

Summary
Generation of human motor neurons (MNs) overcomes the inaccessibility to patient brain tissues and greatly facilitates the research in MN-related diseases. Here, we describe a protocol for generation of neural progenitor cells (NPCs) from human induced pluripotent stem cells (hiPSCs), followed by preparation of functional MNs. The optimized induction condition with the expression of three transcription factors in a single lentiviral vector significantly improved the yield and purity, making it possible to biochemically identify dysregulated factors in diseased neurons.
For complete details on the use and execution of this protocol, please refer to Ding (2021), Ding et al. (2021), and Sepehrimanesh and Ding (2020).
Subject areas: Cell Biology, Cell culture, Molecular Biology, Neuroscience, Stem Cells, Cell Differentiation
Graphical abstract
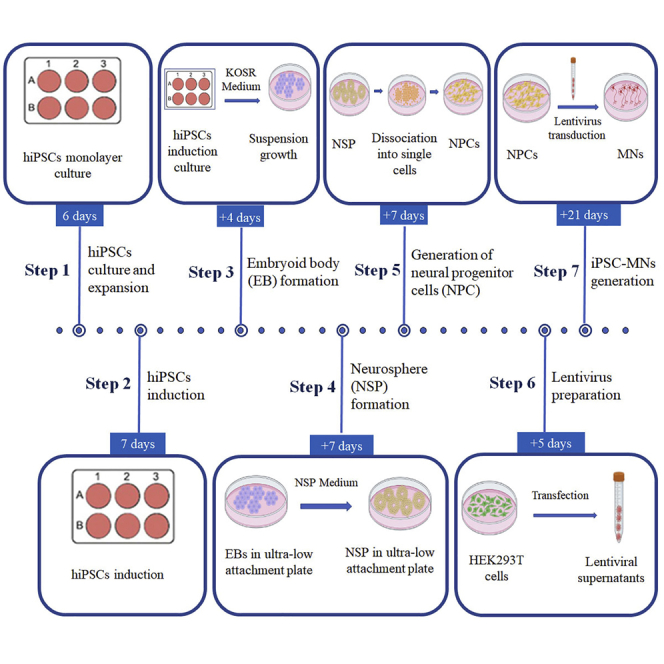
Highlights
-
•
Detailed protocol to generate motor neurons from human induced pluripotent stem cells
-
•
Protocol assesses the identity and efficiency of motor neuron generation in vitro
-
•
Optimized induction condition improves the yield and purity of motor neurons
Generation of human motor neurons (MNs) overcomes the inaccessibility to patient brain tissues and greatly facilitates the research in MN-related diseases. Here, we describe a protocol for generation of neural progenitor cells (NPCs) from human induced pluripotent stem cells (hiPSCs), followed by preparation of functional MNs. The optimized induction condition with the expression of three transcription factors in a single lentiviral vector significantly improved the yield and purity, making it possible to biochemically identify dysregulated factors in diseased neurons.
Before you begin
-
1.
Obtain the permission from institutional animal care and use committee (IACUC) for the animal study. This protocol has been approved by IACUC at University of Louisiana at Lafayette.
-
2.
Order the antibodies and qPCR primers that are listed in the key resources table for quality control and assessment of conversion efficiency.
-
3.
Acquire all critical reagents mentioned in the key resources table.
-
4.
Acquire/thaw hiPSCs and maintain as undifferentiated cultures in mTeSR-based medium.
-
5.
Prepare media using recipes below.
Note: Both the hiPSCs maintenance and differentiation media are good for up to 2 weeks at 4°C. Thus, it is not advisable to prepare all media at once, rather it is better to prepare it when needed at the specific step of the procedure.
CRITICAL: All procedures are performed in a BSL-2 certified laboratory biosafety cabinet with standard aseptic techniques. Cultures are grown and maintained in a humidified incubator at 37°C with 5% CO2.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-SOX2 (1:200) | Santa Cruz | Cat# sc-365823, RRID:AB_10842165 |
| Mouse anti-OCT4 (1:200) | Santa Cruz | Cat# sc-5279, RRID:AB_628051 |
| Mouse anti-SSEA4 (1:200) | Abcam | Cat# ab16287, RRID:AB_778073 |
| Rabbit anti-Nanog (1:100) | Abcam | Cat# ab21624, RRID:AB_446437 |
| Mouse anti-NESTIN (1:500) | Santa Cruz | Cat# sc-23927, RRID:AB_627994 |
| Rabbit anti-TUBB3, monoclonal (1:2000) | BioLegend | Cat# 801201, RRID:AB_2313773 |
| Goat anti-CHAT, monoclonal (1:200) | Millipore | Cat# AB144P, RRID:AB_2079751 |
| Mouse anti-HB9, monoclonal (1:500) | DSHB | Cat# 81.5C10-c |
| Rabbit anti-Islet1 (ISL1), monoclonal (1:250) | Abcam | Cat# ab109517; RRID: AB_10866454 |
| Donkey anti-Mouse IgG (H + L), Alexa Fluor 488 (1:500) | Jackson ImmunoResearch | Cat# 715-545- 150, RRID:AB_2340846 |
| Sheep anti-Mouse IgG (H + L), Alexa Fluor 594 (1:500) | Jackson ImmunoResearch | Cat# 515-515-062, RRID:AB_2340331 |
| Donkey Anti-Rabbit IgG (H+L), Alexa Fluor 488 (1:500) | Jackson ImmunoResearch | Cat# 711-545-152, RRID:AB_2313584 |
| Donkey Anti-Rabbit IgG (H+L), Alexa Fluor 594 (1:500) | Jackson ImmunoResearch | Cat# 711-585- 152, RRID:AB_2340621 |
| Recombinant DNA | ||
| pMDLg/pRRE | (Dull et al., 1998) | RRID: Addgene_12251 |
| pRSV-Rev | (Dull et al., 1998) | RRID: Addgene_12253 |
| pMD2.G | (Dull et al., 1998) | RRID: Addgene_12259 |
| pCSC-NGN2-IRES-GFP-T2A-Sox11 | (Liu et al., 2016) | RRID: Addgene_90214 |
| pCSC-ISL1-T2A-LHX3 | (Liu et al., 2016) | RRID: Addgene_90215 |
| pCSC-Ngn2-IRES-ISL1-T2A-LHX3 | (Sepehrimanesh and Ding, 2020) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| mTeSR1 complete kit (basal medium plus 5× supplement) | STEMCELL Technologies | Cat#85850 |
| Dulbecco's modified Eagle’s medium/nutrient mixture F-12 (DMEM-F12) | Hyclone | Cat#SH30023.02 |
| Dulbecco's Modified Eagle Medium (DMEM) | Gibco | Cat# 11965092 |
| Neurobasal Media | Thermo Fisher Scientific | Cat#21103049 |
| MEM Non-Essential Amino Acids Solution (NEAA) (100×) | Thermo Fisher Scientific | Cat#11140050 |
| Glutamax (100×) | Thermo Fisher Scientific | Cat#35050 |
| N2 supplement (100×) | Thermo Fisher Scientific | Cat#17502-048 |
| B27 supplement (50×) | Thermo Fisher Scientific | Cat#17504-044 |
| Retinoic acid (RA) | MilliporeSigma | Cat#R2625 |
| Valproic acid (VPA) | MilliporeSigma | Cat#99-66-1 |
| 2-Mercaptoethanol (BME) | Thermo Fisher Scientific | Cat#21985023 |
| Heat Stable Recombinant Human Basic Fibroblast Growth Factor (bFGF) | Gibco | Cat#PHG0367 |
| Human EGF Recombinant Protein | Gibco | Cat# PHG0311L |
| Heparin | Thermo Fisher Scientific | Cat#BP252450 |
| bFGF | PeproTech | Cat#100-18B |
| BDNF (Brain Derived Neurotrophic Factor) | PeproTech | Cat#450-02 |
| GDNF (Glial Derived Neurotrophic Factor) | PeproTech | Cat#450-10 |
| NT3 (Neurotrophic factor 3) | PeproTech | Cat# 450-03 |
| Forskolin (FSK) | MilliporeSigma | Cat# F6886 |
| DMSO, Dimethyl Sulfoxide | Thermo Fisher Scientific | Cat# D1391 |
| KnockOut™ Serum Replacement | Thermo Fisher Scientific | Cat#10828010 |
| Penicillin/streptomycin (Pen-Strep) | Thermo Fisher Scientific | Cat#15140122 |
| Dulbecco (d) PBS (without calcium, magnesium) | Thermo Fisher Scientific | Cat#14190250 |
| Triton X-100 | MilliporeSigma | Cat#X100 |
| Accutase (100 mL) | Corning | Cat#25-058-CI |
| Polyethylenimine (PEI) | Polysciences | Cat# 23966-2 |
| Versene | Thermo Fisher Scientific | Cat#A4239101 |
| Matrigel Matrix | Corning | Cat#356234 |
| Bovine serum albumin (BSA) | Thermo Fisher Scientific | Cat# bp9706 |
| Rho kinase (ROCK) inhibitor (Y-27632), 10 mM | MilliporeSigma | Cat#129830-38-2 |
| TRIzol™ Reagent | Invitrogen™ | Cat#15596026 |
| SuperScript™ III First-Strand Synthesis System | Invitrogen™ | Cat#18080051 |
| Antifade Mounting Medium | Vector Laboratories | Cat#H-1400-10 |
| PowerTrack™ SYBR Green Master Mix | Thermo Fisher Scientific | Cat#A46012 |
| Experimental models: Organisms/strains | ||
| Postnatal day 1 (P1) pups | Charles River | C57BL/6(B6) Mouse Inbred 027 |
| Experimental models: Cell lines | ||
| Mice Primary Astrocytes | N/A | N/A |
| iPSC lines | Coriell Institute for Medical Research | Coriell Cat# GM23476, RRID: CVCL_T841 |
| HEK293T cells | ATCC | ATCC Cat# CRL-3216, RRID: CVCL_0063 |
| Oligonucleotides | ||
| See Primer List in Table 1. | (Akter et al., 2021) | See Primer List in Table 1. |
| Other | ||
| 6-well plate | Corning | Cat#490007-412 |
| Petri Dishes with Clear Lid | Thermo Fisher Scientific | Cat#FB0875713 |
| Armadillo PCR Plate, 96-well T | Thermo Fisher Scientific | Cat# AB2496 |
| 24-well cell culture plates | Thermo Fisher Scientific | Cat#142485 |
| 0.22-μm vacuum filter | MilliporeSigma | Cat# SLGP033RB |
| 0.45-μm vacuum filter | MilliporeSigma | Cat# SLHV033RB |
| 15 mL polystyrene Conical tube | Thermo Fisher Scientific | Cat# 14-959-53A |
| 50 mL polystyrene Conical tube | Thermo Fisher Scientific | Cat# 14-432-22 |
Table 1.
Primer list
| Marker | Target | Forward/Reverse primer sequence (5′-3′) |
|---|---|---|
| Pluripotency marker | SOX 2 | AGGATAAGTACACGCTGCCC/TTCATGTGCGCGTAACTGTC |
| Pluripotency marker | NANOG | TGTCTTCTGCTGAGATGCCT/CAGAAGTGGGTTGTTTGCCT |
| Pluripotency marker | KLF4 | TCTCCAATTCGCTGACCCAT/CGGATCGGATAGGTGAAGCT |
| Differentiation marker | PAX6 | GGGCGGAGTTATGATACCTACA/ATATCAGGTTCACTTCCGGGAA |
| Differentiation marker | OTX1 | TACGCCCTCCTCTTCCTACT/GCATGTGGGTGGTGATGATG |
| Differentiation marker | DCN | CTGAAGAACCTTCACGCATTGA/GGCAATTCCTTCAGCTGATTCT |
| Differentiation marker | IGF2 | CAATATGACACCTGGAAGCAGT/GTAGAGCAATCAGGGGACGG |
| Differentiation marker | GATA2 | ACCTGTTGTGCAAATTGTCAGA/ATCCCTTCCTTCTTCATGGTCA |
| Differentiation marker | SOX7 | ACTCCACTCCAACCTCCAAG/TTCATTGCGATCCATGTCCC |
| Differentiation marker | SOX17 | ATCGGGGACATGAAGGTGAA/TCCTTAGCCCACACCATGAA |
| House-Keeping Gene | GAPDH | CAAATTCCATGGCACCGTCA/GGACTCCACGACGTACTCAG |
Materials and equipment
All equipment and materials need to be sterilized or autoclaved. Any reagents or liquids that cannot be autoclaved need filter (0.22 μm) sterilization. We recommend all materials to be prepared freshly and stored at 4°C for no longer than two weeks. Scaling up and down according to the amount of medium needed.
iPSC culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| mTeSR1Basal media | N/A | 400 mL |
| 5× supplement | 1× | 100 mL |
| Pen/Strep (100×) | 1× | 5 mL |
| Total | N/A | 505 mL |
iPSC induction media
| Reagent | Final concentration | Amount |
|---|---|---|
| iPSC culture medium | N/A | ∼50 mL |
| Retinoic acid (RA, 100 mM) | 10 μM | 5 μL |
| Valproic acid (VPA, 1 M) | 0.5 mM | 25 μL |
| Total | N/A | 50 mL |
Resuspension growth/KOSR media
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 | N/A | ∼38.5 mL |
| KOSR (100%) | 20% | 10 mL |
| Glutamax (100×) | 1× | 0.5 mL |
| NEAA (100×) | 1× | 0.5 mL |
| BME (1000×) | 50 μM | 50 μL |
| Pen/Strep (100×) | 1× | 0.5 mL |
| Total | N/A | 50 mL |
Neurosphere (NSP) medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 | N/A | ∼48 mL |
| N2 supplement (100×) | 1× | 0.5 mL |
| Pen/Strep (100×) | 1× | 0.5 mL |
| Glutamax (100×) | 1× | 0.5 mL |
| NEAA (100×) | 1× | 0.5 mL |
| BME (1000×) | 50 μM | 50 μL |
| Heparin (5 mg/mL) | 8 μg/mL | 80 μL |
| bFGF (200 ug/mL) | 10 ng/mL | 2.5 μL |
| EGF (100 ug/mL) | 10 ng/mL | 5 μL |
| Total | N/A | 50 mL |
Neuron progenitor cell (NPC) medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 (1:1) | N/A | 24 mL |
| Neurobasal | N/A | 24 mL |
| Pen/Strep (100×) | 1× | 0.5 mL |
| N2 supplement (100×) | 1× | 0.5 mL |
| B27 supplement (50×) | 1× | 1 mL |
| Glutamax (100×) | 1× | 0.5 mL |
| NEAA (100×) | 1× | 0.5 mL |
| BME (1000×) | 50 μM | 50 μL |
| bFGF (200 ug/mL) | 10 ng/mL | 2.5 μL |
| EGF (100 ug/mL) | 10 ng/mL | 5 μL |
| ROCK inhibitor (Y-27632, 10 mM) | 10 μM | 50 μL |
| Total | N/A | 50 mL |
NPC freezing medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Complete NPC medium | N/A | 30 mL |
| KOSR (100%) | 30% | 15 mL |
| DMSO (100%) | 10% | 5 mL |
| ROCK inhibitor (Y-27632, 10 mM) | 10 μM | 50 μL |
| Total | N/A | 50 mL |
Neuron maturation medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM: F12 (1:1) | N/A | 24 mL |
| Neurobasal media | N/A | 24 mL |
| Pen/Strep (100×) | 1% | 0.5 mL |
| N2 (100×) | 1× | 0.5 mL |
| B27 (50×) | 1× | 1 mL |
| FSK (20 mM) | 5 mM | 12.5 μL |
| BDNF (20 μg/mL) | 10 ng/mL | 25 μL |
| GDNF (20 μg/mL) | 10 ng/mL | 25 μL |
| NT3 (20 μg/mL) | 10 ng/mL | 25 μL |
| Total | N/A | 50 mL |
Note: Medium compositions of NPC medium, Neurosphere medium, and maturation media were adapted from (Akter et al., 2021; Ding et al., 2021; Sepehrimanesh and Ding, 2020).
Astrocyte cell culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Dulbecco’s Modified Eagle Medium (DMEM) | N/A | ∼ 42.5 mL |
| Fetal bovine serum (FBS) | 15% | 7.5 mL |
| Pen/Strep (100×) | 1× | 500 μL |
| Total | N/A | 50 mL |
HEK Cell culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM | N/A | ∼45 mL |
| FBS | 10% | 5 mL |
| Pen/Strep (100×) | 1× | 500 μL |
| Total | N/A | 50 mL |
Store complete medium at 2°C–8°C for up to 2 weeks.
PEI solution stock
| Reagent | Final concentration | Amount |
|---|---|---|
| PEI (transfection grade, 25 kDa, Linear) | 1 μg/μL | 50 mg |
| Milli-Q water | N/A | 50 mL |
| Total | N/A | ∼50 mL |
RT-PCR reaction condition
RT-PCR master mix
| Reagent | Amount |
|---|---|
| PowerTrack™ SYBR™ Green Master Mix | 6.25 μL |
| Forward primers | 0.25 μL |
| Reverse primers | 0.25 μL |
| cDNA | 1 μL |
| Nuclease-free water | 4.25 μL |
| Total RT PCR volume | 12.00 μL |
RT-PCR cycling conditions
| Steps | Temperature | Duration | Cycles |
|---|---|---|---|
| Enzyme activation | 95°C | 2 min | 1 |
| Denature | 95°C | 15 s | 40 |
| Anneal/extend | 60°C | 60 s | |
| Melting curve analysis | Refer to instrument documentation | ||
Step-by-step method details
Part 1: hiPSCs culture and expansion
Timing: 6–10 days
This part permits the expansion of hiPSCs for cryostorage and produces healthy pluripotent colonies suitable for neural differentiation. hiPSCs are cultured under feeder-free conditions and passaged when cells approach about 70% confluency.
-
1.Prepare Matrigel Matrix coated plates before starting the hiPSC culture. One 6-well plate (at 80% confluency, about 1 million cells per well) is sufficient to start the differentiation.
-
a.Thaw Matrigel Matrix at 4°C. Make smaller aliquots using refrigerated pipette tips. Aliquots can be stored at −80°C for long term storage.Note: Matrigel is sensitive to temperature. When aliquoting Matrigel, place Matrigel and microcentrifuge tubes on ice within the biosafety cabinet. We suggest diluting Matrigel in DMEM-F12 (1:1) to prevent gelling during long term storage.
-
b.Dilute Matrigel at 1:100 in cold DMEM-F12 medium. For example, add 100 μL Matrigel in 10 mL DMEM-F12 media.Note: If hiPSCs colonies show enhanced differentiation and low adherence, higher concentration of Matrigel (1:50) could be used for coating plates to facilitate cell attachment.
-
c.Add 1 mL diluted Matrigel matrix to each well of the 6-well plate. Make sure to let Matrigel cover the entire surface of the well and incubate the plate at 37°C for minimum of 1 h.
-
d.Aspirate Matrigel just before seeding cells.Note: Avoid air dry of Matrigel-coated wells before seeding cells.
-
a.
-
2.
Rapidly thaw a hiPSC cryovial by gently swirling the vial in a 37°C water bath until there is just a small bit of ice left in the vial. In a laminar flow hood, transfer cells to a 15 mL conical tube containing 5 mL of pre-warmed iPSC culture medium mTesR1 containing 10 μM ROCK inhibitor.
-
3.
Spin cells at 200×g for 3 min.
-
4.
Remove supernatant and resuspend the cell suspension in mTesR1 medium containing 10 μM ROCK inhibitor and transfer 2 mL of the cell suspension into each well of the Matrigel-coated 6-well plate.
Note: Resuspend cell pellet in a proper volume of culture medium depending on the cell numbers in the frozen stock. We recommend seeding cells ranging from 1 × 104 to 2 × 104 cells/cm2.
-
5.
Change medium every day. Cells will be ready for the first passage in 2–4 days according to the growth of the hiPSC line.
Note: At this point, the healthy hiPSC colonies will appear as tight colonies with smooth edges and are separated from each other (Figure 1A).
-
6.
Once cells reach about 70% confluency, start the first cell passage by preparing another Matrigel-coated 6-well plate. This 6-well plate could be directly used for the iPSC induction.
-
7.Cells are passaged at 1:3 ratio. This way, two wells of the first 6-well plate (starting culture plate) can be passaged into a freshly prepared Matrigel-coated 6-well plate for iPSC induction.
-
a.Remove medium and wash wells with DPBS once.
-
b.Add 1 mL Versene to each well and incubate at 37°C for 6–7 min. The optimized incubation time varies from different hiPSC lines. For example, some hiPSC lines require 5 min while the genetically modified or patient derived hiPSC lines requires variable minutes.
-
c.Aspirate Versene from the wells. Add 2 mL mTesR1 medium to the wells and gently pipette up and down to dislodge the cells. Large aggregates of stem cell colonies should be seen floating in the medium. Gently triturate 2–3 times with a 5 mL pipette to break colonies into smaller pieces.Note: Versene is an EDTA solution to be used as a gentle non-enzymatic cell dissociation reagent and hence can be used for clump passaging. Typically, the cells remain attached at the bottom of the culture plate before adding neutralized medium.CRITICAL: Avoid generating a single-cell suspension because it will result in differentiation and poor cell survival.
-
d.Collect medium containing the cell colonies in a 15 mL tube and add extra mTesR1 medium up to 12 mL.
-
e.Gently mix by pipetting up and down 2 or 3 times. Transfer 2 mL cell suspension to each well of the newly Matrigel-coated 6-well plate.
-
f.Place 6-well plate in incubator and change iPSC culture medium daily.
-
a.
-
8.
The newly passaged 6-well plate should reach 80%–90% confluency in 3–5 days and can be used to start the iPSC induction.
Note: We recommend using hiPSCs culture within 2–4 passages from a frozen stock for this differentiation protocol.
Figure 1.

Images of healthy and differentiated hiPSC colonies
(A) Typical morphology of a healthy hiPSC colony with smooth edges.
(B) Differentiated hiPSC colonies usually appear as clusters of scattered and flat cells (arrowheads). Scale bars, 200 μm.
Part 2: iPSC induction
The next step is to direct hiPSCs toward a neuroectodermal fate, which can be assessed by the generation of NESTIN+/ PAX6+ neural progenitors.
-
9.
Prepare iPSC induction medium containing 10 μM RA and 0.5 mM VPA. Warm up to room temperature before use.
-
10.
Aspirate hiPSCs culture medium from the 6-well plate and add 2 mL of iPSC induction medium to each well.
-
11.
Every day, replace the medium completely with fresh medium for seven days.
Note: As cells are growing over time, more medium can be used to maintain the pH value (e.g., 3 mL per well of 6-well plate).
-
12.
Cells should appear as a monolayer and grow together tightly at the end of day 7 (Figure 2A). High & tight confluency is optimal for resuspension growth in the next step.
Figure 2.

Micrographs of cells at different stages during the induction of hiPSCs to neural progenitor cells (NPCs)
(A) Typical morphology of hiPSCs cultured in induction medium at 7 days.
(B) Embryoid body (EB) formed in resuspension growth medium.
(C) Neurosphere (NSP) in NSP medium.
(D) Neural Progenitor cells cultured in NPC medium.
(E) Some aggregates (white dot) could be noticed in NPC cultures. Scale bars, 100 μm.
Part 3: Embryoid body (EB) formation
This stage requires ultra-low attachment 10 cm Petri Dishes which prevent cells from attaching to the bottom.
Note: Ultra-low attachment plates have a specialized surface coated with a hydrophobic hydrogel which inhibits cell attachment and forces them to remain in the suspension.
-
13.
Prepare resuspension growth medium/KOSR medium with 10 μM Rock inhibitor.
Note: ROCK inhibitor Y-27632 has been shown to significantly inhibit apoptosis and enhance cell survival (Pettinato et al., 2015). The ROCK inhibitor could be added throughout the process for cell lines with poor growth.
-
14.
Warm up resuspension growth medium/KOSR medium at room temperature.
-
15.
Remove iPSC induction medium and wash wells once with DPBS.
-
16.
Add 1 mL Versene to each well and incubate at 37°C for 6–7 min.
-
17.
Remove Versene and add 1 mL of KOSR medium (resuspension medium) to each well. Gently shake the plates to dissociate cells. Scrape cells by using a 5 mL pipette if necessary.
-
18.
Transfer cell suspension to a 15 mL tube.
Note: Triturate the cell suspension 1–2 times using a pipette to break apart the larger cell clusters into smaller aggregates.
-
19.
Add 1 mL KOSR medium to rinse the well and pool together to the 15 mL tube.
Note: For 3 wells of 6-well plate, the total cell suspension volume is ∼6 mL.
-
20.
Add ∼9 mL of KOSR medium to a low attachment Petridish. Transfer 6 mL cell suspension to the Petridish and gently mix.
Note: Check under microscope and most cells should be in aggregates. To let aggregates fully suspended, the total volume should be ∼15 mL in each Petridish.
-
21.
Change resuspension growth medium/KOSR medium every 2 days and culture cell aggregates for 4 days to form embryoid body (EB) (Figure 2B).
-
22.Change medium during suspension growth.
-
a.Gently swirl the plate and transfer cell suspension into a 15 mL conical tube (if more than 15 mL, split into two tubes).
-
b.Allow the cell aggregates to settle to the bottom of the tube (it will take about 10 min).
-
c.Remove supernatant (leaving ∼1 mL) and add ∼14 mL of fresh KOSR medium.
-
d.Carefully transfer cell suspension to the Ultra-low attachment Petridish.
-
e.Gently shake the petri dish to aid uniform distribution of the aggregates and place it back to the incubator.
-
a.
Part 4: Neurosphere (NSP) formation
-
23.
Prepare Neurosphere (NSP) medium and warm it up at room temperature.
-
24.
Transfer cell suspension (embryoid body at 4 days in step 22) to a 15 mL tube (if more than 15 mL, split into two tubes) and let aggregates settle down to the bottom of the tube.
-
25.
Remove supernatant (leaving ∼1 mL liquid) and add ∼14 mL NSP Medium.
-
26.
Carefully transfer cell suspension to the Ultra-low attachment Petridish.
-
27.
Replace with fresh NSP medium every two days as describe in step 22 (∼15 mL NSP medium/10 cm Petridish).
-
28.
Cells are cultured in NSP Medium for 7 days to form neurospheres (Figure 2C).
Part 5: Generation of neural progenitor cells (NPCs)
In this stage, neurospheres will be digested into singular cells and expanded to generate NPCs.
-
29.
Prepare Matrigel-coated 10 cm culture plates as described in step 1.
-
30.
Transfer NSP aggregates to a 15 mL tube and let aggregates sit down to the bottom (∼10 min).
-
31.
Remove supernatant (leaving∼1 mL liquid) and add 5 mL of DPBS to wash the aggregates.
-
32.
Let aggregates sit down to the bottom (∼10 min). Remove the supernatant and add 8 mL Accutase to NSP aggregates.
-
33.
Incubate at 37°C for 15 min. Gently pipetting 5 strokes with a 5 mL pipette in the middle (at ∼7 min) to break the large aggregates.
-
34.
Add ∼3 mL NPC medium to neutralize the digestion.
-
35.
Gently pipette up and down several times to break most of aggregates into singular cells.
-
36.
Spin cells at 200×g for 3 min.
-
37.
Remove supernatant and resuspend cell pellet into appropriate volume of NPC medium.
-
38.
Seed NPCs onto Matrigel coated 10 cm culture plates (Figure 2D).
Note: Neurospheres from one plate can give rise to 2–4 million cells and can be seeded onto four 10 cm plates.
-
39.
Mix well and culture cells in incubator.
-
40.
Change NPC medium every two days until cells are close to full confluency and then prepare NPC frozen stocks in the following stage.
Preparation of NPC frozen stocks
When NPCs reach ∼90% confluency, prepare for frozen stocks. At this stage, some small aggregates could be noticed in the culture plate. Remove these aggregates (white spots) by a filter pipette tip (Figure 2E).
-
41.
Gently shake the medium and remove the supernatant. Wash once with DPBS.
-
42.
Add 3 mL Accutase to each 10 cm plate and incubate at 37°C for 2–3 min. Gently shake the plate and most cells should be dissociated from the plate.
-
43.
Add 3 mL NPC medium with rock inhibitor to neutralize the digestion. Pipette gently to dissociate cells from the plate.
-
44.
Transfer cell suspension to a 15 mL tube. Spin down at 200×g for 3 min.
Note: To estimate the yield, an aliquot (∼20 μL) could be taken from cell suspension for cell counting by a hemocytometer.
-
45.
Remove supernatant and resuspend cell pellets into desired volume of precold (4°C) NPC freeing medium and aliquot quickly into cryovials.
-
46.
Transfer cryovials into a freezing container and place it into a −80°C freezer. On the next day, transfer cryovials to a cryogenic storage container with liquid nitrogen for long-term storage.
Note: It is recommended to freeze NPC at a concentration of 1 × 106/mL/vial. The yield of one 10 cm plate is about 4 × 106 NPCs.
Part 6: Lentivirus preparation for motor neuron (MNs) generation
This part describes the preparation of lentiviruses expressing transcription factors, which are necessary and sufficient to induce NPCs to MNs. To ensure high induction efficiency, it is critical to prepare high-quality supercoiled plasmids and make lentiviruses with good titers. For detailed information about lentivirus preparation and the applications, please see our previous studies (Ding et al., 2013, 2016, 2018; Ding and Kilpatrick, 2013). In this protocol, we used three lentiviral vectors, pCSC-NGN2-IRES-GFP-T2A-Sox11 (Plasmid 1), pCSC-ISL1-T2A-LHX3 (plasmid 2), and pCSC-Ngn2-IRES-ISL1-T2A-LHX3 (plasmid 3) (see key resources table).
Culture and expansion of HEK293T cells
-
47.Quickly thaw a frozen stock of HEK293T cells in a 37°C water bath.
-
a.Transfer the cells to a 15 mL tube with 5 mL DMEM medium and centrifuge at 200×g for 3 min.
-
b.Discard the supernatant and re-suspend the cells in 10 mL fresh HEK Cell Culture medium.
-
a.
-
48.
Seed HEK293T cells into a 10 cm tissue culture plate. Grow cells at 37°C with 5% CO2 in a standard tissue culture incubator.
Note: It is recommended to use low passage number (<20) HEK cells in the preparation of lentiviruses.
-
49.Once cells reach about 90% confluency, split cells for expansion.
-
a.Aspirate media from culture plate and wash once with 1× PBS.
-
b.Incubate cells with 0.5 mL of 0.05% Trypsin (0.25% Trypsin diluted with 1× PBS) for 2–4 min until cells have been fully detached.
-
c.Add 3 mL of HEK cell culture medium to inactivate the dissociation reagent.
-
d.Triturate the cells to create single cell suspension, collect the cells into a 15 mL tube and centrifuge the cells at 200×g for 3 min.
-
e.Remove the supernatant and resuspend the cell pellet into proper volume of HEK culture medium. Split cells in 10 cm plates at the ratio of 1:3 to 1:5.
-
a.
Transfection and lentivirus preparation
The day before transfection, the HEK293T cells must be seeded at a proper cell density. Lentiviral plasmid DNA is delivered by the Polyethylenimine (PEI)-mediated transfection method.
-
50.
Day 0: Seed about 8 × 106 HEK293T cells into each 10 cm culture plate for lentivirus production. Next day, HEK cells should be about 80% confluency for transfection.
-
51.Day 1 (transfection):
-
a.Add the lentiviral plasmid and the packaging vectors to DMEM (no serum) as described in the recipe below. Scale up based on how many plates to be transfected.
-
b.Mix well by pipetting up and down for a few times.
-
c.Add desired amount of PEI into plasmid mixture and immediately mix thoroughly by pipetting up and down for 10 times or rigorously vortex for 15 s.
-
d.Incubate the mixture at room temperature (∼25°C) for 15 min.
-
e.Add the mixture (1 mL/ 10 cm plate) dropwise to each plate with gentle rocking.
-
f.Gently mix by shaking up and down, and then left and right for a few times before putting the plate back into incubator.
-
a.
| Recipe of lentiviral transfection mixture (for one 10 cm plate) | ||
|---|---|---|
| Reagent | Amount | Volume |
| DMEM | N/A | 1 mL |
| pMDLg/pRRE (1 μg/μL) | 2 μg | 2 μL |
| pMD2.G (1 μg/μL) | 1 μg | 1 μL |
| pRSV-Rev (1 μg/μL) | 0.5 μg | 0.5 μL |
| Desired plasmid (1 μg/μL) | 4 μg | 4 μL |
| Polyethylenimine (PEI) | 1 μg/μL | 22.5 μL |
Note: The ratio of PEI to the total amount of DNA is 1:3. For example, the total amount of DNA is 7.5 μg for a 10 cm plate, add 22.5 μg (22.5 μL of 1 μg/μL) PEI into the plasmid mixture in 1 mL DMEM.
-
52.
Day 2: After overnight (∼16 h) incubation, gently replace medium with 10 mL HEK cell culture medium for each plate.
Note: It is recommended to warm up HEK cell culture medium to room temperature and carefully replace the medium to avoid cell detachment.
-
53.
Day 3: Collect viral supernatants at about 48 h post-transfection (first collection). Add back 10 mL fresh HEK cell culture medium for each plate and incubate. Store the viral supernatant at 4°C.
-
54.
Day 4: Collect viral supernatants at about 72 h post-transfection (second collection). Discard the plates in a biohazard bag and trash it after autoclave.
-
55.
Pool two collections of the same lentivirus together and filter the viral supernatants with 0.45 μm vacuum filters. Store lentivirus at 4°C till use (within two weeks), or freeze at −80°C for longer term storage.
Note: Transfection efficiency can be examined under a fluorescence microscope if the lentiviral vector contains GFP reporter (e.g., plasmid #1). At lentivirus collection, most HEK cells (>90%) should be GFP (+) (Figure 3B).
Figure 3.

Transfection efficiency of plasmid with GFP reporter
(A) Micrograph of HEK cells (phase contrast) at virus collection.
(B) Micrograph of HEK cells (GFP) at virus collection (72 h post-transfection). Almost all HEK cells are transfected positive (plasmid #1 with co-expressed GFP). Scale bars, 100 μm.
Lentivirus titration
Before transduction, we recommend to titer the virus to verify the quality. The virus can be titrated based on GFP reporter (plasmid#1) (Figures 4A and 4B) or immunocytochemistry (ICC) of lentivirus expressed factors (plasmid#2 and #3) (Figures 4C–4F) as previously reported (Ding and Kilpatrick, 2013; Sepehrimanesh et al., 2021).
-
56.
Seed HEK cells in a 24 well plate at a density of 8 × 104 cells/well/0.5 mL medium. Cells will reach ∼70% confluency the next day at the time of titration.
-
57.
Make a serial dilution (e.g., 1:10, 1:100 etc.) of viral supernatant with HEK culture medium.
-
58.
Add 50 μL of undiluted and diluted lentivirus to desired wells. Set one well as a negative control without transduction.
-
59.
Change the medium after 8 h or overnight (∼16 h) exposure to lentivirus.
-
60.
Check the percentage of positive cells after 48–72 h.
Note: For lentivirus expressing GFP reporter, examine GFP (+) cells directly under a fluorescence microscope in living cells. For lentivirus that does not express fluorescent reporters, cells need to be fixed and perform ICC to examine lentivirus-expressed factors.
-
61.
Transduced positive cells should be more than 90% in undiluted wells and more than 30% in 10-fold diluted wells (Figure 4).
Figure 4.

Lentivirus titration
(A) Micrograph of HEK cells transduced with undiluted virus expressing GFP reporter (plasmid #1) at 72 h post-transduction. Scale bar, 50 μm.
(B) Micrograph of HEK cells transduced with 1:10 diluted virus expressing GFP reporter (plasmid #1) at 72 h post-transduction. Scale bar, 50 μm.
(C) Micrograph of HEK cells (phase contrast) transduced undiluted virus (plasmid #3) and ICC with ISL1 antibody. Scale bar, 25 μm.
(D) Micrograph of HEK cells (fluorescent) transduced undiluted virus (plasmid #3) and ICC with ISL1 antibody. Scale bar, 25 μm.
(E) Micrograph of HEK cells (phase contrast) transduced 1:10 diluted virus (plasmid #3) and ICC with ISL1 antibody. Scale bar, 25 μm.
(F) Micrograph of HEK cells (fluorescent) transduced 1:10 diluted virus (plasmid #3) and ICC with ISL1 antibody. Scale bar, 25 μm.
Part 7: Lentiviral transduction of NPCs to generate MNs
This part directs NPCs toward MNs by lentiviral delivery of transcription factors. This part can be done in two different ways. One approach is to deliver four transcription factors N-S-I-L (NEUROG2, SOX11, ISL1, and LHX3) with two lentiviruses. Another approach is to use a single lentivirus co-expressing three factors N-I-L (NEUROG2, ISL1, and LHX3). Both approaches can generate mature functional MNs. The second approach tends to generate MNs with higher purity and yield (Figure 6). These two approaches have been systematically examined and compared in our previous study (Sepehrimanesh and Ding, 2020).
-
62.
Prepare Matrigel-coated 6-well culture plate as described in step 1.
-
63.
Rapidly thaw NPC frozen cryovials by gently swirling the vial in a 37°C water bath until there is just a small bit of ice left in the vial. Transfer cells to a 15 mL conical tube containing 5 mL of pre-warmed NPC culture medium in a laminar flow cabinet.
-
64.
Spin down cells at 200×g for 3 min.
-
65.
Gently resuspend cell pellets into proper volume of NPC medium and seed NPCs onto Matrigel-coated plates at a density of 3 × 104 cells/cm2.
Note: One frozen vial with 1 × 106 NPCs is enough to seed 3 wells of a 6-well plate.
-
66.
Next day, the NPC confluency is expected to be about 60%. Add about 300 μL lentiviral supernatant to each well.
Note: At lentivirus transduction, the proper NPC density (50%–60% confluence) is critical to obtain higher yield of MNs. If two lentiviruses are used for transduction, add 300 μL of each lentiviruses at MOI ∼2.
-
67.
NPCs are exposed to lentiviruses for around 6 h to overnight (∼16 h). Then replace the culture medium with Neuronal Maturation Medium.
-
68.
Half change the medium every other day till analysis or replating.
Note: The age of iPSC-MNs is counted from the lentiviral transduction as dpi (days post-viral infection). Neuron-like morphology can be noticed after 3 dpi.
Figure 6.

Optimized induction condition dramatically improved iPSC-MN survival and yield
(A) Phase contrast micrographs of iPSC-MNs induced via indicated combinations of transcription factors at 7 dpi. Scale bars, 100 μm.
(B) Survived neurons under indicated conditions. The number of neurons at 1 wpi (weeks post-viral infection) was set as 100%. N, NGN2; S, mSox11; I, ISL1; L, LHX3. N (neurons) > 3,000 from triplicates. ∗∗ p<0.01; ∗∗∗ p<0.001.
(C) Representative micrographs of iPSC-MNs induced via indicated transcription factors at 3 wpi with immunostaining of TUBB3. Scale bars, 100 μm.
(D) The yield of iPSC-MNs at 3 wpi from 1 million NPCs at transduction. N (biological replicates) = 3. ∗∗∗∗ p<0.0001. Adapted with permission from (Sepehrimanesh and Ding, 2020).
Preparation of coverslips with monolayer of astrocyte cells
Mouse astrocytes were isolated from postnatal day 1 (P1) pups and cultured in DMEM with 15% FBS as reported (Schildge et al., 2013). The Animal Procedure Statement in this study was approved by institutional animal care and use committee (IACUC) at University of Louisiana at Lafayette. The IACUC approval number is 2019-8717-027.
-
69.Preparation of astrocyte cells:
-
a.Prepare Matrigel-coated 10 cm culture plate as described in step 1.
-
b.Rapidly thaw frozen astrocytes in a 37°C water bath. Transfer cells to a 15 mL conical tube containing 5 mL of pre-warmed astrocytes culture medium in a laminar flow cabinet.
-
c.Centrifuge the cells at 200×g for 3 min.
-
d.Add 10 mL of astrocytes culture medium and gently resuspend cell pellet.
-
e.Seed astrocytes onto Matrigel-coated plates at a density of 3 × 104 cells/cm2. One frozen vial with 1 × 106 astrocyte cells can seed a 10 cm plate.
-
a.
-
70.Preparation of coverslips with a monolayer of astrocyte cells:
-
a.Prepare Matrigel-coated 24 wells plate with glass coverslips.
-
b.Once astrocyte cells reach about 90% confluency, aspirate media from astrocytes cell culture plate and wash once with 1× PBS.
-
c.Incubate cells with 3 mL 0.05% Trypsin in PBS for 2–4 min until cells have been fully detached.
-
d.Add 3 mL of astrocyte cell culture medium to inactivate the dissociation reagent.
-
e.Collect the cells into a 15 mL tube and centrifuge at 200×g for 3 min.
-
f.Remove the supernatant and resuspend cells in 12 mL of astrocyte cell culture medium.
-
g.Triturate the cells to create single cell suspension and seed onto Matrigel-coated coverslips (0.5 mL/well).
-
a.
Replating MNs onto coverslips with monolayer astrocytes
At 5 dpi, MNs can be replated and seeded onto coverslips with a mono-layer of astrocytes for long-term culture (more than 14 dpi) and further analysis.
-
71.Dissociate MNs from culture plates.
-
a.Aspirate media from the cell culture plate and wash once with 1× PBS.
-
b.For each well of 6 well plate, incubate cells with 1 mL of dissociation reagent (e.g., accutase) for 2–4 min until most of cells are detached.Note: This step is critical for neuronal survival. Excess of accutase or longer incubation can be detrimental to neurons.
-
c.Add 2 mL of DMEM: F12 medium to neutralize the dissociation reagent.
-
d.Collect the cells into a 15 mL tube and centrifuge at 200×g for 3 min.
-
e.Remove the supernatant and resuspend cells gently in an appropriate volume of neuronal maturation medium.
-
a.
-
72.
Seed at a density of 1 × 103 cells/cm2 dissociated MNs onto coverslips with astrocytes (0.5 mL/well).
-
73.
Change half volume of Neuronal Maturation Medium (1:1) every other day till analysis.
Examination of MN identity
MN identity can be verified by immunostaining of specific markers at different developmental stages. The survival, purity and yield can be calculated as our previous reports (Ding et al., 2021; Sepehrimanesh and Ding, 2020).
Neuronal morphology is examined under inverted microscopy directly in living cultures. Within 7 dpi, neurites will rapidly grow out from the soma displaying neuronal phenotype. At 14 dpi, coverslips are fixed and immunostained with neuron specific markers, such as TUBB3, MAP2, and neurofilament protein (SMI-32) (Figure 5).
Figure 5.

Verification of iPSC-MN identity
(A) Confocal micrographs of MNs at 7 dpi. MAP2 antibody recognizes dendrites and SMI-32 antibody recognizes neurofilaments. Scale bar, 50 μm.
(B) Confocal micrograph of MNs at 10 dpi. Neuron marker TUBB3 shows the soma and neuron processes, HST (Hoechst 33342) stained nuclei, and nuclear HB9 was used as the early MN marker. The rectangle highlighted MN was also shown at a large magnification with separated channels. Scale bars, 50 μm and inset 20 μm.
(C) Confocal micrograph of MNs at 14 dpi. Neuronal marker TUBB3 shows the soma and neuron processes, HST stained nuclei, and choline acetyltransferase (ChAT) was used as a mature MN marker. The rectangle highlighted MN was also shown at a large magnification with separated channels. Scale bars, 50 μm and inset 20 μm. Adapted with permission from (Sepehrimanesh and Ding, 2020).
To verify MN identity, immunostaining for HB9 and/or ChAT (Figure 5) are performed at different maturation stages. HB9 is an early MN marker and highly expressed at 10–20 dpi. ChAT is a mature MN marker gradually increase the expression at 14 dpi or later.
-
74.ICC MN markers:
-
a.Take coverslips from 24-well plates at desired time points (7 dpi, 14 dpi, etc.)
-
b.Fix cultured cells with 4% paraformaldehyde (PFA) in PBS (pH 7.4) and incubate at room temperature for 30 min.
-
c.Wash twice with PBS.
-
d.Incubate fixed cells in blocking buffer (PBS containing 0.2% Triton X-100 and 3% BSA) for 1 h for permeabilization and blocking.
-
e.Incubate fixed cells with primary antibodies in blocking buffer (PBS containing 0.2% Triton X-100 and 3% BSA) at 4°C overnight (∼16 h).Note: The antibodies include anti-TUBB3 and anti-MAP2 as neuronal markers, anti-HB9 as an early MN marker, anti-CHAT as a late MN marker.
-
f.After removal of primary antibodies, wash coverslips with PBS for 10 min × 3.
-
g.Incubate fixed cells with corresponding fluorophore-conjugated secondary antibodies in dark, at room temperature for 2 h.
-
h.Wash coverslips with PBS for 10 min × 3.
-
i.Incubate cells with Hoechst 33342 (1 μg/mL in PBS) for 10 min to stain cell nuclei.
-
j.Briefly wash coverslips with PBS twice.
-
k.Mount coverslips with mounting medium on microslides.
-
l.Air dry the coverslips at room temperature in dark (30 min to a few hours).
-
a.
-
75.
Visualize cells under the confocal microscope.
-
76.
MNs purity could be calculated using the ratios of HB9 (+) cells to TUBB3 (+) cells, and/or CHAT (+) cells to TUBB3 (+) cells as previously reported (Ding et al., 2021; Sepehrimanesh and Ding, 2020).
Expected outcomes
Using this protocol, hiPSCs can be induced to NPCs (Figure 2), and then differentiated into MNs. To validate cells at different stages in culture, immunocytochemistry and/or quantitative RT-PCR analysis can be used to examine the expression of stage-specific genes. For example, hiPSCs highly express pluripotency markers such as OCT4, NANOG, SOX2 and SSEA4 (Akter et al., 2021). NPCs strongly express PAX6 and NESTIN (Liu et al., 2021). Successfully differentiated neurons robustly express generic neuron marker TUBB3, MAP2 and neurofilament protein (SMI-32) (Figure 5). The MN identity can be verified by the early MN marker nuclear HB9 (Figure 5A) and the late MN marker CHAT (Figure 5B). Compared with the use of two lentiviruses expressing four transcription factors (N-S-I-L), the optimized protocol using a single lentivirus expressing three factors (N-I-L) significantly improved the neuronal survival and the final iPSC-MN yield (Figure 6).
Limitations
The generation of iPSC-MNs from hiPSCs following this protocol is easy, reliable, and efficient. Preparation of high quality of hiPSCs and NPCs is prerequisite to successfully generate MNs. Transduction of NPCs with proper amount of lentivirus is critical to obtain MNs with high purity and yield. Using this protocol, we have successfully generated MNs using patient derived iPSC lines (Ding, 2022; Ding et al., 2021). However, the efficiency of this protocol may vary with different hiPSC lines and may require the optimization of transduction and culture conditions.
Troubleshooting
Problem 1
Poor recovery of hiPSC frozen stocks (steps 1–4)
Poor recovery of hiPSC from frozen stocks can be manifested by more floating dead cells and very slow growth in the first several days after seeding.
Potential solution
Before starting the hiPSC culture, use freshly prepared Matrigel Matrix to coat plates at least 30 min at 37°C. Prewarm the culture medium to room temperature. Quickly thaw the frozen stocks in a 37°C water bath. Gently resuspend the cell pellets and transfer cell suspension to culture plates and minimize the physical damage of cells. These measures will improve the recovery of hiPSC from frozen stock.
Problem 2
Poor growth and more differentiation in hiPSC culture (steps 5–8)
Slow growth and substantial differentiated hiPSCs are observed.
Potential solution
To improve the growth and minimize the differentiation, use freshly prepared culture medium. Prewarm the culture medium to room temperature and change the medium daily.
Problem 3
Poor neural differentiation (steps 9–12)
Poor neural differentiation is observed when directing pluripotent hiPSCs toward a neuroectodermal fate.
Potential solution
In our hands, iPSC induction medium containing RA, and VPA is sufficient to direct hiPSCs toward a neuroectodermal fate. However, if poor neural differentiation is observed, SMAD inhibitors e.g., dorsomorphin (BMP signaling inhibitor, 1.5 μM) and SB431542 (TGF beta inhibitor, 10 μM) can be added to the iPSC induction medium. Inhibition of BMP/TGFβ signaling has been shown to enhance neuroectodermal fate commitment in hESCs (Wattanapanitch et al., 2014).
Problem 4
Poor attachment during neuron progenitor cells generation (steps 29–40)
During NPC formation, the single-cell dissociation step can result in poor attachment and poor viability for certain cell lines, even with the addition of ROCK inhibitors. This issue will dramatically affect the efficiency and yield of NPCs and iPSC-MNs.
Potential solution
There are several factors need to be considered. For example, at the NSP stage, making single cells will lead to neuronal death and larger aggregates can lead to poor attachment. In such an instance, gentle pipetting can break down NSP aggregates into small aggregates and also avoid making single cells. Coating plates with appropriate concentration (1:100) of Matrigel is crucial for these steps.
Problem 5
Low titers of lentiviruses (steps 50–61)
Potential solution
This might occur due to multiple reasons. One reason could be poor growth of HEK293T cells. Hence, it is recommended to use low-passage cells and let the cells fully recover from frozen stock (passage at least twice) before setting transfection. Improper HEK cell density (too low or too high) also can result in low transfection efficiency. Secondly, low transfection efficiency of plasmids. It is advisable to check the quality of plasmids and transfection reagents. Proper ratio of helper plasmids, lentiviral vectors, and transfection reagent will improve lentivirus titers.
Resource availability
Lead contact
Further information and requests for resources and reagents can be directed to Dr. Baojin Ding (baojin.ding@lsuhs.edu).
Materials availability
The majority of materials used in this protocol are commercially available. The sequence of qPCR primers used for characterization of hiPSCs and NPCs were listed in Table 1.
Acknowledgments
We thank members of the Ding laboratory for help and discussion. This work was supported by a NIH grant (NIH/NINDS NS112910 to B.D.) and the Department of Defense (DoD) Peer Reviewed Medical Research Program (PRMRP) Discovery Award (W81XWH2010186 to B.D.).
Author contributions
Conceptualization, B.D.; methodology, B.D. and M.A.; investigation, B.D., M.A., H.C., M.S., and M.A.H.; writing, B.D. and M.A.; funding acquisition and supervision, B.D.
Declaration of interests
The authors declare no competing interests.
Data and code availability
This study did not generate/analyze any datasets or code that were submitted to any other repositories.
References
- Akter M., Cui H., Chen Y.H., Ding B. Generation of two induced pluripotent stem cell lines with heterozygous and homozygous GAG deletion in TOR1A gene from a healthy hiPSC line. Stem Cell Res. 2021;56:102536. doi: 10.1016/j.scr.2021.102536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B. Generation of patient-specific motor neurons in modeling movement diseases. Neural Regen. Res. 2021;16:1799–1800. doi: 10.4103/1673-5374.306083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B. Novel insights into the pathogenesis of DYT1 dystonia from induced patient-derived neurons. Neural Regen. Res. 2022;17:561–562. doi: 10.4103/1673-5374.320978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B., Cave J.W., Dobner P.R., Mullikin-Kilpatrick D., Bartzokis M., Zhu H., Chow C.W., Gronostajski R.M., Kilpatrick D.L. Reciprocal autoregulation by NFI occupancy and ETV1 promotes the developmental expression of dendrite-synapse genes in cerebellar granule neurons. Mol. Biol. Cell. 2016;27:1488–1499. doi: 10.1091/mbc.E15-07-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B., Dobner P.R., Mullikin-Kilpatrick D., Wang W., Zhu H., Chow C.W., Cave J.W., Gronostajski R.M., Kilpatrick D.L. BDNF activates an NFI-dependent neurodevelopmental timing program by sequestering NFATc4. Mol. Biol. Cell. 2018;29:975–987. doi: 10.1091/mbc.E16-08-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B., Kilpatrick D.L. Lentiviral vector production, titration, and transduction of primary neurons. Methods Mol. Biol. 2013;1018:119–131. doi: 10.1007/978-1-62703-444-9_12. [DOI] [PubMed] [Google Scholar]
- Ding B., Tang Y., Ma S., Akter M., Liu M.L., Zang T., Zhang C.L. Disease modeling with human neurons reveals LMNB1 dysregulation underlying DYT1 dystonia. J. Neurosci. 2021;41:2024–2038. doi: 10.1523/JNEUROSCI.2507-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B., Wang W., Selvakumar T., Xi H.S., Zhu H., Chow C.W., Horton J.D., Gronostajski R.M., Kilpatrick D.L. Temporal regulation of nuclear factor one occupancy by calcineurin/NFAT governs a voltage-sensitive developmental switch in late maturing neurons. J. Neurosci. 2013;33:2860–2872. doi: 10.1523/JNEUROSCI.3533-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.C., Liu F.Y., Gao X.Y., Chen Y.L., Meng Q.Q., Song Y.L., Li X.H., Bao S.Q. Global transcriptional analyses of the Wnt-induced development of neural stem cells from human pluripotent stem cells. Int. J. Mol. Sci. 2021;22:7473. doi: 10.3390/ijms22147473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.L., Zang T., Zhang C.L. Direct lineage reprogramming reveals disease-specific phenotypes of motor neurons from human ALS patients. Cell Rep. 2016;14:115–128. doi: 10.1016/j.celrep.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinato G., Wen X., Zhang N. Engineering strategies for the formation of embryoid bodies from human pluripotent stem cells. Stem Cells Dev. 2015;24:1595–1609. doi: 10.1089/scd.2014.0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildge S., Bohrer C., Beck K., Schachtrup C. Isolation and culture of mouse cortical astrocytes. J. Vis. Exp. 2013;71:50079. doi: 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepehrimanesh M., Akter M., Ding B. Direct conversion of adult fibroblasts into motor neurons. STAR Protoc. 2021;2:100917. doi: 10.1016/j.xpro.2021.100917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepehrimanesh M., Ding B. Generation and optimization of highly pure motor neurons from human induced pluripotent stem cells via lentiviral delivery of transcription factors. Am. J. Physiol. Cell Physiol. 2020;319:C771–C780. doi: 10.1152/ajpcell.00279.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattanapanitch M., Klincumhom N., Potirat P., Amornpisutt R., Lorthongpanich C., U-pratya Y., Laowtammathron C., Kheolamai P., Poungvarin N., Issaragrisil S. Dual small-molecule targeting of SMAD signaling stimulates human induced pluripotent stem cells toward neural lineages. PLoS One. 2014;9:e106952. doi: 10.1371/journal.pone.0106952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze any datasets or code that were submitted to any other repositories.