

Introduction
Amyotrophic lateral sclerosis (ALS) is a relentlessly progressive degenerative disease of upper and lower motor neurons. Approximately 15% of patients display clinical features consistent with frontotemporal dementia (FTD) and 35% display milder degrees of cognitive and behavioural impairment at some stage during their illness.1 Several genes have been reported to cause both ALS and FTD. Nevertheless, it remains unclear why some patients with ALS develop cognitive impairment, while other cases, often within the same family, remain unaffected.
The GBA gene (OMIM *606463) encodes glucocerebrosidase (GCase), a lysosomal enzyme that converts glucocerebroside into glucose and ceramide. Heterozygous GBA mutations increase the risk of Parkinson’s disease (PD) and the risk of cognitive impairment in patients with PD.2
It is increasingly recognised that variants in genes causing Mendelian neurodegenerative diseases may exhibit pleiotropic effects and impact the phenotypic heterogeneity of those disorders. Moreover, lysosomal dysfunction has recently been associated with both Dementia with Lewy Bodies and FTD spectrum (online supplemental table 1). Based on this, we postulated that GBA variants may influence the cognitive status of patients with ALS.
jnnp-2021-327426supp001.pdf (162.5KB, pdf)
Materials and methods
We examined the GBA variants’ association with the risk of cognitive impairment in 751 patients with ALS from the population-based Piemonte and Valle d’Aosta Register for ALS that had undergone both a detailed neuropsychological evaluation and a whole-genome sequencing screening.3 Patients were classified as ALS with normal cognitive function (ALS-CN), ALS-FTD and ALS with intermediate cognitive deficits. The characteristics of the study population and a detailed description of neuropsychological testing and genetic screening are reported in online supplementarl materials in the Methods section.
A mutational screening of GBA exonic variants was performed and their frequencies were compared with an internal control cohort (see online supplemental methods, Subjects). To assess whether pathogenic rare variants (minor allele frequency<1%) in GBA contribute to cognitive decline risk in ALS, a gene-based rare variants association test was performed as previously described.3 In the following step of the analysis, only variants known to be a risk factor for cognitive decline in PD were considered. First, a binomial test was used to assess the prevalence of GBA mutations across cognitive groups. Then, a linear mixed-effects model was used to test for associations between GBA genotype and cognitive functioning while including the following covariates: sex, age, site of disease onset, bulbar signs at diagnosis, rate of ALS Functional Rating Scale-Revised (ALS-FRS-R) decline and C9orf72 status. Further details on the statistical analysis are reported in online supplementary materials. All statistical analyses were performed in R V.3.6.0 (https://www.r-project.org/).
Results
The gene-based rare variant association test identified an enrichment of rare GBA variants in patients with ALS with intermediate cognitive dysfunction (p SKAT-O=0.000005), but not in ALS-FTD cases (p SKAT-O=0.184) (online supplemental table 2).
We identified one GBA mutation (p.N409S), known to cause Gaucher Disease in homozygous carriers, one likely pathogenic variant (p.R209H) and two GBA polymorphisms that are known to increase the risk of dementia in patients with PD (p.E365K and p.T408M). The remaining identified coding GBA variants are reported in online supplemental table 3. The frequency of GBA variants was not increased in our cohort as compared with healthy controls (online supplemental table 4). Thirteen out of 18 (72.2%) of patients with ALS carrying GBA variants displayed cognitive impairment in the form of FTD or intermediate cognitive phenotypes. In contrast, cognitive impairment was observed among 47.1% (298 out of 733) of patients with ALS not carrying GBA variants (binomial test p value=0.0357). We repeated the analysis excluding C9orf72 expansion carriers and the difference remained significant (p=0.0486). To confirm the effect of GBA variants on cognitive phenotype, we modelled the association between the GBA variants and cognitive impairment using a linear mixed-effects model that controlled for relevant covariates (figure 1). In the mixed-effects model, GBA mutation status was associated with the clinical diagnosis of cognitive impairment (OR=3.74, 95% CI 1.25 to 12.72, p=0.023). This effect was not seen when considering either only the ALS-FTD phenotype or the intermediate phenotype.
Figure 1.
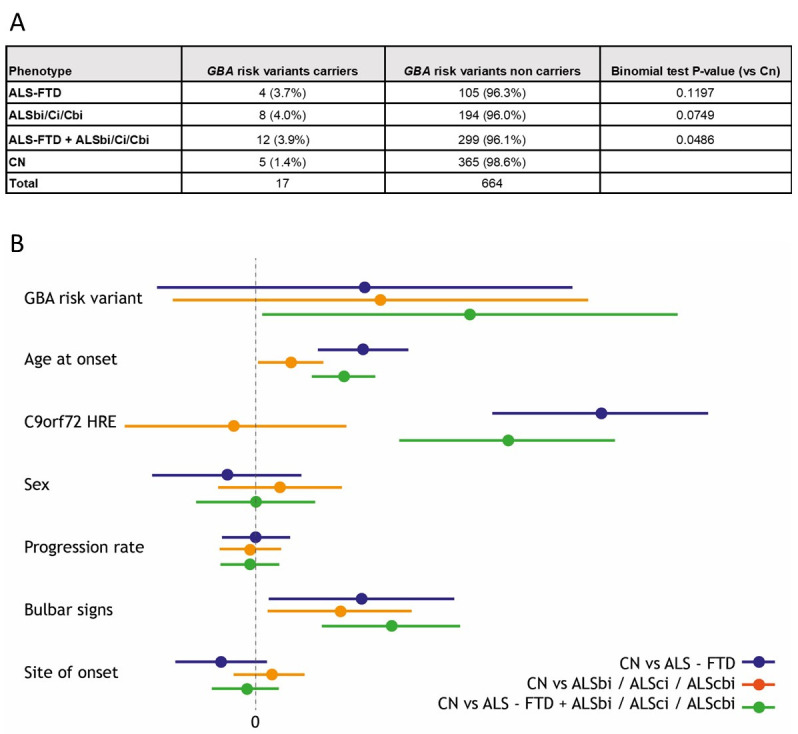
(A) Cognitive phenotype frequencies in GBA risk variant carriers. Given the strong influence of C9orf72 on cognitive status, here we performed the binomial test without C9orf72 expansion carriers to rule out its impact on the results. (B) Results of the linear mixed-effects model, coefficient and 95% CI. Model estimates are expressed as eOR. Colours correspond to the results of the three different models (1) ALS-CN versus ALS-FTD (blue); (2) ALS-CN versus ALSbi+ALSci+ALScbi (red); (3) ALS-CN versus ALS-FTD+ALSbi+ALSci+ALScbi (green). See online supplemental file 1 for further information about cognitive classification. ALS, amyotrophic lateral sclerosis; FTD, frontotemporal dementia.
Discussion
We observed that the burden of rare variants in the GBA gene was associated with cognitive impairment in patients with ALS. Patients carrying known pathogenic GBA variants (p.E365K, p.T408M, p.N409S) were three times more likely to develop cognitive impairment compared with non-carriers, independently of age, sex, site of onset, bulbar involvement, rate of ALS-FRS-R decline and C9orf72 status.
In multivariate analysis, we identified an effect of GBA variants only when ALS-CN cases were compared with patients with FTD and intermediate deficits combined. However, the results of the burden test suggest that this finding is primarily driven by patients with intermediate deficits. The course of cognitive deterioration among patients with ALS may partially explain our findings: recent studies have shown that cognitive impairment may worsen over time and that it is correlated to more severe motor deficits.4 The neuropsychological assessment at diagnosis might have captured an early phase of the trajectory of cognitive deterioration over time. Nonetheless, also the small number of GBA variant carriers may have conditioned such findings.
A possible role of GBA in the neurodegenerative process underlying ALS is suggested by increasing evidence of the involvement of endolysosomal dysfunction in ALS pathogenesis. Several genes causing ALS and FTD, including C9orf72, TBK1, OPTN, SQSTM1 and VCP, are related to lysosomal function and protein degradation. This research field deserves further attention as several therapeutic agents targeting lysosomal pathways have been proposed for neurological diseases.5 Our results expand the spectrum of neurodegenerative diseases for which heterozygous GBA variants represent a detrimental factor. It is possible that such variants are kept in populations because they provide some biological advantage.
As a limitation of our study, we acknowledge that we could not evaluate whether different variants had a variable impact on the risk of cognitive impairment, on the pattern of cognitive deficits and on other clinical characteristics, mostly due to the relatively small number of GBA variant carriers (online supplemental tables 5 and 6).
In conclusion, we found that variants of the GBA gene are associated with an increased risk of cognitive impairment in patients with ALS. Our results broaden the spectrum of genetic factors that modulate the vulnerability of patients with ALS to cognitive dysfunction and strengthen the role of lysosomal impairment in the neurodegenerative process underlying ALS, highlighting that genes can modify not only the risk of ALS but also modulate different aspects of its phenotype. Addressing the gap in our understanding of the role that genetic modifiers play in ALS is essential for diagnosis, prognosis, and therapy development.
Acknowledgments
The Authors thank the Laboratory of Neurogenetics (NIH) staff for their collegial support and technical assistance.
Footnotes
AC and MG contributed equally.
AC and AC contributed equally.
Contributors: AC: study concept and design; data collection; data analysis; drafting of the manuscript; study supervision.MG: study concept and design; data collection; data analysis; drafting of the manuscript; study supervision.CM: data collection; critical revision of the manuscript for important intellectual content; administrative, technical, and material support.BI: data collection; critical revision of the manuscript for important intellectual content; administrative, technical, and material support.LP: data collection; critical revision of the manuscript for important intellectual content; administrative, technical, and material support.SG: critical revision of the manuscript for important intellectual content; administrative, technical, and material support.MB: data collection; critical revision of the manuscript for important intellectual content; administrative, technical, and material support.MB: data collection; critical revision of the manuscript for important intellectual content; administrative, technical, and material support.LS: data collection; critical revision of the manuscript for important intellectual content; administrative, technical, and material support.FP: data collection; critical revision of the manuscript for important intellectual content; administrative, technical, and material support.SC: data collection; critical revision of the manuscript for important intellectual content; administrative, technical, and material support.UM: data collection; critical revision of the manuscript for important intellectual content; administrative, technical, and material support.RV: data collection; critical revision of the manuscript for important intellectual content; administrative, technical, and material support.BJT: study concept and design; drafting of the manuscript; critical revision of the manuscript for important intellectual content; study supervision.LC: critical revision of the manuscript for important intellectual content; administrative, technical, and material support.SD’A: critical revision of the manuscript for important intellectual content; administrative, technical, and material support.LM: critical revision of the manuscript for important intellectual content; administrative, technical, and material support.AC: data collection; drafting of the manuscript; critical revision of the manuscript for important intellectual content; study supervision.AC: study concept and design; drafting of the manuscript; critical revision of the manuscript for important intellectual content; obtained funding; study supervision.
Funding: This work was supported by the Italian Ministry of Health (Ministero della Salute, Ricerca Sanitaria Finalizzata, grant RF-2016–02362405); the Progetti di Rilevante Interesse Nazionale programprogramme of the Ministry of Education, University and Research (grant 2017SNW5MB); the European Commission’s Health Seventh Framework Programme (FP7/2007–2013 under grant agreement 259867); and the Joint Programme–Neurodegenerative Disease Research (Strength, ALS-Care and Brain-Mend projects), granted by Italian Ministry of Education, University and Research. This study was performed under the Department of Excellence grant of the Italian Ministry of Education, University and Research to the “Rita Levi Montalcini” Department of Neuroscience, University of Torino, Italy, and to the Department of Health Sciences, University of Eastern Piedmont, Novara, Italy. This work was also supported by the Intramural Research Programme of the NIH, National Institute on Ageing (Z01-AG000949-02), and by the National Institute of Neurologic Disorders and Stroke (1ZIANS003154).
Disclaimer: The funders had no role in data collection or analysis and did not participate in writing or approving the manuscript.
Competing interests: BJT holds European, Canadian, and American patents on the clinical testing and therapeutic intervention for the hexanucleotide repeat expansion of C9orf72. AC has received a research grant from Cytokinetics.AC serves on scientific advisory boards for Mitsubishi Tanabe, Roche, Denali Pharma, Cytokinetics, and Amylyx.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Ethics statements
Patient consent for publication
Consent obtained directly from patient(s).
Ethics approval
The study was approved by the Ethical Committees of the two institutions involved in the study. Written informed consent was obtained at enrollment. The databases were anonymised according to Italian law for the protection of privacy.
References
- 1. Phukan J, Elamin M, Bede P, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry 2012;83:102–8. 10.1136/jnnp-2011-300188 [DOI] [PubMed] [Google Scholar]
- 2. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361:1651–61. 10.1056/NEJMoa0901281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grassano M, Calvo A, Moglia C. Mutational analysis of known ALS genes in an Italian population-based cohort. Neurology 2020;96. 10.1212/WNL.0000000000011209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beeldman E, Govaarts R, de Visser M, et al. Progression of cognitive and behavioural impairment in early amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2020;91:779–80. 10.1136/jnnp-2020-322992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peng W, Minakaki G, Nguyen M, et al. Correction to: preserving lysosomal function in the aging brain: insights from neurodegeneration. Neurotherapeutics 2019;16:920–1. 10.1007/s13311-019-00756-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jnnp-2021-327426supp001.pdf (162.5KB, pdf)