

SUMMARY
Expression reprogramming directed by transcription factors is a primary gene regulation underlying most aspects of the biology of any organism. Our views of how gene regulation is coordinated are dramatically changing thanks to the advent and constant improvement of high-throughput profiling and transcriptional network inference methods: from activities of individual genes to functional interactions across genes. These technical and analytical advances can reveal the topology of transcriptional networks in which hundreds of genes are hierarchically regulated by multiple transcription factors at systems level. Here we review the state of the art of experimental and computational methods used in plant biology research to obtain large-scale datasets and model transcriptional networks. Examples of direct use of these network models and perspectives on their limitations and future directions are also discussed.
Keywords: gene-regulatory networks, transcriptional regulation, machine learning, plants
INTRODUCTION
Social networks are an important part of our daily lives: we connect with others via personal interactions or remotely in several ways. In biological systems, molecular components (i.e., DNA, RNA and proteins) function by this same creed, physically or indirectly interacting with each other to form complex networks and control many aspects of the biology of an organism. Because the activity of individual genes or gene sets is often insufficient to reprogram global cellular functions in response to developmental and environmental stimuli, gene networks operate as part of a genome-wide system at different biological scales (e.g., individual cells, across cells in tissues, within the organism). Over the course of evolution, plants have developed networking as a means to thrive in a variety of environments that set them apart from other organisms, e.g., compared with animals and fungi, plants generally have more transcription factor (TF) families (Shiu et al., 2005; Yamasaki et al., 2013) and cis-regulatory elements (CREs) that physically interact to regulate and integrate diverse biological processes (O’Malley et al., 2016; Franco-Zorrilla and Solano, 2017). There has also been a considerable proliferation in the number of copies of critical genes in plants by frequent whole-genome duplication events (Van de Peer et al., 2009; Panchy et al., 2016), suggesting highly diversified gene–gene, protein–protein or protein–DNA interaction networks in plants compared with other species.
In the past decade or so, the revolutionary advent of next-generation sequencing (NGS) technologies has resulted in the proliferation of several types of genome-wide data that have enabled the cataloging of genes, gene products and their interactions within the biological context (Goodwin et al., 2016). The plant science community has been a frontier for that matter: Lister et al. (2008) reported on the Arabidopsis epigenome with single-base resolution, which, to our knowledge, is one of the first published papers that used NGS technologies to profile transcriptome in a biological system (Lister et al., 2008). The major challenge in this post-genomics era is to mine large omics datasets effectively for a deeper understanding of the multitude of molecular mechanisms underlying complex biological traits. Such an understanding will ultimately facilitate plant improvement by engineering or germplasm selection through breeding (Evans et al., 2015; Wing et al., 2018; Chen et al., 2019; Gao et al., 2019). An approach that is valuable for this matter is a network-based analysis to integrate global measurements at different molecular levels and derive models describing the biological systems (Ideker et al., 2001; Kitano, 2002; Barabási and Oltvai, 2004; Bonneau, 2008). This approach emphasizes the understanding of interactions between molecular components (e.g., genes and TFs), rather than the function of components alone, to identify processes underlying biological systems. One of the research areas in which network-based approaches are being extensively employed is transcriptional regulation in plants (Figure 1).
Figure 1.
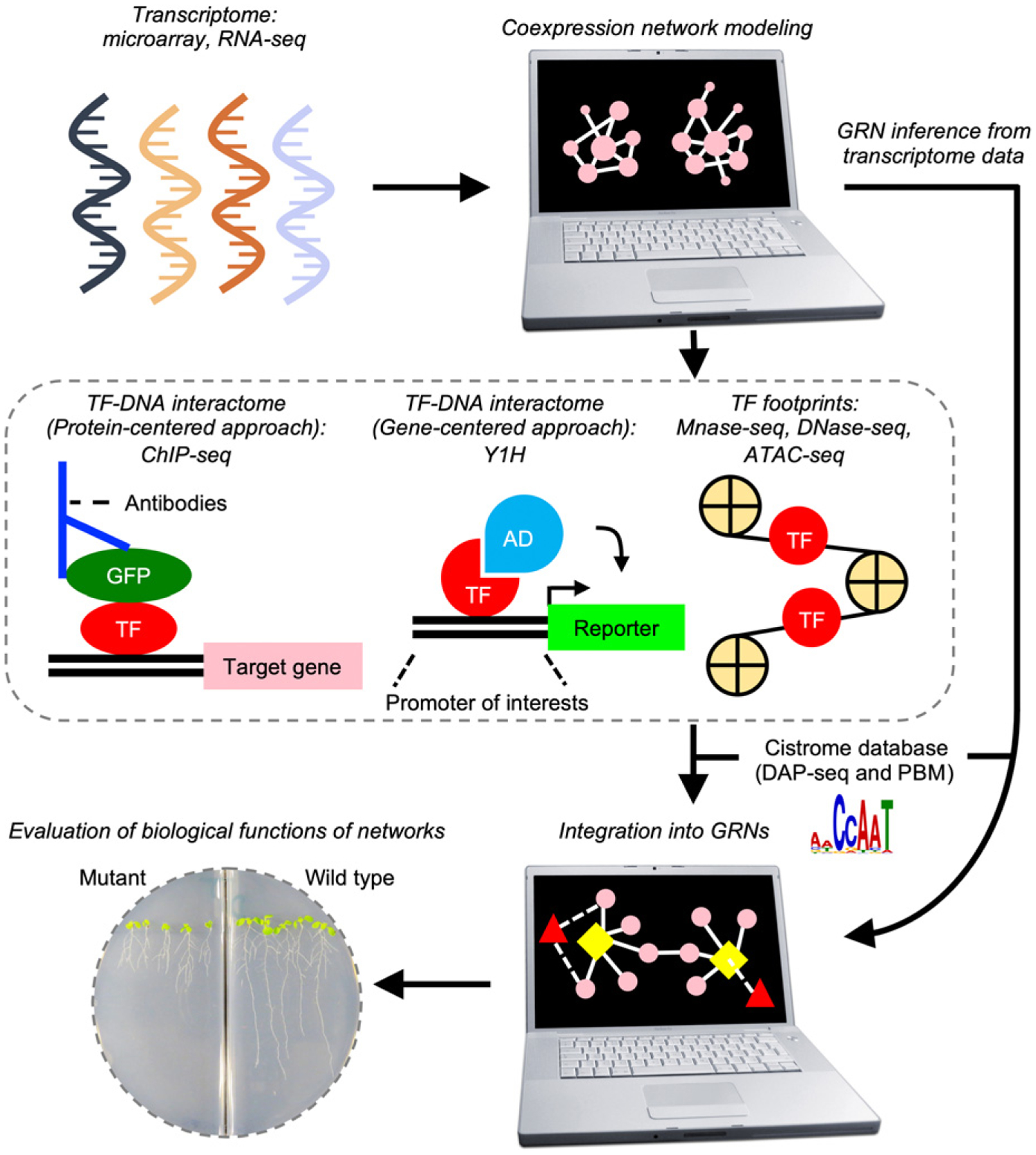
Schematic view of transcriptional network analysis.
Transcriptional network analysis consists of data acquisition, network modeling and assessment of network functions. Gene expression data obtained by high-throughput technologies can be used to construct coexpression networks, which are subsequently either integrated with transcription factor (TF)-DNA interactome data to build gene-regulatory networks (GRNs) or used to infer GRN bypassing the generation of TF-DNA interactome data. Biological functions of resulting GRNs are evaluated through genetic perturbations of predictive regulatory hub genes. Pink circle: genes affiliated in GRNs. Yellow rhombus: predicted regulatory hub genes, Red triangles: TFs. Edges indicated by solid and dot lines, respectively: coexpression interactions. AD, activation domain; ATAC-seq, Assay for Transposase-Accessible Chromatin with high-throughput sequencing; ChIP-seq, chromatin immunoprecipitation sequencing; DAP-seq, DNA affinity purification sequencing; GFP, green fluorescence protein; PBM, protein-binding microarray; Y1H, yeast one-hybrid.
An important prerequisite for a systems-level understanding of transcriptional networks is a high-quality global quantification of molecular components and their interactions. The putative biological functions of the networks inferred through these analyses are routinely tested via genetic perturbations of the hub genes (i.e., most connected genes in the network), which are predicted to be critical for network performance. The resulting networks, hubs and their functional roles can also be remodeled to predict additional hub genes, which are not accessible with the available datasets, so to generate new testable hypotheses. Furthermore, the selection of candidate genes for testing function and genetic modification can be improved using network-based machine learning (ML) approaches. This framework is expected to provide novel insights into complex gene-regulatory networks (GRNs) and allow plant researchers to understand the hierarchical organization of genes controlling several aspects of plant biology, including flowering, growth, development and response to environmental stress (Box 1 and 2).
Box 1. Bullet point summary.
Gene regulation is a complex biological process in which multiple molecular components work together to support the growth, development and stress responses of a biological system.
The systems-level understanding of gene regulation requires high-quality datasets of transcriptome, TF-DNA interactome and TF footprints, which can be effectively collected by the recently developed and improved high-throughput technologies.
The development of cistrome database and diverse online network tools has helped plant biologists mine multi-omics data conveniently and for systematic understanding of transcriptional networks underlying complex biological traits.
Several competing computational approaches, which are here categorized into coexpression network modeling and GRN modeling, have been proposed to infer transcriptional networks, highlighting the complex nature of gene regulation.
Transcriptional network approaches have been successfully applied to understand the interplay between genes, TFs and epigenetic components and dissect functional roles of epigenetic regulation underlying significant biological processes in plants.
In vitro TF-DNA binding database and versatile online tools have led to data-driven systems-level approaches to explore complex TF networks underlying significant biological questions.
ML-based transcriptional network modeling holds a great promise to improve prediction accuracy, interpretability and applicability over the current network modeling in crops in which genomic and experimental data are relatively scarce.
The demand for transcriptional network-based analyses is ever-increasing, yet it has limitations that need to be overcome to infer network models with high accuracy.
Box 2. Open questions.
How do we improve the current high-throughput technologies for transcriptome and TF-DNA interactome to obtain high-quality datasets in a cost-effective manner?
How do we better standardize current transcriptional network modeling methods to broaden the availability of the systems-level analysis in gene regulation?
How can transcriptional network-based analyses integrate highly heterogeneous, noisy, complex and dimensional datasets into accurate network models to provide systems-level biological insights into gene regulation?
In light of the growing importance of network-based approaches to understand gene regulation in plants, in this review, we illustrate recent technical advances in monitoring dynamic transcriptome changes and TF-DNA interactions in different experimental conditions. Next, we describe established computational modeling methods for transcriptional networks and highlight some of the emergent properties identified by various modeling algorithms in plant biology. Finally, we introduce challenges and future directions of systems-level understanding in the gene regulation of plants. This review is particularly aimed at stimulating plant biologists who are curious about recent developments and applications of transcriptional network modeling with little to no relevant knowledge of this approach.
DATA COLLECTION FOR GENE NETWORK INFERENCE
The rapid improvements of high-throughput deep sequencing technologies (e.g., NGS), increasing access to them and the plummeting of their cost have led to a revolution in plant functional genomics that has made systems-level approaches widely available for identifying transcriptional networks. A variety of methods has been employed to measure the abundance and interactions of molecular components of transcriptional networks on a genome-scale (Long et al., 2008; Moreno-Risueno et al., 2010; Gaudinier and Brady, 2016). Here, we highlight a set of approaches for omics data collection that are necessary for modeling transcriptional networks at a systems-level (Figure 1).
Genome assembly and annotation
The genome sequencing of the model plant species Arabidopsis thaliana (Initiative, 2000) has been followed by the sequencing of major crops, such as maize (Schnable et al., 2009) and rice (International Rice Genome Sequencing Project, 2005). Since then, 383 green plant species (Viridiplantae) with a wide range of genome size, complexity and ploidy have been sequenced at a whole-genome level, and 576 genome assemblies now are available (Kersey, 2019). It is expected that the genome sequences of 10 000 plant and algal species will be available soon through the 10 000 Plants Genome Sequencing Project (10 KP) (Cheng et al., 2018).
The high-throughput sequencing and assembly of plant genomes have been possible with the rapid development of sequencing technologies: NGS, long-read sequencing technologies (Goodwin et al., 2016) and physical mapping technology (Liu and Weigel, 2015). Although whole-genome sequencing itself does not serve directly for transcriptional network construction, the unprecedented breadth of well-assembled reference genomes produced from these state-of-the-art sequencing approaches provides exciting opportunities to explore the dynamics of global gene regulation and set the foundations for answering significant biological questions at a mechanistic level in plants.
Transcriptome analysis
Because most biological processes are driven by changes in gene activity, quantification of gene expression has been a frontier to address biological questions in any living organism (Brady et al., 2007; Busch and Lohmann, 2007; Romero et al., 2012). Quantification of global gene expression changes is the most essential part of building transcriptional networks. RNA-sequencing (RNA-seq), a transcriptome profiling approach that uses NGS technologies, offers the most straightforward and unbiased way to investigate transcript abundance with increased speed and depth compared with earlier approaches (e.g., microarrays) (Wang et al., 2009; Metzker, 2010). The power of RNA-seq lies in the fact that a highly standardized experimental and bioinformatics pipeline (Wang et al., 2011; Conesa et al., 2016) can be adapted to any biological system; this has made RNA-seq rapidly evolve and become widely available to plant researchers. For example, the multiplexing RNA-seq technique, which was introduced after the advent of single-lane RNA-seq, has allowed for scaling up the experiment size in a cost-effective manner (Craig et al., 2008). These advantages coupled with technical improvements (i.e., pair-end sequencing, increased yield and read length) have made RNA-seq a most suitable approach for transcriptome profiling. In addition, the recent development of single-cell RNA-seq has opened new avenues for plant biologists who are interested in dynamics of gene expression changes at the individual cell level (Efroni et al., 2016; Denyer et al., 2019; Jean-Baptiste et al., 2019; Nelms and Walbot, 2019; Ryu et al., 2019; Shulse et al., 2019).
TF-DNA interaction profiling
Most of the complex developmental, growth and differentiation processes in eukaryotes are mediated by dynamic TF-DNA interactions that direct gene transcription fate (Wray et al., 2003; Spitz and Furlong, 2012). Genome-wide mapping of TF-DNA interactions is therefore necessary for a comprehensive understanding of the transcriptional regulation underlying various biological processes (Farnham, 2009).
There are mainly three distinct experimental techniques to map binding sites of TFs on a genome-scale: (i) chromatin immunoprecipitation followed by deep sequencing (ChIP-seq); (ii) heterologous expression systems, such as yeast one-hybrid (Y1H) screening; and (iii) mapping open chromatin regions (Table 1). Although powerful, these approaches have caveats (Table 1). For example, a physical binding of TFs to gene promoters that can be found in ChIP-seq analyses does not always indicate gene regulation. Several studies have reported that <20% of plant TF–gene interactions identified in ChIP-seq analyses result in a functional interaction (e.g., alteration of gene expression level) (Gitter et al., 2009; Marchive et al., 2013; Swift and Coruzzi, 2017). Inferring GRNs from open chromatin regions requires global information of TF-DNA binding motifs (Bubb and Deal, 2020), which is not available for most crops. Importantly also, the TF–gene interaction datasets provide static snapshots from mixed cell types from a whole tissue. However, gene regulation is rather the result of spatiotemporal dynamics of TFs binding to gene regions in response to developmental and environmental cues (Kaufmann et al., 2010; Sparks et al., 2013; Vihervaara et al., 2018). Therefore, experimental design should take into consideration the dynamic properties of the activities of TFs in the context of gene regulation to generate meaningful datasets and provide sufficient grounds for interpretation.
Table 1.
Illustration of global TF-DNA interaction and DNA accessibility profiling methods
| ChIP-seq | Y1H screening | DNase-seq | MNase-seq | ATAC-seq | |
|---|---|---|---|---|---|
| Assay type | In vivo protein-DNA interactions | In vitro protein-DNA interactions | In vivo open chromatin regions | In vivo open chromatin regions | In vivo open chromatin regions |
| Major underlying approach | Fragmented genomic DNA bound by TFs of interests are immunoprecipitated by specific antibodies for the TFs. DNA sequences are identified by deep sequencing | Reporter gene driven by a bait promoter is activated when the promoter is bound by a protein (prey) fused to the activation domain of the yeast Gal4 TF in the yeast strain | Non-specific DNase I cleaves within accessible chromatin (i.e., transcriptionally active). The DNA sequences are identified by deep sequencing | Endonuclease/exonuclease MNase both cuts and digests accessible DNA. DNA sequences are then identified by deep sequencing | Hyperactive Tn5 transposase simultaneously cuts accessible chromatin in unfixed nuclei and ligates adapters for NGS. This enables the identification of the cut sequences by deep sequencing |
| Source of potential platform noise | Non-specific binding, GC content bias, chromatin structure affecting fragmentation by sonication (e.g., heterochromatin versus euchromatin) | Activation by endogenous yeast TFs, improper prey protein folding because of the fusion to an activation domain | High dependency of efficiency in identifying TF footprints on fragment size, cleavage in a sequence-dependent manner (three nucleotides on either side of the cleavage site) | Variable enrichment pattern depending on fragment size (fragments of one nucleosome length (147 bp) are typically selected for the sequencing), cleavage in a sequence-dependent manner (preference to AT-rich) | Quality of preparation of isolated nuclei |
| Advantages | Straightforward detection of binding events in vivo, high interpretability | High-throughput and high coverage with robotic mating platform, compatibility with prey libraries, detection of TF isoform-specific binding | Relatively standardized experimental and computational workflows via the ENCODE project, well-understood bias effects | Nucleosome binding information in addition to TF binding information | Much less input materials (<50, 000 nuclei) than DNase-seq and MNase-seq (>1 million), simplified experimental procedures, short time for processing |
| Limitations | Necessity of antibody for TF of interests or transgenic lines expressing epitopetagged versions of proteins of interests, interaction of TF with chromatin does not necessarily indicate a functional interaction in gene regulation | No functional insights into the TF-DNA interactions, high rate of false-positives | Laborious enzyme titration, high amount of input material, time-consuming sample preparation | Laborious enzyme titration, indirect detection of active regulatory regions, high amount of input material | Higher sequencing coverage, prevalence of plastid and mitochondria genome contamination, standardized data analysis pipeline not optimized yet |
| Key references | (Barski et al., 2007; Park, 2009) | (Deplancke et al., 2004; Gaudinier et al., 2011) | (Crawford et al., 2006; Boyle et al., 2008) | (Schones et al., 2008; Pajoro et al., 2018) | (Buenrostro et al., 2013; Buenrostro et al., 2015) |
ChIP-seq.
ChIP-seq is a TF-centered approach (i.e., finding DNA sequences bound by TFs of interests; Table 1), and as such, it is the main tool for global mapping sites of TF-DNA interactions in vivo (Barski et al., 2007; Johnson et al., 2007; Mikkelsen et al., 2007; Robertson et al., 2007; Park, 2009). ChIP was initially developed in cultured Drosophila cells (Solomon et al., 1988) and has been applied to other eukaryotes, including plants. Indeed, nearly 500 ChIP-seq data series published from 2009 to 2020 can be retrieved by searching for (“plants” [Organism]) AND “genome binding/occupancy profiling by high throughput sequencing” [DataSet Type] in the NCBI Gene Expression Omnibus.
In short, to generate a ChIP-seq library, proteins bound to DNA, such as TFs, are cross-linked in vivo by fixatives (e.g., formaldehyde). The protein–DNA complexes are then immunoprecipitated by a specific antibody against the DNA-binding protein of interest or the tag fused with the protein. The DNA is then deep-sequenced using NGS technologies (Saleh et al., 2008; Park, 2009). The ChIP-seq approach is widely used in Arabidopsis (Yu et al., 2016; Chen et al., 2018) but its implementation in crops appears to be more challenging due to technical limitations to prepare goodquality ChIP-seq libraries for crops compared with Arabidopsis. For instance, generating transgenic crop lines with a protein tag fusion of a TF of interests, which may be necessary for weakly expressed TFs or to bypass the production of TF-specific antibodies, may be time-consuming or not feasible at all because of the absence of suitable transformation protocols. Nonetheless, online databases such as Expresso (https://bioinformatics.cs.vt.edu/expresso/) (Aghamirzaie et al., 2017) and the C3C4 project data portal (http://www.epigenome.cuhk.edu.hk/C3C4.html) provide processed data of TF ChIP-seq in plants and may serve as a resource for identifying TF-DNA interactions.
Y1H—
Y1H screening is a “gene-centered” method that allows the identification of TFs binding to DNA sequences of interest (Li and Herskowitz, 1993; Wang and Reed, 1993) (Table 1). The development of high-throughput Y1H screening combined with complete gold-standard TF collections has enabled the identification of thousands of potential interactions at a genome-scale in plants (Gaudinier et al., 2011; Burdo et al., 2014; Pruneda-Paz et al., 2014; Taylor-Teeples et al., 2015; Yang et al., 2017; Gaudinier et al., 2018; Ikeuchi et al., 2018; Li et al., 2018; Smit et al., 2020a; Smit et al., 2020b). Y1H screening comes with the major innate risk of any heterologous expression system where interactions in non-plant cells do not necessarily imply that TFs identified in the screen bind to the DNA sequence in their native environment or under particular experimental conditions.
Mapping open chromatin regions.
In eukaryotes, core histones are generally wrapped by 147 bp of DNA, forming an array of nucleosomes, which is a key structural component of chromatin (Kornberg, 1974; Kaplan et al., 2009). TF-DNA binding repositions nucleosomes on the genome and increases the chromatin accessibility for nuclease enzymes (such as DNase I and micrococcal nuclease [MNase]), which allows defining potential cis-regulatory regions. The combination of the enzyme treatment with high-throughput sequencing such as DNase I sequencing (DNase-seq) and MNase sequencing (MNase-seq) has enabled unbiased global profiling of open chromatin regions bound by TFs (i.e., TF footprints) occurring in different cell types, developmental stages and environmental stresses in several plant species (Zhang et al., 2012a; Zhang et al., 2012b; Sullivan et al., 2014; Rodgers-Melnick et al., 2016; Oka et al., 2017; Burgess et al., 2019) (Table 1). A significant improvement of nuclease-based methods for identifying accessible regions of chromatin and TF binding is the Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq; Table 1), which uses a hyperactive Tn5 transposase to integrate preloaded sequencing adapters into regions of open chromatin (Buenrostro et al., 2013; Buenrostro et al., 2015) and has been increasingly applied to plant research (Wilkins et al., 2016; Maher et al., 2018; Sijacic et al., 2018; Reynoso et al., 2019). ATAC-seq conveniently offers a fast protocol with simple library amplification steps and requires relatively small amounts of starting material (50 000 freshly prepared nuclei for Arabidopsis) (Bajic et al., 2018), making it a vast improvement over MNase-seq and DNase-seq, which require more genetic material and complex sample preparation (Klemm et al., 2019). However, a major drawback of ATAC-seq is that the hyperactive Tn5 transposase also targets the genomes of mitochondria and chloroplasts. This decreases the proportion of sequencing reads that map to the nuclear genome, reducing the amount of information that can be used to infer cis-regulatory regions of the genome in the nucleus. This pitfall can be bypassed by using nuclei enrichment techniques from tissues or specific cell types, such as the approach from Isolation of Nuclei TAgged in specific Cell Types (INTACT) (Deal and Henikoff, 2010; Sijacic et al., 2018). The TFs putatively bound to open chromatin regions can be inferred through de novo motif analysis followed by TF motif mapping, using computational tools such as the MEME suite (Bailey et al., 2009). The DNA binding of the TF candidates can be further tested through parallel approaches such as ChIP or gene expression analyses. The approaches detailed above have the common limitation that TFs with a short residence time on the DNA may not generate detectable footprints on the genome under analysis. Furthermore, it may be challenging to assign a particular TF to a single footprint due to similar DNA-binding motif patterns of several TFs (O’Malley et al., 2016).
In vitro TF-DNA binding databases
Typically, a TF binds to CREs that are proximal to the transcriptional start sites (TSS) of target genes and provide DNA binding specificity to the TFs. Collectively, a global set of CREs populating the transcriptional regulation sites of a gene represents the cistrome in an organism, and are critical elements controlling gene expression. Differences in the nucleotide sequence, frequency or position of the CREs with respect to the TSS within a cistrome contribute to influencing gene expression, e.g., enrichment of the same types of CREs in a cistrome is more likely to occur for more highly differentially regulated genes compared with genes that are less differentially regulated in the same environmental conditions or developmental stages (Liu et al., 2018).
Thus far, there have been two major breakthroughs from in vitro approaches on the cistrome identification in plants, i.e., protein-binding microarray (PBM) (Weirauch et al., 2014) and DNA affinity purification sequencing (DAP-seq) (O’Malley et al., 2016; Galli et al., 2018). Processed data that indicate in vitro DNA-binding motifs for 2913 TFs from different species obtained through PBM, and of 526 Arabidopsis TFs obtained through DAP-seq are available in the CIS-BP Database (http://cisbp.ccbr.utoronto.ca) and the Plant Cistrome Database (http://neomorph.salk.edu/dev/pages/shhuang/dap_web/pages/index.php), respectively. Statistical mapping of CREs to the cistromes can be used to define putative TF-binding sites on promoters of differentially expressed genes or open chromatin regions using computational tools, such as the MEME suite (Bailey et al., 2009) and HOMER (Heinz et al., 2010). These resources and computational tools also allow inferring putative TF co-regulators through the identification of CREs that are bound by other TFs in the DNA region targeted by a specific TF (e.g., ChIP-seq). Thus, the cistrome database is a critical resource for inferring in silico plant GRNs.
NETWORK INFERENCE: MODELING METHODS
The availability of the transcriptome, TF-DNA interactome and cistrome database discussed above has enabled a systems-wide understanding of transcriptional networks underpinning complex biological traits. Transcriptional networks can be used to generate hypotheses by making in silico predictions and provide guidance to execute in planta experiments.
Transcriptional networks, including coexpression networks and GRNs, are generally graphed with nodes and edges similar to other biological networks. Nodes indicate the molecular components (genes or TFs) while edges represent gene–gene, gene–TF, or TF–TF interactions (coexpression or physical binding) (Figure 2). Coexpression networks are elaborated into modules representing gene–gene interaction structures based on expression similarity. The functional linkages among the nodes of the network constitute the network architecture, which can lead to a systematic understanding of the gene-regulatory mechanisms. GRNs, which depict the putative architecture of such gene-regulatory mechanisms, can be computationally inferred from transcriptome data but can also integrate multiple data sources, e.g., GRNs can be generated by integrating TF-DNA interactome networks with corresponding gene coexpression networks in the most straightforward way.
Figure 2.
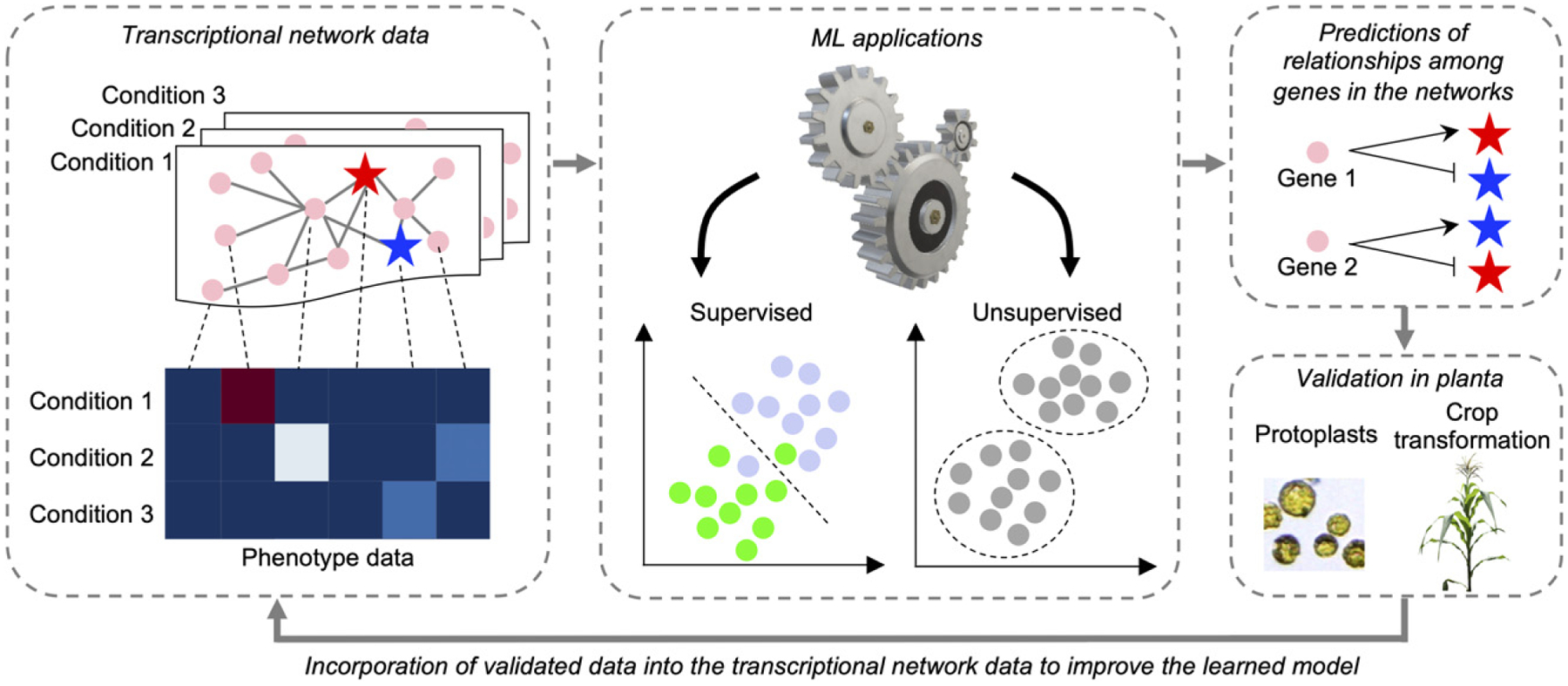
Schematic view of machine learning (ML)-based transcriptional network approaches in crops.
Transcriptional networks, such as coexpression networks or inferred GRNs, are constructed and subsequently integrated with phenotype data available. These networks can be used as input for following ML approaches. In ML methods, data consist of instances and features. In transcriptional networks, instances are genes and features are gene expression levels or phenotype. ML methods are largely categorized into supervised and unsupervised learning. Supervised methods are used when labels are available for input data (e.g., classification) whereas unsupervised methods are applied when the labels on the input data are unknown so that the model can learn only from patterns (e.g., clustering). ML models generate predictions of relationships among genes in the given networks, which need to be functionally tested in planta. Functional validation can be performed in protoplasts (i.e., transient expression analysis) or transgenic crop plants depending on the biological questions. Validated data can be directly incorporated into the input data to improve the performance of the trained model.
GRN inference from transcriptome data is popular because gene expression analyses are generally less technically challenging, time-consuming and costly in many plant species compared using TF-DNA interactome analyses, such as ChIP-seq and Y1H screening. A number of modeling methods for inferring coexpression networks and GRNs in plants have been described in depth previously (Li et al., 2015; Serin et al., 2016; Banf and Rhee, 2017; Haque et al., 2019; Marshall-Colón and Kliebenstein, 2019), and are therefore only briefly summarized here. We illustrate examples of the modeling methods used to explore complex transcriptional networks underlying significant biological traits.
Coexpression network modeling
In recent years, coexpression network modeling has grown in popularity for addressing many biological questions because of the fast development of transcriptomic technologies, publicly available gene expression databases and robust clustering algorithms. This modeling approach allows the simultaneous identification, clustering and exploration of thousands of genes with similar expression patterns across multiple conditions (coexpressed genes). For example, based on “guilt-by-association,” genes that belong to the same gene module (i.e., coexpression module) are considered co-regulated by a common set of regulators, which may be predicted by de novo motif analysis on their promoters (e.g., cistrome analysis Vandepoele et al., 2009; Hickman et al., 2017) (Figure 1). Coexpression network modeling has been powerful to identify novel plant genes with a role in the biosynthesis of specialized metabolites, including anthocyanins, flavonoids, sinapoyl esters, glucosinolates, terpenoids, camalexin and other tryptophan derivatives. To identify genes underlying the production of these metabolites and the mechanisms regulating their production in a shared or lineage-specific manner, Wisecaver et al. (2017) elaborated a single-network approach in which a coexpression network modeling based on Pearson coefficient correlation was applied to a set of 10 transcriptome datasets produced across eight plant species (Wisecaver et al., 2017). This network inference generated a catalog of coexpression gene modules, many of which were linked to known specialized metabolic pathways.
Coexpression analysis can also be applied to study yet uncharacterized metabolic pathways in plants, e.g., an integrative analysis of coexpression clustering and metabolomics enabled the selection of candidate gene clusters for the biosynthetic pathway of a pathogen-responsive lipid compound, falcarindiol, in tomato (Jeon et al., 2020). Furthermore, coexpression analysis of mayapple time-series transcriptome datasets in a wounding experiment designed to trigger the metabolic synthesis of podophyllotoxin, the natural product precursor of therapeutic etoposide aglycone, provided a gold set of 29 candidate genes. These genes were used in combinatorial expression experiments in tobacco, an approach that ultimately identified a six-enzyme pathway for aglycone biosynthesis (Lau and Sattely, 2015). The coexpression network modeling adopted in this study may be applied to any metabolic biosynthesis pathway, fast advancing research in plants. Nonetheless, the accuracy of the predictions and the number of genes potentially involved in the biosynthesis of secondary metabolites that can be functionally assessed may be limited by key regulatory events that do not necessarily happen at transcriptional levels and the strength of the computational frameworks currently available (Yonekura-Sakakibara et al., 2013; Banf and Rhee, 2017).
Coexpression network analyses have been also implemented for the study of pathways other than secondary metabolites, such as hormones. For example, Hickman et al. (2017) investigated the temporal regulatory dynamics of gene expression in conditions of treatment with jasmonic acid (JA), an essential hormone for plant growth, development and stress responses (Wasternack, 2015). They identified 3611 differentially expressed genes (Hickman et al., 2017), and then used SplineCluster, a hierarchical clustering method based on a regression model with a marginal likelihood criterion (Heard et al., 2006), to separate the identified differentially expressed genes into coexpression clusters. This yielded 27 clusters containing distinct expression responses linked to distinct biological pathways. This network analysis ultimately led to a discovery that specific TFs modulate JA-responsive GRN and laid the ground for testing new hypotheses on JA signaling in plants.
In coexpression network modeling, the distances of the coexpression modules can be quantitatively measured and serve to establish a molecular phenotype. For example, the weighted gene coexpression network analysis (WGCNA) is one popular modeling approach that applies unsupervised correlation analysis and soft thresholding to convert gene expression measures in adjacency matrices to a connection weight. This approach generates module eigengenes, which represent quantitatively measured expression values of coexpression modules (Langfelder and Horvath, 2008). WGCNA has been successfully applied to map gene expression dynamics in environmental stress. For example, Greenham et al. (2017) performed transcriptome analysis and various physiological measurements, such as stomatal conductance, photosynthetic rate and photosystem II efficiency, over a 2-day time course of drought stress in Brassica rapa (Greenham et al., 2017). WGCNA was applied to generate coexpression gene modules, which were subsequently associated with the phenotypic traits measured. The innovative coupling of coexpression gene modules and quantitative values of physiological traits identified drought-related modules containing drought-responsive genes associated with a wide range of biological processes. In addition, Lanver et al. (2018) used WGCNA to identify coexpression modules of the genes from the maize smut fungus Ustilago maydis during fungal development (Lanver et al., 2018). The analysis revealed 14 coexpression modules, each of which displayed a unique expression signature of the eigengene and was associated with significant biological processes. This led to the selection of the most likely influential modules for U. maydis virulence in maize.
Coexpression network modeling can take advantage of the temporal dynamics of gene expression changes. Wigwams (identifying genes working across multiple situations) is an efficient statistical method to identify coexpression gene modules in multiple time series of gene expression data (Polanski et al., 2014). Wigwams measures the Pearson correlation coefficient in time-series data for establishing coexpression gene modules. This is particularly useful to reconstruct coexpression networks associated with time-specific modules of co-regulated genes. However, handling large datasets for coexpression network analyses can become very complex and challenge data interpretation (Usadel et al., 2009).
In contrast to GRNs, and because of their static representation, coexpression networks per se do not provide information on the nature of the regulatory relationship of connected genes (i.e., direct or indirect) (Stuart et al., 2003). Careful application of coexpression network analysis tools and strategies is therefore required to maximize the information extraction, disentangle reliable network connections and infer true biological meaning.
GRN modeling
Computational prediction of the TF–gene interactions from transcriptome data in GRN modeling has been a challenging endeavor that requires the development of powerful bioinformatics methods to study the complex architecture of gene regulation (Figure 1). A wide range of network inference methods have been proposed, and can broadly be divided into model-based and model-free methods. Model-based methods construct a computational model of the biological system and subsequently learn the parameters of this model to solve the network inference problems, creating a dynamic model that is optimized for the given dataset. Model-based methods are clearly interpretable and can be used for other gene expression predictions.One of such methods most commonly used is the Bayesian network (BN), a type of probabilistic graphical model in which the known conditional dependence on directed edges is explicitly captured (Pe’er, 2005). BN models have been successfully applied to infer functional relationships between TFs and downstream target genes controlling a number of biological phenomena (Needham et al., 2009; Bechtold et al., 2016; Scofield et al., 2018); e.g., a conditional dependency of SHOOT MERISTEM (STM), a key TF in the development of shoot apical meristem with 56 genes encoding other multifunctional TFs involved in shoot apical meristem formation was inferred using a BN, revealing the topology of the STM-mediated GRN. This analysis led to the hypothesis of a positive transcriptional feedback loop between STM and CUP-SHAPED COTYLEDON 1 (CUC1), which was experimentally validated (Scofield et al., 2018). BN models encompass several algorithmic variants, including the dynamic BN, which has been developed to identify statistically meaningful relationships between time-dependent variables while incorporating noise (Murphy and Mian, 1999). Application of a dynamic BN method, the Metropolis variational Bayesian state-space modeling, on high-resolution time-series transcriptomics data coupled with physiological and metabolic analyses during a transition to drought conditions, has been used to identify AGAMOUS-LIKE22 (AGL22) as a hub gene in the control of water stress responses (Bechtold et al., 2016). Because AGL22 is a TF known to be involved in the transition from vegetative state to flowering (Hartmann et al., 2000), this approach successfully identified a key regulator in common to two seemingly unrelated biological pathways, supporting the strength of BN models to generate new hypotheses and make new discoveries.
Despite the potential of model-based methods, multiple issues have eroded their attractiveness: they tend to be computationally demanding and rely on strong assumptions about the model dynamics. In contrast, model-free methods take a different approach to avoid the pitfalls of model-based methods, i.e., they do not make any assumptions about the nature of gene regulation but rather optimize theoretical measures of co-variation between genes (Margolin et al., 2006; Huynh-Thu et al., 2010). Such methods, including an ML-based regression tree algorithm, the Gene Network Inference with Ensemble of Trees 3 (GENIE3) (Huynh-Thu et al., 2010), typically have good scalability for the network construction, high flexibility due to the absence of a benchmark model (no constrain) and have consistently achieved reconstruction performance that is comparable with other algorithms (Marbach et al., 2012). Shibata et al. (2018) used GENIE3 to infer a GRN of three important TFs (GT-2-LIKE 1 [GTL1], DF1, RHD6-LIKE 4 [RSL4]) and their 36 common target genes in regulating root hair growth based on TF-DNA interactome and transcriptome data (Shibata et al., 2018). The GRN interference not only revealed the topology of GRN underlying root growth, it also identified a negative transcriptional feedback loop of two of those TFs (i.e., GTL1 and RSL4). A regression tree-based pipeline that implements GENIE3, the Regression Tree Pipeline for Spatial, Temporal, And Replicate, has been recently used to integrate time-series transcriptome datasets with phospho-proteome data into systems-level GRN models, and successfully revealed new components for a crosstalk of JA signaling with other signaling pathways (Zander et al., 2020). Despite their demonstrated performance, model-free methods are generally difficult to interpret, which limits their predictive power. A hybrid approach, Jump3 (Huynh-Thu and Sanguinetti, 2015), has been applied to bridge the gap between model-based and model-free methods, using transcriptome networks of murine macrophages (Blanc et al., 2011), in silico and synthetic gene networks of yeast (Cantone et al., 2009; Prill et al., 2010; Marbach et al., 2012). As such, Jump3 shows a competitive performance compared with other existing methods (Huynh-Thu and Sanguinetti, 2019) and may be applied to solve highly complex plant GRNs.
Network visualization and online tools
Inferred gene networks need to be visualized in a way that the interactions are comprehensively recognized. The currently available network visualization tools were summarized in detail earlier (Marshall-Colón and Kliebenstein, 2019). Cytoscape is the most widely utilized tool for network visualization equipped with built-in network topology analysis algorithms. The function of Cytoscape can be expanded by >200 applications available (https://apps.cytoscape.org/apps/all) to improve the network presentation or for critical downstream analyses (Shannon et al., 2003).
There are several online tools for retrieving already predicted or identified interactions of the genes of interests to generate new hypotheses. For example, the VirtualPlant (http://virtualplant.bio.nyu.edu) provides a convenient online platform in which users can identify interactions among genes of interests based on published genome-wide datasets in multiple plant species and that can be visualized (Katari et al., 2010). In additions, the Arabidopsis Interaction Viewer 2.0 (AIV2) (http://bar.utoronto.ca/interactions2/) allows for the visualization of predicted and experimentally validated protein–DNA interactions as well as protein–protein interactions in A. thaliana (Dong et al., 2019).
Arabidopsis regulatory network databases are also available for TFs, e.g., the Plant Regulome database (http://www.plantregulome.org) offers a user-friendly interface for the Arabidopsis regulatory network map of 235 TFs driven by DNase-I seq (Sullivan et al., 2014), and the PlantRegMap (http://plantregmap.cbi.pku.edu.cn) (Tian et al., 2020) visualizes the Arabidopsis Transcriptional Regulatory Map (ATRM, http://atrm.cbi.pku.edu.cn/vis_network.php), which consists of 1431 TF-DNA interactions curated through an extensive literature mining (Jin et al., 2015).
Adding to these tools, the TF2Network (http://bioinformatics.psb.ugent.be/webtools/TF2Network/) predicts potential regulators for coexpressed or functionally related genes with a high accuracy (75%–92%) in A. thaliana (Kulkarni et al., 2018). Databases have been also developed to analyze CREs. For example, the Cistrome (http://bar.utoronto.ca/cistome/cgi-bin/BAR_Cistome.cgi) allows users to explore CREs at different lengths (250, 500 and 1000 bp) of gene promoters using the cistrome dataset or user-provided motifs in A. thaliana (Austin et al., 2016).
Collectively, these online tools serve as a convenient means to mine multi-omics datasets for understanding transcriptional networks without an extensive knowledge of network construction in plant research.
EMERGING FIELDS OF TRANSCRIPTIONAL NETWORK APPLICATIONS
Transcriptional network-based approaches help narrow down hundreds or thousands of genes into relatively small sets of genes responsible for the molecular or physiological traits of interests. Indeed, in addition to studies briefly exemplified in the previous section, the construction of transcriptional networks has been extensively applied to providing testable hypotheses in the development of flowers (Heyndrickx et al., 2014; Ó’Maoiléidigh et al., 2014; Chen et al., 2018), seeds (Xiong et al., 2017) and root (Brady et al., 2011; Moreno-Risueno et al., 2015), as well as in nutrient signaling (Varala et al., 2018), pathogen infection (Windram et al., 2012) and abiotic stresses (Vermeirssen et al., 2014; Wilkins et al., 2016; Van den Broeck et al., 2017). Below, we discuss research areas in plants being increasingly addressed with transcriptional network prediction analyses.
Epigenetic modifications affecting transcriptional networks
TFs dynamically compete or collaborate with chromatin modification markers, such as methylation on DNA or histones, to promote local access to regulatory DNA regions (Gates et al., 2017; Talbert and Henikoff, 2017). While the role of gene body DNA methylation is unclear, promoter DNA methylation is usually accompanied by a reduction of gene transcription (Zhang et al., 2006; Lei et al., 2015; Williams et al., 2015), largely through a reduction of the binding of TFs to the promoter (Domcke et al., 2015; Zhang et al., 2018). DNA methylation, which occurs at cytosine bases in CG, CHG and CHH (H=A, T or C) contexts in plants (Zhang et al., 2006; Lister et al., 2008), alters gene expression in response to adverse environmental conditions and during development (Matzke and Mosher, 2014; Niederhuth and Schmitz, 2017). Histone-associated epigenetic changes are also important in modulating transcriptional networks underpinning many aspects of plant biology. From yeast to plants to humans, trimethylation of histone 3-lysine 27 (H3K27me3) and H3K4me3 has been associated with genes transcribed at low and high levels, respectively, while acetylation of histone 3 lysine 9 and 13 (H3K9ac and H3K14ac) leads to gene activation by reducing an interaction between DNA and core histones (Allis and Jenuwein, 2016). In Arabidopsis, wounding-inducible genes are marked with H3K9ac, H3K14ac and H3K27ac shortly after wounding (Rymen et al., 2019). In addition, Song et al. (2018) demonstrated that an Arabidopsis COMPASS-Like complex, which accumulates H3K4me3 at gene promoters, physically interacts with basic leucine zipper protein (bZIP) 28 (bZIP28) and bZIP60 TFs to regulate expression of the endoplasmic reticulum (ER) stress-responsive genes, providing mechanistic insights into how the epigenetic modifications are deeply embedded into the ER stress-responsive gene networks (Song et al., 2015). Zhang et al. (2017) also showed that rapid transcriptional changes of hundreds of binding target genes of ETHYLENE INSENSITIVE 2 (EIN2), a positive transcriptional regulator of ethylene signaling, is accompanied with increased levels of H3K14ac and H3K23ac at the gene promoters, which are mediated by a physical interaction between EIN2 and EIN2 nuclear-associated protein 1 (ENAP1), a histone-binding protein (Zhang et al., 2017). These examples underscore critical regulatory roles of the epigenetic components in defining the architecture of transcriptional networks operating in plant development and stress responses. Accordingly, Chen et al.(2018) performed a systems-level network analysis using ChIP-seq profiles of 15 key floral TFs and transcriptomes of mRNA and microRNA (miRNA) (Chen et al., 2018), tightly linked with DNA methylation (Matzke and Mosher, 2014). The network inference revealed a prevalence of a feed-forward loop mediated by TFs and miRNAs through integrated GRN modeling and led to an experimental validation that SEPALLATA3 acts as an upstream regulator of miR319a/TCP4 module to regulate petal development.
ML-based transcriptional network modeling
Despite the wide applicability and effectiveness demonstrated in a number of studies, transcriptional network modeling has unavoidable limitations, including that the interpretation of gene-regulatory modules is often based on postulated functions of yet uncharacterized genes. This emphasizes a need for developing new effective approaches to improve network prediction, interpretability and efficiency of data usage. ML is a collection of data algorithms aimed at establishing predictive models from multidimensional datasets and has already been used in a number of advanced analyses in plant biology: predicting new specialized metabolism genes (Moore et al., 2019; Toubiana et al., 2019), putative CREs in abiotic and biotic stress responses (Zou et al., 2011), CREs regulating root cell type-specific gene expression (Uygun et al., 2019), gene annotation (Sartor et al., 2019), phenotyping (Bernotas et al., 2019) and crop yield (Khaki and Wang, 2019; Zhang et al., 2019; Herrero-Huerta et al., 2020) (Figure 2).
A number of ML algorithms are designed to infer transcriptional networks by providing data-driven prediction models from transcriptome profiles in Arabidopsis. In addition to GENIE3 described earlier, such algorithms include State-Space Models (SSMs) and Supervised Inference of Regulatory Networks (SIREN). SSMs, a set of ML-based graphical model formulations, allow the prediction hidden values of gene expression at future time-points based on observed values (Beal et al., 2005). SSMs have been used to predict temporal network models responsive to nitrate and infer a functional GRN from time-series transcriptome data in plants (Krouk et al., 2010). SIREN is an ML-based supervised regulatory interaction network modeling framework (Mordelet and Vert, 2008) that has been used to analyze the information gained from microarrays systematically and infer GRNs in the control of secondary cell wall biosynthesis (Taylor-Teeples et al., 2015).
ML-based approaches have shown a great potential for understanding transcriptional networks in crops in which the depth of transcriptome and protein–DNA interactome datasets is much weaker compared with Arabidopsis and the functions of only a small number of genes have been experimentally validated (Figure 2), e.g., full scanning of the maize reference genome for CREs is expensive, laborious and technically challenging. The shortcoming of genome-wide information and experimental data for CREs and aggregated genomic complexity have urged maize (Zea mays) researchers to predict CREs using ML for transcriptional network mapping. For example, Mejía-Guerra and Buckler (2019) established the architecture of gene-regulatory regions at a k-mer (nucleotide sequence in a certain length) level in maize using an ML-based computational framework in which two trained models were able to make a distinction between regulatory and non-regulatory genomic regions with >90% accuracy (Mejía-Guerra and Buckler, 2019).
The predicting power of ML models can be further improved by training with new layers of genomic information such as the 3D structure of cis-regulatory regions and genomic sequence variation data of the maize diversity panels. Such a framework is highly applicable to other crops. In addition, ML modeling can also aid the identification of key regulators in response to abiotic stress in crops. For example, Gupta et al. (2020) took a network-based supervised ML framework to predict TFs that likely play key roles in response to drought stress in rice (Oryza sativa) (Gupta et al., 2020). In this study, to prioritize TFs, multiple GRNs were inferred from published rice transcriptome datasets, and then the consensus GRN was provided in the ML framework in which the support vector machine, a binary classification algorithm, trained models to learn the regulatory patterns of TFs in responses to drought. By pursuing this approach, the TF OsbHLH148 was predicted to play a key role in drought stress. These predictions were functionally validated by the characterization of an OsbHLH148 knockout (bhlh148), which showed growth defects specifically under drought stress compared with wild type. Furthermore, consistent with a prediction that, in the consensus GRN, the target genes of OsbHLH148 would be enriched with TFs of the WRKY and AP2-EREBP families, the genes annotated to be controlled by these TF families were highly downregulated in the drought-treated bhlh148 but not wild type or well-watered bhlh148. Therefore, this study demonstrates the effectiveness of the ML-based approach to identify genes in biological pathways in plants but also to advance research using predictive modeling in crops. Nonetheless, despite their strong potential to improve the predictive power, accuracy and biological interpretability of gene networks in crops, ML-based approaches require high-quality training datasets for robust and effective learning (Camacho et al., 2018), which may be a limiting factor for their implementation into transcriptional network mapping, particularly in crops.
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
With the rapidly increasing access to NGS technologies and decreased implementation costs, it is clear that new global datasets will continue to be rapidly accumulated and add to the abundant datasets already available. As such, the demand for transcriptional network modeling will exponentially increase (Box 1). However, several limitations of the current analyses could prevent us from maximizing the effectiveness and usefulness of these “big data” and computational frameworks (Box 2). A realistic obstacle lies in a lack of standardized computational pipelines for transcriptional network analyses, particularly of ML-based approaches. This consequently hampers a wider usage of transcriptional network-based approaches for biological questions that are difficult to be addressed via the traditional approaches based on a modification of the expression of a single gene or alteration of a single biological pathway. Another significant bottleneck is the quality input datasets, which undermines the performance of network inference. The recipe for high-quality data consists of correct controls, high signal-to-noise ratio and reproducibility. Datasets can miss one or more of these components with the result of erroneous biological interpretation due to the generation of inaccurate network predictions. Nonetheless, difficulties in generating transcriptional networks can be helpful in evaluating the quality of the input data and understanding where the experimental design needs rethinking.
Because they consist of many nodes of low degree and few nodes of high degree, known as scale-free topology, transcriptional networks are often resistant to perturbations (i.e., buffering effects by other gene members) (Barabási, 2009). Although gene functional redundancy may contribute to the fitness of a biological system, it does not allow validating the phenotypic effects of gene networks by single perturbation (i.e. single loss-of-function mutation). Combinatorial mutations of multiple genes predicted to be crucial for the gene network can overcome this limitation. While such an approach is possible for the model plant Arabidopsis, it may be not as feasible for other plant species. The recent expansion of the genome-editing toolbox, transcription activator-like effector nucleases and the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system, to generate mutations on a combination of multiple gene targets could make this task more feasible, particularly in crops (Čermák et al., 2017).
A significant question we are facing in the post-genomics era is whether we can extract biologically meaningful insights from highly heterogeneous, noisy, complex and dimensional datasets at a reasonable computational cost. Despite facing analytic challenges, rapid advancements in ML-based and artificial intelligence methodologies, such as deep learning (Ching et al., 2018; Eraslan et al., 2019), hold great promise to bypass caveats imposed by traditional statistical algorithms. These state-of-art computational approaches are also expected to help to explore spatiotemporal dynamics of transcriptional networks using single-cell transcriptome data in plants, which are increasingly valuable, by improving data clustering (Kiselev et al., 2019).
The systems-level understanding in gene regulation is still in its infancy, yet its demand is increasing often beyond the capability of individual labs. Both experimental biologists and bioinformaticians need to communicate directly for their own biological, experimental and analytical problems and come up with solutions in collaborative settings such as online or virtual workshops (e.g., the Plant Cell Atlas initiative; Rhee et al., 2019). This will help advance our systems-level understanding of gene regulation in plant biology and build strong research communities.
ACKNOWLEDGMENTS
This study was supported primarily by the National Institutes of Health (GM101038) with contributing support from by the Great Lakes Bioenergy Research Center, US Department of Energy, Office of Science, Office of Biological and Environmental Research (award DE-SC0018409), Chemical Sciences, Geoscience and Biosciences Division, Office of Basic Energy Sciences, Office of Science, US Department of Energy (award number DE-FG02-91ER20021) and MSU AgBioResearch (MICL02598).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- Aghamirzaie D, Raja Velmurugan K, Wu S, Altarawy D, Heath LS and Grene R (2017) Expresso: a database and web server for exploring the interaction of transcription factors and their target genes in. F1000Res 6, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD and Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat. Rev. Genet 17, 487–500. [DOI] [PubMed] [Google Scholar]
- Austin RS, Hiu S, Waese J et al. (2016) New BAR tools for mining expression data and exploring Cis-elements in Arabidopsis thaliana. Plant J 88, 490–504. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW and Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic M, Maher KA and Deal RB (2018) Identification of open chromatin regions in plant genomes using ATAC-Seq. Methods Mol. Biol 1675, 183–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banf M and Rhee SY (2017) Computational inference of gene regulatory networks: approaches, limitations and opportunities. Biochim. Biophys. Acta Gene Regul. Mech 1860, 41–52. [DOI] [PubMed] [Google Scholar]
- Barabási AL (2009) Scale-free networks: a decade and beyond. Science, 325, 412–413. [DOI] [PubMed] [Google Scholar]
- Barabási AL and Oltvai ZN (2004) Network biology: understanding the cell’s functional organization. Nat. Rev. Genet 5, 101–113. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I and Zhao K (2007) High-resolution profiling of histone methylations in the human genome. Cell, 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Beal MJ, Falciani F, Ghahramani Z, Rangel C and Wild DL (2005) A Bayesian approach to reconstructing genetic regulatory networks with hidden factors. Bioinformatics, 21, 349–356. [DOI] [PubMed] [Google Scholar]
- Bechtold U, Penfold CA, Jenkins DJ et al. (2016) Time-series transcriptomics reveals that AGAMOUS-LIKE22 affects primary metabolism and developmental processes in drought-stressed Arabidopsis. Plant Cell, 28, 345–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernotas G, Scorza LCT, Hansen MF, Hales IJ, Halliday KJ, Smith LN, Smith ML and McCormick AJ (2019) A photometric stereo-based 3D imaging system using computer vision and deep learning for tracking plant growth. Gigascience, 8, giz056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Watterson S, Shui G, Lacaze P, Khondoker M, Dickinson P, Sing G and Rodr ıguez-Mart ın S (2011) Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol 9, e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau R (2008) Learning biological networks: from modules to dynamics. Nat. Chem. Biol 4, 658–664. [DOI] [PubMed] [Google Scholar]
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS and Crawford GE (2008) High-resolution mapping and characterization of open chromatin across the genome. Cell, 132, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady SM, Orlando DA, Lee JY, Wang JY, Koch J, Dinneny JR, Mace D, Ohler U and Benfey PN (2007) A high-resolution root spatiotemporal map reveals dominant expression patterns. Science, 318, 801–806. [DOI] [PubMed] [Google Scholar]
- Brady SM, Zhang L, Megraw M et al. (2011) A stele-enriched gene regulatory network in the Arabidopsis root. Mol. Syst. Biol 7, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubb KL and Deal RB (2020) Considerations in the analysis of plant chromatin accessibility data. Curr. Opin. Plant Biol 54, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY and Greenleaf WJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods, 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY and Greenleaf WJ (2015) ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdo B, Gray J, Goetting-Minesky MP et al. (2014) The Maize TFome–development of a transcription factor open reading frame collection for functional genomics. Plant J 80, 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SJ, Reyna-Llorens I, Stevenson SR, Singh P, Jaeger K and Hibberd JM (2019) Genome-wide transcription factor binding in leaves from C3 and C4 grasses. Plant Cell, 31, 2297–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch W and Lohmann JU (2007) Profiling a plant: expression analysis in Arabidopsis. Curr. Opin. Plant Biol 10, 136–141. [DOI] [PubMed] [Google Scholar]
- Camacho DM, Collins KM, Powers RK, Costello JC and Collins JJ (2018) Next-generation machine learning for biological networks. Cell, 173, 1581–1592. [DOI] [PubMed] [Google Scholar]
- Cantone I, Marucci L, Iorio F, Ricci MA, Belcastro V, Bansal M, Santini S, Di Bernardo M, Di Bernardo D and Cosma MP (2009) A yeast synthetic network for in vivo assessment of reverse-engineering and modeling approaches. Cell, 137, 172–181. [DOI] [PubMed] [Google Scholar]
- Čermák T, Curtin SJ, Gil-Humanes J et al. (2017) A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell, 29, 1196–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Yan W, Fu LY and Kaufmann K (2018) Architecture of gene regulatory networks controlling flower development in Arabidopsis thaliana. Nat. Commun 9, 4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wang Y, Zhang R, Zhang H and Gao C (2019) CRISPR/Cas genome editing and precision plant breeding in agriculture. Annu. Rev. Plant Biol 70, 667–697. [DOI] [PubMed] [Google Scholar]
- Cheng S, Melkonian M, Smith SA, Brockington S, Archibald JM, Delaux P-M, Li F-W, Melkonian B, Mavrodiev EV and Sun W (2018) 10KP: a phylodiverse genome sequencing plan. Gigascience, 7, giy013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching T, Himmelstein DS, Beaulieu-Jones BK et al. (2018) Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface, 15, 20170387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL and Zhang X (2016) A survey of best practices for RNA-seq data analysis. Genome Biol 17, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig DW, Pearson JV, Szelinger S, Sekar A, Redman M, Corneveaux JJ, Pawlowski TL, Laub T, Nunn G and Stephan DA (2008) Identification of genetic variants using bar-coded multiplexed sequencing. Nat. Methods, 5, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford GE, Holt IE, Whittle J et al. (2006) Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res 16, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB and Henikoff S (2010) A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev. Cell, 18, 1030–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denyer T, Ma X, Klesen S, Scacchi E, Nieselt K and Timmermans MCP (2019) Spatiotemporal developmental trajectories in the arabidopsis root revealed using high-throughput single-cell RNA sequencing. Dev. Cell, 48, 840–852.e845. [DOI] [PubMed] [Google Scholar]
- Deplancke B, Dupuy D, Vidal M and Walhout AJ (2004) A gateway-compatible yeast one-hybrid system. Genome Res 14, 2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, Bardet AF, Ginno PA, Hartl D, Burger L and Schübeler D (2015) Competition between DNA methylation and transcription factors determines binding of NRF1. Nature, 528, 575–579. [DOI] [PubMed] [Google Scholar]
- Dong S, Lau V, Song R et al. (2019) Proteome-wide, structure-based prediction of protein-protein interactions/new molecular interactions viewer. Plant Physiol 179, 1893–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni I, Mello A, Nawy T, Ip PL, Rahni R, DelRose N, Powers A, Satija R and Birnbaum KD (2016) Root regeneration triggers an embryo-like sequence guided by hormonal interactions. Cell, 165, 1721–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eraslan G, Avsec Ž, Gagneur J and Theis FJ (2019) Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet 20, 389–403. [DOI] [PubMed] [Google Scholar]
- Evans J, Crisovan E, Barry K et al. (2015) Diversity and population structure of northern switchgrass as revealed through exome capture sequencing. Plant J 84, 800–815. [DOI] [PubMed] [Google Scholar]
- Farnham PJ (2009) Insights from genomic profiling of transcription factors. Nat. Rev. Genet 10, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Zorrilla JM and Solano R (2017) Identification of plant transcription factor target sequences. Biochim. Biophys. Acta Gene Regulat. Mech 1860(1), 21–30. [DOI] [PubMed] [Google Scholar]
- Galli M, Khakhar A, Lu Z, Chen Z, Sen S, Joshi T, Nemhauser JL, Schmitz RJ and Gallavotti A (2018) The DNA binding landscape of the maize AUXIN RESPONSE FACTOR family. Nat. Commun 9, 4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Gonda I, Sun H et al. (2019) The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet 51, 1044–1051. [DOI] [PubMed] [Google Scholar]
- Gates LA, Foulds CE and O’Malley BW (2017) Histone marks in the ‘Driver’s Seat’: functional roles in steering the transcription cycle. Trends Biochem. Sci 42, 977–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudinier A and Brady SM (2016) Mapping transcriptional networks in plants: data-driven discovery of novel biological mechanisms. Annu. Rev. Plant Biol 67, 575–594. [DOI] [PubMed] [Google Scholar]
- Gaudinier A, Rodriguez-Medina J, Zhang L et al. (2018) Transcriptional regulation of nitrogen-associated metabolism and growth. Nature, 563, 259–264. [DOI] [PubMed] [Google Scholar]
- Gaudinier A, Zhang L, Reece-Hoyes JS et al. (2011) Enhanced Y1H assays for arabidopsis. Nat. Methods, 8, 1053–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitter A, Siegfried Z, Klutstein M, Fornes O, Oliva B, Simon I and Bar-Joseph Z (2009) Backup in gene regulatory networks explains differences between binding and knockout results. Mol. Syst. Biol 5(1), 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin S, McPherson JD and McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet 17, 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenham K, Guadagno CR, Gehan MA, Mockler TC, Weinig C, Ewers BE and McClung CR (2017) Temporal network analysis identifies early physiological and transcriptomic indicators of mild drought in. Elife, 6, e29655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta C, Ramegowda V, Basu S and Pereira A (2020) Prediction and characterization of transcription factors involved in drought stress response. bioRxiv 10.1101/2020.04.29.068379 [DOI] [Google Scholar]
- Haque S, Ahmad JS, Clark NM, Williams CM and Sozzani R (2019) Computational prediction of gene regulatory networks in plant growth and development. Curr. Opin. Plant Biol 47, 96–105. [DOI] [PubMed] [Google Scholar]
- Hartmann U, Höhmann S, Nettesheim K, Wisman E, Saedler H and Huijser P (2000) Molecular cloning of SVP: a negative regulator of the floral transition in Arabidopsis. Plant J 21, 351–360. [DOI] [PubMed] [Google Scholar]
- Heard NA, Holmes CC and Stephens DA (2006) A quantitative study of gene regulation involved in the immune response of anopheline mosquitoes: an application of Bayesian hierarchical clustering of curves. J. Am. Stat. Assoc 101, 18–29. [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H and Glass CK (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Huerta M, Rodriguez-Gonzalvez P and Rainey K (2020) Yield prediction by machine learning from UAS-based multi-sensor data fusion in soybean. Plant Methods, 16, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyndrickx KS, Van de Velde J, Wang C, Weigel D and Vandepoele K (2014) A functional and evolutionary perspective on transcription factor binding in Arabidopsis thaliana. Plant Cell, 26, 3894–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman R, Van Verk MC, Van Dijken AJH et al. (2017) Architecture and dynamics of the jasmonic acid gene regulatory network. Plant Cell, 29, 2086–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh-Thu VA, Irrthum A, Wehenkel L and Geurts P (2010) Inferring regulatory networks from expression data using tree-based methods. PLoS One, 5, e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh-Thu VA and Sanguinetti G (2015) Combining tree-based and dynamical systems for the inference of gene regulatory networks. Bioinformatics, 31, 1614–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh-Thu VA and Sanguinetti G (2019) Tree-based learning of regulatory network topologies and dynamics with Jump3. Methods Mol. Biol 1883, 217–233. [DOI] [PubMed] [Google Scholar]
- Ideker T, Galitski T and Hood L (2001) A new approach to decoding life: systems biology. Annu. Rev. Genomics Hum. Genet 2, 343–372. [DOI] [PubMed] [Google Scholar]
- Ikeuchi M, Shibata M, Rymen B, Iwase A, Bågman A-M, Watt L, Coleman D, Favero DS, Takahashi T and Ahnert SE (2018) A gene regulatory network for cellular reprogramming in plant regeneration. Plant Cell Physiol. 59, 770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Initiative AG (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature, 408, 796–815. [DOI] [PubMed] [Google Scholar]
- International Rice Genome Sequencing Project. (2005) The map-based sequence of the rice genome. Nature, 436, 793–800. [DOI] [PubMed] [Google Scholar]
- Jean-Baptiste K, McFaline-Figueroa JL, Alexandre CM, Dorrity MW, Saunders L, Bubb KL, Trapnell C, Fields S, Queitsch C and Cuperus J (2019) Dynamics of gene expression in single root cells of A. thaliana. Plant Cell, 31(5), 993–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon JE, Kim JG, Fischer CR, Mehta N, Dufour-Schroif C, Wemmer K, Mudgett MB and Sattely E (2020) A pathogen-responsive gene cluster for highly modified fatty acids in tomato. Cell, 180, 176–187.e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, He K, Tang X, Li Z, Lv L, Zhao Y, Luo J and Gao G (2015) An arabidopsis transcriptional regulatory map reveals distinct functional and evolutionary features of novel transcription factors. Mol. Biol. Evol 32, 1767–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM and Wold B (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science, 316, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD and Widom J (2009) The DNA-encoded nucleosome organization of a eukaryotic genome. Nature, 458, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katari MS, Nowicki SD, Aceituno FF, Nero D, Kelfer J, Thompson LP, Cabello JM, Davidson RS, Goldberg AP and Shasha DE (2010) VirtualPlant: a software platform to support systems biology research. Plant Physiol 152, 500–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K, Pajoro A and Angenent GC (2010) Regulation of transcription in plants: mechanisms controlling developmental switches. Nat. Rev. Genet 11, 830–842. [DOI] [PubMed] [Google Scholar]
- Kersey PJ (2019) Plant genome sequences: past, present, future. Curr. Opin. Plant Biol 48, 1–8. [DOI] [PubMed] [Google Scholar]
- Khaki S and Wang L (2019) Crop yield prediction using deep neural networks. Front. Plant Sci 10, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselev VY, Andrews TS and Hemberg M (2019) Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet 20, 273–282. [DOI] [PubMed] [Google Scholar]
- Kitano H (2002) Systems biology: a brief overview. Science, 295, 1662–1664. [DOI] [PubMed] [Google Scholar]
- Klemm SL, Shipony Z and Greenleaf WJ (2019) Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet 20, 207–220. [DOI] [PubMed] [Google Scholar]
- Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science, 184, 868–871. [DOI] [PubMed] [Google Scholar]
- Krouk G, Mirowski P, LeCun Y, Shasha DE and Coruzzi GM (2010) Predictive network modeling of the high-resolution dynamic plant transcriptome in response to nitrate. Genome Biol 11, R123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni SR, Vaneechoutte D, Van de Velde J and Vandepoele K (2018) TF2Network: predicting transcription factor regulators and gene regulatory networks in Arabidopsis using publicly available binding site information. Nucleic Acids Res 46, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P and Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanver D, Müller AN, Happel P, Schweizer G, Haas FB, Franitza M, Pellegrin C, Reissmann S, Altmüller J and Rensing SA (2018) The biotrophic development of Ustilago maydis studied by RNA-seq analysis. Plant Cell, 30, 300–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau W and Sattely ES (2015) Six enzymes from mayapple that complete the biosynthetic pathway to the etoposide aglycone. Science, 349, 1224–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Zhang H, Julian R, Tang K, Xie S and Zhu J-K (2015) Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc. Natl Acad. Sci. USA, 112, 3553–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Tang M, Nelson A, Caligagan H, Zhou X, Clark-Wiest C, Ngo R, Brady SM and Kliebenstein DJ (2018) Network-guided discovery of extensive epistasis between transcription factors involved in aliphatic glucosinolate biosynthesis. Plant Cell, 30, 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ and Herskowitz I (1993) Isolation of ORC6, a component of the yeast origin recognition complex by a one-hybrid system. Science, 262, 1870–1874. [DOI] [PubMed] [Google Scholar]
- Li Y, Pearl SA and Jackson SA (2015) Gene networks in plant biology: approaches in reconstruction and analysis. Trends Plant Sci 20, 664–675. [DOI] [PubMed] [Google Scholar]
- Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH and Ecker JR (2008) Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell, 133, 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C and Weigel D (2015) Chromatin in 3D: progress and prospects for plants. Genome Biol 16, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MJ, Sugimoto K, Uygun S, Panchy N, Campbell MS, Yandell M, Howe GA and Shiu SH (2018) Regulatory divergence in wound-responsive gene expression between domesticated and wild tomato. Plant Cell, 30, 1445–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long TA, Brady SM and Benfey PN (2008) Systems approaches to identifying gene regulatory networks in plants. Annu. Rev. Cell Dev. Biol 24, 81–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher KA, Bajic M, Kajala K et al. (2018) Profiling of accessible chromatin regions across multiple plant species and cell types reveals common gene regulatory principles and new control modules. Plant Cell, 30, 15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Costello JC, Küffner R et al. (2012) Wisdom of crowds for robust gene network inference. Nat. Methods, 9, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchive C, Roudier F, Castaings L, Bréhaut V, Blondet E, Colot V, Meyer C and Krapp A (2013) Nuclear retention of the transcription factor NLP7 orchestrates the early response to nitrate in plants. Nat. Commun 4, 1–9. [DOI] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R and Califano A (2006) ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics, 7(Suppl 1), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall-Colón A and Kliebenstein DJ (2019) Plant networks as traits and hypotheses: moving beyond description. Trends Plant Sci 24, 840–852. [DOI] [PubMed] [Google Scholar]
- Matzke MA and Mosher RA (2014) RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet 15, 394–408. [DOI] [PubMed] [Google Scholar]
- Mejía-Guerra MK and Buckler ES (2019) A k-mer grammar analysis to uncover maize regulatory architecture. BMC Plant Biol 19, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker ML (2010) Sequencing technologies - the next generation. Nat. Rev. Genet 11, 31–46. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB et al. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature, 448, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BM, Wang P, Fan P, Leong B, Schenck CA, Lloyd JP, Lehti-Shiu MD, Last RL, Pichersky E and Shiu SH (2019) Robust predictions of specialized metabolism genes through machine learning. Proc. Natl Acad. Sci. USA, 116, 2344–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordelet F and Vert J-P (2008) SIRENE: supervised inference of regulatory networks. Bioinformatics, 24, i76–i82. [DOI] [PubMed] [Google Scholar]
- Moreno-Risueno MA, Busch W and Benfey PN (2010) Omics meet networks—using systems approaches to infer regulatory networks in plants. Curr. Opin. Plant Biol 13, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Risueno MA, Sozzani R, Yardımcı GG et al. (2015) Transcriptional control of tissue formation throughout root development. Science, 350, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K and Mian S (1999) Modelling gene expression data using dynamic Bayesian networks: Technical report, Computer Science Division, University of California. [Google Scholar]
- Needham CJ, Manfield IW, Bulpitt AJ, Gilmartin PM and Westhead DR (2009) From gene expression to gene regulatory networks in Arabidopsis thaliana. BMC Syst. Biol 3, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelms B and Walbot V (2019) Defining the developmental program leading to meiosis in maize. Science, 364, 52–56. [DOI] [PubMed] [Google Scholar]
- Niederhuth CE and Schmitz RJ (2017) Putting DNA methylation in context: from genomes to gene expression in plants. Biochim. Biophys. Acta Gene Regul. Mech 1860, 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley RC, Huang SC, Song L, Lewsey MG, Bartlett A, Nery JR, Galli M, Gallavotti A and Ecker JR (2016) Cistrome and epicistrome features shape the regulatory DNA landscape. Cell, 165, 1280–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ó’Maoiléidigh DS, Graciet E and Wellmer F (2014) Gene networks controlling Arabidopsis thaliana flower development. New Phytol 201, 16–30. [DOI] [PubMed] [Google Scholar]
- Oka R, Zicola J, Weber B et al. (2017) Genome-wide mapping of transcriptional enhancer candidates using DNA and chromatin features in maize. Genome Biol 18, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajoro A, Muiño JM, Angenent GC and Kaufmann K (2018) Profiling nucleosome occupancy by MNase-seq: experimental protocol and computational analysis. Methods Mol. Biol 1675, 167–181. [DOI] [PubMed] [Google Scholar]
- Panchy N, Lehti-Shiu M and Shiu SH (2016) Evolution of gene duplication in plants. Plant Physiol 171, 2294–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PJ (2009) ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet 10, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pe’er D (2005) Bayesian network analysis of signaling networks: a primer. Sci. STKE, 2005(281), pl4. [DOI] [PubMed] [Google Scholar]
- Polanski K, Rhodes J, Hill C et al. (2014) Wigwams: identifying gene modules co-regulated across multiple biological conditions. Bioinformatics, 30, 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prill RJ, Marbach D, Saez-Rodriguez J, Sorger PK, Alexopoulos LG, Xue X, Clarke ND, Altan-Bonnet G and Stolovitzky G (2010) Towards a rigorous assessment of systems biology models: the DREAM3 challenges. PLoS One, 5, e9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda-Paz JL, Breton G, Nagel DH, Kang SE, Bonaldi K, Doherty CJ, Ravelo S, Galli M, Ecker JR and Kay SA (2014) A genome-scale resource for the functional characterization of Arabidopsis transcription factors. Cell Rep 8, 622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynoso MA, Kajala K, Bajic M et al. (2019) Evolutionary flexibility in flooding response circuitry in angiosperms. Science, 365, 1291–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SY, Birnbaum KD and Ehrhardt DW (2019) Towards building a plant cell atlas. Trends Plant Sci 24, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M et al. (2007) Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods, 4, 651–657. [DOI] [PubMed] [Google Scholar]
- Rodgers-Melnick E, Vera DL, Bass HW and Buckler ES (2016) Open chromatin reveals the functional maize genome. Proc. Natl Acad. Sci. USA, 113, E3177–E3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IG, Ruvinsky I and Gilad Y (2012) Comparative studies of gene expression and the evolution of gene regulation. Nat. Rev. Genet 13, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymen B, Kawamura A, Lambolez A et al. (2019) Histone acetylation orchestrates wound-induced transcriptional activation and cellular reprogramming in Arabidopsis. Commun. Biol 2, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KH, Huang L, Kang HM and Schiefelbein J (2019) Single-cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiol 179, 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh A, Alvarez-Venegas R and Avramova Z (2008) An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat. Protoc 3, 1018–1025. [DOI] [PubMed] [Google Scholar]
- Sartor RC, Noshay J, Springer NM and Briggs SP (2019) Identification of the expressome by machine learning on omics data. Proc. Natl Acad. Sci. USA, 116, 18119–18125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science, 326, 1112–1115. [DOI] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G and Zhao K (2008) Dynamic regulation of nucleosome positioning in the human genome. Cell, 132, 887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield S, Murison A, Jones A, Fozard J, Aida M, Band LR, Bennett M and Murray JAH (2018) Coordination of meristem and boundary functions by transcription factors in the SHOOT MERISTEMLESS regulatory network. Development, 145, dev157081. 10.1242/dev.157081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serin EA, Nijveen H, Hilhorst HW and Ligterink W (2016) Learning from co-expression networks: possibilities and challenges. Front. Plant Sci 7, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Breuer C, Kawamura A et al. (2018) GTL1 and DF1 regulate root hair growth through transcriptional repression of ROOT HAIR DEFECTIVE 6-LIKE 4 in Arabidopsis. Development, 145, dev159707. 10.1242/dev.159707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiu S-H, Shih M-C and Li W-H (2005) Transcription factor families have much higher expansion rates in plants than in animals. Plant Physiol 139, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulse CN, Cole BJ, Ciobanu D et al. (2019) High-throughput single-cell transcriptome profiling of plant cell types. Cell Rep 27, 2241–2247.e2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijacic P, Bajic M, McKinney EC, Meagher RB and Deal RB (2018) Changes in chromatin accessibility between Arabidopsis stem cells and mesophyll cells illuminate cell type-specific transcription factor networks. Plant J 94, 215–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit ME, Llavata-Peris CI, Roosjen M et al. (2020a) Specification and regulation of vascular tissue identity in the Arabidopsis embryo. Development, 147(8), dev186130. 10.1242/dev.186130 [DOI] [PubMed] [Google Scholar]
- Smit ME, McGregor SR, Sun H, Gough C, Bågman A-M, Soyars CL, Kroon JT, Gaudinier A, Williams CJ and Yang X (2020b) A PXY-mediated transcriptional network integrates signaling mechanisms to control vascular development in Arabidopsis. Plant Cell, 32, 319–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon MJ, Larsen PL and Varshavsky A (1988) Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell, 53, 937–947. [DOI] [PubMed] [Google Scholar]
- Song ZT, Sun L, Lu SJ, Tian Y, Ding Y and Liu JX (2015) Transcription factor interaction with COMPASS-like complex regulates histone H3K4 trimethylation for specific gene expression in plants. Proc. Natl Acad. Sci. USA, 112, 2900–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks E, Wachsman G and Benfey PN (2013) Spatiotemporal signalling in plant development. Nat. Rev. Genet 14, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F and Furlong EE (2012) Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Stuart JM, Segal E, Koller D and Kim SK (2003) A gene-coexpression network for global discovery of conserved genetic modules. Science, 302, 249–255. [DOI] [PubMed] [Google Scholar]
- Sullivan AM, Arsovski AA, Lempe J et al. (2014) Mapping and dynamics of regulatory DNA and transcription factor networks in A. thaliana. Cell Rep 8, 2015–2030. [DOI] [PubMed] [Google Scholar]
- Swift J and Coruzzi GM (2017) A matter of time—How transient transcription factor interactions create dynamic gene regulatory networks. Biochim. Biophys. Acta Gene Regulat. Mech 1860(1), 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB and Henikoff S (2017) Histone variants on the move: sub-strates for chromatin dynamics. Nat. Rev. Mol. Cell Biol 18, 115–126. [DOI] [PubMed] [Google Scholar]
- Taylor-Teeples M, Lin L, de Lucas M et al. (2015) An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature, 517, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F, Yang DC, Meng YQ, Jin J and Gao G (2020) PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res 48, D1104–D1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubiana D, Puzis R, Wen L et al. (2019) Combined network analysis and machine learning allows the prediction of metabolic pathways from tomato metabolomics data. Commun. Biol 2, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usadel B, Obayashi T, Mutwil M, Giorgi FM, Bassel GW, Tanimoto M, Chow A, Steinhauser D, Persson S and Provart NJ (2009) Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ 32, 1633–1651. [DOI] [PubMed] [Google Scholar]
- Uygun S, Azodi CB and Shiu SH (2019) Cis-regulatory code for predicting plant cell-type transcriptional response to high salinity. Plant Physiol 181, 1739–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer Y, Maere S and Meyer A (2009) The evolutionary significance of ancient genome duplications. Nat. Rev. Genet 10, 725–732. [DOI] [PubMed] [Google Scholar]
- Van den Broeck L, Dubois M, Vermeersch M, Storme V, Matsui M and Inzé D (2017) From network to phenotype: the dynamic wiring of an Arabidopsis transcriptional network induced by osmotic stress. Mol. Syst. Biol 13, 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandepoele K, Quimbaya M, Casneuf T, De Veylder L and Van de Peer Y (2009) Unraveling transcriptional control in Arabidopsis using cis-regulatory elements and coexpression networks. Plant Physiol 150, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varala K, Marshall-Colón A, Cirrone J et al. (2018) Temporal transcriptional logic of dynamic regulatory networks underlying nitrogen signaling and use in plants. Proc. Natl Acad. Sci. USA, 115, 6494–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeirssen V, De Clercq I, Van Parys T, Van Breusegem F and Van de Peer Y (2014) Arabidopsis ensemble reverse-engineered gene regulatory network discloses interconnected transcription factors in oxidative stress. Plant Cell, 26, 4656–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara A, Duarte FM and Lis JT (2018) Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet 19, 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Si Y, Dedow LK, Shao Y, Liu P and Brutnell TP (2011) A low-cost library construction protocol and data analysis pipeline for Illumina-based strand-specific multiplex RNA-seq. PLoS One, 6, e26426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MM and Reed RR (1993) Molecular cloning of the olfactory neuronal transcription factor Olf-1 by genetic selection in yeast. Nature, 364, 121–126. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M and Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasternack C (2015) How jasmonates earned their laurels: past and present. J. Plant Growth Regul 34, 761–794. [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I and Cook K (2014) Determination and inference of eukaryotic transcription factor sequence specificity. Cell, 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins O, Hafemeister C, Plessis A et al. (2016) EGRINs (Environmental Gene Regulatory Influence Networks) in rice that function in the response to water deficit, high temperature, and agricultural environments. Plant Cell, 28, 2365–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BP, Pignatta D, Henikoff S and Gehring M (2015) Methylation-sensitive expression of a DNA demethylase gene serves as an epigenetic rheostat. PLOS Genet 11(3), e1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windram O, Madhou P, McHattie S et al. (2012) Arabidopsis defense against Botrytis cinerea: chronology and regulation deciphered by high-resolution temporal transcriptomic analysis. Plant Cell, 24, 3530–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing RA, Purugganan MD and Zhang Q (2018) The rice genome revolution: from an ancient grain to green super rice. Nat. Rev. Genet 19, 505–517. [DOI] [PubMed] [Google Scholar]
- Wisecaver JH, Borowsky AT, Tzin V, Jander G, Kliebenstein DJ and Rokas A (2017) A global coexpression network approach for connecting genes to specialized metabolic pathways in plants. Plant Cell, 29, 944–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV and Romano LA (2003) The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol 20, 1377–1419. [DOI] [PubMed] [Google Scholar]
- Xiong W, Wang C, Zhang X, Yang Q, Shao R, Lai J and Du C (2017) Highly interwoven communities of a gene regulatory network unveil topologically important genes for maize seed development. Plant J 92, 1143–1156. [DOI] [PubMed] [Google Scholar]
- Yamasaki K, Kigawa T, Seki M, Shinozaki K and Yokoyama S (2013) DNA-binding domains of plant-specific transcription factors: structure, function, and evolution. Trends Plant Sci 18, 267–276. [DOI] [PubMed] [Google Scholar]
- Yang F, Li W, Jiang N et al. (2017) A maize gene regulatory network for phenolic metabolism. Mol. Plant, 10, 498–515. [DOI] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Fukushima A and Saito K (2013) Transcriptome data modeling for targeted plant metabolic engineering. Curr. Opin. Biotechnol 24, 285–290. [DOI] [PubMed] [Google Scholar]
- Yu CP, Lin JJ and Li WH (2016) Positional distribution of transcription factor binding sites in Arabidopsis thaliana. Sci. Rep 6, 25164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander M, Lewsey MG, Clark NM et al. (2020) Integrated multi-omics framework of the plant response to jasmonic acid. Nat. Plants, 6, 290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang L, Qi B, Zhao B, Ko EE, Riggan ND, Chin K and Qiao H (2017) EIN2 mediates direct regulation of histone acetylation in the ethylene response. Proc. Natl Acad. Sci. USA, 114, 10274–10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lang Z and Zhu JK (2018) Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol 19, 489–506. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wu Y, Schnable JC, Zeng Z, Freeling M, Crawford GE and Jiang J (2012a) High-resolution mapping of open chromatin in the rice genome. Genome Res 22, 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhang T, Wu Y and Jiang J (2012b) Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin in Arabidopsis. Plant Cell, 24, 2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan S-W-L, Chen H, Henderson IR, Shinn P, Pellegrini M and Jacobsen SE (2006) Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell, 126, 1189–1201. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Jin Y, Chen B and Brown P (2019) California almond yield prediction at the orchard level with a machine learning approach. Front. Plant Sci 10, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, Sun K, Mackaluso JD, Seddon AE, Jin R, Thomashow MF and Shiu S-H (2011) Cis-regulatory code of stress-responsive transcription in Arabidopsis thaliana. Proc. Natl Acad. Sci. USA, 108, 14992–14997. [DOI] [PMC free article] [PubMed] [Google Scholar]