

Abstract
Abnormal protein–membrane attachment is involved in deregulated cellular pathways and in disease. Therefore, the possibility to modulate protein–membrane interactions represents a new promising therapeutic strategy for peripheral membrane proteins that have been considered so far undruggable. A major obstacle in this drug design strategy is that the membrane-binding domains of peripheral membrane proteins are usually unknown. The development of fast and efficient algorithms predicting the protein–membrane interface would shed light into the accessibility of membrane–protein interfaces by drug-like molecules. Herein, we describe an ensemble machine learning methodology and algorithm for predicting membrane-penetrating amino acids. We utilize available experimental data from the literature for training 21 machine learning classifiers and meta-classifiers. Evaluation of the best ensemble classifier model accuracy yields a macro-averaged F1 score = 0.92 and a Matthews correlation coefficient = 0.84 for predicting correctly membrane-penetrating amino acids on unknown proteins of a validation set. The python code for predicting protein–membrane interfaces of peripheral membrane proteins is available at https://github.com/zoecournia/DREAMM.
Keywords: protein–membrane interface, machine learning, membrane-penetrating amino acids, peripheral membrane proteins, protein–membrane regions
Introduction
Membrane proteins are topologically divided into transmembrane proteins that are permanently incorporated to the interior of the membrane, peripheral membrane proteins that associate non-covalently with the surface of the membrane and lipid-anchored proteins that attach to the membrane with a covalent bond [1]. Peripheral membrane proteins are essential in cellular processes, such as transporting substances across the cell membrane, activating proteins and enzymes and regulating signal transduction, and other functions [1, 2]. Abnormal protein–membrane attachment-due to membrane-binding domain mutations and peripheral membrane protein overactivation or underactivation-is involved in deregulated cellular pathways and in disease [1, 3–8]. Hence, the possibility to modulate protein–membrane interactions represents a promising therapeutic strategy for many disease indications and in particular for targeting membrane proteins that have been considered undruggable such as the membrane-anchored KRAS protein, which is implicated in over 30% of cancer types [9, 10]; α-synuclein, which is a main pathological hallmark of Parkinson’s disease [11, 12]; and lipid kinases such as PI3Kα, which is the most frequently mutated kinase and present in a variety of tumors [13] with one of its hotspot mutations, H1047R, acting on altering the protein’s association with the cell membrane [14–17].
The feasibility of targeting protein–membrane interfaces is supported by the fact that peripheral membrane proteins contain a membrane-binding domain with cavities that could be potentially targeted by small molecules [18, 19]. The literature reports the feasibility of targeting the protein–membrane interface, indicating that therapeutic targets binding transiently to the membrane can be targeted with small molecules and that inhibitors of protein–membrane interactions may be identified [18, 20–25]. However, these examples are only limited compared to the overall drug design efforts of the community, indicating that the accessibility of protein–membrane interfaces by small molecules has been so far unexplored possibly due to the complexity of the interface, the limited protein–membrane structural information and the absence of tools and workflows to automate the drug design process at the protein–membrane interface. Moreover, protein–membrane interaction sites of peripheral membrane proteins are usually undiscovered; hence, the first step into modulating the protein–membrane interface is their identification.
Several efforts towards the design of tools that detect protein–membrane regions, domains, and lipid-binding sites have appeared [26–29]; however, these are mainly applied directly to 1D protein sequences without considering the protein structural information, and in many cases, the web links are outdated [26–28]. To our knowledge, only two methodologies, which predict these interaction sites from the 3D protein structure, are currently publically available: the Positioning of Proteins in Membrane (PPM) [30, 31] and the Membrane Optimal Docking Area (MODA) [32]. PPM combines an anisotropic solvent representation of the lipid bilayer, an all atom representation of a solute, and a universal solvation model, calculating rotational and translational positions of transmembrane and peripheral membrane proteins in membranes [30, 31]. MODA is based on the protein–protein interface predictor PIER [33], which builds a set of evenly distributed points at 5 Å from one another and from the protein surface, defining each patch as the set of all protein surface atoms. Then, it calculates a score based on atom solvent-accessible surface area and atom type-specific weights and transfers the patch membrane propensity scores to surface amino acids, thereby predicting which amino acids contact the cell membranes.
Herein, we present an automated prediction algorithm using ensemble machine learning, which identifies membrane-binding interfaces with high accuracy [macro-averaged F1 score = 0.92 and Matthews correlation coefficient (MCC) = 0.84], taking as input the 3D peripheral membrane protein coordinates, and demonstrating improved accuracy compared to existing methods.
Methods
Data preparation
To construct the dataset, we used 54 peripheral membrane proteins with known 3D structures and experimentally known membrane-penetrating amino acids, retrieved from extensive literature search. For the dataset generation, protein structures were prepared by deleting unwanted chains and non-protein atoms, adding missing side chain atoms and converting non-standard amino acids to their standard equivalents using the software High-Throughput Molecular Dynamics (HTMD) [34]. In case of NMR-resolved structures, the first model of the NMR ensemble was kept. Then, the dataset was split into a training set (~85% of the dataset, Supplementary Table S1 available online at http://bib.oxfordjournals.org/) and a validation set (~15% of the dataset, Supplementary Table S2 available online at http://bib.oxfordjournals.org/. Finally, a training set of 12.805 amino acids and a validation set of 2.177 amino acids were assembled. These samples were labeled in two classes, the membrane-penetrating and the non-penetrating amino acids, leading to a highly imbalanced binary classification problem (supervised learning), where the membrane-penetrating amino acids comprise ~1.3% of the total samples in the training set. Before balancing the two classes, feature extraction, and feature and data selection were performed utilizing a variety of techniques. Data selection was performed in both the training and validation sets. For more details, refer to the ``Methods'' section in the Supporting Information.
Using tree-based methods, the most important features were determined such as hydrophobicity and solvent exposure, but also the evolutionary conservation, secondary structure (coil loop or not), flexibility (squared fluctuations calculated with the Gaussian network model, probably associated with the flexible coil loops), dihedral angles (which again might be associated with the secondary structure) and Transferable Atom Equivalent (TAE) descriptors [35] (which are molecular properties related to electron density distribution). These features are critical for the accuracy of the results (more information in the Supporting Information, in Supplementary Figure S1 available online at http://bib.oxfordjournals.org/ and in Supplementary Table S3 available online at http://bib.oxfordjournals.org/).
Next, the initial training set was split in five stratified randomized folds, in order to later perform hyper-parameter optimization using 5-fold cross-validation (see below), and class balancing was performed in each fold with different techniques. The reason for applying class balancing in these five folds separately is to prevent information leak in the validation fold during the 5-fold cross-validation procedure. To balance the two classes, several over- and under-sampling techniques were utilized leading to six different balanced training sets (see Supporting Information for more information and Supplementary Figure S2 available online at http://bib.oxfordjournals.org/).
Furthermore, a test set of 11 peripheral membrane proteins with known 3D structures and experimentally known membrane-penetrating regions (not amino acids) was assembled. As no specific amino acids were experimentally tested, these proteins were assessed qualitatively.
Finally, to ensure that the predictions are unbiased, the percentage of identical amino acids of the pairwise sequence alignment was calculated for all sequence pairs of the dataset. This revealed high percentage identity values (more than 40%) for proteins in the training set, but not in the validation or test sets, ensuring that the predictions in the validation and test sets are unbiased (more information in the Supporting Information and Supplementary Tables S4 and S5 available online at http://bib.oxfordjournals.org/).
Ensemble machine learning methodology
For each one of the six training sets, 21 machine learning classifiers were trained: 19 from the scikit-learn Python package [36], the LightGBM classifier [37] and the XGBoost classifier [38]. The hyperparameters of each classifier were optimized to discover the best hyperparameters that separate the two classes (Supplementary Table S6 available online at http://bib.oxfordjournals.org/) [39, 40]. Specifically, for every training set and every classifier, the randomized search cross-validation technique was performed using 5-fold in a wide range of hyperparameter values, training hundreds of thousands models (Supplementary Table S6 available online at http://bib.oxfordjournals.org/). Subsequently, iteratively exhaustive searches were performed (grid search cross-validation with 5-fold) in a small range of hyperparameter values in the vicinity of the best hyperparameter space determined from the randomized search cross-validation, which led to a set of optimal hyperparameters.
To assess the performance of the classifier models for both the randomized and grid search cross-validation procedures, the macro-averaged harmonic mean of the precision and recall, F1 score, was calculated. Recall expresses the amount of correctly predicted true positives (Equation 1), while precision expresses the predicted positives that are actually true (Equation 2). The general formula of F score is derived based on a positive real variable β, where β determines the importance of recall over precision (Equation 3). When β = 1 (F1 score), recall and precision are weighted equally (Equation 4), when β < 1 more weight is given in precision and when β > 1 recall is favored.
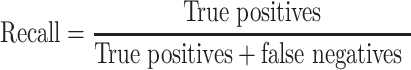 |
(1) |
 |
(2) |
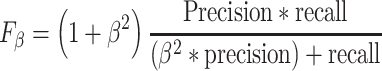 |
(3) |
 |
(4) |
For more information about the hyperparameter tuning process and the performance metrics, please refer to the Supporting Information.
The resulting predictions of the aforementioned classifiers were input to meta-classifiers (ensemble classifier). The voting classifier, which classifies a sample based on the majority voting of the first-level classifiers [41], and the stacking classifier, which trains a classifier based on the output of the first-level classifiers in order to compute the final prediction [42], were employed using the Python library mlxtend [43]. In both meta-classifiers, all possible combinations of the first-level classifiers were examined to discover the best classifier combination. Every classifier combination was tested using the validation set with known protein–membrane amino acids (Supplementary Table S2 available online at http://bib.oxfordjournals.org/) to find the combination with the best performance. Subsequently, considering that not every amino acid in the protein sequence of the datasets was experimentally tested, resulting in membrane-penetrating amino acids marked as non-penetrating, the best models were manually inspected in order to assess their false positives, and the final model was chosen based on F2 score (see Results). A schematic representation of the above procedure is illustrated in Figure 1.
Figure 1.
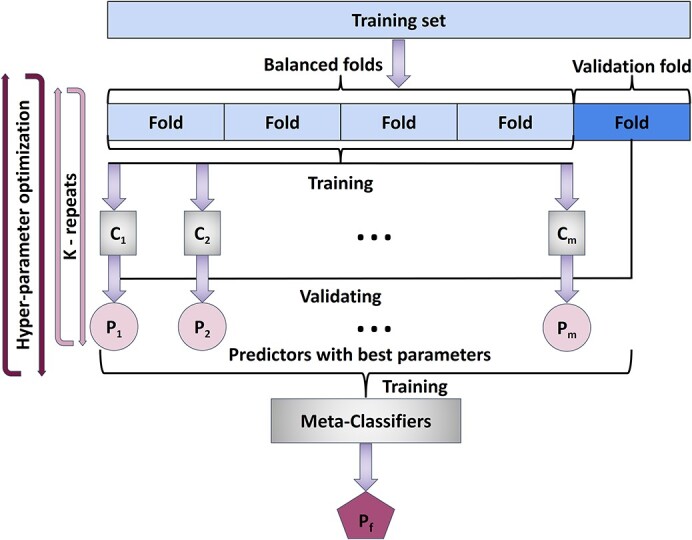
For each of the six datasets, we optimized the hyperparameter space of 21 classifiers using 5-fold cross-validation on the training set. The predictions of the models with the best F1 score from these classifiers were provided as input to meta-classifiers. Given the F2 score on the validation set, the best meta-classifier was kept as the final predictor.
Results
The first-level classifiers providing the most accurate classification were those trained in the initial dataset using weights (See ``Class imbalance problem'' section available online at http://bib.oxfordjournals.org/). For these classifiers and initial dataset, several ensemble classifier models exhibited better performance than the first-level classifiers in terms of F1 score, precision/recall area under the curve (PR AUC), MCC and other scoring metrics. The receiver operating characteristic area under the curve (ROC AUC), which is regularly used in the literature was not considered as it may be misleading for highly imbalanced classification problems such as our case [44, 45]. Then, results from the top ensemble classifier models were subject to manual inspection and the best was selected according to the F2 score to emphasize on recall. Although, seemingly, it is natural to prioritize on precision because in our case false positives are more critical than false negatives, manual inspection of the false positive results of the top meta-classifiers indicated that these could actually be true positive membrane-penetrating amino acids as in many cases they are adjacent to amino acids that are membrane-penetrating or aligned with them in a putative membrane plane (see validation set predictions in Figure 2). Finally, the best performing ensemble classifier was the voting classifier for a combination consisting of five classifiers: the linear discriminant analysis, the logistic regression, the linear support vector classifier, the decision tree classifier and the light gradient boosting machine. Various scoring metrics of the 21 first-level classifiers and the ensemble classifier model for the initial dataset using weights are reported in Supplementary Table S7 available online at http://bib.oxfordjournals.org/.
Figure 2.

Proteins of the validation set. The experimental membrane-penetrating amino acids predicted from the ensemble classifier model are depicted in red, the experimental membrane-penetrating amino acids not predicted from the classifier model are depicted in green and the amino acids predicted from the classifier model that have not been experimentally verified are depicted in purple. The putative membrane plane is depicted as a red dotted line.
The validation set predictions can be viewed in Figure 2 and Supplementary Table S8 available online at http://bib.oxfordjournals.org/, where ~2/3 of the false-positive amino acids are in fact correct predictions as they are located in the protein–membrane interface adjacent to true positives or on adjacent loops defining the membrane plane. For example, in retinoid isomerohydrolase, amino acid F262 lies within 4 Å from the experimentally confirmed membrane-penetrating amino acids; for the glycolipid transfer protein, amino acids I143 and Y153 are next to and aligned with W142. In other examples, i.e. the cholesterol-regulated START protein 4, amino acid M196 although located in a different loop, it is aligned with L124; for the phosphatidylinositol transfer protein beta isoform, M74 resides in a different loop but is aligned with the experimentally confirmed membrane-penetrating amino acids W202 and W203; for the PH domain of the ceramide transfer protein, I37 and W40 are next to W33 and Y36, and F81 is aligned with them in an adjacent loop. Considering that these predicted membrane-penetrating amino acids are in fact located in the plane of the protein–membrane interface, they can be considered as true positives and the macro-averaged F1 score increases from 0.86 to 0.92 and MCC from 0.71 to 0.84.
To compare the protein-membrane interface predictions of our ensemble classifier model with those of other computational tools, we applied the ensemble model to the proteins of the validation set, this time retaining all amino acid types (without data selection). Results showed that several non-hydrophobic amino acids were predicted as membrane penetrating both in the actual protein-membrane interface but also in the solvent exposed regions of the protein (false positives). To restrict such false positive predictions, we incorporated in the algorithm the condition that non-hydrophobic amino acids with center of mass distance of 14 Å from at least one of the predicted hydrophobic amino acids are kept and the rest of the non-hydrophobic amino acid predictions are discarded.
Finally, we compared the protein-membrane interface prediction of our final model with those of two computational tools that predict protein–membrane interfaces from 3D structures: the PPM web-server [46], which also predicts the orientation of proteins in membranes and the MODA web-server [32], using the validation set without performing data selection (Supplementary Table S9 available online at http://bib.oxfordjournals.org/). Specifically, for the retinoid isomerohydrolase homodimer, PPM falsely predicted the membrane orientation (probably affected by missing chains in the PDB structure) and placed the protein in an orientation in which only one monomer is in contact with the membrane instead of the dimer, while our ensemble classifier model and MODA correctly predicted the protein–membrane regions in both chains, but with false-positive predictions for MODA (Supplementary Figure S3 available online at http://bib.oxfordjournals.org/). For the VSTx1 toxin, all three tools predicted the protein–membrane interface. Our ensemble classifier model falsely predicted as membrane-penetrating amino acids near the N- and C-termini and MODA falsely predicted the beta sheet V20–S23, which lies on the opposite side of the protein–membrane interface and the C-terminal region to be membrane-penetrating (Supplementary Figure S4 available online at http://bib.oxfordjournals.org/). For cytotoxin 2, all tools identify the protein-membrane interface with false positives for our model and MODA in the 41-46 region (Supplementary Figure S5 available online at http://bib.oxfordjournals.org/). For sphingomyelinase C, all tools recognized the experimentally verified membrane-penetrating amino acids W284 and F285, with PPM and MODA recognizing amino acids in distant loops that are aligned with the experimentally verified protein–membrane region suggesting a multiregional interaction with the membrane, in accordance with the proposed membrane binding model (Supplementary Figure S6 available online at http://bib.oxfordjournals.org/). For the glycolipid transfer protein, our model and MODA provided similar results correctly identifying the membrane-penetrating α-helix G141–Y153, with MODA falsely predicting the C-terminus and our model amino acid Y81 to be membrane-penetrating amino acids. PPM also suggested the insertion of the membrane-penetrating α-helix G141–Y153 with the addition of the P40-P44 region (Supplementary Figure S7 available online at http://bib.oxfordjournals.org/). For the cholesterol-regulated START protein 4, all tools predicted correctly the experimentally verified amino acid L124. Our ensemble classifier model and PPM additionally predicted the 196-200 region to be membrane penetrating, MODA falsely predicted the C-terminus. Our model also predicted amino acid W91 as membrane penetrating (Supplementary Figure S8 available online at http://bib.oxfordjournals.org/). Finally, for the PH domain of the ceramide transfer protein and the phosphatidylinositol transfer protein beta isoform, the outcome was similar and correct for all tools (Supplementary Figures S9 and S10 available online at http://bib.oxfordjournals.org/).
The performance of the ensemble classifier model was tested with additional protein use cases with known membrane-penetrating regions (test set, Figure 3), and the results were compared with PPM and MODA (Supplementary Table S10 available online at http://bib.oxfordjournals.org/). For the cases of cholesterol oxidase, cytochrome P450 3A4, monoglyceride lipase MGLL, L-amino acid deaminase and intestinal fatty acid binding protein, all tools correctly identified the protein–membrane regions (Supplementary Figures S11, S12, S14, S19 and S20 available online at http://bib.oxfordjournals.org/). For 9-cis-epoxycarotenoid dioxygenase 1, chloroplastic, all tools predicted the protein–membrane regions. Our model predicted the insertion of one of the two parallel amphipathic helices, instead of both (Supplementary Figure S13 available online at http://bib.oxfordjournals.org/). For the dihydroorotate dehydrogenase, all tools predicted the protein–membrane regions, with our model additionally identifying amino acid W362 and MODA falsely identifying the region 245-247 (Supplementary Figure S15 available online at http://bib.oxfordjournals.org/). For phosphatase PTEN, our model successfully identified the protein–membrane region 263-269 of the C2 domain and the L42 amino acid of phosphatase domain in the same membrane plane. MODA also identified the same phosphatase region; however, it falsely identified the opposite side of the C2 domain as a protein–membrane region. PPM also falsely identified the opposite side of the C2 domain suggesting an orientation, which is opposite to the proposed membrane orientation (Supplementary Figure S16 available online at http://bib.oxfordjournals.org/). For (S)-mandelate dehydrogenase, the protein–membrane region was correctly identified by all tools, but our model and MODA also identified amino acids 53–56 to be membrane-penetrating (Supplementary Figure S17 available online at http://bib.oxfordjournals.org/); these amino acids lie actually at the protein–protein interaction interface in the homotetramer of (S)-mandelate dehydrogenase, and are not misclassified if we perform the predictions in the homotetramer biological assembly (Supplementary Figure S18 available online at http://bib.oxfordjournals.org/). For phosphatidylinositol 4,5-bisphosphate 3-kinase alpha (PI3Kα), all tools predicted amino acids 232–233 as a protein–membrane interface, which is not in agreement with experimental results, but belong in fact to the region, where PI3Kα binds to RAS (Ras binding domain). Additionally, our model and MODA successfully identified the p110α 863-872 and the iSH2 512-525 regions, but falsely identified the 498-508 region, which links the C2 domain with the helical domain (Supplementary Figure S21 available online at http://bib.oxfordjournals.org/). The membrane orientation resulting from PPM is different from the proposed one [14]. Finally, for the phosphatidylcholine transfer protein, all three tools provided the same results identifying the experimentally proven membrane-interacting region 184-193 and an adjacent loop, while MODA additionally predicted loop 147-148 to be membrane binding, which is in the same plane with the other two membrane binding regions (Supplementary Figure S22 available online at http://bib.oxfordjournals.org/).
Figure 3.

The predictions of the ensemble classifier model for proteins with known protein–membrane interface regions (test set). The membrane-penetrating amino acids predicted from our model are depicted in red and the experimental membrane-penetrating regions are denoted with red circles. The red dotted lines represent the protein-membrane plane as proposed in the literature.
Furthermore, the ensemble classifier model was applied to the full structure of prothrombin (PDB: 5EDM [47]) and the results of identified membrane–protein interfaces were compared with those of PPM and MODA (Figure 4). All tools predicted the GLA domain 3-5 region as a membrane contacting region, which is obvious for our model because the GLA domain of prothrombin was included in the training set. Additionally, our model predicted amino acids Y93, W398, and V458 and MODA predicted amino acids Y93, Y377, R379 and R484, suggesting an orientation parallel to a putative membrane plane with a different orientation suggested by PPM. Y93 is a key prothrombin amino acid, which is essential for stabilizing the closed form and shields the active site pocket of the protease domain [48]. In the prothrombin closed form (PDB: 6BJR [48]), Y93 inserts its aromatic side chain into the active site of the protease domain engaging W547 (W533 of 5EDM) and forms pi-pi interactions (Supplementary Figure S23 available online at http://bib.oxfordjournals.org/). The results provided by our model and MODA suggest that Y93 may penetrate into the membrane; we thus hypothesize that when prothrombin engages the membrane, the open form is favored with Y93 anchoring the membrane and opening the active site.
Figure 4.

Comparison of the predictions provided by our model, PPM and MODA for the open form of the prothrombin protein. Our model and MODA suggest an orientation parallel to a putative membrane plane, where Y93 inserts in the membrane which in turn opens the active site of the protease domain (Supplementary Figure S23 available online at http://bib.oxfordjournals.org/). In our model the membrane-penetrating predictions are depicted with red, in PPM the non-protein residues are depicted in CPK representation, and in MODA the membrane-penetrating predictions are depicted with red secondary structure.
Finally, the ensemble classifier model was also assessed for the prediction of the protein–membrane regions of nine transmembrane enzymes described in Ref. [49], which include a soluble domain performing extracellular catalysis. In agreement with experimental results, our model predicted amino acids that lie in the hydrophobic lipid bilayer core, along with membrane-interacting extracellular amino acids (93% precision) (Supplementary Figure S24 available online at http://bib.oxfordjournals.org/).
Discussion and Conclusions
Drugging the protein-membrane interface is relatively underexplored due to the complexity of the interface and the lack of a suitable workflows and simulation technology capable of implementing such a drug design strategy. Furthermore, protein-membrane interaction regions of peripheral membrane proteins are commonly unknown, and only a few rational methodologies exist that predict these regions from the 3D protein structure. To assist in protein–membrane interface recognition, a novel ensemble machine learning classifier model is trained using experimental data retrieved from extensive literature search.
The ensemble classifier model results predict correctly the membrane-penetrating amino acids in the validation set, delivering a macro-averaged F1 score = 0.92 and an MCC = 0.84. Additionally, using an independent test set with experimentally known protein–membrane regions, our model correctly identified membrane-penetrating amino acids in these regions with only five false-positive predictions out of 51 (91% precision). In addition, comparative results demonstrated that our model performed similarly, and in some cases better, than the only two available web-servers that predict protein–membrane interaction sites from the 3D protein structure: PPM and MODA. Moreover, our model successfully predicted the membrane-interacting amino acids as well as amino acids that lie in the hydrophobic core of the lipid bilayer in transmembrane proteins containing a soluble catalytic domain.
The features used in this study have a significant impact on the performance of the ensemble classifier model. The fact that apart from hydrophobicity and solvent exposure, other features—such as evolutionary conservation, secondary structure, flexibility, dihedral angles and TAE descriptors—are important in our model decision-making process, offers novel physicochemical insights for the mechanisms with which peripheral membrane proteins contact and attach to the membrane.
During the development of computational tools, several obstacles may emerge. Here, the first obstacle was the low number of peripheral membrane proteins with experimentally known membrane-penetrating amino acids described in the literature. The second and more crucial constraint was the small number of amino acids that were tested experimentally resulting in membrane-penetrating amino acids being marked as non-penetrating, which in turn resulted in misinforming the classifiers during the training process and rendering the selection of the best ensemble classifier strenuous. Moreover, based on the fact that membrane-penetrating amino acids that are not experimentally confirmed are labeled as non-penetrating, the performance metrics (e.g. F scores) do not reflect the actual accuracy of the ensemble classifier, which is higher, and therefore, a direct numerical comparison of our model, PPM and MODA results is not meaningful.
Manual inspection of false-positive results revealed that several amino acids were located near the N- or C-termini, or near missing loops, probably because the area is more solvent exposed. Intriguingly, other amino acids falsely predicted as membrane-penetrating are found to be implicated in protein–protein interactions. For example, in the case of (S)-mandelate dehydrogenase homodimer our classifier model predicts as membrane-penetrating amino acids the amino acids that are located at the homodimer interface forming the tetramer of the protein; when applied to the homotetramer form of the protein all our predictions are correct. Hence, for predicting the protein-membrane interface it is advisable to use the complex protein structure if it is available. The assumption that protein–membrane interactions are similar to protein–protein interactions was also deduced by Kufareva et al. [32], who adapted their protein–protein interaction interface prediction PIER algorithm [33] for MODA.
Also, it should be noted that the predictions depend on structural information; therefore, in case a conformational change is necessary to place membrane-penetrating amino acids towards the membrane, or if the protein is intrinsically disordered, the ensemble classifier model would not be able to predict them. Furthermore, it should be noted that neither our model, nor PPM or MODA are suitable tools for classifying if a protein is a peripheral membrane protein or not. A sequence-/evolutionary-based deep learning classifier would be more appropriate for this purpose [50]. Also, with the recent advancements in protein structure predictions, i.e. AlphaFold2 [51] and RoseTTAFold [52], the structure of unresolved proteins can be predicted with high accuracy; however, in many cases, these models fail to fold the N- or C-terminus or various protein segments. It is thus recommended to remove these regions, i.e. amino acids with confidence score (pLDDT) less than 70, before applying our model, because these unfolded regions are going to affect the prediction accuracy.
Our tool does not currently predict the protein-membrane orientation; henceforth we plan to devise methods that orient peripheral membrane proteins in the membrane according to the predicted membrane-penetrating amino acids. Such an approach could involve the following steps: (i) placing the protein in a model membrane in all possible orientations based on the ensemble classifier predictions, (ii) measuring the protein–membrane interface energy with an energy function specific for this purpose, and (iii) retaining the protein–membrane orientation with the lowest energy. After the appropriate method to define the membrane–protein orientation is devised, designing a web-database with our model predictions, similar to PPM and OPM [46], could also be envisaged. Moreover, modifying the labeling system by partitioning the structures with a well-defined plane into membrane penetrating and not penetrating parts, or by labeling any amino acid within a specific distance from the experimentally known membrane penetrating amino acids as a true label, warrants further investigation. Such a consideration would improve the class imbalance problem, although the definition of a plane and distance may be subjective to generate. Also, incorporating the model for hydrophobic protrusions in our model might improve the predictions, eliminating false positives [53].
Membrane-penetrating amino acids might exert significant allosteric control in enzymes [18, 20–25]. For example, lipid binding is a mechanism that activates PI3Kα, and a hotspot cancer mutation on this enzyme acts by altering the interaction between PI3Kα and the membrane by allosterically enhancing its activity [17]. We strongly believe that allosteric binding pockets at the protein-membrane interface could modulate the activity of protein function representing a novel therapeutic strategy by disrupting the protein-membrane interactions [17, 54].
Key Points
A dataset of peripheral membrane proteins with experimentally known membrane-penetrating amino acids was assembled.
An ensemble machine learning classifier model was trained utilizing thermodynamic, topographic and property-based features.
The ensemble machine learning classifier model yielded a macro-averaged F1 score = 0.92 and an MCC = 0.84 in identifying membrane-penetrating amino acids.
The python code is publically available at https://github.com/zoecournia/DREAMM.
Supplementary Material
Alexios Chatzigoulas is a PhD Student at the Biomedical Research Foundation of the Academy of Athens and in the Department of Informatics and Telecommunications, National and Kapodistrian University of Athens, Athens, Greece.
Zoe Cournia is a Senior Researcher at the Biomedical Research Foundation of the Academy of Athens and an Instructor at the Department of Informatics and Telecommunications, National and Kapodistrian University of Athens, Athens, Greece.
Contributor Information
Alexios Chatzigoulas, Biomedical Research Foundation, Academy of Athens, 4 Soranou Ephessiou, 11527 Athens, Greece; Department of Informatics and Telecommunications, National and Kapodistrian University of Athens, 15784 Athens, Greece.
Zoe Cournia, Biomedical Research Foundation, Academy of Athens, 4 Soranou Ephessiou, 11527 Athens, Greece.
Funding
State Scholarships Foundation (ΙΚΥ) (MIS-5000432 to A.C.); the Hellenic Foundation for Research and Innovation (H.F.R.I.) (1780 to Z.C.); and the high-performance computing resources provided by the National Infrastructures for Research and Technology S.A. (GRNET S.A.) (pr008033_gpu-Mem-Surf, pr008033_thin-Mem-Surf, and pr009022_gpu-Mem-Surf).
References
- 1. Boes DM, Godoy-Hernandez A, McMillan DGG. Peripheral membrane proteins: promising therapeutic targets across domains of life. Membranes 2021;11:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Monje-Galvan V, Klauda JB. Peripheral membrane proteins: tying the knot between experiment and computation. Biochim Biophys Acta Biomembr 2016;1858:1584–93. [DOI] [PubMed] [Google Scholar]
- 3. Segers K, Dahlbäck B, Nicolaes GAF. Coagulation factor V and thrombophilia: background and mechanisms. Thromb Haemost 2007;98:530–42. [PubMed] [Google Scholar]
- 4. Lashuel HA, Overk CR, Oueslati A, et al. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 2013;14:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mirsaeidi M, Gidfar S, Vu A, et al. Annexins family: insights into their functions and potential role in pathogenesis of sarcoidosis. J Transl Med 2016;14:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci 2016;129:1287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Costeira-Paulo J, Gault J, Popova G, et al. Lipids shape the electron acceptor-binding site of the peripheral membrane protein dihydroorotate dehydrogenase. Cell Chem Biol 2018;25:309–17.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mirza FJ, Zahid S. The role of Synapsins in neurological disorders. Neurosci Bull 2018;34:349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cox AD, Fesik SW, Kimmelman AC, et al. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov 2014;13:828–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kessler D, Gmachl M, Mantoulidis A, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A 2019;116:15823–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Oliveira GAP, Silva JL. Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson's disease. Commun Biol 2019;2:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hijaz BA, Volpicelli-Daley LA. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol Neurodegener 2020;15:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang J, Nie J, Ma X, et al. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer 2019;18:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gabelli SB, Huang CH, Mandelker D, et al. Structural effects of oncogenic PI3Kα mutations. Curr Top Microbiol Immunol 2010;347:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gkeka P, Evangelidis T, Pavlaki M, et al. Investigating the structure and dynamics of the PIK3CA wild-type and H1047R oncogenic mutant. PLoS Comput Biol 2014;10:e1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gkeka P, Papafotika A, Christoforidis S, et al. Exploring a non-ATP pocket for potential allosteric modulation of PI3Kα. J Phys Chem B 2015;119:1002–16. [DOI] [PubMed] [Google Scholar]
- 17. Cournia Z, Chatzigoulas A. Allostery in membrane proteins. Curr Opin Struct Biol 2020;62:197–204. [DOI] [PubMed] [Google Scholar]
- 18. Segers K, Sperandio O, Sack M, et al. Design of protein–membrane interaction inhibitors by virtual ligand screening, proof of concept with the C2 domain of factor V. Proc Natl Acad Sci U S A 2007;104:12697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sudhahar CG, Haney RM, Xue Y, et al. Cellular membranes and lipid-binding domains as attractive targets for drug development. Curr Drug Targets 2008;9:603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spiegel PC, Kaiser SM, Simon JA, et al. Disruption of protein-membrane binding and identification of small-molecule inhibitors of coagulation factor VIII. Chem Biol 2004;11:1413–22. [DOI] [PubMed] [Google Scholar]
- 21. Liu Z, Lin L, Yuan C, et al. Trp2313-His2315 of factor VIII C2 domain is involved in membrane binding: structure of a complex between the C2 domain and an inhibitor of membrane binding. J Biol Chem 2010;285:8824–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nicolaes GAF, Kulharia M, Voorberg J, et al. Rational design of small molecules targeting the C2 domain of coagulation factor VIII. Blood 2014;123:113–20. [DOI] [PubMed] [Google Scholar]
- 23. Chen L, Du-Cuny L, Moses S, et al. Novel inhibitors induce large conformational changes of GAB1 pleckstrin homology domain and kill breast cancer cells. PLoS Comput Biol 2015;11:e1004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nawrotek A, Benabdi S, Niyomchon S, et al. PH-domain-binding inhibitors of nucleotide exchange factor BRAG2 disrupt Arf GTPase signaling. Nat Chem Biol 2019;15:358–66. [DOI] [PubMed] [Google Scholar]
- 25. Li Z, Buck M. Computational design of myristoylated cell-penetrating peptides targeting oncogenic K-Ras.G12D at the effector-binding membrane interface. J Chem Inf Model 2020;60:306–15. [DOI] [PubMed] [Google Scholar]
- 26. Scott DL, Diez G, Goldmann WH. Protein-lipid interactions: correlation of a predictive algorithm for lipid-binding sites with three-dimensional structural data. Theor Biol Med Model 2006;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bhardwaj N, Stahelin RV, Langlois RE, et al. Structural bioinformatics prediction of membrane-binding proteins. J Mol Biol 2006;359:486–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sharikov Y, Walker RC, Greenberg J, et al. MAPAS: a tool for predicting membrane-contacting protein surfaces. Nat Methods 2008;5:119. [DOI] [PubMed] [Google Scholar]
- 29. Nastou KC, Tsaousis GN, Papandreou NC, et al. MBPpred: proteome-wide detection of membrane lipid-binding proteins using profile hidden Markov models. Biochim Biophys Acta 2016;1864:747–54. [DOI] [PubMed] [Google Scholar]
- 30. Lomize AL, Pogozheva ID, Lomize MA, et al. Positioning of proteins in membranes: a computational approach. Protein Sci 2006;15:1318–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lomize AL, Pogozheva ID, Mosberg HI. Anisotropic solvent model of the lipid bilayer. 2. Energetics of insertion of small molecules, peptides, and proteins in membranes. J Chem Inf Model 2011;51:930–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kufareva I, Lenoir M, Dancea F, et al. Discovery of novel membrane binding structures and functions. Biochem Cell Biol 2014;92:555–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kufareva I, Budagyan L, Raush E, et al. PIER: protein interface recognition for structural proteomics. Proteins 2007;67:400–17. [DOI] [PubMed] [Google Scholar]
- 34. Doerr S, Harvey MJ, Noe F, et al. HTMD: high-throughput molecular dynamics for molecular discovery. J Chem Theory Comput 2016;12:1845–52. [DOI] [PubMed] [Google Scholar]
- 35. Whitehead CE, Breneman CM, Sukumar N, et al. Transferable atom equivalent multicentered multipole expansion method. J Comput Chem 2003;24:512–29. [DOI] [PubMed] [Google Scholar]
- 36. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: machine learning in Python. J Mach Learn Res 2011;12:2825–30. [Google Scholar]
- 37. Ke G, Meng Q, Finley T, et al. Lightgbm: a highly efficient gradient boosting decision tree. Adv Neural Inf Process Sys 2017;30:3146–54. [Google Scholar]
- 38. Chen T, Guestrin C. XGBoost: A Scalable Tree Boosting System. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, California, USA. 2939785: ACM, New York, NY, USA: Association for Computing Machinery; 2016, 785–94. [Google Scholar]
- 39. Bergstra J, Bengio Y. Random search for hyper-parameter optimization. J Mach Learn Res 2012;13:281–305. [Google Scholar]
- 40. Claesen M, De Moor B. Hyperparameter search in machine learning. arXiv preprint 2015;arXiv:150202127. [Google Scholar]
- 41. Littlestone N, Warmuth MK. The weighted majority algorithm. Inf Comput 1994;108:212–61. [Google Scholar]
- 42. Wolpert DH. Stacked generalization. Neural Netw 1992;5:241–59. [Google Scholar]
- 43. Raschka S. MLxtend: providing machine learning and data science utilities and extensions to Python's scientific computing stack. J Open Source Softw 2018;3:638. [Google Scholar]
- 44. Davis J, Goadrich M. The relationship between Precision-Recall and ROC curves. Proceedings of the 23rd international conference on Machine learning. New York, NY, USA: Association for Computing Machinery, Pittsburgh, Pennsylvania, USA; 2006. p. 233–40. [Google Scholar]
- 45. Saito T, Rehmsmeier M. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One 2015;10:e0118432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lomize MA, Pogozheva ID, Joo H, et al. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 2012;40:D370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pozzi N, Chen Z, Di Cera E. How the linker connecting the two kringles influences activation and conformational plasticity of prothrombin. J Biol Chem 2016;291:6071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chinnaraj M, Chen Z, Pelc LA, et al. Structure of prothrombin in the closed form reveals new details on the mechanism of activation. Sci Rep 2018;8:2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dufrisne MB, Petrou VI, Clarke OB, et al. Structural basis for catalysis at the membrane-water interface. Biochim Biophys Acta Mol Cell Biol Lipids 2017;1862:1368–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guo L, Wang S, Li M, et al. Accurate classification of membrane protein types based on sequence and evolutionary information using deep learning. BMC Bioinf 2019;20:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baek M, DiMaio F, Anishchenko I, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021;373:871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fuglebakk E, Reuter N. A model for hydrophobic protrusions on peripheral membrane proteins. PLoS Comput Biol 2018;14:e1006325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chatzigoulas A, Cournia Z. Rational design of allosteric modulators: challenges and successes. WIREs Comput Mol Sci 2021;11:e1529. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.