonstrated that AjBNIP3 and AjLC3 were pulled down together (Figure 4D). In addition, MST assays were employed to verify the binding kinetics between AjBNIP3 and AjLC3, and the dissociation constant (Kd) of AjBNIP3 and AjLC3 was 2.82 μmol/L (Figure 4E). To determine whether AjBNIP3 binds to AjLC3 at a greater degree during pathogen infection, we examined protein interactions via immunofluorescence and found that LPS treatment increased the colocalization of AjBNIP3 and AjLC3 compared with the control samples (Figure 4F). After AjBNIP3 interference, the interaction was markedly inhibited by LPS challenge in the coelomocytes, as indicated by a decrease in AjBNIP3-AjLC3 colocalization (Figure 4G). Finally, as shown in Figure 4H, the cotransfection experiments in HeLa and HEK 293T cells revealed that mCherry-AjBNIP3 colocalized with GFP-AjLC3, and fluorescence-labelled proteins increased significantly after cotransfection for 36 h compared with cells transfected for 24 h.
Figure 4.

Initiation of mitophagy by AjBNIP3 and AjLC3 interactions
A: Amino acid (aa) sequence analysis of AjBNIP3. Entire deduced aa sequence is depicted using single letter codes above corresponding nucleotide sequence. LIR is marked in green and indicated in bold. BH3 domain is marked in pink and indicated in bold. C-terminal transmembrane domain is marked in red and indicated in bold. B: Predicted 2D structural model of AjBNIP3. C: Predicted combination model of AjBNIP3 and AjLC3. D: Co-immunoprecipitation of AjBNIP3 and AjLC3 after V. splendidus infection for 48 h. E: Microscale thermophoresis (MST) assays of interactions between His-AjBNIP3 and His-AjLC3. Solid curve is the fit of data points to the standard Kd-fit function. Experiments were repeated at least three times. F: Immunofluorescence colocalization analysis of AjBNIP3 and AjLC3 after LPS treatment for 12 h, with DAPI (blue) staining to identify nuclei. G: Immunofluorescence colocalization analysis of AjBNIP3 and AjLC3 in coelomocytes after transfection with AjBNIP3 siRNA and subsequent exposure to LPS for 12 h, with DAPI (blue) staining to identify nuclei. H: Cotransfection of pCMV-N-Cherry-AjBNIP3 and pIRES2-EGFP-AjLC3 in HeLa and HEK 293T cells for 24 and 36 h, with DAPI (blue) staining to identify nuclei.
Protective effects of AjBNIP3-mediated mitophagy on coelomocyte survival
As seen in Figure 5A, AjBNIP3 interference affected coelomocyte survival in the interference group (66%, P<0.01) compared with the control group. Consistently,AjBNIP3 knockdown also significantly increased the apoptosis rate (1.27-fold, P<0.05,Figure 5B). To explain the beneficial effects of AjBNIP3-mediated mitophagy in protecting cell survival, we treated LPS-challenged coelomocytes to different concentrations of lactate, which can suppress BNIP3-mediated mitophagy (Zhu et al., 2019b). As shown in Figure 5C, the AjBNIP3 mRNA expression level increased significantly in the coelomocytes treated with 5 and 10 mmol/L lactate (2.32-fold (P<0.01) and 1.37-fold (P<0.05), respectively), but not in those treated with 15 mmol/L lactate. Similarly, the protein expression levels of AjBNIP3 after LPS challenge were suppressed by high concentrations of lactate. The mitophagy level in coelomocytes stimulated with LPS also increased. While the addition of 5 mmol/L lactate had no significant effect on mitophagy level, high concentrations of lactate inhibited mitophagy (Figure 5D). Subsequently, the apoptosis rate (Figure 5E) in coelomocytes treated with high concentrations of lactate increased significantly. Compared with the untreated group, apoptosis in the coelomocytes treated with 5 mmol/L lactate increased by 1.25-fold (P<0.05), and apoptosis in the coelomocytes treated with a high concentration of lactate increased by 2.65-fold (P<0.01). In contrast, compared with the control group, the survival rate of coelomocytes treated with 5 mmol/L lactate decreased by 32% (P<0.05) and the survival rates of coelomocytes treated with high concentrations of lactate significantly decreased by 78% (P<0.01) (Figure 5F).
Figure 5.

AjBNIP3-mediated mitophagy protects coelomocyte survival by reducing ROS production
A: Cell survival was determined after transfection with AjBNIP3 siRNA and subsequent LPS exposure for 12 h. *: P<0.05;**: P<0.01,n=3. B: Apoptosis rate was determined using an Annexin V-FITC/PI apoptosis assay kit after transfection with AjBNIP3 siRNA and subsequent LPS exposure for 12 h. *: P<0.05;**: P<0.01,n=3. C: Quantification and immunoblots of AjBNIP3 in coelomocytes after treatment with different concentrations of lactate for 24 h. *: P<0.05;**: P<0.01,n=3. D: Detection of mitophagy flux in coelomocytes after treatment with different concentrations of lactate for 24 h. E: Apoptosis rate was determined using an Annexin V-FITC/PI apoptosis assay kit after treatment with different concentrations of lactate for 24 h. *: P<0.05;**: P<0.01,n=3. F: Survival rate was determined using an MTT kit after treatment with different concentrations of lactate for 24 h. *: P<0.05;**: P<0.01,n=3.
DISCUSSION
Although low levels of ROS serve as regulators of cell signaling, high levels of ROS can cause oxidative stress, leading to various diseases and injury, including cancer, neurological disorders, and cell death (Bjelland & Seeberg, 2003). Mitochondria are the principal sites of ROS production in mammalian cells. In dysfunctional mitochondria, ROS production is increased due to disrupted electron transport along the respiratory chain and decreased oxygen formation (Mammucari & Rizzuto, 2010; Zorov et al., 2014). Fortunately, ROS produced by damaged mitochondria can induce mitophagy, which, in turn, eliminates excessive ROS (Scherz-Shouval & Elazar, 2011). As an invertebrate, A. japonicus relies on innate immunity only and uses ROS as the main bactericidal weapon in defense against pathogens (Sun et al., 2020). However, excessive ROS may cause apoptosis and affect coelomocyte survival. Whether mitophagy can be activated to eliminate damaged mitochondria and excessive ROS in A. japonicus remains poorly understood. In this study, we first demonstrated that V. splendidus infection and LPS challenge can induce mitochondrial injury and increase mitophagosomes. We subsequently identified a mitophagy receptor in A. japonicus, i.e., AjBNIP3, located on the mitochondrial membrane. Interestingly, high levels of ROS activated AjBNIP3-mediated mitophagy. Mitochondria-localized AjBNIP3 interacted with AjLC3 on the autophagosome to induce mitochondrial removal and promote coelomocyte survival (Figure 6). To the best of our knowledge, this is the first study to confirm the mechanisms underlying ROS-mediated BNIP3-dependent mitophagy in lower invertebrate sea cucumbers against pathogen infection.
Figure 6.
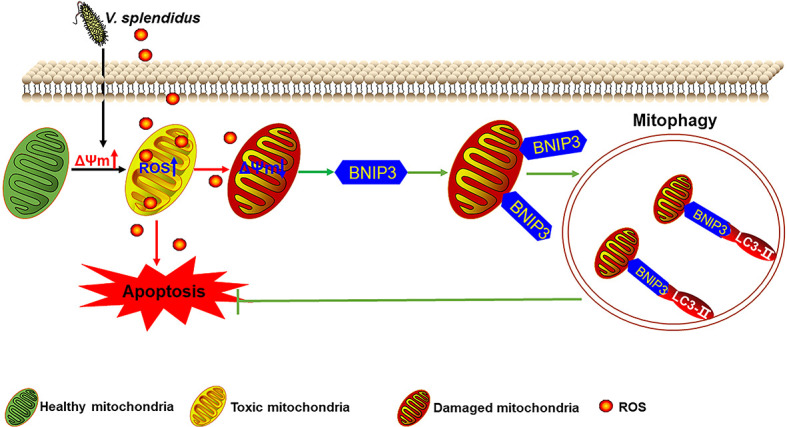
Role of ROS-mediated AjBNIP3-dependent mitophagy towards V. splendidus infection
Vibrio splendidus infection may increase mitochondrial membrane potential (ΔΨm) to produce ROS, which directly kill pathogens and cause mitochondrial injury, thereby decreasing ΔΨm. To eliminate damaged mitochondria, excessive ROS can activate mitophagy receptor BNIP3 expression and degrade damaged mitochondria via binding to LC3 in autophagosomes.
As a selective form of macroautophagy, mitophagy is the only known pathway that removes damaged and dysfunctional mitochondria in response to stresses such as pathogen infection and nutrient deprivation (Zhang et al., 2008). Mitophagy is a critical component of mitochondrial quality control and essential for the maintenance of a healthy mitochondrial population and cell viability. As mitophagy is essential for maintaining cellular functions, mitophagy deficiency is associated with a wide array of disorders, including neurodegenerative diseases (e.g., Parkinson’s and Alzheimer’s disease) and autoimmune diseases (Lou et al., 2019; Thangaraj et al., 2019; Xu et al., 2020; Yang et al., 2020). When mitochondria are damaged, they lose their membrane potential, release additional ROS, and activate mitophagy to ensure their degradation by lysosomes in coelomocytes. (Novikoff & Essner, 1962). In the present study, several lines of evidence supported the occurrence of mitophagy in A. japonicus coelomocytes, including mitophagosome formation in coelomocytes, up-regulation of mitophagosome and lysosome flux during pathogen infection, and removal of damaged mitochondria (Figure 1). Consistent with previous reports (Zhang et al., 2008; Novikoff & Essner, 1962), our findings strongly support the involvement of mitophagy in V. splendidus infection and LPS challenge.
Mitophagy may involve multiple pathways. Several studies have demonstrated that BNIP3 is a significant mitophagy contributor (Hanna et al., 2012; Ney, 2015). In this study, AjBNIP3 was localized to the mitochondria during pathogen infection and AjBNIP3 expression closely mirrored infection severity, with little to no increase in expression under no-infection conditions (Figure 2). Gao et al (2020) reported that BNIP3 is a member of the Bcl-2 family and present in the BH3-only domain and TMD. Of note, Ghavami et al. (2009) reported that the BH3-only domain is essential for the insertion of BNIP3 into the mitochondrial membrane. In addition, research has suggested that BNIP3 is anchored to the outer mitochondrial membrane via its C-terminal TMD, which is essential for targeting BNIP3 to the mitochondria (Chen et al., 1997; Kubli et al., 2008; Ray et al., 2000). However, it is unknown whether AjBNIP3 also interacts with LC3 via its WXXL motif and whether this interaction is important for selective removal of mitochondria. Therefore, after cloning AjBNIP3, we found that AjBNIP3 contained a BH3-only domain in the N-terminal and a TMD in the C-terminal (Figure 4). Moreover, the AjBNIP3 domains shared high similarity with BNIP3s from other species (Supplementary Figures S1). The BH3-only domain and TMD were important for targeting AjBNIP3 to the mitochondrial membrane, suggesting that AjBNIP3 plays an important role in mitochondrial elimination of coelomocytes. It is conceivable that BNIP3-mediated mitophagy failure increases infection-induced mitochondrial damage, leading to further mitochondrial damage and ultimately irreversible cell injury and cell death. However, it is still unclear how an autophagosome knows to remove a particular mitochondrion. It has been reported that interactions between BNIP3 and LC3 are important for promoting the removal of damaged mitochondria (Yurkova et al., 2008). Yurkova et al. (2008) reported that BNIP3 on the outer mitochondrial membrane interacts with processed LC3 on the phagophore membrane to promote mitochondrial sequestration within the autophagosome for degradation. In this study, LPS challenge enhanced the interaction between AjBNIP3 and AjLC3 (Figure 4), and more importantly, AjBNIP3 was found to contain a WXXL motif (LIR) in the N terminus, which was important for binding to LC3. Thus, up-regulated mitophagy may be correlated with enhanced interactions between AjBNIP3 and AjLC3. We also found that AjBNIP3 interference significantly reduced mitophagy by disrupting the interactions between AjBNIP3 and AjLC3. Consistent with Lamy et al. (2003) and Wei et al. (2015), our results indicate that mitochondrial-located AjBNIP3 interacts with AjLC3 and serves to dock mitochondria to the autophagosomes, thereby promoting mitochondrial sequestration and degradation.
BNIP3-dependent mitophagy can limit ROS generation and apoptosis in tumor cells by reducing dysfunctional mitochondria (Chourasia & Macleod, 2015; Sathiyaseelan et al., 2019). BNIP3 is also involved in the inhibition of nerve cell apoptosis (Liu et al., 2019; Zhu et al., 2019a). In contrast, mitophagy defects arising from the mitophagy receptor, BNIP3, accelerate the accumulation of dysfunctional mitochondria and excessive ROS production (Chourasia et al., 2015). Previous studies have indicated that down-regulating BNIP3 expression can increase ROS production and cell damage (Glick et al., 2012; Li et al., 2019). In this study, we investigated the mechanism by which AjBNIP3 promotes mitophagy in cells. We also discovered that inhibiting mitophagy by AjBNIP3 interference blocked ROS elimination and accelerated LPS-induced apoptosis in coelomocytes, thereby affecting cell survival (Figure 5). Zhu et al. (2019b) reported that BNIP3-induced mitophagy plays a protective role against mitochondrial dysfunction under lactate exposure by controlling ROS generation. Here, we found that high concentrations of lactate significantly prevented AjBNIP3 expression and AjBNIP3-mediated mitophagy in coelomocytes, and significantly up-regulated ROS and apoptosis levels (Figure 5). The role of BNIP3 in activating mitophagy to protect cells against apoptosis has also been demonstrated in mouse microglial BV-2 cells (Lei et al., 2018). These results reveal previously unknown mechanisms by which AjBNIP3-mediated mitophagy alleviates LPS-induced apoptosis and promotes survival by reducing mtROS production in coelomocytes.
Consistent with the protective function of mitophagy in coelomocyte survival found in the present study, mitophagy also participates in the protection of other tissues and cells. For example, BNIP3-mediated mitophagy plays a protective role in retinal detachment (Liu et al., 2016) and in mouse granulosa cells in response to follicle-stimulating hormone exposure (Zhou et al., 2017). Abnormal development of mitophagy is also a key contributor to various diseases. For example, inhibition of mitophagy can exacerbate hepatotoxicity (Ni et al., 2012). The therapeutic potential of BNIP3 in diseases associated with secondary mitochondrial dysfunction has also been explored in a variety of clinical trials, as well as in in vitro and in vivo studies, over the past decade (Brocca et al., 2012; Dhingra et al., 2017; Li et al., 2018; Smiles et al., 2016). Thus, accumulating evidence indicates that BNIP3 is strongly associated with mitophagy and mitophagy-related diseases. In addition, mitophagy-mediated cell death induced by BNIP3 is associated with ROS generation via inflammatory responses (Velde et al., 2000; Chung et al., 2019). In the current study, we showed that mtROS is important for activating AjBNIP3-mediated mitophagy in coelomocytes and AjBNIP3 expression is induced in a dose-dependent manner (Figure 3). Furthermore, AjBNIP3-mediated mitophagy during pathogen infection can be suppressed by ROS inhibitors and scavengers. We concluded that excessive ROS induced mitophagy, which, in turn, reduced ROS levels. Based on these findings, various defense mechanisms can be developed to protect cells against oxidative stress, including the up-regulation of antioxidants and removal of damaged mitochondria by mitophagy (Lemasters, 2005). Therefore, BNIP3 may be a promising target for maintaining mitochondrial integrity and function in cells via its important roles in mitophagy. Investigation of the regulatory mechanisms involved in the maintenance of BNIP3-mediated mitophagy homeostasis is of great significance for the prevention and treatment of diseases caused by excessive ROS. Overall, AjBNIP3 mediated mitophagy and promoted coelomocyte survival against V. splendidus infection and excessive ROS, and thus may serve as a new research target for protection against oxidative damage (Figure 6).
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
L.L.S performed the experiments, interpreted the data, and wrote the manuscript; Y.N.S. and C.H.L. participated in the experimental design, interpreted the data, and revised the manuscript. M.X.Y. and C.H.L. contributed new reagents and analytic tools. All authors read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Ping-Ping Zhan and Dr. Chun-Dan Zhang for help in TEM and confocal laser microscopy. In addition, we thank Dr. Yan-Zhen Tian for help in MST analysis.
Funding Statement
This work was supported by the National Natural Science Foundation of China (32073003, 32102825), Natural Science Foundation of Zhejiang Province (LZ19C190001), Key Project from Science Technology Department of Zhejiang Province (2019R52016), and K.C. Wong Magna Fund in Ningbo University
References
- 1.Adam-Vizi V, Chinopoulos C Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends in Pharmacological Sciences. 2006;27(12):639–645. doi: 10.1016/j.tips.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Awan MUF, Hasan M, Iqbal J, Lei RH, Lee WF, Hong M, et al Neuroprotective role of BNIP3 under oxidative stress through autophagy in neuroblastoma cells. Molecular Biology Reports. 2014;41(9):5729–5734. doi: 10.1007/s11033-014-3444-7. [DOI] [PubMed] [Google Scholar]
- 3.Bjelland S, Seeberg E Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2003;531(1-2):37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 4.Brocca L, Cannavino J, Coletto L, Biolo G, Sandri M, Bottinelli R, et al The time course of the adaptations of human muscle proteome to bed rest and the underlying mechanisms. The Journal of Physiology. 2012;590(20):5211–5230. doi: 10.1113/jphysiol.2012.240267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen G, Ray R, Dubik D, Shi LF, Cizeau J, Bleackley RC, et al The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. Journal of Experimental Medicine. 1997;186(12):1975–1983. doi: 10.1084/jem.186.12.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chourasia AH, Macleod KF Tumor suppressor functions of BNIP3 and mitophagy. Autophagy. 2015;11(10):1937–1938. doi: 10.1080/15548627.2015.1085136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, et al Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Reports. 2015;16(9):1145–1163. doi: 10.15252/embr.201540759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung LY, Tang SJ, Wu YC, Yang KC, Huang HJ, Sun GH, et al Platinum-based combination chemotherapy triggers cancer cell death through induction of BNIP3 and ROS, but not autophagy. Journal of Cellular and Molecular Medicine. 2019;24(2):1993–2003. doi: 10.1111/jcmm.14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng H, He CB, Zhou ZC, Liu C, Tan KF, Wang NB, et al. 2009. Isolation and pathogenicity of pathogens from skin ulceration disease and viscera ejection syndrome of the sea cucumber Apostichopus japonicus. Aquaculture, 287(1–2): 18–27.
- 10.Dhingra A, Jayas R, Afshar P, Guberman M, Maddaford G, Gerstein J, et al Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radical Biology and Medicine. 2017;112:411–422. doi: 10.1016/j.freeradbiomed.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 11.Elmore SP, Qian T, Grissom SF, Lemasters JJ The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB Journal. 2001;15(12):2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 12.Fleury C, Mignotte B, Vayssière JL Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84(2-3):131–141. doi: 10.1016/S0300-9084(02)01369-X. [DOI] [PubMed] [Google Scholar]
- 13.Gao AB, Jiang JY, Xie F, Chen LX Bnip3 in mitophagy: novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clinica Chimica Acta. 2020;506:72–83. doi: 10.1016/j.cca.2020.02.024. [DOI] [PubMed] [Google Scholar]
- 14.Ghavami S, Eshraghi M, Kadkhoda K, Mutawe MM, Maddika S, Bay GH, et al Role of BNIP3 in TNF-induced cell death-TNF upregulates BNIP3 expression. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2009;1793(3):546–560. doi: 10.1016/j.bbamcr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Glick D, Zhang WS, Beaton M, Marsboom G, Gruber M, Simon MC, et al BNip3 regulates mitochondrial function and lipid metabolism in the liver. Molecular and Cellular Biology. 2012;32(13):2570–2584. doi: 10.1128/MCB.00167-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo WJ, Sun Y, Liu W, Wu XX, Guo LL, Cai PF, et al Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014;10(6):972–985. doi: 10.4161/auto.28374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. Journal of Biological Chemistry. 2012;287(23):19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubli DA, Quinsay MN, Huang CQ, Lee Y, Gustafsson ÅB Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. American Journal of Physiology:Heart and Circulatory Physiology. 2008;295(5):H2025–H2031. doi: 10.1152/ajpheart.00552.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamy L, Ticchioni M, Rouquette-Jazdanian AK, Samson M, Deckert M, Greenberg AH, et al CD47 and the 19 kDa interacting protein-3 (BNIP3) in T cell apoptosis. Journal of Biological Chemistry. 2003;278(26):23915–23921. doi: 10.1074/jbc.M301869200. [DOI] [PubMed] [Google Scholar]
- 20.Lei QY, Tan J, Yi SQ, Wu N, Wang YL, Wu H Mitochonic acid 5 activates the MAPK-ERK-yap signaling pathways to protect mouse microglial BV-2 cells against TNFα-induced apoptosis via increased Bnip3-related mitophagy. Cellular & Molecular Biology Letters. 2018;23:14. doi: 10.1186/s11658-018-0081-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemasters JJ Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Research. 2005;8(1):3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Qi F, Meng XC, Zhu CP, Gao YT Mst1 regulates colorectal cancer stress response via inhibiting Bnip3-related mitophagy by activation of JNK/p53 pathway. Cell Biology and Toxicology. 2018;34(4):263–277. doi: 10.1007/s10565-017-9417-6. [DOI] [PubMed] [Google Scholar]
- 23.Li XM, Wang QL, Ren YF, Wang XM, Cheng HX, Yang H, et al Tetramethylpyrazine protects retinal ganglion cells against H2O2-induced damage via the microRNA-182/mitochondrial pathway . International Journal of Molecular Medicine. 2019;44(2):503–512. doi: 10.3892/ijmm.2019.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu HW, Huang HT, Li RX, Bi WT, Feng L, E LL, et al Mitophagy protects SH-SY5Y neuroblastoma cells against the TNFα-induced inflammatory injury: Involvement of microRNA-145 and Bnip3. Biomedicine & Pharmacotherapy. 2019;109:957–968. doi: 10.1016/j.biopha.2018.10.123. [DOI] [PubMed] [Google Scholar]
- 25.Liu HY, Zhu H, Li T, Zhang PF, Wang N, Sun XD Prolyl-4-hydroxylases inhibitor stabilizes HIF-1α and increases mitophagy to reduce cell death after experimental retinal detachment. Investigative Ophthalmology & Visual Science. 2016;57(4):1807–1815. doi: 10.1167/iovs.15-18066. [DOI] [PubMed] [Google Scholar]
- 26.Liu HZ, Zheng FR, Sun XQ, Hong XG, Dong SL, Wang B, et al Identification of the pathogens associated with skin ulceration and peristome tumescence in cultured sea cucumbers Apostichopus japonicus (Selenka) . Journal of Invertebrate Pathology. 2010;105(3):236–242. doi: 10.1016/j.jip.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 27.Liu L, Sakakibara K, Chen Q, Okamoto K Receptor-mediated mitophagy in yeast and mammalian systems. Cell Research. 2014;24(7):787–795. doi: 10.1038/cr.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
28.Livak KJ, Schmittgen TD Analysis of relative gene expression data using real-time quantitative PCR and the 2
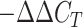 method
. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
method
. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar] - 29.Lou GF, Palikaras K, Lautrup S, Scheibye-Knudsen M, Tavernarakis N, Fang EF Mitophagy and neuroprotection. Trends in Molecular Medicine. 2019;26(1):8–20. doi: 10.1016/j.molmed.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Lu HQ, Li GL, Liu LM, Feng LF, Wang X, Jin HC Regulation and function of mitophagy in development and cancer. Autophagy. 2013;9(11):1720–1736. doi: 10.4161/auto.26550. [DOI] [PubMed] [Google Scholar]
- 31.Ma YX, Xu GR, Chang YQ, Zhang EP, Zhou W, Song LS Bacterial pathogens of skin ulceration disease in cultured sea cucumber Apostichopus japonicus (Selenka) juveniles . Journal of Dalian Fisheries University. 2006;21(1):13–18. [Google Scholar]
- 32.Mammucari C, Rizzuto R Signaling pathways in mitochondrial dysfunction and aging. Mechanisms of Ageing and Development. 2010;131(7-8):536–543. doi: 10.1016/j.mad.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merjaneh M, Langlois A, Larochelle S, Cloutier CB, Ricard-Blum S, Moulin VJ Pro-angiogenic capacities of microvesicles produced by skin wound myofibroblasts. Angiogenesis. 2017;20(3):385–398. doi: 10.1007/s10456-017-9554-9. [DOI] [PubMed] [Google Scholar]
- 34.Métivier D, Dallaporta B, Zamzami N, Larochette N, Susin SA, Marzo I, et al Cytofluorometric detection of mitochondrial alterations in early CD95/Fas/APO-1-triggered apoptosis of Jurkat T lymphoma cells. Comparison of seven mitochondrion-specific fluorochromes. Immunology Letters. 1998;61(2-3):157–163. doi: 10.1016/S0165-2478(98)00013-3. [DOI] [PubMed] [Google Scholar]
- 35.Mills EL, O'Neill LA Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. European Journal of Immunology. 2016;46(1):13–21. doi: 10.1002/eji.201445427. [DOI] [PubMed] [Google Scholar]
- 36.Ney PA Mitochondrial autophagy: origins, significance, and role of BNIP3 and NIX. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2015;1853(10):2775–2783. doi: 10.1016/j.bbamcr.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 37.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55(1):222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Novikoff AB, Essner E Cytolysomes and mitochondrial degeneration. Journal of Cell Biology. 1962;15(1):140–146. doi: 10.1083/jcb.15.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray R, Chen G, Velde CV, Cizeau J, Park JH, Reed JC, et al BNIP3 heterodimerizes with Bcl-2/Bcl-XL and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites . Journal of Biological Chemistry. 2000;275(2):1439–1448. doi: 10.1074/jbc.275.2.1439. [DOI] [PubMed] [Google Scholar]
- 40.Roperto S, De Falco F, Perillo A, Catoi C, Roperto F Mitophagy mediated by BNIP3 and BNIP3L/NIX in urothelial cells of the urinary bladder of cattle harbouring bovine papillomavirus infection. Veterinary Microbiology. 2019;236:108396. doi: 10.1016/j.vetmic.2019.108396. [DOI] [PubMed] [Google Scholar]
- 41.Sathiyaseelan P, Rothe K, Yang KC, Xu J, Chow NS, Bortnik S, et al Diverse mechanisms of autophagy dysregulation and their therapeutic implications: does the shoe fit. Autophagy. 2019;15(2):368–371. doi: 10.1080/15548627.2018.1509609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scherz-Shouval R, Elazar Z Regulation of autophagy by ROS: physiology and pathology. Trends in Biochemical Sciences. 2011;36(1):30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 43.Smiles WJ, Parr EB, Coffey VG, Lacham-Kaplan O, Hawley JA, Camera DM Protein coingestion with alcohol following strenuous exercise attenuates alcohol-induced intramyocellular apoptosis and inhibition of autophagy. American Journal of Physiology-Endocrinology and Metabolism. 2016;311(5):E836–E849. doi: 10.1152/ajpendo.00303.2016. [DOI] [PubMed] [Google Scholar]
- 44.Sun LL, Guo M, Lv ZM, Shao YN, Li CH. 2020. Hypoxia-inducible factor-1α shifts metabolism from oxidative phosphorylation to glycolysis in response to pathogen challenge in Apostichopus japonicus. Aquaculture, 526: 735393.
- 45.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. The EMBO Journal. 2001;20(21):5971–5981. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thangaraj A, Periyasamy P, Guo ML, Chivero ET, Callen S, Buch S Mitigation of cocaine-mediated mitochondrial damage, defective mitophagy and microglial activation by superoxide dismutase mimetics. Autophagy. 2019;16(2):289–312. doi: 10.1080/15548627.2019.1607686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tolkovsky AM Mitophagy. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2009;1793(9):1508–1515. doi: 10.1016/j.bbamcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Velde CV, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, et al BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Molecular and Cellular Biology. 2000;20(15):5454–5468. doi: 10.1128/MCB.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei HF, Liu L, Chen Q Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2015;1853(10):2784–2790. doi: 10.1016/j.bbamcr.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 51.Wende AR, Young ME, Chatham J, Zhang JH, Rajasekaran NS, Darley-Usmar VM Redox biology and the interface between bioenergetics, autophagy and circadian control of metabolism. Free Radical Biology and Medicine. 2016;100:94–107. doi: 10.1016/j.freeradbiomed.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu J, Li XY, Zhu GL, Zhang YX, He M, Zhang J The role of Resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Experimental Cell Research. 2016;341(1):42–53. doi: 10.1016/j.yexcr.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 54.Xu Y, Shen J, Ran ZH Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. 2020;16(1):3–17. doi: 10.1080/15548627.2019.1603547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang X, Pan WN, Xu GS, Chen LX Mitophagy: A crucial modulator in the pathogenesis of chronic diseases. Clinica Chimica Acta. 2020;502:245–254. doi: 10.1016/j.cca.2019.11.008. [DOI] [PubMed] [Google Scholar]
- 56.Youle RJ, Bliek AMVD Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Youle RJ, Narendra DP Mechanisms of mitophagy. Nature Reviews Molecular Cell Biology. 2011;12(1):9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yurkova N, Shaw J, Blackie K, Weidman D, Jayas R, Flynn B, et al The cell cycle factor E2F-1 activates Bnip3 and the intrinsic death pathway in ventricular myocytes. Circulation Research. 2008;102(4):472–479. doi: 10.1161/CIRCRESAHA.107.164731. [DOI] [PubMed] [Google Scholar]
- 59.Zhang HF, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. Journal of Biological Chemistry. 2008;283(16):10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Zhang PJ, Li CH, Shao YN, Chen XC, Li Y, Su XR, et al. 2014. Identification and characterization of miR-92a and its targets modulating Vibrio splendidus challenged Apostichopus japonicus. Fish & Shellfish Immunology, 38(2): 383–388.
- 61.Zhang TM, Xue L, Li L, Tang CY, Wan ZQ, Wang RX, et al BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. Journal of Biological Chemistry. 2016;291(41):21616–21629. doi: 10.1074/jbc.M116.733410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou H, Du WJ, Li Y, Shi C, Hu N, Ma S, et al Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. Journal of Pineal Research. 2018a;64(1):e12450. doi: 10.1111/jpi.12450. [DOI] [PubMed] [Google Scholar]
- 63.Zhou H, Yue Y, Wang J, Ma Q, Chen YD Melatonin therapy for diabetic cardiomyopathy: a mechanism involving Syk-mitochondrial complex I-SERCA pathway. Cellular Signalling. 2018b;47:88–100. doi: 10.1016/j.cellsig.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 64.Zhou JL, Yao W, Li CY, Wu WJ, Li QF, Liu HL Administration of follicle-stimulating hormone induces autophagy via upregulation of HIF-1α in mouse granulosa cells . Cell Death & Disease. 2017;8(8):e3001. doi: 10.1038/cddis.2017.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou RB, Yazdi AS, Menu P, Tschopp J A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 66.Zhu L, Qi BX, Hou DR Roles of HIF1α- and HIF2α-regulated BNIP3 in hypoxia-induced injury of neurons. Pathology-Research and Practice. 2019a;215(4):822–827. doi: 10.1016/j.prp.2019.01.022. [DOI] [PubMed] [Google Scholar]
- 67.Zhu Y, Ji JJ, Yang R, Han XQ, Sun XJ, Ma WQ, et al Lactate accelerates calcification in VSMCs through suppression of BNIP3-mediated mitophagy. Cellular Signalling. 2019b;58:53–64. doi: 10.1016/j.cellsig.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 68.Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. Journal of Biological Chemistry. 2010;285(43):33154–33164. doi: 10.1074/jbc.M110.143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. The Journal of Experimental Medicine. 2000;192(7):1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zorov DB, Juhaszova M, Sollott SJ Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiological Reviews. 2014;94(3):909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data to this article can be found online.