

Abstract
Since numerous RNAs and RBPs prevalently localize to active chromatin regions, many RNA‐binding proteins (RBPs) may be potential transcriptional regulators. RBPs are generally thought to regulate transcription via noncoding RNAs. Here, we describe a distinct, dual mechanism of transcriptional regulation by the previously uncharacterized tRNA‐modifying enzyme, hTrmt13. On one hand, hTrmt13 acts in the cytoplasm to catalyze 2'‐O‐methylation of tRNAs, thus regulating translation in a manner depending on its tRNA‐modification activity. On the other hand, nucleus‐localized hTrmt13 directly binds DNA as a transcriptional co‐activator of key epithelial–mesenchymal transition factors, thereby promoting cell migration independent of tRNA‐modification activity. These dual functions of hTrmt13 are mutually exclusive, as it can bind either DNA or tRNA through its CHHC zinc finger domain. Finally, we find that hTrmt13 expression is tightly associated with poor prognosis and survival in diverse cancer patients. Our discovery of the noncatalytic roles of an RNA‐modifying enzyme provides a new perspective for understanding epitranscriptomic regulation.
Keywords: cancer metastasis, RNA modification, transcription, translation, tRNA
Subject Categories: Cancer; Chromatin, Transcription & Genomics; RNA Biology
hTrmt13 regulates gene expression via translation in the cytoplasm, but also by acting in a non‐catalytic manner as a transcriptional coactivator.
Introduction
Transfer RNAs (tRNAs) are bridges between messenger RNAs (mRNAs) and amino acids to ensure the accurate decoding of genetic information (Ishimura et al, 2014; Hanson & Coller, 2018; Schimmel, 2018). tRNAs undergo various modifications that are crucial for their folding, stability, and decoding (Nedialkova & Leidel, 2015; Liu et al, 2016; Frye et al, 2018). Importantly, tRNA modifications are directly associated with many human diseases, such as diabetes, neurological diseases, and cancers (Torres et al, 2014; Rapino et al, 2018; Barbieri & Kouzarides, 2020). tRNA modifications are responsive to environmental changes, including viral infections, changes in growth medium and temperature, oxidative stress, and the cell cycle (Liu et al, 2016; Frye et al, 2018). These various tRNA modifications have recently been recognized under the umbrella of “tRNA epitranscriptomics”, adding an essential layer of gene regulation to the central dogma of molecular biology.
An increasing number of studies have shown that tRNA not only serves as a transmitter of genetic information but also actively regulates gene expression. For instance, tRNA can promote breast cancer metastasis via enhancing oncogene expression (Goodarzi et al, 2016). In addition, tRNA could further be cleaved into tRNA‐derived fragments (tRFs; Gapp & Miska, 2016). tRFs are a novel class of small RNAs with important biological functions in intergenerational inheritance (Chen et al, 2016; Sharma et al, 2016), cancer development (Goodarzi et al, 2015), and oxidative stress (Thompson & Parker, 2009). tRFs can act as microRNAs (Maute et al, 2013) or interact with other proteins to play regulatory roles (Ivanov et al, 2011; Saikia et al, 2014; Goodarzi et al, 2015). It is worth noting that the procession and function of tRFs are modulated by the original modifications of tRNAs (Zhang et al, 2018).
The identification of tRNA‐modifying enzymes was mainly accomplished in bacteria and yeast but largely remain unclear in more complicated eukaryotes such as human due to technical limitations. Gene mutations on putative human tRNA‐modifying enzymes are associated with various human diseases, suggesting a critical role of tRNA‐modifying enzymes (Nedialkova & Leidel, 2015; Rubio et al, 2017). Interestingly, some recent data indicate that tRNA modifying enzymes may also possess noncanonical functions independent of catalytic activity, for example, the human tRNA‐modifying enzyme PUS10 regulates miRNA processing independent of its catalytic activity (Song et al, 2020). Thus, functional studies of tRNA modifications become more complicated in light of an additional layer of genetic regulation.
Modification at the acceptor stem of tRNAs is rare, but 2'‐O‐methylation at the 4th position (Nm4) is conserved in eukaryotes, implying the importance of this modification (Towns & Begley, 2012; Wang et al, 2017; Endres et al, 2020). In yeast, a special RNA methyltransferase Trm13p, which has no sequence similarity with other known methyltransferases, was identified as the modifying enzyme for Nm4 of tRNA. However, the function of tRNA:Nm4 in yeast remains controversial (Wilkinson et al, 2007). In rice, OsTrm13 was identified as the tRNA: Nm4 methyltransferase and regulated salt stress tolerance by an unknown mechanism (Wang et al, 2017). Moreover, the trm13 mutant of Saccharomyces cerevisiae is sensitive to oxidative stress (Endres et al, 2020). So far, the tRNA:Nm4‐modifying enzyme in human remains elusive.
Here, we identified CCDC76 (coiled‐coil domain‐containing protein 76) as the human tRNA Nm4 methyltransferase (hTrmt13). We first identified the tRNA substrate of hTrmt13. Next, we showed that the tRNA modification by hTrmt13 modulated global protein translation most likely through affecting a specific tRF in the cytoplasm. While in the nucleus, hTrmt13 localized to chromatin, largely at promoter regions, through direct binding of DNA via a CHHC zinc finger domain. Nuclear hTrmt13 acted as a transcriptional co‐factor to modulate the transcription of many genes, especially those that influence cell migration. We validated that hTrmt13 promotes cell migration in cell lines and breast tumor metastatic mice models and found that this function was independent of its tRNA‐modifying catalytic activity. At last, the expression of hTrmt13 was significantly associated with patient survival in breast carcinoma and liver and renal cancer patients. Our findings of a tRNA‐modifying enzyme that modulates transcription are in good agreement with the recent observations that many RBPs are enriched at active chromatin regions (Luo et al, 2016; Xiao et al, 2019). In our case, hTrmt13 modulated transcription independent of its binding with tRNAs. These findings bring new perspectives to understanding gene expression regulation by RNA modifications and relevant epitranscriptomic regulators.
Results
CCDC76 is the human tRNA Nm4 methyltransferase, hTrmt13
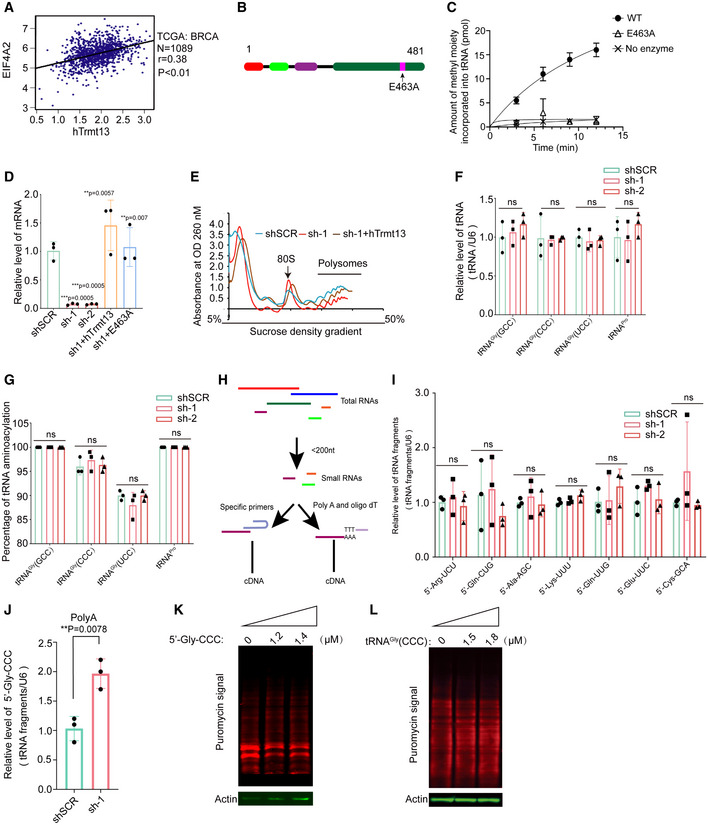
CCDC76 is an uncharacterized human protein containing 481 amino acid residues that shares 27% sequence similarity with yeast Trm13p (Wilkinson et al, 2007; Towns & Begley, 2012). CCDC76 contains two zinc‐finger domains (CCCH, Zn‐1; CHHC, Zn‐2), a coiled‐coil domain, and a SAM‐methyltransferase (MTase) domain (Fig 1A). CCDC76 is the only Trm13 homolog in human; thus, we hypothesized that it is a putative tRNA: Nm4 methyltransferase and named it hTrmt13. In order to know whether hTrmt13 interact with RNA in the cell, we performed UV cross‐linking and immunoprecipitation followed by high‐throughput sequencing (CLIP‐seq) after RNA modifications were removed by AlkBs (Huppertz et al, 2014; Zheng et al, 2015) (Figs 1B and C, and EV1A). Replicate experiments consistently demonstrated that tRNAs were enriched in the hTrmt13‐based CLIP products (˜70% tRNA in total) compared to other RNAs (Fig 1D, Dataset EV1). Previous research has shown that yeast tRNAHis, tRNAPro, and tRNAGly are the substrates of Trm13p (Wilkinson et al, 2007). Our results showed that human cytoplasmic tRNAHis, tRNAPro, and tRNAGly (HctRNAHis, HctRNAPro, and HctRNAGly) were enriched in the hTrmt13‐based CLIP products; however, HctRNAArg, HctRNALys, and HctRNAThr were also present in large quantities (Fig 1E, Dataset EV1).
Figure 1. CCDC76 (hTrmt13) is a tRNA 2'‐O‐methyltransferase of position 4.
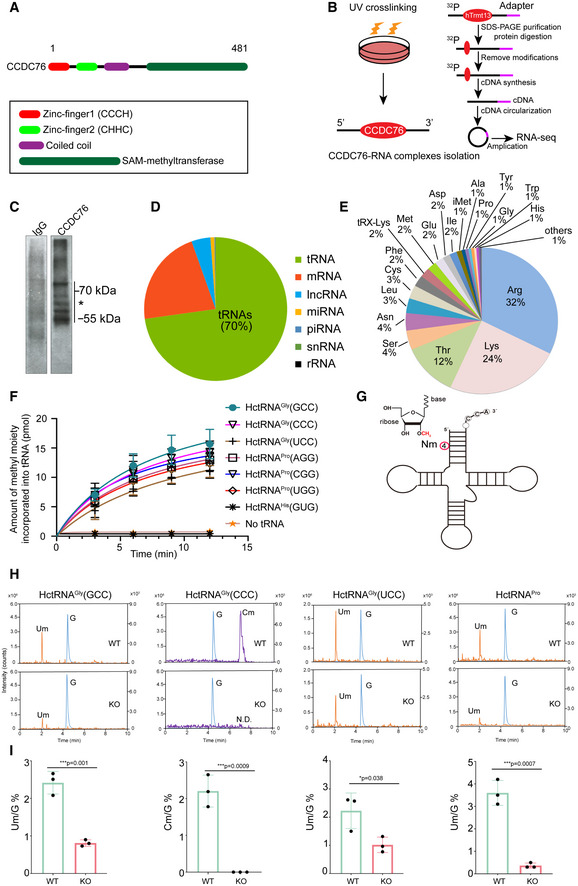
- A representation of the primary structure of human CCDC76 (hTrmt13).
- Schematic of the modified CLIP‐seq approach employed to profile genome‐wide hTrmt13‐RNA interactions.
- Autoradiograph of hTrmt13‐RNA complexes in the MDA‐MB‐231 cells immunoprecipitated with an antibody against hTrmt13 (right lane) and IgG as a negative control (left lane). * RNA residing in the regions between marked lines was recovered for sequencing.
- Pie chart showing distinct categories of RNA species bound to hTrmt13 identified by CLIP‐seq.
- Pie chart showing the different species tRNAs bound to hTrmt13 from CLIP‐seq.
- The capacity of HctRNAs to be methylated by hTrmt13. Error bars represent mean ± SD for at least three independent experiments.
- A representative secondary structure of tRNA with Nm4 modification marked.
- Mass chromatograms of the nucleosides, Um (Q1/Q3 = 259.1/113.1), Cm (Q1/Q3 = 258.1/112.1), and G (Q1/Q3 = 284.1/152.2) of HctRNAGly(GCC), HctRNAGly(CCC), HctRNAGly(UCC), and HctRNAPros isolated from WT and hTrmt13 KO cells. Um, 2′‐O‐methyluridine, Cm, 2′‐O‐methylcytidine. Q1/Q3: the mass of the precursor ion and the mass of the product ion. These endogenous tRNAs were purified separately from total cellular RNAs using sequence‐specific probes.
- Quantification of the Um/G and Cm/G ratios in endogenous tRNAs isolated from WT and hTrmt13 KO cells. Quantification was performed by comparison with the standard curve obtained from pure nucleoside standards running in the same batch. Statistical analysis was performed using t‐tests. Error bars represent mean ± SD for three independent experiments.
Figure EV1. hTrmt13 is a human tRNA 2'‐O‐methyltransferase at position 4.
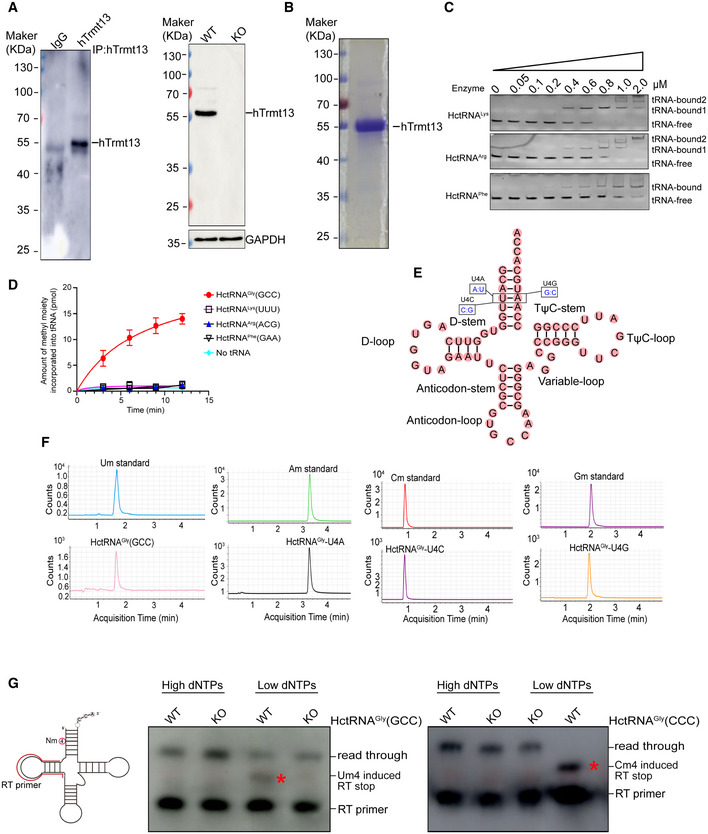
- Verification of the customized anti‐hTrmt13. The ability of anti‐hTrmt13 for immunoprecipitation in MDA‐MB‐231 cells assayed by Western blot (left). The protein level of hTrmt13 in WT and KO MDA‐MB‐231 cells assayed by Western blot using customized antibody of hTrmt13, GAPDH was used as control. A total 20 μg proteins of cell lysates were loaded.
- SDS–PAGE analysis of the purified hTrmt13. Standard molecular weights are shown on the left.
- The binding affinities of hTrmt13 for HctRNALys(UUU), HctRNAArg(ACG), and HctRNAPhe(GAA) analyzed by EMSA. The concentration of tRNAs is 0.15 μM.
- The capacity of HctRNAGly(GCC), HctRNALys(UUU), HctRNAArg(ACG), and HctRNAPhe(GAA) to be methylated by hTrmt13. Error bars represent mean ± SD for at least three independent experiments.
- The secondary structure of HctRNAGly(GCC) summarizing the mutations in A: U (U4A), C: G (U4C), and G:C (U4G) base pairs.
- The Um, Am, Cm, and Gm modification of HctRNAGly(GCC), U4A, and U4C and U4G mutants was detected by mass spectrometry after hTrmt13 incubation.
- Detection of Um4 in endogenous tRNAGly(GCC) or Cm4 in tRNAGly(CCC) using primer‐extension assays. 40 μM or 0.5 μM dNTPs were used in the high or low dNTPs groups. The Nm modifications caused RT stop in low dNTPs groups, and the products (labeled by a star) are shorter than the RT read through products.
To further investigate whether hTrmt13 could methylate these HctRNAs, we designed an in vitro enzymatic assay system (Long et al, 2016). Purified hTrmt13 (Fig EV1B) and the transcribed tRNAs were used for the methylation assays. Our results showed that hTrmt13 can methylate HctRNAGly(GCC), HctRNAGly(CCC), HctRNAGly(UCC), HctRNAPro(AGG), HctRNAPro(CGG), and HctRNAPro(UGG) but not HctRNAHis(GUG) (Fig 1F). Interestingly, other tRNAs that bound to hTrmt13 (Figs 1E and EV1C), such as HctRNAArg, HctRNALys, and HctRNAPhe, could not be methylated (Fig EV1D). We further chose HctRNAGly(GCC) for subsequent enzymatic assays because hTrmt13 exhibited the highest methylation activity on this substrate. Ultra‐performance liquid chromatography (UPLC)‐MS/MS was used to identify the modification type catalyzed by hTrmt13. One peak was detected after the reaction of HctRNAGly(GCC) with hTrmt13 and accurately matched the Um standard (Fig EV1E and F). To further confirm that 2'‐O‐methylation occurred at the 4th position, U4 of HctRNAGly(GCC) was mutated to either A, or C, or G (Figs 1G and EV1E). Consequently, we detected Am, Cm, and Gm in the corresponding mutants after incubation with hTrmt13 (Fig EV1F). Furthermore, primer extension assays showed that Nm‐induced RT arrest products were observed in the low dNTPs groups of tRNAs purified from the WT MDA‐MB‐231 cells, but not in the hTrmt13‐KO cells (CRISPR‐Cas9 system) (Fig EV1G), suggesting that the modification introduced by hTrmt13 is most likely to be Nm modifications, which is consistent with our RNA‐MS results.
The roles of the two conserved zinc‐finger domains of hTrmt13 in methylation and tRNA recognition were investigated next (Fig EV2A). The deletion of the CHHC Zn‐2 completely abolished the catalytic activity of hTrmt13, while the deletion of the CCCH Zn‐1 did not affect (Fig EV2B). Electrophoretic mobility shift assays (EMSA) revealed that the deletion of the CHHC Zn‐2 prevented binding to tRNA, while the mutant deleted CCCH Zn‐1 had almost the same affinity to tRNA as WT hTrmt13 (Fig EV2C). These results suggested that the CHHC Zn‐2 is crucial for hTrmt13 binding of tRNA.
Figure EV2. The CHHC Zn‐2 domain is crucial for hTrmt13 binding to tRNA.
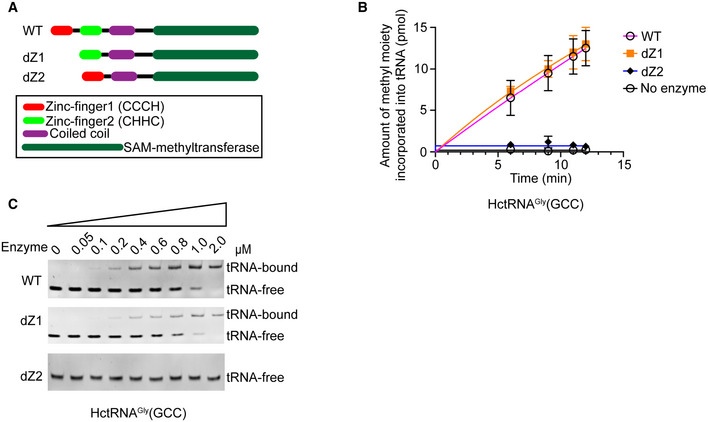
- Schematic diagram of the primary structure of hTrmt13 summarizing the mutations. dZ1 indicates deletion of the CCCH Zn‐1 domain, and dZ2 indicates deletion of the CHHC Zn‐2 domain.
- The methyltransferase activities of hTrmt13 (WT) and its mutants (dZ1 and dZ2). For all experiments, three independent experiments were performed. Error bars represent mean ± SD for three independent experiments.
- The binding affinities of hTrmt13 and its mutants for HctRNAGly(GCC) (0.15 μM) analyzed by EMSA.
The above results showed that standalone hTrmt13 could catalyze tRNA:Nm4 modification in vitro. To verify the activity of hTrmt13 in vivo, the substrate‐tRNAs of hTrmt13, HctRNAGly(GCC), HctRNAGly(CCC), HctRNAGly(UCC), and HctRNAPros (HctRNAPros is the mixture of HctRNAPro(AGG), HctRNAPro(CGG), and HctRNAPro(UGG); their sequence are identical except for the nucleoside at the 34th position), were purified from hTrmt13‐KO and wild‐type (WT) cells, respectively. The purified tRNAs were digested and subsequently subjected to RNA‐mass spectrometry (Fig 1H). Notably, the level of the corresponding Nm modification in all the tRNAs was observed to be significantly decreased in KO cells comparing with WT cells (Fig 1H). The nucleoside at the 4th position of HctRNAGly(GCC), HctRNAGly(UCC), and HctRNAPros is U; accordingly, the Um/G values in these tRNAs from KO cells decreased to 34%, 46%, and 11% of the Um/G values in tRNAs from WT cells (Fig 1H and I); the nucleoside at the 4th position of HctRNAGly(CCC) is C, and the Cm dropped to an undetectable level in this tRNA after KO of hTrmt13 (Fig 1H and I). These results together revealed that hTrmt13 was responsible for the 2'‐O‐methylation at the 4th position of the human tRNAGly and tRNAPro isoacceptors in vivo.
hTrmt13 modulates protein translation depending on its tRNA modification activity
The expression of hTrmt13 is positively correlated with that of eukaryotic initiation factor 4A2 (EIF4A2) in breast cancer patients according to data from The Cancer Genome Atlas (TCGA) database (Fig EV3A). To investigate whether hTrmt13 affects protein synthesis, we monitored global de novo protein synthesis by assaying the incorporation of puromycin, a protein synthesis inhibitor, into nascent polypeptides (Dewe et al, 2017). Both hTrmt13 knockdown cell lines (sh‐1, sh‐2) displayed a lower rate of puromycin incorporation than shSCR, with levels of reduction of ˜15% and ˜13% in sh‐1 and sh‐2 cells, respectively (Fig 2A). The decreased protein synthesis in hTrmt13 knockdown cells could be restored to control levels by stable expression of a wild‐type hTrmt13 allele (Fig 2A). Critically, an enzymatically inactive mutant (E463A) in the proposed catalytic center of hTrmt13 (Tkaczuk, 2010) (Fig EV3B and C) failed to rescue protein synthesis in hTrmt13 knockdown cells (Figs 2A and EV3D). We also performed polysome profiling on a sucrose gradient. Our results suggested that the level of the polysome was reduced in hTrmt13 knockdown MDA‐MB‐231 cells (sh‐1) compared to controls (shSCR), suggesting that protein synthesis was mildly decreased (Fig EV3E). This difference could be rescued by expression of the wild‐type hTrmt13 allele (sh‐1+hTrmt13) (Fig EV3E). These findings thus establish a role for hTrmt13‐mediated 2'‐O‐methylation in modulating translational capacity.
Figure EV3. Identification of catalytic inactive mutation of hTrmt13 and analyses of tRFs.
- Analysis of the TCGA dataset for the mRNA expression of hTrmt13 and EIF4A2 in breast cancer patients. Spearman order correlation analysis was performed.
- Schematic diagram of the primary structure of inactive hTrmt13 (E463A).
- The methyltransferase activities of hTrmt13 and E463A mutant toward HctRNAGly(GCC).
- The mRNA level of hTrmt13 in shSCR, sh‐1, sh‐2, sh+WT, and sh+E463A were detected by RT–qPCR.
- The polysome profiling of control (shSCR), hTrmt13 knockdown MDA‐MB‐231 cells (sh‐1), and sh‐1 with stable expression of hTrmt13 (sh‐1+hTrmt13).
- Statistical analysis of the tRNA steady state levels assayed by northern blot.
- Statistical analysis of the in vivo aminoacylation level of hTrmt13 substrate tRNAs.
- Workflow of obtaining cDNA from tRFs by two methods (stem‐loop method and Poly‐A tail method). Small RNAs were enriched by mirVana™ miRNA Isolation Kit (Thermo Scientific). The reverse transcription primer of the stem‐loop method consists of two parts: (1) universal stem ring structure and (2) reverse complementary bases with the target tRF. For the Poly‐A method, Oligo Dt was used as the reverse transcription primer for Poly‐A tail, which was tagged to small RNAs using QuantiMir™ Kit (SBI).
- RT–qPCR using a stem‐loop primer to quantify the tRFs levels in shSCR, sh‐1, and sh‐2.
- Poly‐A‐based RT–qPCR to quantify the level of 5'‐Gly‐CCC in shSCR and sh‐1.
- De novo protein synthesis in MDA‐MB‐231 cells transfected with different concentrations of 5'‐Gly‐CCC oligos.
- De novo protein synthesis in MDA‐MB‐231 cells transfected with different concentrations of HctRNAGly(CCC).
Data information: In C, D, F, G, I, J, statistical analysis was performed using t‐tests. Error bars indicate mean ± SD for three independent experiments.
Figure 2. hTrmt13 impacts protein biosynthesis depending on its catalytic activity.
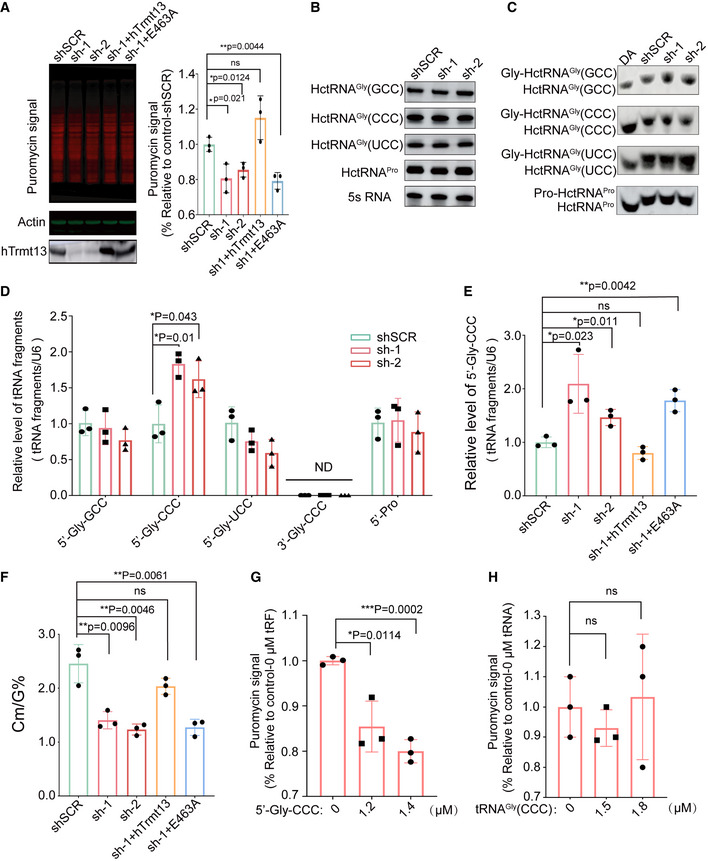
- De novo protein synthesis measured by puromycin incorporation in shSCR, hTrmt13 knockdown cells (sh‐1 and sh‐2), and sh‐1 with stable expression of wild‐type hTrmt13 (sh‐1+hTrmt13) or enzymatically inactive hTrmt13 (sh‐1+E463A). Puromycin‐labeled proteins were detected by immunoblotting. Representative images (left) and statistical analysis (right) are shown. Knockdown and overexpression efficiency of hTrmt13 in MDA‐MB‐231 verified by Western blots. See also Fig EV3D.
- Northern blot analysis of substrate tRNAs of hTrmt13 from control and the hTrmt13 knockdown MDA‐MB‐231 cells. Statistical analysis is shown in Fig EV3F.
- The in vivo aminoacylation level of hTrmt13 substrate tRNAs assayed by northern blot. The samples were also deacylated (DA) by incubating with alkaline buffer (pH 9.0) to obtain fully uncharged tRNA control. Statistical analysis is shown in Fig EV3G.
- RT–qPCR using a stem‐loop primer to quantify the tRFs levels in shSCR, sh‐1, and sh‐2.
- Quantification of the level of 5'‐Gly‐CCC in shSCR, sh‐1, sh‐2, sh‐1+hTrmt13, and sh‐1+E463A assayed by RT–qPCR.
- Quantification of the Cm/G ratios of endogenous HctRNAGly(CCC) isolated from shSCR, sh‐1, sh‐2, sh‐1+hTrmt13, and sh‐1+E463A cells by UPLC‐MS/MS analysis.
- Quantification of de novo protein synthesis in cells treated with different concentration of tRFs as indicated in Fig EV3K.
- Quantification of de novo protein synthesis in cells treated with different concentration of tRNAGly(CCC) as indicated in Fig EV3L.
Data information: In A, D–H: statistical analysis was performed using t‐tests, and error bars represent mean ± SD for three independent experiments.
To understand the mechanism of this tRNA modification in protein translation, we detected the stable level as well as the aminoacylation level of tRNAs. Using Northern blot assays, we found that knockdown of hTrmt13 did not affect the stable level of cellular tRNA substrates including three tRNAGly isoacceptors and tRNAPros (Figs 2B and EV3F). Similarly, the aminoacylation level of substrate tRNAs was almost the same in the control and hTrmt13 knockdown cells (Figs 2C and EV3G). These results suggested that the Nm4 in the acceptor stem did not affect the stability of amino acid charging capability of substrate tRNAs.
Previous studies have shown that some tRNA modifications can tune the production or stability of tRFs (Li et al, 2018), and specific tRFs (e.g., 5'‐tiRNA(Ala), 5'‐tiRNA(Cys), 5'‐tiRNA(Gly), and 3'‐tiRNA(Pro)) can inhibit protein translation (Ivanov et al, 2011). To accurately detect the level of tRFs, we performed a modified quantitative reverse transcription PCR (RT–qPCR) assay using a stem‐loop primer (Chen et al, 2019; Fig EV3H). Thirteen tRFs that could be derived from hTrmt13 tRNA substrates or other tRFs that are known to affect protein synthesis (Ivanov et al, 2011; Anderson & Ivanov, 2014) were detected. The results showed that only the level of 5'‐Gly‐CCC (the 5' tRF of HctRNAGly(CCC)) was increased in hTrmt13 knockdown cells compared to the control, while the levels of ten other detectable tRFs were not changed (Figs 2D and EV3I). We further confirmed the increasing level of 5'‐Gly‐CCC by using another innovative RT–qPCR assay (Fig EV3H and J). To further check whether the level of 5'‐Gly‐CCC was altered in hTrmt13 knockdown (KD) cells as a result of the modification activity of hTrmt13, we performed rescue experiments by over‐expressing wild‐type hTrmt13 or its catalytically inactive mutant (hTrmt13‐E463A). We found that the over‐expression of hTrmt13 decreased the level of 5'‐Gly‐CCC in knockdown cells to a similar degree as in the control cells, while the over‐expression of hTrmt13‐E463A did not affect the level of 5'‐Gly‐CCC (Fig 2E), and the Cm modification level of HctRNAGly(CCC) is negatively correlated with the level of 5'‐Gly‐CCC in these cells (Fig 2F), indicating that the methyltransferase activity of hTrmt13 downregulates the formation of 5'‐Gly‐CCC.
To further test whether 5'‐Gly‐CCC affected protein translation in MDA‐MB‐231 cells as reported in U2OS cells (Ivanov et al, 2011), we performed puromycin incorporation assays in MDA‐MB‐231 cells transduced with 5'‐Gly‐CCC oligos. Our results showed that 5'‐Gly‐CCC decreased protein synthesis when reaching a threshold concentration, and the effect was increased when the level of 5'‐Gly‐CCC was elevated (Figs 2G and EV3K). In contrast, a similar amount of tRNAGly(CCC) did not affect protein synthesis (Figs 2H and EV3L). Together, these results suggest that hTrmt13 modulates protein synthesis depending on its tRNA modification activity and most likely via regulating the level of a specific tRF.
hTrmt13 binds tRNA in cytoplasm and localizes to chromatin via directly binding to promoter DNA in nucleus
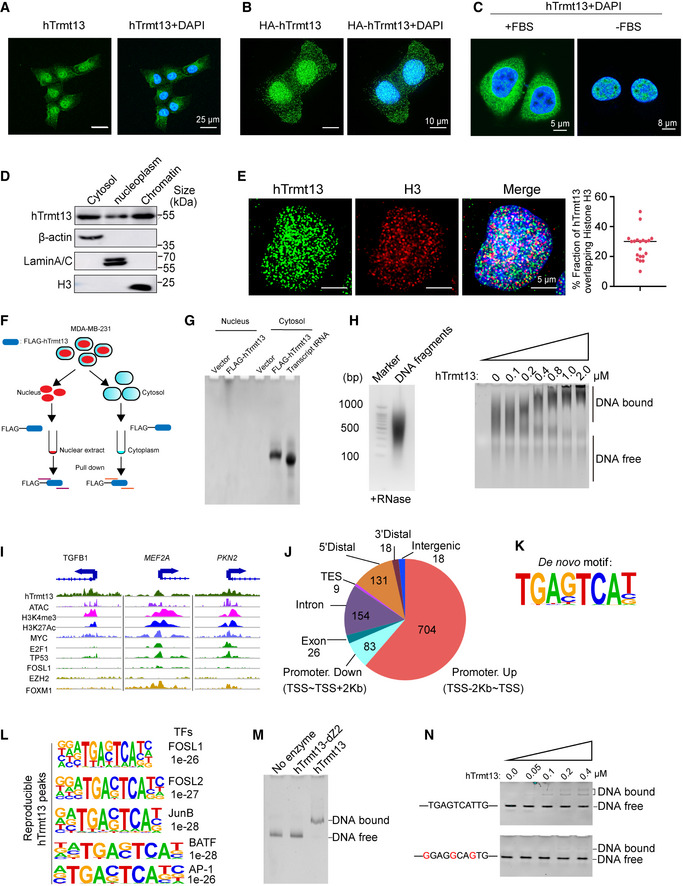
To detect the localization of hTrmt13, we performed immunofluorescence assays (IF). The endogenous hTrmt13 was detected using a highly specific antibody (Fig EV1A, Appendix Fig S1A), and the immunolabeling showed that hTrmt13 localized to both the cytoplasm and nucleus in MDA‐MB‐231 cells (Fig 3A). The cytoplasmic and nuclear localization of hTrmt13 was further confirmed by the stable overexpression of HA‐hTrmt13 using an anti‐HA antibody (Fig 3B), and these patterns were also consistent in other cell lines such as HeLa and HEK293 (Appendix Fig S1B and C). Intriguingly, when deprived of fetal bovine serum (FBS) during cell culture, the distribution of hTrmt13 in cytoplasm almost completely disappeared (Fig 3C), suggesting that cytoplasmic hTrmt13 might be regulated by some signaling pathway. In addition, RNA fluorescence in situ hybridization assay (RNA‐FISH) combined with hTrmt13 IF revealed that HctRNAGly(GCC) and HctRNAGly(CCC) co‐localized with hTrmt13 in the cytoplasm (Appendix Fig S1D), indicating that the tRNA modification function of hTrmt13 is mainly performed in the cytoplasm. Thus, we hypothesized that hTrmt13 might have other functions besides tRNA modification in the nucleus. Subcellular fractionation assays further demonstrated that hTrmt13 co‐localized with chromatin in the nucleus (Fig 3D). Furthermore, we performed confocal microscopy to monitor the co‐localization of hTrmt13 and histone H3. We found that hTrmt13 and H3 (˜27%) were co‐localized in the nucleus (Fig 3E). Next, we performed nucleic acid pull‐downs by co‐immunoprecipitation of hTrmt13 without adding DNase to the nuclear extract and cytoplasm, respectively (Fig 3F), and found that hTrmt13 mainly bound tRNAs in the cytoplasm (Fig 3G). Strikingly, hTrmt13 bound to larger nucleic acids other than tRNAs in the nucleus (Fig 3G).
Figure 3. hTrmt13 binds tRNA in cytoplasm and directly binds DNA in nucleus.
-
A, BRepresentative images showing endogenous hTrmt13 (A) and HA‐tagged hTrmt13 (HA‐hTrmt13, B) subcellular localization by confocal microscopy analysis.
-
CRepresentative confocal microscopy images showing the subcellular localization of hTrmt13 in MDA‐MB‐231 cells cultured with 10% FBS (left) or no FBS (right).
-
DThe distribution of hTrmt13 analyzed by subcellular fraction assay. β‐actin as a marker of cytosol, Lamin A/C as a marker of nucleoplasm, Histone H3 as a marker of chromatin.
-
EConfocal microscopy images of MDA‐MB‐231 cells were co‐immunolabeled with antibodies against hTrmt13 and histone H3 (left). The images were captured by GE DeltaVision OMX SR. Co‐localization of H3 and hTrmt13 was analyzed by ImageJ (right, n = 20 cells, center line, the median of the data).
-
FSchematic diagram illustrating the principle of nucleic acids‐IP performed in nuclear extract and cytoplasm without adding RNase or DNase.
-
GhTrmt13 bound nucleic acids from the cytosol and nuclear extract were stained by GoldView. Transcribed HctRNAGly(GCC) was used as a size marker for tRNAs.
-
HGenomic DNA was sheared to fragments by Biorupt (left). EMSA of hTrmt13 and DNA fragments (15 ng/μl DNA was used).
-
IThe binding profiles of hTrmt13, H3K4me3, H3K27Ac, and other transcription factors (MYC, E2F1, TP53, FOSL1, EZH2, FOXM1) from ChIP‐seq and ATAC‐seq on representative target genes TGFB1, MEF2A, and PKN2. hTrmt13 track was average across 2 biological replicates.
-
JGenomic distribution of hTrmt13 reproducible (N = 2) binding sites determined by ChIP‐seq analysis in MDA‐MB‐231 cells.
-
KhTrmt13 binding de novo motif for reproducible (N = 2) binding sites.
-
LhTrmt13 reproducible (N = 2) binding sites enriched for known motifs for FOSL1, FOSL2, JunB, BATF, and AP‐1.
-
MThe binding affinities of wild‐type and mutant hTrmt13s for synthesized DNA (0.25 μM) analyzed by EMSA. The concentration of hTrmt13‐dZ2 or hTrmt13 used in the system was 1 μM.
-
NEMSA of hTrmt13 for synthesized DNA (0.25 μM) carrying consensus motif or mutated motif.
Data information: In A, B, C and E, DAPI staining was included to visualize the cell nucleus (Blue).
The above results showed that hTrmt13 co‐localized with chromatin and could bind to nucleic acids beyond tRNA. To test whether hTrmt13 could directly bind DNA, we performed in vitro EMSA. Genomic DNA extracted from MDA‐MB‐231 cells was sheared to 200–1,000 base pairs in length (Fig 3H left) to conduct EMSAs with hTrmt13. In order to exclude the influence of any potential RNAs, the sheared DNA was treated with RNase A and RNase H. EMSAs revealed that hTrm13 strongly bound to genomic DNA, and its affinity with DNA was completely lost when the CHHC Zn‐2 domain was deleted (Fig 3H right, Appendix Fig S1E).
To characterize the DNA‐binding landscape of endogenous hTrmt13 in vivo, chromatin immunoprecipitation‐based deep sequencing (ChIP‐seq) was performed in MDA‐MB‐231 cells. Following ChIP, hTrmt13‐associated DNAs were amplified using nonbiased conditions, labeled, and sequenced via HiSeq 2500. We identified 1,143 hTrmt13 reproducible peaks (Dataset EV2). The representative ChIP‐seq peak data are shown in Fig 3I. The DNA sequences associated with these peaks were then cross‐analyzed with publicly available ATAC‐seq and ChIP‐seq datasets for H3K4me3, H3K27Ac, Myc, E2F1, TP53, FOSL1, EZH2, and FOXM1 (Fig 3I, Appendix Fig S2A and B). Interestingly, although genomic distribution analysis showed that hTrmt13 binding was mainly identified in promoter regions (Fig 3J), we observed that cluster 5 from the k‐means analysis was lacking the promoter marker H3K4me3 and was particularly more enriched with the FOSL1 signal (Appendix Fig S2B). Both de novo motif analysis and known motif analysis using homer2 were performed, and a unique DNA motif TGA[GC]TCA[TCG] enriched at hTrmt13 sites was identified (Fig 3K, Dataset EV2). We found that FOSL1, FOSL2, JunB, BATF, and AP‐1 motifs were the most significantly enriched in hTrmt13 binding sites (Fig 3L, Dataset EV2).
Moreover, using EMSA, we found that hTrmt13 bound to synthesized double‐stranded DNA containing a TGAGTCAT motif, and the binding disappeared upon deletion of the CHHC Zn‐2 domain (Fig 3M). We further found that the DNA interaction by hTrmT13 was attenuated when the consensus motif was mutated (Fig 3N), suggesting that the interaction between hTrmt13 and DNA is specific to a certain DNA sequence. Interestingly, we also found that a tRF or single‐stranded DNA containing this motif could not bind hTrmt13 (Appendix Fig S3A and B). Overall, our findings indicate that hTrmt13 directly interacts with specific DNA motifs via the CHHC Zn‐2 in vitro and binds to the promoters of target genes in vivo.
hTrmt13 promotes cell migration independent of its tRNA modification activity
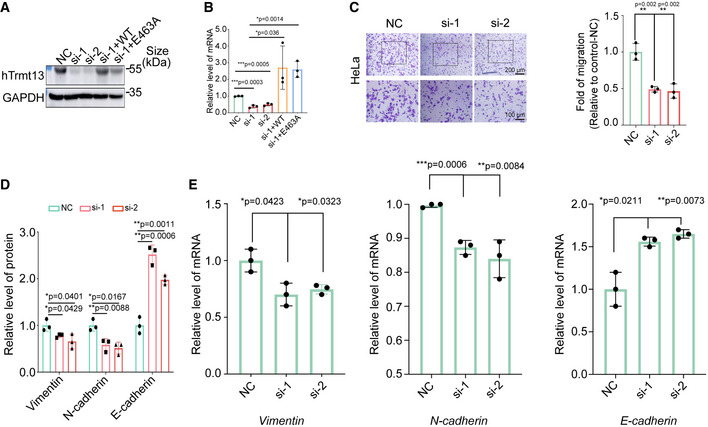
We further investigated the cellular function of hTrmt13. Surprisingly, knockdown of hTrmt13 did not affect cell proliferation (Fig 4A). Critically, hTrmt13 significantly affected cell migration in MDA‐MB‐231 cells, and transwell assays showed that cell migration was significantly attenuated in both hTrmt13 knockdown cells (si‐1 and si‐2) (Figs 4B and EV4A and B). Furthermore, the migration of hTrmt13 knockdown cells was restored to control levels by stable expression of the wild‐type hTrmt13 allele (si‐1+WT) (Fig 4B). Surprisingly, the hTrmt13 catalytically inactive mutant (E463A) could also successfully rescue the ability of cell migration (Fig 4B). These results indicated that hTrmt13 plays a role in cell migration independent of its function as a tRNA methyltransferase. Furthermore, knockdown of hTrmt13 also led to decreased migration potential in another breast cancer cell line HCC38 (Fig 4C) and a cervical cancer cell line HeLa (Fig EV4C). Epithelial–mesenchymal transition (EMT) is considered to be an important process in cancer cell metastasis (Lamouille et al, 2014). Loss of E‐cadherin (encoded by the CDH1 gene) and gain of N‐cadherin (encoded by the CDH2 gene) are phenomena specific to EMT progression (Lamouille et al, 2014). In order to characterize the roles of hTrmt13 in EMT, we investigated the changes of these EMT markers. The mRNA and protein levels of N‐cadherin and Vimentin (encoded by the VIM gene) were decreased in hTrmt13 knockdown MDA‐MB‐231 cells, while E‐cadherin mRNA and protein levels were up‐regulated in hTrmt13 knockdown MDA‐MB‐231 cells (Figs 4D and EV4D and E), indicating that hTrmt13 promotes EMT in MDA‐MB‐231 cells.
Figure 4. hTrmt13 promotes cell migration independent of tRNA modification activity.
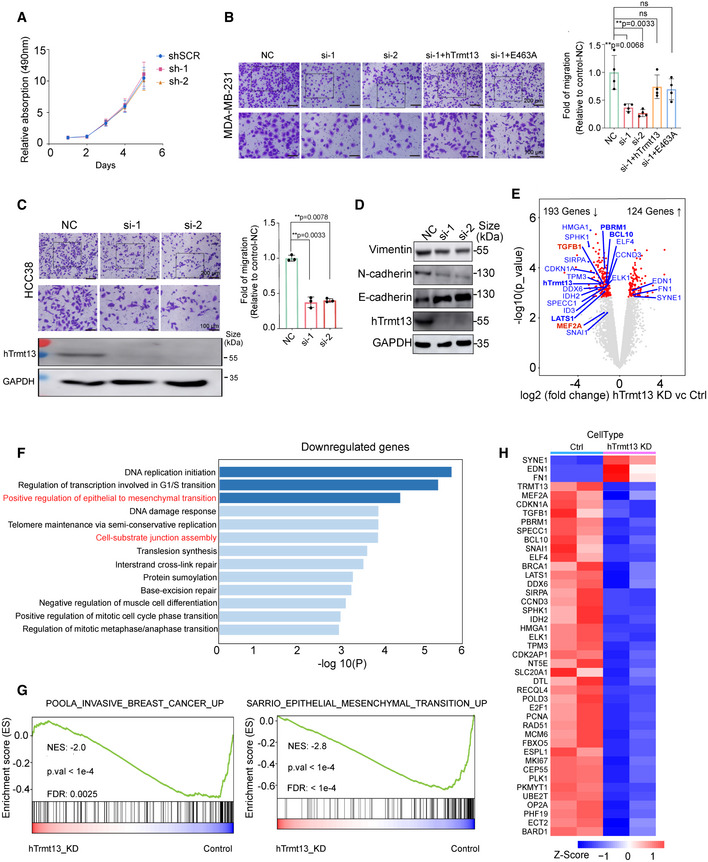
- MTT assay for cell proliferation in MDA‐MB‐231 cells infected with lentiviruses carrying control shRNA (shSCR) or shRNA against hTrmt13 (sh‐1, sh‐2). Error bars represent mean ± SD for three independent experiments.
- Transwell migration assays in MDA‐MB‐231 cells. Control with siRNA scramble (NC), treated with siRNA against hTrmt13 (si‐1, si‐2) and stably expressing wild‐type hTrmt13 (si‐1+WT) or enzymatic inactive hTrmt13 mutant (si‐1+E463A). Representative images (left panel) and statistical analysis (middle panel) are shown.
- Transwell migration assays in HCC38 cells infected with scrambled siRNA (NC) or siRNA against hTrmt13 (si‐1, si‐2).
- Western blot of the expression of Vimentin, N‐cadherin, and E‐cadherin in MDA‐MB‐231 cells with the indicated treatments. See also Fig EV4D.
- Volcano plot of differential expressed gene analysis results for RNA‐seq data comparing hTrmt13 knockdown MDA‐MB‐231 cells (N = 2) to control cells (N = 2), the dots for significant regulated genes (FDR corrected P < 0.05) were colored red, the dots and labels of interesting genes with oncogenic implications have been colored in blue or red.
- Functional categories of downregulated genes in hTrmt13 knockdown MDA‐MB‐231 cells showing the P‐value for the enrichment of biological process GO term.
- GSEA plot showing an interesting gene set from MSigDB were enriched for gene downregulated in hTrmt13 knockdown MDA‐MB‐231 cells (hTrmt13 KD) for POOLA_INVASIVE_BREAST_CANCER_UP and SARRIO_EPITHELIAL_MESENCHYMAL_TRANSITION_UP (extracted from GSE47203).
- Heatmap of expression level for highlighted genes and core enriched genes in EMT gene set.
Data information: In B and C, for each group, five different fields were chosen and counted using a microscope with 20‐fold magnification. Statistical analysis was performed using t‐tests. Error bars represent mean ± SD for four (B) or three (C) independent experiments.
Figure EV4. hTrmt13 regulates cancer cell migration.
-
A, BKnockdown efficiency of hTrmt13 in MDA‐MB‐231 verified by Western blot (A) and by RT–qPCR (B).
-
CTranswell migration assays in HeLa cells infected with control siRNA (NC) or siRNA against hTrmt13 (si‐1, si‐2). Representative images (left) and statistical analysis (right) are shown. For each group, five different fields were chosen and counted using a microscope with 20‐fold magnification.
-
DStatistical analysis of the expression of Vimentin, N‐cadherin, and E‐cadherin in control (NC) and hTrmt13 knockdown cells (si‐1, si‐2).
-
ERT–qPCR analysis of EMT‐associated genes Vimentin (left), N‐cadherin (middle), and E‐cadherin (right) in hTrmt13 knockdown MDA‐MB‐231 cells compared to control.
Data information: In B–E, statistical analysis was performed using t‐tests. Error bars represent mean ± SD for three independent experiments.
To further understand the underlying function and mechanism of hTrmt13, RNA‐seq transcriptomic analysis was performed to detect and quantify the genome‐wide transcriptomic changes in MDA‐MB‐231 cells among the negative control (NC) and hTrmt13 knockdown groups (KD). Compared to the NC groups, 193 genes were significantly downregulated and 124 genes were significantly upregulated more than twofold (Fig 4E, Dataset EV3). Several well‐studied genes involved in EMT, for example, a known EMT inducer TGFβ1 (Encoded by TGFB1) and the core EMT transcription factor Snail1 (Encoded by SNAI1) were downregulated in hTrmt13 knockdown cells compared to the control (Fig 4E). Consistently, gene ontology (GO) analysis found EMT and cell–substrate junction assembly terms as the most highly enriched for downregulated genes (Fig 4F). Next, we performed gene set enrichment analysis (GSEA) and found that the gene sets POOLA_INVASIVE_CANCER_UP and SARRIO_EPITHELIAL_MESENCHYMAL_TRANSITION_UP were significantly enriched for downregulated genes in hTrmt13 knockdown cells (Fig 4G). Heatmaps representing these highlighted genes along with core enriched EMT‐associated genes in hTrmt13 knockdown cells confirmed the consistent trend and reproducibility of our analyses (Fig 4H). Taken together, our data indicate that hTrmt13 promotes cell migration in cancer cell lines independent of catalytic activity and regulates the expression of EMT‐associated genes in MDA‐MB‐231 cells.
hTrmt13 promotes transcription of key cell migration‐related factors
By integrating our ChIP‐seq and RNA‐seq data, we found many of the differentially expressed genes also exhibit hTrmt13 binding at the promoter (Dataset EV4). Among these genes, many have been reported to play a key role in cell migration, such as TGFB1, PKN2, and MEF2A. TGFB1 is a potent inducer of EMT (Xu et al, 2009). Silencing of PKN2 resulted in decreased migration in head and neck cancer cell lines (Rajagopalan et al, 2018). MEF2A regulates the expression of matrix metalloproteinases which are associated with tumor invasion, metastasis, and endopeptidases (Ishikawa et al, 2010). To check whether TGFB1, PKN2, and MEF2A are true targets of hTrmt13 in vivo, we performed ChIP‐qPCR analysis for both endogenous and stably overexpressed hTrmt13. The results showed a strong enrichment of hTrmt13 and FLAG‐hTrmt13 on the promoters of TGFB1, PKN2, and MEF2A (Fig 5A). Also, we analyzed the mRNA levels of these genes by RT–qPCR assays. The results showed that the downregulation of hTrmt13 resulted in reductions of TGFB1, PKN2, and MEF2A expression (Fig 5B).
Figure 5. hTrmt13 promotes transcription of TGFB1, PKN2, and MEF2A .
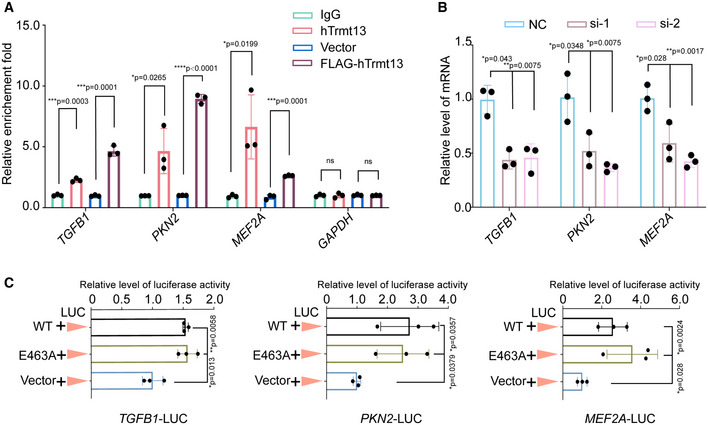
- Verification of ChIP‐seq results by ChIP‐qPCR analysis of the indicated genes.
- The mRNA level of the indicated genes was measured by RT–qPCR.
- Luciferase‐based reporter system to monitor the transcriptional level changes of indicated genes under overexpression of hTrmt13; MDA‐MB‐231 cells were co‐transfected with TGFB1‐Luc (left), PKN2‐Luc (middle), or MEF2A‐Luc (right) and expression construct for wild‐type hTrmt13 (WT) or catalytic inactive mutant (E463A) compared with control (Vector). Relative luciferase activity was calculated as firefly luciferase activity divided by renilla luciferase activity and shown relative to the control.
Data information: In A–C, statistical analysis was performed using t‐tests, and the error bars indicate mean ± SD for three independent experiments.
We next ligated the promoters of TGFB1, PKN2, and MEF2A with a luciferase reporter to investigate whether hTrmt13 can promote their transcription in MDA‐MB‐231 cells. We found that hTrmt13 promoted the activity of all the three promoters (Fig 5C) and that the catalytically inactive mutant hTrmt13‐E463A was also able to trigger promoter activity for these three genes (Fig 5C). TGFβ1 is a known EMT inducer that promotes the expression of some EMT core transcription factors, such as Snail1and ZEB1 (Joseph et al, 2014; Wei et al, 2017). The results from RT–qPCR assays and Western blots revealed that both the mRNA and protein levels of SNAI1 and ZEB1 were decreased in hTrmt13 knockdown MDA‐MB‐231 cells (Appendix Fig S4A and B); however, those of SNAI2 were not affected (Appendix Fig S4A and B). These data suggest that hTrmt13 modulates TGFβ1‐Snail1/ZEB1 axis during the EMT process in MDA‐MB‐231 cells. Taken together, we found that hTrmt13 targets and promotes the gene expression of cell migration‐related genes, such as TGFB1, PKN2, and MEF2A, independent of its catalytic activity.
hTrmt13 interacts with FOSL1, E2F1, and USF1 to regulate transcription
We further sought to understand the molecular basis of hTrmt13 in transcription. Our ChIP‐seq results showed that nuclear hTrmt13 was mainly targeted to promoters (Fig 3J). Using published ChIP‐seq and ATAC‐seq data in MDA‐MB‐231 cells, we found that ˜35% of hTrmt13 binding sites were also bound by FOSL1 and ˜78% of genes were also bound by E2F1, while 92% of hTrmt13 and FOSL1 co‐binding sites also exhibited E2F1 binding (Fig 6A). We next ask whether hTrmt13 could interact with these known transcription factors in vivo. First, we performed co‐immunoprecipitation of FLAG‐hTrmt13 and found that both FOSL1 and E2F1 could be pulled down by hTrmt13 (Fig 6B). Furthermore, we performed affinity purification and mass spectrometry to explore the in vivo hTrmt13 interactome. Interestingly, in the list of putative hTrmt13‐bound proteins (Dataset EV5), we identified many transcription factors including the upstream stimulatory factor 1 (USF1) which was previously reported to interact with FOSL1 (Pognonec et al, 1997; Luck et al, 2020) and E2F1 (Denechaud et al, 2016). Furthermore, the interaction of USF1 and hTrmt13 was confirmed by Western blotting (Fig 6B). It is worth noting that FOSL1, E2F1, and USF1 have all been reported to play roles in EMT or cancer cell migration. Specifically, FOSL1 promoted EMT by modulating TGFβ1 and ZEB1/2 expression in mouse breast cancer cells (Bakiri et al, 2015). E2F1 modulated EMT in non‐small cell lung cancer cells (Pillai et al, 2015). USF1 promotes melanoma invasiveness (Noguchi et al, 2018) and regulates EMT in human colon cancer cells (Ikeda et al, 2014). The interaction between endogenous hTrmt13 and FOSL1 or E2F1 or USF1 was further confirmed by co‐immunoprecipitation assay (Fig 6C). Proximity ligation assays (Duolink PLA technology, PLAs) allow for in situ detection of interactions between two proteins within 40 nm (Avin et al, 2017) (Fig 6D). PLAs were applied to detect the protein–protein interactions between hTrmt13 and FOSL1 or E2F1 or USF1 in MDA‐MB‐231 cells. The resulting images indicate that hTrmt13 colocalizes with FOSL1, E2F1, and USF1 predominantly in the nucleus (Fig 6E).
Figure 6. hTrmt13 interacts with FOSL1, E2F1 and USF1 to regulate transcription.
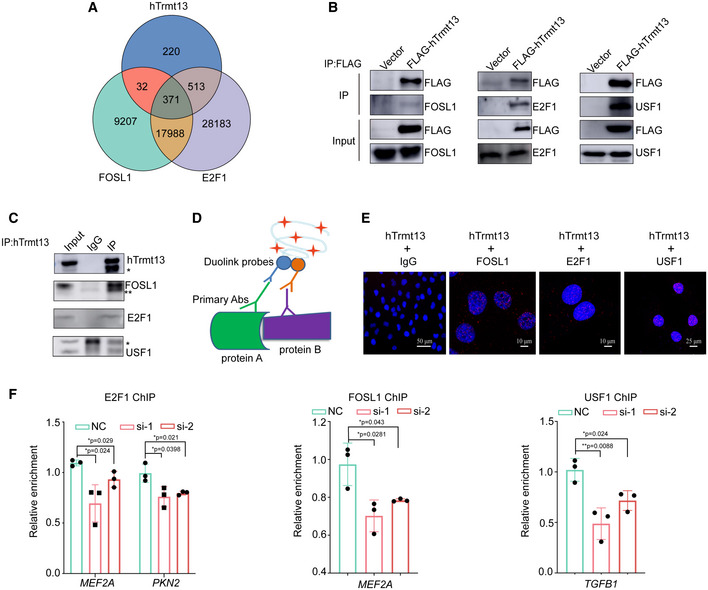
- The Venn diagram of overlapping genes targeted by hTrmt13, FOSL1, and E2F1 in MDA‐MB‐231 cells. Among 1136 (220 + 32 + 371 + 513 = 1136) hTrmt13 binding sites, ˜35% were also bound by FOSL1 (32 + 371 = 403), ˜78% were also bound by E2F1 (513 + 371 = 884), while 92% of hTrmt13 and FOSL1 co‐binding sites (403) also have E2F1 binding (371).
- Interaction of FLAG‐hTrmt13 with endogenous FOSL1, E2F1, and USF1. Whole‐cell lysates from MDA‐MB‐231 cells stably expressing FLAG‐hTrmt13 were prepared and immunoprecipitation was performed with anti‐FLAG followed by immunoblotting with antibodies against indicated proteins.
- Interaction of endogenous hTrmt13 with FOSL1, E2F1, and USF1.Whole‐cell lysates from MDA‐MB‐231 cells were immunoprecipitated with antibodies against hTrmt13 or IgG followed by immunoblotting with the antibodies against the indicated proteins. * and ** are nonspecific bands.
- A graphical scheme illustrating PLA of protein–protein interaction.
- PLA showing representative images obtained from imaging confocal microscopy of the interaction of hTrmt13 and FOSL1, hTrmt13 and E2F1, hTrmt13 and USF1. Red dots indicate that the detected protein interacts with hTrmt13, and DAPI staining was included to visualize the cell nucleus (Blue). The anti‐hTrmt13 and IgG was used as a negative control.
- ChIP‐qPCR analysis of the selected promoters was performed using antibodies against indicated proteins in control and hTrmt13 knockdown MDA‐MB‐231 cells. Results are represented as fold change over control IgG. Statistical analysis was performed using t‐tests. Error bars indicate mean ± SD for three independent experiments.
To further understand the biological significance of the interaction between hTrmt13 and transcription factors, ChIP experiments were performed for FOSL1, E2F1, and USF1, respectively, in WT and hTrmt13 knockdown cells. The enrichment of the co‐target genes (such as MEF2A, PKN2A, and TGFB1) between hTrmt13 and each transcription factor were detected by ChIP‐qPCR. Knockdown of hTrmt13 expression resulted in a remarkable reduction of recruitment of the corresponding transcription factor at the promoters of the target genes (Fig 6F). Altogether, our results indicated that hTrmt13 enhances the transcriptional regulatory activity of transcription factors such as FOSL1, E2F1, and USF1.
hTrmt13 directly interacts with transcription factors to exclude binding with tRNAs in the nucleus
tRNAs have both cytoplasmic and nuclear localization. However, it remains unclear why hTrmt13 prefers to bind DNA instead of tRNAs in the nucleus. To address this question, we attempted to characterize the biochemical properties of hTrmt13. First, we conducted pull‐down assays to determine whether hTrmt13 directly interacts with the above‐listed transcription factors. Unfortunately, among FOSL1, E2F1, and USF1, only USF1 could be purified in vitro. Our results showed that recombinant USF1 could be co‐purified with hTrmt13 from a bacterial co‐expression system (Fig 7A), suggesting a direct interaction between hTrmt13 and USF1. To further confirm the direct interaction between hTrmt13 and USF1, we purified each protein separately and incubated them together before gel filtration analysis; the chromatography results show that hTrmt13 and USF1 formed a higher molecular weight complex than their standalone proteins (Fig 7B). These results suggest a strong and direct interaction between hTrmt13 and USF1 and that their interaction is independent of DNA or tRNA.
Figure 7. hTrmt13 directly interacts with USF1 to exclude binding with tRNAs in nucleus.
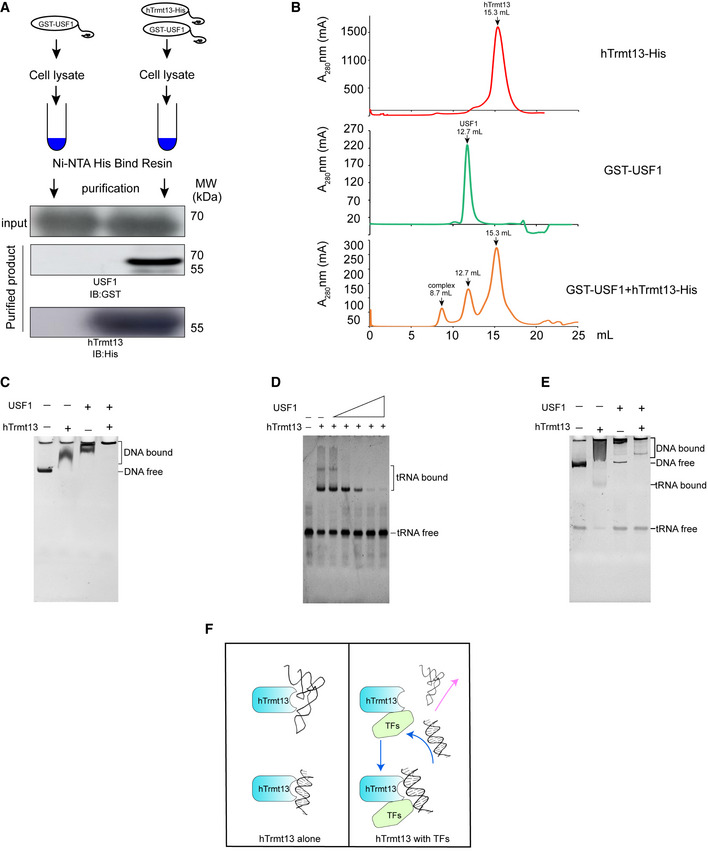
- In vitro pull‐down assay of hTrmt13 with USF1. USF1 with an N‐terminal GST tag (GST‐USF1) co‐purified by hTrmt13 with a C‐terminal His tag (hTrmt13‐His) using Ni‐NTA resin. GST‐USF1 and hTrmt13‐His were expressed separately in E. coli BL21.
- The gel filtration chromatography of hTrmt13‐His or USF1‐GST or their mixture on Superdex 200 Increase.
- The binding shift assays of hTrmt13, USF1, and their mixture for synthesized DNA from the TGFB1 promoter. In the reaction system, 10 ng/μl DNA was used, and 1 μM hTrmt13 or 1 μM USF1 or a mixture (0.5 μM hTrmt13 + 0.5 μM USF1) was applied.
- The binding shift assays of hTrmt13 for HctRNAGly(GCC) with the presence of various concentrations of USF1. In the reaction system, 0.3 μM HctRNAGly(GCC) and 0.5 μM hTrmt13 were used, and a varied amount of USF1 (0, 0.5, 1, 2, 3, 4 μM) were used.
- The binding shift assays of hTrmt13 (0.5 μM), or USF1 (0.5 μM) or the hTrmt13‐USF1 complex (0.5 μM) for both tRNA (0.1 μM) and DNA (15 ng/μl) in the same reaction.
- A proposed model based on the above results showing that standalone hTrmt13 binds either to tRNA or synthesized DNA, while the complex of hTrmt13 and TFs (e.g., USF1) excludes the binding with tRNA but not DNA.
We further performed binding shift assays of hTrmt13 for DNA and tRNA with or without USF1. Interestingly, we found that USF1 enhanced the binding of hTrmt13 to synthesized double‐stranded DNA containing a TGAGTCAT motif (Fig 7C), while USF1 attenuated or even inhibited the binding of hTrmt13 to tRNA (Fig 7D). In an attempt to mimic the situation in the nucleus, we added equimolar amounts of DNA and tRNA to the binding assay reaction and found that hTrmt13 alone bound to both DNA and tRNA, while the hTrmt13‐USF1 complex could only bind with DNA but not tRNA (Fig 7E). These results suggest that the interaction of hTrmt13 with transcription factors such as USF1 in the nucleus might exclude its binding to tRNAs (Fig 7F); therefore, nuclear hTrmt13 is more likely to modulate transcription than tRNA modification.
hTrmt13 regulates in vivo cancer progression
We have shown that hTrmt13 promotes the transcription of TGFB1, PKN2, and MEF2A in cell lines. Using the TCGA database, we unveiled a positive correlation between the production of hTrmt13 and PKN2, hTrmt13, and MEF2A in breast cancer patients (Fig 8A). To further investigate whether the working mechanism of hTrmt13 is widely applicable, we utilized the TCGA online portal (https://www.cbioportal.org/) for an hTrmt13‐centered analysis. We selected the largest breast cancer cohort with hTrmt13 RNA‐seq data (Breast Invasive Carcinoma TCGA, Firehose Legacy, 1090 patients, http://bit.ly/brca_htrmt13) and then separated them by quartile expression of hTrmt13. In line with our hypothesis, the groups of hTrmt13 expression level showed significant association with breast cancer subtypes (Fig 8B) (P = 5.7e‐5, chi‐square test), such as the hTrmt13 high group contained more invasive lobular breast carcinoma patients that were reported to have worse outcomes and more frequent metastases to bones. We also noticed that MMP1 (matrix metalloproteinase 1) was more highly expressed in the hTrmt13 low group, while CDH1 was also significantly higher (Fig 8A). These observations are consistent with both the MMP1 high group and the CDH1 high group containing less invasive lobular breast carcinoma patients along with more invasive ductal breast carcinoma patients (Fig 8B). We further ran GSEA on these data and found many of the enriched terms further emphasized the importance of hTrmt13 expression in breast cancer invasiveness. For example, SMID_BREAST_CANCER_ERBB2_UP (Smid et al, 2008) was enriched for the hTrmt13 low group (Fig EV5A), and this is consistent with lower expression of BRCA2. The enrichment of SOTIRIOU_BREAST_CANCER_GRADE_1_VS_3_UP (genes highly expressed in grade 1 of invasive breast cancer versus grade 3; Sotiriou et al, 2006) for the hTrmt13 low group (Fig 8C) suggested hTrmt13 might have a role as a marker for grading invasive breast cancer. Both TURASHVILI_BREAST_DUCTAL_CARCINOMA_VS_DUCTAL_NORMAL_DN and TURASHVILI_BREAST_LOBULAR_CARCINOMA_VS_LOBULAR_NORMAL_UP (Turashvili et al, 2007) were enriched for the hTrmt13 high group further strengthen the notion hTrmt13 high group more correlated with lobular breast carcinoma subtype rather than ductal breast carcinoma subtype (Figs 8C and EV5B).
Figure 8. hTrmt13 regulates in vivo cancer progression.
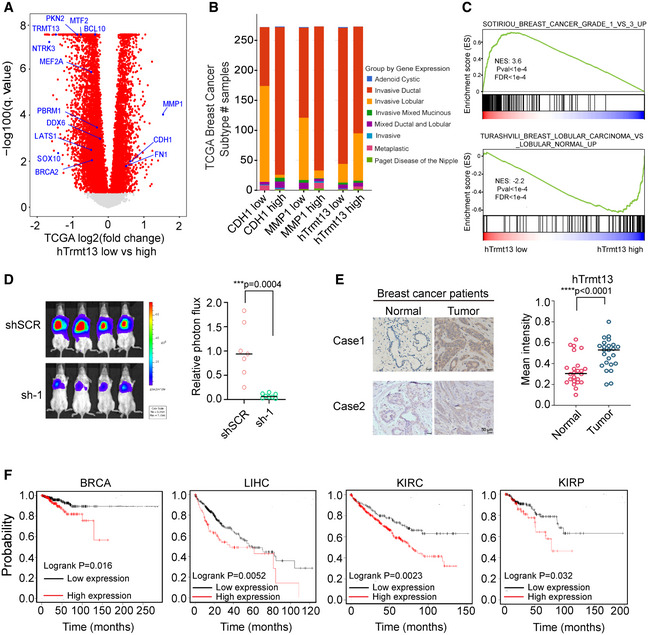
- Volcano plot of differential expressed gene analysis results for RNA‐seq data on TCGA comparing hTrmt13 expression low group patients (first quartile) versus high group patients (fourth quartile). Significant differentially expressed genes (FDR corrected P < 0.05) were colored by red, interesting genes with oncogenic implications are labeled in blue. Pseudo values 1e‐15 were added to FDR correlated P‐value for better visualization.
- Stacked bar plot of patients number diagnosed as various breast cancer subtype in TCGA group by the expression level of CDH1 or MMP1 or hTrmt13 (low as the first quartile, high as the fourth quartile).
- GSEA plot showing an interesting gene set from MSigDB were enriched for genes expressed higher in hTrmt13 low group for SOTIRIOU_BREAST_CANCER_GRADE_1_VS_3_UP (upper) and enriched for genes expressed higher in hTrmt13 high group for TURASHVILI_BREAST_LOBULAR_CARCINOMA_VS_LOBULAR_NORMAL_UP (down).
- Lung metastasis assay in mice model. MDA‐MB‐231‐Luc‐D3H2LN cells infected with lentivirus carrying control (shSCR) or shRNA against hTrmt13 (sh‐1) were injected through the tail vein of 6‐week‐old female SCID mice (n = 8). Lung metastasis was monitored by bioluminescent imaging after three weeks of injection. Representative in vivo bioluminescent images are shown (left); bioluminescent quantitation of lung metastasis (right). Statistical analysis was performed using t‐tests.
- Immunohistochemical staining of tissue arrays containing 24 breast carcinoma samples paired with adjacent normal mammary tissues for hTrmt13 expression. Representative images are shown (left), and the mean staining intensity was calculated using Image‐Pro Plus software (right). Statistical analysis was performed using t‐tests.
- Kaplan–Meier survival analysis of Kaplan–Meier plotter (http://kmplot.com/analysis/) for the relationship between the relapse‐free survival (RFS) or overall survival time (OS) of breast cancer patients (BRCA, n = 349 (low)/205 (high)), liver cancer patients (LIHC, n = 270 (low)/100 (high)), patients with kidney renal clear cell carcinoma (KRIC, n = 156 (low)/374 (high)), and patients with kidney renal papillary cell carcinoma (KIRP, n = 208 (low)/79 (high)) and their expression of hTrmt13.
Figure EV5. hTrmt13 regulates in vivo cancer progression.
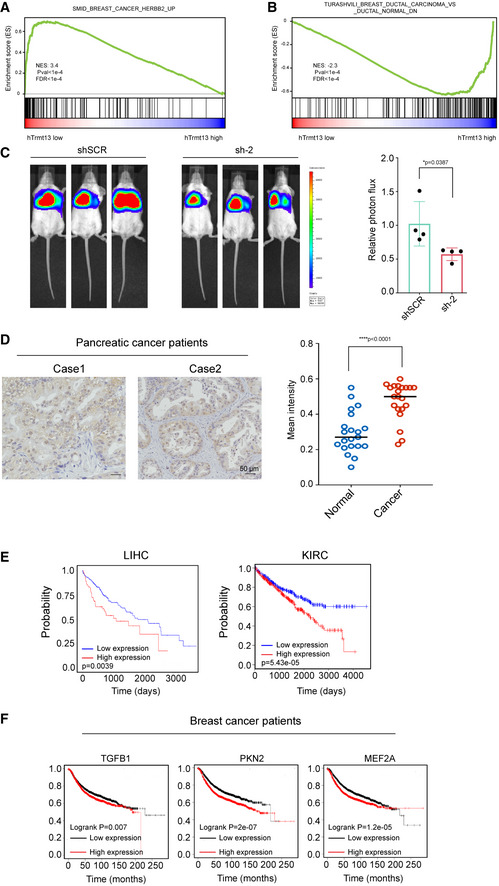
-
A, BGSEA plot showing interesting gene sets from MSigDB that were enriched for gene expressed higher in hTrmt13 low group for SMID_BREAST_CANCER_HERBB2_UP (A) and enriched for gene expressed higher in hTrmt13 high group for TURASHVILI_BREAST_DUCTAL_CARCINOMA_VS_DUCTAL_NORMAL_UP (B).
-
CLung metastasis assay in mice model. MDA‐MB‐231‐Luc‐D3H2LN cells infected with lentivirus carrying control (shSCR) or shRNA against hTrmt13 (sh‐2) were injected through the tail vein of 6‐week‐old female SCID mice (n = 4). Lung metastasis was monitored by bioluminescent imaging after three weeks of injection. Representative in vivo bioluminescent images are shown (left); bioluminescent quantitation of lung metastasis (right). Statistical analysis was performed using t‐tests. Error bars represent mean ± SD from mice of shSCR or sh‐2 groups.
-
DImmunohistochemical staining of 21 pancreatic carcinoma samples for hTrmt13 expression. Representative images are shown (left), and the mean staining intensity was analyzed by Image‐Pro Plus software (right). Statistical analysis was performed using t‐tests. Error bars represent mean ± SD from patient samples.
-
ETCGA analysis of the relationship between the overall survival time (OS) of liver cancer patients (LIHC, left, n = 277 (low)/88 (high), http://ualcan.path.uab.edu/analysis.html), patients with kidney renal clear cell carcinoma (KRIC, right, n = 260 (low)/259 (high), http://linkedomics.org/admin.php), and their expression of hTrmt13 using TCGA online analysis website.
-
FKaplan–Meier survival analysis (http://kmplot.com/analysis/) of Kaplan–Meier plotter for the relationship between relapse free survival (RFS) of breast cancer patients and their expression of TGFB1 (n = 2537 (low)/2392 (high)), PKN2 (n = 3025 (low)/1904 (high)), or MEF2A (n = 1307 (low)/3622 (high)).
To investigate whether hTrmt13 can affect cancer cell metastasis in vivo, we examined the effect of hTrmt13 loss‐of‐function on breast cancer cell dissemination in a mouse model. MDA‐MB‐231 cells that were engineered to stably express a gene encoding firefly luciferase were infected with lentiviruses carrying control shRNA (shSCR) or hTrmt13 shRNA. These cells were then injected into NOD/SCID mice (n = 8) intravenously and lung metastatic lesions were measured by quantitative bioluminescence imaging after three weeks of injection. The hTrmt13‐silenced cells rarely metastasized to the lung (Figs 8D and EV5C). Next, we examined hTrmt13 protein levels via immunohistochemical staining of human tissues of 24 breast carcinoma samples compared with normal tissues. The expression of hTrmt13 was significantly elevated in breast carcinoma samples (Fig 8E). We also found that hTrmt13 was also elevated in pancreatic cancer patients (Fig EV5D). Importantly, Kaplan–Meier survival analysis (https://kmplot.com/analysis/) for expression of hTrmt13 and the clinical behaviors of different cancers suggested that a high level of hTrmt13 was significantly correlated with poor prognosis of breast cancer (BRCA), liver cancer (LIHC), and renal cancers (KIRC, KIRP) (Fig 8F). These results were consistent with a parallel analysis of the TCGA database (Fig EV5E). Consistently, elevated hTrmt13 target genes TGFB1, PKN2, or MEF2A were significantly correlated with a poor prognosis of BRCA (Fig EV5F).
Together, our data indicate that hTrmt13 promotes breast cancer development, putatively by transcriptionally activating TGFB1, PKN2, and MEF2A, and that the expression of hTrmt13 is significantly correlated with patient survival in various cancer patients.
Discussion
tRNA research is expanding once again thanks to the development of new research techniques (Cozen et al, 2015; Zheng et al, 2015). Besides its central role in protein synthesis, many unexpected regulatory roles have been identified for tRNAs and tRNA‐derived tRFs (Kirchner & Ignatova, 2015). tRFs have diverse functions in many aspects of cell biology, especially translation inhibition process, while their comprehensive mechanisms are still remained to be deciphered (Thompson & Parker, 2009; Ivanov et al, 2011; Goodarzi et al, 2015; Gapp & Miska, 2016; Schimmel, 2018). Furthermore, the biogenesis of tRFs, especially the role of tRNA modifications in this process, is barely understood. Previous studies demonstrate that tRNA:m5C38 by Dnmt2 inhibits global angiogenin‐mediated tRF generation (Zhang et al, 2018). Here, we report tRNA: Nm4 by hTrmt13 modulates only the level of 5'‐Gly‐CCC but no other tRFs, suggesting that the effect of tRNA modification on the generation of tRFs is highly specific. tRNA modifications may work as a marker to recruit/exclude ANG or other RNA endonuclease directly, or via unknown reader proteins. In addition, the structures of tRNA are generally similar, whether modifications could introduce subtle structural differences and then affect the formation of tRF is also a question worthy of consideration. Furthermore, the modifications on tRNA could be delivered to tRF, whether they affect the structure or stability of tRF is also worth to be investigated.
The functions of tRNA‐modifying enzymes in higher eukaryotes are largely unknown. Very recently, the non‐catalytic functions of a few tRNA‐modifying enzymes became apparent. For instance, bacterial TruB can act as a tRNA chaperone independent of its pseudouridylation activity (Keffer‐Wilkes et al, 2016). Also, human PUS10 promotes pre‐miRNA processing independent of its tRNA pseudouridylation activity (Song et al, 2020). These findings suggest that the effect of tRNA‐modifying enzymes on cell phenotypes cannot be explained by modifications or catalytic activity alone. More importantly, aberrant expression and mutations of tRNA‐modifying enzymes have been reported to be closely associated with diseases, especially cancers, so it is vital to distinguish their catalytic and non‐catalytic functions. Our study reveals that hTrmt13 promotes the migration of breast cancer cells independent of its catalytic activity, emphasizing the importance to consider additional functions of enzymes outside of the traditional one protein–one function model. Moreover, hTrmt13 could bind many non‐substrate tRNAs (Figs 1E and EV1C and D) without an unknown function. Interestingly, this phenomenon is also observed in some other tRNA‐modifying enzymes, such as Nsun6 and PUS7 (Haag et al, 2015; Guzzi et al, 2018), which is worthy of attention in future studies.
Large‐scale analyses revealed that RNA‐binding proteins (RBP) are enriched in active chromatin regions in the human genome (Xiao et al, 2019). Generally, these RBPs are thought to regulate transcription via noncoding RNAs (Xiao et al, 2019). Our study demonstrated that chromatin‐associated hTrmt13 directly interacts with promoter DNA while excluding tRNA binding to promote transcription. DNA and RNA are quite different in structure, but here we showed that hTrmt13 bound to either DNA or tRNA with high affinity via the same CHHC zinc‐finger. This CHHC Zn‐finger was only identified in the Trmt13 family, U11/U12 small nuclear ribonucleoprotein 48 kDa protein (SNRNP48), and gametocyte‐specific factors (GTSF1 and GTSF1L; Andreeva & Tidow, 2008). The CHHC Zn‐finger mainly binds double‐strand RNA in SNRNP48 (Tidow et al, 2009), while in GTSF1, it interacts with protein (Almeida et al, 2018). In our work, the CHHC Zn‐finger is essential for hTrmt13 to bind DNA and the double‐stranded region of tRNA. However, due to the lack of structural information, we currently still cannot accurately characterize the molecular mechanism of how hTrmt13 and the CHHC Zn‐finger recognize DNA and tRNA. Interestingly, previous studies showed that the structural fold of the CHHC Zn‐finger from SNRNP48 is generally similar to the classical C2H2 Zn‐finger (Tidow et al, 2009). Particularly, it is most similar to Zn‐finger 5 of Transcription Factor IIIA (TFIIIA; Tidow et al, 2009). This Zn‐finger 5 was reported to bind DNA and RNA in different ways. Specifically, it interacts with the backbone of a double helix of RNAs, while binding to the major groove of its target DNA (Hansen et al, 1993; Lu et al, 2003).
Interestingly, tRNA‐modifying enzymes performing functions in transcription might be more prevalent than previously expected. For example, the human tRNA m1A methyltransferase hTrmt6 was reported to interact with the acetylated histone H4 (Lang et al, 2013). The human elongator complex responsible for tRNA modifications functions as a histone acetyltransferase that modulates gene transcription (Kim et al, 2002; Bento‐Abreu et al, 2018). Therefore, tRNA‐modifying enzymes act as a large group of RBPs, and their function(s) in transcription deserves more attention. It is worth noting that the regulatory role of chromatin‐associated modifying enzymes of other RNAs (e.g., mRNA:m6A writer, Mettl3) is also beginning to be realized (Liu et al, 2020; Chelmicki et al, 2021; Xu et al, 2021).
A growing number of studies have shown that tRNA‐modifying enzymes are associated with various cancers (Orioli, 2017). Two main mechanisms of tRNA modification linked to cancer progression have been identified: (i) Some aberrant tRNA modifications induce the production of tRFs to modulate translation (Guzzi & Bellodi, 2020) and (ii) some modifications on the anticodon loop modulates codon‐specific translation of oncogenes (Delaunay et al, 2016; Rapino et al, 2017, 2018). Here, we show that hTrmt13 promotes breast cancer metastasis through a novel mechanism by transcriptional regulation of key epithelial–mesenchymal transition factors instead of relying on its tRNA modification activity. Importantly, the expression of hTrmt13 is significantly correlated to patient survival in many different cancer types such as liver cancer, renal cancer, and breast carcinoma (Figs 8F and EV5E and F). Cancer stages are a widely applicable indicator of prognosis. The degree to which tumors infiltrated nearby tissues or other organs is one of the main determinants of the cancer stage and is highly correlated with the ability of a cell to metastasis. EMT enables the initial detachment of cells from the primary tumor by altering the phenotype of cancer cells and improving their ability to migrate. As a well‐studied signaling pathway in EMT, TGF‐β was considered as a good target for treatment. However, although many reports have found that TGF‐β inhibitors perform well in cell culture (in vitro) and animal models (in vivo), results in clinical trials are poor or inconsistent (Teixeira et al, 2020). Thus, a better understanding of the comprehensive EMT regulator pathway remains valuable as a means for cancer treatment. Here, we revealed the novel player hTrmt13 that likely acts upstream of the TGF‐β pathway, our results also provide a molecular basis with which to target hTrmt13 for EMT and emphasizes the importance of targeting the transcriptional regulation ability over the tRNA methyltransferase ability for therapeutic purposes.
Materials and Methods
Purification of hTrmt13 and USF1
The cDNA of hTrmt13 (NM_019083.2) and the cDNA of USF1 (NM_007122.5) were inserted between NotI and XhoI in pET22b (along with a DNA sequence encoding a C‐terminal 6xHis tag) and pGEX‐6P‐1 (along with a DNA sequence encoding a N‐terminal GST tag), respectively. The constructs were expressed in Escherichia coli Rosetta. Recombinant hTrmt13 was purified by affinity chromatography on nickel‐nitrilotriacetic acid Superflow resin, followed by gel filtration chromatography on a Superdex™ 200 column. Recombinant USF1 was purified by affinity chromatography on Glutathione Sepharose™ 4B and gel filtration chromatography using Superdex™ 200.
tRNA methyl transfer assays
The reactions were performed at 37°C under the conditions in a 25 μl reaction mixture containing 50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mg/ml BSA, 200 μM [methyl‐3H] SAM, 5 μM tRNAs, and 5 mM DTT. Reactions were initiated by the addition of 300 nM hTrmt13 or its mutants. At time intervals ranging between 3 and 12 min, 5 μl aliquots were removed to glass fiber filter discs and soaked in 5% trichloroacetic acid to precipitate the labeled methylated tRNAs. The amount of radioactive [methyl‐3H] tRNA on each disc was measured in a Beckman Ls6500 scintillation counting apparatus. The steady‐state kinetics were measured under the same conditions with a range of 50–500 nM hTrmt13 and a range of 0.1–100 μM tRNA, and the reaction time was 2 or 5 min. The K m and k cat values of hTrmt13 were calculated from the methyl transfer reaction using Lineweaver–Burk plots.
Mass spectrometry analysis of tRNA modifications in vitro
The tRNA and corresponding mutants were incubated with hTrmt13 at 37°C for 2 h. 10 μg of tRNAs were hydrolyzed by phosphodiesterase I, benzonase, and bacterial alkaline phosphatase, respectively, in a 60 μl reaction containing 50 mM NaCl, 20 mM Tris–HCl, pH 8.0, 2 mM MgCl2. The methylated tRNA was completely hydrolyzed at 37°C for 24 h. 1 μl of the solution was applied to UPLC‐MS/MS. The nucleosides were separated by UPLC on a C18 column (Agilent Zorbax Eclipse Plus C18, 2.1 × 50 mm, 1.8 μm) and then detected by a triple‐quadrupole mass spectrometer (Agilent 6495 QQQ) in the positive ion multiple reaction‐monitoring mode. The nucleosides were quantified using the nucleoside‐to‐base ion mass transitions of 282.1–136.2 (Am), 298.1–152.1 (Gm), 258.1–112.1 (Cm), 259.1–113.1 (Um).
Isolation of a given tRNA by biotinylated DNA probes
Total RNA was extracted from cells using TRIzol (Invitrogen). The endogenous tRNAs used in this study were isolated by their own biotinylated DNA probe and were purified by Streptavidin Agarose Resin (Thermo Fisher Scientific). 30 μl of high‐capacity streptavidin‐conjugated agarose beads were washed with buffer A (10 mM Tris–HCl, pH 7.5) and suspended in buffer B (100 mM Tris–HCl, pH 7.5). Subsequently, 200 μM biotinylated oligonucleotides were mixed and incubated at room temperature for 100 min. After the incubation, the oligonucleotide‐coated beads were then washed three times in buffer A and equilibrated in 6× NTE solution (1× NTE: 200 mM NaCl, 5 mM Tris–HCl, pH 7.5, 2.5 mM EDTA). The oligonucleotide‐coated beads and total RNAs in 6× NTE solutions were heated for 5 min at 70°C and cooled down to 30°C. Then, the beads were washed with 3× NTE for three times and 1× NTE for twice. The specific tRNA retained on the beads was eluted with 0.1× NTE at 70°C and precipitated using 75% ethanol.
Probe of HctRNAGly(GCC): 5'biotin‐GCGAGAATTCTACCACTGAACCACCCA‐3'
Probe of HctRNAGly(CCC): 5'biotin‐TCTTGCATGATACCACTACACCAGCGG‐3'
Probe of HctRNAGly(UCC): 5'biotin‐CTATGCTAACCACTATACCACCAACGC‐3'
Probe of HctRNAPro: 5'biotin‐AGAATCATACCCCTAGACCAACGAGCC‐3'
RNA‐mass spectrometry analysis of cellular tRNA modifications
200 ng of specific tRNAs purified by the biotinylated DNA probes was digested with 0.3 μl benzonase, 0.3 μl phosphodiesterase I, and 0.3 μl bacterial alkaline phosphatase in a 20 μl solution including 4 mM NH4OAc at 37°C overnight. After complete hydrolysis, 1 μl of the solution was applied to ultra‐performance liquid chromatography–mass spectrometry/mass spectrometry (UPLC‐MS/MS). The nucleosides were separated on an Atlantis® HILIC Silica column (3 μm, 2.1 × 150 mm) and then detected by a mass spectrometer (AB Sciex QTRAP 6500+) in the positive ion multiple reaction monitoring (MRM) mode. The ratio of Cm and Um to the sum of G was determined based on the calculated concentrations.
Electrophoretic mobility shift assays (EMSA)
Purified proteins (hTrmt13, USF1), tRNA, or DNA or cellular total DNA fragments were incubated in 20 μl buffer (20 mM Tris–HCl (pH 7.5), 200 mM NaCl, 5 mM MgCl2, and 15% glycerol) at 4°C for 20 min. The final concentration of tRNA or DNA used was from 0.1 to 0.3 μM, and the final concentration of hTrmt13 proteins varied from 0 to 2 μM. After incubation, 4 μl loading solution (0.25% bromophenol blue, 0.25% xylene cyanide, and 60% glycerol) was added into each sample and loaded immediately on a 6% polyacrylamide native gel. Electrophoresis was carried out on ice at a constant voltage of 90 V for 50 min. Ethidium bromide or SYBR™ Gold Nucleic Acid Gel Stain and Coomassie brilliant dye were used to stain the gel to detect RNA or DNA and protein, respectively.
shRNA and lentivirus‐mediated knockdown
Lentiviruses were generated in HEK293 cells by Lipofectamine 2000‐mediated co‐transfection of lentiviral‐based shRNA plasmids and the pCMV‐VSVG (envelope) and pCMV‐dR8.9 (packaging) plasmids. MDA‐MB‐231 cells were transduced with virus in media supplemented with polybrene (10 μg/ml). Cells were incubated 24 h with viruses and then cultured in fresh media for 24 h, and then, cells were cultured in media supplemented with 1 μg/ml puromycin for another 48 h. When using recombinant hTrmt13 in the rescue experiments, the codon sequences of htrmt13 have been modified in the gene by keeping the same amino acid readout, while these mutations on mRNA could avoid the targeting by these two shRNAs.
Northern blotting
Total RNAs were electrophoresed through a 12% polyacrylamide 8 M urea gel and then transferred to nylon membrane (MILLIPORE). Membrane was crosslinked at 254 nM, with 150 mJ/cm2 using UV Stratalinker 1800. The membrane was then pretreated with ULTRAhyb Ultrasensitive Hybridization Buffer (Thermo Fisher Scientific) at 55°C for 2 h. Digoxigenin (DIG)‐labeled DNA probes were denatured and added into the hybridization buffer to hybridize at 55°C for 12 h. The membrane was washed at 55°C and then incubated in blocking buffer (Roche) for 1 h at room temperature (RT). DIG antibody (Roche) was added and incubated for 30 min. The membrane was washed with wash buffer (100 mM Maleic acid, 150 mM NaCl, 0.3%(v/v) Tween 20, pH 7.5) and incubated in detection buffer (100 mM NaCl, 100 mM Tris–HCl 9.5). Lastly, the membrane was incubated with CDP‐Star (Roche) and signal intensities were detected by Amersham Imager 680 (GE).
Probe of HctRNAGly(GCC): 5'DIG‐GCGAGAATTCTACCACTGAACCACCCA‐3'
Probe of HctRNAGly(CCC): 5'DIG‐TCTTGCATGATACCACTACACCAGCGG‐3'
Probe of HctRNAGly(UCC): 5'DIG‐CTATGCTAACCACTATACCACCAACGC‐3',
Probe of HctRNAPro: 5'DIG‐AGAATCATACCCCTAGACCAACGAGCC‐3'
Cellular tRNA aminoacylation level assays
Total RNAs under acid conditions were electrophoresed at 4°C through an acid (pH 5.2) 10% polyacrylamide, 8 M urea gel. The gels were then electroblotted onto nylon membrane for the hybridization analysis with DIG‐labeled oligonucleotide probe as described for northern blotting. To distinguish non‐aminoacylated tRNA from aminoacylated tRNA, tRNAs were deacylated by being incubated for 30 min at 37°C (pH 9.0) and then run in parallel.
Reverse transcription and RT–qPCR
For quantification of tRFs, small RNAs (< 200 nt) were enriched from cells using the mirVana™ miRNA Isolation Kit (Invitrogen). The small RNAs were reverse‐transcribed using specific primers (Dataset EV6) by Superscript III (Invitrogen) and used for RT–qPCR using a miScript SYBR Green PCR kit (Qiagen). For the poly‐A‐based RT–qPCR method, poly‐A was tagged to small RNAs using the QuantiMir™ Kit (SBI) before reverse transcription and an oligo‐dT‐adapter primer was used. For quantification of tRFs, all RT reaction was performed under 37°C without deacylation or demodification treatment. For quantification of mRNA, total RNAs were reverse‐transcribed using Promega GoScript (Promega) followed by RT–qPCR using the Eastep® qPCR Master Mix (Promega).
Transwell cell migration assay
Cells with the indicated treatment were starved in serum‐free DMEM for 16 h. 24‐well 8 μM pore transwell polycarbonate membrane inserts from Corning were used. For each insert, 1 × 104 cells were seeded in serum‐free media in the upper chamber, and the chamber was then transferred to a well containing complete DMEM containing 10% FBS. Cells were incubated at 37°C for 36 h. The membranes were stained with crystal violet (Sigma) and the remaining cells were counted. Five fields of view were counted for each membrane.
MTT assay for cell proliferation
1 × 103 cells were seeded in 24‐well culture plates and assayed every 24 h. After the incubation period, serum‐free media with MTT labeling reagent were added into the cells and incubated for 3 h at 37°C. Then, we added DMSO (Sigma) into each well and shook plates on an orbital shaker to completely dissolve the purple crystals. Lastly, we measured absorbance at OD490 (490 nm) and determined the degree of change as a percentage of the control after background subtraction.
RNA‐seq
2 μg total RNA was subjected to two rounds of rRNA removal via the Truseq® Stranded RNA LT Kit (Illumina). Then, RNA was used to construct the library by NEBNext Ultra RNA Library Prep kit. Each library was sequenced using TruSeq SBS kit v4‐HS, in paired‐end mode with a read length of 2 × 150 bp.
Immunofluorescence
Cells were briefly washed in PBS and then fixed in PBS containing 4% paraformaldehyde (Sigma) for 10 min at RT. Fixed cells were washed in PBS and were then permeabilized in PBS with 0.2% Triton X‐100 for 5 min at RT. Cells were blocked in PBS with 4% BSA (Sigma). Primary and secondary antibody incubations were performed in PBS with 1% BSA overnight at 4°C and 1 h at RT, respectively. Slides were imaged on a Leica TCS sp8 STED or GE DeltaVision OMX SR microscope. Antibodies used include mouse anti‐HA (CST), rabbit anti‐hTrmt13, and mouse anti‐Histone 3 (Abcam).
Pull‐down assays
The pET22b‐hTrmt13‐His and pGEX‐6P‐1‐GST‐USF1 constructs were expressed in Escherichia coli (Rosetta). Cells were cultured at 37°C to OD600 = 0.6 and induced by 0.1 mM IPTG (Sangon Biotech) at 16°C for overnight. The cells were collected and resuspended with lysis buffer (200 mM NaCl, 20 mM Tris–HCl, pH 7.5, 10% glycerol, 2 mM DTT) and then sonicated by ultrasonic crusher. The lysate containing GST‐USF1 and hTrmt13‐His were mixed and then incubated with nickel‐nitrilotriacetic acid Superflow resin. The resin was then washed 3 times using the lysis buffer with 20 mM imidazole. The complex was eluted by elution buffer (200 mM NaCl, 20 mM Tris–HCl, pH 7.5, 10% glycerol, 2 mM DTT, and 250 mM imidazole). The complex was loaded on SDS–PAGE and analyzed by Western blotting.
Separation of cytoplasmic and nuclear extracts from cells
Separation of cytoplasmic and nuclear extracts was performed on MDA‐MB‐231 cells using NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific). The chromatin was disrupted by Covaris M220.
Immunoprecipitation (IP) and identification of protein interactome of hTrmt13
MDA‐MB‐231 cellular extracts were prepared by incubating the cells in IP lysis buffer (150 mM NaCl, 50 mM Tris–HCl, pH 7.5, 0.5% NP40, 1 mM EDTA, 10% glycerol) for 20 min at 4°C followed by centrifugation at 13,800 g for 10 min at 4°C. The chromatin was resuspended by digest buffer (5 mM MgCl2, 20 mM Tris–HCl, pH 7.5 150 mM NaCl) and digested by DNase I for 30 min at 4°C. This was followed by centrifugation at 13,800 g for 5 min at 4°C. The supernatant together with the whole cell lysate was used for immunoprecipitation. 1 mg total protein was incubated with 5 μl Anti‐FLAG® M2 Magnetic Beads or specific antibodies (10 μg) for 12 h at 4°C with constant rotation; 30 μl of protein A/G agarose beads was then added and the incubation was continued for an additional 3 h. Beads were then washed five times using the IP lysis buffer. The IP complex was then eluted by elution buffer (150 mM NaCl, 20 mM Tris–HCl, pH 7.5, 1% SDS). The eluted IP samples were loaded on SDS–PAGE followed by Western blot analysis. To identify the protein interactome of hTrmt13, the eluted IP samples of FLAG‐hTrmt13 were digested into peptides and then subjected to LC‐MS/MS for protein identification.
Proximity ligation in situ assay
The in situ interaction was measured using Duolink™ In Situ kit (Sigma) according to the manufacturer’s protocol. The following antibodies were used: mouse anti‐E2F1 (Santa Cruz), mouse anti‐USF1 (Santa Cruz), mouse anti‐FOSL1 (Santa Cruz), and anti‐hTrmt13 from rabbit. The slides were imaged on a Leica TCS sp8 STED microscope.
In vivo metastasis
MDA‐MB‐231 cells that had been transfected to stably express firefly luciferase (PerkinElmer) were infected with lentiviruses carrying control shRNA or shhTrmt13. 3 × 106 cells were injected into the lateral tail vein of 6‐week‐old female SCID mice. After three weeks, mice were injected abdominally with 200 mg/g of D‐luciferin (YEASEN) in PBS. 10 min after injection, mice were anesthetized and bioluminescence was imaged with a charge‐coupled device camera (IVIS; Xenogen). Photon flux was normalized to the background that was defined from a relative optical intensity drawn over a mouse that was not given an injection of luciferin. Mice were maintained in a specific pathogen‐free animal facility with ad libitum access to food and water. The procedures for care and use of animals were approved by Institutional Animal Care and use Committee (China, SIBCB‐S209‐1705‐007‐c2), and all applicable institutional and governmental regulations (e.g., ARRIVE guidelines) concerning the ethical use of animals were followed.
Immunohistochemistry
The paraffin sections of human cancer tissues (Servicebio, slides) were placed in xylene (100%) and incubated at 55°C for 2 h. Slides then underwent a series of washes (3 min per step at RT): twice in xylene, xylene:ethanol (1:1), twice in 100% ethanol, twice in 95% ethanol, 70% ethanol, and 50% ethanol. Slides were then rinsed gently in distilled water for 5 min at RT. The antigen was retrieved by high‐pressure cooking in 10 mM sodium citrate buffer. Then, the samples were blocked in PBS with 10% normal goat serum and incubated overnight at 4°C in primary antibody solution buffer with anti‐hTrmt13 (1:100). The slides were washed with 10 mM PBS buffer, the samples were incubated with polymer HRP goat anti‐mouse and rabbit IgG (GBI) at RT for 30 min, developed with DAB (3,3'‐diaminobenzidine tetrahydrochloride), and counterstained with hematoxylin (Sigma). The mean staining intensity was calculated using Image‐Pro Plus software.
Statistics
Statistics was performed using GraphPad Prism v8. All plots with sample sizes show individual data points.
More details about procedures and reagents used in this study are provided in the Appendix Materials and Methods.
Author contributions
R‐JL, E‐DW, and HL conceived the ideas for this work. HD performed in vivo Metastasis assay. BX performed bioinformatics analysis. HL, Q‐PX, and Z‐XH generated plasmids. C‐TL performed tRF assays. W‐QY performed pull‐down assay. HL performed all the other experiments with technical helps from R‐JL, Q‐YZ, and JL. R‐JL, E‐DW, HL, and BX wrote the manuscript, which was reviewed by all authors.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Acknowledgments
This work was supported by National Key Research and Development Program of China (2020YFA0803401, 2017YFA0504000); Natural Science Foundation of China (91940302, 32022040, 31971230, 31870811, 31770842); Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19010203). We thank the National Center for Protein Sciences at Peking University in Beijing, China, for assistance with the UPLC‐MS/MS work. We thank Dr. Chao Peng at the National Facility for Protein Science in Shanghai (NFPS), Zhangjiang Lab, China, for technical support in MS analysis of protein identification. We thank professors Dangsheng Li, Hanhui Ma, and Jilong Liu for their helpful discussions on the manuscript. We thank professors Hong Cheng and Yi Zeng in SIBCB for sharing experimental techniques. We thank the Molecular and Cell Biology Core Facility and the Multi‐Omics Core Facility at the SLST of Shanghai Tech University for providing technical support.
Conflicts of Interests
The authors declare that they have no conflict of interest.
The EMBO Journal (2022) 41: e108544.
Contributor Information
En‐Duo Wang, Email: edwang@sibcb.ac.cn.
Ru‐Juan Liu, Email: liurj@shanghaitech.edu.cn.
Data availability
All data are available in the main text or the appendix data. Raw and processed data have been deposited at NCBI GEO Super‐Series under accession GSE166278 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166278) including CLIP‐Seq: GEO GSE166274 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166274); RNA‐Seq: GEO GSE166276 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166276); ChIP‐Seq: GEO GSE166270 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166270).
References
- Almeida MV, Dietz S, Redl S, Karaulanov E, Hildebrandt A, Renz C, Ulrich HD, Konig J, Butter F, Ketting RF (2018) GTSF‐1 is required for formation of a functional RNA‐dependent RNA polymerase complex in Caenorhabditis elegans . EMBO J 37: e99325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Ivanov P (2014) tRNA fragments in human health and disease. FEBS Lett 588: 4297–4304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva A, Tidow H (2008) A novel CHHC Zn‐finger domain found in spliceosomal proteins and tRNA modifying enzymes. Bioinformatics 24: 2277–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avin A, Levy M, Porat Z, Abramson J (2017) Quantitative analysis of protein‐protein interactions and post‐translational modifications in rare immune populations. Nat Commun 8: 1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakiri L, Macho‐Maschler S, Custic I, Niemiec J, Guio‐Carrion A, Hasenfuss SC, Eger A, Muller M, Beug H, Wagner EF (2015) Fra‐1/AP‐1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFbeta expression. Cell Death Differ 22: 336–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I, Kouzarides T (2020) Role of RNA modifications in cancer. Nat Rev Cancer 20: 303–322 [DOI] [PubMed] [Google Scholar]
- Bento‐Abreu A, Jager G, Swinnen B, Rué L, Hendrickx S, Jones A, Staats KA, Taes I, Eykens C, Nonneman A et al (2018) Elongator subunit 3 (ELP3) modifies ALS through tRNA modification. Hum Mol Genet 27: 1276–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelmicki T, Roger E, Teissandier A, Dura M, Bonneville L, Rucli S, Dossin F, Fouassier C, Lameiras S, Bourc'his D (2021) m6A RNA methylation regulates the fate of endogenous retroviruses. Nature 591: 312–316 [DOI] [PubMed] [Google Scholar]
- Chen QI, Yan M, Cao Z, Li X, Zhang Y, Shi J, Feng G‐H, Peng H, Zhang X, Zhang Y et al (2016) Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 351: 397–400 [DOI] [PubMed] [Google Scholar]
- Chen Z, Qi M, Shen B, Luo G, Wu Y, Li J, Lu Z, Zheng Z, Dai Q, Wang H (2019) Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA‐derived small RNAs. Nucleic Acids Res 47: 2533–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozen AE, Quartley E, Holmes AD, Hrabeta‐Robinson E, Phizicky EM, Lowe TM (2015) ARM‐seq: AlkB‐facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat Methods 12: 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay S, Rapino F, Tharun L, Zhou Z, Heukamp L, Termathe M, Shostak K, Klevernic I, Florin A, Desmecht H et al (2016) Elp3 links tRNA modification to IRES‐dependent translation of LEF1 to sustain metastasis in breast cancer. J Exp Med 213: 2503–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denechaud P‐D, Lopez‐Mejia IC, Giralt A, Lai Q, Blanchet E, Delacuisine B, Nicolay BN, Dyson NJ, Bonner C, Pattou F et al (2016) E2F1 mediates sustained lipogenesis and contributes to hepatic steatosis. J Clin Invest 126: 137–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewe JM, Fuller BL, Lentini JM, Kellner SM, Fu D (2017) TRMT1‐catalyzed tRNA modifications are required for redox homeostasis to ensure proper cellular proliferation and oxidative stress survival. Mol Cell Biol 37: e00214–e217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres L, Rose RE, Doyle F, Rahn T, Lee B, Seaman J, McIntyre WD, Fabris D (2020) 2'‐O‐ribose methylation of transfer RNA promotes recovery from oxidative stress in Saccharomyces cerevisiae . PLoS One 15: e0229103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Harada BT, Behm M, He C (2018) RNA modifications modulate gene expression during development. Science 361: 1346–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gapp K, Miska EA (2016) tRNA fragments: novel players in intergenerational inheritance. Cell Res 26: 395–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H, Liu X, Nguyen HC, Zhang S, Fish L, Tavazoie SF (2015) Endogenous tRNA‐derived fragments suppress breast cancer progression via YBX1 displacement. Cell 161: 790–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF (2016) Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 165: 1416–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzzi N, Bellodi C (2020) Novel insights into the emerging roles of tRNA‐derived fragments in mammalian development. RNA Biol 17: 1214–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzzi N, Cieśla M, Ngoc PCT, Lang S, Arora S, Dimitriou M, Pimková K, Sommarin MNE, Munita R, Lubas M et al (2018) Pseudouridylation of tRNA‐derived fragments steers translational control in stem cells. Cell 173: 1204–1216 [DOI] [PubMed] [Google Scholar]
- Haag S, Warda AS, Kretschmer J, Gunnigmann MA, Hobartner C, Bohnsack MT (2015) NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. RNA 21: 1532–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen PK, Christensen JH, Nyborg J, Lillelund O, Thøgersen HC (1993) Dissection of the DNA‐binding domain of Xenopus laevis TFIIIA. Quantitative DNase I footprinting analysis of specific complexes between a 5 S RNA gene fragment and N‐terminal fragments of TFIIIA containing three, four or five zinc‐finger domains. J Mol Biol 233: 191–202 [DOI] [PubMed] [Google Scholar]
- Hanson G, Coller J (2018) Codon optimality, bias and usage in translation and mRNA decay. Nat Rev Mol Cell Biol 19: 20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppertz I, Attig J, D'Ambrogio A, Easton LE, Sibley CR, Sugimoto Y, Tajnik M, König J, Ule J (2014) iCLIP: protein‐RNA interactions at nucleotide resolution. Methods 65: 274–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda R, Nishizawa Y, Tajitsu Y, Minami K, Mataki H, Masuda S, Furukawa T, Akiyama S, Yamada K, Takeda Y (2014) Regulation of major vault protein expression by upstream stimulating factor 1 in SW620 human colon cancer cells. Oncol Rep 31: 197–201 [DOI] [PubMed] [Google Scholar]
- Ishikawa F, Miyoshi H, Nose K, Shibanuma M (2010) Transcriptional induction of MMP‐10 by TGF‐beta, mediated by activation of MEF2A and downregulation of class IIa HDACs. Oncogene 29: 909–919 [DOI] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P, Senju S, Nishimura Y, Chuang JH, Ackerman SL (2014) RNA function. Ribosome stalling induced by mutation of a CNS‐specific tRNA causes neurodegeneration. Science 345: 455–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P (2011) Angiogenin‐induced tRNA fragments inhibit translation initiation. Mol Cell 43: 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JV, Conroy S, Tomar T, Eggens‐Meijer E, Bhat K, Copray S, Walenkamp AM, Boddeke E, Balasubramanyian V, Wagemakers M et al (2014) TGF‐beta is an inducer of ZEB1‐dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis 5: e1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keffer‐Wilkes LC, Veerareddygari GR, Kothe U (2016) RNA modification enzyme TruB is a tRNA chaperone. Proc Natl Acad Sci USA 113: 14306–14311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lane WS, Reinberg D (2002) Human Elongator facilitates RNA polymerase II transcription through chromatin. Proc Natl Acad Sci USA 99: 1241–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner S, Ignatova Z (2015) Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat Rev Genet 16: 98–112 [DOI] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 15: 178–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang D, Schumann M, Gelato K, Fischle W, Schwarzer D, Krause E (2013) Probing the acetylation code of histone H4. Proteomics 13: 2989–2997 [DOI] [PubMed] [Google Scholar]
- Li S, Xu Z, Sheng J (2018) tRNA‐derived small RNA: A novel regulatory small non‐coding RNA. Genes 9: 246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, Wang X, Hao Z, Dai Q, Zheng G et al (2016) ALKBH1‐mediated tRNA demethylation regulates translation. Cell 167: 816–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, Zhao S, Shen B, Gao Y, Han D et al (2020) N6‐methyladenosine of chromosome‐associated regulatory RNA regulates chromatin state and transcription. Science 367: 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long T, Li J, Li H, Zhou M, Zhou XL, Liu RJ, Wang ED (2016) Sequence‐specific and Shape‐selective RNA recognition by the human RNA 5‐methylcytosine methyltransferase NSun6. J Biol Chem 291: 24293–24303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Ma S, Klug A (2003) Crystal structure of a zinc‐finger‐RNA complex reveals two modes of molecular recognition. Nature 426: 96–100 [DOI] [PubMed] [Google Scholar]
- Luck K, Kim D‐K, Lambourne L, Spirohn K, Begg BE, Bian W, Brignall R, Cafarelli T, Campos‐Laborie FJ, Charloteaux B et al (2020) A reference map of the human binary protein interactome. Nature 580: 402–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Lu J, Liu L, Yin Y, Chen C, Han X, Wu B, Xu R, Liu W, Yan P et al (2016) Divergent lncRNAs regulate gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell 18: 637–652 [DOI] [PubMed] [Google Scholar]
- Maute RL, Schneider C, Sumazin P, Holmes A, Califano A, Basso K, Dalla‐Favera R (2013) tRNA‐derived microRNA modulates proliferation and the DNA damage response and is down‐regulated in B cell lymphoma. Proc Natl Acad Sci USA 110: 1404–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedialkova DD, Leidel SA (2015) Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161: 1606–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi K, Dincman TA, Dalton AC, Howley BV, McCall BJ, Mohanty BK, Howe PH (2018) Interleukin‐like EMT inducer (ILEI) promotes melanoma invasiveness and is transcriptionally up‐regulated by upstream stimulatory factor‐1 (USF‐1). J Biol Chem 293: 11401–11414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orioli A (2017) tRNA biology in the omics era: Stress signalling dynamics and cancer progression. BioEssays 39: 1600158 [DOI] [PubMed] [Google Scholar]
- Pillai S, Trevino J, Rawal B, Singh S, Kovacs M, Li X, Schell M, Haura E, Bepler G, Chellappan S (2015) Beta‐arrestin‐1 mediates nicotine‐induced metastasis through E2F1 target genes that modulate epithelial‐mesenchymal transition. Cancer Res 75: 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pognonec P, Boulukos K, Aperlo C, Fujimoto M, Ariga H, Nomoto A, Kato H (1997) Cross‐family interaction between the bHLHZip USF and bZip Fra1 proteins results in down‐regulation of AP1 activity. Oncogene 14: 2091–2098 [DOI] [PubMed] [Google Scholar]
- Rajagopalan P, Nanjappa V, Patel K, Jain AP, Mangalaparthi KK, Patil AH, Nair B, Mathur PP, Keshava Prasad TS, Califano JA et al (2018) Role of protein kinase N2 (PKN2) in cigarette smoke‐mediated oncogenic transformation of oral cells. J Cell Commun Signal 12: 709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapino F, Delaunay S, Zhou Z, Chariot A, Close P (2017) tRNA modification: is cancer having a wobble? Trends Cancer 3: 249–252 [DOI] [PubMed] [Google Scholar]
- Rapino F, Delaunay S, Rambow F, Zhou Z, Tharun L, De Tullio P, Sin O, Shostak K, Schmitz S, Piepers J et al (2018) Codon‐specific translation reprogramming promotes resistance to targeted therapy. Nature 558: 605–609 [DOI] [PubMed] [Google Scholar]
- Rubio MA, Gaston KW, McKenney KM, Fleming IM, Paris Z, Limbach PA, Alfonzo JD (2017) Editing and methylation at a single site by functionally interdependent activities. Nature 542: 494–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikia M, Jobava R, Parisien M, Putnam A, Krokowski D, Gao XH, Guan BJ, Yuan Y, Jankowsky E, Feng Z et al (2014) Angiogenin‐cleaved tRNA halves interact with cytochrome c, protecting cells from apoptosis during osmotic stress. Mol Cell Biol 34: 2450–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel P (2018) The emerging complexity of the tRNA world: mammalian tRNAs beyond protein synthesis. Nat Rev Mol Cell Biol 19: 45–58 [DOI] [PubMed] [Google Scholar]
- Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, Belleannee C, Kucukural A, Serra RW, Sun F et al (2016) Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 351: 391–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, Klijn JG, Foekens JA, Martens JW (2008) Subtypes of breast cancer show preferential site of relapse. Cancer Res 68: 3108–3114 [DOI] [PubMed] [Google Scholar]
- Song J, Zhuang Y, Zhu C, Meng H, Lu B, Xie B, Peng J, Li M, Yi C (2020) Differential roles of human PUS10 in miRNA processing and tRNA pseudouridylation. Nat Chem Biol 16: 160–169 [DOI] [PubMed] [Google Scholar]
- Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe‐Kains B et al (2006) Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst 98: 262–272 [DOI] [PubMed] [Google Scholar]
- Teixeira AF, Ten Dijke P, Zhu HJ (2020) On‐target anti‐TGF‐beta therapies are not succeeding in clinical cancer treatments: What are remaining challenges? Front Cell Dev Biol 8: 605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DM, Parker R (2009) Stressing out over tRNA cleavage. Cell 138: 215–219 [DOI] [PubMed] [Google Scholar]
- Tidow H, Andreeva A, Rutherford TJ, Fersht AR (2009) Solution structure of the U11–48K CHHC zinc‐finger domain that specifically binds the 5' splice site of U12‐type introns. Structure 17: 294–302 [DOI] [PubMed] [Google Scholar]
- Tkaczuk KL (2010) Trm13p, the tRNA:Xm4 modification enzyme from Saccharomyces cerevisiae is a member of the Rossmann‐fold MTase superfamily: prediction of structure and active site. J Mol Model 16: 599–606 [DOI] [PubMed] [Google Scholar]
- Torres AG, Batlle E, Ribas de Pouplana L (2014) Role of tRNA modifications in human diseases. Trends Mol Med 20: 306–314 [DOI] [PubMed] [Google Scholar]
- Towns WL, Begley TJ (2012) Transfer RNA methytransferases and their corresponding modifications in budding yeast and humans: activities, predications, and potential roles in human health. DNA Cell Biol 31: 434–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turashvili G, Bouchal J, Baumforth K, Wei W, Dziechciarkova M, Ehrmann J, Klein J, Fridman E, Skarda J, Srovnal J et al (2007) Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 7: 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li D, Gao J, Li X, Zhang R, Jin X, Hu Z, Zheng B, Persson S, Chen P (2017) The 2'‐O‐methyladenosine nucleoside modification gene OsTRM13 positively regulates salt stress tolerance in rice. J Exp Bot 68: 1479–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Kim TJ, Peng DH, Duan D, Gibbons DL, Yamauchi M, Jackson JR, Le Saux CJ, Calhoun C, Peters J et al (2017) Fibroblast‐specific inhibition of TGF‐beta1 signaling attenuates lung and tumor fibrosis. J Clin Invest 127: 3675–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson ML, Crary SM, Jackman JE, Grayhack EJ, Phizicky EM (2007) The 2'‐O‐methyltransferase responsible for modification of yeast tRNA at position 4. RNA 13: 404–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R, Chen J‐Y, Liang Z, Luo D, Chen G, Lu ZJ, Chen Y, Zhou B, Li H, Du X et al (2019) Pervasive chromatin‐RNA binding protein interactions enable RNA‐based regulation of transcription. Cell 178: 107–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lamouille S, Derynck R (2009) TGF‐beta‐induced epithelial to mesenchymal transition. Cell Res 19: 156–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Li J, He C, Wen J, Ma H, Rong B, Diao J, Wang L, Wang J, Wu F et al (2021) METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature 591: 317–321 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang X, Shi J, Tuorto F, Li X, Liu Y, Liebers R, Zhang L, Qu Y, Qian J et al (2018) Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non‐coding RNAs. Nat Cell Biol 20: 535–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, Lambowitz AM, Pan T (2015) Efficient and quantitative high‐throughput tRNA sequencing. Nat Methods 12: 835–837 [DOI] [PMC free article] [PubMed] [Google Scholar]