

Abstract
Promoters are crucial regulatory DNA regions for gene transcriptional activation. Rapid advances in next-generation sequencing technologies have accelerated the accumulation of genome sequences, providing increased training data to inform computational approaches for both prokaryotic and eukaryotic promoter prediction. However, it remains a significant challenge to accurately identify species-specific promoter sequences using computational approaches. To advance computational support for promoter prediction, in this study, we curated 58 comprehensive, up-to-date, benchmark datasets for 7 different species (i.e. Escherichia coli, Bacillus subtilis, Homo sapiens, Mus musculus, Arabidopsis thaliana, Zea mays and Drosophila melanogaster) to assist the research community to assess the relative functionality of alternative approaches and support future research on both prokaryotic and eukaryotic promoters. We revisited 106 predictors published since 2000 for promoter identification (40 for prokaryotic promoter, 61 for eukaryotic promoter, and 5 for both). We systematically evaluated their training datasets, computational methodologies, calculated features, performance and software usability. On the basis of these benchmark datasets, we benchmarked 19 predictors with functioning webservers/local tools and assessed their prediction performance. We found that deep learning and traditional machine learning–based approaches generally outperformed scoring function–based approaches. Taken together, the curated benchmark dataset repository and the benchmarking analysis in this study serve to inform the design and implementation of computational approaches for promoter prediction and facilitate more rigorous comparison of new techniques in the future.
Keywords: machine learning, deep learning, promoter identification, performance evaluation
Introduction
Promoters are noncoding regions located close to the transcription start sites (TSSs) in the genomic DNA (i.e. gDNA) sequences, serving as a ‘switch’ to facilitate the transcription and to determine the activities of particular genes [1, 2]. Promoter regions contain short conserved DNA sequences, namely core promoter elements, as binding sites for RNA polymerase and transcription factors [3, 4]. It is well known that the promoter structure and binding complexity of regulating gene expression vary between prokaryotic and eukaryotic genomes. In prokaryotic cells, a collection of different 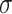 subunit factors of RNA holoenzyme are responsible for binding to the specific promoter regions during gene transcription [5]. Accordingly, the types of prokaryotic promoters are determined by a variety of
subunit factors of RNA holoenzyme are responsible for binding to the specific promoter regions during gene transcription [5]. Accordingly, the types of prokaryotic promoters are determined by a variety of 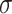 factor types labeled based on their molecular weights, for example
factor types labeled based on their molecular weights, for example 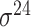 ,
, 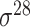 ,
, 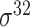 ,
, 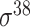 ,
, 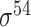 and
and 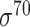 -promoter in E. coli and
-promoter in E. coli and 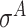 (equivalently,
(equivalently, 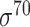 -promoter in E. coli),
-promoter in E. coli), 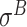 ,
, 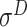 ,
, 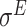 ,
, 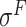 ,
, 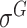 ,
, 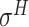 ,
, 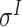 ,
, 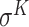 ,
, 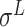 ,
, 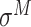 ,
, 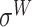 ,
, 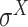 and
and 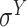 -promoter in B. subtilis [6]. Promoters in bacteria commonly contain two short DNA sequences: Pribnow box (i.e. −10 box) with consensus nucleic trait ‘TATAAT’ around −10 bp upstream of TSS, and −35 box with consensus ‘TTGACA’ around −35 bp upstream of TSS [7].
-promoter in B. subtilis [6]. Promoters in bacteria commonly contain two short DNA sequences: Pribnow box (i.e. −10 box) with consensus nucleic trait ‘TATAAT’ around −10 bp upstream of TSS, and −35 box with consensus ‘TTGACA’ around −35 bp upstream of TSS [7].
Transcription in eukaryotic cells, on the other hand, commences only when the competent pre-initiation complex (PIC) composed of RNA polymerase (RNA pol) and several general transcription factors are recruited to the promoters [8, 9]. Three types of RNA polymerase promoters have been reported responsible for transcribing different subsets of genes due to different locations of RNA pol in the nucleus, including (i) RNA pol I promoters that transcribe genes encoding rRNAs; (ii) RNA pol II promoters that transcribe genes encoding mRNAs, long noncoding RNAs (lncRNAs) and small nuclear RNAs (snRNAs); and (iii) RNA pol III promoters that transcribe genes encoding tRNAs and other small RNAs [10]. As only RNA pol II can transcribe mRNA precursors and translate them into proteins after processing and maturation, the RNA pol II promoter is our main focus in this study. Eukaryotic cells require a minimum of seven transcription factors (i.e. TATA box-binding protein (TBP), pol II-associated TF (TF II D, TF II A, TF II B, TF II F, TF II E and TF II H)) to be involved in transcription initiation [11]. Eukaryotic promoters are significantly more complex and diverse than prokaryotic promoters, spanning a wide range of DNA sequences, including the TATA box located about 25–35 base pairs upstream of the TSS with consensus sequence of ‘TATAAA’, and the upstream activating sequence located at −40 to −110 bp containing the CAAT box and GC box, which control the rate of transcription initiation [12, 13].
A variety of sequencing techniques have been developed for the identification and characterization of promoter sequences [14], including genomic sequencing coupled with full-length cDNA capture and ascertainments [15, 16], such as CAGE [17], 3PEAT [18] and RAMPAGE [19]. Such advanced techniques have led to rapid proliferation of experimental evidence of both prokaryotic and eukaryotic promoter sequences in the post-genomic era, which underpins the design and implementation of computational approaches for promoter prediction. During the past two decades, a number of computational approaches have been developed for predicting prokaryotic and eukaryotic promoters and can be generally categorized into three groups according to their computational methodologies (Figure 1A) including (i) sequence-scoring function–based methods; (ii) traditional machine learning–based methods, and (iii) deep learning–based methods. Several surveys of promoter prediction have been published [2, 13, 20–28] (Figure 1B); however, all focus on tools published prior to 2010 (excluding more recent tools such as TSSPlant [29], CNNProm [30], iProEP [31] and DeePromoter [32]) and moreover do not systematically cover prokaryotic promoter identification tools (40 such approaches). More importantly, most published surveys did not systematically assess the prediction performance of compared approaches by conducting extensive and independent benchmarking tests.
Figure 1.

A timeline of (A) the details of computational approaches for predicting prokaryotic and eukaryotic promoters. Font colors are used to distinguish algorithm types and stick colors indicate the prediction targets; and (B) historical reviews and assessments of these methods.
With the goal to address the above issues, in this study, we provide a systematic and comparative analysis by surveying the most up-to-date research progress for predicting both eukaryotic and prokaryotic promoters. To this end, we have manually curated 58 large-scale, reliable, and up-to-date benchmark datasets accompanying this comprehensive survey analysis, which are created to assist the community to assess the relative functionality of alternative approaches and support future research on promoters in both prokaryotes and eukaryotes. A total number of 106 computational approaches based on our literature mining using the papers collected from PubMed and Web of Science with the key words ‘promoter identification’ or ‘promoter prediction’, including 61 for eukaryotic promoter, 40 for prokaryotic promoter and 5 for both, are carefully assessed, benchmarked and extensively discussed in terms of the model construction, performance evaluation strategy, webserver and software usability. More importantly, using the 58 independent test datasets that cover diverse species from a variety of databases, we have systemically assessed the prediction performance of investigated approaches with available webservers or locally executable tools. We anticipate that the comparative analysis in this study serves as a critical analysis of state-of-the-art approaches and represents a stepping-stone toward future development for accurate identification of both prokaryotic and eukaryotic promoters.
Systematic comparison of computational approaches for prokaryotic and eukaryotic promoter prediction
Existing approaches for prokaryotic and eukaryotic promoter prediction
Among the 106 computational approaches for promoter prediction analyzed in this study, 40 were designed and implemented for predicting prokaryotic promoters only, including E. coli, B. subtilis, Cyanobacteria and Chlamydia [33–72]. These methods are systematically described and summarized in Table 1, in terms of algorithm, selected features, evaluation strategy, webserver availability and targeted species. The majority of predictors for prokaryotic promoters can be categorized into three types, including (i) specifically for E. coli 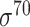 -promoter; (ii) generally for E. coli promoters (such as
-promoter; (ii) generally for E. coli promoters (such as 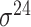 ,
, 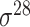 ,
, 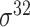 ,
, 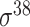 ,
, 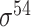 and
and 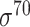 ) and (iii) generally for B. subtilis promoters. For eukaryotic promoter prediction, we collected 61 computational prediction tools for H. sapiens, M. musculus, A. thaliana and D. melanogaster [29, 32, 73–131] etc., and further summarized these methods in Table 2. Another five frameworks, including Rani et al.-I [132], Rani et al.-II [133], IPMD [134], CNNProm [30] and iProEP [31], are capable of accurately identifying both prokaryotic and eukaryotic promoters and have been summarized in Table 3. Figure 2 illustrates a flowchart describing four generic steps used by these computational approaches for identifying prokaryotic and eukaryotic promoters, which are discussed in detail in the following sections. An annual breakdown (from 2000 to 2020) of the numbers of publications of predicting prokaryotic and eukaryotic promoters have been provided in Figure 3A.
) and (iii) generally for B. subtilis promoters. For eukaryotic promoter prediction, we collected 61 computational prediction tools for H. sapiens, M. musculus, A. thaliana and D. melanogaster [29, 32, 73–131] etc., and further summarized these methods in Table 2. Another five frameworks, including Rani et al.-I [132], Rani et al.-II [133], IPMD [134], CNNProm [30] and iProEP [31], are capable of accurately identifying both prokaryotic and eukaryotic promoters and have been summarized in Table 3. Figure 2 illustrates a flowchart describing four generic steps used by these computational approaches for identifying prokaryotic and eukaryotic promoters, which are discussed in detail in the following sections. An annual breakdown (from 2000 to 2020) of the numbers of publications of predicting prokaryotic and eukaryotic promoters have been provided in Figure 3A.
Table 1.
A comprehensive list of the reviewed methods/tools for the prediction of prokaryotic promotersa
| Framework | Toolb | Year | Webserver/toolc | Features/Motifs | Scoring function /Algorithm | Evaluation strategy | Promoter typed | Speciese | Sequence length (bp)f |
|---|---|---|---|---|---|---|---|---|---|
| Deep learning–based | Le et al. [67] | 2019 | Yes* | FastText n-grams | CNN | 5-fold CV | Strong and weak;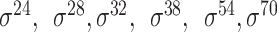 and unknow and unknow |
E. coli | 81 |
| iPromoter-BnCNN [70] | 2020 | Decommissioned | Monomer, trimer and DSP | CNN | 5-fold CV and independent test |
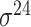 ,
, 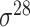 , , 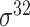 , , 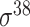 , , 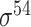 and and 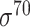
|
E. coli | 81 | |
| Traditional machine learning–based | Leo Gordon et al. [33] | 2003 | Decommissioned | SAK | SVM | 50% train, 50% test | σ 70 | E. coli | 80 |
| Monteiro et al. [36] | 2005 | No | – | Comparative study of NBC, DT, SVM and ANN | Leave-one-out | – | B. subtilis and E. coli | 117, 57 | |
| da Silva et al. [38] | 2006 | No | – | Comparative study of KNN, NBC, DT, SVM and ANN | 10-fold CV | – | B. subtilis, B. licheniformis, B. cereus, B. megaterium, B. thuringiensis, and B. firmus | 111 | |
| Wang et al. [40] | 2006 | No | DSP1, −10 motif scores | Fisher LDA | Independent test | – | E. coli and B. subtilis | 100 | |
| J. J. Gordon et al. [41] | 2006 | Decommissioned | 5-mer tagged with its location, −10 and −35 hexamers | committee-SVM | 10-fold CV | σ 70 | E. coli | 200 | |
| Towsey et al.-I [42] | 2006 | No | 5-mer tagged with its location, −10 and −35 hexamers | SVM | 10-fold CV | σ 70 | E. coli | 200 | |
| pHMM-ANN [39] | 2007 | No | UP element, −10, −35 elements pHMMs | ANN | Independent test | – | E. coli | – | |
| Towsey et al.-II [44] | 2007 | No | Similarity score of candidate TSS, −10, −35 scores, TSS-GSS distance, DSP2 | C4.5 | 10-fold CV | σ 70 | E. coli | 250 | |
| TSS-PREDICT [47] | 2008 | No | −10 and −35 hexamers, 5-mer tagged with its location, TSS- GSS distribution |
Ensemble-SVM | Independent test | σ 70; σ43; σ66 | E. coli, B. subtilis and C. trachomatis | 200 | |
| N4 [48] | 2009 | No | DDS | ANN | Leave-one-out | – | E. coli | 414 | |
| Polat et al. [49] | 2009 | No | 57 sequential DNA nucleotide attributes | Fuzzy-AIRS | 10-fold CV | – | E. coli | 57 | |
| Song et al. [53] | 2012 | Yes* | vw Z-curve | PLS | 10-fold CV |
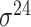 ,
, 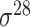 , , 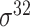 , , 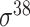 , , 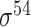 and and 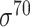 ; ; 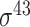 , , 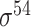 , , 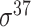 etc. etc. |
E. coli and B. subtilis | 80 | |
| iPro54-PseKNC [55] | 2014 | Yes* | PseKNC | SVM | 10-fold CV and leave-one-out | σ 54 | E. coli | 81 | |
| de Avila e Silva et al. [56] | 2014 | No | DDS | ANN | 2,3,10-fold CV |
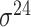 ,
, 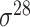 , , 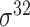 , , 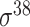 , , 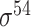 and and 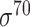
|
E. coli | 80 | |
| bTSSfinder [57] | 2017 | Decommissioned | PE, DPE, k-mer, TFBSD, PCP | ANN | Independent test |
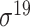 ,
, 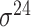 , , 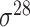 , , 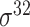 , , 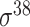 , , 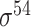 and and 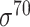
|
E. coli, S. elongatus, Nostoc, and Synechocystis | 251, 1101 | |
| iPromoter-2L [58] | 2018 | Yes | PseKNC | RF | 5-fold CV |
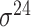 ,
, 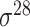 , , 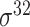 , , 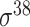 , , 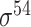 and and 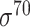
|
E. coli | 81 | |
| 70ProPred [59] | 2018 | Yes | PSTNPSS/PSTNPDS, PseEIIP | SVM | 5-fold CV and leave-one-out | σ 70 | E. coli | 81 | |
| IBPP-SVM [60] | 2018 | Yes* | ‘image’ | SVM | Independent test | σ 70 | E. coli | 81 | |
| BacSVM+ [61] | 2018 | Decommissioned | – | SVM | – |
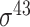 ,
, 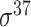 , , 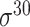 , , 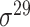 , , 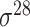 and and 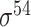
|
B. subtilis | 80 | |
| iPro70-PseZNC [62] | 2019 | Yes | PseZNC | SVM | 5-fold CV | σ 70 | E. coli | 81 | |
| iPromoter-FSEn [63] | 2019 | Yes | k-mer, g-gapped k-mer, NSM, ASPC, PSO, DN | SVM, LDA, LR | 10-fold CV and leave-one-out | σ 70 | E. coli | 81 | |
| iPro70-FMWin [64] | 2019 | Yes | k-mer, g-gapped k-mer, NSM, ASPC, PSO | LR | 10-fold CV | σ 70 | E. coli | 81 | |
| iPSW(2L)-PseKNC [65] | 2019 | Yes | General PseKNC | SVM | 5-fold CV | Strong and weak;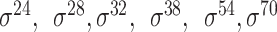 and unknow and unknow |
E. coli | 81 | |
| MULTiPly [66] | 2019 | Yes | BPB, KNN, KNC, DAC | SVM | 5-fold CV, leave-one-out and independent test |
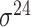 ,
, 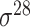 , , 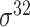 , , 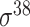 , , 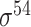 and and 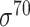
|
E. coli | 81 | |
| iPromoter-2L2.0 [68] | 2019 | Yes | k-mer, PseKNC | SVM, EL | 5-fold CV |
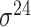 ,
, 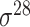 , , 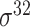 , , 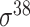 , , 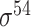 and and 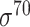
|
E. coli | 81 | |
| SELECTOR [69] | 2020 | Yes | CKSNAP, PCPseDNC, PSTNPss and DNA strand | RF, AdaBoost, GBDT, LightGBM, XGBoost | 5-fold CV and independent test |
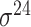 ,
, 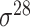 , , 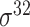 , , 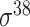 , , 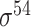 and and 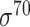
|
E. coli | 81 | |
| Scoring function–based | Huerta et al. [34] | 2003 | No | −10 and −35 box, spacer between −10 and −35 box | PWM | Independent test | σ 70 | E. coli | 250 |
| TLS-NNPP [35] | 2005 | No | TSS-TLS distance, the results from NNPP2.2 | Probability 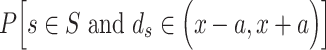
|
Independent test | – | E. coli | 500 | |
| Kanhere et al. [37] | 2005 | No | DDS | DE | Independent test | – | E. coli, B. subtilis and C. glutamicum | 1000 | |
| Li et al. [43] | 2006 | No | Hexamer sequence conservation | PCSF | 10-fold CV | σ 70 | E. coli | 81 | |
| Beagle [72] | 2006 | Decommissioned | UP element, −10, −35 and extended −10 elements, and TSS-GSS gap | PWM | 10-fold CV | σ 70 | E. coli and B. subtilis | 250 | |
| Footy [45] | 2007 | Decommissioned | −10 and −35 hexamers | PWM | Independent test | σ 66 | C. trachomatis, C. pneumoniae, C. caviae and C. muridarum | – | |
| Rangannan et al. [46] | 2007 | No | DDS | DE | Independent test | – | E. coli and B. subtilis | 101, 1001 | |
| PromPredict [50] | 2009 | Yes* | DDS | DE | Independent test | – | E. coli, B. subtilis and M. tuberculosis | 1001 | |
| PromPredict [51] | 2010 | Yes* | DDS, GC content | DE | Independent test | – | 913 bacteria in PromBase [183] | 1001 | |
| BacPP [52] | 2011 | Yes | Rules extracted from neural networks | Weighting promoter prototypes | 2, 3, 10-fold CV |
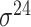 ,
, 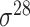 , , 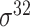 , , 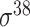 , , 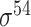 and and 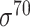
|
E. coli | 80 | |
| Todt et al. [54] | 2012 | No | −10, −35 and extended −10 elements | PWM | Independent test | σ 70 | L. plantarum | 100 | |
| G4PromFinder [71] | 2018 | Yes* | AT-rich element and G-quadruplex motif | – | Independent test | – | S. coelicolor and P. aeruginosa | 251 |
aAbbreviations: CNN—convolutional neural network; CV—cross-validation; DSP—DNA structural property; SAK—sequence alignment kernel; SVM—support vector machine; TSS-TLS distance—the distance between the transcription start site (TSS) and the translation start site (TLS); TDNN—time-delay neural network; NBC—naïve Bayes classifier; DT—decision tree; ANN—artificial neural network; KNN—k-nearest neighbor; DSP1—SIDD, curvature, deformability, thermodynamic stability; SIDD—stress-induced DNA duplex destabilization; LDA—linear discriminant analysis; committee-SVM—DGS, PWM and ensemble SVM; DGS—the distribution of TSS distance to gene start; PWM—position weight matrix; pHMMs—profile hidden Markov models; DSP2—DNA curvature, SIDD, stacking energy; DDS—DNA duplex stability; Fuzzy-AIRS—Artificial Immune Recognition System with Fuzzy resource allocation mechanism; vw Z-curve—variable-window Z-curve; PLS—partial least squares; PseKNC—pseudo–K-tuple nucleotide composition; PE—promoter elements including −10, −35, −15 and AT-rich UP elements, together with the new TSS motifs by the authors; DPE—distances (d) between promoter elements (contains d(−10/−35), d(−10/TSS) and d(−15/−10)); TFBSD—TFBSs density; PCP—physico-chemical properties (i.e. free energy, base stacking, entropy and melting temperature); RF—random forest; PSTNPSS/PSTNPDS—position-specific trinucleotide propensity based on single-stranded or double-stranded characteristic, PseEIIP—electron–ion interaction pseudo-potentials of trinucleotide; PseZNC—pseudo–multi-window Z-curve nucleotide composition; NSM—nucleotide statistical measure; ASPC—approximate signal pattern count; PSO—position specific occurrences; DN—distribution of nucleotides; LR—logistic regression; BPB—bi-profile Bayesian signatures; KNC—k-tuple nucleotide composition; DAC—dinucleotide-based auto-covariance; EL—ensemble learning; CKSNAP—composition of k-spaced nucleic acid pairs; PCPseDNC—parallel correlation pseudo-dinucleotide composition; GBDT—gradient boosting decision tree; DE—relative stability (the difference in free energy); PCSF—position-correlation scoring function.
bThe URL addresses for the listed tools: iPro54-PseKNC—http://lin-group.cn/server/iPro54-PseKNC; iPromoter-2L—http://bioinformatics.hitsz.edu.cn/iPromoter-2L/; 70ProPred—http://server.malab.cn/70ProPred/; iPro70-PseZNC—http://lin-group.cn/server/iPro70-PseZNC; iPro70-FMWin—http://ipro70.pythonanywhere.com/server; iPromoter-FSEn—http://ipromoterfsen.pythonanywhere.com/server; iPSW(2L)-PseKNC—http://www.jci-bioinfo.cn/iPSW(2L)-PseKNC; MULTiPly—http://flagshipnt.erc.monash.edu/MULTiPly/; iPromoter-2L2.0—http://bliulab.net/iPromoter-2L2.0/; SELECTOR—http://SELECTOR.erc.monash.edu/; PromPredict— http://nucleix.mbu.iisc.ernet.in/prompredict/prompredict.html; BacPP—http://bacpp.bioinfoucs.com/home.
cYes—The approach is accompanied with a webserver/tool and it is still working; Decommissioned—The webserver/tool is no longer available; No—The approach has no webserver or tool; Yes*—The server/tool was not involved in our performance comparison due to the unavailable pretrained model, unavailable latest test data or the unmatched sequence length.
dWe listed the detailed prokaryotic promoter types based on the description in the papers. ‘–’ demonstrates such information is not present in the paper.
eThe species information of the sequences used in corresponding studies was directly extracted from the studies. For some species, the Latin names have been provided according to the predictors; for other species, based on the information provided in the papers, we just provided the general names of the species when their Latin names are not available.
f‘–’ demonstrates that no clearly length information is provided in the paper.
Table 2.
A comprehensive list of the reviewed methods/tools for the prediction of eukaryotic promotersa.
| Method type | Toolb | Year | Webserver/toolc | Features/Motifs | Scoring function /Algorithm | Evaluation strategy | Promoter typed | Speciese | Sequence length (bp)f |
|---|---|---|---|---|---|---|---|---|---|
| Deep learning–based | Qian et al. [127] | 2018 | No | – | CNN | 10-fold CV | – | H. sapiens | 300 |
| DeePromoter [32] | 2019 | Yes | CNN, BiLSTM | 5-fold CV | TATA-containing, TATA-less |
H. sapiens and M. musculus | 300 | ||
| DeeReCT-PromID [129] | 2019 | Decommissioned | CNN | 90% train; 10% test | TATA-containing, TATA-less |
H. sapiens | 10000 | ||
| Depicter [130] | 2020 | Yes | CNN and capsule network | 5-fold CV and independent test | TATA-containing, TATA-less |
H. sapiens, M. musculus, D. melanogaster and A. thaliana | 300 | ||
| Traditional machine learning–based | CpG_promoter [74] | 2000 | Decommissioned | CpG island parameters | QDA | 70% train, 30% test | CpG-related, non-CpG-related | H. sapiens | – |
| Ohler et al.-I [75] | 2000 | Decommissioned | Upstream, TATA box and Inr/downstream regions | SSM | 5-fold CV | – |
H. sapiens and D. melanogaster |
300 | |
| McPromoter [76] | 2000 | Decommissioned | Segmental and structural profile features | ANN | Independent test | – | D. melanogaster | 300 | |
| FirstEF [77] | 2001 | Decommissioned | Hexamer and pentamer frequencies, the CpG and G+C percentage | QDA | Independent test | CpG-related, non-CpG-related | H. sapiens | 570 | |
| Hannenhalli et al. [78] | 2001 | No | 5-mer and TFBSD | LDA | Independent test | – | H. sapiens | 1200 | |
| NNPP2.2 [79] | 2001 | Yes | TATA box and Inr | TDNN | 4-fold CV and independent test | – |
H. sapiens and D. melanogaster |
– | |
| Eponine [81] | 2002 | Decommissioned | – | RVM | Independent test | – | H. sapiens and M. musculus | – | |
| CpGProD [82] | 2002 | Yes* | CpG island | GLM | – | CpG-related | H. sapiens and M. musculus | – | |
| Ohler et al.-II [83] | 2002 | No | – | ANN | 5-fold CV and independent test | – | D. melanogaster | 300 | |
| DPF [84] | 2002 | Decommissioned | PWMs of pentamers | ANN | Independent test | – | Vertebrates in EPD [184] | – | |
| DRAGON [87] | 2003 | Decommissioned | PWMs of pentamers | ANN | Independent test | – | Vertebrates in EPD [184] | 250 | |
| PromH [88] | 2003 | Decommissioned | PEs | LDA | Independent test | TATA-containing, TATA-less |
H. sapiens and M. musculus | – | |
| DragonGSF [89] | 2003 | Decommissioned | CpG island, TSS location, DPE | ANN | Independent test | – | H. sapiens | 10000 | |
| ProGA [90] | 2003 | Decommissioned | DNC | GA | 80% train; 20% test | TATA-containing, DPE-containing |
D. melanogaster | 400 | |
| Kasabov et al. [91] | 2004 | No | Similarity reflexed on the promoter vocabulary | TSVM | 3-fold CV | – | Vertebrates in EPD [184] | 250 | |
| Prometheus [93] | 2005 | No | k-mer, GC%, Lyapunov component and Tsallis entropy | SVM | 10-fold CV and independent test | – | H. sapiens | 300 | |
| TSSP-TCM [94] | 2005 | Decommissioned | Content and signal features | SVM | Independent test | TATA-containing, TATA-less |
Plant in PlantProm [185] | 351 | |
| BayesProm [95] | 2005 | Decommissioned | Oligonucleotide positional density | NBC | Independent test | – | H. sapiens | 600 | |
| PromoterExplorer [96] | 2006 | No | Local distribution of pentamers, positional CpG island features and digitized DNA sequence | AdaBoost | Independent test | – | Vertebrates in EPD [184] | 250 | |
| ARTS [99] | 2006 | Decommissioned | WDs, spectrum kernel, twisting angles and stacking energies | SVM | Independent test | – | H. sapiens | 2000 | |
| FProm [100] | 2006 | Yes | Motif density, triplets, hexaplets, position triplet matrix, CpG content, TATA box, similarity, Protein-DNA-twist, Protein-induced deformability | LDA | Independent test | TATA-containing, TATA-less |
H. sapiens | 250 | |
| Ohler et al.-III [102] | 2006 | No | TATA box, Inr, DPE, MTE, M1/6 | ANN | 5-fold CV and independent test | – | D. melanogaster | 300 | |
| CoreBoost [103] | 2007 | Decommissioned | PEs, TFBSs, flexibility scores, Markovian scores, and k-mer | LogitBoost | 5-fold CV and leave-one-out | CpG-related, non-CpG-related | H. sapiens | 300 | |
| TSS-AMOSA [104] | 2007 | No | PEs, TFBSs, flexibility scores, Markovian scores and k-mer | LDA | 5-fold CV | CpG-related, non-CpG-related | H. sapiens | 300 | |
| MetaProm [107] | 2007 | No | Predictions from PSPA, FirstEF, McPromoter, DragonGSF, DragonPF, and FProm along with their features | ANN | 10-fold CV | CpG-related, non-CpG-related | H. sapiens | – | |
| PromMachine [108] | 2008 | No | 4-mer | SVM | 7-fold CV | – | D. melanogaster, plant in PlantProm [185], H. sapiens, M. musculus and R. norvegicus | 251 | |
| IDQD [109] | 2008 | No | 4, 5, 6-mer, G+C content | IDQD | Independent test | – | H. sapiens | 2000 | |
| Yang et al. [113] | 2008 | No | Z curve analysis, CTD, EDP | LDA | 50% train, 50% test | – | H. sapiens | 600 | |
| CoreBoost_HM [115] | 2009 | Decommissioned | PEs, TFBSs, flexibility scores, Markovian scores and k-mer | LogitBoost | 10-fold CV | CpG-related, non-CpG-related | H. sapiens | 300 | |
| RBF-TSS [116] | 2009 | Yes* | k-mer | RBFNN | Independent test | – | H. sapiens | 2400 | |
| SCS [117] | 2010 | No | Signal, context and structure features | C4.5 | 3-fold CV | – | H. sapiens | 251 | |
| DDM [118] | 2010 | Decommissioned | k-mer | Multi-staged daisy-chain filtering | 4-fold CV | – | H. sapiens and M. musculus | 200, 1600 | |
| PromoBot [119] | 2011 | No | Hexamer frequency, RTP | SVM | 5-fold CV | – | Plant in PlantProm [185] | 251 | |
| Zuo et al. [120] | 2011 | No | KPCS, KIOCD, GCSS, DNA geometric flexibility | SVM | 10-fold CV, independent test | TATA-containing, TATA-less |
Plant in PlantProm [185] | 251 | |
| GPMiner [123] | 2012 | Decommissioned | CpG islands, nucleotide composition and DNA stability | SVM | 5-fold CV | – | Human, mouse, rat, chimpanzee and dog in DBTSS | 6000 | |
| ProMT [125] | 2014 | No | DSP | MCM | Independent test | – | H. sapiens | 1000 | |
| TSSPlant [29] | 2017 | Yes | PE, d(PE, TSS), k-mer, TFBSD, CG skew, AC skew | ANN | Independent test | TATA-containing, TATA-less |
A. thaliana, O. sativa | 251, 1101 | |
| DCDE-MSVMs [128] | 2019 | Decommissioned | k-mer, DCDE | SVM, BD | k-fold CV | – | H. sapiens | 251 | |
| Scoring function–based | PromoterInspector [73] | 2000 | Decommissioned | IUPAC groups with wildcards | – | 3-fold CV | – | Vertebrates in EPD [184] | 100 |
| Levitsky et al. [80] | 2001 | Decommissioned | DNC | TATA box weight matrix |
Leave-one-out | TATA-containing, TATA-less |
D. melanogaster | 400 | |
| CONPRO [85] | 2002 | Decommissioned | – | Combining the results of TSSG, TSSW, NNPP, PROSCAN and PromFD methods | Independent test | – | H. sapiens and M. musculus | – | |
| PromoSer [86] | 2003 | Decommissioned | – | Alignments of partial and full-length mRNA sequences to genomic DNA |
– | – | H. sapiens, M. musculus and R. norvegicus | – | |
| Ma et al. [92] | 2004 | No | TFBSs | PSSD | Independent test | – | Vertebrates in EPD [184] | 300 | |
| PSPA [97] | 2006 | Decommissioned | Position specific k-mers | PSPA | 10-fold CV | CpG-related, non-CpG-related | H. sapiens | 200 | |
| PromAn [98] | 2006 | Decommissioned | GC distribution, TSS location and TFBS predictions | Phylogenetic footprinting | – | – | Species in EPD [184] and DBTSS | – | |
| Pandey et al. [101] | 2006 | No | A+T content, relative entropy, periodicity, DNA curvature | PWM | Independent test | TATA-containing, TATA-less |
Plant in PlantProm [185], yeast and E. coli | 100 | |
| Wu et al. [105] | 2007 | No | k-mer | PWM | Independent test | – | H. sapiens | 250 | |
| ProStar [106] | 2007 | Decommissioned | Dinucleotide flexibility parameters | MD | Independent test | – | H. sapiens | 500 | |
| EP3 [110] | 2008 | Yes | GC content and DSP1 profiles | – | Independent test | – | H. sapiens, M. musculus, P. falciparum, O. pacifica, O. tauri, A. thaliana, O. sativa, P. trichocarpa, S. cerevisiae, S. pombe, C. elegans, D. melanogaster, T. nigroviridis | 400 | |
| EnsemPro [111] | 2008 | No | – | Combining the results of TSSG, TSSW, NNPP, Proscan, FirstEF, Dragon, Eponine, Promoter2.0 methods | fold CV, independent test | – | H. sapiens | – | |
| Akan et al. [112] | 2008 | No | DSP2, CpG counts and ATG codon count | PWM | – | CpG-related, non-CpG-related | H. sapiens and M. musculus | 1201 | |
| TSSer [114] | 2009 | No | Positional frequency of 5’ EST matches on genomic DNA | – | Independent test | – | A. thaliana and M. musculus | 400 | |
| PromPredict [121] | 2011 | Yes | DDS | DE | Independent test | – | A. thaliana, O. sativa | 1001 | |
| Fang et al. [122] | 2011 | No | TFBSs-SV, TFBSs-PV | IWM | Independent test | – | H. sapiens, vertebrate | 300 | |
| NPEST [124] | 2013 | Decommissioned | DEST | NPML | – | – | A. thaliana | 3000 | |
| Datta et al [131] | 2013 | No | CpG islands and CpG counts, PEs, DSP3 | CFG rules | Independent test | – | H. sapiens | 1201 | |
| PromPredict [126] | 2018 | Yes | DDS, GC content | DE | Independent test | – | 48 eukaryotic species | 1001 |
aAbbreviations: CNN—convolutional neural network; CV—cross-validation; BiLSTM—bidirectional long short-term memory; CpG island parameters—length, C+G mononucleotide content and ratio of observed to expected CpG content; QDA—quadratic discriminant analysis; Inr—initiator; SSM—stochastic segment models; ANN—artificial neural network; TFBSD—TFBS density; TDNN—time-delay neural network; RVM—relevance vector machine; PWMs—positional weight matrices; PEs—promoter elements; LDA—linear discriminant analysis; DPE—downstream promoter element; DNC—dinucleotide composition; GA—genetic algorithm; TSVM—transductive support vector machines; NBC—naïve Bayes classifier; WDs—weighted degree kernel with shifts; MTE—motif 10 element; TFBSs—transcription factor binding sites; IDQD—increment diversity with quadratic discriminant analysis; CTD—composition–transition–distribution; EDP—entropy density profile; RBFNN—radial basis function neural network; RTP—random triplet-pair; KPCS—k-mer position correlation score; KIOCD—k-mer increment of overlap content diversity; GCSS—GC-Skew score; DSP—DNA structural properties; MCM—Markov chain model; d(PE, TSS)—distance between promoter element and TSS; TFBSD—TFBS density; CG skew and AC skew—variations in base frequencies along sequence; DCDE—deep convolutional divergence encoding; BD—bilayer decision model; PSSD—TFBS pair scoring system with distance; PSPA—position-specific propensity analysis; MD—Mahalanobis distance; DSP1—base-stacking property, bendability and duplex stability-free energy; DSP2—propeller twist angle, bendability and nucleosome positioning preference; DE—relative stability (the difference in free energy); TFBSs-SV—transcription factor binding sites structural variability; TFBSs-PV—transcription factor binding sites positional variability; IWM—interval weight matrix; DEST—the distribution of expressed sequence tags; NPML—nonparametric maximum likelihood; DSP3—propeller twist, bendability, nucleosome position, DNA denaturation, zDNA, base staking energy, bDNAtwist and Aphilicity; CFG—context-free grammar.
bThe URL addresses for the listed tools are DeePromoter—https://home.jbnu.ac.kr/NSCL/deepromoter.htm; Depicter—https://depicter.erc.monash.edu/; TSSPlant—http://www.softberry.com/berry.phtml?topic=tssplant&group=programs&subgroup=promoter; PromPredictvhttp://nucleix.mbu.iisc.ac.in/prompredict/prompredict.html; CpGProD—http://pbil.univ-lyon1.fr/software/cpgprod.html; FProm—http://www.softberry.com/berry.phtml?topic=fprom&group=programs&subgroup=promoter; NNPP2.2—https://www.fruitfly.org/seq_tools/promoter.html.
cYes—The approach is accompanied with a webserver/tool and it is still working; Decommissioned—The webserver/tool is no longer available; No—The approach has no webserver or tool; Yes*—The server/tool was not involved in our performance comparison due to the fact that the outputs of the predictor need extra steps to interpret.
dWe listed the detailed eukaryotic promoter types based on the description in the papers. ‘–’ demonstrates such information is not present in the paper.
eThe species information of the sequences used in corresponding studies was directly extracted from the papers. For some species, the Latin names have been provided according the predictors; for other species, based on the information provided in the papers, we just provided the general names of the species when their Latin names are not available.
f‘–’ demonstrates that no clearly length information is provided in the paper.
Table 3.
A comprehensive list of the reviewed methods/tools for prediction of prokaryotic and eukaryotic promotersa
| Method type | Toolb | Year | Webserverc | Features/Motifs | Scoring function /Algorithm | Evaluation strategy | Species and promoter type | Sequence length (bp) |
|---|---|---|---|---|---|---|---|---|
| Deep learning–based | CNNProm [30] | 2017 | Yes | – | CNN | 70% train, 20% test, 10% validation |
H. sapiens (TATA-containing and TATA-less), M. musculus (TATA-containing and TATA-less), A. thaliana (TATA-containing and TATA-less), E. coli (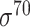 ) and B. subtilis ) and B. subtilis
|
81, 251 |
| Traditional machine learning–based | Rani et al.-I [132] | 2007 | No | DNC | ANN | 5-fold CV and independent test |
E. coli ( ), and D. melanogaster ), and D. melanogaster
|
80, 241 |
| Rani et al.-II [133] | 2009 | No | n-gram (n=2,3,4,5) | ANN | 5-fold CV and independent test |
E. coli (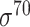 ) and D. melanogaster ) and D. melanogaster
|
80, 300 | |
| iProEP [31] | 2019 | Yes | PseKNC and PCSF | SVM | 5, 10-fold CV |
D. melanogaster, H. sapiens, C. elegans, E. coli (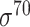 ) and B. subtilis ( ) and B. subtilis (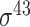 ) ) |
81, 300 | |
| Scoring function–based | IPMD [134] | 2010 | No | PCSF and ID | Modified MD | 10-fold CV and independent test |
D. melanogaster, H. sapiens, C. elegans, E. coli (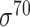 ) and B. subtilis ( ) and B. subtilis (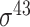 ) ) |
81, 300 |
aAbbreviations: CNN—convolutional neural network; DNC—dinucleotide composition; ANN—artificial neural network; CV—cross-validation; PseKNC—pseudo–K-tuple nucleotide composition; PCSF—position-correlation scoring function; SVM—support vector machine; ID—increment of diversity; MD—Mahalanobis Discriminant.
bThe URL addresses for the predictors with functioning webserver: CNNProm—http://www.softberry.com/berry.phtml?topic=cnnprom&group=programs&subgroup=deeplearn; iProEP—http://lin-group.cn/server/iProEP/pages/download.php.
cYes—The approach is accompanied with a webserver/tool and it is still working; Decommissioned—The webserver/tool is no longer available; No—The approach has no webserver or tool.
Figure 2.

A graphical illustration of four common steps for the construction and evaluation of computational approaches for predicting prokaryotic and eukaryotic promoters, including data collection, feature engineering, classification algorithm selection and performance evolution and webserver development.
Figure 3.

A statistical analysis of current computational approaches for promoter prediction. (A) The numbers of annual publications for prokaryotic and eukaryotic promoter prediction since 2000. Different colors represent different types of predictors, including promoter prediction tools for prokaryote, eukaryote and both. (B) Length distribution of prokaryotic and eukaryotic promoter sequences from a variety of training datasets. (C) Comparison of frequency of computational approaches applied promoter prediction for prokaryote and eukaryote. (D) Pie charts demonstrating the types and availability of prokaryotic and eukaryotic promoter prediction tools.
Construction of training datasets
Four public databases, including the Eukaryotic Promoter Database (EPDnew) [135], DBTSS [136], RegulonDB [137] and DBTBS [138], have been used as mainstream data resources for constructing the training datasets for prokaryotic and eukaryotic promoter predictors. In addition, Ensembl [139], BioMart [140] and UCSC Genome Browser [141] have been used for retrieving and extracting the promoter datasets as well. A brief summary of these databases is provided in Table 4. EPDnew is the major database that provides a nonredundant collection of eukaryotic pol II promoters from a variety of species, including animals, plants, fungi and invertebrates, while DBTSS is a database documenting the biological information of transcription start sites. For prokaryotic promoters, RegulonDB and DBTBS are considered as the major hubs containing 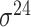 ,
, 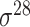 ,
, 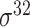 ,
, 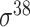 ,
, 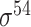 and
and 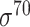 -promoter data from E. coli and
-promoter data from E. coli and 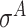 ,
, 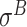 ,
, 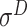 ,
, 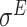 ,
, 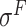 ,
, 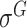 ,
, 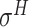 ,
, 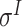 ,
, 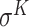 ,
,  ,
, 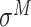 ,
, 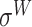 ,
, 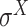 and
and 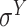 -promoter data from B. subtilis, respectively. We collected the training sequences from all available predictors for analyzing the length distribution of prokaryotic and eukaryotic promoters, respectively. The sequence length in the prokaryotic promoter training data ranges from 57 to 1101 bp, while the sequence length ranges from 100 bp to 10000 bp in the eukaryotic promoter training data. As shown in Figure 3B, most of the prokaryotic promoters are 81 bp long and less than 251 bp, while the vast majority of the eukaryotic promoters are within 251–300 bp long. The maximum length for the eukaryotic promoter sequences in Figure 3B has been restricted to 3000 bp for a better representation due to the highly skewed data.
-promoter data from B. subtilis, respectively. We collected the training sequences from all available predictors for analyzing the length distribution of prokaryotic and eukaryotic promoters, respectively. The sequence length in the prokaryotic promoter training data ranges from 57 to 1101 bp, while the sequence length ranges from 100 bp to 10000 bp in the eukaryotic promoter training data. As shown in Figure 3B, most of the prokaryotic promoters are 81 bp long and less than 251 bp, while the vast majority of the eukaryotic promoters are within 251–300 bp long. The maximum length for the eukaryotic promoter sequences in Figure 3B has been restricted to 3000 bp for a better representation due to the highly skewed data.
Table 4.
The databases extensively applied to collect multifarious promoter sequences
| Type | Database | Latest version | Feature | Website (URL) |
|---|---|---|---|---|
| Prokaryotic | RegulonDB [137] | 10.8 | RegulonDB is the primary database on transcriptional regulation in Escherichia coli K-12 containing knowledge manually curated from original scientific publications, complemented with high-throughput datasets and comprehensive computational predictions. | http://regulondb.ccg.unam.mx/ |
| DBTBS [138] | 5.0 | DBTBS is a database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. | http://dbtbs.hgc.jp/ | |
| Eukaryotic | EPDnew [135] | latest | EPDnew is an annotated nonredundant collection of eukaryotic POL II promoters, for which the transcription start site has been determined experimentally. | https://epd.epfl.ch//EPD_database.php |
| DBTSS [136] | 10.1 | DBTSS is a database of transcription start sites. | https://dbtss.hgc.jp/ |
Construction of independent test datasets
In order to objectively compare the prediction performance of current existing approaches, we constructed in total 58 balanced and unbalanced independent test datasets by cross-referencing public databases and removing the overlapping sequences with the training datasets of compared approaches. The resulting independent test datasets contain sequences from a variety of species, including E. coli, B. subtilis, H. sapiens, R. norvegicus, A. thaliana, D. melanogaster and Z. mays. Herein we describe the detailed procedures for curating the independent test datasets (Table 5).
Table 5.
The balanced independent test datasets for seven species
| Type | Species | Dataset name | TATA | Number of promoters | Number of non-promoters | Location | Length |
|---|---|---|---|---|---|---|---|
| Prokaryotic |
E. coli 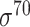
|
E. coli-I | – | 1089 | 1089 | [−60, +20] | 81 |
| E. coli | E. coli-II | 48 | 48 | [−60, +20] | 81 | ||
| B. subtilis | B. subtilis | 283 | 283 | [−60, +20] | 81 | ||
| Eukaryotic | H. sapiens | H. sapiens-I | TATA-containing | 229 | 229 | [−200, +50], [−249, +50], [−500, +500] | 251, 300, 1001 |
| H. sapiens-II | TATA-less | 1328 | 1328 | [−200, +50], [−249, +50], [−500, +500] | 251, 300, 1001 | ||
| R. norvegicus | R. norvegicus-I | TATA-containing | 456 | 456 | [−200, +50], [−249, +50], [−500, +500] | 251, 300, 1001 | |
| R. norvegicus-II | TATA-less | 3834 | 3834 | [−200, +50], [−249, +50], [−500, +500] | 251, 300, 1001 | ||
| D. melanogaster | D. melanogaster-I | TATA-containing | 207 | 207 | [−249, +50], [−500, +500] | 300, 1001 | |
| D. melanogaster-II | TATA-less | 1153 | 1153 | [−249, +50], [−500, +500] | 300, 1001 | ||
| A. thaliana | A. thaliana-I | TATA-containing | 474 | 474 | [−200, +50], [−249, +50], [−500, +500] | 251, 300, 1001 | |
| A. thaliana-II | TATA-less | 1073 | 1073 | [−200, +50], [−249, +50], [−500, +500] | 251, 300, 1001 | ||
| Z. mays | Z. mays-I | TATA-containing | 3051 | 3051 | [−200, +50], [−249, +50], [−500, +500] | 251, 300, 1001 | |
| Z. mays-II | TATA-less | 13813 | 13813 | [−200, +50], [−249, +50], [−500, +500] | 251, 300, 1001 |
Based on the three types of predictors for prokaryotic promoters (refer to ‘Existing approaches for prokaryotic and eukaryotic promoter prediction’), three balanced independent test datasets were constructed for evaluating the prediction performance of these predictors (Table 5). We collected all types of promoter sequences from E. coli with 81 bp length from RegulonDB [137] (version 10.8, last update on 10 December 2020). After removing those sequences with pairwise sequence identities greater than 80% using CD-HIT-EST [142, 143], 3028 sequences were retained (including 488 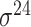 , 137
, 137 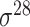 , 294
, 294 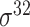 , 232
, 232 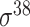 , 99
, 99 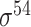 and 1778
and 1778 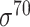 -promoter sequences). As DNA sequences only contain four letters, it is possible to achieve high similarity between unrelated sequences. In general, comparing DNAs at low identity <80% may not be very effective. Such threshold for sequence redundancy removal is a stringent cutoff, which is consistent with the threshold used in the previous studies [31, 58, 66, 69]. Non-promoter sequences (i.e. negative samples) refer to the DNA sequences/regions that do not contain promoter regions (i.e. the 0-th positions of sequences that are confirmed not to be TSSs). According to the methods for selecting negative samples in the majority of the literature works, non-promoter sequences for B. subtilis and E. coli include the coding sequences and convergent intergenic sequences. Thus, we extracted 4695 E. coli K12 code-region sequences from RegulonDB. All possible subsequences with 81 bp length were then generated, and 20 000 sequences were randomly selected. Then the similar sequences were removed using similarity of 80% by CD-HIT-EST, leaving 18244 non-promoter sequences retained. For the available predictors designed specifically for
-promoter sequences). As DNA sequences only contain four letters, it is possible to achieve high similarity between unrelated sequences. In general, comparing DNAs at low identity <80% may not be very effective. Such threshold for sequence redundancy removal is a stringent cutoff, which is consistent with the threshold used in the previous studies [31, 58, 66, 69]. Non-promoter sequences (i.e. negative samples) refer to the DNA sequences/regions that do not contain promoter regions (i.e. the 0-th positions of sequences that are confirmed not to be TSSs). According to the methods for selecting negative samples in the majority of the literature works, non-promoter sequences for B. subtilis and E. coli include the coding sequences and convergent intergenic sequences. Thus, we extracted 4695 E. coli K12 code-region sequences from RegulonDB. All possible subsequences with 81 bp length were then generated, and 20 000 sequences were randomly selected. Then the similar sequences were removed using similarity of 80% by CD-HIT-EST, leaving 18244 non-promoter sequences retained. For the available predictors designed specifically for 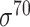 -promoter (iPro70-PseZNC, 70ProPred, iPromoter-FSEn, iPro70-FMWin), we removed those sequences greater than 80% similarity by comparing the 741
-promoter (iPro70-PseZNC, 70ProPred, iPromoter-FSEn, iPro70-FMWin), we removed those sequences greater than 80% similarity by comparing the 741 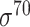 -promoter with our newly collected 1778
-promoter with our newly collected 1778 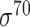 -promoter sequences. For negative samples (i.e. non-promoter sequences), the similar procedure was applied to the 18 244 non-promoter sequences, and the equal number of non-promoter sequences was randomly selected. The resulting independent test dataset E. coli-I contains 1089
-promoter sequences. For negative samples (i.e. non-promoter sequences), the similar procedure was applied to the 18 244 non-promoter sequences, and the equal number of non-promoter sequences was randomly selected. The resulting independent test dataset E. coli-I contains 1089 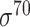 -promoter and 1089 non-promoter sequences. Similarly, the independent test dataset E. coli-II was generated by removing the similar sequences using CD-HIT-EST compared to the training datasets of available predictors for generic E. coli promoters (including BacPP, iPSW(2L)-PseKNC, iPromoter-2L, MULTiPly, iPromoter-2L 2.0, SELECTOR, iProEP and CNNProm). The resulting E. coli-II dataset contained 48 promoter and 48 non-promoter sequences after removing the similar sequences using CD-HIT-EST. For B. subtilis, the promoter sequences were downloaded from DBTBS [138] Release 5. After removing redundant sequences and aligning with the combined B. subtilis training datasets from CNNProm and iProEP, 283 promoter sequences remained after similarity removal by CD-HIT-EST, constituting the positive samples of the independent test dataset B. subtilis. Over 100 coding-region sequences of B. subtilis were manually extracted from NCBI Nucleotide database (accession number NC_000964.3). Similarly, all possible subsequences with 81 bp length were extracted. After applying CD-HIT-EST, 7314 non-promoter sequences remained, 283 out of which were randomly selected to constitute the negative samples of the B. subtilis dataset. As iProEP can predict general promoters of both E. coli and B. subtilis, it was compared and assessed using all the three independent test datasets.
-promoter and 1089 non-promoter sequences. Similarly, the independent test dataset E. coli-II was generated by removing the similar sequences using CD-HIT-EST compared to the training datasets of available predictors for generic E. coli promoters (including BacPP, iPSW(2L)-PseKNC, iPromoter-2L, MULTiPly, iPromoter-2L 2.0, SELECTOR, iProEP and CNNProm). The resulting E. coli-II dataset contained 48 promoter and 48 non-promoter sequences after removing the similar sequences using CD-HIT-EST. For B. subtilis, the promoter sequences were downloaded from DBTBS [138] Release 5. After removing redundant sequences and aligning with the combined B. subtilis training datasets from CNNProm and iProEP, 283 promoter sequences remained after similarity removal by CD-HIT-EST, constituting the positive samples of the independent test dataset B. subtilis. Over 100 coding-region sequences of B. subtilis were manually extracted from NCBI Nucleotide database (accession number NC_000964.3). Similarly, all possible subsequences with 81 bp length were extracted. After applying CD-HIT-EST, 7314 non-promoter sequences remained, 283 out of which were randomly selected to constitute the negative samples of the B. subtilis dataset. As iProEP can predict general promoters of both E. coli and B. subtilis, it was compared and assessed using all the three independent test datasets.
For eukaryotic promoter prediction, given that some models divided the eukaryotic promoters into TATA-containing promoters (i.e. promoter sequences with the TATA box element) and TATA-less promoters (i.e. promoter sequences without the TATA box element) [32, 88, 129, 130, 144], we downloaded the eukaryotic promoter sequences of four species (i.e. H. sapiens, R. norvegicus, A. thaliana and D. melanogaster), based on the presence of core promoter elements, i.e. TATA box, from the EPDnew [135] database (last update October 2019). These sequences are 251 bp long from 200 bp upstream to 50 bp downstream regions of TSS (i.e. [−200, +50]), with the TSS referred to as the 0-th site. Subsequently, for each species, the CD-HIT-EST program was employed to remove sequence redundancy with the sequence identity threshold of 80%. A similar sequence removal procedure was applied to each species and promoter type, resulting in eight balanced independent test datasets (Table 5). Note that DeePromoter provides both human and mouse models trained by the M. musculus and H. sapiens promoter sequences, respectively. However, there is no update of M. musculus or H. sapiens promoter sequences in EPDnew. We therefore used sequences of R. norvegicus to perform a cross-species evaluation for DeePromoter, considering that M. musculus and R. norvegicus are murids and the sequences among these two species share high sequence identity. We also did not compare the similarity between the training human promoter sequences of DeePromoter with our dataset. There are many unidentified nucleotides in the R. norvegicus promoter sequences that cannot be processed by some tools, and these sequences with unidentified nucleotides were removed. Note that different predictors require different lengths of input sequences. In general, three different sequence lengths are needed, including 251, 300 and 1001 bp. The input for the corresponding predictors was then adjusted to the requested sequence length (251: [−200, +50]; 300: [−249, +50] and 1001: [−500, +500]). The non-promoter sequences of four species, containing introns and exons, were extracted from Exon-Intron Database (EID) (http://bpg.utoledo.edu/~afedorov/lab/eid.html). The initially collected non-promoter sequences were processed as above and then we randomly selected the same number as the promoter sequences. The details on the numbers and location ranges of promoter and non-promoter sequences for each species in the independent test datasets are given in Table 5.
For each of the six species, in addition to the balanced test datasets, we have constructed unbalanced independent test datasets, which include a small number of positive samples (i.e. promoter regions/sequences) and a large number of negative samples (i.e. non-promoter regions/sequences). Specifically, the ratios of promoters to non-promoters (i.e. positive: negative) are 1:2, 1:3, 1:4 and 1:5 for E. coli, B. subtilis, H. sapiens, R. norvegicus, A. thaliana and D. melanogaster, respectively. The numbers of non-promoter sequences in the unbalanced independent test datasets for six species are shown in Table 6. Apart from the commonly used dicot plant (i.e. A. thaliana) promoter dataset, we further extracted the monocot plant (i.e. Z. mays) promoter dataset from the EPDnew [135] database, which contained 3051 TATA-containing promoters and 13813 TATA-less promoters after removing the redundant sequence and those sequences with ‘N’. As sufficient samples are available, for each promoter type, we constructed the balanced dataset (positive: negative = 1:1) for performance evaluation. We also have added the 5′ and 3′ UTR sequences as the non-promoter sequences into the unbalanced eukaryotic independent test datasets.
Table 6.
The unbalanced independent test datasets of six species
| Type | Species | Dataset name | TATA | 1:1a | 1:2 | 1:3 | 1:4 | 1:5 |
|---|---|---|---|---|---|---|---|---|
| Prokaryotic |
E. coli 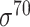
|
E. coli-I | – | 1089 | 2178 | 3267 | 4356 | 5445 |
| E. coli | E. coli-II | 48 | 96 | 144 | 192 | 240 | ||
| B. subtilis | B. subtilis | 283 | 566 | 849 | 1132 | 1415 | ||
| Eukaryotic | H. sapiens | H. sapiens-I | TATA-containing | 229 | 458 | 687 | 916 | 1145 |
| H. sapiens-II | TATA-less | 1328 | 2656 | 3984 | 5312 | 6640 | ||
| R. norvegicus | R. norvegicus-I | TATA-containing | 456 | 912 | 1368 | 1824 | 2280 | |
| R. norvegicus-II | TATA-less | 3834 | 7668 | 11502 | 15336 | 19170 | ||
| D. melanogaster | D. melanogaster-I | TATA-containing | 207 | 414 | 621 | 828 | 1035 | |
| D. melanogaster-II | TATA-less | 1153 | 2306 | 3459 | 4612 | 5765 | ||
| A. thaliana | A. thaliana-I | TATA-containing | 474 | 948 | 1422 | 1896 | 2370 | |
| A. thaliana-II | TATA-less | 1073 | 2146 | 3219 | 4292 | 5365 |
aPositive:negative ratio.
In addition, we downloaded the human chromosome 22 DNA sequence that is short and manageable, from the UCSC Genome Browser [141] (http://genome.ucsc.edu/) as an independent test dataset to evaluate the performance of EP3 [110] and PromPredict [126], which are two human promoter prediction methods and can predict promoters on the genome level. To avoid a biased evaluation of currently available promoter predictors, performance comparison was conducted on the CAGE dataset, which is based on the cap analysis gene expression (CAGE) technique [17]. It covers the whole human genome more widely and is retrieved from the FANTOM3 project (http://fantom.gsc.riken.go.jp/). As described in EP3 [110], only tag clusters with at least two mapped tags on the same genomic location were considered to be real TSSs. After mapping these tags to the human genome sequence, we obtained 181,047 unique human TSSs. The whole human genome (hg17) was retrieved using the UCSC Genome Browser [141].
Feature engineering and representation
To build a reliable and robust computational model for promoter prediction, features representing the DNA sequences should be carefully designed and extracted. Four major classes of features have been widely applied in the 106 approaches we revisited in this study, including signal-, context-, structure-based and integrated features.
Signal-based features
Signal-based features mainly contain the information of core promoter elements, specifically TATA box and mammalian CpG islands, which reflect the salient biological signals [145, 146]. The prokaryotic promoter regions may contain a Pribnow box (i.e. −10 box), which serves as a homolog compared to the eukaryotic TATA box with consensus sequence TATAAT, and −35 box [7]. Another important element is the AT-rich UP element, which serves as binding site for the alpha subunit of RNA polymerase [147, 148]. Given that the exact number and the location of individual promoter elements vary, it is expedient to define a promoter region by calculating the distance between the element and the TSS [35, 44, 47]. Eukaryotic promoters, on the other hand, typically contain seven core promoter elements, including TATA box, BREu and BREd (TF II B recognition element), Inr (initiator element), TCT (polypyrimidine), MTE (motif ten element) and DPE (downstream promoter element). Located within 25–30 bp upstream of the TSS with consensus sequence of ‘TATAAA’ [149], the TATA box appears in approximately 30–50% of all known promoters, except for its absence from the promoters of some particular genes, such as housekeeping and photosynthesis genes [150]. These promoters are therefore referred to as TATA-less promoters. Two BRE motifs are located in either upstream (BREu) or downstream (BREd) of a subset of TATA box elements [151]. The TCT motif, which serves as a key component of an RNA polymerase II system, spans from −2 to +6 relative to the TSS [152], while the MTE and DPE motifs are adjacent and both appear to be in the proximity to the TF II D subunits TAF6 and TAF9 [153]. In addition, CpG islands are unmethylated DNA segments with length of at least 200 bp [154]. The CpG island is featured by its GC pairs that account for more than 50% of the content, and its observed-to-expected CpG ratio (>60%). Approximately, half of the promoters in mammalian genomes have the CpG islands close to the starts of genes [146]. Therefore, the presence of a CpG island may represent a useful global signal for locating the promoters across genomes [74, 82, 89, 96].
Context-based features
Context-based features are extracted to describe the genomic context of the promoter sequences. We classify these context-based features into three groups: (i) DNA primary sequence-derived features, such as k-mer composition [68, 118], g-gapped k-mer composition [64], nucleotide statistical measure (NSM) [63] and variable-window Z-curve [53]; (ii) nucleotide physicochemical properties including pseudo–k-tuple nucleotide composition (PseKNC) [58], general PseKNC [65], pseudo–multi-window Z-curve nucleotide composition (PseZNC) [62], electron–ion interaction pseudo-potentials of trinucleotide (PseEIIP) [59] and dinucleotide-based auto-covariance (DAC) [66]; and (iii) position-specific scoring matrices, such as bi-profile Bayes (BPB) [66], position-correlation scoring function (PCSF) [31] and position-specific trinucleotide propensity based on single-stranded or double-stranded characteristic of DNA (PSTNPSS/PSTNPDS) [69]. Among these features, k-mer composition score, which describes the k-length substring of all possible combinations of A, C, G and T, has been extensively used in computational biology [155–157]. Most of the features above can be easily calculated by state-of-the-art bioinformatics pipelines, such as Pse-in-One [158], iFeature [159], iLearn [160] and iLearnPlus [161]. Compared to earlier published models for prokaryotic promoters (i.e. before 2010), which tended to use signal- or structure-based feature encoding schemes rather than context-based features, a majority of machine learning–based methods for prokaryotic promoter after 2010 favored context-based features for identifying promoter sequences. On the other hand, context-based features have been constantly the widely applied feature type for eukaryotic promoter prediction.
Structure-based features
Structure-based features are calculated based on the DNA 3D structures that characterize promoters [162]. It has been reported that the local changes of structural features around the classical TATA box and the TSS are contributive to differentiating the promoters from non-promoter sequences [112]. In light of this, several structure-based features have been introduced and demonstrated to be effective in boosting the prediction performance for both scoring function–based and traditional machine learning–based prokaryotic and eukaryotic promoter predictors. There are four major types of structure-based features, including (i) DNA curvature, which describes the extent of the DNA deviation due to the interaction of adjacent base pairs [163]; (ii) DNA bendability that describes the anisotropic bending of duplex DNA, which is closely related to DNase I cutting [164]; (iii) DNA duplex stability (DDS), which is used to describe the ability of DNA to open up or melt, depending on its hydrogen bonding and base pair stacking [56, 126]; (iv) stress-induced DNA duplex destabilization (SIDD), which is the required incremental free energy (kcal/mole) to keep the base pair open under the assumed superhelicity [40, 44]; and (v) G-quadruplex, which is a DNA secondary structure motif consisting of multiple vertically stacked guanine tetrads [71, 164].
Integrated features
Rather than using the single type of features described above, many promoter predictors combined different types of features [165], seeking to further improve the prediction performance. For example, by combining signal-, context- and structure-based features using decision trees, SCS [117] reached the highest sensitivity and specificity, while bTSSfinder [57] used oligomer frequencies, promoter elements and physicochemical properties to describe bacterial promoter regions.
Predictive algorithms employed
There are three types of major prediction algorithms applied in prokaryotic and eukaryotic promoter prediction (Figure 1 and Tables 1–3), including (i) scoring function–based algorithms, such as Position-Correlation Scoring Function (PCSF) [43], Modified Mahalanobis Discriminant (Modified MD) [134], Positional Weight Matrix (PWM) [34, 45, 54, 105] and Relative Stability (DE) [50, 126]; (ii) traditional machine learning–based algorithms, such as Support Vector Machine (SVM) [33, 55, 60, 65, 66, 123], Fisher’s Linear Discriminant Analysis (LDA) [40, 78, 88, 104], Logistic Regression (LR) [64], Random Forest (RF) [58], Naive Bayes Classifier (NBC) [95], Decision Tree (DT) [44] and Artificial Neural Network (ANN) [39, 48, 57, 76, 133]; and (iii) deep learning–based frameworks such as Convolutional Neural Network (CNN) [30, 32, 67, 127, 129].
Scoring function–based approaches
The statistical methods applied in the scoring function–based approaches [34, 43, 50, 134] are straightforward and simplified, such as sequence similarity and consensus patterns derived from the training data (i.e. the validated promoter sequences). Consequently, scoring function–based methods are particularly efficient for the identification of promoter sequences in a high-throughput manner. The mathematical descriptions of the commonly used scoring function–based algorithms are provided in the Supplementary Methods, available online at http://bib.oxfordjournals.org/.
Traditional machine learning–based algorithms
Among the surveyed predictors in this study, SVM, LDA and ANN are the most extensively used machine learning algorithms (Figure 3C). Previously, Monteiro and da Silva et al. [36, 38] only reported an empirical comparison of machine learning techniques for prokaryotic promoter prediction, including NBC, DT, SVM and ANN. According to the performance comparison, SVM outperformed other compared algorithms on an independent test dataset from B. subtilis. In addition to the individual machine learning techniques, ensemble strategies have also been successfully applied in both DNA (prokaryotic and eukaryotic) and noncoding RNA promoter prediction [63, 69, 166], as well as mRNA subcellular localization prediction [167]. For example, a feature subspace ensemble involving three different classifiers (i.e. SVM, LDA and LR) was applied to develop iPromoter-FSEn [63] for the identification of E. coli σ70-promoter sequences. Li et al. [69] constructed the stacking ensemble-learning model, namely SELECTOR, for the identification of promoters in E. coli by employing five popular individual and ensemble-learning algorithms, including RF, AdaBoost [168], GBDT [169], XGBoost [170] and LightGBM [171].
Deep learning–based approaches
Evolving from traditional ANN frameworks, deep learning techniques have been reported to achieve significantly boosted prediction performance across a variety of research fields, including natural language processing [172], drug design [173] and medical image analysis [174]. Among the different deep learning techniques, convolutional neural network (CNN) has drawn significant attention and has been widely applied to both prokaryotic and eukaryotic promoter prediction. A number of predictors have been developed using CNN and CNN-based deep learning techniques [30, 32, 67, 70, 127, 129] in combination with other strategies, such as bidirectional long short-term memory (BiLSTM) [175].
Strategies and measures for performance assessment
K-fold cross-validation (CV) test, jackknife validation test (i.e. leave-one-out CV) and independent test have been used to evaluate the prediction performance for the predictors surveyed in this study (Tables 1–3). Among these performance evaluation strategies, k-fold CV (k = 5 or 10) is the most frequently used strategy to construct performance matrices. In the k-fold CV test, the dataset is randomly split into k equally sized subsets. Each of the subsets is used as a test dataset once to evaluate the performance of the predictor trained by the remaining k − 1 subsets. The training and testing procedures are then conducted k times, and the average performance of the k combinations is usually reported. The jackknife test, on the other hand, is considered as a special case of k-fold CV, where k is set to the number of samples in the dataset. To make an objective and effective assessment and comparison with previous existing methods, the independent test is usually implemented using a newly assembled dataset with no overlap or low similarity with the sequences in the training datasets of compared predictors. A variety of widely applied performance measures in the field of bioinformatics [176–178], including area under the curve (AUC), sensitivity, specificity, accuracy (Acc), Matthew’s correlation coefficient [179] (MCC), precision and F1 score, have been used to quantitatively estimate the prediction performance. These measures are defined as follows:
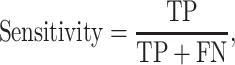 |
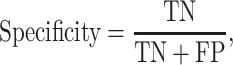 |
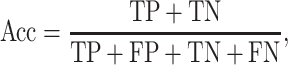 |
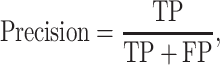 |
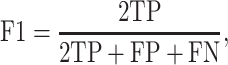 |
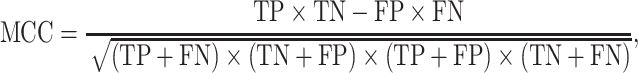 |
where TP, TN, FP and FN denote the numbers of true positives, true negatives, false positives and false negatives, respectively.
Webserver/software availability and usability
A user-friendly webserver and/or a locally executable tool can significantly facilitate the use of the proposed prediction tool for both prokaryotic and eukaryotic promoters. In total, 59.4% (63 out of 106) of the surveyed predictors have available websites/tools and 46.0% (29 out of 63) of these are still active (Figure 3D). Based on our investigation of the predictors for high-throughput prokaryotic promoter prediction in this study, 24 predictors out of 40 (60.0%) were implemented with webservers and/or stand-alone software (Table 1). For eukaryotic promoter prediction (Table 2), 37 out of 61 (60.7%) predictors provide webservers and/or stand-alone program; however, more than half unfortunately are offline. Among the predictors that can predict both prokaryotic and eukaryotic promoters, iProEP and CNNProm provide executable webservers.
All the webservers mentioned above allow users to submit their sequences of interest in the FASTA format. However, certain servers have some special requirements in terms of number and length of the submitted sequences. For example, BacPP only allows up to 2000 characters of input sequences; the minimum length of submitted sequences is 1001 bp for PromPredict; iPSW(2L)-PseKNC only allows no more than 100 sequences submitted each time; the allowed length for each sequence is 251–100 000 bps for TSSPlant; DeePromoter and iProEP allow fixed 300 bp of each sequence, while CNNProm allows for submitted sequences with 251 bp. Most prokaryotic promoter predictors require fixed 81 bp long sequences. In particular, BacPP was designed to analyze sequences with 80 nucleotides. Longer sequences will be sliced by a sliding window for further processing.
A well-designed output format is critical for users to understand and interpret the prediction results. Most servers display the instantaneous prediction results on their webpages. MULTiPly, SELECTOR and Depicter also allow users to retrieve previous prediction results using the assigned job IDs. The output generally contains predicted labels of query sequences (e.g. promoter or non-promoter). Some methods such as iPro70-PseZNC, iPro70-FMWin, iPromoter-FSEn, iProEP, CNNProm, NNPP2.2 and SELECTOR offer the probability scores of promoters or non-promoters as well. TSSPlant provides predicted positions and scores of TSS and TATA box, while NNPP2.2 and PromPredict can identify more than one promoter sequence with the start and end positions.
Experimental results
Performance comparison of prokaryotic promoter prediction tools
Three balanced independent test datasets were used to assess the prediction performance of the predictors for prokaryotic promoter, including E. coli-I (i.e. for 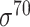 promoters), E. coli-II (i.e. for general E. coli promoters) and B. subtilis (i.e. for general B. subtilis promoters). See ‘Construction of independent test datasets’ for the detailed dataset construction procedures. Note that only predictors with available webservers/tools were assessed (bolded in Tables 1 and 3). The prediction performance is shown in Figure 4 and Supplementary Table 1A available online at http://bib.oxfordjournals.org/. Among the available predictors, iPro70-PseZNC, 70ProPred, iPromoter-FSEn and iPro70-FMWin were designed for σ70 promoters only while iProEP was implemented for generic prokaryotic promoters. The Acc, AUC and MCC values for σ70 promoters are shown in Figure 4A. iPro70-FMWin outperformed all the predictors by achieving Acc of 86.46%, AUC of 0.936 and MCC of 0.733. For prediction of general E. coli promoters, eight available predictors were employed, including BacPP, iPSW(2L)-PseKNC, iPromoter-2L, MULTiPly, iPromoter-2L2.0, SELECTOR, iProEP and CNNProm. Among these methods, iPSW(2L)-PseKNC achieved the best prediction performance in terms of all the measures except specificity and precision (Figure 4B and Supplementary Table 1A available online at http://bib.oxfordjournals.org/). In addition, Le et al. and iPSW(2L)-PseKNC further categorized the predicted promoters into two types, namely ‘strong’ and ‘weak’, which is a unique feature of the two methods compared to others. For B. subtilis, there is no available specific computational pipeline, and we therefore assessed the prediction performance of two general predictors, including iProEP and CNNProm (Figure 4C and Supplementary Table 1A available online at http://bib.oxfordjournals.org/). Despite that iProEP achieved higher AUC (0.828), CNNProm demonstrated its prediction ability by higher Acc (77.03%) and MCC (0.557). In addition, we plotted two heatmaps to illustrate the precision and F1 score of all the compared predictors using the three datasets (Figure 4E and F). iProEP is the only predictor that is applicable to all the three datasets. While iProEP has not been ranked as the top predictor, it has achieved satisfactory prediction performance on all the three datasets and therefore may be a useful tool to classify promoter sequences with unknown species or promoter type. When evaluated on the unbalanced independent test datasets for E. coli-I (i.e. for
promoters), E. coli-II (i.e. for general E. coli promoters) and B. subtilis (i.e. for general B. subtilis promoters). See ‘Construction of independent test datasets’ for the detailed dataset construction procedures. Note that only predictors with available webservers/tools were assessed (bolded in Tables 1 and 3). The prediction performance is shown in Figure 4 and Supplementary Table 1A available online at http://bib.oxfordjournals.org/. Among the available predictors, iPro70-PseZNC, 70ProPred, iPromoter-FSEn and iPro70-FMWin were designed for σ70 promoters only while iProEP was implemented for generic prokaryotic promoters. The Acc, AUC and MCC values for σ70 promoters are shown in Figure 4A. iPro70-FMWin outperformed all the predictors by achieving Acc of 86.46%, AUC of 0.936 and MCC of 0.733. For prediction of general E. coli promoters, eight available predictors were employed, including BacPP, iPSW(2L)-PseKNC, iPromoter-2L, MULTiPly, iPromoter-2L2.0, SELECTOR, iProEP and CNNProm. Among these methods, iPSW(2L)-PseKNC achieved the best prediction performance in terms of all the measures except specificity and precision (Figure 4B and Supplementary Table 1A available online at http://bib.oxfordjournals.org/). In addition, Le et al. and iPSW(2L)-PseKNC further categorized the predicted promoters into two types, namely ‘strong’ and ‘weak’, which is a unique feature of the two methods compared to others. For B. subtilis, there is no available specific computational pipeline, and we therefore assessed the prediction performance of two general predictors, including iProEP and CNNProm (Figure 4C and Supplementary Table 1A available online at http://bib.oxfordjournals.org/). Despite that iProEP achieved higher AUC (0.828), CNNProm demonstrated its prediction ability by higher Acc (77.03%) and MCC (0.557). In addition, we plotted two heatmaps to illustrate the precision and F1 score of all the compared predictors using the three datasets (Figure 4E and F). iProEP is the only predictor that is applicable to all the three datasets. While iProEP has not been ranked as the top predictor, it has achieved satisfactory prediction performance on all the three datasets and therefore may be a useful tool to classify promoter sequences with unknown species or promoter type. When evaluated on the unbalanced independent test datasets for E. coli-I (i.e. for 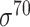 promoters), iPro70-FMWin constantly outperformed all other predictors. The prediction performance is shown in Supplementary Table 1B–E available online at http://bib.oxfordjournals.org/. For general E. coli promoter prediction, iProEP and CNNProm performed exceptionally well in terms of specificity and precision. While for B. subtilis, iProEP achieved higher specificity and precision while CNNProm obtained higher Acc and MCC. As shown in Figure 4 and Supplementary Table 1 available online at http://bib.oxfordjournals.org/, deep learning–based methods have not achieved satisfactory prediction performance for prokaryotic promoters. One possible reason is that the datasets used for training and testing the prokaryotic promoter predictors are very limited, while deep learning models usually require a larger volume of training samples to enable the construction of reliable and accurate models. In contrast, traditional machine learning–based methods developed based on handcrafted features can competently deal with small datasets and can achieve more robust performance.
promoters), iPro70-FMWin constantly outperformed all other predictors. The prediction performance is shown in Supplementary Table 1B–E available online at http://bib.oxfordjournals.org/. For general E. coli promoter prediction, iProEP and CNNProm performed exceptionally well in terms of specificity and precision. While for B. subtilis, iProEP achieved higher specificity and precision while CNNProm obtained higher Acc and MCC. As shown in Figure 4 and Supplementary Table 1 available online at http://bib.oxfordjournals.org/, deep learning–based methods have not achieved satisfactory prediction performance for prokaryotic promoters. One possible reason is that the datasets used for training and testing the prokaryotic promoter predictors are very limited, while deep learning models usually require a larger volume of training samples to enable the construction of reliable and accurate models. In contrast, traditional machine learning–based methods developed based on handcrafted features can competently deal with small datasets and can achieve more robust performance.
Figure 4.
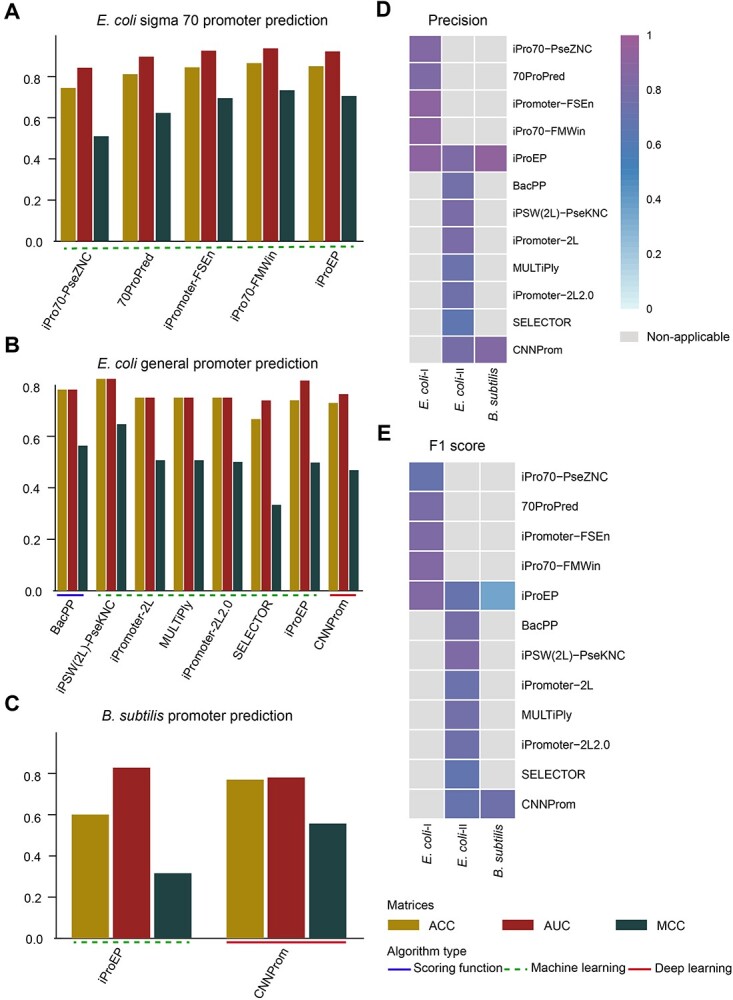
Prediction performance of prokaryotic promoter predictors on three independent test datasets, including E. coli-I, E. coli-II and B. subtilis in terms of Acc, AUC and MCC for (A) E. coli 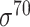 promoters (E. coli-I dataset), (B) E. coli promoters (E. coli-II dataset) and (C) B. subtilis promoters (B. subtilis dataset). Two heatmaps were plotted to illustrate (D) the Precision and (E) the F1 scores of assessed predictors using all the three datasets. The gray grids in the heatmaps (D) and (E) mean the current method is non-applicable to this dataset.
promoters (E. coli-I dataset), (B) E. coli promoters (E. coli-II dataset) and (C) B. subtilis promoters (B. subtilis dataset). Two heatmaps were plotted to illustrate (D) the Precision and (E) the F1 scores of assessed predictors using all the three datasets. The gray grids in the heatmaps (D) and (E) mean the current method is non-applicable to this dataset.
Performance comparison of eukaryotic promoter prediction tools
Ten balanced independent test datasets were constructed for two major types of eukaryotic promoters (i.e. TATA-containing and TATA-less) for five species including A. thaliana, Z. mays, D. melanogaster, R. norvegicus and H. sapiens. In total, eight available predictors (bolded in Tables 2 and 3) were benchmarked using these datasets. The prediction results are illustrated in Figure 5 and Supplementary Tables 2A, 3, 4A, 5A and 6A available online at http://bib.oxfordjournals.org/. Note that different methods request different lengths of submitted sequences. There are three major lengths of submitted sequences, including 251 bp ([−200, +50]), 300 bp ([−249, +50]) and 1001 bp ([−500, +500]). For these methods, we submitted the promoter sequences with different lengths of extended upstream and downstream of TSS.
Figure 5.
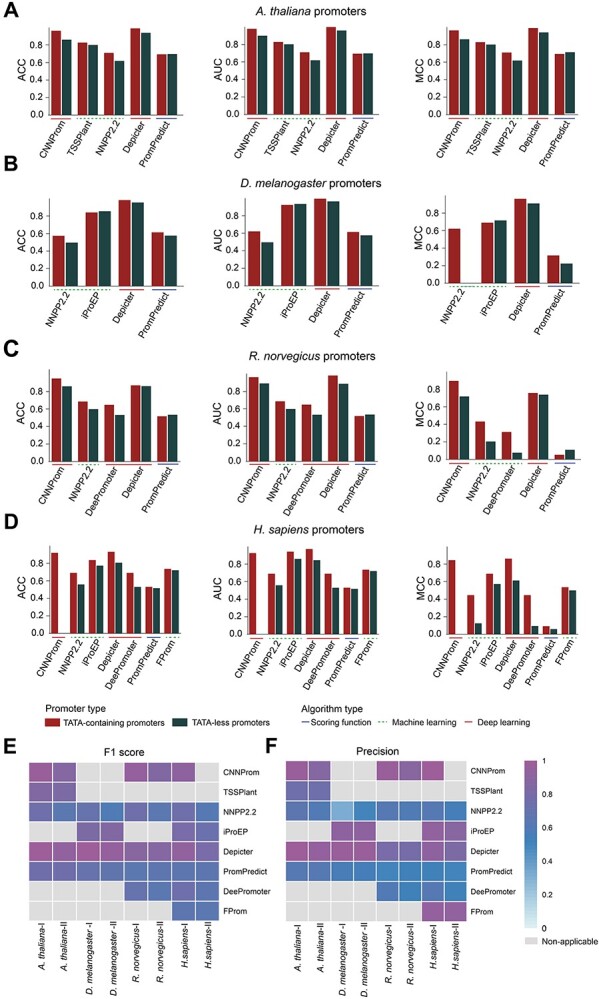
Prediction performance of different predictors on eight eukaryotic promoter independent test datasets in terms of AUC, Acc and MCC on the (A) A. thaliana, (B) D. melanogaster, (C) R. norvegicus and (D) H. sapiens datasets. In addition, two heatmaps demonstrate the (E) F1 and (F) Precision scores of all the compared predictors on these datasets. The gray grids in the heatmaps (E) and (F) mean the current method is non-applicable to this dataset.
Five available tools, including CNNProm, TSSPlant, NNPP2.2, Depicter and PromPredict (2018 version), were designed for recognition of A. thaliana promoters. These predictors allow submissions with 251, 300 and 1001 bp sequence extension flanking the TSS. Due to the unexpected unavailability of the CNNProm server, we were unable to include it in the performance comparison on the unbalanced datasets. As shown in Figure 5A, E, F and Supplementary Table 2A available online at http://bib.oxfordjournals.org/, Depicter achieved the best prediction performance for predicting both TATA-containing (MCC of 0.977; F1 of 0.989) and TATA-less promoters (MCC of 0.876; F1 of 0.937). CNNProm was ranked the second-best predictor based on the performance measures. Note that both Depicter and CNNProm are deep learning–based approaches and the prediction results have clearly demonstrated the improved performance powered by deep learning techniques, compared to scoring function–based and traditional machine learning–based approaches. Tested on the unbalanced independent test datasets of A. thaliana, similar performance results described above can be observed in the Supplementary Table 2B–E available online at http://bib.oxfordjournals.org/. We further examined the methods designed for recognition of A. thaliana promoters using the additional test dataset of monocot plant (i.e. Z. mays), including CNNProm, TSSPlant, NNPP2.2, Depicter and PromPredict. The predictive performance is provided in Supplementary Table 3 available online at http://bib.oxfordjournals.org/. It can be seen that for the TATA-containing promoters, Depicter achieved optimal performance metrics except sensitivity, while for the TATA-less promoters, TSSPlant performed overall best with MCC (0.442) and F1 (0.730).
NNPP2.2, iProEP, Depicter and PromPredict were benchmarked for performance comparison on D. melanogaster promoters (Figure 5B, E, F and Supplementary Table 4A available online at http://bib.oxfordjournals.org/). Depicter again outperformed other baselines by achieving AUC of 0.993, MCC of 0.962 and Acc of 98.07% for TATA-containing promoter and AUC of 0.963, MCC of 0.909 and Acc of 95.40% for TATA-less promoter, respectively. It is also noticeable that iProEP was ranked as the second-best predictor, outperforming both NNPP2.2 and PromPredict. The prediction results suggest that using only DNA structural properties or signal-based features is not sufficient to accurately predict promoters. Rather, the addition of context-based features such as k-mer and PseKNC can assist the predictor to obtain more accurate and reliable prediction results. The difference in the context between promoters and non-promoters (from coding regions) may be the main reason for this result. Tested on the unbalanced independent test datasets of D. melanogaster, iProEP achieved higher specificity and precision for TATA-containing promoter and outperformed the other tools in terms of all the measures except sensitivity with the increase of negative samples (Supplementary Table 4B–E available online at http://bib.oxfordjournals.org/).
CNNProm, NNPP2.2, DeePromoter, Depicter and PromPredict can be used to predict M. musculus promoters. However, there has not been a major update for M. musculus promoters in EPDnew and we therefore could not assemble an independent test dataset from M. musculus. Instead, we generated two promoter datasets (i.e. TATA-containing and TATA-less) from R. norvegicus sequences documented in EPDnew (see ‘Construction of independent test datasets’). In general, CNNProm outperformed other predictors for R. norvegicus TATA-containing promoters more accurately with MCC of 0.895 and F1 of 0.946 (Figure 5C, E, F and Supplementary Table 5A available online at http://bib.oxfordjournals.org/). While for R. norvegicus TATA-less promoters, Depicter achieved the highest MCC of 0.738 and F1 of 0.874, and CNNProm achieved highest specificity (0.891) and precision (0.883). CNNProm and Depicter still performed well, separately for TATA-containing and TATA-less promoters on the unbalanced independent test datasets of R. norvegicus (Supplementary Table 5B–E available online at http://bib.oxfordjournals.org/). The prediction results suggest that these predictors can be reliably applied to R. norvegicus sequences, despite that they were trained using the sequences from M. musculus.
Seven predictors were used to assess the performance on H. sapiens promoters. As shown in Figure 5D, E, F and Supplementary Table 6A available online at http://bib. oxfordjournals.org/, Depicter again achieved the highest AUC (0.972), Acc (93.01%) and MCC (0.862) for TATA-containing promoters. For TATA-less promoters, Depicter achieved highest Acc and MCC values, while iProEP attained the highest AUC (0.859). CNNProm achieved satisfactory prediction performance for TATA-containing promoters, but unfortunately it cannot process TATA-less promoter sequences. Interestingly, NNPP2.2, DeePromoter and PromPredict achieved low specificity values, suggesting that they tend to misclassify the non-promoters as promoters, thereby leading to high false positive rate. iProEP performed best in terms of all measures except for sensitivity for TATA-less promoters (Supplementary Table 6B–E available online at http://bib.oxfordjournals.org/) on the unbalanced independent test datasets of H. sapiens.
Among the available eukaryotic promoter prediction tools, EP3 and PromPredict are able to recognize promoters on the whole-genome scale with rapid computing speed. Thus, we compared these two methods on the task of human chromosome 22 genome promoter prediction. The prediction performance was evaluated with the reference to the annotated TSS set of the CAGE dataset. Specifically, we set the maximum allowed mismatch values as 2000, 1000 and 500 bp used in EP3, between the prediction and the true TSS. If a predicted promoter region is within the maximum allowed mismatch from a true TSS, it is then regarded as a true TSS hit (i.e. true positive, TP). If a region contains a true TSS but is not predicted as a promoter region, it is then regarded as a false negative (FN). If a predicted promoter region is further than the maximum allowable distance from a true TSS, it is then considered as a false positive (FP). The performance of EP3 and PromPredict in terms of sensitivity, precision and F1 score based on the three maximum allowed mismatch thresholds is shown in Supplementary Figure 1 available online at http://bib.oxfordjournals.org/. For each maximum allowed mismatch threshold, PromPredict achieved a considerably high sensitivity but low precision, thereby leading to a low F1 score. On the contrary, EP3 showed a high precision and low sensitivity, but its balance of precision and sensitivity appeared to be better than PromPredict.
Discussion
Promoters play critical roles in regulating DNA transcription, and precise recognition of promoter regions can therefore assist biologists to further improve genome annotation and guide experimental design for elucidation of gene transcriptional regulation mechanisms. Benefiting from the advances of sequencing techniques, more experimentally validated species-specific promoter sequences have become publicly available, thereby providing reliable training data to construct computational tools for predicting prokaryotic and eukaryotic promoters. Before 2012, most of the computational studies for promoter prediction focused on eukaryotic cells, while more tools for prokaryotic promoter prediction were published after 2012, due to the rapid accumulation of prokaryotic sequencing data (Figure 3A). Earlier predictive models for promoter prediction are usually based on scoring functions, which are trained using the underlying sequence patterns of known promoters. Compared to traditional machine learning and deep learning techniques, scoring function–based approaches usually predict rapidly with lower prediction performance. Despite the recent efforts, there is large room for in silico methods to further improve their prediction performance. Almost all current available methods are species-specific. As such, they are trained only on one species and can predict promoters for that particular species. In addition, many state-of-the-art tools limit the length of the test sequence. We also note that the webservers of a number of promoter predictors developed in the past two decades are not well maintained, as shown in Figure 3D. This has significantly impacted their usability and applicability. As user friendly and publicly accessible webservers represent the future direction for developing practically useful models, it is highly encouraged that source codes and webservers can be publicly accessible to ensure the best practice of data reproducibility. For eukaryotic promoter prediction, some models [32, 88, 100, 129] divided the eukaryotic promoters into TATA-containing promoters and TATA-less promoters based on the presence of core promoter elements, i.e. TATA box. Other classes of promoter elements such as Inr [9], TCT [152] and MTE [153] are also significantly conserved in eukaryotes; however, there is a lack of computational tools for such classes of eukaryotic promoters.
There are three major prediction strategies applied to the promoter identification, namely, scoring function, traditional machine learning–based techniques and deep learning–based techniques. Based on the independent tests using 58 datasets, deep learning–based models, which usually require more time and computational resource to train, have achieved outstanding prediction performance, while scoring function–based methods, which are straightforward to implement, usually demonstrate less desirable performance. Traditional machine learning–based methods, on the other hand, balance the computational burden and algorithm complexity. It is therefore at developers’ discretion to determine the most cost-effective prediction strategy, based on the training data. Scoring function–based tools tend to predict a number of false positives or have poor sensitivity when being applied to predefined promoter sequences. Nevertheless, the prediction outputs by these methods can still be helpful for drawing some meaningful biological conclusions. For example, PromPredict can identify predicted regions that have lower stability, higher curvature and less bendability, which favors transcription initiation (which may act as promoters). DDM finds that on average more than 40% of the human genome can be highly unlikely to initiate transcription based on the analysis of human chromosomes 4, 21 and 22. While Wang et al.’s work shows that the propensity for SIDD is closely associated with specific promoter regions. The features used to represent the promoter sequences are another important performance-determining factor for both traditional machine learning– and deep learning–based approaches. We summarized the major feature types of the surveyed predictors, namely signal-, context-, structure-based and integrated features. Comparing to individual feature type, combining all the three types of features (i.e. integrated features) can significantly improve the prediction performance. For example, SCS [117] combining signal, context and structure features by the decision trees reached the highest sensitivity and specificity. On the other hand, it is possible that the dimensionality of calculated feature vectors is high and feature selection methods should be therefore widely applied to reduce the dimensionality by filtering out the misleading features, thereby further improving the prediction performance [62, 64, 67]. Another problem that needs to be considered is the unbalanced amount of positive and negative sequences, as the non-promoter sequences significantly outnumber the promoter sequences. To build a balanced classifier, it is practical to randomly select a pool of negative sequences with the same numbers of positive sequences. Ideally, such random selection should be repeated multiple times, and the average results should be reported. In addition, various effective resampling algorithms such as Safe-Level-SMOTE [180] oversampling, and most distant undersampling techniques, have been developed to address the data imbalanced problem [181]. Therefore, such methods can be considered in the model construction when the datasets are significantly unbalanced.
Conclusion
In this study, we have surveyed 106 state-of-the-art computational approaches for predicting prokaryotic and eukaryotic promoter sequences and benchmarked 19 predictors with available and functioning webservers/local tools. To our best knowledge, our study represents the most comprehensive and large-scale benchmarking test of promoter predictors. A wide range of aspects in detail were summarized, including employed algorithms, calculated features, performance evaluation strategies and software usability. By curating 58 independent test datasets for various species and promoter types, we extensively benchmarked 19 available predictors and demonstrated their prediction performance. The prediction results show that traditional machine learning–based and deep learning–based methods generally outperform scoring function–based methods for predicting species-specific promoters. One should bear in mind that the performance of the surveyed predictors is subject to change in cases where different test datasets and new data are used to train the models [182]. In addition, we have discussed several issues and provided some insights to potentially useful strategies that may be used to further enhance the performance of promoter prediction. We expect this analysis to be a steppingstone toward design and implementation of more accurate predictors for both prokaryotic and eukaryotic promoters in the future.
Data Availability
All the 58 independent test datasets used for benchmarking various predictors in this study are available at https://github.com/chenli-bioinfo/promoter/.
Author Contributions
J.S., L.J.M.C. and Q.Z. conceived and designed the project. M.Z., C.J., F.L. and C.L. conducted the data analysis and independent tests and drafted the manuscript. T.A. and G.I.W. provided critical comments and useful insight to improve the scientific quality of the work. Q.Z., L.J.M.C. and J.S. revised the manuscript, which has been approved by all the other authors.
Key Points
We curated 58 comprehensive, up-to-date, benchmark datasets for 7 different species to assist the research community to assess the relative functionality of alternative approaches and support future research on both prokaryotic and eukaryotic promoters.
We revisited 106 predictors published since 2000 for promoter identification (40 for prokaryotic promoter, 61 for eukaryotic promoter, and 5 for both).
We found that deep learning and traditional machine learning–based approaches generally outperformed scoring function–based approaches.
We have assessed the prediction performance of 19 available prokaryotic and eukaryotic promoter predictors using our newly curated 58 benchmark datasets.
Supplementary Material
Meng Zhang received her MS degree from Dalian Maritime University, China. She is currently a PhD student in Nanjing University of Aeronautics and Astronautics. Her research interests are bioinformatics, computational biology and machine learning.
Cangzhi Jia is an associate professor in the College of Science, Dalian Maritime University. She obtained her PhD degree in the School of Mathematical Sciences from Dalian University of Technology in 2007. Her major research interests include mathematical modeling in bioinformatics and machine learning.
Fuyi Li is currently a Bioinformatics Research Officer in the Department of Microbiology and Immunology, the Peter Doherty Institute for Infection and Immunity, the University of Melbourne, Australia. His research interests are bioinformatics, computational biology, machine learning and data mining.
Chen Li is a Research Fellow in the Biomedicine Discovery Institute and Department of Biochemistry of Molecular Biology, Monash University. He is currently a CJ Martin Early Career Research Fellow, supported by Australian National Health and Medicine Research Council (NHMRC). His research interests include systems proteomics, immunopeptidomics, personalized medicine and experimental bioinformatics.
Yan Zhu is currently pursuing her MS degree in the School of Science, Dalian Maritime University, China. Her research interests are bioinformatics, deep learning and machine learning.
Tatsuya Akutsu has been a professor in Bioinformatics Center, Institute for Chemical Research, Kyoto University since 2001. He obtained Dr Eng. degree from University of Tokyo in 1989. His research interests include bioinformatics, complex networks and discrete algorithms.
Geoffrey I. Webb received his PhD degree in Computer Science in 1987 from La Trobe University. He is the research director of the Monash Data Futures Institute and a professor in the Faculty of Information Technology at Monash University. His research interests include machine learning, data mining, computational biology and user modeling.
Quan Zou is a professor at University of Electronic Science and Technology of China. He is a senior member of IEEE and ACM. His research interests include bioinformatics, machine learning and algorithms.
Lachlan J.M. Coin is a professor and group leader in the Department of Microbiology and Immunology at the University of Melbourne. He is also a member of the Department of Clinical Pathology, University of Melbourne. His research interests are bioinformatics, machine learning, transcriptomics and genomics.
Jiangning Song is an associate professor and group leader in the Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia. He is also affiliated with the Monash Centre of Data Science, Faculty of Information Technology, Monash University. His research interests include bioinformatics, computational biology, machine learning, data mining and pattern recognition.
Contributor Information
Meng Zhang, School of Science, Dalian Maritime University, Dalian 116026, China.
Cangzhi Jia, School of Science, Dalian Maritime University, Dalian 116026, China.
Fuyi Li, Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; The Peter Doherty Institute for Infection and Immunity, The University of Melbourne, VIC, Australia.
Chen Li, Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia.
Yan Zhu, School of Science, Dalian Maritime University, Dalian 116026, China.
Tatsuya Akutsu, Bioinformatics Center, Institute for Chemical Research, Kyoto University, Kyoto 611-0011, Japan.
Geoffrey I Webb, Department of Data Science and Artificial Intelligence, Monash University, Melbourne, VIC 3800, Australia; Monash Data Futures Institute, Monash University, Melbourne, VIC 3800, Australia.
Quan Zou, Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, China.
Lachlan J M Coin, The Peter Doherty Institute for Infection and Immunity, The University of Melbourne, VIC, Australia.
Jiangning Song, Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Monash Data Futures Institute, Monash University, Melbourne, VIC 3800, Australia.
Funding
National Natural Science Foundation of China (62071079), the National Health and Medical Research Council of Australia (NHMRC) (APP1127948 and APP1144652), the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965), a Major Inter-Disciplinary Research project awarded by Monash University, and the collaborative research program of the Institute for Chemical Research, Kyoto University (#2021-28). J.S. is supported by the JSPS Invitational Fellowship (L21503). L.J.M.C. is supported by an NHMRC career development fellowship (1103384), as well as an NHMRC-EU project grant GNT1195743. C.L. is currently supported by an NHMRC CJ Martin Early Career Research Fellowship (1143366).
References
- 1. Butler JEF, Kadonaga JT. The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev 2002;16:2583–92. [DOI] [PubMed] [Google Scholar]
- 2. Werner T. Models for prediction and recognition of eukaryotic promoters. Mamm Genome 1999;10:168–75. [DOI] [PubMed] [Google Scholar]
- 3. Juven-Gershon T, Kadonaga JT. Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev Biol 2010;339:225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shen ZJ, Lin Y, Zou Q. Transcription factors-DNA interactions in rice: identification and verification. Brief Bioinform 2020;21:946–56. [DOI] [PubMed] [Google Scholar]
- 5. Browning DF, Busby SJW. The regulation of bacterial transcription initiation. Nat Rev Microbiol 2004;2:57–65. [DOI] [PubMed] [Google Scholar]
- 6. Helmann JD, Chamberlin MJ. Structure and function of bacterial sigma factors. Annu Rev Biochem 1988;57:839–72. [DOI] [PubMed] [Google Scholar]
- 7. Hawley DK, McClure WR. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res 1983;11:2237–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burley SK, Roeder RG. Biochemistry and structural biology of transcription factor IID (TFIID). Annu Rev Biochem 1996;65:769–99. [DOI] [PubMed] [Google Scholar]
- 9. Smale ST, Jain A, Kaufmann J, et al. The initiator element: a paradigm for core promoter heterogeneity within metazoan protein-coding genes. Cold Spring Harb Symp Quant Biol 1998;63:21–31. [DOI] [PubMed] [Google Scholar]
- 10. Greenblatt J. RNA polymerase II holoenzyme and transcriptional regulation. Curr Opin Cell Biol 1997;9:310–9. [DOI] [PubMed] [Google Scholar]
- 11. Coulombe B, Li J, Greenblatt J. Topological localization of the human transcription factors IIA, IIB, TATA box-binding protein, and RNA polymerase II-associated protein 30 on a class II promoter. J Biol Chem 1994;269:19962–7. [PubMed] [Google Scholar]
- 12. Smith AD, Sumazin P, Xuan ZY, et al. DNA motifs in human and mouse proximal promoters predict tissue-specific expression. Proc Natl Acad Sci U S A 2006;103:6275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang MQ. Computational analyses of eukaryotic promoters. BMC Bioinformatics 2007;8:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Triska M, Solovyev V, Baranova A, et al. Nucleotide patterns aiding in prediction of eukaryotic promoters. Plos One 2017;12:e0187243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carninci P, Waki K, Shiraki T, et al. Targeting a complex transcriptome: The construction of the mouse full-length cDNA encyclopedia. Genome Res 2003;13:1273–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alexandrov NN, Brover VV, Freidin S, et al. Insights into corn genes derived from large-scale cDNA sequencing. Plant Mol Biol 2009;69:179–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shiraki T, Kondo S, Katayama S, et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc Natl Acad Sci U S A 2003;100:15776–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morton T, Petricka J, Corcoran DL, et al. Paired-end analysis of transcription start sites in arabidopsis reveals plant-specific promoter signatures. Plant Cell 2014;26:2746–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Batut P, Gingeras TR. RAMPAGE: promoter activity profiling by paired-end sequencing of 5′-complete cDNAs. Curr Protoc Mol Biol 2013;104:Unit 25B.11–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fickett JW, Hatzigeorgiou AC. Eukaryotic promoter recognition. Genome Res 1997;7:861–78. [DOI] [PubMed] [Google Scholar]
- 21. Pedersen AG, Baldi P, Chauvin Y, et al. The biology of eukaryotic promoter prediction—a review. Comput Chem 1999;23:191–207. [DOI] [PubMed] [Google Scholar]
- 22. Ohler U, Niemann H. Identification and analysis of eukaryotic promoters: recent computational approaches. Trends Genet 2001;17:56–60. [DOI] [PubMed] [Google Scholar]
- 23. Werner T. The state of the art of mammalian promoter recognition. Brief Bioinform 2003;4:22–30. [DOI] [PubMed] [Google Scholar]
- 24. Bajic VB, Tan SL, Suzuki Y, et al. Promoter prediction analysis on the whole human genome. Nat Biotechnol 2004;22:1467–73. [DOI] [PubMed] [Google Scholar]
- 25. Bajic VB, Brent MR, Brown RH, et al. Performance assessment of promoter predictions on ENCODE regions in the EGASP experiment. Genome Biol 2006;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abeel T, Van de Peer Y, Saeys Y. Toward a gold standard for promoter prediction evaluation. Bioinformatics 2009;25:I313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zeng J, Zhu S, Yan H. Towards accurate human promoter recognition: a review of currently used sequence features and classification methods. Brief Bioinform 2009;10:498–508. [DOI] [PubMed] [Google Scholar]
- 28. Singh S, Kaur S, Goel N. A review of computational intelligence methods for eukaryotic promoter prediction. Nucleosides Nucleotides Nucleic Acids 2015;34:449–62. [DOI] [PubMed] [Google Scholar]
- 29. Shahmuradov IA, Umarov RK, Solovyev VV. TSSPlant: a new tool for prediction of plant Pol II promoters. Nucleic Acids Res 2017;45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Umarov RK, Solovyev VV. Recognition of prokaryotic and eukaryotic promoters using convolutional deep learning neural networks. Plos One 2017;12:e0171410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lai H-Y, Zhang Z-Y, Su Z-D, et al. iProEP: a computational predictor for predicting promoter. Mol Ther Nucleic Acids 2019;17:337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oubounyt M, Louadi Z, Tayara H, et al. DeePromoter: robust promoter predictor using deep learning. Front Genet 2019;10:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gordon L, Chervonenkis AY, Gammerman AJ, et al. Sequence alignment kernel for recognition of promoter regions. Bioinformatics 2003;19:1964–71. [DOI] [PubMed] [Google Scholar]
- 34. Huerta AM, Collado-Vides J. Sigma70 promoters in Escherichia coli: specific transcription in dense regions of overlapping promoter-like signals. J Mol Biol 2003;333:261–78. [DOI] [PubMed] [Google Scholar]
- 35. Burden S, Lin YX, Zhang R. Improving promoter prediction improving promoter prediction for the NNPP2.2 algorithm: a case study using Escherichia coli DNA sequences. Bioinformatics 2005;21:601–7. [DOI] [PubMed] [Google Scholar]
- 36. Monteiro MI, de Souto MCP, Goncalves LMG, et al. Machine learning techniques for predicting Bacillus subtilis promoters. In: Setubal JC, Verjovski Almeida S (eds). Advances in Bioinformatics and Computational Biology, Proceedings. Springer, 2005, 77–84.
- 37. Kanhere A, Bansal M. A novel method for prokaryotic promoter prediction based on DNA stability. BMC Bioinformatics 2005;6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. da Silva KP, Monteiro MI, de Souto MCP, et al. In silico prediction of promoter sequences of Bacillus species. In: 2006 IEEE International Joint Conference on Neural Network Proceedings. IEEE, Vol. 1–10, 2006, 2319. [Google Scholar]
- 39. Mann S, Li J, Chen Y-PP. A pHMM-ANN based discriminative approach to promoter identification in prokaryote genomic contexts. Nucleic Acids Res 2007;35:e12–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang HQ, Benham CJ. Promoter prediction and annotation of microbial genomes based on DNA sequence and structural responses to superhelical stress. BMC Bioinformatics 2006;7:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gordon JJ, Towsey MW, Hogan JM, et al. Improved prediction of bacterial transcription start sites. Bioinformatics 2006;22:142–8. [DOI] [PubMed] [Google Scholar]
- 42. Towsey MW, Gordon JJ, Hogan JM. The prediction of bacterial transcription start sites using SVMs. Int J Neural Syst 2006;16:363–70. [DOI] [PubMed] [Google Scholar]
- 43. Li Q-Z, Lin H. The recognition and prediction of Sigma(70) promoters in Escherichia coli K-12. J Theor Biol 2006;242:135–41. [DOI] [PubMed] [Google Scholar]
- 44. Towsey M, Hogan JM, Mathews S, et al. The in silico prediction of promoters in bacterial genomes. In: Ng SK, Mamitsuka H, Wong L (eds). Genome Informatics 2007: Genome Informatics Series, Vol. 19, 2007, 178–89. [PubMed]
- 45. Grech B, Maetschke S, Mathews S, et al. Genome-wide analysis of chlamydiae for promoters that phylogenetically footprint. Res Microbiol 2007;158:685–93. [DOI] [PubMed] [Google Scholar]
- 46. Rangannan V, Bansal M. Identification and annotation of promoter regions in microbial genome sequences on the basis of DNA stability. J Biosci 2007;32:851–62. [DOI] [PubMed] [Google Scholar]
- 47. Towsey M, Timms P, Hogan J, et al. The cross-species prediction of bacterial promoters using a support vector machine. Comput Biol Chem 2008;32:359–66. [DOI] [PubMed] [Google Scholar]
- 48. Askary A, Masoudi-Nejad A, Sharafi R, et al. N4: a precise and highly sensitive promoter predictor using neural network fed by nearest neighbors. Genes Genet Syst 2009;84:425–30. [DOI] [PubMed] [Google Scholar]
- 49. Polat K, Gunes S. A new method to forecast of Escherichia coli promoter gene sequences: integrating feature selection and Fuzzy-AIRS classifier system. Expert Syst Appl 2009;36:57–64. [Google Scholar]
- 50. Rangannan V, Bansal M. Relative stability of DNA as a generic criterion for promoter prediction: whole genome annotation of microbial genomes with varying nucleotide base composition. Mol Biosyst 2009;5:1758–69. [DOI] [PubMed] [Google Scholar]
- 51. Rangannan V, Bansal M. High-quality annotation of promoter regions for 913 bacterial genomes. Bioinformatics 2010;26:3043–50. [DOI] [PubMed] [Google Scholar]
- 52. de Avila e Silva S, Echeverrigaray S, GJL G. BacPP: Bacterial promoter prediction—a tool for accurate sigma-factor specific assignment in enterobacteria. J Theor Biol 2011;287:92–9. [DOI] [PubMed] [Google Scholar]
- 53. Song K. Recognition of prokaryotic promoters based on a novel variable-window Z-curve method. Nucleic Acids Res 2012;40:963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Todt TJ, Wels M, Bongers RS, et al. Genome-wide prediction and validation of Sigma70 promoters in Lactobacillus plantarum WCFS1. Plos One 2012;7:e45097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lin H, Deng E-Z, Ding H, et al. iPro54-PseKNC: a sequence-based predictor for identifying Sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res 2014;42:12961–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. de Avila e Silva S, Forte F, ITS S, et al. DNA duplex stability as discriminative characteristic for Escherichia coli Sigma(54)- and Sigma(28)-dependent promoter sequences. Biologicals 2014;42:22–8. [DOI] [PubMed] [Google Scholar]
- 57. Shahmuradov IA, Razali RM, Bougouffa S, et al. bTSSfinder: a novel tool for the prediction of promoters in cyanobacteria and Escherichia coli. Bioinformatics 2017;33:334–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu B, Yang F, Huang D-S, et al. iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics 2018;34:33–40. [DOI] [PubMed] [Google Scholar]
- 59. He W, Jia C, Duan Y, et al. 70ProPred: a predictor for discovering Sigma70 promoters based on combining multiple features. BMC Syst Biol 2018;12:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang S, Cheng X, Li Y, et al. Image-based promoter prediction: a promoter prediction method based on evolutionarily generated patterns. Sci Rep 2018;8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Coelho RV, de Avila e Silva S, Echeverrigaray S, et al. Bacillus subtilis promoter sequences data set for promoter prediction in Gram-positive bacteria. Data Brief 2018;19:264–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lin H, Liang Z-Y, Tang H, et al. Identifying Sigma70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans Comput Biol Bioinform 2019;16:1316–21. [DOI] [PubMed] [Google Scholar]
- 63. Rahman MS, Aktar U, Jani MR, et al. iPromoter-FSEn: identification of bacterial Sigma(70) promoter sequences using feature subspace based ensemble classifier. Genomics 2019;111:1160–6. [DOI] [PubMed] [Google Scholar]
- 64. Rahman MS, Aktar U, Jani MR, et al. iPro70-FMWin: identifying Sigma70 promoters using multiple windowing and minimal features. Mol Genet Genomics 2019;294:69–84. [DOI] [PubMed] [Google Scholar]
- 65. Xiao X, Xu Z-C, Qiu W-R, et al. iPSW(2L)-PseKNC: a two-layer predictor for identifying promoters and their strength by hybrid features via pseudo K-tuple nucleotide composition. Genomics 2019;111:1785–93. [DOI] [PubMed] [Google Scholar]
- 66. Zhang M, Li F, Marquez-Lago TT, et al. MULTiPly: a novel multi-layer predictor for discovering general and specific types of promoters. Bioinformatics 2019;35:2957–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nguyen Quoc Khanh L, EKY Y, Nagasundaram N, et al. Classifying promoters by interpreting the hidden information of DNA sequences via deep learning and combination of continuous fasttext N-grams. Front Bioeng Biotechnol 2019;7:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu B, Li K. iPromoter-2L2.0: identifying promoters and their types by combining smoothing cutting window algorithm and sequence-based features. Mol Ther Nucleic Acids 2019;18:80–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li F, Chen J, Ge Z, et al. Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework. Brief Bioinform 2021;22:2126–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Amin R, Rahman CR, Ahmed S, et al. iPromoter-BnCNN: a novel branched CNN based predictor for identifying and classifying sigma promoters. Bioinformatics 2020;36:4869–75. [DOI] [PubMed] [Google Scholar]
- 71. Di Salvo M, Pinatel E, Tala A, et al. G4PromFinder: an algorithm for predicting transcription promoters in GC-rich bacterial genomes based on AT-rich elements and G-quadruplex motifs. BMC Bioinformatics 2018;19:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Maetschke S, Towsey M, Hogan J. Bacterial promoter modeling and prediction for E. coli and B. subtilis with Beagle. In: Proceedings of the 2006 Workshop on Intelligent Systems for Bioinformatics. Australian Computer Society Inc, 2006, 9–13.
- 73. Scherf M, Klingenhoff A, Werner T. Highly specific localization of promoter regions in large genomic sequences by PromoterInspector: a novel context analysis approach. J Mol Biol 2000;297:599–606. [DOI] [PubMed] [Google Scholar]
- 74. Ioshikhes IP, Zhang MQ. Large-scale human promoter mapping using CpG islands. Nat Genet 2000;26:61–3. [DOI] [PubMed] [Google Scholar]
- 75. Ohler U, Stemmer G, Harbeck S, et al. Stochastic segment models of eukaryotic promoter regions. Pac Symp Biocomput 2000;380–91. [DOI] [PubMed] [Google Scholar]
- 76. Ohler U, Niemann H, Liao G, et al. Joint modeling of DNA sequence and physical properties to improve eukaryotic promoter recognition. Bioinformatics (Oxford, England) 2001;17(Suppl 1):S199–206. [DOI] [PubMed] [Google Scholar]
- 77. Davuluri RV, Grosse I, Zhang MQ. Computational identification of promoters and first exons in the human genome. Nat Genet 2001;29:412–7. [DOI] [PubMed] [Google Scholar]
- 78. Hannenhalli S, Levy S. Promoter prediction in the human genome. Bioinformatics (Oxford, England) 2001;17(Suppl 1):S90–6. [DOI] [PubMed] [Google Scholar]
- 79. Reese MG. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem 2001;26:51–6. [DOI] [PubMed] [Google Scholar]
- 80. Levitsky VG, Katokhin AV. Computer analysis and recognition of Drosophila melanogaster gene promoters. Mol Biol 2001;35:826–32. [PubMed] [Google Scholar]
- 81. Down TA, Hubbard TJP. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res 2002;12:458–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ponger L, Mouchiroud D. CpGProD: identifying CpG islands associated with transcription start sites in large genomic mammalian sequences. Bioinformatics 2002;18:631–3. [DOI] [PubMed] [Google Scholar]
- 83. Ohler U, Liao G-C, Niemann H, et al. Computational analysis of core promoters in the Drosophila genome. Genome Biol 2002;3:RESEARCH0087–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bajic VB, Seah SH, Chong A, et al. Dragon promoter finder: recognition of vertebrate RNA polymerase II promoters. Bioinformatics 2002;18:198–9. [DOI] [PubMed] [Google Scholar]
- 85. Liu RX, States DJ. Consensus promoter identification in the human genome utilizing expressed gene markers and gene modeling. Genome Res 2002;12:462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Halees AS, Leyfer D, Weng ZP. PromoSer: a large-scale mammalian promoter and transcription start site identification service. Nucleic Acids Res 2003;31:3554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bajic VB, Brusic V. Computational detection of vertebrate RNA polymerase II promoters. In: Adhya S, Garges S (eds). Methods in enzymology 2003;370:237–50. [DOI] [PubMed]
- 88. Solovyev VV, Shahmuradov IA. PromH: promoters identification using orthologous genomic sequences. Nucleic Acids Res 2003;31:3540–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bajic VB, Seah SH. Dragon gene start finder: an advanced system for finding approximate locations of the start of gene transcriptional units. Genome Res 2003;13:1923–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Levitsky VG, Katokhin AV. Recognition of eukaryotic promoters using a genetic algorithm based on iterative discriminant analysis. In Silico Biol 2003;3:81–7. [PubMed] [Google Scholar]
- 91. Kasabov N, Pang S. Transductive support vector machines and applications in bioinformatics for promoter recognition. In: International Conference on Neural Networks and Signal Processing, 2003. Proceedings of the 2003. Nanjing, China: IEEE, 2003, 1–6. [Google Scholar]
- 92. Ma XT, Qian MP, Tang HX. Predicting polymerase II core promoters by cooperating transcription factor binding sites in eukaryotic genes. Acta Biochim Biophys Sin 2004;36:250–8. [DOI] [PubMed] [Google Scholar]
- 93. Gangal R, Sharma P. Human pol II promoter prediction: time series descriptors and machine learning. Nucleic Acids Res 2005;33:1332–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shahmuradov IA, Solovyev VV, Gammerman AJ. Plant promoter prediction with confidence estimation. Nucleic Acids Res 2005;33:1069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Narang V, Sung WK, Mittal A. Computational modeling of oligonucleotide positional densities for human promoter prediction. Artif Intell Med 2005;35:107–19. [DOI] [PubMed] [Google Scholar]
- 96. Xie X, Wu S, Lam K-M, et al. PromoterExplorer: an effective promoter identification method based on the AdaBoost algorithm. Bioinformatics 2006;22:2722–8. [DOI] [PubMed] [Google Scholar]
- 97. Wang J, Hannenhalli S. A mammalian promoter model links cis elements to genetic networks. Biochem Biophys Res Commun 2006;347:166–77. [DOI] [PubMed] [Google Scholar]
- 98. Lardenois A, Chalmel F, Bianchetti L, et al. PromAn: an integrated knowledge-based web server dedicated to promoter analysis. Nucleic Acids Res 2006;34:W578–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sonnenburg S, Zien A, Raetsch G. ARTS: accurate recognition of transcription starts in human. Bioinformatics 2006;22:E472–80. [DOI] [PubMed] [Google Scholar]
- 100. Solovyev V, Kosarev P, Seledsov I, et al. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol 2006;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pandey SP, Krishnamachari A. Computational analysis of plant RNA Pol-II promoters. Biosystems 2006;83:38–50. [DOI] [PubMed] [Google Scholar]
- 102. Ohler U. Identification of core promoter modules in Drosophila and their application in accurate transcription start site prediction. Nucleic Acids Res 2006;34:5943–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhao X, Xuan Z, Zhang MQ. Boosting with stumps for predicting transcription start sites. Genome Biol 2007;8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wang X, Bandyopadhyay S, Xuan Z, et al. Prediction of transcription start sites based on feature selection using AMOSA. Comput Syst Bioinformatics Conf 2007;6:183–93. [PubMed] [Google Scholar]
- 105. Wu S, Xie X, Liew AW-C, et al. Eukaryotic promoter prediction based on relative entropy and positional information. Phys Rev E 2007;75:041908. [DOI] [PubMed] [Google Scholar]
- 106. Goni JR, Perez A, Torrents D, et al. Determining promoter location based on DNA structure first-principles calculations. Genome Biol 2007;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wang J, Ungar LH, Tseng H, et al. MetaProm: a neural network based meta-predictor for alternative human promoter prediction. BMC Genomics 2007;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Anwar F, Baker SM, Jabid T, et al. Pol II promoter prediction using characteristic 4-mer motifs: a machine learning approach. BMC Bioinformatics 2008;9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lu J, Luo L. Prediction for human transcription start site using diversity measure with quadratic discriminant. Bioinformation 2008;2:316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Abeel T, Saeys Y, Bonnet E, et al. Generic eukaryotic core promoter prediction using structural features of DNA. Genome Res 2008;18:310–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Won H-H, Kim M-J, Kim S, et al. EnsemPro: an ensemble approach to predicting transcription start sites in human genomic DNA sequences. Genomics 2008;91:259–66. [DOI] [PubMed] [Google Scholar]
- 112. Akan P, Deloukas P. DNA sequence and structural properties as predictors of human and mouse promoters. Gene 2008;410:165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Yang J-Y, Zhou Y, Yu Z-G, et al. Human Pol II promoter recognition based on primary sequences and free energy of dinucleotides. BMC Bioinformatics 2008;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Troukhan M, Tatarinova T, Bouck J, et al. Genome-wide discovery of cis-elements in promoter sequences using gene expression. OMICS 2009;13:139–51. [DOI] [PubMed] [Google Scholar]
- 115. Wang X, Xuan Z, Zhao X, et al. High-resolution human core-promoter prediction with CoreBoost_HM. Genome Res 2009;19:266–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Mahdi RN, Rouchka EC. RBF-TSS: identification of transcription start site in human using radial basis functions network and oligonucleotide positional frequencies. Plos One 2009;4:e4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zeng J, Zhao X-Y, Cao X-Q, et al. SCS: signal, context, and structure features for genome-wide human promoter recognition. IEEE/ACM Trans Comput Biol Bioinform 2010;7:550–62. [DOI] [PubMed] [Google Scholar]
- 118. Schaefer U, Kodzius R, Kai C, et al. High sensitivity TSS prediction: estimates of locations where TSS cannot occur. Plos One 2010;5:e13934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Azad AKM, Shahid S, Noman N, et al. Prediction of plant promoters based on hexamers and random triplet pair analysis. Algorithms Mol Biol 2011;6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zuo Y-C, Li Q-Z. Identification of TATA and TATA-less promoters in plant genomes by integrating diversity measure, GC-Skew and DNA geometric flexibility. Genomics 2011;97:112–20. [DOI] [PubMed] [Google Scholar]
- 121. Morey C, Mookherjee S, Rajasekaran G, et al. DNA free energy-based promoter prediction and comparative analysis of arabidopsis and rice genomes. Plant Physiol 2011;156:1300–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Fang R, Wu S, Zhang W, et al. A new algorithm of promoter prediction and identification. In: The Fourth International Workshop on Advanced Computational Intelligence. Wuhan, China: IEEE, 2011, 236–41. [Google Scholar]
- 123. Lee T-Y, Chang W-C, Hsu JB-K, et al. GPMiner: an integrated system for mining combinatorial cis-regulatory elements in mammalian gene group. BMC Genomics 2012;13:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Tatarinova T, Kryshchenko A, Triska M, et al. NPEST: a nonparametric method and a database for transcription start site prediction. Quant Biol (Beijing, China) 2013;1:261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Xiong D, Liu R, Xiao F, et al. ProMT: effective human promoter prediction using markov chain model based on DNA structural properties. IEEE Trans Nanobioscience 2014;13:374–83. [DOI] [PubMed] [Google Scholar]
- 126. Yella VR, Kumar A, Bansal M. Identification of putative promoters in 48 eukaryotic genomes on the basis of DNA free energy. Sci Rep 2018;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Qian Y, Zhang Y, Guo B, et al. An improved promoter recognition model using convolutional neural network. In: 2018 IEEE 42nd Annual Computer Software and Applications Conference (COMPSAC). Tokyo, Japan: IEEE, 2018, 471–6. [Google Scholar]
- 128. Xu W, Zhu L, Huang D-S. DCDE: an efficient deep convolutional divergence encoding method for human promoter recognition. IEEE Trans Nanobioscience 2019;18:136–45. [DOI] [PubMed] [Google Scholar]
- 129. Umarov R, Kuwahara H, Li Y, et al. Promoter analysis and prediction in the human genome using sequence-based deep learning models. Bioinformatics 2019;35:2730–7. [DOI] [PubMed] [Google Scholar]
- 130. Zhu Y, Li F, Xiang D, et al. Computational identification of eukaryotic promoters based on cascaded deep capsule neural networks. Brief Bioinform 2021;22:bbaa299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Datta S, Mukhopadhyay S. A composite method based on formal grammar and DNA structural features in detecting human polymerase II promoter region. Plos One 2013;8:e54843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Rani TS, Bhavani SD, Bapi RS. Analysis of E. coli promoter recognition problem in dinucleotide feature space. Bioinformatics 2007;23:582–8. [DOI] [PubMed] [Google Scholar]
- 133. Rani TS, Bapi RS. Analysis of n-gram based promoter recognition methods and application to whole genome promoter prediction. In Silico Biol 2009;9:S1–16. [PubMed] [Google Scholar]
- 134. Lin H, Li Q-Z. Eukaryotic and prokaryotic promoter prediction using hybrid approach. Theory Biosci 2011;130:91–100. [DOI] [PubMed] [Google Scholar]
- 135. Dreos R, Ambrosini G, Perier RC, et al. The eukaryotic promoter database: expansion of EPDnew and new promoter analysis tools. Nucleic Acids Res 2015;43:D92–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Yamashita R, Sugano S, Suzuki Y, et al. DBTSS: DataBase of Transcriptional Start Sites progress report in 2012. Nucleic Acids Res 2012;40:D150–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Santos-Zavaleta A, Salgado H, Gama-Castro S, et al. RegulonDB v 10.5: tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12. Nucleic Acids Res 2019;47:D212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sierro N, Makita Y, de Hoon M, et al. DBTBS: a database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. Nucleic Acids Res 2008;36:D93–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Howe KL, Achuthan P, Allen J, et al. Ensembl 2021. Nucleic Acids Res 2021;49:D884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Smedley D, Haider S, Durinck S, et al. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res 2015;43:W589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006;22:1658–9. [DOI] [PubMed] [Google Scholar]
- 143. Zou Q, Lin G, Jiang X, et al. Sequence clustering in bioinformatics: an empirical study. Brief Bioinform 2020;21:1–10. [DOI] [PubMed] [Google Scholar]
- 144. Zou Q, Wan S, Ju Y, et al. Pretata: predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst Biol 2016;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Annu Rev Biochem 2003;72:449–79. [DOI] [PubMed] [Google Scholar]
- 146. Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A 2002;99:3740–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ross W, Gosink KK, Salomon J, et al. A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase. Science (New York, NY) 1993;262:1407–13. [DOI] [PubMed] [Google Scholar]
- 148. Fyfe JAM, Davies JK. An AT-rich tract containing an integration host factor-binding domain and two UP-like elements enhances transcription from the pilEp(1) promoter of Neisseria gonorrhoeae. J Bacteriol 1998;180:2152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Smale ST. Transcription initiation from TATA-less promoters within eukaryotic protein-coding genes. Biochim Biophys Acta Gene Struct Express 1997;1351:73–88. [DOI] [PubMed] [Google Scholar]
- 150. Basehoar AD, Zanton SJ, Pugh BF. Identification and distinct regulation of yeast TATA box-containing genes. Cell 2004;116:699–709. [DOI] [PubMed] [Google Scholar]
- 151. Kadonaga JT. Perspectives on the RNA polymerase II core promoter. Wiley Interdiscip Rev Dev Biol 2012;1:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Parry TJ, Theisen JWM, Hsu J-Y, et al. The TCT motif, a key component of an RNA polymerase II transcription system for the translational machinery. Genes Dev 2010;24:2013–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Theisen JWM, Lim CY, Kadonaga JT. Three key subregions contribute to the function of the downstream RNA polymerase II core promoter. Mol Cell Biol 2010;30:3471–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Cross SH, Bird AP. CpG islands and genes. Curr Opin Genet Dev 1995;5:309–14. [DOI] [PubMed] [Google Scholar]
- 155. Liu Q, Chen J, Wang Y, et al. DeepTorrent: a deep learning-based approach for predicting DNA N4-methylcytosine sites. Brief Bioinform 2021;22:bbaa124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chen W, Lv H, Nie F, et al. i6mA-Pred: identifying DNA N-6-methyladenine sites in the rice genome. Bioinformatics 2019;35:2796–800. [DOI] [PubMed] [Google Scholar]
- 157. Zhang Y, Xie R, Wang J, et al. Computational analysis and prediction of lysine malonylation sites by exploiting informative features in an integrative machine-learning framework. Brief Bioinform 2019;20:2185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Liu B, Liu F, Wang X, et al. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res 2015;W65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Zhen C, Pei Z, Li F, et al. iFeature: a python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018;34:2499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Zhen C, Pei Z, Fuyi L, et al. iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief Bioinform 2020;21:1047–57. [DOI] [PubMed] [Google Scholar]
- 161. Chen Z, Zhao P, Li C, et al. iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res 2021;49:e60–e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Pedersen AG, Baldi P, Chauvin Y, et al. DNA Structure in Human RNA Polymerase II Promoters. Journal of molecular biology 1998;281:663–73. [DOI] [PubMed] [Google Scholar]
- 163. Kozobay-Avraham L, Hosid S, Bolshoy A. Involvement of DNA curvature in intergenic regions of prokaryotes. Nucleic Acids Res 2006;34:2316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yella VR, Bansal M. DNA structural features of eukaryotic TATA-containing and TATA-less promoters. Febs Open Bio 2017;7:324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Yella VR, Bansal M. In silico identification of eukaryotic promoters. In: Singh V, Dhar PK (eds). Systems and Synthetic Biology. Dordrecht: Springer Netherlands, 2015, 63–75. [Google Scholar]
- 166. Tang Q, Nie F, Kang J, et al. ncPro-ML: an integrated computational tool for identifying non-coding RNA promoters in multiple species. Comput Struct Biotechnol J 2020;18:2445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Tang Q, Nie F, Kang J, et al. mRNALocater: enhance the prediction accuracy of eukaryotic mRNA subcellular localization by using model fusion strategy. Mol Ther 2021;29:2617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Freund Y, Schapire RE. Experiments with a new boosting algorithm. In: Proceedings of the Thirteenth International Conference (ICML '96). Morgan Kaufmann Publishers Inc., 1996, 148–56.
- 169. Friedman JH. Greedy function approximation: a gradient boosting machine. Ann Stat 2001;29:1189–232. [Google Scholar]
- 170. Chen T, Guestrin C, Assoc CM. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining, 2016;785–94. [Google Scholar]
- 171. Ke G, Meng Q, Finley T, et al. LightGBM: a highly efficient gradient boosting decision tree. Advances in Neural Information Processing Systems 30 2017;30:3146–54. [Google Scholar]
- 172. Wu S, Roberts K, Datta S, et al. Deep learning in clinical natural language processing: a methodical review. J Am Med Inform Assoc 2020;27:457–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Popova M, Isayev O, Tropsha A. Deep reinforcement learning for de novo drug design. Sci Adv 2018;4:eaap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Krizhevsky A, Sutskever I, Hinton GE. ImageNet classification with deep convolutional neural networks. Commun ACM 2017;60:84–90. [Google Scholar]
- 175. Schuster M, Paliwal KK. Bidirectional recurrent neural networks. IEEE Trans Signal Process 1997;45:2673–81. [Google Scholar]
- 176. Li F, Li C, Marquez-Lago TT, et al. Quokka: a comprehensive tool for rapid and accurate prediction of kinase family-specific phosphorylation sites in the human proteome. Bioinformatics 2018;34:4223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Li F, Chen J, Leier A, et al. DeepCleave: a deep learning predictor for caspase and matrix metalloprotease substrates and cleavage sites. Bioinformatics 2020;36:1057–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Yang S, Li H, He H, et al. Critical assessment and performance improvement of plant-pathogen protein-protein interaction prediction methods. Brief Bioinform 2019;20:274–87. [DOI] [PubMed] [Google Scholar]
- 179. Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta 1975;405:442–51. [DOI] [PubMed] [Google Scholar]
- 180. Bunkhumpornpat C, Sinapiromsaran K, Lursinsap C. Safe-level-SMOTE: safe-level-synthetic minority over-sampling technique for handling the class imbalanced problem. In: 13th Pacific-Asia Conference on Knowledge Discovery and Data Mining. Bangkok, Thailand: IEEE, 2009, 475–82. [Google Scholar]
- 181. Jia C, Zhang M, Fan C, et al. Formator: predicting lysine formylation sites based on the most distant undersampling and safe-level synthetic minority oversampling. IEEE/ACM Trans Comput Biol Bioinform 2021;18:1937–45. [DOI] [PubMed] [Google Scholar]
- 182. Buchka S, Hapfelmeier A, Gardner PP, et al. On the optimistic performance evaluation of newly introduced bioinformatic methods. Genome Biol 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Rangannan V, Bansal M. PromBase: a web resource for various genomic features and predicted promoters in prokaryotic genomes. BMC Res Notes 2011;4:257–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Perier RC, Praz V, Junier T, et al. The eukaryotic promoter database (EPD). Nucleic Acids Res 2000;28:302–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Shahmuradov IA, Gammerman AJ, Hancock JM, et al. PlantProm: a database of plant promoter sequences. Nucleic Acids Res 2003;31:114–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the 58 independent test datasets used for benchmarking various predictors in this study are available at https://github.com/chenli-bioinfo/promoter/.