

Abstract
A cytosolic iron chaperone poly(rC)-binding protein 1 (PCBP1) is a multifunctional RNA-binding protein involving gene transcription, RNA regulation, and iron loading to ferritins. PCBP1 is also known to repress autophagy, but the role of PCBP1 in ferritinophagy and ferroptosis remains unrevealed. Therefore, we examined the role of PCBP1 in ferritinophagy-mediated ferroptosis in head and neck cancer (HNC) cells. The effects of system xc– cystine/glutamate antiporter (xCT) inhibitors and PCBP1 gene silencing/overexpression were tested on HNC cell lines and mouse tumor xenograft models. These effects were analyzed by assessing cell viability and death, lipid reactive oxygen species and iron production, lipid, malondialdehyde, mRNA/protein expression, and autophagy flux assays. Interaction between PCBP1 and BECN1 mRNA was also examined by luciferase and RNA-protein pull-down assays. PCBP1 gene silencing increased autophagosome generation and autophagic flux. Conversely, PCBP1 upregulation inhibited autophagy activation via direct binding to the CU-rich elements on the 3′-untranslated region (3′-UTR) of BECN1 mRNA. The internal deletion or mutation of the 3′-UTR F2 region recovered BECN1 mRNA stability repressed by PCBP1, resulting in enhanced ferritinophagy-mediated ferroptosis. Besides, PCBP1 knockdown promoted polyunsaturated fatty acid peroxidation by increasing ALOX15 expression. Further, excess iron accumulation caused mitochondrial dysfunction in PCBP1-suppressed cells. A ferroptosis inducer sulfasalazine significantly suppressed tumor growth in mice with the transplantation of PCBP1-silenced HNC. Our data suggest that the dual functions of PCBP1 repressing BECN1 and ALOX15 mRNAs contribute to attenuating cancer susceptibility to ferroptosis inducers.
Keywords: Ferroptosis, poly(rC)-binding protein 1, Ferritinophagy, Iron, Lipid peroxidation
Graphical abstract
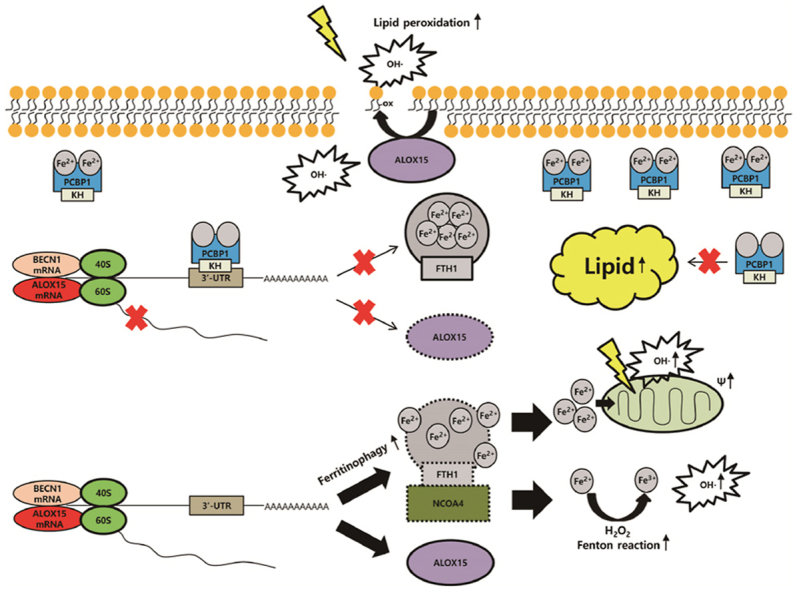
The suppression of poly(rC)-binding protein 1 (PCBP1) in cancer cells promotes ferroptosis. PCBP1 negatively controls BECN1 mRNA, ALOX15 mRNA, and lipogenesis. Knockdown of PCBP1 enhances ferritinophagy, increasing labile iron pool. Ferrous iron accumulation promotes mitochondrial dysfunction producing hydroxyl radical (OH·), and ALOX15 accelerates oxidized polyunsaturated fatty acid (PUFA). Oxidized PUFA reacts with hydroxyl radical, which produces lipid peroxidation and ferroptosis.
Highlights
-
•
PCBP1 expression is related to head and neck cancer survival and response to ferroptosis inducers.
-
•
Knockdown of PCBP1 increased ferroptosis sensitivity by inducing an increased labile iron pool.
-
•
PCBP1 negatively regulated ferritinophagy by the 3′-UTR binding of BECN1 mRNA.
-
•
Knockdown of PCBP1 increased lipid peroxidation by ALOX15 expression.
-
•
PCBP1 depletion promoted ferroptosis susceptibility in vitro and in vivo.
Abbreviations
- CCK-8
cell counting kit-8
- DMSO
dimethyl sulfoxide;
- DMT1
divalent metal transporter 1
- EMT
epithelial-mesenchymal transition
- FFA
free fatty acid
- GSH
glutathione
- GPX4
Glutathione peroxidase 4
- HNC
head and neck cancer
- 4-HNE
4-hydroxynonenal
- KH
K homology
- LC3
microtubule–associated proteins 1A/1B-light chain 3
- LIP
labile iron pool
- 15-LOX
ALOX15, 15-lipoxygenase
- MDA
malondialdehyde
- MMP
mitochondrial membrane potential
- MUFA
monounsaturated fatty acid
- PCBP1
Poly(rC)-binding protein 1
- PCBP2
Poly(rC)-binding protein 2
- PUFA
polyunsaturated fatty acid
- RPA
rhodamine B-[(1,10-phenanthroline-5-yl)-aminocarbonyl]benzyl ester
- ROS
reactive oxygen species
- TMRE
tetramethylrhodamine ethyl ester
- xCT
system xc– cystine/glutamate antiporter
- 3′-UTR
3′-untranslated region
1. Introduction
Ferritinophagy is an autophagic phenomenon that involves iron-storing ferritin to release intracellular free iron by nuclear receptor coactivator 4 (NCOA4) [1,2]. This process is accompanied by autophagosome formation of microtubule-associated proteins 1A/1B-light chain 3 (LC3) and is completed when combined with lysosomes [3]. Mounting evidence also shows that redox reaction between ferrous iron and polyunsaturated fatty acid (PUFA) leads to distinct non-apoptotic cell death, ferroptosis [4]. Ferroptosis is an autophagic process based on the iron-dependent accumulation of excessive lipid peroxidation [5]. PUFAs are essential sources to produce lipid peroxidation by multiple inner bonds of reactive oxygen species (ROS) [4]. Consequently, excessive lipid peroxidation disturbs cellular membrane integrity and causes cell death [6]. Glutathione peroxidase 4 (GPX4), a key regulator of ferroptosis, is a seminal cellular antioxidant for eliminating lipid ROS by reducing and oxidizing glutathione (GSH) [7]. System xc– cystine/glutamate antiporter (xCT) is another central regulator of ferroptosis as a substrate of GSH synthesis contributing to lipid ROS detoxification [7].
Poly(rC)-binding protein 1 (PCBP1) is a multifunctional RNA-binding protein that plays various roles in gene transcription, RNA regulation, and iron chaperone [8,9]. PCBP1 is a major iron chaperon facilitating iron loading to ferritins and an RNA-binding protein interacting with other target protein BolA2 independently at its different faces [10]. PCBP1-lacked mice in bone marrow showed impaired heme synthesis by iron flux dysfunction [11]. PCBP1 works as an adapter protein containing three conserved K homology (KH) domains enabling interactions with RNA and binding multiple types of ligands such as iron-GSH complexes [12]. Further, PCBP1 is associated with regulating 15-lipoxygenase (15-LOX, ALOX15) translation via binding the 3ʹ-untranslated region (3′-UTR) regulatory complex controlling 80s ribosome subunit assembly [13,14]. ALOX15 is a non-heme iron-containing protein known for generating PUFAs, a source of lipid peroxidation [15].
PCBP1 represses autophagy, an essential process of ferroptosis, but the role of PCBP1 in ferritinophagy and ferroptosis remains unknown. The present study has newly found the therapeutic possibility of PCPB1 depletion promoting ferroptosis in cancer cells. Here, we examined the role of PCBP1 in ferritinophagy-mediated ferroptosis in head and neck cancer (HNC) cells.
2. Methods
2.1. Cell viability and death assays
After exposure to cyst(e)ine deprivation, erastin, or sulfasalazine, cell viability and death were assessed using cell counting kit-8 (CCK-8) (Dojindo Molecular Technologies, Inc., Tokyo, Japan) and SYTOX Green (Thermo Fisher Scientific, Waltham, MA, USA) staining, respectively, according to the manufacturer's protocol.
2.2. Measurements of lipid and mitochondrial ROS
Cellular lipid and mitochondrial ROS were measured by adding 5 μM BODIPY™ C11 (Thermo Fisher Scientific) and 5 μM mitoSOX™ Red (Thermo Fisher Scientific). Image quantification was performed using ImageJ software.
2.3. Measurements of intracellular iron
Labile iron pool (LIP) assay was measured by using calcein acetoxymethyl ester (Corning Inc., Corning, NY, USA) and iron chelator, deferoxamine (Abcam, Cambridge, UK). Intracellular iron contents were also quantified using an iron assay kit (Sigma-Aldrich, St. Louis, MO, USA). FerroOrange (Dojindo) was used to stain intracellular ferrous iron using a 5 μM FerroOrange solution. The mitochondrial ferrous iron level was assayed using rhodamine B-[(1,10-phenanthroline-5-yl)-aminocarbonyl]benzyl ester (RPA) assay (Squarix GmbH, Marl, Germany) and Mito-FerroGreen (Dojindo) staining.
2.4. Measurements of autophagic flux
The LC3-GFP-puncta was assessed using the ZEISS LSM 880 confocal microscope in the cells treated with erastin, sulfasalazine, cyst(e)ine deprivation with or without 30 μM bafilomycin A1. Co-localization of LC3-GFP-puncta and lysosome was also examined with Lysotracker™ Red DND-99 (Thermo Fisher Scientific) staining.
2.5. Measurements of free fatty acid and malondialdehyde
Intracellular free fatty acid was measured using the PicoSens™ Free Fatty Acid Quantification Kit (BIOMAX, Seoul, Republic of Korea). Cellular malondialdehyde (MDA) level was quantified using a lipid peroxidation (MDA) assay kit (Abcam, Cambridge, UK).
2.6. RNA interference, gene transfection, plasmid construction, and RNA-protein pull-down assays
Supplementary methods refer to details on target gene silencing, expression induction, and plasmid construction. Interaction between PCBP1 and BECN1 mRNA was examined by luciferase and RNA-protein pull-down assays.
2.7. Tumor xenograft
HN12 cells with transfection of shPCBP1 or control vector and HN2 cells with PCBP1 overexpression vector or control vector were subcutaneously injected into the bilateral flank of six-week-old athymic BALB/c male nude mice (nu/nu) that were subjected to the treatment of vehicle or sulfasalazine (250 mg/kg daily per intraperitoneal route).
2.8. Statistical analyses
The expression levels of PCBP1 mRNA were obtained from the normal mucosa (n = 44) and HNC (n = 526) datasets of TCGA and compared using a t-test. The Kaplan–Meier and log-rank tests were used to determine and statistically compare the survival rates in the HNC cohort, respectively. The statistically significant differences between the treatment groups were assessed using Mann–Whitney U test or analysis of variance (ANOVA) with the Bonferroni post-hoc test. All statistical tests were two-sided, and a P-value of <0.05 was statistically significant.
Additional details on the data analyses and experimental procedures are provided in Supplementary Methods.
3. Results
PCBP1 expression was associated with cancer survival and ferroptosis sensitivity. The mean mRNA expression levels of PCBP1 of normal mucosa and HNC from the TCGA did not significantly differ (P = 0.770, Fig. S1A). The best cutoff of tumor PCBP1 expression was 99.89. Five-year overall survivals of patients with high and low PCBP1 expression were 40.8 ± 3.7% and 62.9 ± 5.9%, respectively (P = 0.016, Fig. S1B). HN2, HN5, and HN10 had relatively low PCPB1 expression, but HN3, HN6, and HN12 had high PCBP1 expression (Figs. S1D–E). The cells with low PCBP1 expression were more sensitive to erastin than those with high PCBP1 expression, accompanied by significantly higher lipid peroxidation and LIP accumulation (Fig. S1C, S1F–I). These were attenuated by ferrostatin-1 or deferoxamine treatment (Fig. S1J). These findings indicate that PCBP1 expression is closely related to HNC survival and ferroptosis sensitivity.
Next, our data showed that PCBP1 depletion promoted iron accumulation and ferroptosis. We examined whether the modulation of PCBP1 expression affected ferroptosis sensitivity in cancer cells. Knockdown of PCBP1 (shPCBP1) in the HN3, HN6, and HN12 with high PCPB1 expression significantly increased intracellular total or ferrous iron, LIP and ROS levels, and cell death compared to vector control when exposed to erastin, sulfasalazine, or cyst(e)ine deprivation (P < 0.001), which was reduced by PCBP1 re-expression (PCBP1res) (Fig. 1A–J and S2). The findings were attenuated by ferrostatin-1 or deferoxamine treatment (Figs. S2B–C). On the contrary, PCBP1 overexpression in HN2 cells with low PCBP1 expression (PCBP1 plasmid) decreased cell death and intracellular iron and lipid ROS levels (Fig. S3). These results were consistent with a previous study: excess iron accumulation might lead to ferroptosis [4]. PCBP1 knockdown induced increased ferrous and total iron accumulation, while introducing the PCBP1 gene decreased intracellular iron levels, and PCBP1 overexpression lowered the ferrous and total iron levels after treatment with erastin, sulfasalazine, or cyst(e)ine deprivation (Fig. 1 and Fig. S3). The expression levels of iron homeostasis-related molecules were changed along with the knockdown of PCBP1: FTH1 and FPN were downregulated, whereas DMT1 and TFRC were upregulated (Fig. 1K–L). Taken together, these findings indicate that PCBP1 depletion promotes the sensitivity to ferroptosis inducers via intracellular ferrous iron accumulation.
Fig. 1.

Knockdown of PCBP1 increases ferroptosis sensitivity.
(A) The iron assay measured the total iron level in HN12 with transfection of vector, shPCBP1, or shPCBP1 plus PCBP1 cDNA plasmid (PCBP1res). The shPCBP1 #1 and #2 were prepared by inserting two different target sequences for PCBP1 into the shRNA vector. (B–G) Cell viability (B) and cell death (C–D), immunoblotting (E), lipid peroxidation (F), and labile iron pool (LIP) (G) assays in HN12 cells with vector, shPCBP1, or PCBP1res transfection. Cell viability and death were measured by using cell counting kit-8 (CCK-8) and SYTOX Green staining, respectively, after exposure to ferroptosis inducers, e.g., 10 μM erastin, 1 mM sulfasalazine (SAS), or cyst(e)ine deprivation (cys (−)) for 48 h. The SYTOX Green-stained cells were quantified using ImageJ software (C), and the mean positive fractions were compared with those of the control or different group (D). Lipid peroxidation was measured by adding 5 μM BODIPY C11 for 30 min, and LIP was measured using calcein acetoxymethyl ester and iron chelator, deferoxamine, in the cells after exposure to the ferroptosis inducers for 6 h. The error bars represent standard errors from three independent experiments. Original magnification, × 200. Scale bar, 50 μm. (H–J) Images of lipid peroxidation cells (H–I) and cellular iron amounts (J). Non-oxidized (red) and oxidized (green) were measured in cells exposed to DMSO or 10 μM erastin for 6 h by adding 5 μM BODIPY C11. Image quantification was performed using ImageJ software (H), and the ratios of oxidized cells to total cells were calculated (I). Iron levels were measured by iron assay after 10 μM erastin, 1 mM SAS, or cys (−) for 6 h. Original magnification, × 200. Scale bar, 50 μm. (K–L) mRNA (K) and protein (L) expression levels of PCBP1, PCBP2, DMT1, FTH1, FPN, and TFRC in HN12 cells were confirmed by RT-qPCR and immunoblotting, respectively. The error bars represent standard errors from three independent experiments. ns; no significance, *P < 0.05, **P < 0.01, ***P < 0.001 among different cells or treatments. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Next, this study examined that PCBP1 was functionally connected to the molecules related to ferritinophagy. PCBP1 has a seminal role in translation as an RNA regulator [16]. ELAVL1/HuR, an RNA-binding protein, increases BECN1 mRNA stability while activating ferritinophagy [17]. We analyzed the sequence of BECN1 mRNA by using the RBP map to find a putative binding region of PCBP1. Our data showed that a putative binding region of PCBP1 was located in the 3′-UTR of BECN1 mRNA (Fig. 2A). The fragments of BECN1 mRNA for luciferase assay were produced from this finding (Fig. 2B and C). With the introduction of pcDNA 3.1-PCBP1, luciferase activity decreased in pLFL (full-length fragment) and pLF2 (including the PCBP1-binding region) (Fig. 2D). The RNA-protein direct interaction was confirmed by RNA-protein pull-down assay (Fig. 2E). Therefore, our data suggested that PCBP1 negatively controls BECN1 mRNA.
Fig. 2.

PCBP1 destabilizes BECN1 mRNA.
(A) BENC1 mRNA 3′-UTR sequence and putative PCBP1 binding region were analyzed using RBP map and UCSC genome browser. (B–C) The fragments of BECN1 3′-UTR mRNA were produced by deletion and site-directed mutation PCR and inserted into the pGL4.54 vector. (D) Luciferase activity was measured in the HEK293FT cell line transfected with BECN1 fragment inserted vector and pcDNA3.1-PCBP1 overexpression vector. (E) mRNA affinity isolation assay was performed with the biotinylated transcripts of the BECN1 mRNA 3′-UTR, and then PCBP1 binding to the different fragments of the BECN1 mRNA 3′-UTR was determined by immunoblotting. The error bars represent standard errors from three independent experiments. ns; not significant, *P < 0.05, **P < 0.01, ***P < 0.001 between vector and PCBP1 plasmid.
We also examined whether the knockdown of PCBP1 promotes ferritinophagy. LC3-GFP vector was transfected into cells, and LC3 expression via GFP was observed after erastin treatment. Bafilomycin A1 was also used to determine autophagic flux. The number of puncta (and co-stained puncta with ferrous iron) increased, cell viability decreased, and intracellular ferrous iron increased in the PCBP1-silenced cells more than in vector control (Fig. 3 and Fig. S2F). LC3B-II, Atg5, and 4-HNE increased, but p62, NCOA4, and ferritin decreased in the PCBP1-silenced cells, whereas Bafilomycin A1 increased the number of puncta and the expression of LC3B-II and p62 when the cells were treated with the ferroptosis inducers (Fig. 3D). Knockdown of PCBP1 significantly increased the cellular contents of ferrous iron and the number of co-stained puncta when cells were exposed to ferroptosis inducers (Fig. 3E–F, Fig. S2F). However, PCBP1 overexpression in HN2 cells decreased autophagy and ferritinophagy when treated with the ferroptosis inducers (Fig. S4). Therefore, these findings indicate that PCBP1 hindered iron release from ferritinophagy via repression of BECN1 mRNA. Further, our study showed that iron overload into mitochondria could cause mitochondrial dysfunction [18]. In addition, TMRE significantly decreased, but mitochondrial superoxide and ferrous iron staining intensities significantly increased in PCBP1-inhibited cells after exposure to the ferroptosis inducers (P < 0.01), which was recovered by re-introduction of PCBP1 (Fig. S5). These findings indicate that the knockdown of PCBP1 induced mitochondrial dysfunction by increasing iron overload into mitochondria.
Fig. 3.

Knockdown of PCBP1 enhances ferritinophagy induction.
(A–B) The LC3 puncta were measured using immunofluorescence in HN12 cells with stable transfection of pEGFP-LC3 after 10 μM erastin treatment for 6 h and then treated with or without 30 μM bafilomycin (Baf) A1 for 2 h. The number of puncta was quantified by ImageJ. Original magnification, × 400. Scale bar, 25 μm. (C) Cell viability was measured 48 h after erastin treatment with or without bafilomycin A1. (D) Autophagic flux and ferroptotic cell death were determined by immunoblotting. (E) Cytoplasmic ferrous iron was measured using FerroOrange staining after exposure to ferroptosis inducers for 6 h, and then all samples were quantified by ImageJ. (F) Co-staining of LC3-GFP puncta and lysosomes was observed by immunofluorescence after 10 μM erastin treatment for 6 h. The yellow color indicates co-stained regions. Original magnification, × 400. Scale bar, 25 μm. The error bars represent standard errors from three independent experiments. ns; no significance, *P < 0.05, **P < 0.01, ***P < 0.001 among different groups. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
PCBP1 is also known to specifically regulate translation from the 3′ end of ALOX15, a critical molecule for oxidized PUFA generation [14]. Therefore, we examined whether the knockdown of PCBP1 contributed to oxidized PUFA increase. Knockdown of PCBP1 increased cellular lipid, oxidized PUFA, free fatty acid levels, and its related molecule expression, and the cellular level of MDA and 4-HNE, the end products of oxidized PUFA (Fig. S6). Cell viability significantly decreased in cells with linoleic acid (10 μM) compared to cells with oleic acid (10 μM) when exposed to erastin or sulfasalazine (Fig. S6F). FASN, FADS2, SCD1, and SREBP1 increased in the cells with PCBP1 knockdown and decreased in the cells with PCBP1 overexpression (P < 0.001) (Fig. S6H). These findings suggest that the knockdown of PCBP1 promoted PUFA generation and lipid peroxidation by activating fatty acid synthesis and ALOX15 expression.
Finally, we examined whether PCBP1 depletion increased ferroptosis susceptibility in vivo. Tumor growth was faster in both the PCBP1-suppressed group (HN12 shPCBP1) and the PCBP1-overexpressing group (HN2 PCBP1 plasmid) than the vector control (P < 0.001) (Fig. 4A–C). Further, tumor growth was significantly suppressed by sulfasalazine administration: more significantly in PCPB1 silencing (HN12 shPCBP1) or low expressing tumors (HN2 vtr) by sulfasalazine treatment (P < 0.001). Cellular iron contents, lipid amounts, and lipid peroxidation accumulation were relatively high in PCBP1-suppressing tumors and increased in those with sulfasalazine treatment (Fig. 4D–F). ACSL4, ALOX15, 4-HNE, and PTGS2 expression increased in the PCBP1-suppressed or low expression group with sulfasalazine treatment, whereas FTH1 and NCOA4 expression decreased, which was reversed in PCBP1-overexpressing cells (Figs. 4G and S7). Therefore, the findings suggest that PCBP1 suppression contributes to the increased ferroptosis sensitivity in vivo.
Fig. 4.

PCBP1 depletion increases tumor ferroptosis sensitivity in vivo.
(A–C) Representative images (A), in vivo growth (B), and weights (C) of tumors treated with or without daily intraperitoneal injection of SAS (250 mg/kg) for 20 days. Each group included seven mice. (D) Cellular iron contents were measured using iron assay kits in tumor tissues. (E–F) Malondialdehyde (MDA) assays (E) and oil red staining (F) in tumor tissues. (G) Immunoblotting of PCBP1, ACSL4, ALOX15, NCOA4, FTH1, 4-HNE, and PTGS2 in HN12 tumors. The error bars represent standard errors from tumors of different groups. ns; no significance, *P < 0.05, **P < 0.01, ***P < 0.001 between different groups. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
The present study showed that PCBP1 was a seminal ferroptosis regulator by inhibiting iron-mediated ferritinophagy and lipid peroxidation. High expression of PCBP1 was closely related to poor survival outcomes of the HNC patients and decreased response to xCT inhibitors. Knockdown of PCBP1 caused an iron depletion response inducing enhanced ferritinophagy to replenish iron in cancer cells. PCBP1 repressed autophagy through direct binding to the 3′-untranslated region of BECN1 mRNA. Knockdown of PCBP1 increased fatty acid synthesis and ALOX15 mRNA expression as well as ferritinophagy-mediated ferroptosis. Knockdown of PCBP1 increased susceptibility to ferroptosis inducers in vitro and in vivo. Therefore, our data suggest that PCBP1 negatively regulated ferroptosis, and its depletion promoted ferroptosis.
PCBP1 depletion led to iron accumulation and increased ferroptosis sensitivity. Blocking PCBP1 function as an iron carrier induced the increased levels of cellular free iron that could predispose iron-dependent cell death, ferroptosis. A recent study suggested iron accumulation and suppression of tumor growth by PCBP1 depletion via blocking p62 degradation [19]. Iron accumulation in cells with PCBP1 suppression appears not to be interrupted by a little increased expression of PCBP2 that carries out the supplement function of iron efflux via ferroportin [9]. The cellular iron levels are modulated by molecules related to iron homeostasis [20]. Suppression of PCBP1 made the cells perceive a lack of cellular iron by a failure of its iron- and RNA-binding activity and caused insufficient iron supply into chaperons requiring iron for a proper role [10,12]. The depletion of PCBP1 may hamper cytosolic iron-sulfur cluster assembly, such as the PCBP1-BolA2 and BolA2-Glrx3 chaperone complexes [10,12]. This may induce a reaction similar to the iron starvation response seen in cancer cells with suppression of NFS1 or Glrx5 when the proteins are not properly supplied with iron: this increased iron import and decreased iron storage and efflux [21,22]. Our results underline that PCBP1 plays a vital role as a defender in ferroptosis by blocking an iron depletion response. Therefore, knockdown of PCBP1 may be a potential target to promote ferroptosis from enhanced cellular iron levels.
The current results showed that PCBP1 had different functions to regulate autophagy, an essential process of ferroptosis, and fine-tune cellular iron homeostasis. In this context, a potential link between ferritinophagy and PCBP1 needed to be studied since cellular iron accumulation and ferroptosis were related to ferritinophagy [1,2]. The previous study suggests that ELAVL1/HuR protein promotes ferritinophagy by stabilizing BECN1 mRNA [17]. PCBP1 can also inhibit autophagy by downregulating LC3B and cancer cell survival [23]. However, the study did not elucidate any mechanism linking PCBP1 and LC3B. The present study showed that PCBP1 repressed ferritinophagy via direct binding of the CU-rich elements on BECN1 mRNA 3′-UTR. On the contrary, knockdown of PCBP1 activated ferritinophagy, promoting ferroptosis sensitivity to xCT inhibitors. Interestingly, the knockdown of PCBP1 led to the upregulation of ELAVL1/HuR. We assumed that ELAVL1 and PCBP1 competitively bound the BECN1 mRNA 3′-UTR since the ELAVL1/HuR-binding region was close to the PCBP1-binding region [17]. The BECN1-activated autophagy by PCBP1 knockdown enhanced NCOA4-mediated ferritinophagy in the cells with exposure to xCT inhibitors. The regulation of PCBP is directly related to cellular iron metabolism. It is a more effective means of enhancing ferroptotic cell death since cancer cells commonly require more cellular iron levels than normal cells [20]. Further, PCBP1 knockdown induced cellular and mitochondrial iron overload, boosting mitochondrial dysfunction by ferroptosis inducers [18]. Our study suggests that PCBP1 negatively regulates ferritinophagy-mediated ferroptosis by destabilizing BECN1 mRNA.
Lipid peroxidation is the main component of pillars consisting of ferroptosis [3]. Suppression of PCBP1 activated lipid synthesis and changed cellular lipid composition, which set up a favorable condition to cause lipid peroxidation and ferroptosis in cancer cells. A recent study indicates that PCBP1 knockdown promotes lipid synthesis and peroxidation [24]. Besides, other previous studies suggest that PCBP1 negatively regulates ALOX15 [13,14]. The upregulation of ALOX15 by knockdown of PCBP1 leads to accelerated oxidized PUFA generation, which helps to induce ferroptosis. Although a clear link between PCBP1 and lipid synthesis is still elusive, the changes of cellular lipid contents might cause epithelial-mesenchymal transition (EMT) as PCBP1 functions as a negative regulator of EMT [25]. Transforming growth factor-β2 might also activate partial EMT and fatty acid metabolism by increasing lipid uptake, promoting cellular membrane fluidity and mobility [26,27]. The changes might promote ferroptosis susceptibility in cancer cells [28]. Our results indicate that PCBP1 negatively regulates ferroptosis via repressing lipid synthesis and peroxidation.
The present study has shown that the knockdown of PCBP1 is a promising approach to promoting ferroptosis in cancer cells. However, this study has several limitations. First, PCBP1 plays diverse roles in cell differentiation, iron delivery, and transcription. Knockdown of PCBP1 may affect the functions of normal cells and cancer cells. Second, we did not use any specific inhibitors of PCBP1 because these have not been developed up to now. The absence of pharmacological PCBP1 inhibition may make this method hardly used in the clinical fields of cancer patient treatment. This may call the urgent development of PCBP1-suppressing agents. Third, this study could not in detail explain that in vivo tumor growth was enhanced in both the PCBP1-suppressed group (HN12 shPCBP1) and the PCBP1 overexpressing group (HN2 PCBP1 plasmid) compared to the vector controls. This may be because PCBP1 plays a role in accelerating tumor growth [29] as well as a role of a tumor suppressor [30], which needs more research. Nevertheless, changes in the amount of intracellular iron and lipid composition according to the regulation of PCBP1 expression affected the tumor-suppressive effect of sulfasalazine. Therefore, PCBP1 depletion provides novel mechanisms of ferritinophagy induction and lipid peroxidation. Our findings may provide aid to the advanced understanding of ferroptosis mechanisms.
In conclusion, the present study suggests that the dual functions of PCBP1 inhibiting BECN1 and ALOX15 mRNAs contribute to attenuating cancer susceptibility to ferroptosis. The tumor expression level of PCBP1 might be a predictor of post-treatment outcomes and response to cancer treatment using ferroptosis inducers. The role of PCBP1 as a negative regulator of autophagy through its direct binding to the 3′-untranslated region of BECN1 is also spotlighted in regulating ferroptosis in cancer cells. Suppression of PCBP1 might be a potential strategy to kill iron-avid recalcitrant cancer cells via promoting their ferroptosis susceptibility.
Author contributions
J.L. and J.-L.R. conceived and designed the experiments. J.L., J.H·Y., and J.-L.R. performed the experiments. J.L. and J.-L.R. analyzed the data. J.L. and J.H.Y. contributed reagents/materials/analysis tools; J.L. and J.-L.R. wrote the draft and checked and revised it. All authors approved to submit this version to this publication.
Declaration of competing interest
The authors have declared no competing interests.
Acknowledgments
This study was supported by the National Research Foundation of Korea (NRF) grant, funded by the Ministry of Science and ICT (MSIT), The Government of Korea (No. 2019R1A2C2002259).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102276.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Gao M., Monian P., Pan Q., Zhang W., Xiang J., Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hou W., Xie Y., Song X., Sun X., Lotze M.T., Zeh H.J., 3rd, Kang R., Tang D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Latunde-Dada G.O. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017;1861:1893–1900. doi: 10.1016/j.bbagen.2017.05.019. [DOI] [PubMed] [Google Scholar]
- 4.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., Morrison B., 3rd, Stockwell B.R. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J., Fulda S., Gascon S., Hatzios S.K., Kagan V.E., Noel K., Jiang X., Linkermann A., Murphy M.E., Overholtzer M., Oyagi A., Pagnussat G.C., Park J., Ran Q., Rosenfeld C.S., Salnikow K., Tang D., Torti F.M., Torti S.V., Toyokuni S., Woerpel K.A., Zhang D.D. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng H., Stockwell B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16 doi: 10.1371/journal.pbio.2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angeli J.P.F., Shah R., Pratt D.A., Conrad M. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol. Sci. 2017;38:489–498. doi: 10.1016/j.tips.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Shi H., Bencze K.Z., Stemmler T.L., Philpott C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science. 2008;320:1207–1210. doi: 10.1126/science.1157643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Philpott C.C., Jadhav S. The ins and outs of iron: escorting iron through the mammalian cytosol. Free Radic. Biol. Med. 2019;133:112–117. doi: 10.1016/j.freeradbiomed.2018.10.411. [DOI] [PubMed] [Google Scholar]
- 10.Patel S.J., Protchenko O., Shakoury-Elizeh M., Baratz E., Jadhav S., Philpott C.C. Vol. 118. Proceedings of the National Academy of Sciences of the United States of America; 2021. (The Iron Chaperone and Nucleic Acid-Binding Activities of poly(rC)-Binding Protein 1 Are Separable and Independently Essential). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryu M.S., Zhang D., Protchenko O., Shakoury-Elizeh M., Philpott C.C. PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J. Clin. Invest. 2017;127:1786–1797. doi: 10.1172/JCI90519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel S.J., Frey A.G., Palenchar D.J., Achar S., Bullough K.Z., Vashisht A., Wohlschlegel J.A., Philpott C.C. A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat. Chem. Biol. 2019;15:872–881. doi: 10.1038/s41589-019-0330-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostareck D.H., Ostareck-Lederer A., Shatsky I.N., Hentze M.W. Lipoxygenase mRNA silencing in erythroid differentiation: the 3'UTR regulatory complex controls 60S ribosomal subunit joining. Cell. 2001;104:281–290. doi: 10.1016/s0092-8674(01)00212-4. [DOI] [PubMed] [Google Scholar]
- 14.Ostareck D.H., Ostareck-Lederer A., Wilm M., Thiele B.J., Mann M., Hentze M.W. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3' end. Cell. 1997;89:597–606. doi: 10.1016/s0092-8674(00)80241-x. [DOI] [PubMed] [Google Scholar]
- 15.Hassannia B., Vandenabeele P., Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35:830–849. doi: 10.1016/j.ccell.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X., Di C., Chen Y., Wang J., Su R., Huang G., Xu C., Chen X., Long F., Yang H., Zhang H. Multilevel regulation and molecular mechanism of poly (rC)-binding protein 1 in cancer. Faseb. J.: Off. Public. Federation Am. Soc.Exper. Biol. 2020;34:15647–15658. doi: 10.1096/fj.202000911R. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z., Yao Z., Wang L., Ding H., Shao J., Chen A., Zhang F., Zheng S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14:2083–2103. doi: 10.1080/15548627.2018.1503146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sumneang N., Siri-Angkul N., Kumfu S., Chattipakorn S.C., Chattipakorn N. The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch. Biochem. Biophys. 2020;680:108241. doi: 10.1016/j.abb.2019.108241. [DOI] [PubMed] [Google Scholar]
- 19.Zhang W., Zhang S., Guan W., Huang Z., Kong J., Huang C., Wang H., Yang S. Poly C binding protein 1 regulates p62/SQSTM1 mRNA stability and autophagic degradation to repress tumor progression. Front. Genet. 2020;11:930. doi: 10.3389/fgene.2020.00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torti S.V., Torti F.M. Iron: the cancer connection. Mol. Aspect. Med. 2020;75:100860. doi: 10.1016/j.mam.2020.100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alvarez S.W., Sviderskiy V.O., Terzi E.M., Papagiannakopoulos T., Moreira A.L., Adams S., Sabatini D.M., Birsoy K., Possemato R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639–643. doi: 10.1038/nature24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee J., You J.H., Shin D., Roh J.L. Inhibition of glutaredoxin 5 predisposes Cisplatin-resistant head and neck cancer cells to ferroptosis. Theranostics. 2020;10:7775–7786. doi: 10.7150/thno.46903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang W., Shi H., Zhang M., Liu B., Mao S., Li L., Tong F., Liu G., Yang S., Wang H. Poly C binding protein 1 represses autophagy through downregulation of LC3B to promote tumor cell apoptosis in starvation. Int. J. Biochem. Cell Biol. 2016;73:127–136. doi: 10.1016/j.biocel.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Protchenko O., Baratz E., Jadhav S., Li F., Shakoury-Elizeh M., Gavrilova O., Ghosh M.C., Cox J.E., Maschek J.A., Tyurin V.A., Tyurina Y.Y., Bayir H., Aron A.T., Chang C.J., Kagan V.E., Philpott C.C. Iron chaperone poly rC binding protein 1 protects mouse liver from lipid peroxidation and steatosis. Hepatology. 2020 doi: 10.1002/hep.31328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y., Gai L., Liu J., Cui Y., Zhang Y., Feng J. Expression of poly(C)-binding protein 1 (PCBP1) in NSCLC as a negative regulator of EMT and its clinical value. Int. J. Clin. Exp. Pathol. 2015;8:7165–7172. [PMC free article] [PubMed] [Google Scholar]
- 26.Morandi A., Taddei M.L., Chiarugi P., Giannoni E. Targeting the metabolic reprogramming that controls epithelial-to-mesenchymal transition in aggressive tumors. Front. Oncol. 2017;7:40. doi: 10.3389/fonc.2017.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corbet C., Bastien E., Santiago de Jesus J.P., Dierge E., Martherus R., Vander Linden C., Doix B., Degavre C., Guilbaud C., Petit L., Michiels C., Dessy C., Larondelle Y., Feron O. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat. Commun. 2020;11:454. doi: 10.1038/s41467-019-14262-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu J., Minikes A.M., Gao M., Bian H., Li Y., Stockwell B.R., Chen Z.N., Jiang X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572:402–406. doi: 10.1038/s41586-019-1426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tripathi V., Sixt K.M., Gao S., Xu X., Huang J., Weigert R., Zhou M., Zhang Y.E. Direct regulation of alternative splicing by SMAD3 through PCBP1 is essential to the tumor-promoting role of TGF-β. Mol. Cell. 2016;64:549–564. doi: 10.1016/j.molcel.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y., Meng L., Xiao L., Liu R., Li Z., Wang Y.L. The RNA-binding protein PCBP1 functions as a tumor suppressor in prostate cancer by inhibiting mitogen activated protein kinase 1, Cellular physiology and biochemistry. Int. J Experiment. Cellular Physiol. Biochem. Pharmacol. 2018;48:1747–1754. doi: 10.1159/000492315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.