

Abstract
Background
Abiotic stresses affect plants in several ways and as such, phytohormones such as abscisic acid (ABA) play an important role in conferring tolerance towards these stresses. Hence, to comprehend the role of ABA and its interaction with receptors of the plants, a thorough investigation is essential.
Aim
The current study aimed to identify the ABA receptors in Oryza sativa, to find the receptor that binds best with ABA and to examine the mutations present to help predict better binding of the receptors with ABA.
Methods
Protein sequences of twelve PYL (Pyrabactin resistance 1) and seven PP2C (type 2C protein phosphatase) receptors were retrieved from the Rice Annotation Project database and their 3D structures were predicted using RaptorX. Protein-ligand molecular docking studies between PYL and ABA were performed using AutoDock 1.5.6, followed by 100ns molecular dynamic simulation studies using Desmond to determine the acceptable conformational changes after docking via root mean square deviation RMSD plot analysis. Protein-protein docking was then carried out in three sets: PYL-PP2Cs, PYL-ABA-PP2C and PYL(mut)-ABA-PP2C to scrutinize changes in structural conformations and binding energies between complexes. The amino acids of interest were mapped at their respective genomic coordinates using SNP-seek database to ascertain if there were any naturally occurring single nucleotide polymorphisms (SNPs) responsible for triggering rice PYLs mutations.
Results
Initial protein-ligand docking studies revealed good binding between the complexes, wherein PYL6-ABA complex showed the best energy of -8.15 kcal/mol. The 100ns simulation studies revealed changes in the RMSD values after docking, indicating acceptable conformational changes. Furthermore, mutagenesis study performed at specific PYL-ABA interacting residues followed by downstream PYL(mut)-ABA-PP2C protein-protein docking results after induction of mutations demonstrated binding energy of -8.17 kcal/mol for PP2C79-PYL11-ABA complex. No naturally occurring SNPs that were responsible for triggering rice PYL mutations were identified when specific amino acid coordinates were mapped at respective genomic coordinates.
Conclusion
Thus, the present study provides valuable insights on the interactions of ABA receptors in rice and induced mutations in PYL11 that can enhance the downstream interaction with PP2C.
Keywords: Abscisic acid, Oryza sativa, PYL, PP2C, docking studies, molecular dynamics simulation, mutagenesis
1. INTRODUCTION
Plants are exposed to a wide variety of stresses, including biotic and abiotic stresses. Abiotic stress conditions
comprise heat, cold, drought and salinity while biotic stress conditions occur majorly due to bacteria, viruses, fungi, insects and nematodes. Adaptation to such adverse situations has led to the evolution of certain plant mechanisms which aid plants to respond well to such stressful conditions. Production of phytohormones is one such way where plants respond and adapt to adverse environmental conditions, especially during abiotic stresses. Some of the major hormones that plants produce during abiotic stress include auxins, cytokinins, gibberellins, abscisic acid (ABA), salicylic acid, ethylene, jasmonates, strigolactones and brassinosteroids. Among these, abscisic acid plays a crucial role in meditating defence responses of plants against abiotic stresses [1]. Studies have demonstrated that stress conditions such as salinity, heat, cold, drought and even plant injury have shown to augment the production of abscisic acid levels [2]. Moreover, ABA is not only involved in stress response, but also in plant growth by regulating the gene responsible for certain physiological processes ranging from stomatal opening to storage of proteins [3].
The receptors involved in ABA signalling were first discovered in Arabidopsis and it involves three major components- receptor PYR/PYL/RCAR (PYL-Pyrabactin resistance 1) protein family, positive regulator class III SNF-1-related protein kinase 2 (SnRK2) and negative regulator type 2C protein phosphatase (PP2C) [4]. PYR proteins are responsible for carrying out proper ABA signal transduction in Arabidopsis [5] and PP2Cs function as negative regulators in ABA-dependent pathways [6]. The chief targets of PP2Cs are associated with protein kinases and among these, class III SNF-1 is most incriminated in the positive regulation of ABA signal transduction [7]. When ABA is present, the formation of complex PYL-PP2C leads to PP2C activity inhibition, which in turn allows for the activation of SnRK2. Furthermore, substrate proteins that are downstream, such as transcription factors, are then phosphorylated by activated SnRK2, thereby facilitating the transcription of ABA-responsive genes [8]. Conversely, the absence of ABA in plants allows the binding of PP2C molecule to SnRK2, leading to the absence of phosphorylation of the molecules and enzymes responsible for ABA response.
The main aim of plant breeding is to boost the productivity of food and to make plants adaptable to conditions of stress. Therefore, to increase the plant tolerance towards abiotic stresses, a thorough investigation is required regarding the role of abscisic acid and its interaction with plant receptors. Currently, studies on abiotic stress tolerance are more focused on Arabidopsis than in other agricultural crops, and very little is known regarding the ABA signal transduction cascade in Oryza sativa, the most widely consumed staple food crop by the largest human population [9, 10]. Therefore, the current study aimed to detect the ABA receptors in Oryza sativa, with a focus on PYLs and to find the receptor that binds best to abscisic acid. Additionally, the study also aimed to analyse and comprehend the mutations present, to help predict better binding of the receptors with ABA. Prediction of the specific amino acids at their respective genome coordinates was also carried out to gain further understanding of the mutational study via SNP detection. Thus, the present study provides valuable insights into the interactions of abscisic acid with important PYL receptors in Oryza sativa, which allows a better understanding of the stress tolerance mechanisms in rice.
2. MATERIALS AND METHODS
2.1. Retrieval of ABA Receptors in Rice, Structure Modelling, and Phylogenetic Analysis
To find the interactions between the ABA receptors in rice, a literature survey was carried out to identify the PYL receptors involved in ABA signalling cascade in Oryza sativa. According to the literature, 12 PYL receptors were identified [9]. Protein sequences of all twelve PYL receptors were obtained using their gene IDs (OsPYL1-12: Os10g573400, Os06g0562200, Os02g0226801, Os01g0827800, Os05g0473000, Os03g0297600, Os06g0526400, Os06g0527800, Os06g0528300, Os02g0255500, Os05g0213500 and Os02g0255300 respectively) from He et al., 2014 and retrieved from RAP-DB (Rice Annotation Project - Database) in FASTA format. Structure modelling of the twelve retrieved receptors was then carried out using RaptorX tool, which uses a statistical approach for template-dependant modelling of protein. RaptorX augments the accuracy of the alignment via structural information exploitation in single or multiple templates [11]. The modelled structures, obtained as .pdb files were then checked for stability by analyzing the Ramachandran plot [12] of the structures using SAVES v 5.0 [13]. Once the structure stabilities were found to be acceptable, an evolutionary analysis was carried out for all PYL receptors. Thus, to determine the evolutionary relationship of the receptors, Clustal Omega [14] was first utilized for performing multiple sequence alignment for the twelve retrieved receptors. Clustering was performed using Neighbour Joining (NJ) method [15] and the default number of iterations was set to 16. Further analysis of these was carried out in MEGA-X, where a phylogenetic tree was developed using the NJ statistical method to study the evolutionary relationship [16]. The results obtained were further scrutinized.
2.2. Docking and Simulation Studies of Os PYLs with ABA
To determine which PYL receptor binds best to the ABA molecules, a docking study was carried out with 12 PYLs as the receptors and ABA as the ligand. The ligand molecule, ABA (Pubchem ID: 5280896) was downloaded from NCBI Pubchem in .sdf format and converted to the required file format such as .pdbqt for the purposes of docking. Protein-ligand docking between ABA molecule as the ligand and PYL as the receptor was performed using AutoDock tools 1.5.6 [17]. AutoDock tools permit reliable computational docking of flexible ligands along with a dozen torsional degrees of freedom, making it a robust tool for protein-ligand docking. Additionally, the empirical free energy force field predicts the binding energies accurately (±2 kcal/mol) with a ±30-fold variation in the binding constants [18]. Due to these reasons, docking was carried out using AutoDock tools. The outputs were obtained as docked complexes in .pdbqt format. The best docking conformation in each case was selected based on the minimum binding energy, in terms of kcal/mol. The protein-ligand interactions of the docked complexes were studied using Discovery Studio [19] to identify the types of bonds between the docked PYL and ABA molecules. To determine how ligand binding induces changes in the protein, simulation studies for all docked complexes were carried out using the Maestro workspace in Desmond. Desmond achieves high scalability, has the latest graphics processing unit, high accuracy, provides realistic simulations and has an interface that is easy to use [20, 21]. Due to these reasons, Desmond was used for simulating the best docked complexes in the present study.
Protein pre-processing was employed on the interaction complex and parameters such as checking for potential errors in the structure, file, minimization for 500 steps using the steepest decent algorithm, and removing waters. Simulation box/environment was constructed using the system builder. TIP3P solvent model was selected and the conditions of the boundary were defined by the orthorhombic box having a minimized volume compressing the complex 10 Å of each of the axis. OPLS3e was the force field applied. Neutralization of the system was performed by adding Cl- or Na+ ions depending on the system’s total charge. During H-bond assignment, the PropKA was set to 7.0 pH. This results in imparting specific protonation states for residues at respective pH and simulation conditions. To perform the molecular dynamics simulation, the system was imported from the maestro workspace. Simulation was performed for 100 ns, with 0.1ns set as the trajectory recording interval. Ensemble class was set to NVT for the protein under conformational changes (thermodynamic parameters of constant pressure, temperature, and variable volume). The simulation protocol involves the following steps- 1. Brownian dynamics, NVT, T = 10k, small time steps, and restraint on solute heavy atoms for 100ps. 2. Brownian dynamics NVT, T = 50k, H2O barrier, membrane restrained in z protein restrained for 50ps. 3. NPT, T = 50k, H2O barrier, protein restrained for 50ps. 4. NVT production and all constraints removed for 200ps. 5. NVT production and all constraints removed for 500ps. 6. Final simulation for 200ns with time step set to 2 femto seconds (fs) and temperature of 310K. The cut-off short range method having a radius of 9.0 Å was employed and the restraints were not pre-defined. Furthermore, a simulation interaction diagram tool was utilized for scrutinizing the outcomes of simulations, including changes in RMSD values and protein-ligand contacts.
For every docking, the Root Mean Square Deviation (RMSD) was calculated that measured the average change in the displacement of a certain selection of atoms for a specific frame with respect to a reference frame [22]. This was calculated for all frames in the trajectory.
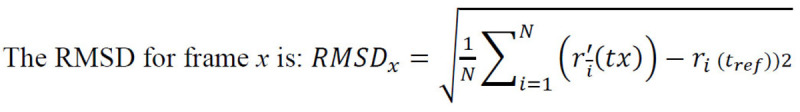
where N is the total number of atoms in atom selection; tref is the reference time (normally, the first frame is the reference and is regarded as time t=0); and r' depicts the selected atom positions in frame x post superimposition to the reference frame, where frame x is recorded at time tx. This method was repeated for each frame in the trajectory of the simulation [20]. Therefore, the docked complexes (protein-ABA) in .pdb format are provided as the input, while the simulation outputs were analysed in terms of the RMSD plots. The effectiveness of the binding of ABA with its respective receptors was examined by studying the RMSD plots to compare the protein before docking with the protein-ligand complex obtained after docking. PyMoL (Python Molecular Viewer) [23] was used for visualization and to assess the changes in RMSD.
2.3. Structure Prediction Studies for Os PP2Cs
To examine the interaction of the best docked complex with PP2C, the protein sequences of PP2C were initially identified via a thorough literature survey [24, 25]. Seven PP2C sequences from Oryza sativa were identified and considered for structure prediction studies and the protein sequences for all seven PP2Cs (PP2C09- Os01t0552300-01, PP2C10- Os01t0583100-01, PP2C12- Os01t0656200-01, PP2C48- Os03t0268600-01, PP2C76- Os05t0457200-01, PP2C79- Os05t0572700-02 and PP2C108- Os09t0325700-01) were retrieved from RAP-DB in FASTA format. The prediction of the structures was then performed using RaptorX online tool [11]. The modelled structures were obtained in .pdb format and these were validated using Ramachandran plot from the SAVES 5.0 server, as mentioned above in section 2.1. The validated structures were then utilized for protein-protein docking studies.
2.4. Protein-Protein Docking and Analysis
To further comprehend the interactions of the best docked complex with PP2C, protein-protein docking was performed. The seven modelled structures of PP2C were docked against two sets- the docked complex of ABA-PYL and with PYL receptor alone to determine the change in interactions before the binding of ABA and after its binding to PYL. Protein-protein docking was carried out using Hex 8.0.0 [26], which is known to have very good computational efficiency when compared to other protein-protein docking tools due to its use of spherical polar Fourier correlations [27] to speed up the calculations. Hex also takes a lesser amount of time to run the docking using the default geometric scoring functions [28]. The best conformations were then chosen based on the predicted binding energy. Furthermore, to analyze the interactions between the docked complexes, PP-check online server was used [29]. This webserver is mainly used for quantifying the strength of a protein-protein interface, to predict the hotspots and distinguish plausible native conformations from non-native ones as attained from protein-protein docking studies [30]. The subsequent outcomes obtained were further examined.
2.5. Mutagenesis Study of Os PYL Receptors
To examine how a mutation in PYLs affects its interaction with ABA, a mutagenesis study of Oryza sativa PYL receptors was performed. Therefore, to predict the effects of mutations on the stability of the proteins, an online tool called SDM (Site Directed Mutator) was used [31]. The SDM server offers a rapid and precise method for evaluating the effect that a specific mutation can have on the structure and stability of a protein. The tool provides a 3-dimensional view of the mutant and wild-type residues and is a beneficial tool for the detection of plausible deleterious SNPs at the genome level [32, 33].
Thus, the resulting suitable mutations were identified. Every residue with hydrogen bond interaction was replaced with nineteen other amino acids to determine the stability of interactions. The most stable mutation was selected and induced. Moreover, the changes in the proteins were created using PyMoL as the sequence editor. The .pdb files of mutated PYLs were retrieved and the edited PYLs of Oryza sativa were docked using AutoDock 1.5.6 with ABA molecule, and subsequently, its binding energy and interactions obtained were further analysed. The best conformations for each docked structure were noted based on the binding energies. Depending on the outcomes obtained, molecular docking of the mutated PYL11-ABA with all seven PP2Cs was carried out and the corresponding results obtained were analyzed further for interactions in the PP-check server. A comparative study between the binding energies of OsPyL-ABA complex and (OsPyL-ABA)-PP2C complex with the original and mutated forms was performed and the outcomes analysed.
2.6. Mapping Amino Acids of Interest to its Respective Genomic Coordinates
To understand the change in the amino acids at the genomic level, mapping the amino acids of interest to its respective genomic coordinates was carried out for the complex which showed a better binding energy when mutated. For this purpose, Rice SNP-Seek database [34] was used. The SNP-Seek database consists of identified 20 million rice SNPs that offer easy and fast retrieval of the SNP alleles for different rice varieties. The database also permits appropriate querying and visualization of the detected SNPs [35]. Therefore, rice_Rp and 3k databases were set as parameters for identifying the SNPs in the database. Furthermore, the gene locus IDs were provided as the inputs (in this case, PYL11 gene ID was used, since it showed the best binding energy when mutated). The search results thus obtained were downloaded in .csv format and the outcomes were analysed thoroughly to find the change in the amino acids in the particular genome after mutation.
3. RESULTS
3.1. Phylogenetic Analysis and Docking Studies of ABA with Os PYLs
Twelve receptors of ABA in rice were successfully retrieved from the RAPD-DB and from phylogenetic analysis, it was observed that the protein sequences of the receptors PYL1-3 belonged to one clade, PYL4-6 belonged to another clade, PYL7-12 to another and all these were closely associated with each other (Fig. 1). Furthermore, the structures were successfully modelled using RaptorX and from the Ramachandran plots, it was observed that all structures were found to be stable. Molecular docking studies showed that all docked complexes displayed negative binding energy, indicating a good binding between the two complexes (Figs. 2A-F, 3A-F, Table 1). Furthermore, among the twelve docked complexes obtained, PYL6-ABA docked complex showed the best binding energy of -8.15 kcal/mol, implying that this structure docked well with ABA.
Fig. (1).

Phylogenetic tree of 12 OsPYL receptors of Oryza sativa, developed using MEGA-X. Three clades were noted in the evolutionary study. PYL 1, 2, 3 is represented in blue, which belongs to one clade, PYL 4, 5, 6 is represented in green which is another clade, and PYL 7-12 is depicted in red, which represents another clade. It was observed that the PYLs from each clade were closely related (A higher resolution / colour version of this figure is available in the electronic copy of the article).
Fig. (2).

2D docked complexes of PYL receptors with ABA molecule, showing the amino acid interactions between proteins and ligand, as viewed in LigPlot +. (A) PYL1-ABA complex (binding energy -6.62 kcal/mol). (B) PYL2-ABA complex (-7.02 kcal/mol). (C) PYL3-ABA complex (binding energy -7.66 kcal/mol). (D). PYL4-ABA complex (binding energy -5.71 kcal/mol). (E) PYL5-ABA (binding energy -7.25 kcal/mol). (F) PYL6-ABA complex (binding energy -8.15 kcal/mol). PYL6 had the best binding energy prior to inducing mutation (A higher resolution / colour version of this figure is available in the electronic copy of the article).
Furthermore, simulation studies demonstrated that all twelve docked complexes showed a change in the RMSD value after docking (Fig. 4, Table 1). The docked complex PYL8-ABA showed the highest RMSD value of 16 Å, PYL4 and PYL9 showed an RMSD of 14 Å, PYL10 of 12 Å, and that of PYL11 was 10.5 Å. When the best conformations of each docking were subjected to molecular dynamic simulation study to analyse the molecular mechanisms involved in the protein-ligand interactions, the amino acid residues causing Vanderwaals interactions, hydrogen bond interactions, alkyl and Pi-alkyl interactions as well as the Pi-sigma bonds, which all play a vital role in the protein-ligand stability observed during docking were revealed (Table 2). For PYL11, it was noted that residues Arg99, Lys80, Val101, Val103, Leu137, and Ser112 showed vanderwaals interactions; Asn187, Glu114 and Ala109 displayed hydrogen bond associations and Val183, Phe82, Leu107, His135, Phe130 and Tyr140 were involved in Alkyl+Pi-Alkyl interactions. No Pi-sigma bonds were observed for PYL11.
Fig. (4).

Protein Root Mean Square Deviation (RMSD) values for all twelve docked complexes after molecular dynamic simulation studies. The x-axis shows the number of frames (ns), while the y-axis depicts the RMSD values (Å). It was noted that the RMSD peaks reached a constancy towards the end indicating the conformation stability and an appropriate simulation for all docked complexes. The protein RMSD values for all evidenced that the docked complexes were stable and could therefore be used for further studies (A higher resolution / colour version of this figure is available in the electronic copy of the article).
3.2. Structure Prediction of Os PP2Cs and Protein-Protein Docking
Since the results were not conclusive for the structures that were predicted for OsPP2Cs, a simulation study aided in obtaining the structures of OsPP2Cs. Protein-protein docking between PYL-ABA and PP2C showed significantly better scores when compared to docking PP2C with PYL alone. This was observed mainly as a result of changes in the energy values of hydrogen bonding, stabilizing energy, hydrophobic interactions, Vanderwaal pairs, and salt bridges. The total stabilizing (-358.39 KJ/mol) and hydrogen bond energy (-140.69 KJ/mol) for PYL4 docked with the OsPP2Cs was found to be higher than other PYL receptors (Table 3). Additionally, the total stabilizing energy (-410.82 KJ/mol) and hydrogen bond energy (-118.72 KJ/mol) were found to be highest when PYL4-ABA complex was docked with PP2C10, PP2C12 and PP2C48. It was also noted that the total stabilizing and hydrogen bond energies for all docked complexes of PYL11-ABA with PP2Cs were found to have consistent values. For receptor PYL11, docking with ABA and with ABA-PP2C complexes seemed to produce consistent values for hydrogen bond energy and total stabilizing energy (Table 4).
Table 3.
Hydrogen bond and total stabilizing energies of all OsPyl receptors docked against OsPP2Cs.
| Type of Pyl-ABA complex | OsPP2Cs | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PP2C79 | PP2C76 | PP2C9 | PP2C108 | PP2C10 | PP2C12 | PP2C48 | |||||||||
| Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | ||
| Pyl1 | -39.56 | -11.03 | -39.56 | -11.03 | -39.56 | -11.03 | -39.56 | -11.03 | -39.56 | -11.03 | -39.56 | -11.03 | -39.56 | -11.03 | |
| Pyl2 | -25.42 | -37.95 | -25.42 | -37.95 | -25.42 | -37.95 | -25.42 | -37.95 | -25.42 | -37.95 | -25.42 | -37.95 | -25.42 | -37.95 | |
| Pyl3 | 0 | 201.44 | 0 | 201.44 | 0 | 201.44 | 0 | 201.44 | -25.42 | -37.95 | 0 | 201.44 | 0 | 201.44 | |
| Pyl4 | -140.69 | -358.39 | -140.69 | -358.39 | -140.69 | -358.39 | -140.69 | -358.39 | -140.69 | -358.39 | -140.69 | -358.39 | -140.69 | -358.39 | |
| Pyl5 | 0 | 25.31 | 0 | 25.31 | 0 | 25.31 | 0 | 25.31 | 0 | 25.31 | 0 | 25.31 | 0 | 25.31 | |
| Pyl6 | 0 | 21.55 | 0 | 21.55 | 0 | 21.55 | 0 | 21.55 | 0 | 21.55 | 0 | 21.55 | 0 | 21.55 | |
| Pyl7 | -14.08 | 8.78 | -14.08 | 8.78 | -14.08 | 8.78 | -14.08 | 8.78 | -14.08 | 8.78 | -14.08 | 8.78 | -14.08 | 8.78 | |
| Pyl8 | -27.82 | 37.82 | -27.82 | 37.82 | -27.82 | 37.8 | -27.82 | 37.82 | -27.82 | 37.82 | -27.82 | 37.82 | -27.82 | 37.82 | |
| Pyl9 | -29.93 | 20.95 | -29.93 | 20.95 | -29.93 | 20.95 | -29.93 | 20.95 | -29.93 | 20.95 | -29.93 | 20.95 | -29.93 | 20.95 | |
| Pyl10 | -5.04 | 27.57 | -5.04 | 27.57 | -5.04 | 27.57 | -5.04 | 27.57 | -5.04 | 27.57 | -5.04 | 27.57 | -5.04 | 27.57 | |
| Pyl11 | -38.06 | 33.68 | -38.06 | 33.68 | -38.06 | 33.68 | -38.06 | 33.68 | -38.06 | 33.68 | -38.06 | 33.68 | -38.06 | 33.68 | |
| Pyl12 | -95.56 | -215.41 | -95.56 | -215.41 | -95.56 | -215.41 | -95.56 | -215.41 | -95.56 | -215.41 | -95.56 | -215.41 | -95.56 | -215.41 | |
Table 4.
Hydrogen bond and total stabilizing energies of all OsPyl-ABA complexes docked against OsPP2Cs.
| Type of Pyl-ABA Complex | OsPP2Cs | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PP2C79 | PP2C76 | PP2C9 | PP2C108 | PP2C10 | PP2C12 | PP2C48 | |||||||||
| Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | Hydrogen Bond Energy (kJ/mol) | Total Stabilizing Energy (kJ/mol) | ||
| Pyl1-ABA | 0 | -107.62 | 0 | -44.95 | 0 | -30.02 | 0 | 25.42 | -39.56 | -11.03 | -39.56 | -11.03 | -39.56 | -11.03 | |
| Pyl2-ABA | 0 | -4.66 | 0 | -13.36 | 0 | -25.05 | NA | NA | -11.04 | -54.68 | -11.04 | -54.68 | -11.04 | -54.68 | |
| Pyl3-ABA | 0 | -4.91 | NA | NA | NA | NA | 0 | -2.64 | -22.06 | -23.6 | -22.06 | -23.73 | -22.06 | -23.73 | |
| Pyl4-ABA | NA | NA | NA | NA | 0 | -1.29 | 0 | -26.98 | -118.72 | -410.82 | -118.72 | -410.82 | -118.72 | -410.82 | |
| Pyl5-ABA | 0 | -64.93 | NA | NA | NA | NA | 0 | 174.04 | 0 | 134.9 | 0 | 131.76 | 0 | 134.22 | |
| Pyl6-ABA | 0 | -19.36 | NA | NA | NA | NA | 0 | 118.73 | 0 | 87.05 | 0 | 87.05 | 0 | 87.05 | |
| Pyl7-ABA | NA | NA | 0 | -0.6 | 0 | 15.43 | 0 | -35.13 | -2.63 | 70.37 | 0 | 72.96 | 0 | 71.82 | |
| Pyl8-ABA | 0 | -0.77 | 0 | -4.15 | NA | NA | 0 | -12 | -41.57 | -38.64 | -41.57 | -38.64 | -41.57 | -38.64 | |
| Pyl9-ABA | NA | NA | 0 | -35.26 | 0 | -4.45 | 0 | -18.94 | -12.84 | -6.12 | -12.84 | -5.68 | -12.84 | -5.68 | |
| Pyl10-ABA | 0 | -0.04 | 0 | -35.26 | 0 | -15.24 | NA | NA | 0 | 117 | 0 | 117 | 0 | 117 | |
| Pyl11-ABA | -25.84 | -22.69 | -25.84 | -22.69 | 0 | -22.69 | -25.84 | -22.69 | -25.84 | -22.69 | -25.84 | -22.69 | -25.84 | -22.69 | |
| Pyl12-ABA | 0 | -0.03 | 0 | -25.96 | 0 | -22.68 | NA | NA | 0 | 76.65 | 0 | 76.7 | 0 | 75.03 | |
NA- Not applicable (no interactions observed).
3.3. Mutagenesis Study of Os PYL Receptors
When the mutagenesis study was carried out by altering the amino acids in the receptor molecules and then utilizing the mutated receptors for docking, the binding energy showed a considerable difference before and after mutation. Interestingly, PYL11-ABA showed a better binding score of -8.17 kcal/mol after mutating Ala109 to Ile109 when compared to the docked scores of other complexes (Table 5). The predicted delta delta G (DDG) value, after inducing the mutation was found to be 1.23 and an increased stability of the protein was observed for the same. A dramatic change in the binding energy after mutation induction was observed for all 7 PP2C molecules docked to OsPYL11-ABA. PYL11-ABA-PP2C48 post mutation showed a binding energy of -20.41 kcal/mol, higher than other PYLs (Table 6). Furthermore, a change in amino acid interactions was also noted between PYL-ABA post mutation induction. The PYL11-ABA interaction at the residual level suggests that the mutation induced leads to more hydrophobic and h-bond interactions. Prior to mutation, the residues involved in h-bond interactions were Lys 80, Ser 112, and Glu 114. Post mutation of Ala 109 to Ile 109, h-bond interactions were observed with residues His 135, Arg 136, Tyr 140, and Glu 187.
Table 5.
Binding energies of OsPYLs against ABA before and after mutation.
| OsPYLs | Binding Energy (Before Mutation) kcal/mol | Binding Energy (After Mutation) kcal/mol |
|---|---|---|
| pyl1 | -6.62 | -5.8 |
| pyl2 | -7.02 | -6.07 |
| pyl3 | -7.66 | -6.77 |
| pyl4 | -5.71 | -5.74 |
| pyl5 | -7.25 | -7.17 |
| pyl6 | -8.15 | - |
| pyl7 | -5.72 | - |
| pyl8 | -6.26 | - |
| pyl9 | -6.71 | -6.42 |
| pyl10 | -7.05 | -6.97 |
| pyl11 | -6.09 | -8.17 |
| pyl12 | -5.88 | -5.46 |
Table 6.
Binding energies and hydrogen bond energies of PYL11-ABA complex docked against all seven PP2C molecules before and after mutagenesis study.
| OsPP2Cs | H-bond (After Mutation) | Binding Energy (After Mutation) kcal/mol | Binding Energy (Before Mutation) kcal/mol |
|---|---|---|---|
| PP2C9 | 0 | -7.57 | -22.69 |
| PP2C10 | - | - | -22.69 |
| PP2C12 | 0 | -7.57 | -22.69 |
| PP2C48 | 0 | -20.41 | -22.69 |
| PP2C76 | 0 | 9.23 | -22.69 |
| PP2C79 | -28.56 | -2.45 | -22.69 |
| PP2C108 | -28.56 | -2.45 | -22.69 |
3.4. Mapping Amino Acids of Interest to its Respective Genomic Coordinates
When the amino acids of interest Glu114 and Ala109 were mapped to their respective genomic coordinates, it was observed that no naturally known SNPs were found which were close to the two amino acids of interest.
4. DISCUSSION
The present research analysed the phylogeny of the twelve PYL receptors, with a focus only on Oryza sativa. Previous studies, however, have been carried out on ABA receptors in a cotton variety called Gossypium to identify the possible number of receptors involved and the expression analysis of the same. The study constructed a phylogenetic tree using the PYL protein sequences from four cotton species, rice, and Arabidopsis, and their outcomes demonstrated that PYLs of Gossypium clustered closely with cocoa PYLs than with PYLs in Oryza sativa [36]. Another recent study identified ten PYL receptors in Glycyrrhiza and the evolutionary study showed that the receptors clustered together within the same family and subfamily [37]. Moreover, a study carried out in 2012 involved reviewing the structural properties of ABA receptors and PP2Cs [38]. The study showed that the docked complexes of ABA-PYL had hydrogen bond interactions with Glu94, Glu141, Ser122, and Tyr120. Despite there being several similar studies, the present study differs slightly in the outcomes obtained and the approach followed. In the present study, molecular docking was studied between Os, PYLs, and ABA, and it was observed that PYL6-ABA docking complex showed better binding by displaying the highest negative docking score among the rest. The amino acid interactions between the docked complexes were found to be with aspartic acid, glutamine, lysine, and arginine. This implies that there is a good interaction between the PYL receptors with the ABA molecules, which can in turn induce ABA response in plants.
Furthermore, recently, an in silico study by Gupta et al., 2020, was carried out on the ABA receptors in the rice variety N22 and compared with receptors involved in Arabidopsis thaliana. The study involved analysing the molecular dynamics between OsPYL2-ABA and OsPYL3-ABA complexes for 25ns to determine the time frame. The present study involved studying the molecule dynamic properties of all docked complexes using 100 ns simulation studies that ensured better accuracy and precision. Since the docked complexes displayed good stability throughout the simulation, it implied that the poses generated while molecular docking was intact and the binding between the ligand and protein was robust [39].
Additionally, hydrogen bonds play a noteworthy role in ligand binding. It is essential to take into account the hydrogen bonding attributes in drug design due to their firm influence on the drug specificity, adsorption, and metabolization. These H-bonds between a ligand and a protein can further be of four subtypes: backbone donor and acceptor, side chain donor, and acceptor. The hydrophobic contacts are subtypes p-p, p-cation, non-specific, and other interactions. Typically, these interactions include an aromatic/aliphatic ligand group and a hydrophobic amino acid. Therefore, modifications in the amino acid interactions via hydrogen and hydrophobic contacts in the PYL receptors will further aid in binding of the ABA molecule during adverse situations, which in turn will help the plants in gaining tolerance. Previously, an in vitro study on rice revealed that by creating mutations in the ABA receptors of rice, i.e., PYL, we showed better binding with the molecules ABA and PP2C molecules [40]. More recently, a study utilized CRISPR/cas9 technology to mutate PYL genes of Oryza sativa to promote its productivity and growth [41]. Therefore, the results obtained in the current study can be substantiated by previous research, where we induced mutations in the PYL receptors of rice and further docked against the complexes (ABA and PP2C), which revealed a better binding score, especially in the PYL11 receptor molecule.
Furthermore, by mapping amino acids of interest to their respective genomic coordinates, an understanding of their interactions was revealed. Therefore, when no naturally occurring SNPs were found when mapped, it implies that the in silico induced mutation resulted in a more stable interaction between PYL11 and ABA. Previously, a study where molecular mapping of the genes that confer aluminium resistance to Oryza sativa has been carried out where certain DNA markers were identified which are important for understanding the genes which may contribute to the improvement of rice productivity [42]. Furthermore, another study performed a large-scale detection and mapping of SNPs in Arabidopsis thaliana, and the study noted that a considerable portion of the SNPs caused changes in the encoded amino acids [42]. Albeit these evidence relate to the present research, the current study presents interesting outcomes as demonstrated via comprehensive interaction analysis.
CONCLUSION
Since currently, there is a paucity in the existing knowledge of abscisic acids and their mechanism of stress tolerance in rice; the present study focused on finding the ABA receptors in Oryza sativa and to detect the PYL receptor that binds best with abscisic acid. Protein-ligand docking revealed PYL6-ABA with the best binding energy and the 100ns simulation analysis further revealed the docked structure stability. Docking among PYL11-ABA-PP2C79 was found to be the best structure allowing further mutagenesis studies to be performed. The mutagenesis study on the PYL receptors suggested that mutated PYL11 showed better binding than non-mutated PYL11. The absence of any SNPs when amino acids of interest were mapped to their respective genomic coordinates indicated that there were no naturally occurring mutant varieties of rice. Thus, the current study elucidates the interactions of abscisic acid with important receptors in Oryza sativa and allows for a clearer comprehension of the stress tolerance mechanism in rice. Furthermore, the outcomes presented in the study demonstrate the direct impact of ABA on rice and its productivity. Using advanced recombinant DNA technology such as CRISPR, inducing mutations in the PYL receptors will in turn, aid in ABA signalling and is prospective future work. Additionally, the results presented in the current study act as preliminary data in furthering our understanding of ABA signalling, which helps in tackling abiotic stress conditions better.
Fig. (3).

2D docked complexes of PYL receptors with ABA molecule, showing the amino acid interactions between proteins and ligand, as viewed in LigPlot +. (A) PYL7-ABA complex (binding energy -5.72 kcal/mol). (B) PYL8-ABA complex (binding energy -6.26 kcal/mol). (C) PYL9-ABA complex (binding energy -6.71 kcal/mol). (D) PYL10-ABA complex (binding energy -7.05 kcal/mol). (E) PYL11-ABA complex (binding energy -6.09 kcal/mol). (F) PYL12-ABA complex (binding energy -5.88 kcal/mol) (A higher resolution / colour version of this figure is available in the electronic copy of the article).
Table 1.
Docking scores of the twelve PYL receptors with abscisic acid (ABA) in Oryza sativa and RMSD values obtained for the docked complexes of PYLs with ABA after docking studies.
| OsPYLs | Binding Energy After Docking (kcal/mol) | RMSD: C Alpha |
|---|---|---|
| pyl1 | -6.62 | 4.2 |
| pyl2 | -7.02 | 4.2 |
| pyl3 | -7.66 | 4.4 |
| pyl4 | -5.71 | 14 |
| pyl5 | -7.25 | 3.5 |
| pyl6 | -8.15 | 6.4 |
| pyl7 | -5.72 | 8 |
| pyl8 | -6.26 | 16 |
| pyl9 | -6.71 | 14 |
| pyl10 | -7.05 | 12 |
| pyl11 | -6.09 | 10.5 |
| pyl12 | -5.88 | 5.6 |
Table 2.
List of Protein-Ligand interactions showing Vanderwaals interactions, Hydrogen bonds, Alkyl+Pi-Alkyl and Pi-sigma bonds between the twelve PYLs and the ABA molecule obtained in AutoDock.
| Interactions (PYL-ABA) | Docking Score | Vander-Waals | Conventional-H Bond | Alkyl+Pi-Alkyl | Pi-Sigma |
|---|---|---|---|---|---|
| pyl1 | -6.62 | Glu112, Phe174, Pro106, Val178, His71, Lys70 | Asn182, Phe72, Ala107 | Val99, Leu105, Val101, Ile73, Leu135 | - |
| pyl2 | -7.02 | Val77, Arg94, Phe123, Ser107, Ala104 | Glu109, Lys74, His130, Leu132, Arg131 | Val185, Phe180, Val125, Val184, Tyr135 | - |
| pyl3 | -7.66 | Pro75, Phe128, Ala109, Val130, Val186, Phe81 | Arg99, Lys79, Glu114, Ser112, His135, Leu137 | Val103, Val101, Ile82, | Tyr140 |
| pyl4 | -5.71 | Val147, Phe174, Asp231, Phe127, Ser158 | Lys125, Glu160, Arg182 | Ile128, Ile176, His181, Leu183, Phe223, Ile227, Val149 | - |
| pyl5 | -7.25 | Val113, Ile194, Phe89, Leu149, Val176 | Asn198, Lys87, Glu126, Ser124 | Ile90, Val115, Phe140, Ala121, His147, Val142, Tyr152 | - |
| pyl6 | -8.15 | Arg158, Ile205, Val202, Tyr162, Val152, Ser134, Glu136 | - | Pro130, Leu129, Phe201, Leu159, His157, Ala131, Val125 | - |
| pyl7 | -5.72 | Gly137, Asn180, Lys85, Pro86, Arg 115, Ala113, Ser116 | Glu118 | Val107, Val88, Ile105, Phe87, Leu176 | Phe134 |
| pyl8 | -6.26 | Glu118, Asn191, Leu188, Ala113, Val107, Arg115, Ser116, Lys84 | - | Leu141, Tyr191, Ile184, Val187, Leu111, phe183 | Phe134, His139 |
| pyl9 | -6.71 | Leu188, Ile184, Ser116, Pro112, Val107 | Lys84, Asn191, Ala113 | Phe86, Leu111, Phe183, Leu141, Phe134, Val187 | - |
| pyl10 | -7.05 | Phe78, Asn182, Leu178, Leu132, Arg131, Phe174, Ser107, Asn97, Val79, Arg94 | Lys76, Glu109 | Phe125, His130, Leu102, Ala104, Val98, Val96 | - |
| pyl11 | -6.09 | Arg99, Lys80, Val101, Val103, Leu137, Ser112 | Asn187, Glu114, Ala109 | Val183, Phe82, Leu107, His135, Phe130, Try140 | - |
| pyl12 | -5.88 | Phe148, Pro137, His104, Ala78 | Arg105, Thr145 | Phe76, Pro77, Val149, Leu106 | - |
ACKNOWLEDGEMENTS
We would like to acknowledge Dr. Shobha G, Professor, Department of Computer Science and Engineering, RV College of Engineering, Bangalore, for providing us with QuADro GV100 GPU for performing computational analysis.
LIST OF ABBREVIATIONS
- ABA
Abscisic Acid
- MEGA
Molecular Evolutionary Genetics Analysis
- Mut
Mutated
- NVT
Normal Volume Temperature
- NPT
Normal Pressure Temperature
- OsPYLs
Oryza sativa Pyrabactin resistance 1 proteins
- PP2C
Type 2C Protein Phosphatase
- PropKA
Protein pKa values
- PYL
Pyrabactin resistance 1
- SNP
Single Nucleotide Polymorphism
- SnRK2
positive Regulator class III SNF-1-related protein kinase 2
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The authors confirm that the data supporting the findings of this study are available within the article.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Nakashima K., Yamaguchi-Shinozaki K. ABA signaling in stress-response and seed development. Plant Cell Rep. 2013;32(7):959–970. doi: 10.1007/s00299-013-1418-1. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J., Jia W., Yang J., Ismail A.M. Role of ABA in integrating plant responses to drought and salt stresses. Field Crops Res. 2006;97(1):111–119. doi: 10.1016/j.fcr.2005.08.018. [DOI] [Google Scholar]
- 3.Sah S.K., Reddy K.R., Li J. Abscisic acid and abiotic stress tolerance in crop plants. Front. Plant Sci. 2016;7:571. doi: 10.3389/fpls.2016.00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duarte K.E., de Souza W.R., Santiago T.R., Sampaio B.L., Ribeiro A.P., Cotta M.G., da Cunha B.A.D.B., Marraccini P.R.R., Kobayashi A.K., Molinari H.B.C. Identification and characterization of core abscisic acid (ABA) signaling components and their gene expression profile in response to abiotic stresses in Setaria viridis. Sci. Rep. 2019;9(1):4028. doi: 10.1038/s41598-019-40623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma Y., Szostkiewicz I., Korte A., Moes D., Yang Y., Christmann A., Grill E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science. 2009;324(5930):1064–1068. doi: 10.1126/science.1172408. [DOI] [PubMed] [Google Scholar]
- 6.Komatsu K., Nishikawa Y., Ohtsuka T., Taji T., Quatrano R.S., Tanaka S., Sakata Y. Functional analyses of the ABI1-related protein phosphatase type 2C reveal evolutionarily conserved regulation of abscisic acid signaling between Arabidopsis and the moss Physcomitrella patens. Plant Mol. Biol. 2009;70(3):327–340. doi: 10.1007/s11103-009-9476-z. [DOI] [PubMed] [Google Scholar]
- 7.Fujii H., Verslues P.E., Zhu J.K. Identification of two protein kinases required for abscisic acid regulation of seed germination, root growth, and gene expression in Arabidopsis. Plant Cell. 2007;19(2):485–494. doi: 10.1105/tpc.106.048538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirayama T., Umezawa T. The PP2C-SnRK2 complex: The central regulator of an abscisic acid signaling pathway. Plant Signal. Behav. 2010;5(2):160–163. doi: 10.4161/psb.5.2.10460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He Y., Hao Q., Li W., Yan C., Yan N., Yin P. Identification and characterization of ABA receptors in Oryza sativa. PLoS One. 2014;9(4):e95246. doi: 10.1371/journal.pone.0095246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ben-Ari G. The ABA signal transduction mechanism in commercial crops: Learning from Arabidopsis. Plant Cell Rep. 2012;31(8):1357–1369. doi: 10.1007/s00299-012-1292-2. [DOI] [PubMed] [Google Scholar]
- 11.Peng J., Xu J. RaptorX: Exploiting structure information for protein alignment by statistical inference. Proteins. 2011;79(S10) Suppl. 10:161–171. doi: 10.1002/prot.23175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hooft R.W.W., Sander C., Vriend G. Objectively judging the quality of a protein structure from a Ramachandran plot. Comput. Appl. Biosci. 1997;13(4):425–430. doi: 10.1093/bioinformatics/13.4.425. [DOI] [PubMed] [Google Scholar]
- 13.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26(2):283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 14.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J.D., Higgins D.G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7(1):539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saitou N., Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 16.Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodsell DS. Computational docking of biomolecular complexes with AutoDock. Spring Harb. Protoc. 2009;2009(5):pdb-rot5200. doi: 10.1101/pdb.prot5200. [DOI] [PubMed] [Google Scholar]
- 19. Biovia, DS Discovery Studio Visualizer; Available at: https://discover.3ds.com/discovery-studio-visualizer-download .
- 20.Bowers K.J., Chow D.E., Xu H., Dror R.O., Eastwood M.P., Gregersen B.A., Klepeis J.L., Kolossvary I., Moraes M.A., Sacerdoti F.D., Salmon J.K. Scalable algorithms for molecular dynamics simulations on commodity clusters.; In: SC'06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; 2006 November 11-17; Tampa, FL, USA. p. 43. [DOI] [Google Scholar]
- 21.Uttarkar A., Niranjan V. Re-profiling of natural inhibitor via combinatorial drug screening: Brefeldin A variant design as an effective antagonist leading to EPAC2 structure modification and antibody design for identification. bioRxiv. 2021 doi: 10.1101/2021.03.31.437986. [DOI] [Google Scholar]
- 22.Cohen F.E., Sternberg M.J.E. On the prediction of protein structure: The significance of the root-mean-square deviation. J. Mol. Biol. 1980;138(2):321–333. doi: 10.1016/0022-2836(80)90289-2. [DOI] [PubMed] [Google Scholar]
- 23.PyMOL. The PyMOL molecular graphics system, version 2.0 Schrödinger LL. . Available from: https://pymol.org/2/support.html? [Google Scholar]
- 24.Singh A., Giri J., Kapoor S., Tyagi A.K., Pandey G.K. Protein phosphatase complement in rice: Genome-wide identification and transcriptional analysis under abiotic stress conditions and reproductive development. BMC Genomics. 2010;11(1):435. doi: 10.1186/1471-2164-11-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian X., Wang Z., Li X., Lv T., Liu H., Wang L., Niu H., Bu Q. Characterization and functional analysis of pyrabactin resistance-like abscisic acid receptor family in rice. Rice (N. Y.) 2015;8(1):28. doi: 10.1186/s12284-015-0061-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ritchie D.W., Kemp G.J. Protein docking using spherical polar Fourier correlations. Proteins. 2000;39(2):178–194. doi: 10.1002/(SICI)1097-0134(20000501)39:2<178::AID-PROT8>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 27.Ritchie D.W., Grudinin S. Spherical polar Fourier assembly of protein complexes with arbitrary point group symmetry. J. Appl. Cryst. 2016;49(1):158–167. doi: 10.1107/S1600576715022931. [DOI] [Google Scholar]
- 28.Huang S-Y. Exploring the potential of global protein-protein docking: An overview and critical assessment of current programs for automatic ab initio docking. Drug Discov. Today. 2015;20(8):969–977. doi: 10.1016/j.drudis.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Sukhwal A, Sowdhamini R. PPCheck: A webserver for the quantitative analysis of protein-protein interfaces and prediction of residue hotspots. Bioinform. Biol. Insights. 2015;9:BBI-S25928. doi: 10.4137/BBI.S25928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sukhwal A., Sowdhamini R. Oligomerisation status and evolutionary conservation of interfaces of protein structural domain superfamilies. Mol. Biosyst. 2013;9(7):1652–1661. doi: 10.1039/c3mb25484d. [DOI] [PubMed] [Google Scholar]
- 31.Pandurangan A.P., Ochoa-Montaño B., Ascher D.B., Blundell T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017;45(W1):W229–W235. doi: 10.1093/nar/gkx439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Worth C.L., Preissner R., Blundell T.L. SDM--a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39(Suppl. 2):W215-22. doi: 10.1093/nar/gkr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uttarkar A., Niranjan V., Pandit S., Subash S. Study of SARS-nCoV2 Indian isolates gaining insights into mutation frequencies, protein stability and prospective effect on pathogenicity. Coronaviruses. 2021;2:12–20. [Google Scholar]
- 34.Mansueto L., Fuentes R.R., Borja F.N., Detras J., Abriol-Santos J.M., Chebotarov D., Sanciangco M., Palis K., Copetti D., Poliakov A., Dubchak I., Solovyev V., Wing R.A., Hamilton R.S., Mauleon R., McNally K.L., Alexandrov N. Rice SNP-seek database update: New SNPs, indels, and queries. Nucleic Acids Res. 2017;45(D1):D1075–D1081. doi: 10.1093/nar/gkw1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexandrov N., Tai S., Wang W., Mansueto L., Palis K., Fuentes R.R., Ulat V.J., Chebotarov D., Zhang G., Li Z., Mauleon R., Hamilton R.S., McNally K.L. SNP-Seek database of SNPs derived from 3000 rice genomes. Nucleic Acids Res. 2015;43:D1023–D1027. doi: 10.1093/nar/gku1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang G., Lu T., Miao W., Sun L., Tian M., Wang J., Hao F. Genome-wide identification of ABA receptor PYL family and expression analysis of PYLs in response to ABA and osmotic stress in Gossypium. PeerJ. 2017;5:e4126. doi: 10.7717/peerj.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui Y.X., Xu Z.C., Chen X.L., Nie L.P., Wu L.W., Wang Y., Song J.Y., Yao H. Genome-wide identification of abscisic acid (ABA) receptor pyrabactin resistance 1-like protein (PYL) family members and expression analysis of PYL genes in response to different concentrations of ABA stress in Glycyrrhiza uralensis. Chin. J. Nat. Med. 2020;18(8):606–611. doi: 10.1016/S1875-5364(20)30072-8. [DOI] [PubMed] [Google Scholar]
- 38.Santiago J., Dupeux F., Betz K., Antoni R., Gonzalez-Guzman M., Rodriguez L., Márquez J.A., Rodriguez P.L. Structural insights into PYR/PYL/RCAR ABA receptors and PP2Cs. Plant Sci. 2012;182:3–11. doi: 10.1016/j.plantsci.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 39.Gupta M.K., Sharma V., Lenka S.K., Chinnusamy V. In silico study revealed major conserve architectures and novel features of pyrabactin binding to Oryza sativa ABA receptors compare to the Arabidopsis thaliana. J. Biomol. Struct. Dyn. 2020;38(11):3211–3224. doi: 10.1080/07391102.2019.1654922. [DOI] [PubMed] [Google Scholar]
- 40.Miao C., Xiao L., Hua K., Zou C., Zhao Y., Bressan R.A., Zhu J.K. Mutations in a subfamily of abscisic acid receptor genes promote rice growth and productivity. Proc. Natl. Acad. Sci. USA. 2018;115(23):6058–6063. doi: 10.1073/pnas.1804774115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen V.T., Burow M.D., Nguyen H.T., Le B.T., Le T.D., Paterson A.H. Molecular mapping of genes conferring aluminum tolerance in rice (Oryza sativa L.). Theor. Appl. Genet. 2001;102(6-7):1002–1010. doi: 10.1007/s001220000472. [DOI] [Google Scholar]
- 42.Schmid K.J., Sörensen T.R., Stracke R., Törjék O., Altmann T., Mitchell-Olds T., Weisshaar B. Large-scale identification and analysis of genome-wide single-nucleotide polymorphisms for mapping in Arabidopsis thaliana. Genome Res. 2003;13(6A):1250–1257. doi: 10.1101/gr.728603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article.