

Abstract
Background
Recent genome-wide association meta-analysis for melanoma doubled the number of previously identified variants. We assessed the performance of an updated polygenic risk score (PRS) in a population of older individuals, where melanoma incidence and cumulative ultraviolet radiation exposure is greatest.
Methods
We assessed a PRS for cutaneous melanoma comprising 55 variants in a prospective study of 12 712 individuals in the ASPirin in Reducing Events in the Elderly Trial. We evaluated incident melanomas diagnosed during the trial and prevalent melanomas diagnosed preenrolment (self-reported). Multivariable models examined associations between PRS as a continuous variable (per SD) and categorical (low-risk [0%-20%], medium-risk [21%-80%], high-risk [81%-100%] groups) with incident melanoma. Logistic regression examined the association between PRS and prevalent melanoma.
Results
At baseline, mean participant age was 75 years; 55.0% were female, and 528 (4.2%) had prevalent melanomas. During follow-up (median = 4.7 years), 120 (1.0%) incident cutaneous melanomas occurred, 98 of which were in participants with no history. PRS was associated with incident melanoma (hazard ratio = 1.46 per SD, 95% confidence interval [CI] = 1.20 to 1.77) and prevalent melanoma (odds ratio [OR] = 1.55 per SD, 95% CI = 1.42 to 1.69). Participants in the highest-risk PRS group had increased risk compared with the low-risk group for incident melanoma (OR = 2.51, 95% CI = 1.28 to 4.92) and prevalent melanoma (OR = 3.66, 95% CI = 2.69 to 5.05). When stratifying by sex, only males had an association between the PRS and incident melanoma, whereas both sexes had an association between the PRS and prevalent melanoma.
Conclusions
A genomic risk score is associated with melanoma risk in older individuals and may contribute to targeted surveillance.
Melanoma incidence increases with age, and the majority of diagnoses occur in people aged 60 years and older (1,2). Age-specific incidence rates are highest among individuals aged 80 years and older, who represent the highest proportion of deaths from melanoma (2). Therefore, the role of genetics in an older population has clinical relevance. Advancing age is associated with thicker melanomas, poorer disease-specific and overall survival, and greater recurrence risk (3,4). Yet mechanisms underlying disease trajectory in older individuals are unclear. Melanomagenesis is a multi-step process resulting from interplay between environmental, host, and genetic factors (5). Understanding etiological pathways underpinning pathogenesis, including the influence of heritable genes that may modulate risk, may help optimize surveillance and preventive strategies through more accurate risk assessment and stratification. Melanoma risk prediction models typically focus on phenotypic features but have recently incorporated markers of genetic susceptibility, with the goal of improving risk stratification (6-8).
Genome-wide association studies (GWAS) provide insights into the biology of polygenic diseases such as melanoma, where risk is influenced by many common variants or single nucleotide polymorphisms (SNPs) across the genome. Although the contributions of individual SNPs to disease risk may be small, in combination they can have a considerable effect on risk, and therefore increased predictive value. This has led to the development of polygenic risk scores (PRSs), which aggregate the effects of SNPs across the genome into a single score (9). Previous studies have attempted to predict risk using a PRS for melanoma (10-12) but have been limited by small numbers of identified variants at the time (13-16). Many of these studies involved analyses of population-based cohorts with diversity of age, where incidence of melanoma overall is low (8,11,17), or familial melanoma cohorts, where incidence is particularly high (18,19). Melanoma PRS performance in older populations has not been evaluated.
The “divergent pathways” of the melanoma hypothesis describes melanoma development in older people as more ultraviolet radiation (UV) dependent (less genetically determined) compared with younger people, who require fewer UV insults to initiate tumorigenesis (20). This hypothesis suggests that genetic factors play a greater role in younger age groups. However, to our knowledge, no studies have assessed a melanoma PRS in a cohort of older individuals, which is representative of a clinically relevant target population due to the higher risk of melanoma with age.
Recently, the largest ever GWAS meta-analysis of melanoma was published, identifying 56 loci associated with clinically confirmed melanoma at genome-wide significance (P < 5 × 10−8) (21). These newly associated genetic variants can now be used to calculate an improved PRS for melanoma. Yet independent validation of this new PRS is challenging, given most of the large genetic studies of melanoma—including the UK Biobank—were used to derive the score within this meta-analysis. Further, many population-based cohorts do not contain large numbers of older individuals (aged >75 years) to model melanoma risk in the age range where the majority of melanomas occur. Therefore, it remains unclear the extent to which a PRS would be associated with melanoma in an older subgroup, who would have high cumulative lifetime UV exposure.
Here, we perform an independent validation of a newly derived melanoma PRS in a well-characterized cohort of older individuals followed prospectively. The ASPirin in Reducing Events in the Elderly (ASPREE) trial population represents a large sample of healthy older individuals from Australia and the United States enrolled in an aspirin primary prevention trial (22-24). All incident melanomas were adjudicated as secondary trial endpoints (25). Our study helps assess the future clinical utility of genomic risk prediction for melanoma in older individuals.
Methods
Study Design and Population
The study population comprised genotyped participants of the ASPREE trial. Study design (26) and trial results (22,23) have been published previously. ASPREE was a randomized placebo-controlled clinical trial to determine whether daily aspirin extended disability-free survival in healthy older individuals with no history of diagnosed cardiovascular events, dementia, or physical disability at enrolment. Participants provided informed consent for genetic research. The study was approved by local ethics committees and registered on Clinicaltrials.gov (NCT01038583). Genotyping was performed on 14 052 participants, with a median follow-up time of 4.7 years (interquartile range = 2.1 years) per participant.
Endpoints
Endpoints used were primary invasive cutaneous melanoma and metastatic melanoma with unknown primary location occurring during the trial (incident). Metastatic recurrence was excluded. Incident melanomas were confirmed by an expert panel using histopathology, imaging of metastasis, or other clinical evidence. If a participant had 2 events during the trial, the time of the first event was used. Prevalent melanomas occurring pretrial were self-reported by participants but not confirmed by review of medical records and assumed to be invasive. Age at diagnosis for self-reported melanomas was reported as either younger or older than 50 years.
Risk Model, Genotyping, and Polygenic Score
Relevant phenotypic information from ASPREE included age, sex, and melanoma family history (parent, sibling, or child). Information on skin pigmentation and number of nevi was not available. Genotyping was performed using the Axiom 2.0 Precision Medicine Diversity Research Array (Thermo Fisher) following standard protocols. Variant calling used a custom pipeline aligned to hg38. We limited our study to participants with European ancestry to mitigate population stratification bias in polygenic scoring, given the PRS was derived from individuals of European descent (21). To define genetic descent, we performed principal component analysis using the 1000 Genomes reference population, excluding ASPREE samples that did not overlap the non-Finnish European 1000 Genomes cluster (Supplementary Figure 1, available online) (27). Samples from 12 712 participants passed the following filters: unrelated (identity-by-descent to third-degree relative); non-Finnish European descent; minimum age at random assignment 70 years. Of these, 12 081 (95%) were from Australian and 631 (5%) from US participants. Imputation was performed using the haplotype reference consortium, European samples (Michigan server) (28). Postimputation QC removed variants with low imputation quality (r2 < 0.3). We calculated the PRS by using 56 variants associated with confirmed melanoma cases (21), 55 of which were present in the ASPREE data (one low-quality imputation failure). Plink v1.9 calculated the weighted sum of the log odds ratios (ORs) reported for the effect alleles for each variant. PRS SNPs and effect sizes are listed in Supplementary Table 1 (available online).
Statistical Analyses
A multivariable Cox proportional hazards regression model was used to evaluate the association between PRS as a continuous variable with incident melanoma, reporting cause-specific hazard ratios (HRs), adjusting for sex, melanoma family history, treatment (aspirin or placebo), age at enrolment, and interaction between PRS and treatment. In a separate model, PRS was categorized into 3 groups; low (0%-20%) medium (21%-80%), and high (81%-100%). We used variance inflation factor to check independence of terms in regression models. The discriminative ability of the model for incident melanoma was measured using the c-index. Competing risks estimates of the cumulative incidence were calculated and plotted for each group using the survfit function from the survival R package. Prevalence analysis of self-reported melanoma before enrolment used a logistic regression model, including sex and family history, reporting the odds ratio of the PRS. Goodness-of-fit for the logistic regression was assessed using the tail-based max-test-statistic (29). DeLong’s test was used to compare between 2 correlated receiver operating characteristic (ROC) curves for logistic regression (30,31). Analyses were performed using R v3.6.1 (32), tidyverse 1.3.0 (33), survival 3.1.12 (34), survminer 0.4.8, and pROC 1.16.2 (35). A z-test was used for the Cox proportional hazards multivariable model and the logistic regression model and to determine if there were differences between the PRS between age groups in which prevalent melanoma occurred. All statistical tests are 2-sided, and a P value of less than .05 was considered statistically significant.
Results
Baseline Characteristics
The mean age of the 12 712 genotyped participants included in the analyses was 75 years (SD = 4.2), with the majority aged 70-79 years (Table 1). Overall, 6990 (55.0%) participants were female, 391 (3.1%) were current smokers, and 528 (4.2%) self-reported a preenrolment melanoma. Of the prevalent cases, 98 participants (37 male, 61 female) reported melanoma before the age of 50 years. The PRS showed an approximately normal distribution in the population with a mean −7.3 (SD = 0.55) (Supplementary Figure 2, available online). We scaled the PRS to have a mean of 0 (SD = 1) for the following analyses.
Table 1.
Baseline characteristics of the ASPREE trial populationa
| Characteristic | No. (%) |
|---|---|
| Genotyped participants, No. | 12 712 |
| Sex, female | 6990 (55.0) |
| Mean age at random assignment (SD), y | 75.06 (4.22) |
| Age group, y | |
| 70-74 | 7725 (60.8) |
| 75-79 | 3198 (25.2) |
| 80-84 | 1389 (10.9) |
| 85+ | 400 (3.1) |
| Current or former smoker | 5232 (41.2) |
| Diabetes | 1178 (9.3) |
| Random assignment to aspirin | 6340 (49.9) |
| Mean body mass index (SD), kg/m2 | 27.97 (4.55) |
| Current alcohol consumer | 10 132 (79.7) |
| Family history of melanoma | 371 (2.9) |
| Mean polygenic risk score (SD) | −7.3 (0.55) |
| Prevalent melanoma (%) self-reported at enrolment | |
| None | 12 129 (95.8) |
| <49 y | 98 (0.8) |
| 50+ y | 430 (3.4) |
ASPREE = ASPirin in Reducing Events in the Elderly.
Association of PRS With Incident Melanoma
During follow-up, 120 (1.0%) incident cutaneous melanomas occurred, of which 110 participants were diagnosed with primary invasive melanoma and 10 with metastatic melanoma with unknown primary location. Four participants had 2 melanomas diagnosed during follow-up. Twenty-two participants with incident cases also had a prevalent melanoma reported at enrolment. All prevalent cases were removed from the incidence analysis, leaving 98 cases (Supplementary Table 2, available online).
PRS as a continuous variable in the model was associated with incident melanoma with a hazard ratio of 1.46 (95% confidence interval [CI] = 1.20 to 1.77, P < .001) per SD of the PRS. Female sex had a hazard ratio for melanoma of 0.49 (95% CI = 0.32 to 0.73, P < .001) compared with men. The variance inflation factor for each term in the multivariable model was less than 1.1, indicating independence of the terms. The c-index for the model with PRS as a continuous variable was 0.643 (95% CI = 0.584 to 0.702), and the c-index for the model excluding the PRS was 0.590 (95% CI = 0.530 to 0.648). We found no statistically significant interaction effect between aspirin treatment and PRS.
We further assessed the effect of the PRS by categorizing the PRS distribution into low- (1%-20%), medium- (21%-80%), and high-risk (81%-100%) groups. When considering associations with incident melanoma using the low-risk group as a reference, there was no statistically significant difference in risk of incident melanoma in the medium- and low-risk groups, but individuals in the high-risk PRS group had increased risk of incident melanoma vs the low-risk group (HR = 2.51, 95% CI = 1.28 to 4.92, P = .007) (Table 2). Supplementary Table 2 (available online) shows the distribution of incident cases by PRS group, stratified by sex. Individuals in the high-risk group had greater cumulative incidence than those in the low- and medium-risk groups (Figure 1, A).
Table 2.
Multivariable Cox regression model for melanoma incidence in the ASPREE trial (clinically confirmed cases, excluding all prevalent cases)a
| Covariate | PRS as continuous variable (per SD) |
PRS as categorical variable (low, medium, high) |
||
|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |
| Sex, female | 0.49 (0.32 to 0.73) | <.001 | 0.48 (0.35 to 0.72) | <.001 |
| Family historya | 1.57 (0.58 to 4.29) | .38 | 1.60 (0.62 to 3.72) | .36 |
| Age at enrolment | 0.99 (0.95 to 1.04) | .83 | 1.00 (0.95 to 1.04) | .84 |
| Treatment, aspirin | 0.88 (0.59 to 1.30) | .52 | 0.88 (0.59 to 1.22) | .53 |
| PRS per SD | 1.46 (1.20 to 1.77) | <.001 | — | — |
| Low PRS, 0%-20% (n = 14) | — | — | 1.00 (Reference) | — |
| Medium PRS, 20%-80% (n = 64) | — | — | 1.61 (0.86 to 2.99) | .14 |
| High PRS, 80%-100% (n = 42) | — | — | 2.51 (1.28 to 4.92) | .007 |
A total of 120 incident melanomas occurred during the ASPREE trial, of which 98 had no pretrial (prevalent) melanoma. A total of 528 participants had self-reported prevalent melanoma at baseline, and a total of 626 participants had melanoma. ASPREE = ASPirin in Reducing Events in the Elderly; CI = confidence interval; HR = hazard ratio; PRS = polygenic risk score.
Figure 1.
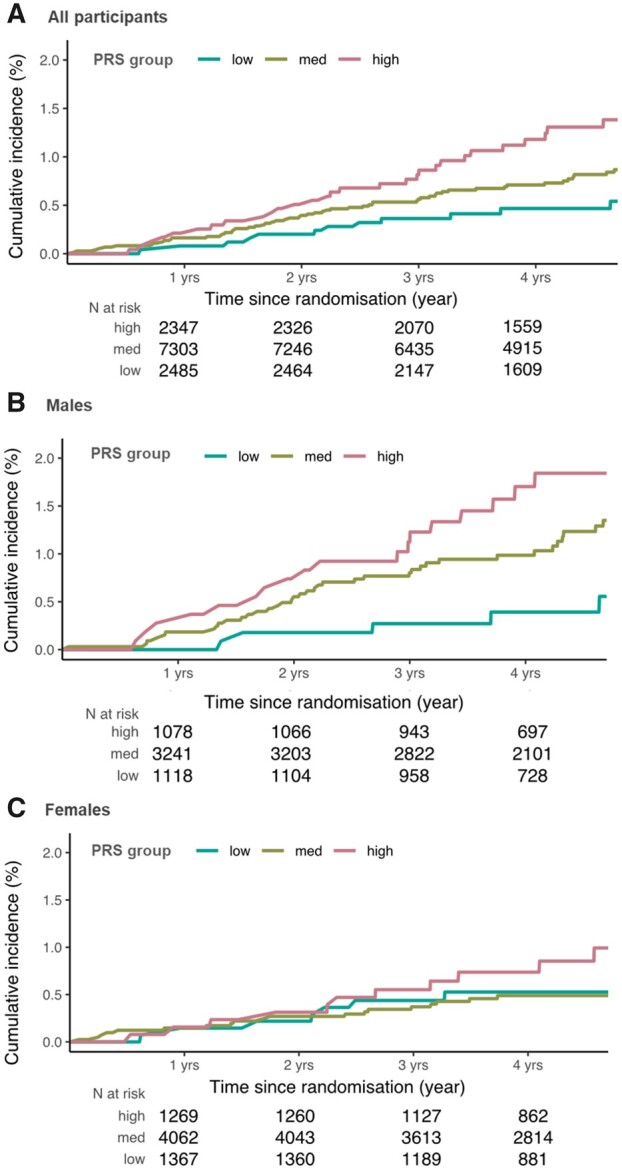
Cumulative incidence of primary cutaneous melanoma in (A) all participants, (B) male participants, and (C) female participants. Competing risks model of the cumulative incidence of primary cutaneous melanoma in the ASPirin in Reducing Events in the Elderly cohort, stratified by a polygenic risk score (PRS) for cutaneous melanoma, categorized into low-risk (0%-20%), medium-risk (20%-80%), and high-risk (80%-100%) groups.
For males, both the medium- and high-risk PRS groups had greater risk of incident melanoma than the low-risk group (HR = 2.61, 95% CI = 1.03 to 6.63, P = .04 and HR = 3.70, 95% CI = 1.37 to 9.98, P = .009, respectively). For females, neither medium- nor high-risk groups had statistically significantly higher melanoma incidence than the low-risk group. Cumulative incidence of melanoma was higher in males than females when accounting for competing mortality risk (Figure 1, B and C).
Association of PRS With Prevalent Melanoma
Prevalent (pre-enrolment) melanomas were self-reported as diagnosed at younger than age 50 years or older than 50 years. No difference was observed in the distribution of PRS between these 2 age categories (z-test, P = .40). The PRS was associated with prevalent melanoma when controlling for female sex, with an odds ratio of 1.55 (95% CI = 1.42 to 1.69, P < .001) per SD (Table 3). The tail-based max-test-statistic did not indicate a lack of goodness-of-fit (P > .05). Individuals in the high-risk PRS group had a statistically significantly higher risk of prevalent melanoma vs the low-risk group (OR = 3.66, 95% CI = 2.69 to 5.05, P < .001). As with incident melanoma, females had lower risk of prevalent melanoma compared with males (OR = 0.82, 95% CI = 0.69 to 0.98, P = .03).
Table 3.
Logistic regression model for prevalent melanoma in participants enrolled in the ASPREE trial (pretrial self-reported cases)a
| Covariate | PRS as continuous variable (per SD) |
PRS as categorical variable (low, medium, high) |
||
|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | |
| Sex, female | 0.82 (0.69 to 0.98) | .03 | 0.82 (0.69 to 0.98) | .02 |
| Family history | 2.36 (1.61 to 3.35) | <.001 | 2.38 (1.63 to 3.37) | <.001 |
| PRS per SD | 1.55 (1.42 to 1.69) | <.001 | — | — |
| Low PRS, 0%-20% (n = 64) | — | — | 1.00 (Reference) | — |
| Medium PRS, 20%-80% (n = 349) | — | — | 1.88 (1.41 to 2.56) | <.001 |
| High PRS, 80%-100% (n = 213) | — | — | 3.66 (2.69 to 5.05) | <.001 |
A total of 120 incident melanomas occurred during the ASPREE trial, of which 98 had no pre-trial (prevalent) melanoma. A total of 528 participants had self-reported prevalent melanoma at baseline. In total: 626 participants had melanoma. ASPREE = ASPirin in Reducing Events in the Elderly; CI = confidence interval; OR = odds ratio; PRS = polygenic risk score.
Family history of melanoma was associated with increased melanoma prevalence (OR = 2.36, 95% CI = 1.61 to 3.35, P < .001) (Table 2). The AUC for the model with continuous PRS was 0.64 (95% CI = 0.62 to 0.66), which was a statistically significant improvement (P < .001 DeLong test) compared with the model without the PRS, based only on family history and sex, for which the AUC was 0.54 (95% CI = 0.52 to 0.57). The ROC curve with PRS is shown in Supplementary Figure 3 (available online).
When stratifying the logistic regression by sex, the association between PRS and prevalent melanoma was statistically significant for both males and females; the odds ratio in females was 1.72 (95% CI = 1.53 to 1.94) and in males was 1.39 (95% CI = 1.23 to 1.57). Prevalent melanoma is shown by PRS risk group and sex in Supplementary Table 2 (available online), and the multivariable logistic regression stratified by sex is shown in Supplementary Table 3 (available online).
Discussion
We assessed the performance of a PRS for melanoma in a population of older individuals followed prospectively. We demonstrated a strong association between the PRS and incident melanoma risk in this cohort, and found meaningfully different rates of melanoma between low-, medium-, and high-risk PRS groups. Our study highlights the potential use of genomic risk scores to improve the prediction of melanoma risk in older populations, where the burden of disease is particularly high. PRS may improve risk stratification for melanoma, towards targeted screening or surveillance for those at greatest risk. Our study population represents an age group at distinctly high risk in which the majority of melanoma incidence and mortality occur (36). Our study validates a newly derived PRS from a recent GWAS meta-analysis, confirming the PRS is associated with incident and prevalent melanoma, even in a population of older individuals mostly residing in a country (Australia) with high ambient UV.
Previous studies have examined PRS performance in younger cohorts, using scores containing fewer SNPs, yet report similar performance for melanoma (8,12,37,38). For example, a previous study examining the performance of a 45-SNP PRS in 2 population-based case-control studies from Australia and the United Kingdom found the odds ratio per SD of the PRS was 1.75 for Australia and 1.63 for the United Kingdom (8), and a three-fold higher risk of melanoma for those in the highest versus lowest PRS tertile. These PRS effects are comparable with our findings (OR = 1.5 per SD, 2.5-fold increase from low to high PRS). In another study using a 21-SNP PRS involving 19 102 post-menopausal women from the Women’s Health Initiative cohort, women in the highest tertile were 1.9 times more likely to develop melanoma compared with the lowest tertile (12). In our study, using a more recent PRS based on a larger number of SNPs, we have demonstrated a similarly robust association with incident melanoma in older adults. Our study has important clinical implications, providing evidence that genetic predisposition (rather than chronic UV damage alone) continues to play an important role in older age groups.
We observed a lower number of incident melanomas in females than males, consistent with previous reports (39,40). Although female sex was protective in both incidence and prevalence analyses, the effect was stronger in the incidence analysis. Both sexes had statistically significant association of PRS with prevalent melanoma. Other studies have demonstrated sex differences in melanoma incidence, mortality, and survival, with a female advantage generally reported (41,42). Whether this stems from differences in hormonal factors, immunological responses, behavioral tendencies, genetics, or a combination of these remains unclear. The low number of melanoma events in females in our study, however, does raise the possibility of power limitations in the assessment of PRS in females. Larger studies may reveal an association of the PRS for incident cases in females. In the self-reported prevalent cases, a retrospective evaluation of incidence across younger ages, the PRS calculated for females was higher than males (1.72 vs 1.39). Incidence of melanoma in females is reported to exceed incidence in men up to the age of 49-50 years in the Australian population (43), which may contribute to the higher PRS score when self-reported cases including those younger than 49 years are included. The extent to which sex differences in melanoma risk are driven by genetic factors warrants further investigation.
A strength of our study is the well-characterized, older population followed prospectively. All melanoma events that occurred during the ASPREE trial were adjudicated by an expert panel. Previous PRS studies have focused on examining younger cohorts and familial clusters. The median age at follow-up was 78 years, allowing observation of melanoma events in the most clinically relevant age group, with melanomas reported across the spectrum of close to a lifetime.
Limitations of our study include the unavailability of some key phenotypic and clinical risk factors associated with melanoma, such as UV exposure, naevi count, and Fitzpatrick skin phototype, which were not collected as part of the trial. The effects of incorporating our PRS into a conventional phenotype-based model, therefore, could not be assessed. Nevertheless, previous studies have shown a measurable increase in predictive ability of adding an earlier PRS to traditional melanoma risk factors, such as skin phototype (8). A lack of efficacy towards the primary ASPREE endpoint led to early termination and a shorter follow-up period than originally intended, limiting the total number of incident melanoma events, especially in females. In addition, melanoma history before trial enrolment was self-reported and not verified through histopathology or supporting documentation, and the study only included participants who were relative healthy at age 70 years and older - and hence excluded cases in the population whose melanoma progressed at younger ages. The reduced reliability of self-report for melanoma compared with other cancers (44) (eg, due to confusion with basal and squamous cell carcinomas) may have contributed to an overestimation of prevalent melanoma events. Our analysis only included participants of European genetic descent, limiting our ability to extrapolate findings to other ethnic backgrounds.
Given our analysis focused on individuals in a country with high ambient UV, it would be informative to conduct a comparable PRS study in a cohort with a similar ethnic background in a country with lower ambient UV. The melanoma PRS includes SNPs associated with a variety of biological processes, including pigmentary characteristics, naevus development, cell adhesion, immune regulation, and DNA repair (21). Although differences in UV exposure may modify PRS performance across different populations, we note that many of the variants reported in the GWAS were unrelated to the classic cutaneous melanoma risk phenotypes (eg, naevus count, hair color, etc) and may not relate to UV exposure (21). The exact mechanisms by which these SNPs influence melanoma development require further functional analyses.
Currently, melanoma risk prediction models are largely based on phenotypic or clinical risk factors. To date, studies have shown that the addition of genomic risk scores based on previously identified SNPs to melanoma risk models containing traditional risk factors only modestly improves discriminatory power (8). Such small incremental improvements offered by PRS may suggest only limited clinical potential. However, as our understanding of the genetic basis of melanoma evolves and the performance of PRS improves, genetic testing may have an increasingly important role in future clinical practice for melanoma. Not only can a PRS potentially serve to improve risk prediction and risk stratification and help personalize surveillance strategies, but it can also shed light on the genetic architecture of melanoma and enhance our understanding of the etiological pathways underpinning melanomagenesis.
In conclusion, we present the first external validation of a newly derived PRS for melanoma in a cohort of older individuals followed prospectively. Our study demonstrates that the PRS is a statistically significant discriminator of incident melanoma events in an older population, with potential clinical implications for risk stratification, surveillance, and prevention in this age group. Further studies are required to assess more rigorously the clinical utility of genetic risk scores for melanoma, when combined with conventional risk phenotype information, and to determine the appropriate clinical context for their use.
Funding
This work was supported by an ASPREE Flagship cluster grant (including the Commonwealth Scientific and Industrial Research Organisation, Monash University, Menzies Research Institute, Australian National University, University of Melbourne); by grants (U01AG029824 and U19AG062682) from the National Institute on Aging and the National Cancer Institute at the National Institutes of Health, and from the National Health and Medical Research Council of Australia (334047 and 1127060); and by Monash University and the Victorian Cancer Agency. A.E.C. receives a NHMRC Career Development Fellowship (1147843). H.P.S. holds an NHMRC MRFF Next Generation Clinical Researchers Program Practitioner Fellowship (APP1137127). V.M. is supported by an NHMRC Early Career Fellowship (1160757). P.L. is supported by a National Heart Foundation Future Leader Fellowship (102604).
Notes
Role of the funder : No sponsor had any role in the study design, data collection, analysis, interpretation, the writing, and decision to submit the manuscript.
D isclosures : HPS is a shareholder of MoleMap NZ Limited and E-derm Consult GmbH and undertakes teledermatological reporting for both companies. HPS is a Medical Consultant for Canfield Scientific Inc. and a Medical Advisor for First Derm and Revenio Research Oy. ATC has previously consulted for Bayer Pharma AG on aspirin-related topics. VM has received honoraria from Merck, Bristol-Myers-Squibb and Novartis. No other conflicts are declared for the other authors.
Author contributions : AB: Conceptualization, Data curation, Formal Analysis, Investigation, Software, Writing—original draft. MY: Data curation, Formal Analysis, Investigation, Writing—original draft. MR: Conceptualization, Data curation, Formal Analysis, Investigation, Software. GP: Data curation, Formal Analysis. SGO: Resources, Data curation, Investigation, Project administration. JT: Investigation, Writing—original draft. RW: Data curation, Formal Analysis, Investigation. AJ: Conceptualization, Methodology. YC: Conceptualization, Methodology. AML: Conceptualization, Validation. TY: Methodology, Validation. MJ: Conceptualization, Validation. HPS: Conceptualization, Validation. AEC: Conceptualization, Validation. MHL: Conceptualization, Data curation. PG: Conceptualization, Resources, Data curation, Methodology. CM: Resources, Data curation, Formal Analysis, Methodology. ATC: Resources, Funding acquisition, Project administration, Supervision. JJM: Resources, Funding acquisition, Project administration, Supervision. VJM: Conceptualization, Methodology, Project administration, Supervision, Writing—original draft. PL: Conceptualization, Data curation, Funding acquisition, Methodology, Supervision, Writing—original draft. All authors were involved in writing (review and editing).
Acknowledgements: We thank the trial staff in Australia and the United States, the participants who volunteered for this trial, and the general practitioners and staff of the medical clinics who cared for the participants.
Supplementary Material
Contributor Information
Andrew Bakshi, Department of Epidemiology and Preventive Medicine, School of Public Health and Preventive Medicine, Monash University, Melbourne, Australia.
Mabel Yan, Department of Epidemiology and Preventive Medicine, School of Public Health and Preventive Medicine, Monash University, Melbourne, Australia.
Moeen Riaz, Department of Epidemiology and Preventive Medicine, School of Public Health and Preventive Medicine, Monash University, Melbourne, Australia.
Galina Polekhina, Department of Epidemiology and Preventive Medicine, School of Public Health and Preventive Medicine, Monash University, Melbourne, Australia.
Suzanne G Orchard, Department of Epidemiology and Preventive Medicine, School of Public Health and Preventive Medicine, Monash University, Melbourne, Australia.
Jane Tiller, Department of Epidemiology and Preventive Medicine, School of Public Health and Preventive Medicine, Monash University, Melbourne, Australia.
Rory Wolfe, Department of Epidemiology and Preventive Medicine, School of Public Health and Preventive Medicine, Monash University, Melbourne, Australia.
Amit Joshi, Clinical and Translational Epidemiology Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA; MGH Cancer Center, Boston, MA, USA.
Yin Cao, Division of Public Health Sciences, Department of Surgery, Washington University School of Medicine, St Louis, MO, USA; Alvin J. Siteman Cancer Center, Washington University School of Medicine, St Louis, MO, USA.
Aideen M McInerney-Leo, The University of Queensland Diamantina Institute, The University of Queensland, Dermatology Research Centre, Brisbane, QLD, USA.
Tatiane Yanes, The University of Queensland Diamantina Institute, The University of Queensland, Dermatology Research Centre, Brisbane, QLD, USA.
Monika Janda, The University of Queensland Diamantina Institute, The University of Queensland, Dermatology Research Centre, Brisbane, QLD, USA; Centre of Health Services Research, Faculty of Medicine, The University of Queensland, Brisbane, Queensland, Australia.
H Peter Soyer, The University of Queensland Diamantina Institute, The University of Queensland, Dermatology Research Centre, Brisbane, QLD, USA.
Anne E Cust, Sydney School of Public Health and Melanoma Institute Australia, Faculty of Medicine and Health, The University of Sydney, Sydney, Australia.
Matthew H Law, Statistical Genetics Lab, QIMR Berghofer Medical Research Institute, Herston, Queensland, Australia; School of Biomedical Sciences, Faculty of Health, and Institute of health and Biomedical Innovation, Queensland University of Technology, Kelvin Grove, Queensland, Australia, Personalised Oncology Division, Walter and Eliza Hall Institute Medical Research and Faculty of Medicine University of Melbourne, Australia.
Peter Gibbs, Department of Anatomical Pathology, Alfred Hospital, Melbourne, Victoria, Australia.
Catriona McLean, Department of Anatomical Pathology, Alfred Hospital, Melbourne, Victoria, Australia.
Andrew T Chan, Clinical and Translational Epidemiology Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA; MGH Cancer Center, Boston, MA, USA.
John J McNeil, Department of Epidemiology and Preventive Medicine, School of Public Health and Preventive Medicine, Monash University, Melbourne, Australia.
Victoria J Mar, Department of Epidemiology and Preventive Medicine, School of Public Health and Preventive Medicine, Monash University, Melbourne, Australia; Victorian Melanoma Service, Alfred Health, Melbourne, Australia.
Paul Lacaze, Department of Epidemiology and Preventive Medicine, School of Public Health and Preventive Medicine, Monash University, Melbourne, Australia.
Data Availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
- 1.National Cancer Institute. Cancer stat facts: melanoma of the skin; 2020.
- 2.Australian Institute of Health and Welfare (AIHW). Skin Cancer in Australia; 2017.
- 3. Lasithiotakis K, Leiter U, Meier F, et al. Age and gender are significant independent predictors of survival in primary cutaneous melanoma. Cancer. 2008;112(8):1795–1804. [DOI] [PubMed] [Google Scholar]
- 4. Macdonald JB, Dueck AC, Gray RJ, et al. Malignant melanoma in the elderly: different regional disease and poorer prognosis. J Cancer. 2011;2:538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leonardi GC, Falzone L, Salemi R, et al. Cutaneous melanoma: from pathogenesis to therapy (review). Int J Oncol. 2018;52(4):1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vuong K, Armstrong BK, Drummond M, et al. Development and external validation study of a melanoma risk prediction model incorporating clinically assessed naevi and solar lentigines. Br J Dermatol. 2020;182(5):1262–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Usher-Smith JA, Emery J, Kassianos AP, Walter FM. Risk prediction models for melanoma: a systematic review. Cancer Epidemiol Biomarkers Prev. 2014;23(8):1450–1463. [DOI] [PubMed] [Google Scholar]
- 8. Cust AE, Drummond M, Kanetsky PA, et al. ; Leeds Case-Control Study Investigators. Assessing the incremental contribution of common genomic variants to melanoma risk prediction in two population-based studies. J Invest Dermatol. 2018;138(12):2617–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17(10):1520–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roberts MR, Asgari MM, Toland AE. Genome‐wide association studies and polygenic risk scores for skin cancer: clinically useful yet? Br J Dermatol. 2019;181(6):1146–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gu F, Chen T-H, Pfeiffer RM, et al. Combining common genetic variants and non-genetic risk factors to predict risk of cutaneous melanoma. Hum Mol Genet. 2018;27(23):4145–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cho HG, Ransohoff KJ, Yang L, et al. Melanoma risk prediction using a multilocus genetic risk score in the Women's Health Initiative cohort. J Am Acad Dermatol. 2018;79(1):36–41.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Amos CI, Wang L-E, Lee JE, et al. ; AMFS Investigators. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum Mol Genet. 2011;20(24):5012–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barrett JH, Iles MM, Harland M, et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat Genet. 2011;43(11):1108–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Macgregor S, Montgomery GW, Liu JZ, et al. Genome-wide association study identifies a new melanoma susceptibility locus at 1q21.3. Nat Genet. 2011;43(11):1114–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ransohoff KJ, Wu W, Cho HG, et al. Two-stage genome-wide association study identifies a novel susceptibility locus associated with melanoma. Oncotarget. 2017;8(11):17586–17592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fritsche LG, Beesley LJ, VandeHaar P, et al. Exploring various polygenic risk scores for skin cancer in the phenomes of the Michigan genomics initiative and the UK Biobank with a visual catalog: PRSWeb. PLoS Genet. 2019;15(6):e1008202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Potjer TP, van der Grinten TWJ, Lakeman IMM, et al. Association between a 46-SNP polygenic risk score and melanoma risk in Dutch patients with familial melanoma [published online ahead of print]. J Med Genet. 2020; jmedgenet-2020-107251. doi: 10.1136/jmedgenet-2020-107251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kasparian NA, Meiser B, Butow PN, Simpson JM, Mann GJ. Genetic testing for melanoma risk: a prospective cohort study of uptake and outcomes among Australian families. Genet Med. 2009;11(4):265–278. [DOI] [PubMed] [Google Scholar]
- 20. Whiteman DC, Watt P, Purdie DM, Hughes MC, Hayward NK, Green AC. Melanocytic nevi, solar keratoses, and divergent pathways to cutaneous melanoma. J Natl Cancer Inst. 2003;95(11):806–812. [DOI] [PubMed] [Google Scholar]
- 21. Landi MT, Bishop DT, MacGregor S, et al. ; GenoMEL Consortium. Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat Genet. 2020;52(5):494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McNeil JJ, Wolfe R, Woods RL, et al. Effect of aspirin on cardiovascular events and bleeding in the healthy elderly. N Engl J Med. 2018;379(16):1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McNeil JJ, Woods RL, Nelson MR, et al. Effect of aspirin on disability-free survival in the healthy elderly. N Engl J Med. 2018;379(16):1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McNeil JJ, Nelson MR, Woods RL, ASPREE Investigator Group, et al. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. 2018;379(16):1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McNeil JJ, Gibbs P, Orchard SG, et al. Effect of aspirin on cancer incidence and mortality in older adults. J Natl Cancer Inst. 2020;113(3):258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.ASPREE Investigator Group. Study design of ASPirin in Reducing Events in the Elderly (ASPREE): a randomized, controlled trial. Contemp Clin Trials. 2013;36(2):555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Genomes PC, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Das S, Forer L, Schonherr S, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song M, Kraft P, Joshi AD, Barrdahl M, Chatterjee N. Testing calibration of risk models at extremes of disease risk. Biostatistics. 2015;16(1):143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44(3):837–845. [PubMed] [Google Scholar]
- 31. Sun X, Xu W. Fast implementation of DeLong’s algorithm for comparing the areas under correlated receiver operating characteristic curves. IEEE Signal Process Lett. 2014;21(11):1389–1393. [Google Scholar]
- 32.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria.
- 33. Wickham H, Averick M, Bryan J, et al. Welcome to the {tidyverse}. J Open Source Software. 2019;4(43):1686–1686. [Google Scholar]
- 34. Therneau TM. A Package for Survival Analysis in R; 2020.
- 35. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011;12:77–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Welfare AIoHa. Skin cancer in Australia; 2016.
- 37. Fang S, Han J, Zhang M, et al. Joint effect of multiple common SNPs predicts melanoma susceptibility. PLoS One. 2013;8(12):e85642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kypreou KP, Stefanaki I, Antonopoulou K, et al. Prediction of melanoma risk in a southern European population based on a weighted genetic risk score. J Invest Dermatol. 2016;136(3):690–695. [DOI] [PubMed] [Google Scholar]
- 39. Curchin DJ, Harris VR, McCormack CJ, Smith SD. Changing trends in the incidence of invasive melanoma in Victoria, 1985–2015. Med J Aust. 2018;208(6):265–269. [DOI] [PubMed] [Google Scholar]
- 40. Liu F, Bessonova L, Taylor TH, Ziogas A, Meyskens FL Jr, Anton-Culver H. A unique gender difference in early onset melanoma implies that in addition to ultraviolet light exposure other causative factors are important. Pigment Cell Melanoma Res. 2013;26(1):128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joosse A, Collette S, Suciu S, et al. Sex is an independent prognostic indicator for survival and relapse/progression-free survival in metastasized stage III to IV melanoma: a pooled analysis of five European organisation for research and treatment of cancer randomized controlled trials. J Clin Oncol. 2013;31(18):2337–2346. [DOI] [PubMed] [Google Scholar]
- 42. de Vries E, Nijsten TEC, Visser O, et al. Superior survival of females among 10,538 Dutch melanoma patients is independent of Breslow thickness, histologic type and tumor site. Ann Oncol. 2008;19(3):583–589. [DOI] [PubMed] [Google Scholar]
- 43. Olsen CM, Thompson JF, Pandeya N, Whiteman DC. Evaluation of sex-specific incidence of melanoma. JAMA Dermatol. 2020;156(5):553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Loh V, Harding J, Koshkina V, Barr E, Shaw J, Magliano D. The validity of self-reported cancer in an Australian population study. Aust N Z J Public Health. 2014;38(1):35–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.