

Abstract
Ketone bodies play significant roles in organismal energy homeostasis, serving as oxidative fuels, modulators of redox potential, lipogenic precursors, and signals, primarily during states of low carbohydrate availability. Efforts to enhance wellness and ameliorate disease via nutritional, chronobiological, and pharmacological interventions have markedly intensified interest in ketone body metabolism. The two ketone body redox partners, acetoacetate and D-β-hydroxybutyrate, serve distinct metabolic and signaling roles in biological systems. We discuss the pleiotropic roles played by both of these ketones in health and disease. While enthusiasm is warranted, prudent procession through therapeutic applications of ketogenic and ketone therapies is also advised, as a range of metabolic and signaling consequences continue to emerge. Organ-specific and cell-type-specific effects of ketone bodies are important to consider as prospective therapeutic and wellness applications increase.
Keywords: ketones and cancer, ketones and neurodegenerative disease, ketones and the gut, ketones and SGLT inhibitors, ketones and heart failure, ketones and fatty liver disease
1. INTRODUCTION
Ketone bodies are endogenously synthetized metabolites that become significant contributors to energy metabolism in mammals during multiple physiological periods such as long-term starvation, short-term fasting, the neonatal period, pregnancy, or adherence to low-carbohydrate, high-fat diets. Attention to ketone body metabolism has intensified recently, as intermittent fasting, time-restricted eating, adherence to low-carbohydrate ketogenic diets, or application of ingested exogenous ketones are all under investigation in clinical studies, with the objective of enhancing wellness and performance, improving health, combatting disease, and offsetting the effects of aging (18, 43, 72, 127, 159). Recent studies highlight roles for ketone bodies in mammalian cell metabolism, homeostasis, and signaling under a wide variety of physiological and pathological states. Apart from serving as energy fuels for extrahepatic tissues such as brain, heart, or skeletal muscle, ketone bodies play pivotal roles as signaling mediators, drivers of protein posttranslational modification (PTM), and modulators of inflammation or oxidative stress. In this review, we provide both classical and modern views of the pleiotropic roles of ketone bodies and their metabolism.
2. KETONE BODY METABOLISM
2.1. Overview of Organismal Physiology and Turnover
In mammals, ketone body turnover involves interorgan and intercellular shuttles governed by integrated regulatory mechanisms (Figure 1). Ketone bodies are considered alternative energy sources, particularly for the nervous system, during periods of diminished glucose availability, since circulating free fatty acids have limited ability to fuel neurons. Skeletal muscle, heart, and kidney also contribute to ketone body disposal. Ketone body turnover consists of two components: production from fatty acids in liver hepatocytes, and uptake and disposal by extrahepatic tissues (6).
Figure 1.
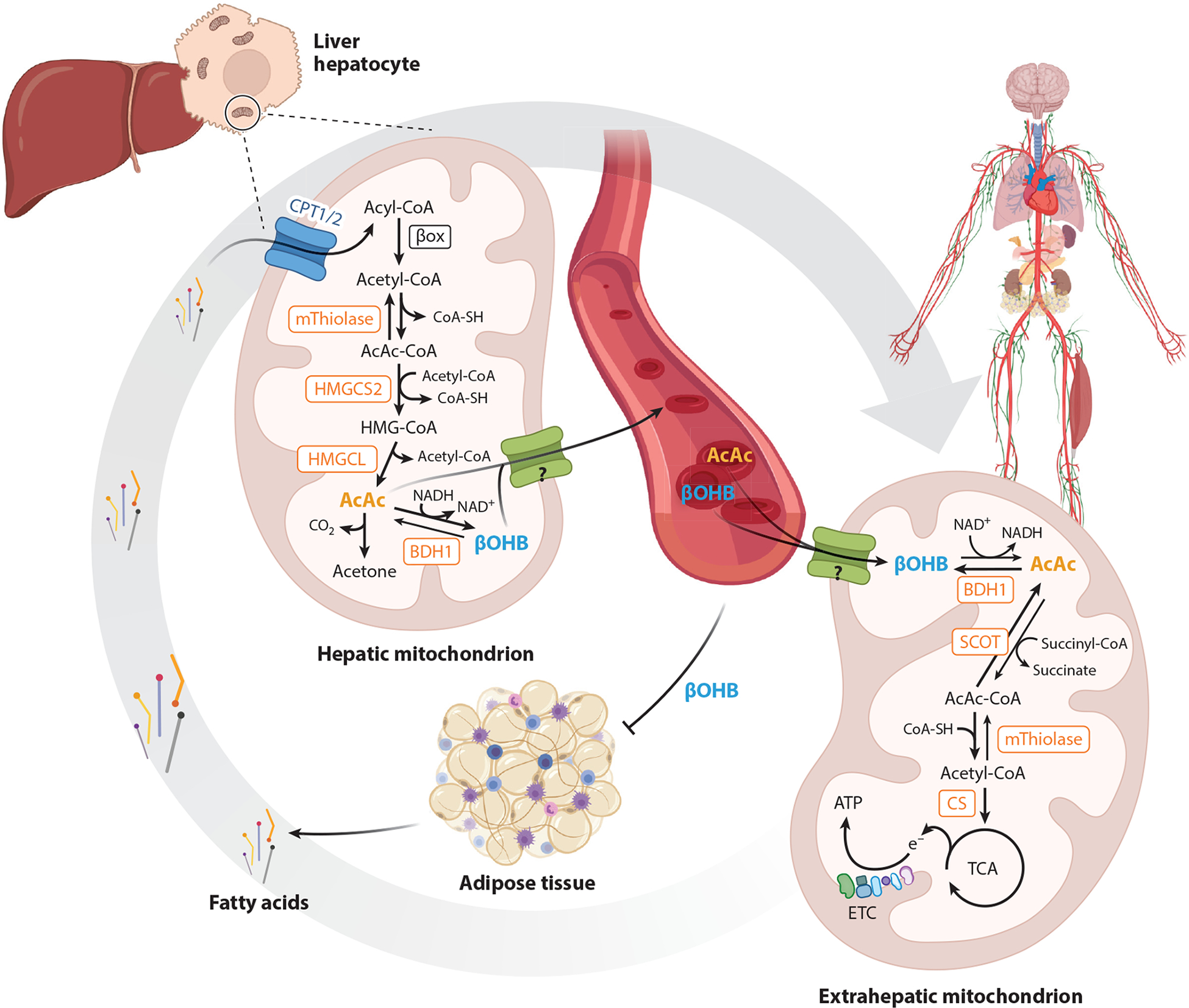
Overview of organismal physiology and turnover. Ketone bodies are primarily generated within hepatic mitochondria from fatty acid–derived acetyl-CoA by the sequence of metabolic reactions requiring the fate-committing enzyme HMGCS2. AcAc and βOHB are transported in circulation to extrahepatic tissue for terminal oxidation through reactions requiring the enzyme SCOT. Adipose tissue lipolysis is inhibited by βOHB, creating a negative feedback loop. Question marks represent the uncertain molecular identity of mitochondrial ketone transporter(s). Abbreviations: AcAc, acetoacetate; ATP, adenosine triphosphate; BDH1, D-β-hydroxybutyrate dehydrogenase 1; βOHB, β-hydroxybutyrate; βox, β-oxidation; CoA, coenzyme A; CoA-SH, CoA sodium salt hydrate; CPT, carnitine palmitoyltransferase; e−, electron; CS, citrate synthase; ETC, electron transport chain; HMGCL, 3-hydroxymethylglutaryl-CoA lyase; HMGCS2, 3-hydroxymethylglutaryl-CoA synthase 2; mThiolase, mitochondrial thiolase; SCOT, succinyl-CoA:3-oxoacid-CoA transferase; TCA, tricarboxylic acid. Figure adapted from images created with BioRender.com.
Adipose tissue–derived fatty acids are released from triacylglycerols (TAGs) by lipolysis and are transported to liver and into the hepatocyte mitochondrial matrix via carnitine palmitoyltransferase 1 (CPT1). Most ketone bodies are produced in hepatocytes from fatty acid β-oxidation-derived acetyl coenzyme A (acetyl-CoA) and transported to extrahepatic tissues for terminal oxidation. In addition to adipocyte lipolysis, ketogenesis rates are governed by hepatic carbohydrate fluxes, mitochondrial redox potential, β-oxidation rates, concentration of tricarboxylic acid (TCA) cycle intermediates, and glucagon and insulin balance (67, 108, 131, 186). Ketogenesis is also regulated at transcriptional and posttranslational levels (see Section 2.3), although finely tuned and cell autonomous regulatory mechanisms remain incompletely resolved.
In healthy adults, the diurnal circadian concentration of ketones oscillates between 50 and 250 μM, contributing to as little as ~5% of total energy expenditure in the fed state and increasing up to ~20% in the fasted and starved states (6, 19, 36). Greater oscillations in circulating ketone body levels occur upon fasting; during adherence to low-carbohydrate, high-fat diets; acutely after exercise; in the neonatal period; or during late pregnancy (136). Prolonged exercise or a 24-h fast can increase circulating ketone body levels to nearly 1 mM, but in pathological states (diabetic ketoacidosis), ketone body concentrations can increase to 20 mM (19, 136). In physiological ketotic states, ketone production and consumption are balanced, typically yielding steady state circulating total ketone body concentrations of 4–5 mM. Ketone body consumption by extrahepatic tissues is rapid, until saturation of import and oxidation occur. The majority of ketones are oxidized, proportionate to circulating concentrations, and only a small fraction of ketones is disposed in the urine (6). Urinary ketone concentrations do rise in ketotic states in which circulating ketones exceed 1 mM, and ketonuria and the sweet odor derived from spontaneous decarboxylation of acetoacetate (AcAc) to acetone can serve as a proxy to detect ketoacidosis in humans (>7 mM) (121, 136). In overnight fasted adult humans, ketogenesis rates are ~0.25 mmol/min, rising to 1–2 mmol/min after 5 days of fasting (6). In extrahepatic tissues, AcAc is directed toward either terminal oxidation or anabolic pathways (Figures 1 and 2), though the capacity of terminal oxidation is tenfold higher than for anabolic disposal pathways, and loss of oxidative capacity yields ketoacidosis, confirming that anabolic disposal alone is insufficient to support efficient ketone turnover (33, 51, 184). Although circulating AcAc and D-β-hydroxybutyrate (D-βOHB) exchange in near equilibrium, the two do not equilibrate entirely, as AcAc fate also includes its disposal (6, 136).
Figure 2.
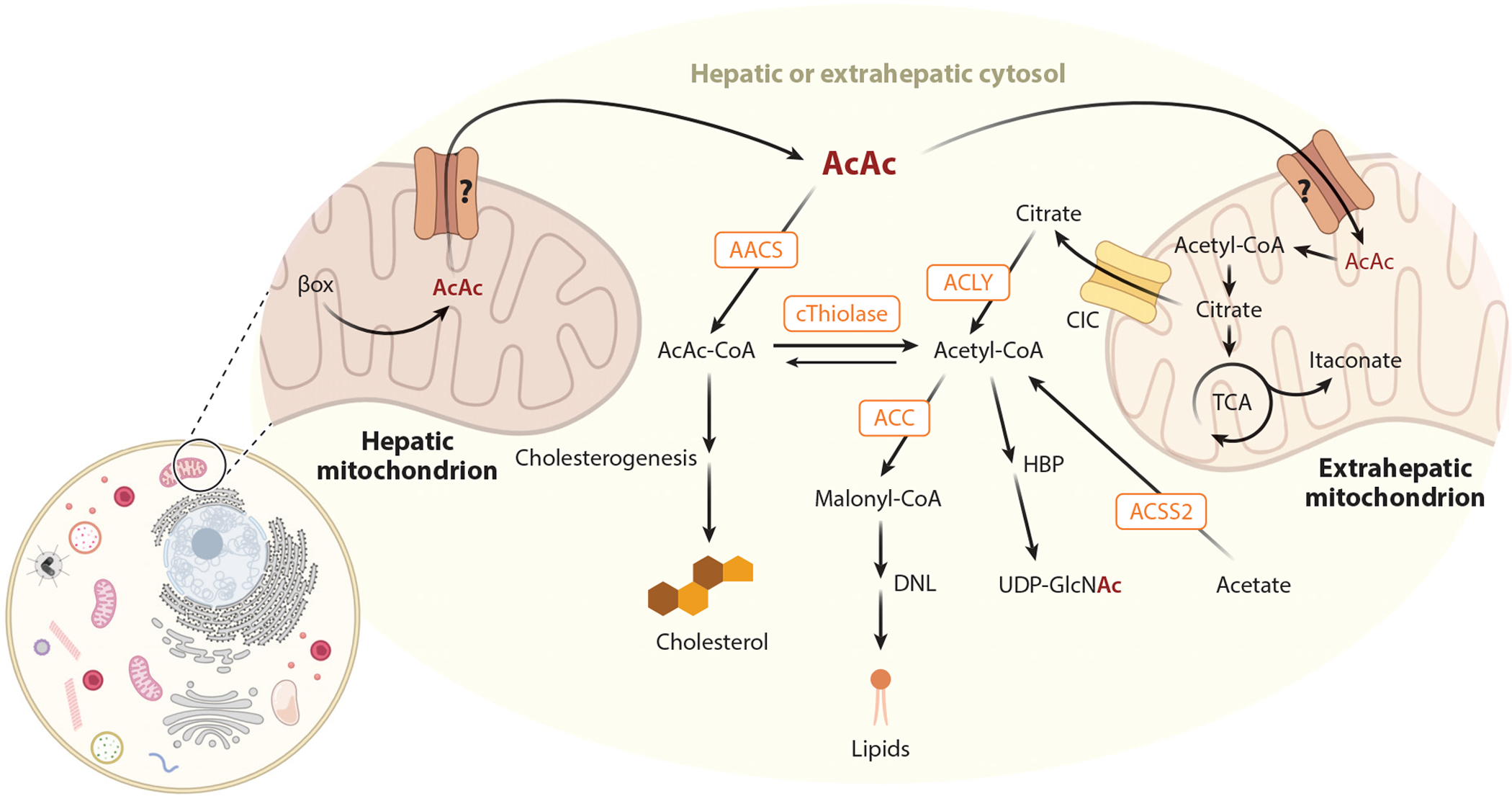
Ketone body metabolic fates beyond terminal oxidation. Ketone body metabolism in hepatic or extrahepatic cytosol is integrated with nonoxidative pathways such as cholesterogenesis and DNL. In extrahepatic cells, AcAc carbon converges with itaconate and HBPs in mitochondrial and cytosolic compartments, respectively. Question marks represent the uncertain molecular identity of mitochondrial ketone transporter(s). Abbreviations: AACS, AcAc-CoA synthetase; AcAc, acetoacetate; ACC, acetyl-CoA carboxylase; ACLY, adenosine triphosphate citrate lyase; ACSS2, acetyl-CoA synthetase; βox, β-oxidation; CIC, citrate transporter; CoA, coenzyme A; cThiolase, cytoplasmic thiolase; DNL, de novo lipogenesis; HBP, hexosamine biosynthetic pathway; TCA, tricarboxylic acid; UDP-GlcNAc, uridine diphospho-N-acetylglucosamine. Figure adapted from images created with BioRender.com.
Despite a more rapid induction in response to fasting than humans, mice remain an excellent model of human ketone turnover, with similar excursions in circulating concentrations over the fed-fast cycle, as well as comparable maximal rates of ketone body appearance and disposal of 1–2 mmol/min/1.73 m2 (see also the sidebar titled Nutritional Provocation of Endogenous Ketogenesis) (6, 161). The transition from the perinatal to the neonatal period is associated with a large increase in ketogenic capacity in both mice and humans (33, 67), although ketogenic latencies in response to duration of fasting tend to increase with progression from childhood to adulthood (19). The effect of older aging is less clear, as reports of variation of ketogenesis or circulating ketones in aging mice and humans have been inconsistent (68, 94, 104, 148). In addition, despite evidence for sex-dependent differences in circulating free fatty acids, consistent evidence for variation of ketogenic rate by gender in humans is also lacking (14, 126).
NUTRITIONAL PROVOCATION OF ENDOGENOUS KETOGENESIS.
There is great interest in the application of nutritional interventions that provoke endogenous ketogenesis to improve health and mitigate the effects of disease, including intermittent fasting, fasting mimicking diet, time-restricted eating, caloric restriction, and adherence to low-carbohydrate ketogenic diets (18, 43, 109, 125). It is important to consider that physiological effects attributable to these interventions may or may not be transduced through ketone bodies themselves. Specifically, diets reducing carbohydrate content usually replace its contribution to the overall kilocalorie pool ingested with fat, which also substantially diminishes circulating insulin concentrations. Rodent studies add an additional set of considerations, because to achieve ketosis through a low-carbohydrate diet, a more extreme macronutrient distribution, particularly restricting both carbohydrate and protein, is required (e.g., 1% kcal carbohydrate, <10% kcal protein). In contrast, many humans can achieve ketosis through adherence to diets that include 5–10% kcal carbohydrate and ≥20% kcal protein. Rodent ketogenic diet formulations are also often choline restricted, and such restriction increases susceptibility to liver injury and to increased ketogenesis (87, 124, 145).
At low circulating concentrations, ketone bodies are imported and oxidized by extrahepatic tissues at the rate of delivery, but at higher concentrations, metabolic clearance rates diminish, as uptake and oxidation are saturable. Thus, the relationship between ketone body production and circulating concentration is exponential; the rise in circulating concentrations amplifies as ketogenesis rates increase, with a maximum clearance rate reached when the circulating concentration is ~6 mM (6). Moreover, the relationship between metabolic clearance rate and delivered ketone concentration may vary by tissue. Skeletal muscle rapidly consumes ketones at lower circulating concentrations, diminishing this rate at high concentrations. Conversely, the brain may maintain a more consistent clearance rate across ketone concentrations, which may be adaptive during starvation. As ketone bodies increase in the circulation, they also inhibit adipose tissue lipolysis by activating the cell surface receptor GPR109A, decreasing the availability of circulating free fatty acids in a negative feedback loop (162) (also see Section 2.5). This pathway is not dominant, however, because in diabetic ketoacidosis, reduced or absent insulin, dehydration, and sympathetic activity cause unrestrained adipocyte fatty acid lipolysis, increasing ketogenesis while ketone disposal rates are curtailed, yielding a mismatch that causes massive hyperketonemia (> 20 mM) and acidosis. The acidosis itself may be attributable to multiple unbuffered sources of protons that emerge through the course from fatty acid hydrolysis through ketogenesis (56).
2.2. Enzymatic Mediators of Ketone Turnover
In liver hepatocytes, ketogenesis occurs at rates proportional to mitochondrial fatty acid β-oxidation, after acetyl-CoA availability has reached maximal citrate synthase activity and/or a low oxaloacetate (OAA) pool limits condensation of acetyl-CoA to form citrate (186). In addition to fatty acid–derived acetyl-CoA, the catabolism of select amino acids, especially leucine, can contribute to up to 4% of ketone body carbon in the postabsorptive state (164). In postabsorptive states, carbohydrate availability is low and pyruvate supplies the TCA cycle by adenosine triphosphate (ATP)-dependent carboxylation to OAA via the pyruvate carboxylase enzyme, contributing to anaplerosis, in a manner that is allosterically activated by β-oxidation-derived acetyl-CoA. Because most hepatic mitochondrial pyruvate does not undergo oxidative decarboxylation to acetyl-CoA, the contribution of glucose or pyruvate to ketone body carbon is insignificant. Acetyl-CoA also allosterically activates pyruvate dehydrogenase kinase, which phosphorylates and inhibits pyruvate dehydrogenase, further enhancing anaplerotic flow of pyruvate into the TCA cycle. Furthermore, conversion of mitochondrial acetyl-CoA to citrate augments the cytoplasmic pool of acetyl-CoA, which inhibits fatty acid oxidation: Acetyl-CoA carboxylase (ACC) catalyzes the conversion of acetyl-CoA to malonyl-CoA, the lipogenic substrate and allosteric inhibitor of mitochondrial CPT1 (108). Thus, the mitochondrial acetyl-CoA pool both regulates and is regulated by ketogenesis, which orchestrates key aspects of hepatic intermediary metabolism.
In hepatocytes, the ketogenesis fate-committing mitochondrial matrix enzyme 3-hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) generates 3-hydroxymethylglutaryl-CoA (HMG-CoA) by condensing β-oxidation-derived acetoacetyl-CoA (AcAc-CoA) and acetyl-CoA (Figure 1). Flux modeling studies show that HMGCS2 activity exerts a strong influence on overall ketogenic flux, perhaps even greater than β-oxidation rate-limiting CPT1 (131). CPT1-independent regulation of ketogenesis in normal physiology was also evident in rats infused with the medium-chain fatty acid octanoic acid, which does not require CPT1 to cross the inner mitochondrial membrane. In these experiments, livers from starved rats generated more octanoate-derived ketones than those from fed rats, suggesting regulation of ketogenesis through mechanisms restricted to mitochondrial matrix (107). HMGCS2-derived HMG-CoA is cleaved to acetyl-CoA and AcAc by HMG-CoA lyase (HMGCL), an enzyme that also supports leucine catabolism into ketones. Mitochondrial phosphatidylcholine-dependent, inner mitochondrial membrane–associated D-β-hydroxybutyrate dehydrogenase 1 (BDH1) reduces AcAc to D-βOHB in an NAD+/NADH coupled reaction (100). The AcAc/D-βOHB ratio is proportional to mitochondrial NAD+/NADH equilibrium (185). Although the molecular identities of the mitochondrial ketone transporter(s) remain unconfirmed, ketones are transported across the hepatocyte plasma membrane via monocarboxylate transporters 1 and 2 (MCT1 and MCT2) (see Section 2.4) into the circulation and then imported via MCTs into extrahepatic tissues (33, 60).
In extrahepatic mitochondria, BDH1 catalyzes the first reaction to convert D-βOHB back to AcAc (Figure 1) (100). Cytosolic D-β-hydroxybutyrate dehydrogenase 2 (BDH2) has only 20% sequence identity to BDH1, has much higher Km values for ketone bodies than does BDH1, and governs iron homeostasis (42). AcAc is converted to AcAc-CoA through a near equilibrium reaction catalyzed by succinyl-CoA:3-oxoacid-CoA transferase (SCOT, encoded by Oxct1) through a CoA exchange with succinyl-CoA. Greater free energy is released by the hydrolysis of AcAc-CoA than of succinyl-CoA, favoring AcAc formation; thus, ketone oxidation occurs due to mass action, with high AcAc substrate supply and rapid turnover of downstream acetyl-CoA via citrate synthase activity, together pulling AcAc toward the AcAc-CoA fate. SCOT-derived AcAc-CoA yields two acetyl-CoA molecules via a reversible AcAc-CoA thiolase reaction catalyzed by one of four mitochondrial thiolases, with acetyl-CoA acetyltransferase 1 (ACAT1) taking the largest role in ketone oxidation (10, 184). SCOT is excluded from hepatocytes, ensuring net efflux of ketones by the liver.
In mammalian extrahepatic tissues, ketone bodies can be terminally oxidized and then diverted into lipid synthesis, sterol synthesis, or other metabolic pathways (Figure 2) (6, 19). The fates of 13C-labeled AcAc in polarized macrophages were recently mapped using isotope tracing untargeted metabolomics (128, 129). Most AcAc-derived carbon labels the TCA cycle but can also contribute to itaconate. AcAc-derived citrate is also exported to the cytosol, notably contributing to the hexosamine biosynthetic pathway. AcAc can also be converted to AcAc-CoA via an ATP-dependent reaction catalyzed by cytoplasmic acetoacetyl-CoA synthetase (AACS). AcAc-CoA is converted to acetyl-CoA by cytoplasmic thiolases, contributing to sterol synthesis through the action of cytosolic 3-hydroxymethylglutaryl-CoA synthase 1 (HMGCS1), or carboxylated by ACC to malonyl-CoA, contributing to fatty acid synthesis (10, 181). The AACS gene is a target sterol regulatory element binding protein 2 (SREBP-2), a master transcriptional regulator of cholesterol synthesis, and knockdown of AACS leads to a decreased serum cholesterol level in mice (64). Thus, it is tempting to link ketone body sourcing of cholesterol to increased lipoprotein outcomes observed in clinical trials using low-carbohydrate ketogenic diets. Specifically, numerous studies have linked ketogenic diets to increased circulating lipoproteins, particularly low-density lipoprotein (LDL) cholesterol (134). However, this effect appears to be population dependent, and LDL particle size may be an important variable to consider, as ketogenic diets may selectively increase the abundance of larger, less pathogenic LDL particles (77). It is important to underscore that the effects of ketogenic diets are pleiotropic (see the sidebar titled Nutritional Provocation of Endogenous Ketogenesis), and it has not been formally demonstrated that ketogenic diet-induced ketones actually source cholesterol or whether the ketogenic diet milieu influences other aspects of complex lipoprotein homeostasis.
2.3. Transcriptional and Posttranslational Regulation of Ketogenic Mediators
In most mammals, HMGCS2 is most robustly expressed in hepatocytes and colonic epithelium. Loss-of-function HMGCS2 mutations in humans cause bouts of hypoketotic hypoglycemia and coma, as well as fatty liver (117), and hepatic knockdown of Hmgcs2 in adult mice causes hepatic injury and remodeled lipidome under high-fat-diet conditions (34, 40). Transcription of the HMGCS2 gene is silenced in fetal liver and nonketogenic tissues by methylation (Figure 3), which is reversed at birth, allowing the gene to become responsive to hormonal regulators (67). Post-natally, both insulin and glucagon regulate hepatic ketogenesis, whereby insulin inhibits adipose tissue lipolysis and thus removes the primary ketogenic substrate from the circulation and also suppresses the transcription of the Hmgcs2 gene by phosphatidylinositol-3-kinase/AKT-dependent inhibition of forkhead transcriptional factor FOXA2 (67, 187). Glucagon stimulates transcription of the Hmgcs2 gene through the cAMP-p300-FOXA2 signaling pathway (174). Free fatty acids or glucocorticoid induction of peroxisome proliferator activated receptor alpha (PPARα), and its target fibroblast growth factor 21 (FGF21), can also augment Hmgcs2 gene transcription (5). Suppression of PPARα and thus ketogenesis is also promoted by tumor-derived interleukin 6 (IL-6) expression (49). Fasting-induced ketogenesis may also be controlled by mammalian target of rapamycin complex 1 (mTORC1), attenuating PPARα signaling (148). The Hmgcs2 gene is also regulated by the circadian oscillator PER2 (24). However, it should be noted that HMGCS2 gene transcription and messenger RNA (mRNA) abundance are often directly correlated with ketogenesis, but causal relationships to ketogenesis, especially in extrahepatic tissues, are frequently not constructed in these studies. Importantly, dynamic expression of HMGCS2 protein in organs canonically associated with high ketone oxidative capacity, such as kidney or heart, likely does not support contribution of those organs to circulating ketone bodies, but it is possible that extrahepatic ketogenesis supports intercellular signaling shuttles (see Sections 2.5 and 4) (167, 182, 183, 193).
Figure 3.
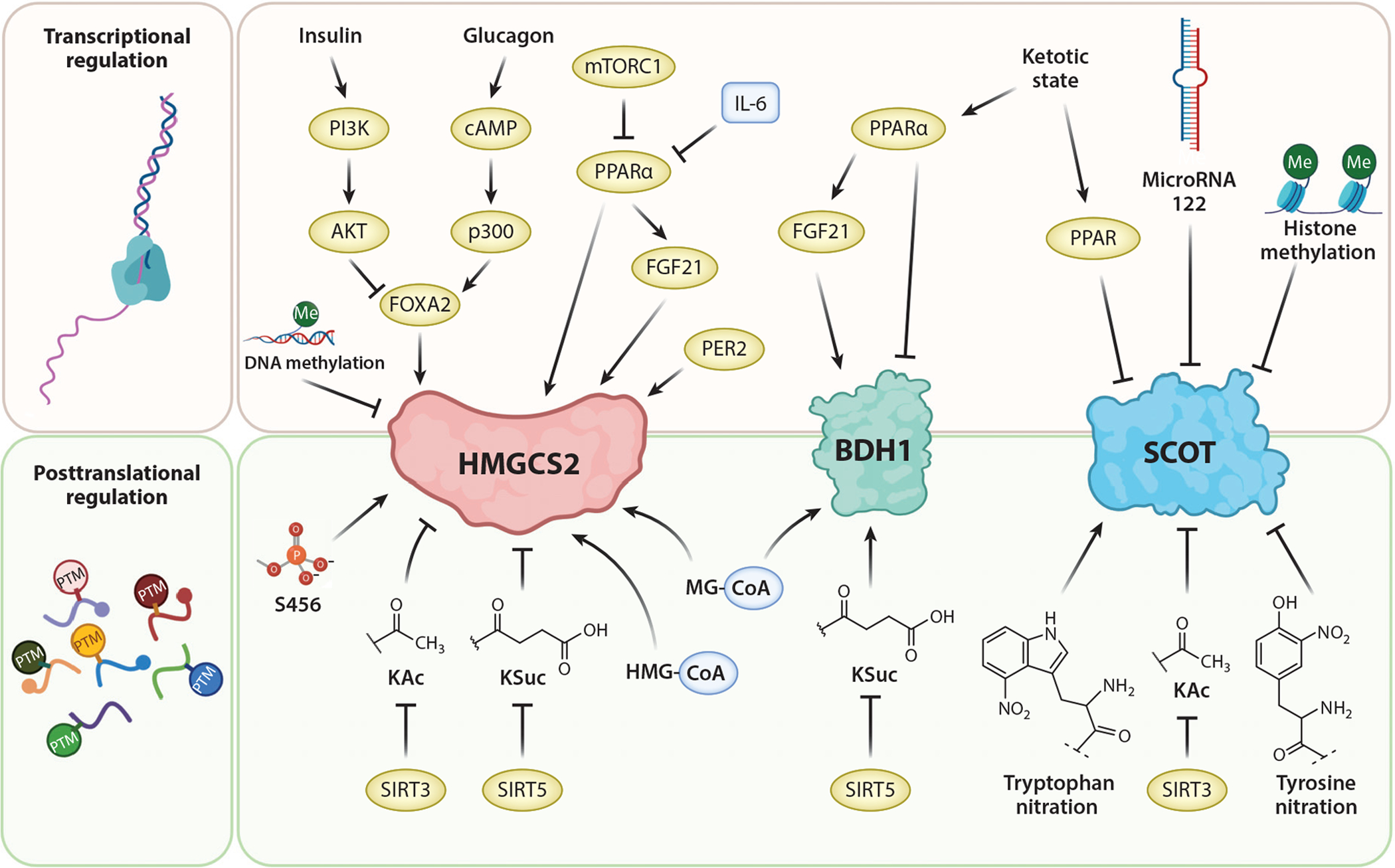
Regulatory mechanisms for key mitochondrial drivers of ketone body metabolism. HMGCS2 is a fate-committing ketogenic enzyme abundant in hepatocytes. BDH1 catalyzes the reduction/oxidation between AcAc and D-βOHB in hepatic and extrahepatic cells. SCOT is required for extrahepatic mitochondrial ketolysis. Abbreviations: AcAc, acetoacetate; BDH1, D-β-hydroxybutyrate dehydrogenase 1; D-βOHB, D-β-hydroxybutyrate; FGF21, fibroblast growth factor 21; HMG-CoA, 3-hydroxymethylglutaryl-CoA; HMGCS2, 3-hydroxymethylglutaryl-CoA synthase 2; IL-6, interleukin 6; KAc, lysine-acetylation; KSuc, lysine-succinylation; MG-CoA, 3-methylglutaryl-CoA; mTORC1, mammalian target of rapamycin complex 1; PI3K, phosphatidylinositol-3-kinase; PPARα, peroxisome proliferator activated receptor alpha; PTM, posttranslational modification; SCOT, succinyl-CoA:3-oxoacid-CoA transferase; SIRT, sirtuin. Figure adapted from images created with BioRender.com.
In contrast to HMGCS2, SCOT, encoded by the Oxct1 gene, is expressed in all mammalian mitochondria except those of hepatocytes. Inborn errors in SCOT function present early in life and result in severe ketoacidosis, lethargy, vomiting, and coma, requiring therapeutic intervention (51). Germline SCOT knockout (KO) mice are unable to oxidize ketones, which results in hyper-ketonemic hypoglycemia, causing neonatal death (33). Selective SCOT KO models do not cause lethality but do generate aberrant phenotypes (2, 35, 53, 129, 146). In hepatocytes, Oxct1 gene expression is suppressed during the fetal to neonatal transition by microRNA 122 and H3K27 histone methylation. Loss of Oxct1 expression in this transition is primarily attributable to the egress of Oxct1 mRNA-rich hematopoietic progenitors from the liver, rather than to diminishing Oxct1 gene expression in hepatocytes (116, 165). In persistently ketotic states, Oxct1 mRNA, SCOT protein, and activity in heart and muscle significantly diminish, possibly in a PPAR-dependent manner, decreasing ketone body disposal.
Loss of BDH1 function in humans has not been reported, suggesting either mild physiological or, less likely, severe developmental consequences. Global BDH1 deficiency in mice does not cause lethality but leads to subtle impairment of fasting liver lipid content (117). Through undefined transcriptional or posttranscriptional mechanisms, macrophages lack BDH1 expression and thus do not oxidize D-βOHB (129). Beyond macrophages, the range of cell types that selectively oxidize only AcAc remains unknown. Experimental loss of BDH1 that is selective in cardiac myocytes exacerbates heart failure in various challenges, suggesting an adaptive and protective function of D-βOHB oxidation in these cells (73) (see Section 3). As with Hmgcs2, Bdh1 in the liver is responsive to a ketogenic diet in a PPARα-FGF21-dependent manner (5), and in heart and skeletal muscle under ketotic states, Bdh1 gene expression becomes diminished, as does expression of Oxct1 (58).
HMGCS2, SCOT, and BDH1 are marked by various PTMs (Figure 3). In response to ketogenesis, catalytic activity of HMGCS2 is enhanced by serine phosphorylation (57). Sirtuin 3 (SIRT3) deacetylates lysine residues and may activate HMGCS2, while lysine residues of HMGCS2, HMGCL, and BDH1 are targets for succinylation in hepatic mitochondria (44, 133, 150). Hyper-succinylation or hyperacetylation of HMGCS2 in SIRT5 or SIRT3 KO mice, respectively, were correlated with lower βOHB production in fasted states, though physiological mechanistic connections were not studied (133, 150). Hyperacetylated SCOT in brains of SIRT3 KO mice was correlated with diminished ketolytic ability (44). Succinyl-CoA, 3-methylglutaryl-CoA, glutaryl-CoA, and HMG-CoA acylation nonenzymatically modify HMGCS2 and BDH1 (176). However, physiological regulatory roles of these PTMs all remain incompletely defined, and indeed the global impact of mitochondrial protein acylation has recently been questioned (47). Among other nonenzymatic modifications to SCOT are tyrosine or tryptophan nitration, which attenuates or augments enzymatic activity, respectively (12, 169).
2.4. Cellular Transport of Ketones
Concentrations of circulating ketone bodies are higher than those in extrahepatic tissues, indicating that ketone bodies are transported down a concentration gradient (63). A rapid, efficient and scalable method that quantifies both AcAc and βOHB in serum and tissues, and also differentiates D-enantiomers from L-enantiomers of βOHB, has recently been developed (130). AcAc and βOHB are released from cells predominantly through MCTs [in mammals, MCT1 and MCT2, also known as solute carrier (SLC) 16A family members 1 and 7, respectively, which also transport lactate] and transported through the circulation to extrahepatic tissues for terminal oxidation (33, 60). Loss-of-function mutations in MCT1 cause spontaneous bouts of ketoacidosis (170), which suggests a primary role for MCT1 in extrahepatic ketone body import but not an obligate role for ketone body efflux from hepatocytes. For both MCT1 and MCT2, no selectivity for AcAc versus βOHB has been observed (61). While dynamic expression of MCTs in varying physiological states has been posed to regulate ketone body turnover, mechanisms that dynamically regulate MCT abundance have not yet been proven to be either necessary or sufficient to augment ketone efflux or import (8). One study showed the requirement for MCT2 expression for the permissive role of βOHB in driving breast cancer tumorigenesis (74), but the relationship between MCT2 expression and tumorigenesis in breast cancer has not been observed uniformly (105).
Unlike ketone transport across the plasma membrane, mechanisms supporting ketone body transport across the inner mitochondrial membrane remain unknown (Figures 1, 2, and 4). While MCT localization to mitochondria has been posed (65), this possibility also raises controversy around the autonomy of the mitochondrial matrix to oxidize lactate (26). A fascinating property of AcAc is its ability to stimulate activity of the mitochondrial pyruvate carrier (MPC) complex, characterized long before revelation of the molecular identity of the MPC complex, SLC54A1–2 (13, 119). Mitochondrial efflux of AcAc stimulates pyruvate import, an effect that is abrogated by the MPC inhibitor α-cyanocinnamate and can be overcome by high pyruvate concentrations (198). It is very unlikely that ketone efflux proceeds through the MPC complex itself (106). Thus, whether ketones are transported across the inner mitochondrial membrane via SLC16A-dependent or orphan mitochondrial SLC-dependent mechanisms remains to be determined.
Figure 4.
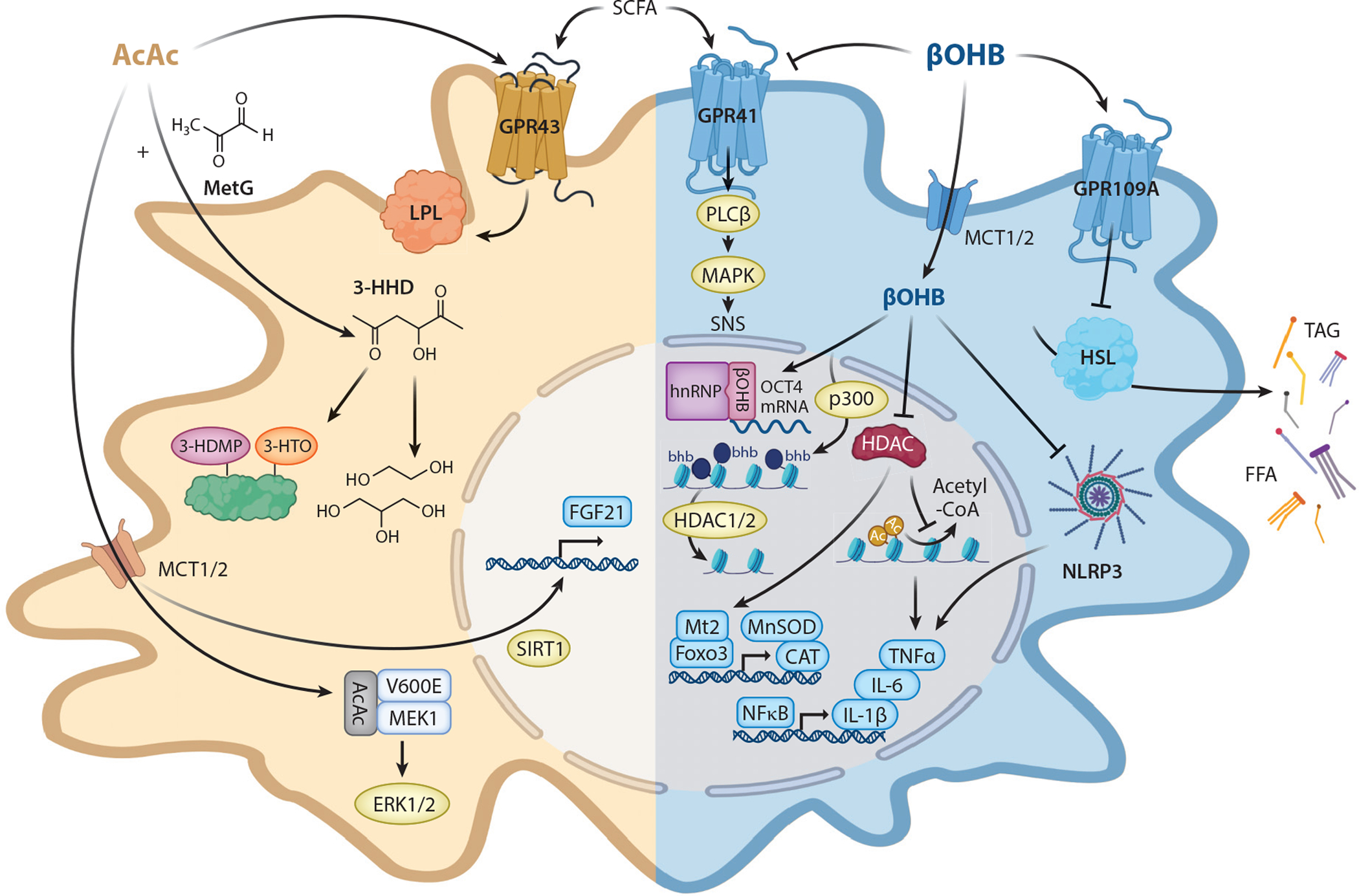
Noncanonical cellular responses to AcAc and βOHB. Ketone bodies exert different cellular effects through changes transduced by epigenetic, posttranslational, and cell surface signaling mechanisms. Abbreviations: 3-HDMP, 3-hydroxypyrrole-derivative; 3-HHD, 3-hydroxyhexane-2,5-dione; 3-HTO, 3-hexane-2,3,4-trione; AcAc, acetoacetate; bhb, lysine β-hydroxybutyrylation; βOHB, β-hydroxybutyrate; ERK, extracellular signal–regulated kinase; FFA, free fatty acid; FGF, fibroblast growth factor; GPR, G-protein coupled receptor; HDAC, histone deacetylase; HSL, hormone-sensitive lipase; IL, interleukin; LPL, lipoprotein lipase; MAPK, mitogen-activated protein kinase; MCT, monocarboxylate transporter; MEK, MAPK/ERK kinase; MetG, methylglyoxal; NFκB, nuclear factor kappa B; PLCβ, phospholipase C-β; SCFA, short-chain fatty acid; SIRT, sirtuin; SNS, sympathetic nervous system; TAG, triacylglycerol; TNF, tumor necrosis factor. Figure adapted from images created with BioRender.com.
2.5. Ketones and Signaling
AcAc and/or βOHB interact with nuclear ribonucleoproteins, inhibit histone deacetylases (HDACs), modify histones and other proteins posttranslationally, influence oxidative stress, inhibit the NLRP3 inflammasome, and modulate G-protein coupled receptor (GPR) signaling (Figure 4). βOHB, but not AcAc, interacts with nuclear ribonucleoprotein hnRNP A1, leading to upregulation of octamer binding transcriptional factor OCT4. This upregulation causes increased expression of Lamin B1, a key DNA senescence protective factor in vascular cells (62). Administration of exogenous βOHB (but not AcAc), fasting, or calorie restriction all increase histone acetylation by inhibiting class I HDACs, driving a shift in the transcriptome, including upregulation of oxidative stress-responsive genes (151). βOHB-mediated inhibition of HDAC-3-activated Fgf21 transcription in liver during the early neonatal period, and in cardiac muscle, increases H3K41 acetylation, attenuating cardiac endothelial hyperpermeability stimulated by high glucose (101, 132). Despite these observations, the impact of βOHB as a potent HDAC inhibitor may be substantially less than that of the classical HDAC inhibitor butyrate (31).
βOHB also covalently modifies histone lysine residues in a process termed β-hydroxybutyrylation that may occur through turnover cycles catalyzed by p300 and HDAC1/2 (76, 189). This was observed in cultured cells and in livers of animals subjected to prolonged fasting or streptozotocin-induced diabetes, with βOHB influencing a select pool of genes responsive to metabolic starvation. A comprehensive quantification of eight distinct histone acyl-PTMs showed that lysine β-hydroxybutyrylation marks emerge as a small fraction of total histone acylations, occupying only 2–5% of the total acylation pool in human histones (154).
AcAc has a distinct repertoire of signaling functions. Endogenously produced or exogenously provided AcAc promotes binding between the BRAF V600E isoform and MEK1, activating MEK-ERK signaling in BRAF-V600E+ cancer cells, promoting tumor growth (83, 188) (see Section 4). AcAc, but not βOHB, induces the expression of FGF21 in HepG2 cells in a SIRT1-dependent manner (173). Chemical products of AcAc may also covalently modify lysine residues. Methylglyoxal (MetG) is a reactive metabolite of glycolytic intermediates that accumulates, especially in diabetes. Via a nonenzymatic aldol reaction, AcAc buffers MetG, producing 3-hydroxyhexane-2,5-dione (3-HHD) (139). AcAc-derived 3-HHD is present as a significant fraction of the MetG pool in blood, and while 3-HHD is a nonglycating metabolite, it can be reduced to diols and triols in blood cells or lead to lysine modifications of human serum albumin. The physiological importance of these phenomena awaits investigation.
Both βOHB and AcAc modulate signaling through GPRs. βOHB, but not AcAc, suppresses sympathetic nervous system activity by antagonizing GPR41-expressed sympathetic ganglia, resulting in reduced energy expenditure and heart rate (90). Conversely, AcAc, but not βOHB, activates GPR43, a receptor expressed in adipocytes, whose deficiency inhibits fasting-induced lipid utilization, impairs plasma lipoprotein lipase activity, and decreases energy expenditure during fasting (110). βOHB, but not AcAc, exerts an antilipolytic effect through activation of the GPR109A receptor on adipocytes, creating a negative feedback loop in which ketosis inhibits adipocyte lipolysis, decreasing circulating free fatty acids, the prime ketogenic substrates (162). GPR41 and GPR43 receptors are also activated by short-chain fatty acids, but βOHB is the only known endogenous ligand of the GPR109A receptor, which is also the niacin receptor (162). Activation of GPR109A in retina suppresses secretion of proinflammatory markers in diabetes (52). Intriguingly, both mature adipocytes and retinal pigment epithelium have been proposed to increase local fatty acid oxidation and engage extrahepatic ketogenesis for paracrine signaling (1, 178). Indeed, the activation of GPR109A by βOHB has a relatively high threshold (EC50 ~ 0.8 mM), but it is possible that locally produced ketones may provide a concentrated signal.
Ketotic states and ketone bodies modulate inflammation and oxidative stress. Oxidative stress, common in inflammatory states, is caused by an imbalance between the abundance and elimination of reactive oxygen and nitrogen species. Prolonged nutrient deprivation or calorie restriction also reduce inflammation, but pathological ketosis is actually a proinflammatory state (191). Mechanistically, βOHB exerts anti-inflammatory effects on macrophages through activation of the GPR109A receptor as well as via inhibition of the NLRP3 inflammasome (9, 191). Inhibition of the NLRP3 inflammasome by βOHB—but not AcAc, butyrate, or acetate—reduces proinflammatory IL-1β, IL-18, and caspase-1 activation, independent of GPR109A (191). In diabetic retina, βOHB and niacin suppress tumor necrosis factor alpha (TNFα) secretion through GPR109A-dependent mechanisms (52). βOHB also suppresses inflammatory signatures via NLRP3 inflammasome inhibition in kidney, decreasing hypertension; in the central nervous system (CNS), supporting antidepressant effects, attenuating neurodegeneration, and improving spinal cord injury; in heart, improving pathogenic signatures of atherosclerosis and impairing the development of heart failure; and in liver, modulating oxidative stress response (17, 23, 81, 89, 152). Understandably, interest has recently emerged in using ketogenic therapies for anti-inflammatory COVID-19 therapeutics (160).
Adipose tissue is another target for ketone bodies. As previously mentioned, βOHB inhibits adipocyte lipolysis through the GPR109A receptor, while AcAc may favor adipocyte lipolysis via the GPR43 receptor (110, 162, 168). In addition, adipocytes exposed to cold or a β3-adrenergic agonist secrete βOHB in a PRDM16-dependent manner, promoting adipocyte beiging in aged animals (178). Interestingly, βOHB possibly produced through extrahepatic ketogenesis and paracrine signaling decreased adipose tissue fibrogenesis. Conversely, administration of exogenous βOHB induced, while AcAc inhibited, hepatic fibrosis, independent of inflammation (129). Moreover, not all studies support βOHB inhibition of the NLRP3 inflammasome (113), and high βOHB concentrations induce the oxidative stress markers malondialdehyde, nitric oxide, inducible nitric oxide synthase, and other nuclear factor kappa B (NFκB)-dependent proinflammatory cytokines (TNFα, IL-6, IL-1β) in calf hepatocytes, while decreasing the antioxidant scavenging response (149). Taken together, these studies highlight the necessity to correlate the specific ketone body studied with its effect. Moreover, those measured effects will be influenced by dosing levels, exposure time, and the specific cellular target of interest.
Finally, many studies underscore that the mechanisms underlying the salutary roles of ketone bodies—as signals versus as metabolic substrates—are not easily parsed. For example, administration of ketone body precursors or sodium glucose cotransporter 2 inhibitors (SGLT2i), which can increase circulating ketone bodies (see Section 3), suppressed kidney injury and attenuated hypertension (23, 79). In an ApoE KO model of diabetic kidney disease, SGLT2i ameliorated kidney damage due to mTORC1 hyperactivation (167). While these benefits appeared to be attributable to ketone bodies, future studies will be important to reveal mechanisms explaining these exciting observations.
3. KETONES AND HEART FAILURE
Under normal conditions, the heart flexibly burns available fuel sources, but diabetic or pathologically remodeled hearts (e.g., due to hypertension or myocardial infarction) become metabolically inflexible and abnormal structurally and hemodynamically (21, 84). Hearts normally oxidize ketone bodies in proportion to their delivery, in direct competition with fatty acid and glucose utilization, and myocardium is the highest ketone body consumer per unit mass (55, 73, 146, 171, 183). Ketone bodies have been posed as more energetically efficient than fatty acids, yielding more energy available for ATP synthesis per molecule of oxygen invested (85). However, experimental evidence supporting this energetic benefit has been inconsistent (70, 71).
Relationships between ketone bodies and heart failure have been recognized for decades, but interest in them has recently intensified. Circulating ketone body concentrations are increased in heart failure patients, in direct proportion to cardiac filling pressures, but the mechanism and significance of these observations remain unknown (7, 103, 138, 157). Although heart failure is associated with insulin resistance (135), insulin resistance alone does not provoke ketosis: Pre-diabetic states in the absence of heart failure are actually linked to diminished ketosis (48, 155). The failing heart oxidizes less fat (21, 96), potentially resulting in the diversion of ketogenic free fatty acids to the liver. Indeed, ketone body utilization is increased in failing hearts of mice (4) and humans (7, 112, 138, 157, 175), compared with normal hearts. Competence of the heart to perform ketone oxidation is likely adaptive, since mice with selective SCOT deficiency in cardiomyocytes exhibit accelerated pathological remodeling in response to pressure overload injury (146), and cardiomyocyte-selective BDH1 KO mice are also more susceptible to accelerated heart failure (73).
The presence of ketosis, and increased myocardial ketone oxidation, in heart failure suggests that augmented ketone body delivery could improve heart failure outcomes. This has stimulated interest in leveraging endogenous hepatic ketogenesis or exogenous ketone administration to treat heart failure (73, 115, 147) (see the sidebars titled Nutritional Provocation of Endogenous Ketogenesis and Nutritional Provocation with Exogenous Ketones). Exogenous βOHB decreases systemic vascular resistance, and increases cardiac output, in both control and heart failure human subjects. As such, it is not yet clear whether acute D-βOHB delivery would improve cardiac function through salutary effects on myocardial energetics, reduced afterload via relaxed vascular tone, or a combination of both (115). Myocardial energetic benefits of exogenous ketones to isolated working hearts have not been observed in all experimental settings (70, 71, 172).
NUTRITIONAL PROVOCATION WITH EXOGENOUS KETONES.
Ketosis can be achieved through ingestible ketone body precursors. Exogenously delivered D-βOHB and AcAc themselves are rapidly turned over (129, 136, 181), and ingestion or infusion of βOHB salts can provoke an untoward sodium load. R/S-1,3-butanediol is a relatively nontoxic dialcohol readily oxidized in the liver, yielding stable circulating D/L-βOHB concentrations of up to 5 mM within 2 h of administration for an additional 3 h (23). Other ketone esters, including monoester of R-1,3-butanediol and D-βOHB (i.e., R-3-hydroxybutyl R-βOHB), glyceryltris-βOHB, and R,S-1,3-butanediol acetoacetate diester, have similar kinetics that provide moderately more durable circulating ketone concentrations than do ingested ketones themselves (36, 159). Cyclic βOHB oligomers and encapsulated βOHB nanoparticles have also been used, but their physiological properties have not been as deeply characterized (28). The distinct formulations supporting exogenous ketone delivery influence rates of turnover, the balance of metabolic versus noncanonical signaling effects, and redox potential. Another important consideration is that exogenous ketone therapies will provoke ketone body import into hepatocyte mitochondria, a nonphysiological event. The consequences of net ketone body import into hepatocyte mitochondria have not been fully defined, but the impact of exogenous ketone therapies on liver health merit consideration (93, 111, 125, 129, 160, 198).
A potential link between myocardial ketone metabolism and pathological ventricular remodeling was proposed through the mortality and heart failure–related hospitalization benefits of patients taking SGLT2i for the management of type 2 diabetes (118). Selective SGLT2i benefits do not proceed through protection from myocardial ischemic insults and cannot be attributed to glycemic control since they have been observed in participants without diabetes. Indeed, while the molecular targets of SGLT2i are limited, and SGLT2 is not expressed in cardiomyocytes, these agents provoke pleiotropic physiological consequences (192). The systemic shift to fatty acid oxidation and increase in hepatic ketogenesis indicate that at least some of the cardiovascular mortality benefit observed with SGLT2i may be attributable to increased myocardial ketone metabolism. Indeed, SGLT2i treatment increased myocardial ketone utilization in a pig model of heart failure (141). However, ketosis is very modest in SGLT2i-treated subjects without diabetes, a population in which SGLT2i-driven cardiovascular mortality benefit is also observed (46). Irrespective of the impact of SGLT2i on myocardial ketone utilization, experiments in animal models have shown either increased cardiac work or left ventricular ejection fraction when SGLT2i are used to treat models of heart failure (15, 16, 172). Thus, while ketosis, myocardial ketone oxidation, and SGLT2 inhibition are all associated, these should not be conflated. SGLT2i-derived hemodynamic and clinical benefit may correlate with increased ketosis, and ketones may exert benefits to an energetically challenged heart—but this correlation does not prove causation extending from SGLT2i through ketone bodies to the heart. SGLT2i clinical benefit is likely multifactorial.
4. KETONES AND TUMORIGENESIS
The roles of ketone bodies in cancer have been of long-standing interest that has recently intensified. To support tumor biomass expansion, cancer cells primarily metabolize glucose through glycolysis—the Warburg effect—and also activate the pentose phosphate pathway and lipogenesis, usually limiting mitochondrial respiration (83, 153). Cancer cells deprived of glucose shift to alternative fuel sources such as acetate, glutamine, or even fatty acids (32, 158, 190). Whether the plasticity of cancer cells applies to ketone bodies remains under investigation.
Hepatic ketogenic insufficiency is associated with a more severe nonalcoholic fatty liver disease (NAFLD) phenotype (34, 40, 180) (see Section 6). In addition, insufficient AcAc oxidation in liver macrophages leads to steatosis-independent, but high-fat-diet-induced, hepatic fibrosis, an early stage of progression toward hepatocellular carcinoma (also known as hepatoma) development (129). Hepatocellular carcinoma is associated with hepatocyte loss of ketogenic capacity, as BDH1 and HMGCS2 are decreased (179, 180, 196), while Oxct1/SCOT, normally excluded from hepatocytes, is augmented, enabling ketone oxidation (75, 196). Loss of HMGCS2 expression is correlated with poor prognosis in both primary mammalian ketogenic organs, the liver and the colon (20, 25, 197) (see Section 5). In hepatocellular carcinoma, disease progression correlates with both HMGCS2 expression and ketones (179), while in colorectal cancer and oral squamous cell carcinoma, the role of HMGCS2 is ketone independent, suggesting a moonlighting function of HMGCS2 in those cancers (25).
Therapeutic roles for ketone metabolism have also drawn attention for malignancies outside the liver and colon. βOHB has been shown to either support (74, 83, 137, 188) or inhibit (38) cancer cell growth and tumorigenesis. Screening of human cancer cells based on the expression of BDH1/OXCT1 distinguished cancer cells with inhibited (PANC-1) or accelerated (HeLa) cell growth rate upon βOHB treatment (194). Proliferation of HeLa cells through βOHB supplementation was BDH1 and OXCT1 dependent, because knockdown of both BDH1 and OXCT1 decreased tumor growth rate in the presence of a ketogenic diet in vivo.
Ketone bodies may influence tumorigenesis and cancer cell growth through fulfillment of cell energy demands or signaling mechanisms (74, 83, 137, 188). The metabolic fate of ketone bodies may influence their ultimate impact on cancer cells; for example, a βOHB paradox model suggests that cells less capable of oxidizing ketones may be more responsive to HDAC inhibition (137). Intriguingly, oncogenic BRAF-V600E+ melanoma cells upregulate HMGCL in an OCT1-dependent manner, leading to local AcAc synthesis in cells incapable of its oxidation, which facilitates V600E-MEK1 binding and induces MEK-ERK signaling events that stimulate cell proliferation and tumor growth (see also Section 2.5) (83, 188).
A local ketone shuttle model poses that stromal cells produce ketone bodies to fulfill energy demands of cancer cells (74, 105). This model could also be consistent with hepatocellular carcinoma development, in which ketogenic hepatocytes support the growth of SCOT-expressing (pre)hepatoma cells (196). Similar models have also been proposed for breast cancer, whereby either adjacent fibroblast-derived or mammary gland–derived adipocyte metabolism enables extrahepatic ketogenesis (74, 105). A βOHB shuttle between mammary gland adipocytes and neighboring MCT2+ breast cancer cells promoted tumorigenesis in a mouse model by increasing global histone H3K9 acetylation that induced IL-1β and LCN2, potentially driving poorer prognosis (74). Characterization of stimuli that rewire cellular programming of nonhepatic stromal cells to activate extrahepatic ketogenesis is worthy of exploration as a potential therapeutic target.
The responses of cancer in vivo to ketogenic diet interventions vary (see the sidebar titled Nutritional Provocation of Endogenous Ketogenesis). High-fat, low-carbohydrate ketogenic diets can either inhibit (72, 153) or accelerate (72, 158, 188, 195) tumor growth. Indeed, while obesity is generally associated with poorer cancer prognosis, different dietary factors can influence tumor growth and its environment (102). As described above, a ketogenic diet promotes BRAF-V600E+ melanoma tumor growth. Conversely, a ketogenic diet inhibited pancreatic tumor growth (153, 194) but enhanced cervical cancer (HeLa) growth in a BDH1/OXCT1-dependent manner (194). In addition, the efficacy of phosphatidylinositol-3-kinase (PI3K) inhibitors is augmented by a ketogenic diet. PI3K-inhibitor-induced hyperglycemia and hyperinsulinemia are often hypothesized to drive tumor resistance (72). A ketogenic diet enhanced PI3K-inhibitor drug efficacy not by modulating glucose levels but by inhibiting the activation of tumor insulin receptors. The authors noted that a ketogenic diet alone induced variable effects, in some instances even accelerating tumor growth. In other studies, endocrine therapy for breast cancer was enhanced by fasting and a fasting mimicking diet through the lowering of circulating insulin growth factor 1 (IGF1) and insulin (18). Elucidation of the comprehensive and systematic impacts of dietary metabolic signatures is required to understand how a ketogenic diet impacts both tumor cancer and stromal cells and whether these effects are transduced through ketone bodies themselves.
5. KETONES, THE GUT, AND THE MICROBIOME
Ketone bodies are generated in the mammalian gut epithelium through HMGCS2 (69). In both small and large intestine, HMGCS2 mRNA and protein are regulated by the microbiome, butyrate content, and the diet (29, 30). Although both small and large intestinal epithelia undergo continuous turnover, HMGCS2 is expressed in very distinct cell types between these two organs, suggesting diverse functions along the gastrointestinal tract (Figure 5) (20, 28, 29, 177). In the large intestine, HMGCS2 expression exhibits spatial variation from the proximal to distal colon and is predominantly expressed in differentiated (not proliferating) colonocytes, and signals that induce cell proliferation downregulate HMGCS2 (20, 69, 177). Conversely, in the small intestine, HMGCS2 is enriched in self-renewing Lgr5+ intestinal stem cells in the intestinal crypt (28).
Figure 5.
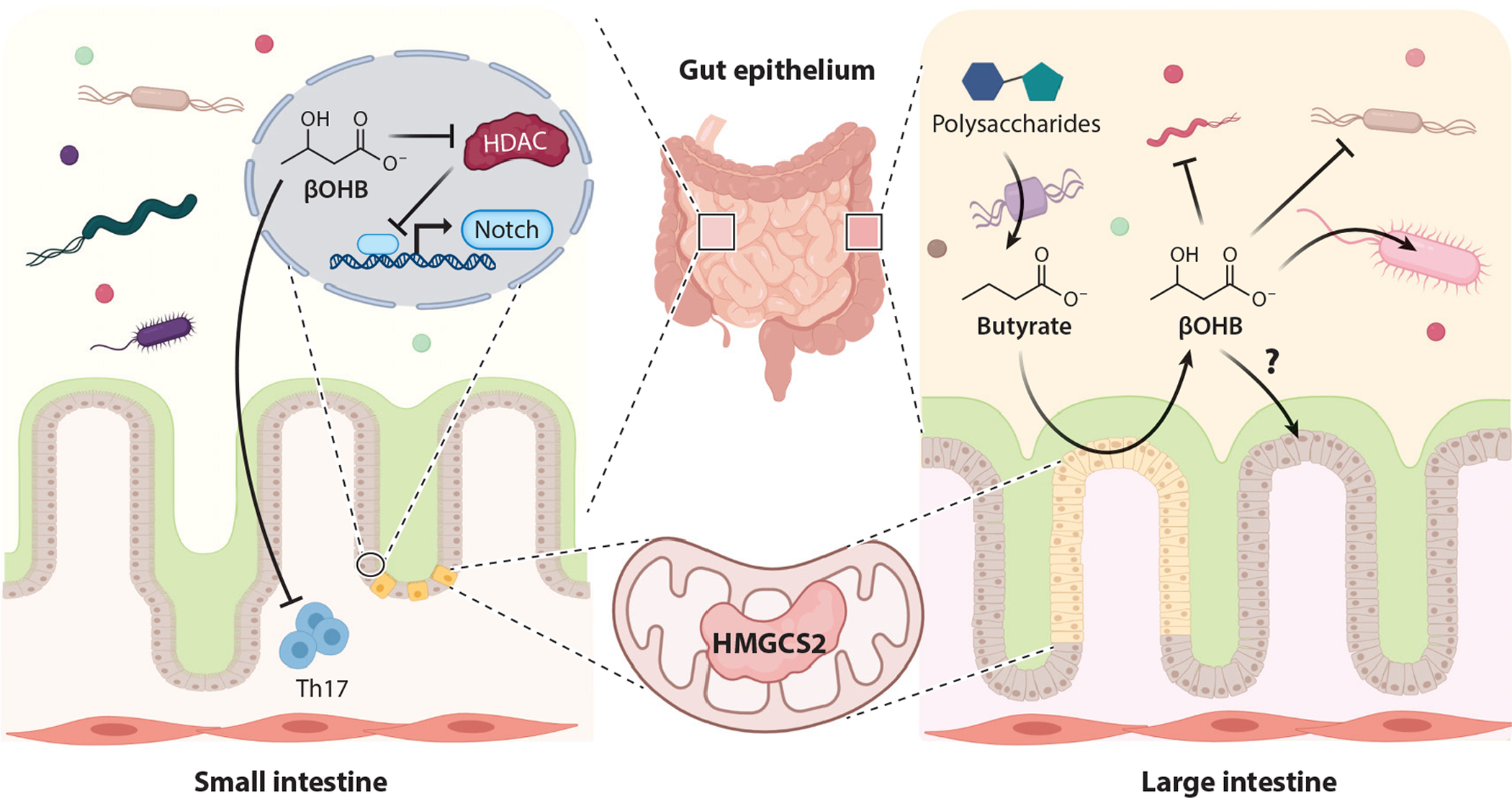
Overview of ketone body metabolism in small and large intestine. HMGCS2 distribution and the roles of βOHB in the small and large intestine vary. In small intestine, βOHB triggers Notch signaling and Th17 immunomodulatory changes. In large intestine, anaerobic bacterial fermentation yields butyrate, a source of βOHB that influences the gut community. Abbreviations: βOHB, β-hydroxybutyrate; HDAC, histone deacetylase; HMGCS2, 3-hydroxymethylglutaryl-CoA synthase. Figure adapted from images created with BioRender.com.
Cecal mucosa converts butyrate, derived from microbial fermentation of polysaccharides, to ketone bodies (69). Conversion of butyrate into ketone bodies correlates with enzymatic activity of HMGCS2. Although ketogenic substrates in small intestine have not been defined, small intestinal HMGCS2 is required for postnatal gut development and for the response to intestinal injury, as loss of HMGCS2 in Lgr5+ progenitor cells redirects them toward a secretory fate (28). In cultured colonocytes, loss or gain of HMGCS2 leads to attenuated or promoted differentiation, respectively (177). In both small intestine and colonocytes, loss of HMGCS2 function is rescued at least partially by administration of exogenous βOHB or a ketogenic diet, suggesting ketogenesis-dependent HMGCS2 function (28, 179). HMGCS2-dependent small intestinal stem cell function regulates the Notch signaling axis through HDAC inhibition (28).
High-fat ketogenic diets are composed of low carbohydrate contents (see the sidebar titled Nutritional Provocation of Endogenous Ketogenesis). Carbohydrates are the primary source of gut-microbially derived short-chain fatty acids, and adherence to a ketogenic diet in mice markedly changes the microbial taxonomy along the gastrointestinal tract (3, 37). When rigorously controlled for protein content, a progressive decline in carbohydrate content gradually increased circulating ketones, and this increase was associated with decreases in Bifidobacterium and Lactobacillus abundances. Circulating insulin and glucose concentrations were not provided in this study, and it remains possible that subtle micronutrient variations among test diets could play roles in community structure (see the sidebar titled Nutritional Provocation of Endogenous Ketogenesis). However, in vitro and in vivo studies supported the notion that growth of discrete bacterial strains are modulated by the presence of βOHB or ketone esters (3, 143), and ketone esters or a ketogenic diet modulated not only the growth of selected bacterial strains but also intestinal proinflammatory Th17 cells in small intestine (3).
It remains unclear whether gut-derived ketone bodies reach the systemic circulation or whether locally sourced ketone bodies function solely in the intestinal mucosa. Another outstanding question is the extent to which liver-derived ketones might directly impact gut mucosal immunity and microbial ecology. Enterocyte-selective loss of HMGCS2 function will be required to address these questions, as will assessment of the role of SCOT-dependent ketone oxidation: Though these factors have not been studied extensively, some studies suggest that small intestine mucosa could have machinery supporting ketolysis (28). βOHB also may serve as an HDAC inhibitor, but given its diminished potency compared with butyrate (31)—and the fact that the concentration of butyrate in colonic lumen reaches ~10 mM (69)—the autonomous role of HDAC inhibition by βOHB, at least in the colon, will need to be evaluated cautiously. Other signaling roles are possible (see Section 2.5) in governing growth and fitness of specific bacterial clades. Finally, while the majority of gut ketogenic literature focuses on the role of βOHB, a significant amount of AcAc is also produced in the gut (30, 69), and the expression and role of BDH1 in the gut are unknown.
6. KETONES, OBESITY, AND NONALCOHOLIC FATTY LIVER DISEASE
Twenty-five percent of the United States’ population has NAFLD, a multiorgan condition including excessive hepatic TAG storage alone (simple steatosis) or steatosis plus inflammation, liver cell injury, and liver cell death [nonalcoholic steatohepatitis (NASH)]. Cirrhosis due to NASH develops in 2% of all Americans, driven through failed regeneration of healthy liver, promoting hepatic stellate cell differentiation into myofibroblasts and execution of fibrogenesis.
Specific roles of mitochondrial dysfunction remain unclear in NAFLD pathogenesis. While not all studies have been concordant (123), four independent methodologies report increased hepatic TCA cycle flux, anaplerosis, and/or gluconeogenesis in obesity and/or NAFLD (45, 78, 91, 144, 161). Oxidative flux provides a fate for a portion of excess fat but does not fully compensate for the hepatocyte load, contributing to ectopic fat accumulation. Moreover, while increased fat oxidation could mitigate accumulation of toxic lipid species, oxidation may also increase super-oxide production by the electron transport chain (144). Over the evolution from simple steatosis to NASH, hepatic mitochondrial oxidative fluxes may decrease, but sentinel predictors, drivers, and consequences remain unresolved (91, 122).
An opposed model of NAFLD pathogenesis contends that mitochondrial fat oxidation is diminished, or inappropriately normal, and that clearing liver fat via therapeutic increases in fat oxidation could confer therapeutic benefit. Indeed, interest is high in the use of agents that augment liver fat oxidation, including ketogenic diets, intermittent fasting, and ketogenic pharmacotherapies (e.g., ACC inhibitors, FGF21, and SGLT2i) to treat obesity, insulin resistance, and NAFLD (54, 88, 140, 142). However, numerous examples exist in skeletal muscle (114) and in liver (99) whereby decreasing fuel mitochondrial supply optimizes energy supply/demand balance in a manner that may not decrease steatosis but could diminish tissue injury and insulin resistance. Ketogenesis provides an index of hepatic fat oxidation, but it is an incomplete reporter: Independent quantifications of both ketogenesis and TCA cycle flux are required to provide a complete index of hepatic fat oxidation. A recent study confirmed that fasting hepatic ketogenesis rates are inversely correlated with the severity of hepatic steatosis in human NAFLD, while fasting TCA cycle flux is directly correlated with hepatic steatosis (48). Decreasing fasting ketogenesis with progressive NAFLD is consistent with prior reports comparing human liver biopsies, which show that hepatic HMGCS2 and BDH1 protein decrease in progressive human NASH, with commensurate decreases in fasting circulating ketone body concentrations (82). Moreover, programmed loss of hepatic ketone metabolism is linked to increased liver injury and fibrosis, both through hepatocyte-autonomous ketogenic mechanisms and through a hepatocyte-hepatic sinusoidal macrophage exchange (an intrahepatic ketone shuttle), in which local hepatic macrophages oxidize ketone bodies produced by neighboring hepatocytes (34, 129). Future studies are needed to independently dissect β-oxidation, ketogenesis, TCA cycle flux, de novo lipogenesis, and the influence of hepatocyte BDH1 over mitochondrial redox potential and signaling in NAFLD pathogenesis.
7. KETONES AND THE NERVOUS SYSTEM
7.1. Feeding Behavior and Energy Expenditure
The brain, to which more than 20% of the body’s energy expenditure is allocated, avidly oxidizes ketones, and much of its energy needs can be sustained by ketone bodies (19). The rationale for leveraging ketone metabolism to support brain health originates from the benefits of ketogenic diets or starvation for seizure disorders (156). Interest in the brain-ketone relationship in regulating energy homeostasis emerged when it was recognized that sustained infusion of βOHB into the third ventricle of rats caused weight loss without a reduction in food intake (Figure 6) (41). Intracerebroventricular ketone infusion potentiates leptin signaling in the hypothalamus (120) and injection of βOHB into the paraventricular or ventromedial hypothalamic nuclei increases sympathetic neuron-firing rate. Long-term infusion and systemic administration of ketones may suppress food intake (95), but acute infusion of ketone bodies via the carotid artery caused hyperphagia with increased hypothalamic peptides NPY and AgRP (22). Moreover, liver-derived ketone bodies may contribute to a food-entrainable oscillator (food anticipation) in communication with the brain (24).
Figure 6.
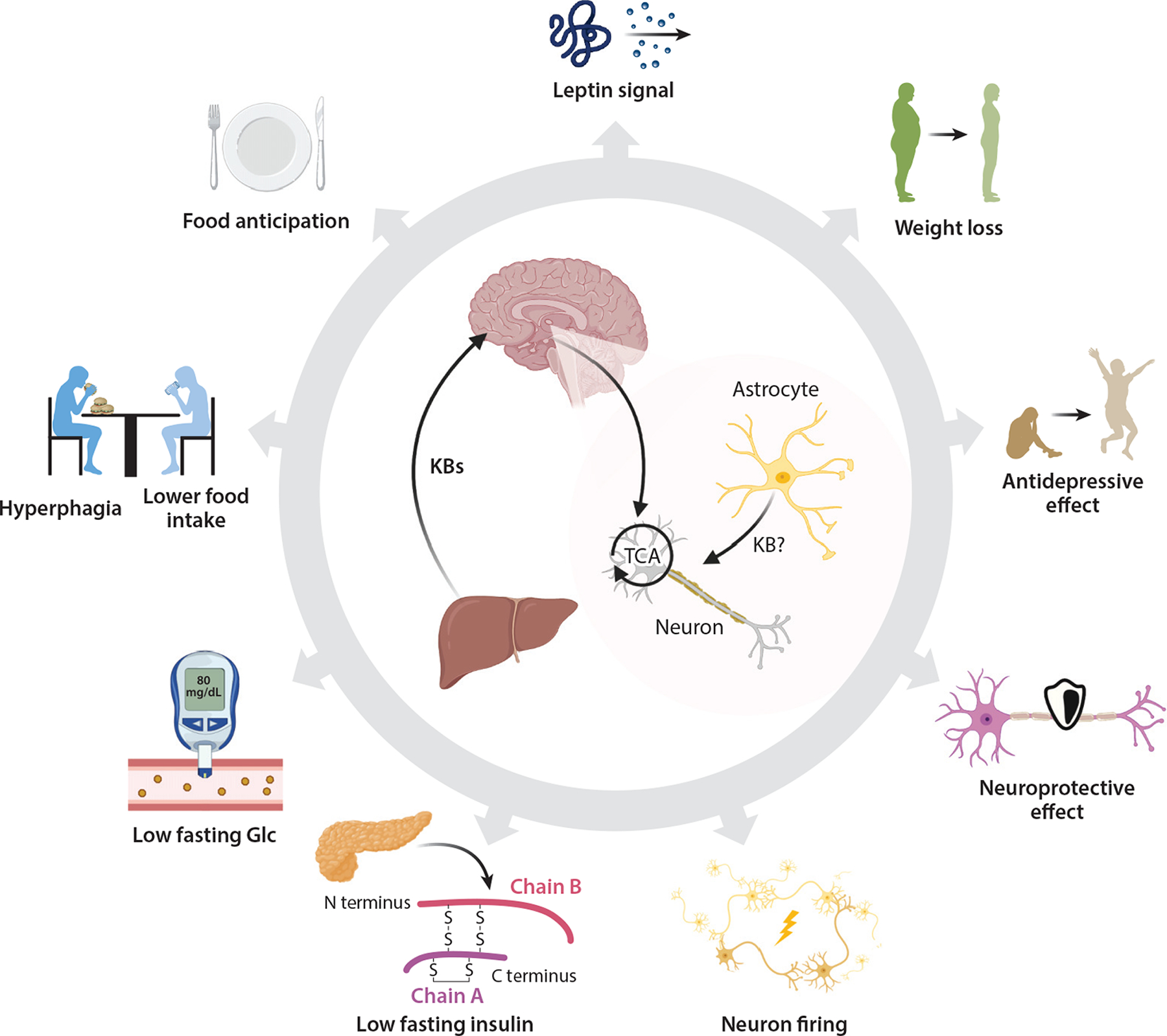
Integrative role of ketone bodies in the nervous system. Mechanisms supporting the pleiotropic effects of ketone bodies or a ketogenic diet in the nervous system still require elucidation. However, effects on feeding behavior, energy expenditure, mood and behavior, and neuroprotection have all been observed. Abbreviations: Glc, glucose; KB, ketone body; TCA, tricarboxylic acid. Figure adapted from images created with BioRender.com.
Although ketone bodies are avidly oxidized by the brain, the requirement for ketone oxidation to transduce the signals that modulate energy homeostasis has not been proven, and nonmetabolic signaling mechanisms may play a contributing role (50, 127). FGF21-driven augmentation of neuronal ketone oxidation suggests a potential role for ketone oxidation in states promoting metabolic health (86). FGF21 is an endocrine hormone produced primarily by the liver (11), signaling through the CNS to regulate hepatic gluconeogenesis and ketogenesis. Administration of FGF21 to obese animal models increases energy expenditure and causes weight loss, and extended administration of FGF21 analogs to obese humans reduces fasting glucose and insulin levels as well as body weight. These effects of FGF21 are also mediated through actions on the CNS through only partially identified mechanisms (80). Although the association between induction of FGF21 and hepatic ketogenesis is understood (5, 173), and ketones and FGF21 both have prominent roles in signaling in the brain, very little is known about crosstalk between ketone and FGF21 signaling in the brain.
Due to ketogenic capacity, the primary source of ketones for the brain is the liver. However, endogenous ketogenesis in astrocytes has been postulated as a local source for neurons (59), and local astrocyte-derived ketogenesis has been posed to mediate these hypothalamic signaling effects (97, 163). However, adult astrocytes do not appear to express HMGCS2 (163). Moreover, ketone bodies are oxidized by astrocytes. Taken together, the drivers, CNS nuclear and cell sub-population targets, outcomes, and mechanisms of ketone signaling in the brain all remain only preliminarily defined. The recently demonstrated ability of astrocyte mitochondrial integrity to coordinate adaptive feedback mechanisms in the regulation of central energy and glucose homeostasis provides a prospective opportunity to assess the integrative role of ketone metabolism within the astrocyte cellular compartment (166).
7.2. Therapies for Diseases of the Nervous System: Seizure Disorders and Neurodegeneration
Abnormalities of glucose metabolism in the brain that accompany, and possibly drive, neurodegeneration have been met with interest in leveraging ketone bodies as a fuel alternative to glucose (39, 92). Patients with mild cognitive impairment or Alzheimer’s disease also have impaired insulin sensitivity and VO2 max compared with controls, factors both linked to mitochondrial function. However, ketone body metabolism may remain intact, and cognitive impairments linked to diminished glucose metabolism may improve with ketone bodies. Moreover, the fact that caloric restriction improves cognition in aged animals raises the hypothesis that this effect is at least partially attributable to ketone metabolism. A recent study suggests that a ketogenic diet, fasting, or ingestion of exogenous R-3-hydroxybutyl R-βOHB (ketone ester) (see the sidebar titled Nutritional Provocation with Exogenous Ketones) improved functional communication among brain regions by changing the predominant dietary fuel from glucose to ketones (111). This communication was mapped via functional magnetic resonance imaging and was termed network stability, whereby brain network destabilization correlates with decreased brain activity and cognitive acuity. This provocative finding will require mechanistic dissection, because while D-βOHB is a highly efficient fuel, its acute exogenous delivery also increases cerebral blood flow by nearly 40% (66).
Multiple prospective mechanisms have linked ketone bodies to preservation of cognitive function. One study found that ketone ester administration increased neuronal expression of the mitochondrial NAD+-dependent deacetylase SIRT3, thereby supporting the survival of GABAergic interneurons and reducing neuronal network hyperexcitability and seizure-related death in a mouse model of Alzheimer’s disease (27). Other experiments have linked neuroprotection in a different Alzheimer’s mouse model to the ability of βOHB to inhibit the NLRP3 inflammasome (152). Finally, low plasma βOHB has been associated with social impairments, depression, and brain white matter alterations among patients with alcohol use disorders (98). The authors of this study posed a relationship among ethanol-producing gut microbiota, signatures of diminished lipolysis, and a loss of βOHB, which eliminated a source of neuroprotection. Nutritional oscillations of carbohydrate content, exercise, and the microbiome all may confer salutary effects on cognitive and emotional well-being. However, key questions remain whether oscillations in ketogenic flux primarily drive these benefits, and if so, whether this occurs through changes in metabolic fuel dynamics, improving mitochondrial energetics, or through signaling effects on mitophagy, mitochondrial biogenesis, gene transcription, and neuroinflammation.
8. PROSPECTUS
The next five years of investigation into the roles of ketone metabolism will reveal additional mechanisms governing ketone body production and their diverse roles in cell, tissue, organ, and organismal homeostasis. The most impactful findings will reveal whether observed phenotypes proceed through the ability of ketone bodies to serve as fuel substrates; as modulators of redox potential; as modulators of substrate supply/demand balance; or as signals transmitted through both cell surface and intracellular targets, perhaps as metabolized products or adducts. These experiments will parse the driver mechanisms proceeding independently through AcAc, D-βOHB, or L-βOHB and assess the prospect of locally produced ketones communicating signals via local shuttles. The role of BDH1, governed by its spatial and temporal cell type specificity, will open new territories in mitochondrial and downstream signaling. Mediators that support ketone transit across mitochondrial membranes hold promise as gatekeepers with both metabolic and signaling consequences. Key processes and targets to be addressed will be in the spaces of mucosal immunity, heart failure, neurodegeneration, and neoplastic disease. Large animal and human participant clinical trials should continue to complement rodent studies, to account for variation that may imperfectly replicate in humans. Finally, whether ketone body metabolism has an autonomous ability to orchestrate the salutary effects of oscillating diet regimens and exercise on wellness remains an unanswered question of great interest.
ACKNOWLEDGMENTS
The authors are grateful for support from NIH (grants DK091538 and AG069781). The authors regret that space limitations precluded inclusion of many impactful contributions.
Footnotes
DISCLOSURE STATEMENT
P.A.C. has served as an external consultant for Pfizer, Inc., Abbott Laboratories, and Janssen Research & Development.
LITERATURE CITED
- 1.Adijanto J, Du J, Moffat C, Seifert EL, Hurley JB, Philp NJ. 2014. The retinal pigment epithelium utilizes fatty acids for ketogenesis: implications for metabolic coupling with the outer retina. J. Biol. Chem 289:20570–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al Batran R, Gopal K, Capozzi ME, Chahade JJ, Saleme B, et al. 2020. Pimozide alleviates hyperglycemia in diet-induced obesity by inhibiting skeletal muscle ketone oxidation. Cell Metab. 31:909–19. e8 [DOI] [PubMed] [Google Scholar]
- 3.Ang QY, Alexander M, Newman JC, Tian Y, Cai J, et al. 2020. Ketogenic diets alter the gut microbiome resulting in decreased intestinal Th17 cells. Cell 181:1263–75. e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, et al. 2016. The failing heart relies on ketone bodies as a fuel. Circulation 133:698–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. 2007. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5:426–37 [DOI] [PubMed] [Google Scholar]
- 6.Balasse EO, Fery F. 1989. Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes Metab. Rev 5:247–70 [DOI] [PubMed] [Google Scholar]
- 7.Bedi KC Jr., Snyder NW, Brandimarto J, Aziz M, Mesaros C, et al. 2016. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation 133:706–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beland-Millar A, Takimoto M, Hamada T, Messier C. 2020. Brain and muscle adaptation to high-fat diets and exercise: metabolic transporters, enzymes and substrates in the rat cortex and muscle. Brain Res. 1749:147126. [DOI] [PubMed] [Google Scholar]
- 9.Benyó Z, Gille A, Kero J, Csiky M, Suchánková MC, et al. 2005. GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J. Biol. Chem 115:3634–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergstrom JD, Wong GA, Edwards PA, Edmond J. 1984. The regulation of acetoacetyl-CoA synthetase activity by modulators of cholesterol synthesis in vivo and the utilization of acetoacetate for cholesterogenesis. J. Biol. Chem 259:14548–53 [PubMed] [Google Scholar]
- 11.BonDurant LD, Potthoff MJ. 2018. Fibroblast growth factor 21: a versatile regulator of metabolic homeostasis. Annu. Rev. Nutr 38:173–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bregere C, Rebrin I, Gallaher TK, Sohal RS. 2010. Effects of age and calorie restriction on tryptophan nitration, protein content, and activity of succinyl-CoA:3-ketoacid CoA transferase in rat kidney mitochondria. Free Radic. Biol. Med 48:609–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, et al. 2012. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Browning JD, Baxter J, Satapati S, Burgess SC. 2012. The effect of short-term fasting on liver and skeletal muscle lipid, glucose, and energy metabolism in healthy women and men. J. Lipid Res 53:577–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byrne NJ, Matsumura N, Maayah ZH, Ferdaoussi M, Takahara S, et al. 2020. Empagliflozin blunts worsening cardiac dysfunction associated with reduced NLRP3 (nucleotide-binding domain-like receptor protein 3) inflammasome activation in heart failure. Circ. Heart Fail 13:e006277. [DOI] [PubMed] [Google Scholar]
- 16.Byrne NJ, Parajuli N, Levasseur JL, Boisvenue J, Beker DL, et al. 2017. Empagliflozin prevents worsening of cardiac function in an experimental model of pressure overload-induced heart failure. JACC Basic Transl. Sci 2:347–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrne NJ, Soni S, Takahara S, Ferdaoussi M, Al Batran R, et al. 2020. Chronically elevating circulating ketones can reduce cardiac inflammation and blunt the development of heart failure. Circ. Heart Fail 13:e006573. [DOI] [PubMed] [Google Scholar]
- 18.Caffa I, Spagnolo V, Vernieri C, Valdemarin F, Becherini P, et al. 2020. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 583:620–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cahill GF Jr. 2006. Fuel metabolism in starvation. Annu. Rev. Nutr 26:1–22 [DOI] [PubMed] [Google Scholar]
- 20.Camarero N, Mascaró C, Mayordomo C, Vilardell F, Haro D, Marrero PF. 2006. Ketogenic HMGCS2 is a c-Myc target gene expressed in differentiated cells of human colonic epithelium and down-regulated in colon cancer. Mol. Cancer Res 4:645–53 [DOI] [PubMed] [Google Scholar]
- 21.Carley AN, Taegtmeyer H, Lewandowski ED. 2014. Mechanisms linking energy substrate metabolism to the function of the heart. Circ. Res 114:717–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carneiro L, Geller S, Fioramonti X, Hébert A, Repond C, et al. 2016. Evidence for hypothalamic ketone body sensing: impact on food intake and peripheral metabolic responses in mice. Am. J. Physiol. Endocrinol. Metab 310:E103–15 [DOI] [PubMed] [Google Scholar]
- 23.Chakraborty S, Galla S, Cheng X, Yeo JY, Mell B, et al. 2018. Salt-responsive metabolite, β-hydroxybutyrate, attenuates hypertension. Cell Rep. 25:677–89. e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chavan R, Feillet C, Costa SS, Delorme JE, Okabe T, et al. 2016. Liver-derived ketone bodies are necessary for food anticipation. Nat. Commun 7:10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen SW, Chou CT, Chang CC, Li YJ, Chen ST, et al. 2017. HMGCS2 enhances invasion and metastasis via direct interaction with PPARα to activate Src signaling in colorectal cancer and oral cancer. Oncotarget 8:22460–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YJ, Mahieu NG, Huang X, Singh M, Crawford PA, et al. 2016. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol 12:937–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng A, Wang J, Ghena N, Zhao Q, Perone I, et al. 2020. SIRT3 haploinsufficiency aggravates loss of GABAergic interneurons and neuronal network hyperexcitability in an Alzheimer’s disease model. J. Neurosci 40:694–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng CW, Biton M, Haber AL, Gunduz N, Eng G, et al. 2019. Ketone body signaling mediates intestinal stem cell homeostasis and adaptation to diet. Cell 178:1115–31. e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cherbuy C, Andrieux C, Honvo-Houeto E, Thomas M, Ide C, et al. 2004. Expression of mitochondrial HMGCoA synthase and glutaminase in the colonic mucosa is modulated by bacterial species. Eur. J. Biochem 271:87–95 [DOI] [PubMed] [Google Scholar]
- 30.Cherbuy C, Darcy-Vrillon B, Morel MT, Pegorier JP, Duee PH. 1995. Effect of germfree state on the capacities of isolated rat colonocytes to metabolize n-butyrate, glucose, and glutamine. Gastroenterology 109:1890–99 [DOI] [PubMed] [Google Scholar]
- 31.Chriett S, Dabek A, Wojtala M, Vidal H, Balcerczyk A, Pirola L. 2019. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep 9:742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Comerford SA, Huang Z, Du X, Wang Y, Cai L, et al. 2014. Acetate dependence of tumors. Cell 159:1591–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cotter DG, d’Avignon DA, Wentz AE, Weber ML, Crawford PA. 2011. Obligate role for ketone body oxidation in neonatal metabolic homeostasis. J. Biol. Chem 286:6902–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cotter DG, Ercal B, Huang X, Leid JM, d’Avignon DA, et al. 2014. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig 124:5175–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cotter DG, Schugar RC, Wentz AE, d’Avignon DA, Crawford PA. 2013. Successful adaptation to ketosis by mice with tissue-specific deficiency of ketone body oxidation. Am. J. Physiol. Endocrinol. Metab 304:E363–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, et al. 2016. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 24:256–68 [DOI] [PubMed] [Google Scholar]
- 37.Crawford PA, Crowley JR, Sambandam N, Muegge BD, Costello EK, et al. 2009. Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. PNAS 106:11276–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui W, Luo W, Zhou X, Lu Y, Xu W, et al. 2019. Dysregulation of ketone body metabolism is associated with poor prognosis for clear cell renal cell carcinoma patients. Front. Oncol 9:1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, et al. 2020. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov 19:609–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.d’Avignon DA, Puchalska P, Ercal B, Chang Y, Martin SE, et al. 2018. Hepatic ketogenic insufficiency reprograms hepatic glycogen metabolism and the lipidome. JCI Insight 3(12):e99762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis JD, Wirtshafter D, Asin KE, Brief D. 1981. Sustained intracerebroventricular infusion of brain fuels reduces body weight and food intake in rats. Science 212:81–83 [DOI] [PubMed] [Google Scholar]
- 42.Davuluri G, Song P, Liu Z, Wald D, Sakaguchi TF, et al. 2016. Inactivation of 3-hydroxybutyrate dehydrogenase 2 delays zebrafish erythroid maturation by conferring premature mitophagy. PNAS 113:E1460–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Cabo R, Mattson MP. 2019. Effects of intermittent fasting on health, aging, and disease. N. Engl. J. Med 381:2541–51 [DOI] [PubMed] [Google Scholar]
- 44.Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, Fotuhi Siahpirani A, et al. 2015. SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. 21:637–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Felig P, Wahren J, Hendler R, Brundin T. 1974. Splanchnic glucose and amino acid metabolism in obesity. J. Biol. Chem 53:582–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, et al. 2016. Shift to fatty substrate utilization in response to sodium-glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes 65:1190–95 [DOI] [PubMed] [Google Scholar]
- 47.Fisher-Wellman KH, Draper JA, Davidson MT, Williams AS, Narowski TM, et al. 2019. Respiratory phenomics across multiple models of protein hyperacylation in cardiac mitochondria reveals a marginal impact on bioenergetics. Cell Rep. 26:1557–72. e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fletcher JA, Deja S, Satapati S, Fu X, Burgess SC, Browning JD. 2019. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 5:e127737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, et al. 2016. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 24:672–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu SP, Liu BR, Wang JF, Xue WJ, Liu HM, et al. 2015. β-Hydroxybutyric acid inhibits growth hormone-releasing hormone synthesis and secretion through the GPR109A/extracellular signal-regulated 1/2 signalling pathway in the hypothalamus. J. Neuroendocrinol 27:212–22 [DOI] [PubMed] [Google Scholar]
- 51.Fukao T, Lopaschuk GD, Mitchell GA. 2004. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot. Essent. Fatty Acids 70:243–51 [DOI] [PubMed] [Google Scholar]
- 52.Gambhir D, Ananth S, Veeranan-Karmegam R, Elangovan S, Hester S, et al. 2012. GPR109A as an anti-inflammatory receptor in retinal pigment epithelial cells and its relevance to diabetic retinopathy. Investig. Ophthalmol. Vis. Sci 53:2208–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.García-Caballero M, Zecchin A, Souffreau J, Truong ACK, Teuwen LA, et al. 2019. Role and therapeutic potential of dietary ketone bodies in lymph vessel growth. Nat. Metab 1:666–75 [DOI] [PubMed] [Google Scholar]
- 54.Goedeke L, Peng L, Montalvo-Romeral V, Butrico GM, Dufour S, et al. 2019. Controlled-release mitochondrial protonophore (CRMP) reverses dyslipidemia and hepatic steatosis in dysmetabolic nonhuman primates. Sci. Transl. Med 11:eaay0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gormsen LC, Svart M, Thomsen HH, Sondergaard E, Vendelbo MH, et al. 2017. Ketone body infusion with 3-hydroxybutyrate reduces myocardial glucose uptake and increases blood flow in humans: a positron emission tomography study. J. Am. Heart Assoc 6:e005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Green A, Bishop RE. 2019. Ketoacidosis—Where do the protons come from? Trends Biochem. Sci 44:484–89 [DOI] [PubMed] [Google Scholar]
- 57.Grimsrud PA, Carson JJ, Hebert AS, Hubler SL, Niemi NM, et al. 2012. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 16:672–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grinblat L, Pacheco Bolanos LF, Stoppani AO. 1986. Decreased rate of ketone-body oxidation and decreased activity of D-3-hydroxybutyrate dehydrogenase and succinyl-CoA:3-oxo-acid CoA-transferase in heart mitochondria of diabetic rats. Biochem. J 240:49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guzman M, Blazquez C. 2001. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol. Metab 12:169–73 [DOI] [PubMed] [Google Scholar]
- 60.Halestrap AP. 2012. The monocarboxylate transporter family—structure and functional characterization. IUBMB Life 64:1–9 [DOI] [PubMed] [Google Scholar]
- 61.Halestrap AP, Wilson MC. 2012. The monocarboxylate transporter family—role and regulation. IUBMB Life 64:109–19 [DOI] [PubMed] [Google Scholar]
- 62.Han YM, Bedarida T, Ding Y, Somba BK, Lu Q, et al. 2018. β-Hydroxybutyrate prevents vascular senescence through hnRNP A1-mediated upregulation of Oct4. Mol. Cell 71:1064–78. e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harrison HC, Long CNH. 1940. The distribution of ketone bodies in tissues. J. Biol. Chem 133:209–18 [Google Scholar]
- 64.Hasegawa S, Noda K, Maeda A, Matsuoka M, Yamasaki M, Fukui T. 2012. Acetoacetyl-CoA synthetase, a ketone body-utilizing enzyme, is controlled by SREBP-2 and affects serum cholesterol levels. Mol. Genet. Metab 107:553–60 [DOI] [PubMed] [Google Scholar]
- 65.Hashimoto T, Masuda S, Taguchi S, Brooks GA. 2005. Immunohistochemical analysis of MCT1, MCT2 and MCT4 expression in rat plantaris muscle. J. Physiol 567:121–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hasselbalch SG, Madsen PL, Hageman LP, Olsen KS, Justesen N, et al. 1996. Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. Am. J. Physiol. Endocrinol. Metab 270:E746–51 [DOI] [PubMed] [Google Scholar]
- 67.Hegardt FG. 1999. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem. J 338(Part 3):569–82 [PMC free article] [PubMed] [Google Scholar]
- 68.Hennebelle M, Courchesne-Loyer A, St-Pierre V, Vandenberghe C, Castellano CA, et al. 2016. Preliminary evaluation of a differential effect of an α-linolenate-rich supplement on ketogenesis and plasma omega-3 fatty acids in young and older adults. Nutrition 32:1211–16 [DOI] [PubMed] [Google Scholar]
- 69.Henning SJ, Hird FJ. 1972. Ketogenesis from butyrate and acetate by the caecum and the colon of rabbits. Biochem. J 130:785–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho KL, Karwi QG, Wagg C, Zhang L, Vo K, et al. 2020. Ketones can become the major fuel source for the heart but do not increase cardiac efficiency. Cardiovasc. Res 117(4):1178–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ho KL, Zhang L, Wagg C, Al Batran R, Gopal K, et al. 2019. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res 115:1606–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hopkins BD, Pauli C, Du X, Wang DG, Li X, et al. 2018. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560:499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, et al. 2019. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 4:e124079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang CK, Chang PH, Kuo WH, Chen CL, Jeng YM, et al. 2017. Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via β-hydroxybutyrate. Nat. Commun 8:14706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang D, Li T, Wang L, Zhang L, Yan R, et al. 2016. Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell Res. 26:1112–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang H, Zhang D, Weng Y, Delaney K, Tang Z, et al. 2021. The regulatory enzymes and protein substrates for the lysine β-hydroxybutyrylation pathway. Sci. Adv 7:eabe2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hyde PN, Sapper TN, Crabtree CD, LaFountain RA, Bowling ML, et al. 2019. Dietary carbohydrate restriction improves metabolic syndrome independent of weight loss. JCI Insight 4(12):e128308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Iozzo P, Bucci M, Roivainen A, Nagren K, Jarvisalo MJ, et al. 2010. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology 139:846–56. e6 [DOI] [PubMed] [Google Scholar]
- 79.Ishimwe JA, Garrett MR, Sasser JM. 2020. 1,3-Butanediol attenuates hypertension and suppresses kidney injury in female rats. Am. J. Physiol. Ren. Physiol 319:F106–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jensen-Cody SO, Flippo KH, Claflin KE, Yavuz Y, Sapouckey SA, et al. 2020. FGF21 signals to glutamatergic neurons in the ventromedial hypothalamus to suppress carbohydrate intake. Cell Metab. 32:273–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kajitani N, Iwata M, Miura A, Tsunetomi K, Yamanashi T, et al. 2020. Prefrontal cortex infusion of beta-hydroxybutyrate, an endogenous NLRP3 inflammasome inhibitor, produces antidepressant-like effects in a rodent model of depression. Neuropsychopharmacol. Rep 40:157–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kakehashi A, Stefanov VE, Ishii N, Okuno T, Fujii H, et al. 2017. Proteome characteristics of non-alcoholic steatohepatitis liver tissue and associated hepatocellular carcinomas. Int. J. Mol. Sci 18(2):434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kang HB, Fan J, Lin R, Elf S, Ji Q, et al. 2015. Metabolic rewiring by oncogenic BRAF V600E links ketogenesis pathway to BRAF-MEK1 signaling. Mol. Cell 59:345–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Karwi QG, Biswas D, Pulinilkunnil T, Lopaschuk GD. 2020. Myocardial ketones metabolism in heart failure. J. Card. Fail 26:998–1005 [DOI] [PubMed] [Google Scholar]
- 85.Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell DA, et al. 1994. Control of glucose utilization in working perfused rat heart. J. Biol. Chem 269:25502–14 [PubMed] [Google Scholar]
- 86.Katsu-Jimenez Y, Gimenez-Cassina A. 2019. Fibroblast growth factor-21 promotes ketone body utilization in neurons through activation of AMP-dependent kinase. Mol. Cell. Neurosci 101:103415. [DOI] [PubMed] [Google Scholar]
- 87.Kennedy AR, Pissios P, Otu H, Xue B, Asakura K, et al. 2007. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am. J. Physiol. Endocrinol. Metab 292:E1724–39 [DOI] [PubMed] [Google Scholar]
- 88.Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, et al. 2017. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 26:394–406. e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim SR, Lee SG, Kim SH, Kim JH, Choi E, et al. 2020. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun 11:2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, et al. 2011. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). PNAS 108:8030–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, et al. 2015. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 21:739–46 [DOI] [PubMed] [Google Scholar]
- 92.Koppel SJ, Swerdlow RH. 2018. Neuroketotherapeutics: a modern review of a century-old therapy. Neurochem. Int 117:114–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Koutnik AP, Poff AM, Ward NP, DeBlasi JM, Soliven MA, et al. 2020. Ketone bodies attenuate wasting in models of atrophy. J. Cachexia Sarcopenia Muscle 11:973–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kuhla A, Hahn S, Butschkau A, Lange S, Wree A, Vollmar B. 2014. Lifelong caloric restriction reprograms hepatic fat metabolism in mice. J. Gerontol. A Biol. Sci. Med. Sci 69:915–22 [DOI] [PubMed] [Google Scholar]
- 95.Laeger T, Metges CC, Kuhla B. 2010. Role of β-hydroxybutyric acid in the central regulation of energy balance. Appetite 54:450–55 [DOI] [PubMed] [Google Scholar]
- 96.Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, et al. 2014. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ. Heart Fail 7:1022–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Le Foll C, Levin BE. 2016. Fatty acid-induced astrocyte ketone production and the control of food intake. Am. J. Physiol. Regul. Integr. Comp. Physiol 310:R1186–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Leclercq S, Le Roy T, Furgiuele S, Coste V, Bindels LB, et al. 2020. Gut microbiota-induced changes in β-hydroxybutyrate metabolism are linked to altered sociability and depression in alcohol use disorder. Cell Rep. 33:108238. [DOI] [PubMed] [Google Scholar]
- 99.Lee J, Choi J, Selen Alpergin ES, Zhao L, Hartung T, et al. 2017. Loss of hepatic mitochondrial long-chain fatty acid oxidation confers resistance to diet-induced obesity and glucose intolerance. Cell Rep. 20:655–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lehninger AL, Sudduth HC, Wise JB. 1960. D-β-Hydroxybutyric dehydrogenase of mitochondria. J. Biol. Chem 235:2450–55 [PubMed] [Google Scholar]
- 101.Li B, Yu Y, Liu K, Zhang Y, Geng Q, et al. 2021. β-Hydroxybutyrate inhibits histone deacetylase 3 to promote claudin-5 generation and attenuate cardiac microvascular hyperpermeability in diabetes. Diabetologia 64:226–39 [DOI] [PubMed] [Google Scholar]
- 102.Lien EC, Vander Heiden MG. 2019. A framework for examining how diet impacts tumour metabolism. Nat. Rev. Cancer 19:651–61 [DOI] [PubMed] [Google Scholar]
- 103.Lommi J, Kupari M, Koskinen P, Naveri H, Leinonen H, et al. 1996. Blood ketone bodies in congestive heart failure. J. Am. Coll. Cardiol 28:665–72 [DOI] [PubMed] [Google Scholar]
- 104.London ED, Margolin RA, Duara R, Holloway HW, Robertson-Tchabo EA, et al. 1986. Effects of fasting on ketone body concentrations in healthy men of different ages. J. Gerontol 41:599–604 [DOI] [PubMed] [Google Scholar]
- 105.Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Lisanti MP, Sotgia F. 2012. Ketone bodies and two-compartment tumor metabolism: Stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 11(21):3956–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, et al. 2015. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab. 22:682–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McGarry JD, Foster DW. 1971. The regulation of ketogenesis from octanoic acid. The role of the tricarboxylic acid cycle and fatty acid synthesis. J. Biol. Chem 246:1149–59 [PubMed] [Google Scholar]
- 108.McGarry JD, Foster DW. 1980. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem 49:395–420 [DOI] [PubMed] [Google Scholar]
- 109.Miller VJ, LaFountain RA, Barnhart E, Sapper TS, Short J, et al. 2020. A ketogenic diet combined with exercise alters mitochondrial function in human skeletal muscle while improving metabolic health. Am.J. Physiol. Endocrinol. Metab 319:E995–1007 [DOI] [PubMed] [Google Scholar]
- 110.Miyamoto J, Ohue-Kitano R, Mukouyama H, Nishida A, Watanabe K, et al. 2019. Ketone body receptor GPR43 regulates lipid metabolism under ketogenic conditions. PNAS 116:23813–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mujica-Parodi LR, Amgalan A, Sultan SF, Antal B, Sun X, et al. 2020. Diet modulates brain network stability, a biomarker for brain aging, in young adults. PNAS 117:6170–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, et al. 2020. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 370:364–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Neudorf H, Myette-Cote E, Little JP. 2020. The impact of acute ingestion of a ketone monoester drink on LPS-stimulated NLRP3 activation in humans with obesity. Nutrients 12(3):854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Neufer PD. 2019. Cutting fuel offers new clues in diabetic mystery. J. Biol. Chem 294:12328–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nielsen R, Moller N, Gormsen LC, Tolbod LP, Hansson NH, et al. 2019. Cardiovascular effects of treatment with the ketone body 3-hydroxybutyrate in chronic heart failure patients. Circulation 139:2129–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Orii KE, Fukao T, Song XQ, Mitchell GA, Kondo N. 2008. Liver-specific silencing of the human gene encoding succinyl-CoA: 3-ketoacid CoA transferase. Tohoku J. Exp. Med 215:227–36 [DOI] [PubMed] [Google Scholar]
- 117.Otsuka H, Kimura T, Ago Y, Nakama M, Aoyama Y, et al. 2020. Deficiency of 3-hydroxybutyrate dehydrogenase (BDH1) in mice causes low ketone body levels and fatty liver during fasting. J. Inherit. Metab. Dis 43:960–68 [DOI] [PubMed] [Google Scholar]
- 118.Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, et al. 2020. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med 383:1413–24 [DOI] [PubMed] [Google Scholar]
- 119.Paradies G, Papa S. 1976. Substrate regulation of the pyruvate-transporting system in rat liver mitochondria. FEBS Lett. 62:318–21 [DOI] [PubMed] [Google Scholar]
- 120.Park S, Kim DS, Daily JW. 2011. Central infusion of ketone bodies modulates body weight and hepatic insulin sensitivity by modifying hypothalamic leptin and insulin signaling pathways in type 2 diabetic rats. Brain Res. 1401:95–103 [DOI] [PubMed] [Google Scholar]
- 121.Pedersen KJ. 1929. The ketonic decomposition of beta-keto carboxylic acids. J. Am. Chem. Soc 51:2098–107 [Google Scholar]
- 122.Peng KY, Watt MJ, Rensen S, Greve JW, Huynh K, et al. 2018. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res 59:1977–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Petersen KF, Befroy DE, Dufour S, Rothman DL, Shulman GI. 2016. Assessment of hepatic mitochondrial oxidation and pyruvate cycling in NAFLD by 13C magnetic resonance spectroscopy. Cell Metab. 24:167–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pissios P, Hong S, Kennedy AR, Prasad D, Liu FF, Maratos-Flier E. 2013. Methionine and choline regulate the metabolic phenotype of a ketogenic diet. Mol. Metab 2:306–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Poff AM, Koutnik AP, Egan B. 2020. Nutritional ketosis with ketogenic diets or exogenous ketones: features, convergence, and divergence. Curr. Sports Med. Rep 19:251–59 [DOI] [PubMed] [Google Scholar]
- 126.Pramfalk C, Pavlides M, Banerjee R, McNeil CA, Neubauer S, et al. 2015. Sex-specific differences in hepatic fat oxidation and synthesis may explain the higher propensity for NAFLD in men. J. Clin. Endocrinol. Metab 100:4425–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Puchalska P, Crawford PA. 2017. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 25:262–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Puchalska P, Huang X, Martin SE, Han X, Patti GJ, Crawford PA. 2018. Isotope tracing untargeted metabolomics reveals macrophage polarization-state-specific metabolic coordination across intracellular compartments. iScience 9:298–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Puchalska P, Martin SE, Huang X, Lengfeld JE, Daniel B, et al. 2019. Hepatocyte-macrophage acetoacetate shuttle protects against tissue fibrosis. Cell Metab. 29:383–98. e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Puchalska P, Nelson AB, Stagg DB, Crawford PA. 2021. Determination of ketone bodies in biological samples via rapid UPLC-MS/MS. Talanta 225:122048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Quant PA, Robin D, Robin P, Girard J, Brand MD. 1989. Control of acetoacetate production from exogenous palmitoyl-CoA in isolated rat liver mitochondria. Biochem. Soc. Trans 17:1089–90 [DOI] [PubMed] [Google Scholar]
- 132.Rando G, Tan CK, Khaled N, Montagner A, Leuenberger N, et al. 2016. Glucocorticoid receptor-PPARα axis in fetal mouse liver prepares neonates for milk lipid catabolism. eLife 5:e11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, et al. 2013. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18:920–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Retterstol K, Svendsen M, Narverud I, Holven KB. 2018. Effect of low carbohydrate high fat diet on LDL cholesterol and gene expression in normal-weight, young adults: a randomized controlled study. Atherosclerosis 279:52–61 [DOI] [PubMed] [Google Scholar]
- 135.Riehle C, Abel ED. 2016. Insulin signaling and heart failure. Circ. Res 118:1151–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Robinson AM, Williamson DH. 1980. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev 60:143–87 [DOI] [PubMed] [Google Scholar]
- 137.Rodrigues LM, Uribe-Lewis S, Madhu B, Honess DJ, Stubbs M, Griffiths JR. 2017. The action of β-hydroxybutyrate on the growth, metabolism and global histone H3 acetylation of spontaneous mouse mammary tumours: evidence of a β-hydroxybutyrate paradox. Cancer Metab. 5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ruiz M, Labarthe F, Fortier A, Bouchard B, Thompson Legault J, et al. 2017. Circulating acylcarnitine profile in human heart failure: a surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am. J. Physiol. Heart Circ. Physiol 313:H768–81 [DOI] [PubMed] [Google Scholar]
- 139.Salomón T, Sibbersen C, Hansen J, Britz D, Svart MV, et al. 2017. Ketone body acetoacetate buffers methylglyoxal via a non-enzymatic conversion during diabetic and dietary ketosis. Cell Chem. Biol 24:935–43. e7 [DOI] [PubMed] [Google Scholar]
- 140.Samuel VT, Shulman GI. 2018. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 27:22–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, et al. 2019. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J. Am. Coll. Cardiol 73:1931–44 [DOI] [PubMed] [Google Scholar]
- 142.Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, et al. 2019. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 392:2705–17 [DOI] [PubMed] [Google Scholar]
- 143.Sasaki K, Sasaki D, Hannya A, Tsubota J, Kondo A. 2020. In vitro human colonic microbiota utilises D-β-hydroxybutyrate to increase butyrogenesis. Sci. Rep 10:8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Satapati S, Kucejova B, Duarte JA, Fletcher JA, Reynolds L, et al. 2015. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Biol. Chem 125:4447–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schugar RC, Huang X, Moll AR, Brunt EM, Crawford PA. 2013. Role of choline deficiency in the fatty liver phenotype of mice fed a low protein, very low carbohydrate ketogenic diet. PLOS ONE 8:e74806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schugar RC, Moll AR, d’Avignon DA, Weinheimer CJ, Kovacs A, Crawford PA. 2014. Cardiomyocytespecific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol. Metab 3:754–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Selvaraj S, Kelly DP, Margulies KB. 2020. Implications of altered ketone metabolism and therapeutic ketosis in heart failure. Circulation 141:1800–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini D. 2010. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468:1100–4 [DOI] [PubMed] [Google Scholar]
- 149.Shi X, Li X, Li D, Li Y, Song Y, et al. 2014. β-Hydroxybutyrate activates the NF-κB signaling pathway to promote the expression of pro-inflammatory factors in calf hepatocytes. Cell. Physiol. Biochem 33:920–32 [DOI] [PubMed] [Google Scholar]
- 150.Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, et al. 2010. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12:654–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, et al. 2013. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339:211–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shippy DC, Wilhelm C, Viharkumar PA, Raife TJ, Ulland TK. 2020. β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J. Neuroinflamm 17:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P. 2014. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Simithy J, Sidoli S, Yuan ZF, Coradin M, Bhanu NV, et al. 2017. Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun 8:1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Soeters MR, Sauerwein HP, Faas L, Smeenge M, Duran M, et al. 2009. Effects of insulin on ketogenesis following fasting in lean and obese men. Obesity 17:1326–31 [DOI] [PubMed] [Google Scholar]
- 156.Sondhi V, Agarwala A, Pandey RM, Chakrabarty B, Jauhari P, et al. 2020. Efficacy of ketogenic diet, modified Atkins diet, and low glycemic index therapy diet among children with drug-resistant epilepsy: a randomized clinical trial. JAMA Pediatr. 174:944–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Song JP, Chen L, Chen X, Ren J, Zhang NN, et al. 2020. Elevated plasma β-hydroxybutyrate predicts adverse outcomes and disease progression in patients with arrhythmogenic cardiomyopathy. Sci. Transl. Med 12:eaay8329. [DOI] [PubMed] [Google Scholar]
- 158.Sperry J, Condro MC, Guo L, Braas D, Vanderveer-Harris N, et al. 2020. Glioblastoma utilizes fatty acids and ketone bodies for growth allowing progression during ketogenic diet therapy. iScience 23:101453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Stubbs BJ, Cox PJ, Evans RD, Santer P, Miller JJ, et al. 2017. On the metabolism of exogenous ketones in humans. Front. Physiol 8:848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stubbs BJ, Koutnik AP, Goldberg EL, Upadhyay V, Turnbaugh PJ, et al. 2020. Investigating ketone bodies as immunometabolic countermeasures against respiratory viral infections. Med 1:43–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sunny NE, Parks EJ, Browning JD, Burgess SC. 2011. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 14:804–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, et al. 2005. (D)-β-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem 280:26649–52 [DOI] [PubMed] [Google Scholar]
- 163.Thevenet J, De Marchi U, Domingo JS, Christinat N, Bultot L, et al. 2016. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J. 30:1913–26 [DOI] [PubMed] [Google Scholar]
- 164.Thomas LK, Ittmann M, Cooper C. 1982. The role of leucine in ketogenesis in starved rats. Biochem. J 204:399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Thorrez L, Laudadio I, Van Deun K, Quintens R, Hendrickx N, et al. 2011. Tissue-specific disallowance of housekeeping genes: the other face of cell differentiation. Genome Res. 21:95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Timper K, Del Rio-Martin A, Cremer AL, Bremser S, Alber J, et al. 2020. GLP-1 receptor signaling in astrocytes regulates fatty acid oxidation, mitochondrial integrity, and function. Cell Metab. 31:1189–205. e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tomita I, Kume S, Sugahara S, Osawa N, Yamahara K, et al. 2020. SGLT2 inhibition mediates protection from diabetic kidney disease by promoting ketone body-induced mTORC1 inhibition. Cell Metab. 32:404–19. e6 [DOI] [PubMed] [Google Scholar]
- 168.Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, et al. 2003. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat. Med 9:352–55 [DOI] [PubMed] [Google Scholar]
- 169.Turko IV, Marcondes S, Murad F. 2001. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoA-transferase. Am. J. Physiol. Heart Circ. Physiol 281:H2289–94 [DOI] [PubMed] [Google Scholar]
- 170.van Hasselt PM, Ferdinandusse S, Monroe GR, Ruiter JP, Turkenburg M, et al. 2014. Monocarboxylate transporter 1 deficiency and ketone utilization. N. Engl. J. Med 371:1900–7 [DOI] [PubMed] [Google Scholar]
- 171.Vanoverschelde JL, Wijns W, Kolanowski J, Bol A, Decoster PM, et al. 1993. Competition between palmitate and ketone bodies as fuels for the heart: study with positron emission tomography. Am. J. Physiol. Heart Circ. Physiol 264:H701–7 [DOI] [PubMed] [Google Scholar]
- 172.Verma S, Rawat S, Ho KL, Wagg CS, Zhang L, et al. 2018. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC Basic Transl. Sci 3:575–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vila-Brau A, De Sousa-Coelho AL, Mayordomo C, Haro D, Marrero PF. 2011. Human HMGCS2 regulates mitochondrial fatty acid oxidation and FGF21 expression in HepG2 cell line. J. Biol. Chem 286:20423–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.von Meyenn F, Porstmann T, Gasser E, Selevsek N, Schmidt A, et al. 2013. Glucagon-induced acetylation of Foxa2 regulates hepatic lipid metabolism. Cell Metab. 17:436–47 [DOI] [PubMed] [Google Scholar]
- 175.Voros G, Ector J, Garweg C, Droogne W, Van Cleemput J, et al. 2018. Increased cardiac uptake of ketone bodies and free fatty acids in human heart failure and hypertrophic left ventricular remodeling. Circ. Heart Fail 11:e004953. [DOI] [PubMed] [Google Scholar]
- 176.Wagner GR, Bhatt DP, O’Connell TM, Thompson JW, Dubois LG, et al. 2017. A class of reactive acyl-CoA species reveals the non-enzymatic origins of protein acylation. Cell Metab. 25:823–37. e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang Q, Zhou Y, Rychahou P, Fan TWM, Lane AN, et al. 2017. Ketogenesis contributes to intestinal cell differentiation. Cell Death Differ. 24:458–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang W, Ishibashi J, Trefely S, Shao M, Cowan AJ, et al. 2019. A PRDM16-driven metabolic signal from adipocytes regulates precursor cell fate. Cell Metab. 30:174–89. e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wang YH, Liu CL, Chiu WC, Twu YC, Liao YJ. 2019. HMGCS2 mediates ketone production and regulates the proliferation and metastasis of hepatocellular carcinoma. Cancers 11:1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang YH, Suk FM, Liao YJ. 2020. Loss of HMGCS2 enhances lipogenesis and attenuates the protective effect of the ketogenic diet in liver cancer. Cancers 12:1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Webber RJ, Edmond J. 1977. Utilization of L(+)-3-hydroxybutyrate, D(−)-3-hydroxybutyrate, acetoacetate, and glucose for respiration and lipid synthesis in the 18-day-old rat. J. Biol. Chem 252:5222–26 [PubMed] [Google Scholar]
- 182.Weidemann MJ, Krebs HA. 1969. The fuel of respiration of rat kidney cortex. Biochem. J 112:149–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wentz AE, d’Avignon DA, Weber ML, Cotter DG, Doherty JM, et al. 2010. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J. Biol. Chem 285:24447–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Williamson DH, Bates MW, Page MA, Krebs HA. 1971. Activities of enzymes involved in acetoacetate utilization in adult mammalian tissues. Biochem. J 121:41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Williamson DH, Lund P, Krebs HA. 1967. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J 103:514–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Williamson JR, Scholz R, Browning ET. 1969. Control mechanisms of gluconeogenesis and ketogenesis.II. Interactions between fatty acid oxidation and the citric acid cycle in perfused rat liver. J. Biol. Chem 244:4617–27 [PubMed] [Google Scholar]
- 187.Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M. 2004. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 432:1027–32 [DOI] [PubMed] [Google Scholar]
- 188.Xia S, Lin R, Jin L, Zhao L, Kang HB, et al. 2017. Prevention of dietary-fat-fueled ketogenesis attenuates BRAF V600E tumor growth. Cell Metab. 25:358–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Xie Z, Zhang D, Chung D, Tang Z, Huang H, et al. 2016. Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol. Cell 62:194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yang L, Achreja A, Yeung TL, Mangala LS, Jiang D, et al. 2016. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab. 24:685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, et al. 2015. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med 21:263–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zelniker TA, Braunwald E. 2020. Mechanisms of cardiorenal effects of sodium-glucose cotransporter 2 inhibitors: JACC state-of-the-art review. J. Am. Coll. Cardiol 75:422–34 [DOI] [PubMed] [Google Scholar]
- 193.Zhang D, Yang H, Kong X, Wang K, Mao X, et al. 2011. Proteomics analysis reveals diabetic kidney as a ketogenic organ in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab 300:E287–95 [DOI] [PubMed] [Google Scholar]
- 194.Zhang J, Jia PP, Liu QL, Cong MH, Gao Y, et al. 2018. Low ketolytic enzyme levels in tumors predict ketogenic diet responses in cancer cell lines in vitro and in vivo. J. Lipid Res 59:625–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhang S, Xie C. 2017. The role of OXCT1 in the pathogenesis of cancer as a rate-limiting enzyme of ketone body metabolism. Life Sci. 183:110–15 [DOI] [PubMed] [Google Scholar]
- 196.Zhang WW, Churchill S, Lindahl R, Churchill P. 1989. Regulation of D-β-hydroxybutyrate dehydrogenase in rat hepatoma cell lines. Cancer Res. 49:2433–37 [PubMed] [Google Scholar]
- 197.Zou K, Hu Y, Li M, Wang H, Zhang Y, et al. 2019. Potential role of HMGCS2 in tumor angiogenesis in colorectal cancer and its potential use as a diagnostic marker. Can. J. Gastroenterol. Hepatol 2019:8348967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zwiebel FM, Schwabe U, Olson MS, Scholz R. 1982. Role of pyruvate transporter in the regulation of the pyruvate dehydrogenase multienzyme complex in perfused rat liver. Biochemistry 21:346–53 [DOI] [PubMed] [Google Scholar]