

Summary
This protocol represents an optimized proteomics-based protocol for the endogenous protein enrichment and protein-protein interaction analysis. This 2-step protocol consists of: 1) co-immunoprecipitation of the bait protein; 2) the bait-protein interactions analysis using LC-MS/MS. Here, we used Dynabeads® for the enrichment of the target protein (the bait) and its interactors. We have tested the protocol using several different cell lines. Our conclusion is that the protocol is applicable to different cell lines and species.
For complete details on the use and execution of this protocol, please refer to Lagundžin et al. (2019).
Subject areas: Protein Biochemistry, Proteomics, Protein expression and purification, Mass Spectrometry
Graphical abstract
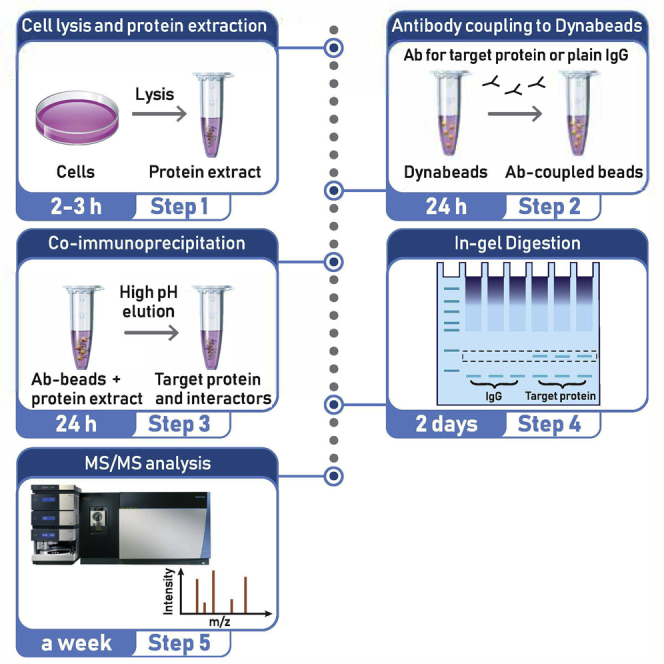
Highlights
-
•
Protocol to determine endogenous protein-protein interactions in cell lines
-
•
Scaled to utilize co-IP to purify protein complexes suitable for LC-MS/MS analysis
-
•
Describes peptide processing steps for LC-MS/MS analysis
-
•
Details conditions for LC-MS/MS analysis of protein complexes
This protocol represents an optimized proteomics-based protocol for the endogenous protein enrichment and protein-protein interaction analysis. This 2-step protocol consists of: 1) co-immunoprecipitation of the bait protein; 2) the bait-protein interactions analysis using LC-MS/MS. Here, we used Dynabeads® for the enrichment of the target protein (the bait) and its interactors. We have tested the protocol using several different cell lines. Our conclusion is that the protocol is applicable to different cell lines and species.
Before you begin
This protocol was used in a recent publication by Lagundžin and co-authors (Lagundzin et al., 2019), in which a proteomics-based approach was employed to study the role of Fanconi Anemia Complementation Group A (FANCA) in pancreas β islet cells. We initially tested the protocol using human embryonal kidney 293 FT, human pancreatic islet EndoC-βH3 and human pancreatic cancer MiaPaCa-2 cell lines. This protocol has been used at our Mass Spectrometry and Proteomics Core Facility, at the University of Nebraska Medical Center, as a standard protocol for protein enrichment and interactome analysis. Besides the human cell lines, the protocol has been tested in experiments with mouse, rat and pig tissues and physiological fluids.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FANCA antibody (Note: The antibody targeting the protein of interest should be determined by the end user. FANCA is only given as an example here.) | Bethyl Laboratories | Cat#A301-980A |
| Rabbit polyclonal IgG (Note: This control IgG antibody should originate from the same species as the specific antibody targeting the user’s protein of interest.) | Santa Cruz Biotechnology | n/a |
| Chemicals, peptides, and recombinant proteins | ||
| 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) | Fisher Scientific | Cat#AAJ1692630 |
| Phosphate Buffered Saline | Fisher Scientific | Cat#10010023 |
| Protease inhibitors | Sigma-Aldrich | Cat#P8340-5ML |
| Phosphatase inhibitors | Fisher Scientific | Cat#A32957 |
| Sodium Chloride, NaCl | Fisher Scientific | Cat#18612546 |
| Potassium Chloride, KCl | Fisher Scientific | Cat#18612546 |
| Magnesium Chloride, MgCl2 | Fisher Scientific | Cat#44-261-5500GM |
| Nonidet P-40 (NP-40) (CAS no.: 9002-93-1) | Spectrum Chemicals | Cat#T1279-100ML |
| Tween-20 | Fisher Scientific | Cat#AAJ20605AP |
| Ammonium Hydroxide, NH4OH | Fisher Scientific | Cat#A470-250 |
| Ethylenediaminetetraacetic acid, EDTA | Fisher Scientific | Cat#60-018-15 |
| Bovine serum albumin, BSA | Fisher Scientific | Cat#BP1600100 |
| Glacial acetic acid (HPLC grade) | Fisher Scientific | Cat#A35-500 |
| Methanol, LC/MS-grade | Fisher Scientific | Cat#A456212 |
| HPLC grade water | Fisher Scientific | Cat#7732-18-5 |
| Acetonitrile (ACN) | Fisher Scientific | Cat#85188 |
| Tris(2-carboxyethyl)phosphine (TCEP) | Thermo Fisher Scientific | Cat#77720 |
| Ammonium bicarbonate (AmBic) | Fisher Scientific | Cat#A643-500 |
| Iodoacetamide (IAA) | Thermo Fisher Scientific | Cat#A39271 |
| Pierce™ trypsin protease, MS-grade | Thermo Fisher Scientific | Cat#90057 or Cat#90058 |
| Trifluoroacetic acid (TFA), LC/MS-grade | Fisher Scientific | Cat#85183 |
| 0.1% Formic acid (FA) in water, LC/MS-grade | Fisher Scientific | |
| Critical commercial assays | ||
| BCA™ Protein Assay kit | Fisher Scientific | Cat#23225 |
| Dynabeads® Antibody Coupling Kit | Fisher Scientific | Cat#14311D |
| 10% Criterion XT Precast Bis-Tris gel | Bio-Rad | Cat#3450111 |
| XT MOPS (3-(N-morpholino)propanesulfonic acid) running buffer | Bio-Rad | Cat#1610788 |
| XT Sample Buffer 4× | Bio-Rad | Cat#1610791 |
| XT Reducing Agent 20× | Bio-Rad | Cat#1610792 |
| Coomassie Brilliant Blue G-250 dye | Fisher Scientific | Cat#20279 |
| Instant Blue | Sigma-Aldrich | Cat#ISB1L |
| Deposited data | ||
| The raw mass spectrometry data files generated for this project have been deposited to the ProteomeXchange Consortium via the PRIDEpartner repository (http://proteomecentral.proteomexchange.org) | Lagundzin et al., 2019 |
https://www.ebi.ac.uk/pride/archive/projects/PXD010589 and https://www.ebi.ac.uk/pride/archive/projects/PXD010570 |
| Software and algorithms | ||
| Proteome Discoverer | Thermo Fisher Scientific | n/a |
| Significance Analysis of INTeractome | http://saint-apms.sourceforge.net/Main.html | n/a |
| Other | ||
| 1.5 mL microcentrifuge tubes | Eppendorf | Cat#022363212 |
| Fisherbrand™ Easy Reader™ conical polypropylene centrifuge tubes | Fisher Scientific | Cat#0553912 |
| Cell scraper | n/a | n/a |
| pH strips | n/a | n/a |
| Pierce® C18 tips | Thermo Fisher Scientific | Cat#87784 |
| Feather™ Disposable Scalpel Set #11 | Electron Microscopy Sciences | Cat#7204211 |
| Criterion staining/blotting trays | Bio-Rad | Cat#3459921 |
| Pipettes | n/a | n/a |
| SpeedVac | n/a | n/a |
| pH meter | n/a | n/a |
| Orbital rotator | n/a | n/a |
| Rocking platform | n/a | n/a |
| Incubator | n/a | n/a |
| Odyssey Fc imaging system | LI-COR | n/a |
| ThermoMixer | Eppendorf | n/a |
| Criterion™ Cell and PowerPac™ Basic Power Supply | Bio-Rad | Cat#1656019 |
| MagJET Separation Rack, 12 × 1.5 mL tube | Thermo Fisher Scientific | Cat#MR02 |
| Nanodrop one microvolume UV-Vis Spectrometer | Thermo Fisher Scientific | Cat#8402274100 |
| Nanoflow liquid chromatography system | n/a | n/a |
| High resolution mass spectrometer | n/a | n/a |
Materials and equipment
Lysis Buffer
| Reagent | Final concentration |
|---|---|
| HEPES, pH 7.4 | 10 mM |
| KCl | 10 mM |
| Nonidet P-40 | 0.05% (v/v) |
| Protease inhibitors | 1 mL/10 mL |
| Phosphatase inhibitors | 1 tablet/10 mL |
| ddH2O | n/a |
Make without protease and phosphatase inhibitors and store at 4°C for up to one month. After adding protease and phosphatase inhibitors, store at −20°C for up to one week.
Destain solution
| Reagent | Final concentration |
|---|---|
| Methanol | 10% (v/v) |
| Glacial acetic acid | 5% (v/v) |
| ddH2O | 85% (v/v) |
Store at 25°C for up to one week.
PBS + BSA
| Reagent | Final concentration |
|---|---|
| Phosphate buffered saline | 1× |
| BSA | 0.1% (w/v) |
Store at 4°C for up to one week.
Bead washing buffer A (BW-A)
| Reagent | Final concentration |
|---|---|
| HEPES, pH 7.4 | 10 mM |
| KCl | 10 mM |
| NaCl | 50 mM |
| MgCl2 | 1 mM |
| Nonidet P-40 | 0.05% (v/v) |
| ddH2O | n/a |
Store at 4°C for up to one month.
Bead washing buffer B (BW-B)
| Reagent | Final concentration |
|---|---|
| HEPES, pH 7.4 | 10 mM |
| KCl | 10 mM |
| Nonidet P-40 | 0.07% (v/v) |
| ddH2O | n/a |
Store at 4°C for up to one month.
Sample elution buffer (SE)
| Reagent | Final concentration |
|---|---|
| NH40H, pH 11.0 | 0.5 M |
| EDTA | 0.5 mM |
| ddH2O | n/a |
Store at 4°C for up to one month.
TCEP reduction solution
| Reagent | Final concentration |
|---|---|
| TCEP (20 mM) | 10% (v/v) |
| AmBic (50 mM) | 90% (v/v) |
Always make fresh and use within 4 h. Do not store.
IAA alkylation solution
| Reagent | Final concentration |
|---|---|
| IAA (375 mM) | 10% (v/v) |
| AmBic (50 mM) | 90% (v/v) |
Always make fresh and use within 4 h. Do not store.
Peptide extraction buffer (PXB)
| Reagent | Final concentration |
|---|---|
| ACN | 50% (v/v) |
| TFA | 0.1% (v/v) |
| ddH2O | 49.9% (v/v) |
Store at 25°C for up to one week.
Step-by-step method details
Extraction of protein from cell lines
Timing: 2 – 3 h
Start with a mammalian cell line that reached confluency of around 80%. For this protocol, around 2.5 mg of total protein is needed per sample to be analyzed by LC-MS/MS. Growth kinetics, total protein concentration and appropriate amount of cell culture plates/flasks of the chosen cell line should be determined before the start of the experiment. For illustrative purpose, the cells are assumed to be harvested from 100 mm plates.
-
1.
Cells are grown in plates/flasks to around 80% confluency in recommended cell culture medium. For our adherent cell lines, we use plates.
-
2.Cells are prepared for lysis.
-
a.Culture medium is removed by aspiration and discarded.
-
b.Cells are washed with 10 mL ice-cold PBS.
-
c.The PBS used for washing is discarded.
-
d.2 mL of ice-cold PBS is added to each 100 mm plate.
-
e.Cells are detached from the plate using a cell scraper.
-
f.The cell suspension in PBS is transferred to a conical centrifuge tube with a pipette.
-
g.Centrifuge the sample for 10 min at 4°C at 1,000×g.
-
h.The supernatant is discarded, and the cell pellet is used to obtain protein lysates in the following steps.
-
a.
-
3.The soluble protein lysate is isolated.
-
a.The cell pellet is lysed by adding an amount of lysis buffer that is 3 times the pellet size (v/v).Note: Tris-based buffer may be used as well, instead of HEPES-based one, that we used.
-
b.The cell pellet is disrupted using lysis buffer and incubated on ice for 30 min in Eppendorf tubes.
-
c.The lysate is centrifuged for 15 min at 4°C at 16,000×g.
-
d.The supernatant containing the soluble protein extract is transferred to a new Eppendorf tube.
-
a.
-
4.
The protein concentration is determined using BCA Protein Assay kit. Using this kit, only 1 μL of a sample is needed for the protein concentration measurement.
-
5.
Aliquots containing 2.5 mg of protein in Eppendorf tubes are made and are stored at −80°C until use.
Generation of beads and antibody coupling
The commercially available Dynabeads® Antibody Coupling Kit, which contains 2.8 μm magnetic M-270 Epoxy beads, was used for antibody coupling. All the reagents (C1, C2, HB, LB, SB) are provided in the kit. All the tubes used were 1.5-mL Eppendorf tubes.
-
6.
For each sample, 3 mg of Dynabeads® M-270 Epoxy beads are weighed and transferred into a microcentrifuge tube.
Note: This amount of beads was optimized for our mass spectrometry experiments to efficiently immunoprecipitate the bait protein with minimum background interference.
-
7.
To begin washing the beads, 1 mL of C1 is added and slowly pipetted up and down.
-
8.
The tubes containing the Dynabeads are placed inside of the tube rack with magnets for 1 min to allow the beads to accumulate.
-
9.
The supernatant is removed and discarded.
Note: Care should be taken to not disturb the accumulated beads. We use gel loading tips for the supernatant removal. This is to avoid accidentally removing the beads during this process.
-
10.
The tubes containing the Dynabeads are removed from the magnetic rack.
-
11.
50 μg of the antibody (Ab) specific for the protein of interest (the bait protein) is added to the tube containing the Dynabeads.
-
12.
In a separate tube, 50 μg of normal IgG is added, which acts as a negative control.
Note: For protein-protein interaction analysis, it is crucial to prepare a negative control, such as normal IgG, to help identify adventitious proteins so that false-positive interactions can be annotated. The IgG control should be sourced from the same species as the Ab used for the protein of interest.
-
13.
The appropriate volume of C1 is added to each of the tubes (from steps 10 and 11) to reach a final volume of 150 μL.
-
14.
150 μL of C2 is added to each of the tubes.
-
15.
The tubes are then incubated at 37°C for around 18 h on an orbital rotator.
CRITICAL: The volume of mix Ab + C1 + C2 should be 300 μL for 3 mg of beads, to mix efficiently. The appropriate volumes of antibodies used in these steps is dependent upon the concentration of the antibody supplied by the vendor, which can vary widely. If the antibody is dilute and a larger volume is required, start with more beads at step 5 in order to achieve the proper ratio for incubation.
Note: Make sure the beads are mixing well, as they can settle at the bottom of the microcentrifuge tube. Use a higher rotation speed for the first 30 min, if possible, and then incubate around 18 h with gentle rotation.
-
16.
The tubes are placed on a magnet for 1 min and the supernatants are carefully removed and discarded.
-
17.
After removing the tubes from the magnet, the beads are washed by adding 800 μL of solution HB and gently pipetting up and down gently at least 10 times.
-
18.
The tubes are placed on a magnet for 1 min and the supernatants are carefully removed and discarded.
-
19.
After removing the tubes from the magnetic rack, the beads are washed by adding 800 μL of solution LB and gently pipetting up and down gently at least 10 times.
-
20.
The tubes are placed on a magnet for 1 min and the supernatants are carefully removed and discarded.
-
21.
After removing the tubes from the magnet, the beads are washed by adding 800 μL of solution SB and gently pipetting up and down gently at least 10 times.
-
22.
The tubes are placed on a magnet for 1 min and the supernatants are carefully removed and discarded.
-
23.
Repeat steps 20 and 21.
-
24.
800 μL of SB is added to the beads and incubated on orbital rotator for 15 min at 25°C.
-
25.
The tubes are placed on a magnet for 1 min and the supernatants are carefully removed and discarded.
-
26.
After removing the tubes from the magnet, 800 μL of PBS + BSA is added to the beads.
-
27.
The slurry is gently mixed using a rotator for 5 min at 25°C.
-
28.
The tubes are placed on a magnet for 1 min and the supernatants are carefully removed and discarded.
-
29.
The tubes containing the antibody conjugated beads are placed on ice and used immediately in the following steps.
Note: The beads are now covalently coupled with Ab.
Co-immunoprecipitation (Co-IP)
At this stage, the cell lysates are incubated with the covalently coupled beads to enrich the bait protein and its interactors (Figure 1).
-
30.
The cell lysate containing 2.5 mg of the total proteins (from step 5, defrosted) is added to the antibody-coupled beads in the Eppendorf tube from step 28.
-
31.
The samples are incubated for 18–24 h with orbital rotation, at 4°C.
-
32.
The tubes are placed on a magnet for 1 min and the supernatants are carefully removed and discarded.
-
33.
After removing the tubes from the magnet, BW-A buffer is added to the beads and gently pipetted up and down at least 10 times.
-
34.
The tubes are placed on a magnet for 1 min and the supernatants are carefully removed and discarded.
-
35.
Steps 32 and 33 are repeated an additional 3 times.
-
36.
After removing the tube from the magnet, BW-B buffer is added.
-
37.
The samples are placed on an orbital rotator for 5 min at 25°C.
-
38.
The tubes are placed on a magnet for 1 min and the supernatants are carefully removed and discarded.
-
39.
500 μL of an SE buffer is added to the beads.
-
40.
The samples are placed on an orbital rotator and incubated for 10 min at 25°C to elute the proteins from the antibody-conjugated beads.
Note: The elution with a high pH buffer does not disrupt the antibody attached to the beads, which avoids problems associated with the heavy and light IgG chains interfering with the results. A high abundance of IgG in the sample can impair the mass spectrometry detection of peptides/proteins that interact with the bait protein. As an alternative to the high-pH elution buffer, a low-pH elution buffer may be used instead. For example, a glycine-HCl buffer can be used, however we didn’t test it in our protocol.
-
41.
The supernatants (which will be around 500 μL) are collected and transferred to new Eppendorf tubes.
-
42.
The samples are then concentrated using a SpeedVac to volume around 50 μL.
-
43.
The pH of the sample should be checked with a pH strip and neutralized with acetic acid, if necessary.
Pause point: The samples can be stored at −80°C until further use.
Figure 1.

Overview of the bait protein enrichment workflow prior to mass spectrometry analysis
The Coomassie-stained gel was visualized using an Odyssey Fc system, using 700 nm channel and green color, for better visualization.
Sample preparation for LC-MS/MS
At this stage, the bait protein and its interacting protein partners are being separated on a gel, digested to peptides, and analyzed by LC-MS/MS. Alternatively, an in-solution digestion protocol can be used instead of this in-gel digestion protocol. In-gel digestion protocol might be useful here, however, as it allows protein visualization before further steps and can reveal if the previous Co-IP was successful or not. The in-gel digestion method remains as an efficient and reproducible method for this type of analysis. 1.5-mL Eppendorf tubes are used throughout the steps, as they are very compatible with mass spectrometry (low leaching effect).
-
44.
40 μL of the protein immunoprecipitate from step 42 is mixed with 10 μL of XT Sample Buffer (with added XT Reducing Agent).
Note: We used commercially available premixed loading buffer, compatible with our gel-separating system and buffers.
-
45.
The samples are incubated for 7 min at 95°C using a ThermoMixer.
Note: All incubation steps on ThermoMixer were conducted at 800 rpm.
-
46.
45–50 μL of the samples are loaded on a 10% Criterion XT Bis-Tris gel, and the electrophoresis is stopped when the dye front moves around 2 inches from the base of the well.
Note: We recommend leaving free lanes between samples to avoid potential cross-contamination that can occur if the wells are overloaded.
-
47.
The gel is washed with water after removing it from its plates.
-
48.
Coomassie Brilliant Blue G-250 is used to stain the gel for 2 h.
-
49.
The gel is destained for around 18–24 h.
Note: To save time, instead of Coomassie staining, use InstantBlue Stain Solution. This allows gel staining/destaining to be completed in less than 20 min. Avoid traditional silver staining, as it might cause chemical crosslinking of the proteins. There are some commercially available MS-compatible silver stains.
-
50.
Cut out the 2-inch lane into 3 or more slices with a disposable scalpel, and transfer into a 1.5-mL Eppendorf tube.
Note: When reusing the scalpel for cutting multiple gel slices from different samples, take care to thoroughly wash it with water, then methanol, then water again, between each slice excision to reduce cross-contamination.
-
51.
100 μL of HPLC-grade water is added to each tube containing a gel slice.
-
52.
The tubes are agitated on the ThermoMixer for 10 min.
-
53.
The supernatant is removed and discarded.
-
54.
100 μL of (ACN) is added to the gel slices and incubated for 5–10 min.
-
55.
The supernatant is removed and discarded.
Note: If gel pieces are still blue at the end of step 54, another 100 μL of ACN is added, and shaking is continued for another 15 min or until the blue stain is removed from the gel slice.
-
56.
For each gel slice, 100 μL of TCEP reduction solution is made by combining 10 μL of 20 mM tris(2-carboxyethyl)phosphine (TCEP) with 90 μL of 50 mM ammonium bicarbonate (AmBic).
-
57.
100 μL of the TCEP solution is added to each gel slice.
-
58.
The tubes are incubated at 37°C for 30 min on the ThermoMixer.
Note: DTT may be used instead of TCEP.
-
59.
100 μL of ACN is added to each tube.
-
60.
The tubes are agitated on the ThermoMixer for 15 min at 25°C.
-
61.
The supernatant is removed and discarded.
-
62.
100 μL of ACN is added to each tube.
-
63.
The tubes are agitated on the ThermoMixer for 5 min at 25°C.
-
64.
The supernatant is removed and discarded.
-
65.
For each gel slice, IAA alkylation solution is made by combining 10 μL of 375 mM IAA with 90 μL of 50 mM AmBic.
-
66.
100 μL of IAA solution is added to each gel slice.
-
67.
The samples are incubated for 20 min at 25°C in the dark.
-
68.
The supernatant is removed and discarded.
-
69.
100 μL of ACN is added to each gel slice and incubated for 10 min.
-
70.
The supernatant is removed and discarded.
-
71.
The gel slices are allowed to air-dry for 10 min before proceeding.
-
72.
A solution of 15 ng/μL trypsin is added in an amount to cover gel slices (usually between 20-50 μL).
-
73.
The samples are incubated on ice for 30 min.
-
74.
Any excess trypsin solution is removed and discarded.
-
75.
100 μL of 25 mM AmBic is added to each gel slice.
-
76.
Incubate the samples for around 18 h at 37°C.
Note: The 25 mM AmBic solution should cover the gel slice completely. After 15 min, check to ensure that the AmBic still covers the gel slices. More AmBic can be added if necessary.
-
77.
Fresh peptide extraction buffer (PXB) is prepared.
-
78.
New Eppendorf tubes are washed with 100 μL of PXB and labeled.
-
79.
Transfer extracts obtained in the following steps to these PXB washed and labeled tubes.
-
80.
100 μL of PXB is added to each gel slice.
-
81.
Samples are incubated in PXB for 15 min at 25°C to extract the trypsin digested peptides.
-
82.
Extracted peptides in PXB are transferred to the appropriately labeled tubes from step 77.
-
83.
Steps 79–81 are repeated, and the 2 extraction fractions for each gel slice are collected in the same tube.
-
84.
Samples are then completely dried using the SpeedVac.
Note: The time for samples to dry the samples is variable depending upon the volume of the sample and the capabilities of the speed-vac instrument. We normally need around 2 h to dry our samples.
-
85.
The dried peptide pellet is resuspended in 100 μL of 0.1% trifluoroacetic acid (TFA) in water.
-
86.
Purification of peptides to remove salts and detergents is performed using Pierce C18 tips (100 μL bed).
Note: C18 tips are ready-to-use pipette tips that capture and concentrate peptides from the sample and remove salts and detergents. Instead of tips, other commercially available C18 cleaning spin columns can be used as well. The tips we used in our protocol have the capacity to bind 80 μg of peptides. We used the User Guide provided by the manufacturer to conduct this step.
-
87.
After peptide purification is complete, the samples are dried with SpeedVac.
-
88.
The peptide pellet is resuspended with 0.1% formic acid (FA) in water, so that the final concentration of the peptides is around 500 ng/μL (check concentration with nanodrop).
-
89.
Now, the samples are ready to be submitted for LC-MS/MS analysis.
LC-MS/MS
-
90.Liquid chromatography is used to separate the isolated peptides prior to MS acquisition.
-
a.Around 1 μg of the peptides are injected into the UltiMate 3000 UHPLC system equipped with C18 analytical LC column (2 μm, 100 Å, 75 μm i.d. × 50 cm, nanoViper, Thermo Scientific), directly connected to Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific).
-
b.After injection, peptides are loaded onto a trap C18 column and washed with Mobile phase A (0.1% formic acid in water), at a flow rate of 4 μL/min.
-
c.Peptides are separated on the analytical C18 column using a step gradient of 4%–25% Mobile phase B (80% ACN/19.9% water/0.1% formic acid) from 10-100 min, and 25%–45% Mobile phase B from 100-130 min, at the flow rate of 300 nL/min, and 50°C.
-
a.
-
91.Eluted peptides are then analyzed by high-resolution mass spectrometry, in data-dependent acquisition mode (DDA).
-
a.A survey full scan MS is set for acquiring in the Orbitrap mode, for a scan range from m/z 350 to 1800, at a resolution of 120,000.
-
b.The automatic gain control (AGC) target for MS1 is set to 4 × 105, with an ion filling time of 100 ms.
-
c.The most intense parent ions with charge states of 2–6 are selected to be isolated in 3 s cycles.
-
d.Parent ions are fragmented using higher-energy collisional dissociation (HCD) with 35% normalized collision energy.
-
e.Fragmented ions are detected at a mass resolution of 30,000 at 200 m/z.
-
f.The AGC target for MS2 is set to 5 × 104, with an ion filling time of 60 ms. Dynamic exclusion is set for 30 s with a 10-ppm mass window.
-
a.
Note: Other nanoLC and high-resolution mass spectrometer systems with similar capabilities may be used. If the experiment is done in triplicate for each sample, a label-free quantitation workflow can be incorporated into the analysis with software such as Proteome Discoverer. Consult the vendor’s manual for detailed instruction. There are also algorithms such as Significance Analysis of INTeractome (SAINT; http://saint-apms.sourceforge.net/Main.html) that can differentiate the bona fide interactors from the adventitious non-specific proteins. Consult the developer’s website for installation and analysis guidelines. We recommend that these experiments be performed in duplicate or triplicate biological replicates to confirm the reproducibility of the results. In addition, validation with orthogonal experimental methods such as co-IP of the identified prey followed by detection by western blot of the original bait or yeast/mammalian two-hybrid systems are important to confirm individual interactions.
Expected outcomes
This protocol is optimized for the enrichment of endogenous bait protein, and the identification and analysis of its interacting partners using mass spectrometry. The bait protein is expected to be one of the top-ranking proteins in the database search results. There should also be an intense band at the molecular weight corresponding to the bait protein after the gel Coomassie staining. Non-specific binders (contaminants) such as keratins and similar are expected to be found to some extent in all samples but should not affect the final list of specific interactors, as they can be excluded based on the results of the negative control IgG probe. The number of proteins detected as interactors depends on several factors, including the cell line and the efficiency, specificity of the antibody, and natural variation in protein interactions between different bait proteins. In our experiment with 293FT cell line, we identified 137 FANCA interactors, whereas in the experiment with EndoC-βH3, more than 200 unique FANCA interactors were found (Lagundzin et al., 2019). The use of this protocol provided new insights into the role of FANCA specific to pancreatic β cells.
Limitations
Studying the protein interactome is very challenging, as some of the interactions might be dependent upon specific stimuli or transient nature, such as enzyme-substrate interactions. In protocols where co-immunoprecipitation is employed, the challenge starts with the cell lysis steps, as this may obstruct the detection of weak protein interactions (Sinder et al., 2015; Dunham et al., 2012). It was reported that sometimes protein interactors cannot be detected at all if the target protein is a part of large protein complexes that require their natural environment in the cell to maintain their association (Miernik and Thelen, 2008; Dunham et al., 2012; Smirle et al., 2013; Sinder et al., 2015). The experimental outcome using this protocol depends on the efficiency of the antibody to immunoprecipitate the bait protein and is improved by use of the appropriate negative control, IgG. If some protein interactions are known for the bait, the presence of these detected by either western blot or LC-MS/MS following Co-IP in the specific Ab pulldown but not the IgG control can act as a validation that the purification procedure is working, protein interactions are maintained, and the Co-IP conditions (namely washing steps) were appropriate. The specificity of the control IgG is of high importance, as it should not bind to the bait protein. Having a good negative control is crucial, as sometimes a portion of non-specific proteins remain in the eluate, despite many washing steps.
We tested the protocol using different cell lines, and although we didn’t test it yet for tissue samples, Zafar et al. (2017) used Dynabeads® to study the protein interactome in human tissue samples and reported promising results.
Troubleshooting
Problem 1
Bait protein was not detected with Western blot (see recommendations in the CRITICAL section following step 42).
Potential solution
We report that the starting protein amount should be 2.5 mg. However, you can try to increase the protein amount in your sample. You may also try increasing the amount of antibody. Finally, it is possible that the expression of the target protein is weak in your cell line.
Problem 2
Bait protein is detected by Western blot and/or LC-MS/MS but is also identified in the IgG sample.
Potential solution
This problem is most likely due to issues arising in the Co-IP steps. We noticed this problem mostly when IgG used was of a mouse origin. However, Andolino et al. (2018) reported no issue using mouse IgG. Some proteins that could be used as bait are naturally adventitious and the appropriate experimental procedures must be optimized to ensure the bait is not also present in the negative control. Utilizing a different normal IgG source, changing the type of beads, or switching to an affinity epitope tagging system for the bait could help reduce non-specific binding of the bait in the negative control, as you cannot proceed with the experiment if IgG is non-specifically binding the bait protein. Additional wash steps during the Co-IP can also decrease non-specific interactions. In some cases the bait protein is found in the IgG control detected by LC-MS/MS, this could be the result of carryover contamination when the LC system is not adequately flushed between injection of samples. It is recommended that a blank sample containing a peptide standard be run between analytical samples. This can help identify potential carryover contaminants as well as flush the system to prevent this problem.
Problem 3
There are too many interactors detected with mass spectrometry.
Potential solution
It is unusual that thousands of proteins represent the real interactors to any protein. This happens when non-specific proteins remain bound to the target protein and create a ‘background’ on the spectra. This can also occur if many non-specific proteins bind to the antibody-coupled beads. This can usually be avoided by increasing the number of washing steps during Co-IP (steps 31–34). Also, instead of the 18–24 h Co-IP suggested in step 30, you may try decreasing the time available for adventitious proteins to bind to the beads by decreasing the time of bait protein capture to around 4–8 h.
Problem 4
There are too many contaminants, such as keratins and polymers detected by mass spectrometry.
Potential solution
Make sure that all reagents and solution used are ‘mass spectrometry’ grade, and that specific attention was given to clean lab practices, such as using gloves and thoroughly cleaning surfaces. This should eliminate many common contaminants.
Problem 5
There are too few interactors detected.
Potential solution
Make sure that all reagents and solution used are freshly made prior to each step. Special attention should be given to the peptide extraction buffer (see recipe for PXB) (used in steps 76–81) and all the solutions used to clean the samples before LC-MS/MS, to avoid any possible peptide loss.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nicholas T. Woods (nicholas.woods@unmc.edu)
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This research was supported by the Fanconi Anemia Research Fund and the NIH through the awards P20GM121316, P30CA036727, and 1P50CA127297.
Author contributions
Conceptualization, D.L. and N.T.W.; Methodology, D.L., H.C.L., and N.T.W.; Investigation, D.L., K.L.K., and C.H.L.; Writing – Original Draft, D.L., H.C.L., K.L.K., and N.T.W.; Visualization, D.L. and H.C.L.; Writing – Review & Editing, D.L., H.C.L., K.L.K., and N.T.W.; Funding Acquisition, N.T.W.; Resources, N.T.W.; Supervision, D.L. and N.T.W.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Dragana Lagundžin, Email: dragana.lagundzin@unmc.edu.
Nicholas T. Woods, Email: nicholas.woods@unmc.edu.
Data and code availability
The raw mass spectrometry data files generated for this project have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the database identifiers PRIDE: PXD010589 and PRIDE: PXD010570.
References
- Andolino C., Hess C., Prince T., Williams H., Chernin M. Drug-induced keratin 9 interaction with Hsp70 in bladder cancer cells. Cell Stress Chaperones. 2018;23:1137–1142. doi: 10.1007/s12192-018-0913-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham W., Mullin M., Gringas A. Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics. 2012;12:1576–1590. doi: 10.1002/pmic.201100523. [DOI] [PubMed] [Google Scholar]
- Lagundžin D., Hu W.-F., Law H.C.H., Krieger K.L., Qiao F., Clement E.J., Drincic A., Nedic O., Naldrett M.J., Alvarez S., Woods N.T. Delineating the role of FANCA in glucose-stimulated insulin secretion in beta cells through its protein interactome. PLoS One. 2019;14:e0220568. doi: 10.1371/journal.pone.0220568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miernik J., Thelen J. Biochemical approaches for discovering protein-protein interactions. Plant J. 2008;53:597–609. doi: 10.1111/j.1365-313X.2007.03316.x. [DOI] [PubMed] [Google Scholar]
- Sinder J., Kotlyar M., Saraon P., Yao Z., Jurisica I., Stagljar I. Fundamentals of protein interaction network mapping. Mol. Syst. Biol. 2015;11:848. doi: 10.15252/msb.20156351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirle J., Au C., Jain M., Dejgaard K., Nillson T., Bergeron J. Cell biology of the endoplasmatic reticulum and the Golgi apparatus through proteomics. Cold Spring Harb. Perspect. Biol. 2013;5:a015073. doi: 10.1101/cshperspect.a015073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafar S., Shafiq M., Younas N., Schmitz M., Ferrer I., Zerr I. Prion protein interactome: identifying novel targets in slowly and rapidly progressive forms of Alzheimer’s disease. J. Alzheimers Dis. 2017;59:265–325. doi: 10.3233/JAD-170237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw mass spectrometry data files generated for this project have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the database identifiers PRIDE: PXD010589 and PRIDE: PXD010570.