

Abstract
The spinal N-methyl-D-aspartate (NMDA) receptor, and particularly its NR2B subunit, plays a pivotal role in neuropathic pain. However, the role of peripheral NMDA receptor in neuropathic pain is less well understood. We first treated cultured human keratinocytes, HaCaT cells with NMDA or NR2B-specific antagonist, ifenprodil and evaluated the level of total and phosphorylated NR2B at 24 h using Western blot. Next, using the chronic post-ischemia pain (CPIP) model, we administered NMDA or ifenprodil subcutaneously into the hind paws of male rats. Nociceptive behaviors were assessed by measuring mechanical and thermal withdrawal thresholds. Expression and phosphorylation of NR2B on keratinocyte were analyzed at 6, 12, 18, and 24 h on day 1 (initiation of pain) as well as day 2, 6, 10 and 14 (development and maintenance of pain) after the ischemia. The level of peripheral sensitization-related proteins (nuclear factor-κB (NF-κB), extracellular regulated protein kinases (ERK), and interleukin-1β (IL-β)) in epidermis and dorsal root ganglion (DRG) were evaluated by immunofluorescence and western blot. Central sensitization-related C-fos induction, as well as astrocytes and microglia activation in the spinal cord dorsal horn (SDH) were studied using immunofluorescence. Administration of NMDA upregulated NR2B phosphorylation on HaCaT cells. CPIP-induced mechanical allodynia and thermal hyperalgesia were intensified by NMDA and alleviated by ifenprodil. CPIP resulted in an early upregulation of NR2B (peaked at 24 h) and late phosphorylation of NR2B (peaked at 14d) in hindpaw keratinocytes. CPIP led to an upregulation and phosphorylation of NF-κB and ERK, as well as an increased IL-1β production in the ipsilateral skin and DRG. CPIP-associated c-fos induction in SDH persisted from acute to chronic stages after ischemia, while microglia and astrocyte activation were only observed in chronic phase. These CPIP-induced changes were also suppressed by ifenprodil administered subcutaneously in the hind paw. Our findings reveal a previously unrecognized role of keratinocyte NMDA receptor subunit 2B in peripheral and central nociceptive sensitization induced by CPIP.
Keywords: Chronic post-ischemia pain, Complex regional pain syndrome, NMDA receptor subunit 2B, Keratinocyte, Peripheral sensitization, Central sensitization, Animal model
1. Introduction
Limb ischemia-reperfusion injury commonly occurs with tissue injury and tissue injury (from ischemia, fracture, immobilization, and/or peripheral nerve injury etc.) is one of the most common triggers for the development of complex regional pain syndrome (CRPS) (Bruehl, 2010; Marinus et al., 2011). Clinical treatment for CRPS is often insufficient largely due to a lack of understanding of the pathophysiology of this syndrome. A chronic post-ischemia pain (CPIP) model has been developed (Coderre et al., 2004) and widely utilized to study CRPS (Bruehl, 2010; Ragavendran et al., 2014; Tang et al., 2018). Both peripheral and central sensitization have been reported to be involved in the development of CRPS (Bruehl, 2010; Marinus et al., 2011; David Clark et al., 2018). The N-methyl-d-aspartic (NMDA) receptor plays a crucial role in central sensitization (Woolf and Thompson, 1991). Using the CPIP model, we and others have studied spinal microglial activation (central sensitization) in CRPS (Tang et al., 2018; Xu et al., 2016; Luo et al., 2016). However, the role of the NMDA receptor and peripheral sensitization is less well defined in this model.
Recent studies revealed that keratinocytes could be activated by neuropeptide and norepinephrine secreted by nerve terminals (Hou et al., 2011). Subsequent release of proinflammatory cytokines leads to nociceptive activation and peripheral sensitization (Birklein et al., 2014; Guo et al., 2012; Li et al., 2010; Li et al., 2009). Additional research indicated that selective keratinocyte stimulation is sufficient to induce noxious stimuli transmission from the peripheral to the central nervous system (Pang et al., 2015). Furthermore, glutamate NMDA receptors have been found to be expressed on keratinocytes, but the role of these receptors in nociception has not been fully defined (Fischer et al., 2004; Nahm et al., 2004; Morhenn et al., 2004).
NMDA receptor (NR) is structurally composed of four subunits: two NR1 subunits together with either two NR2 subunits or a combination of NR2 and NR3 subunits (Traynelis et al., 2010). At least four types of NR2 subunits have been discovered in the central nervous system, among which NR2B plays a significant role in neuropathic pain (Paoletti et al., 2013; Zhuo, 2009). In the present study, we tested the hypothesis that activation of NR2B on keratinocytes is essential for peripheral and central sensitization as well as the development of acute and chronic pain after ischemia–reperfusion injury. Our work reveals a previously unrecognized role of keratinocyte NR2B in peripheral and central nociceptive sensitization induced by CPIP.
2. Material and methods
2.1. Cell culture and drug administration
The human keratinocyte cell line HaCaT was a kind gift from Dr. Nora Singer (Case Western Reserve University, OH, USA). These cells were grown in 100-mm plastic tissue culture dishes (BD Labware) in a 3:1 mixture of Dulbecco’s Modified Eagle Medium (DMEM, BioWhittaker, Walkersville, MD) and Ham’s F12 (Invitrogen Life Technologies, Grand Island, NY), supplemented with 10% FBS, 1.2% L-glutamine, 1% penicillin/streptomycin, 1% nonessential amino acids (BioWhittaker), and 1% Na-pyruvate (Invitrogen Life Technologies). The HaCaT cells were passaged using 0.25% trypsin/1 mM ethylenediaminetetraacetic acid (EDTA, Invitrogen Life Technologies). Cells were treated with NMDA (Sigma-Aldrich Company, USA) (200 μM) or normal saline (NS, control) for 24 h. Cell lysate were collected for Western Blot analysis following an established protocol (Colombo et al., 2017) (see below).
2.2. The CPIP model of rats
Adult male Sprague-Dawley rats aged 6–7 weeks weighing 200–220 g were purchased from the National Institute for Food and Drug Control in China. We only used male rats because the CPIP model was developed in male rats (Coderre et al., 2004); and microglial signaling in pain is sex-dependent (Sorge et al., 2015). The animals were kept in standard conditions (3 rats/cage) under a 12-h light/dark cycle and an ambient temperature of 25 ± 3 °C, with ad libitum food and water. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Chinese Academy of Medical Sciences, Peking Union Medical College Hospital, and conformed to the guidelines set forth by the Committee for Research and Ethical Issues of the International Association for the Study of Pain (IASP).
The CPIP model was induced according to Coderre et al. (2004); and rats were anesthetized with an intraperitoneal injection of sodium pentobarbital 60 mg/kg, followed by 20 mg/kg every 60 min during the 3 h ischemic period, depending on the response to anesthesia in each animal. A Nitrile 70 Durometer O- ring (Kangda Chemical Company, China), whose internal diameter was 0.56 cm, was placed tightly around the rat’s right (ipsilateral) hindlimb just proximal to its medial malleolus. The O-ring was then left on the limb for 3 h and cut before the rat emerged from anesthesia to allow blood reperfusion. Sham rats received the same procedures, except that O-rings were cut and only loosely surrounded their ankle joints.
2.3. Animal study design and drug administration
Rats were randomly assigned to the CPIP and sham groups. The CPIP group was further divided into three treatment groups (9–10 rats/group): NMDA 1 mM 100 μl, NR2B-specific antagonist ifenprodil (IFEN, Sigma-Aldrich Company, USA) 0.1 mM 100 μl, and NS 100 μl. Drugs were subcutaneously injected into the ventral side of the rat’s hind paw on the side subjected to ischemia–reperfusion injury.
Each treatment group was further divided into acute and chronic pain phases after ischemia (Klafke et al., 2016). In the acute pain groups, rats received drugs for 3 consecutive days before ischemia and reperfusion injury (CPIP modeling). Behavior assessment was performed before drug administration (baseline), before modeling and every 6 h for 24 h. Ipsilateral hind paw plantar skin, dorsal root ganglion (DRG) and spinal dorsal horn (SDH) (L4–6) were harvested at 24 h (Fig. 1A). Animals in the chronic pain groups were given drugs once daily from day 8 to day 14 after modeling. Behavior assessment was conducted before modeling (baseline) and every 4 days from day 2 to day 14. Tissues were harvested on day 14 (Fig. 1B).
Fig. 1.
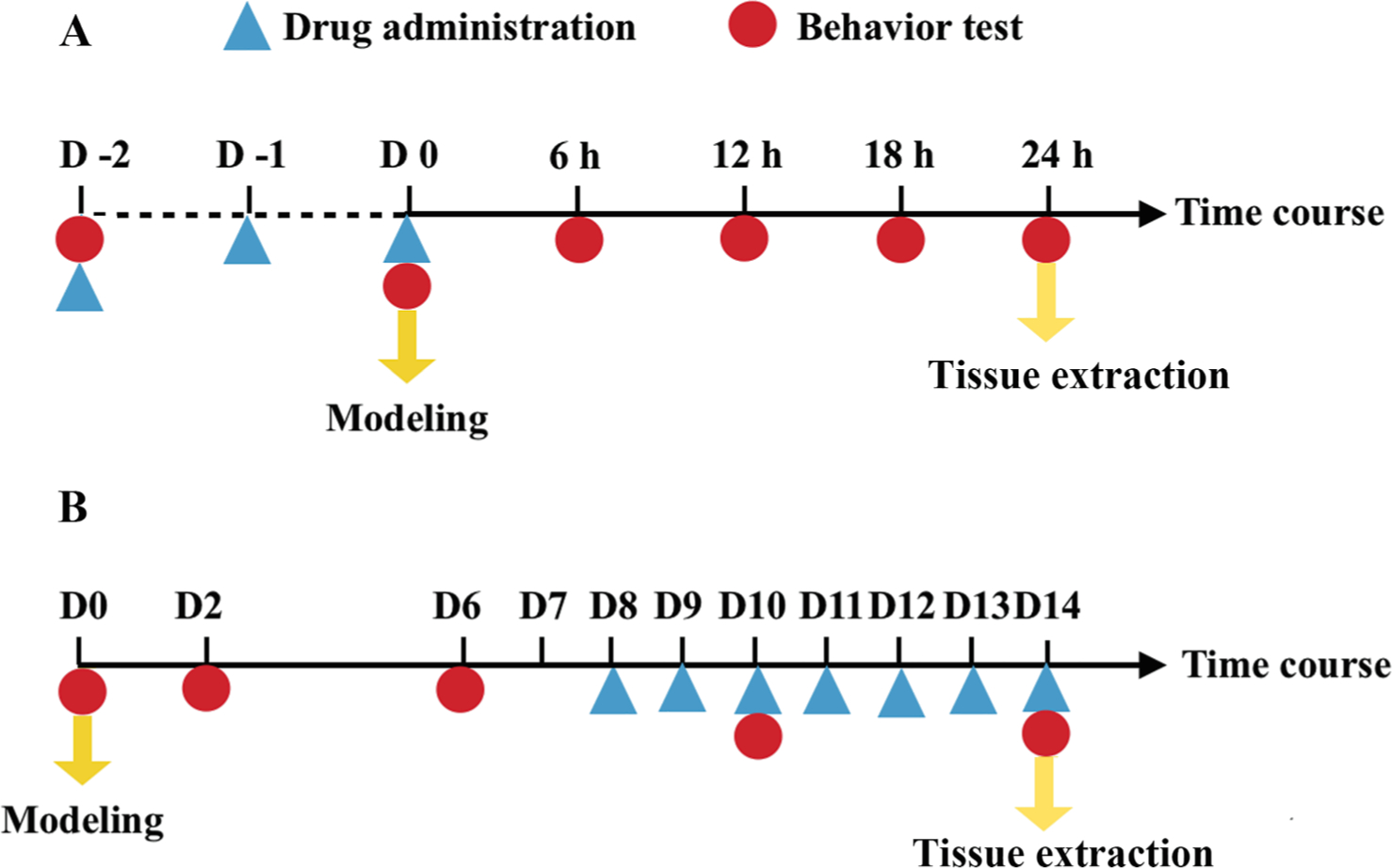
Timeline. In acute groups (A), rats received drugs for three consecutive days before modeling (CPIP induction). Behavior tests were performed before drug administration (baseline), before modeling and every 6 h after modeling till the 24th hour when ipsilateral hindpaw plantar skin, L4-6 SDH and DRGs were harvested. Rats in chronic groups (B) were given drugs once daily from day 8 to 14. Behavioral tests were conducted before modeling (baseline) and every 4 days from day 2 to 14. On day 14, Tissue samples were collected. Abbreviations: SDH, spinal dorsal horn; DRG, dorsal root ganglion
To investigate the change in NR2B expression in the epidermis during CPIP progression, we also built a CPIP model in another group of rats (n = 40). Five of them were sacrificed for ipsilateral hind paw plantar skin collection every 6 h on day 1 and every 4 days from day 2 to 14.
2.4. Behavioral testing
To evaluate nociceptive behavior, hind paw mechanical allodynia and thermal hyperalgesia were tested. Rats were acclimated to the measurement environment for 1 h before behavioral tests. Behavioral measurements were carried out by an investigator blinded to the experimental design and drug administration.
2.4.1. Hindpaw mechanical allodynia
Hindpaw mechanical allodynia was evaluated by the up-down method using a calibrated electronic von Frey apparatus (IITC, USA). Rats were placed in suspended transparent cages with a wire-mesh floor. After acclimatization, the probe was stabbed perpendicularly to the hind paw at a force with no acceleration and held for 6–8 s. A withdrawal of the hind paw was considered to be a positive response. The hind paw withdrawal threshold was repeatedly measured 3 times at approximately 10-minute intervals, and the results were averaged.
2.4.2. Hindpaw thermal hyperalgesia
Hindpaw thermal hyperalgesia was assessed using the heat-plate method. Rats were placed in transparent plastic cages on a glass plate maintained at a constant temperature of 25 °C. After acclimatization, light from a radiant heat device (BME-410A, Chinese Institute of Biomedical Engineering, China) was focused on the plantar surface of the ipsilateral hind paw. The hind paw withdrawal latency was repeatedly tested 3 times at 10-minute intervals, and the measurements were averaged.
2.5. Immunofluorescence staining
To examine the expression of NR2B and its phosphorylation on keratinocytes, double-labeling immunofluorescence with the keratinocyte marker pan-keratin and NR2B or pNR2B was performed. The expression of IL-1β in keratinocytes was also studied by double-labeling. Microglial and astrocyte activation were evaluated by staining ionized calcium-binding adaptor molecule 1 (Ibal) and glial fibrillary acidic protein (GFAP), respectively. The expression of the neuronal activation marker c-fos was stained in the ipsilateral SDH.
Rats were anesthetized by intraperitoneal injection of sodium pentobarbital (40 mg/kg) and perfused with saline followed by fresh 4% paraformaldehyde. The plantar skin of the ipsilateral hind paw and SDH was harvested, fixed in 30% formaldehyde for 4 h, and then dehydrated in 30% sucrose overnight at 4 °C. Samples were embedded in optimal cutting temperature compound (OCT) (Sakura Finetek, Japan), frozen at −20 °C for 2 h, cut into frozen sections with a thickness of 9 μm for skin tissues and 12 μm for spinal cord by a cryostat, and finally mounted onto Superfrost Plus microscope slides (Shitai, China). The slides were dried at 37 °C, blocked with phosphate buffer solution (PBS), and permeabilized with 0.2% Triton X-100 (Jiangchen, China). Sample sections were blocked in 10% normal goat serum for 1 h at room temperature and then incubated overnight at 4 °C in 1% bovine serum albumin (BSA) (Sigma-Aldrich, USA) containing mixed primary antibodies (Table 1). After washing with PBS 3 times, sample sections were incubated with the appropriate secondary antibodies (Table 1). The slides were then washed in PBS 3 times and cover-slipped with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector, USA). Images were captured by a laser scanning microscope (FV1000, Olympus, USA). Immunofluorescence intensity (for epidermal section) or the number of positively stained cells within unit area (for SDH section) was calculated and statistically analyzed by ImageJ software (National Institutes of Health, USA). At least 10 fields of view (×200) from 5 sections in each sample were examined.
Table 1.
Primary and Secondary Antibodies for Immunofluorescent Staining.
| Antibody | Host | Company | Catalog Number | Dilution |
|---|---|---|---|---|
|
| ||||
| NR2B | Rabbit | Abcam | ab65783 | 1:250 |
| pNR2B | Rabbit | Abcam | ab18532 | 1:250 |
| IL-1β | Rabbit | Abcam | ab9722 | 1:300 |
| pan-keratin | Mouse | Abcam | ab8068 | 1:50 |
| c-fos | Rabbit | Abcam | ab209794 | 1:50 |
| GFAP | Goat | Abcam | ab53554 | 1:200 |
| Iba1 | Goat | Abcam | ab107159 | 1:100 |
| Alexa Fluor 488-conjugated anti-goat IgG | Donkey | Abcam | ab150129 | 1:300 |
| Alexa Fluor 596-conjugated anti-rabbit IgG | Donkey | Abcam | ab150076 | 1:300 |
| Alexa Fluor 488-conjugated anti-rabbit IgG | Goat | Zhongshan Jinqiao | 1:300 | |
| Alexa Fluor 488-conjugated anti-mouse IgG | Goat | Zhongshan Jinqiao | 1:300 | |
| Alexa Fluor 596-conjugated anti-mouse IgG | Goat | Zhongshan Jinqiao | 1:300 | |
Abbreviation: NR2B, N-methyl-d-aspartate receptor subunit 2B; pNR2B, phosphorylated N-methyl-d-aspartate receptor subunit 2B; IL-1β, interleukine 1 β; NF-κB, nuclear factor-κB; pNF-κB, phosphorylated nuclear factor-κB; GFAP, glial fibrillary acidic protein; Iba1, ionized calcium binding adaptor molecule 1.
2.6. Western blot
HaCaT cells were washed once with cold phosphate-buffered saline and then quickly detached by gently scraping. Cell pellets were collected by centrifugation for 2 min at 4500 rpm and stored at −80 °C. Whole-protein lysate buffer was prepared using 50 mM Tris-HCl buffer, pH 6.8, with 2% SDS. Cell pellets were dissolved in the lysate buffer by pipetting the suspension up and down and then boiled at 95 °C for 5 min. The cell lysate was cleared by centrifugation at 13,000 rpm for 2 min, and the clear supernatant fractions were subjected to Western blot assays.
The ipsilateral hind paw plantar skin and L4-6 DRG were collected from rats deeply anesthetized by sodium pentobarbital (40 mg/kg) and snap-frozen in liquid nitrogen. Tissues were cut into fine pieces and homogenized by an ultrasonic cell disruptor in ice-cold RIPA lysis buffer containing a cocktail of phenylmethanesulfonyl fluoride (PMSF), protease inhibitor and phosphatase inhibitor (Sigma-Aldrich, USA).
The protein concentration of supernatants was estimated by the bicinchoninic acid (BCA) method (with reagents from Suolaibao, China), according to which the total protein content between samples was equalized. Next, protein lysates were denatured with loading buffer (0.25 M Tris-HCl, 52% glycerol, 6% sodium dodecyl sulfate (SDS), 5% β-mercaptoethanol, and 0.1% bromophenol blue) for 10 min at 100 °C. Denatured lysates (20 μg) were then separated by SDS-polyacrylamide gel electrophoresis gels and transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare Life Sciences, USA). After blocking with 5% skim milk for 1 h at room temperature, membranes were incubated in Tris-buffered saline with 0.1% Tween 20 (TBST) overnight at 4 °C with primary antibodies (Table 2). After 3 washes with TBST, the membranes were incubated with secondary antibodies (Table 2) diluted with 5% skim milk in TBST for 1 h at room temperature. Bands were reacted with enhanced chemiluminescence reagents (ECL, CWBio, China) and developed by an ImageQuant LAS 4000 mini Chemiluminescence imaging analysis system (GE Healthcare Life Sciences, USA). The optical density (OD) of bands was statistically analyzed by ImageJ software.
Table 2.
Primary and Secondary Antibodies for Western Blot.
| Antibody | Host | Company | Catalog Number | Dilution |
|---|---|---|---|---|
|
| ||||
| NR2B (HaCaT) | Rabbit | Abcam | ab65783 | 1:1000 |
| pNR2B (HaCaT) | Rabbit | Abcam | ab18532 | 1:500 |
| NR2B (tissue) | Rabbit | Abcam | ab65783 | 1:500 |
| pNR2B (tissue) | Rabbit | Abcam | ab18532 | 1:500 |
| IL-1β | Rabbit | Abcam | ab9722 | 1:1000 |
| ERK | Rabbit | CST | #4370 | 1:1000 |
| pERK | Rabbit | CST | #4695 | 1:1000 |
| NF-κB | Rabbit | CST | #8242 | 1:1000 |
| pNF-κB | Rabbit | CST | #3033 | 1:1000 |
| β-actin | Mouse | Abcam | ab8226 | 1:1000 |
| Anti-rabbit IgG HRP | Goat | Abcam | ab6721 | 1:3000 |
| Anti-mouse IgG HRP | Goat | Abcam | ab6789 | 1:3000 |
Abbreviation: NR2B, N-methyl-D-aspartate receptor subunit 2B; pNR2B, phosphorylated N-methyl-D-aspartate receptor subunit 2B; IL-1β, interleukine 1 β; ERK, extracellular signal-regulated protein kinase; pERK, phosphorylated extracellular signal-regulated protein kinas; NF-κB, nuclear factor-κB; pNF-κB, phosphorylated nuclear factor-κB; HRP, horseradish peroxidase.
2.7. Statistical analysis
All statistical analysis was conducted with SPSS 22.0. Immunofluorescence and Western blot data were analyzed by one-way analysis of variance (ANOVA). Behavioral data were analyzed by two-way repeated-measures ANOVA followed by the Newman-Keuls multiple comparison test. All data are presented as the mean ± standard deviation, and error bars represent the standard error of the mean. The criterion for statistical significance was p < 0.05.
3. Results
3.1. NR2B is expressed on human keratinocytes, and its phosphorylation is upregulated by NMDA
We first studied whether NR2B is expressed on the human keratinocyte cell line HaCaT and whether it can be activated by NMDA. Western blot (Fig. 2) showed NR2B expression on HaCaT cells. The phosphorylation of NR2B was increased after 24-h NMDA treatment (p < 0.01). Treatment with NMDA did not cause changes in the expression of total NR2B. These results confirm that NR2B is expressed on human keratinocytes and can be activated by NMDA receptor agonists.
Fig. 2.
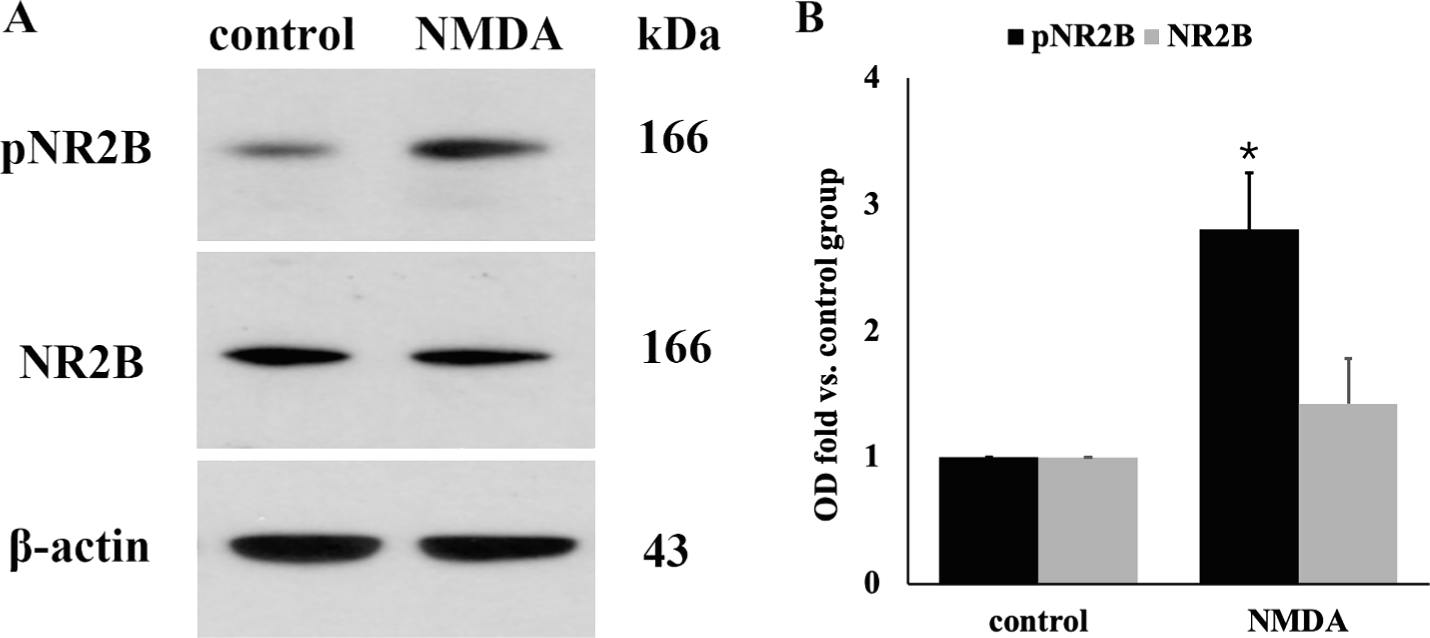
NR2B is expressed on cultured human keratinocytes HaCaT cells and can be activated by NMDA. A: Western blot image; B: Quantitative analysis of the relative optical density of NR2B and pNR2B (n = 3/group). One-way ANOVA, * p < 0.05 versus control. Western blot demonstrated the expression of NR2B and phosphorylated NR2B on HaCaT cells. The phosphorylation of NR2B was significantly increased after 24-h treatment with NMDA. There was no significant difference in the expression of total NR2B after NMDA treatment.
3.2. CPIP induces the upregulation and phosphorylation of NR2B on keratinocytes in the acute and chronic phases after ischemia–reperfusion injury
We then investigated whether ischemia-reperfusion injury could lead to NR2B activation in the skin where the keratinocytes are localized and the peripheral sensitization is initiated. Double immunofluorescence labeling of the ipsilateral skin tissue (Fig. 3A–E) showed that NR2B and pNR2B were colocalized in keratin-positive cells. The immunofluorescence staining intensity of NR2B and pNR2B was significantly increased in the CPIP groups in both acute (Fig. 3A, B and E) and chronic phases (Fig. 3C–E) compared with the sham groups (p < 0.001 for both NR2B and pNR2B). Similarly, Western blot of the ipsilateral skin tissue (Fig. 3F–G) showed increased expression of NR2B starting at 12 h, peaking at 18–24 h, and maintained until the last time point (14 d) tested in this study. Activation (phosphorylation) of NR2B (pNR2B) occurred as early as 6 h and persisted until day 14 after CPIP induction. (6 h: p = 0.024 for NR2B and p < 0.001 for pNR2B; 12 h, 18 h, 24 h, 2 d, 6 d, 10 d, 14 d: p < 0.001 for both NR2B and pNR2B. Fig. 3F–G). These results indicate that CPIP results in persistent activation of NR2B in keratinocytes of the skin ipsilateral to the injury.
Fig. 3.
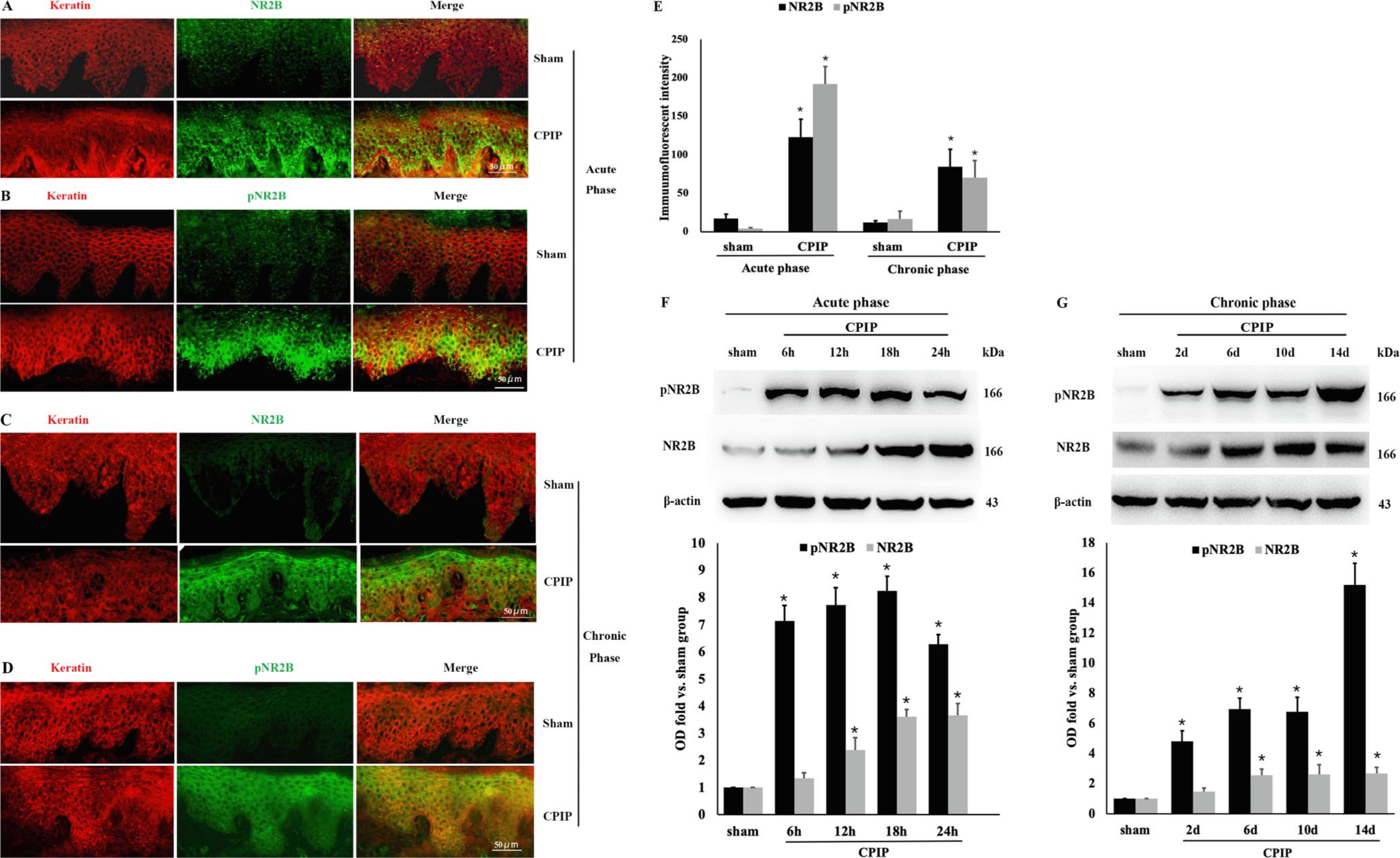
CPIP induces acute and chronic upregulation and phosphorylation of NR2B on keratinocytes. A–D: Double-labeling immunofluorescence of NR2B or pNR2B (green) and keratin (red) in ipsilateral epidermal tissue. Representative images showing that NR2B and pNR2B were co-localized in keratin-positive cells. The immunoreactivity of NR2B and pNR2B was significantly increased in CPIP groups than sham groups in both acute (24 h after CPIP, A, B) and chronic (14 d after CPIP, C, D) phases (n = 5/group). E: Quantitative analysis of the immunofluorescent intensity results from A–D. One-way ANOVA, * p < 0.05 versus sham. F, G: Western blot showed that the expression of NR2B in ipsilateral epidermal tissue was upregulated starting at 12 h, peaked at 18–24 h, and maintained till 14d. Phosphorylation of NR2B (pNR2B) occurred as early as 6 h and persisted for 14d after CPIP expression in acute (F) and chronic (G) phases. One-way ANOVA, * p < 0.05 versus sham. Abbreviations: NR2B, NMDA receptor subunit 2B; pNR2B, phosphorylated NMDA receptor subunit 2B; CPIP, chronic post-ischemia pain
3.3. Selective inhibition of peripheral NR2B activation attenuates CPIP-induced nociceptive behavior
Next, we examined whether NR2B is essential for CPIP-mediated nociceptive hypersensitivity. Compared with the sham treatment, CPIP induced a significant decrease in hind paw mechanical withdrawal threshold and thermal withdrawal latency (CPIP + NS vs. sham: p < 0.001. Fig. 4). Subcutaneous injection of NMDA into the ipsilateral plantar aggravated mechanical allodynia and thermal hyperalgesia in the acute phase (CPIP + NMDA vs. CPIP + NS: p < 0.001 at 12, 18, and 24 h, Fig. 4A, C) and chronic phase (CPIP + NMDA vs. CPIP + NS: p < 0.001 on 10 d and 14 d, Fig. 4B, D). Daily intraplantar injection of normal saline (100 μl) from day 8 to 14 did not change mechanical allodynia or thermal hyperalgesia (Supplementary Fig. 1). CPIP-induced mechanical and thermal hypersensitivity were mitigated by ipsilateral intraplantar administration of ifenprodil in both acute and chronic groups (Fig. 4A–D). These findings suggest that peripheral NR2B is required for mechanical and thermal hypersensitivity induced by CPIP.
Fig. 4.
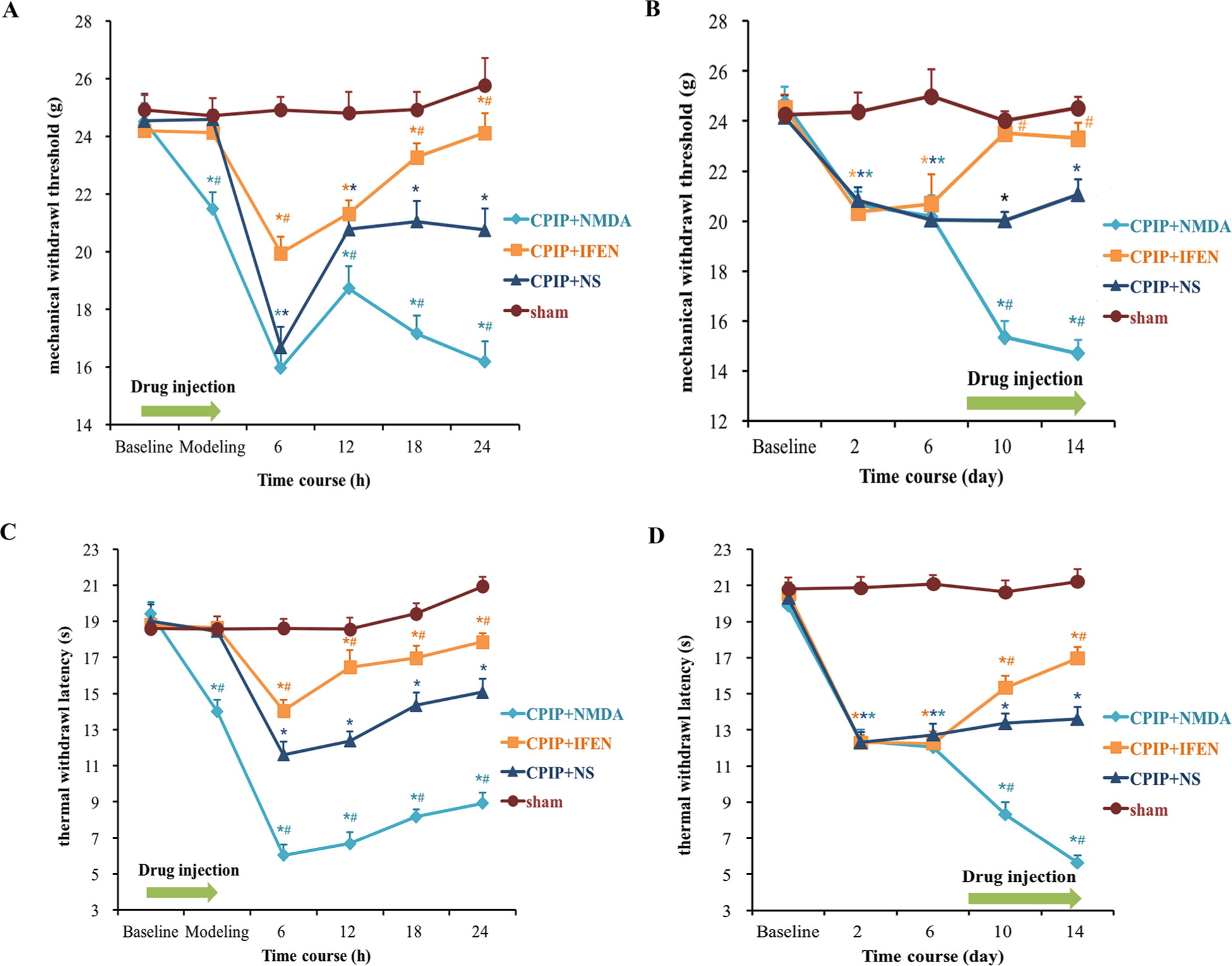
Peripheral NR2B mediates CPIP-induced mechanical allodynia and thermal hyperalgesia. (A) acute mechanical allodynia; (B) chronic mechanical allodynia; (C) acute thermal hyperalgesia; (D) chronic thermal hyperalgesia (n = 10/group). CPIP induced a significant decrease in hind paw mechanical withdrawal threshold and heat withdrawal latency in both acute and chronic phase. Behavioral hypersensitivity was exacerbated by NMDA and alleviated by ifenprodil, the selective NR2B inhibitor. Two-way repeated-measure ANOVA, * p < 0.05 versus sham; # p < 0.05 versus CPIP + NS. Abbreviations: CPIP, chronic post-ischemia pain; NS, normal saline; NMDA, N-methyl-D-aspartate; IFEN, ifenprodil.
3.4. Peripheral NR2B mediates CPIP-induced activation of NF-κB p65 and ERK 1/2 and production of IL-1β in epidermal tissue and DRG – role of NR2B in peripheral sensitization
As NF-κB p65, ERK 1/2, and IL-1β are key players in nociceptive peripheral sensitization, we next studied whether peripheral NR2B is required for the activation or production of these molecules in the ipsilateral hindpaw skin after CPIP. Western blot analysis showed that CPIP significantly upregulated the expression of NF-κB p65, phosphorylated NF-κB p65, ERK 1/2 and phosphorylated ERK 1/2 in the acute phase (24 h) (CPIP + NS vs. sham: p = 0.022 for pNF-κB, p = 0.026 for NF-κB, p < 0.001 for ERK and pERK, Fig. 5A, B). These changes persisted in the chronic phase (14 d) (CPIP + NS vs. sham: p < 0.001 for each of them, Fig. 5C, D). Such acute and chronic increases were exacerbated by NMDA (CPIP + NMDA vs. CPIP + NS: acute phase: p < 0.001 for each of them, Fig. 5A, B; chronic phase: p < 0.001 for pNF-κB, NF-κB and pERK, p = 0.008 for ERK, Fig. 5C, D) and abolished by ifenprodil (CPIP + IFEN vs. CPIP + NS: acute phase: p = 0.019 for pNF-κB, p < 0.001 for NF-κB and pERK, p = 0.024 for ERK, Fig. 5A, B; chronic phase: p = 0.023 for pNF-κB, p = 0.004 for NF-κB, p < 0.001 for pERK, p = 0.028 for ERK, Fig. 5C, D).
Fig. 5.
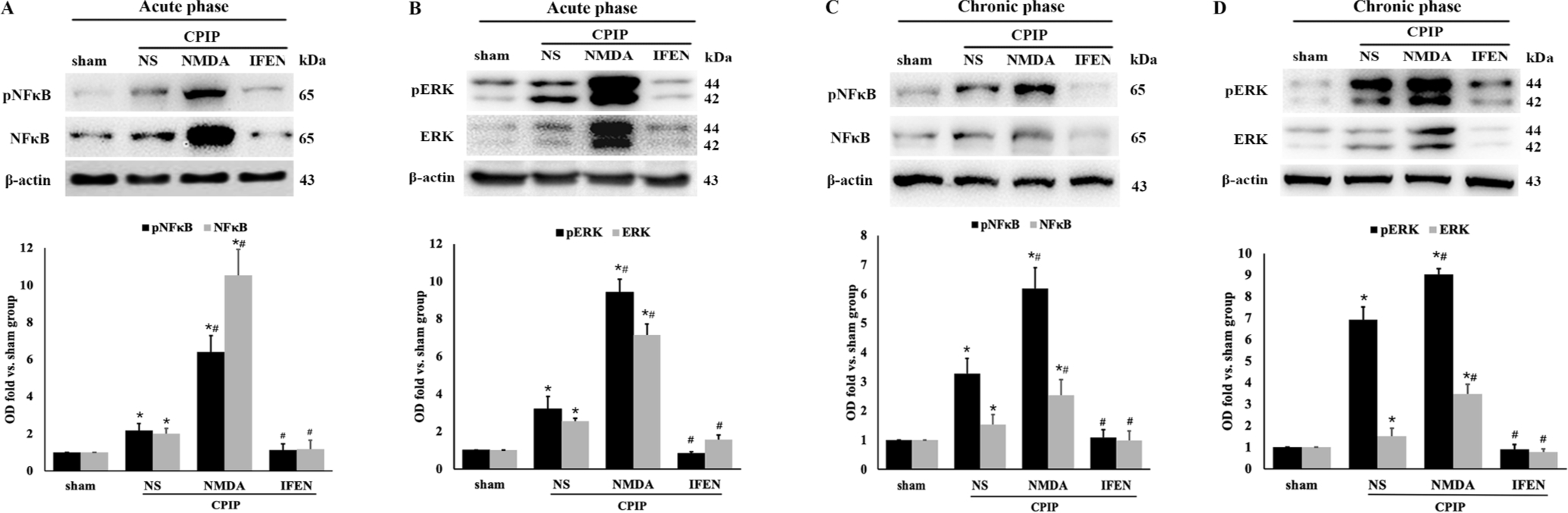
Peripheral NR2B mediates CPIP-induced phosphorylation of NF-κB and ERK in ipsilateral hindpaw skin. NF-κB p65 and pNF-κB p65 in acute (A) and chronic (C) phase; ERK 1/2 and pERK 1/2 in acute (B) and chronic (D) phase. Western blot results shown on the top; quantification analysis (relative to sham group) shown on the bottom (n = 5/group). One-way ANOVA, * p < 0.05 versus sham, # p < 0.05 versus CPIP. Abbreviations: CPIP, chronic post-ischemia pain; NS, normal saline; NMDA, N-methyl-D-aspartate; IFEN, ifenprodil. NF-κB, nuclear factor-κB; pNF-κB, phosphorylated nuclear factor-κB; ERK, extracellular regulated protein kinases; pERK, phosphorylated extracellular regulated protein kinases.
To examine whether IL-1β is produced in the keratinocytes, we stained for IL-1β and keratin in the hindpaw skin tissue. Immunofluorescence assays illustrated the colocalization of IL-1β and keratin at 24 h (Fig. 6A, acute phase) and 14 d (Fig. 6B, chronic phase) after ischemia-reperfusion injury (Fig. 6A, B), indicating that IL-1β was upregulated in keratinocytes immediately and persistently after CPIP (CPIP + NS vs. sham: p < 0.001, Fig. 6C). CPIP-induced IL-1β upregulation was aggravated by NMDA (CPIP + NMDA vs. CPIP + NS: p < 0.001, Fig. 6A–C) and abolished by ifenprodil (CPIP + IFEN vs. CPIP + NS: p < 0.001, Fig. 6A–C) in both the acute and chronic phases. Western blot also showed that IL-1β production was significantly increased after CPIP in both acute (CPIP + NS vs. sham: p < 0.001, Fig. 6D) and chronic phases (CPIP + NS vs. sham: p = 0.036, Fig. 6E). NMDA administration further increased IL-1β production (CPIP + NMDA vs. CPIP + NS: acute phase: p = 0.023, Fig. 6D; chronic phase: p < 0.001, Fig. 6E), while ifenprodil suppressed IL-1β production (CPIP + IFEN vs. CPIP + NS: acute phase: p < 0.001, Fig. 6D; chronic phase: p = 0.011, Fig. 6E).
Fig. 6.
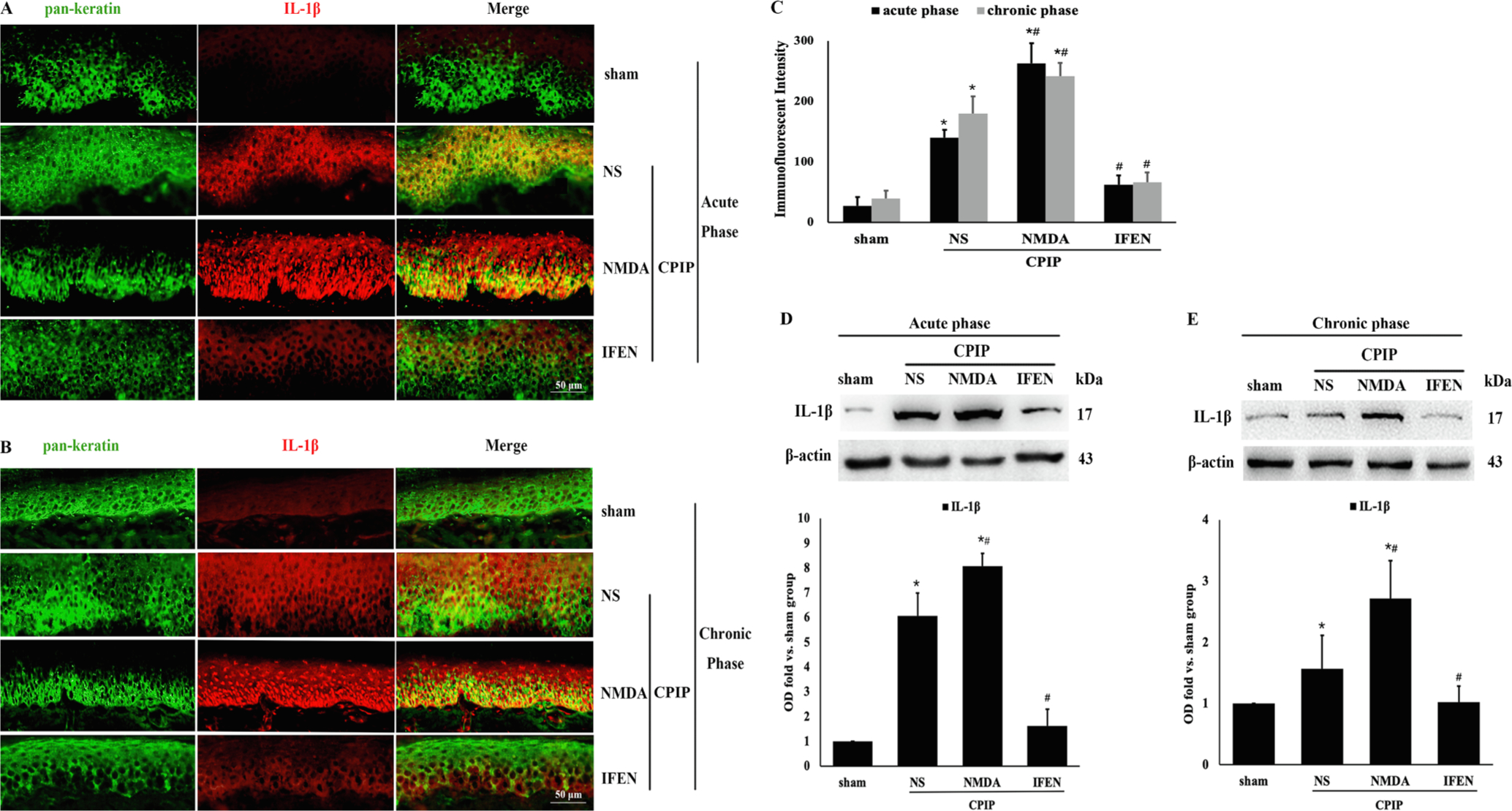
Peripheral NR2B mediates CPIP-induced IL-1β production in ipsilateral hindpaw skin. Double-labeling immunofluorescence of IL-1β (red) and keratin (green) in epidermal tissue in acute (Fig. 6 A) and chronic (Fig. 6 B) phase after CPIP. IL-1β was co-localized in keratin-positive cells. C: Quantitative analysis of the immunofluorescent intensity results from A and B. D–E: Western bolt of IL-1β in ipsilateral epidermal tissue in acute (D) and chronic (E) phase after CPIP. Western blot results shown on the top; quantification analysis (relative to sham group) on the bottom (n = 5 in each). One-way ANOVA, * p < 0.05 versus sham, # p < 0.05 versus CPIP. Abbreviations: CPIP, chronic post-ischemia pain; NS, normal saline; NMDA, N-methyl-D-aspartate; IFEN, ifenprodi; IL-lβ, interleukin lβ
Peripheral sensitization involves increased excitability of DRG neurons (Scholz and Woolf, 2007). We then investigated whether NR2B is also required for sensitization in DRG after CPIP. Western blot of ipsilateral DRG revealed no significant change in the expression of NR2B after CPIP (Fig. 7A, B). However, in both the acute and chronic phases, NR2B phosphorylation, i.e., activation, (Fig. 7A, B) was significantly induced by CPIP (CPIP + NS vs. sham: p < 0.001 in the acute phase, p = 0.014 in the chronic phase), which was intensified by NMDA (CPIP + NMDA vs. CPIP + NS: p < 0.001 in both phases) and alleviated by ifenprodil (CPIP + IFEN vs. CPIP + NS: p < 0.001 in both phases). The expression of NF-κB p65, pNF-κB p65, ERK 1/2, pERK 1/2 and IL-1β in ipsilateral DRG was also upregulated after CPIP (CPIP + NS vs. sham: acute phase: p = 0.038 for ERK and p < 0.001 for others; chronic phase: p < 0.001 for all; CPIP + NMDA vs. CPIP + NS: p < 0.001 for all in both phases; CPIP + IFEN vs. CPIP + NS: acute phase: p = 0.043 for pERK, p = 0.005 for ERK and p < 0.001 for others; chronic phase: p = 0.014 for pERK, p = 0.022 for ERK and p < 0.001 for others; Fig. 7C–H). Again, CPIP-induced upregulation of these proinflammatory markers in DRG was blocked by ifenprodil, similar to the changes found in the ipsilateral skin. Taken together, the skin and DRG results strongly suggest that NR2B is required for peripheral sensitization after CPIP.
Fig. 7.
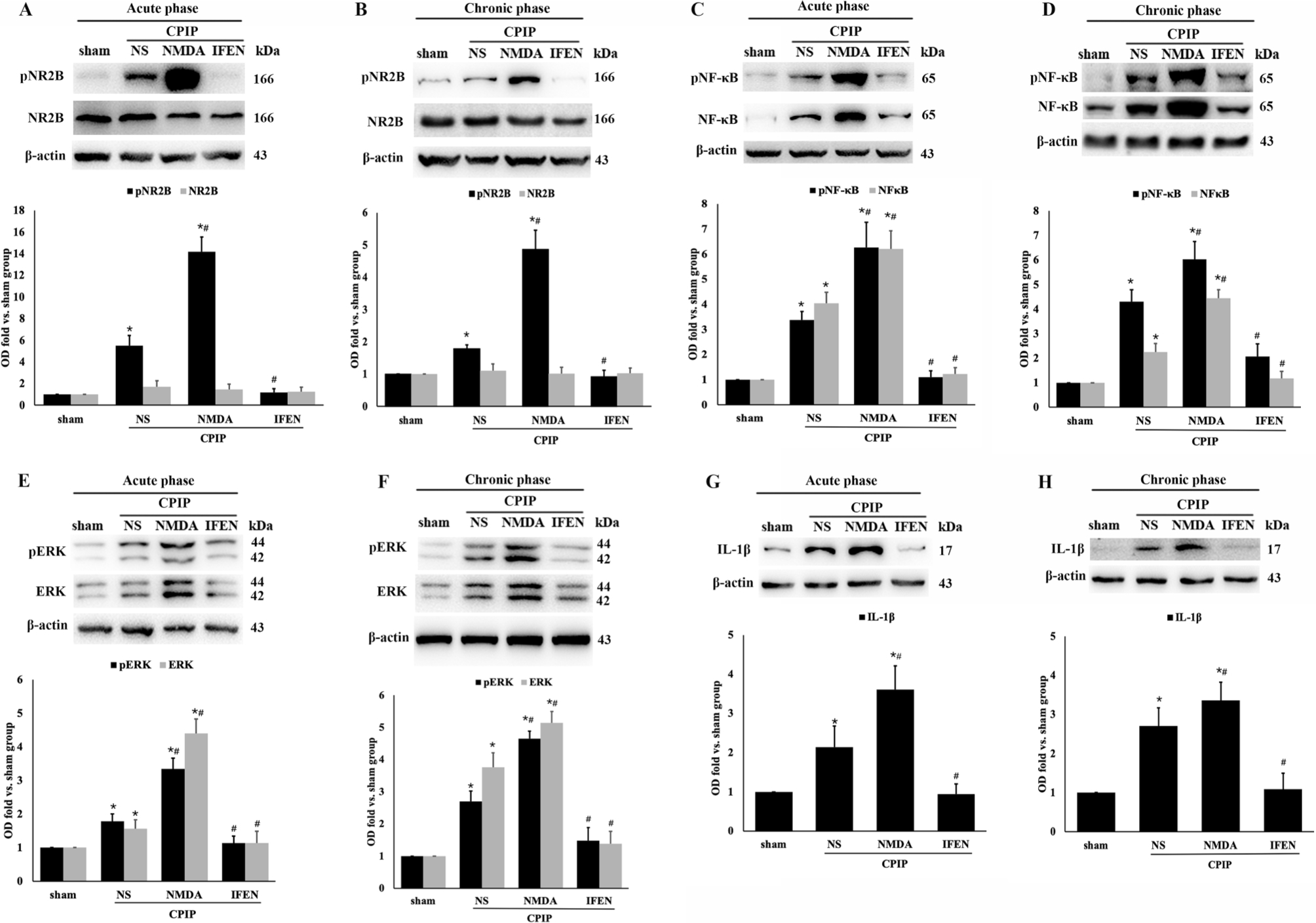
Peripheral NR2B mediates CPIP-induced phosphorylation of NR2B, NF-κB p65, and ERK 1/2, as well as production of IL-1β in DRG. Expression of NR2B and pNR2B in acute (A) and chronic (B) phase; NF-κB p65 and pNF-κB p65 in acute (C) and chronic (D) phase; ERK 1/2 and pERK 1/2 in acute (E) and chronic (F) phase; IL-1β in acute (G) and chronic (H) phase. Western blot results are shown on the top and quantification analysis (relative to sham group) on the bottom (n = 5 in each). One-way ANOVA, * p < 0.05 versus sham, # p < 0.05 versus CPIP. Abbreviations: CPIP, chronic post-ischemia pain; NS, normal saline; NMDA, N-methyl-D-aspartate; IFEN, ifenprodil; NF-κB, nuclear factor-κB; pNF-κB, phosphorylated nuclear factor-κB; ERK, extracellular regulated protein kinases; pERK, phosphorylated extracellular regulated protein kinases; IL-lβ, interleukin 1-β.
3.5. Peripheral NR2B regulates c-fos induction and glia activation in SDH – role of NR2B in central sensitization
Robust evidence suggests that activated glia in the spinal cord dorsal horn (SDH) release abundant proinflammatory mediators (e.g., cytokines and chemokines) and enhance neuronal excitability (e.g., induction of c-fos), which eventually lead to central nociceptive sensitization (Scholz and Woolf, 2007; Ji et al., 2013). To determine the role of NR2B in central sensitization after CPIP, we examined c-fos induction and glial activation in the ipsilateral SDH in both the acute and chronic phases after ischemia and reperfusion injury. Immunofluorescence staining (Fig. 8A, B) showed that the number of c-fos-positive neurons in SDH was markedly elevated by CPIP (CPIP + NS vs. CPIP + sham: p < 0.001) and further augmented by NMDA (CPIP + NMDA vs. CPIP + NS: p = 0.013). Induction of c-fos was significantly reduced by ifenprodil treatment (CPIP + IFEN vs. CPIP + NS: p < 0.001) in both acute and chronic phases (Fig. 8A, B).
Fig. 8.
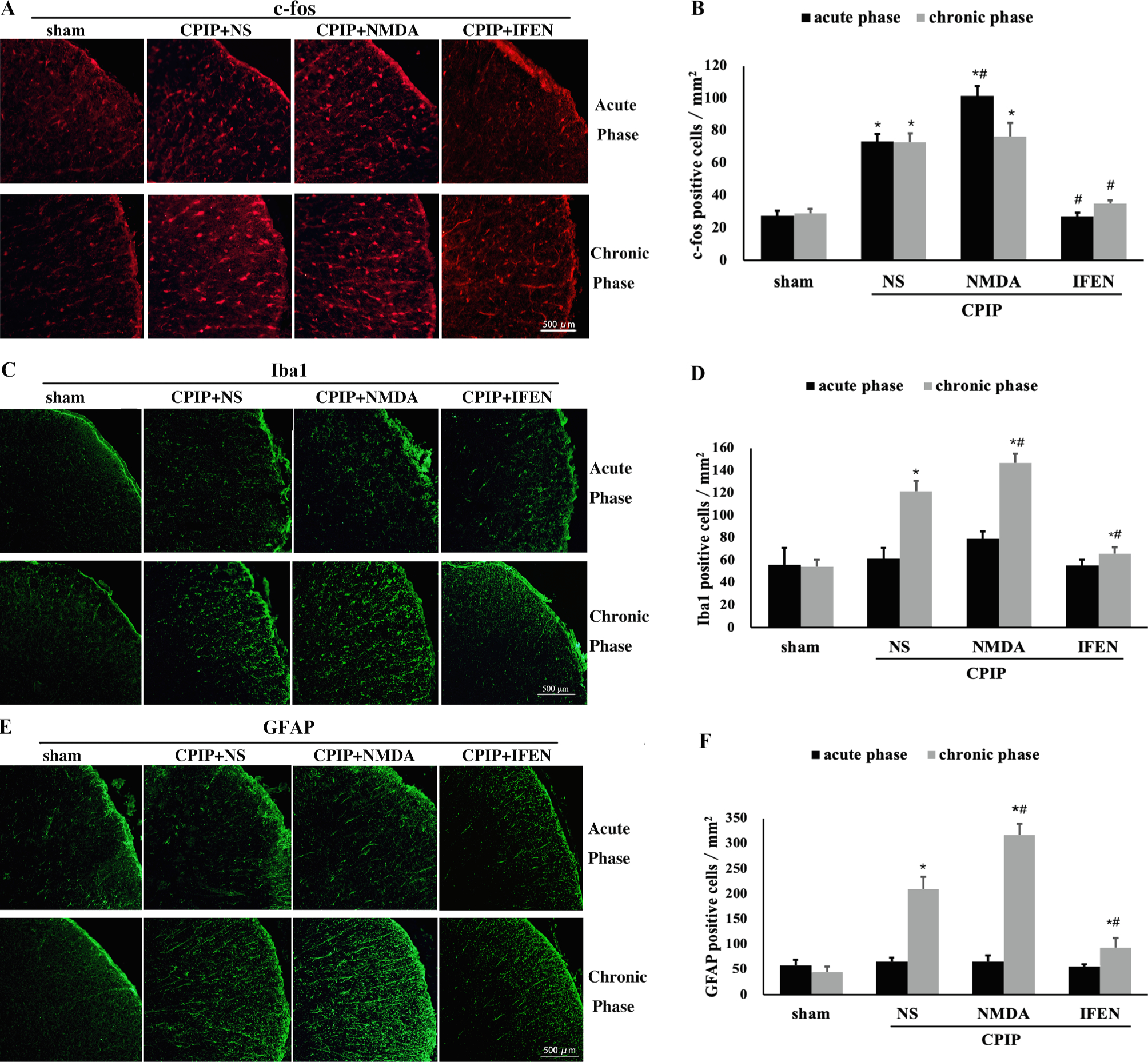
Peripheral NR2B mediates CPIP-induced c-fos induction and glial activation in SDH. (A) Immunofluorescence showed c-fos induction through acute to chronic phase after CPIP which was abolished by ifenprodil. (B) Quantification of results from A (n = 5 in each). Significant microglia (Iba 1 staining) (C, D) and astrocyte activation (GFAP staining) (E, F) were observed only in chronic but not acute phase. One-way ANOVA, * p < 0.05 versus sham, # p < 0.05 versus CPIP. Abbreviations: SDH, spinal cord dorsal horn; CPIP, chronic post-ischemia pain; NS, normal saline; NMDA, N-methyl-D-aspartate; IFEN, ifenprodil; Iba1, ionized calcium binding adaptor molecule 1; GFAP, glial fibrillary acidic protein.
An upregulation of both Iba1 and GFAP (for microglial and astrocyte activation, respectively) in the ipsilateral SDH was observed in the chronic but not acute phase after CPIP (CPIP vs. CPIP + NS: p < 0.001 in the chronic phase, Fig. 8C–F). Again, glial activation was further aggravated by NMDA (CPIP + NMDA vs. CPIP + NS: p = 0.004 for Iba1, p < 0.001 for GFAP, Fig. 8C–F) and suppressed by ifenprodil (CPIP + IFEN vs. CPIP + NS: p < 0.001 for both. Fig. 8C–F). These results suggest that NR2B is crucial for glial activation after CPIP.
4. Discussion
In the present study, we investigated the expression and activation of NMDA receptor subunit 2B on keratinocytes, as well as its roles in peripheral and central nociceptive sensitization induced by ischemia-reperfusion injury in a rat model of CRPS. We found that NR2B was expressed and could be activated by NMDA on cultured human keratinocyte cells. CPIP-induced hypersensitivity was associated with persistent upregulation and phosphorylation of NR2B on keratinocytes. In response to CPIP, there was significant activation of NF-κB p65 and ERK 1/2 and increased production of IL-1β in ipsilateral hindpaw skin and DRG, which may underlie the peripheral sensitization after CPIP. Spinal c-fos induction was upregulated in both acute and chronic stages, while spinal glial activation was found only in the chronic phase of CPIP. Both c-fos induction and glial activation are indicators of central sensitization. The above changes were aggravated by intraplantar injection of NMDA and alleviated by the NR2B-specific antagonist ifenprodil. Our work sheds light on a previously unknown yet essential role of NR2B on keratinocytes in peripheral and central sensitization in CPIP.
Recent studies have reported that keratinocytes play a critical role in nociception (Shi et al., 2011; Baumbauer et al., 2015; Huang et al., 2008; Zhao et al., 2008; Radtke et al., 2010; Ritter-Jones et al., 2016). Keratinocytes are in close contact with the peripheral nociceptive nerves and the frontline of sensory system (Finch et al., 2009). Stimulation of keratinocytes can directly elicit action potentials (Baumbauer et al., 2015) and is sufficient to induce nociceptive activation (Pang et al., 2015; Radtke et al., 2010). Neuropeptides (Shi et al., 2011), transient receptor potential vanilloid 3 (Huang et al., 2008), and voltage-gated sodium ion channels (Zhao et al., 2008) have been reported to contribute to nociceptive transduction in keratinocytes. In the present study, we found that the NMDA receptor NR2B subunit on epidermal keratinocytes is essential for the hypersensitivity induced by ischemia and reperfusion injury. Previous studies also reported that intraplantar injection of NMDA could initiate mechanical allodynia (Zhou et al., 1996); which could be relieved by an NMDA receptor antagonist (Taniguchi et al., 1997). However, this nociceptive effect was attributed to NMDA receptors on peripheral nerves rather than on keratinocytes (Carlton et al., 1995; Ma and Hargreaves, 2000). NR2B is associated with nociceptive transmission in a series of pain models (Qu et al., 2009; Liao and Xu, 2017; Nozaki et al., 2011; Xu et al., 2018). The expression of NR2B in the forebrain (Zhuo, 2009; Li et al., 2009) and spinal cord (Boyce et al., 1999) has been found to enhance central pain potentiation. Activation of NR2B in DRG has been found to be responsible for nociceptive hypersensitivity in visceral and peripheral pain (Norcini et al., 2016; Chen et al., 2016). Expression of NR2B has been reported in the skin of patients with fibromyalgia (Kim et al., 2006). However, the exact location of NR2B in skin tissue and its mechanistic role in nociceptive hypersensitivity were not clear.
To explore the role of keratinocyte NR2B in peripheral and central sensitization, we first confirmed the expression and activation of NR2B on cultured HaCaT human keratinocytes (Fig. 2) and on rat epidermal keratinocytes (Fig. 4) using the CPIP pain model. We then studied the expression and activation of NF-κB p65 (Liu et al., 2018; Arruri et al., 2017; Zhang et al., 2017; Xu et al., 2014) and ERK (Zhang et al., 2017; Liu et al., 2016; Hensellek et al., 2007), key intracellular mediators for peripheral neuronal sensitization. As expected, we found that both NF-κB and ERK were activated after CPIP (Fig. 5). The activation was aggravated by NMDA and alleviated by the NR2B-specific antagonist ifenprodil, both of which were administered in the ipsilateral epidermal tissue of the ischemia-reperfusion injury. Activation of NF-κB and ERK signaling cascades leads to the production of proinflammatory cytokines, predominantly IL-1β (Bessler et al., 2006; Wolf et al., 2003; Hollo et al., 2017), and the development of hypersensitivity. The production of IL-1β is increased in local cutaneous tissues in patients with CRPS (Birklein et al., 2014; Walker and Drummond, 2011; Uceyler et al., 2007). IL-1β production in keratinocytes is increased in the rat tibia fracture CRPS model (Li et al., 2009). Peripheral sensitization induced by the neuropeptide substance P and calcitonin gene-related peptide is enhanced by IL-1β production in keratinocytes (Shi et al., 2011). Preclinical studies also indicate the efficacy of systemic IL-1 receptor antagonist treatment in acute CRPS (Guo et al., 2014; Wei et al., 2016). In this study, we discovered that increased IL-1β production in keratinocytes after CPIP was abolished by epidermal administration of ifenprodil (Fig. 6), indicating that the upregulation of this proinflammatory cytokine was mediated by NR2B. Peripheral sensitization in the skin transmits to ipsilateral DRG (Basbaum et al., 2009). We found that activation of NR2B, NF-κB, and ERK and production of IL-1β in the ipsilateral DRG after CPIP were also abolished by epidermal administration of ifenprodil (Fig. 7), further confirming the essential role of NR2B in peripheral sensitization.
Peripheral nociceptive signals project to the dorsal horn of the spinal cord, where c-fos is induced and glial cells are activated, which further aggravate hyperexcitability, a phenomenon known as central sensitization (Basbaum et al., 2009). We found that epidermal NR2B was required for persistent neuronal excitability (c-fos induction) and late activation of glial cells (microglial and astrocytes) in the spinal cord after CPIP, indicating its role in central sensitization. Spinal c-fos induction was found in the tibia fracture-immobilization model of CRPS (Guo et al., 2014). Glial activation has been extensively studied in the pathogenesis of chronic pain (Ji et al., 2013; Watkins et al., 2001). Glial activation is characterized by increased expression of Iba1 on microglia and GFAP on astrocytes. Spinal microglial activation is rapid, while astrocyte activation is delayed and persistent (Ji et al., 2018). Our data demonstrated different activation states of neuronal and glial cells during the course of CPIP. Spinal neurons were activated early, and the activation persisted to the chronic phase (Fig. 8A, B), whereas glial activation in SDH was not observed until the chronic phase (Fig. 8C–F). These findings are consistent with previous studies (Klafke et al., 2016; Wei et al., 2016; Tajerian et al., 2015), which showed significant peripheral changes in acute stage and featured spinal involvement in chronic stage. One possible explanation is that sensory dysfunction is initiated by peripheral inflammation in the early stage (Klafke et al., 2016; Wei et al., 2016; Tajerian et al., 2015) and maintained by central sensitization afterwards (Wei et al., 2016; Tajerian et al., 2015). It is still unclear how nociceptive stimuli were transmitted from keratinocytes to the DRG and SDH, and further studies to investigate the interaction between keratinocytes and peripheral nociceptive nerve endings are warranted.
Our study has some limitations. First, the specific NR2B inhibitor ifenprodil can also interact with G protein-activated inwardly rectifying K+ (GIRK) channels, α1-adrenergic receptors and serotonin receptors (Kobayashi et al., 2006); some of which are located in the peripheral nervous system. Therefore, intraplantar injection of ifenprodil may affect neuronal function besides acting on NR2B. Second, although immunofluorescence staining studies showed that NR2B was expressed remarkably in the keratinocytes (Fig. 3), other local cells, including macrophages and Langerhans cells, might also be involved in the inflammatory response in this model. Finally, local injection of the medication can lead to systemic absorption, which may affect NR2B receptors located other than in epidermis. Further studies are warranted to test these hypotheses.
5. Conclusions
Collectively, our findings reveal the expression and activation of NMDA receptor subunit 2B (NR2B) in cultured human keratinocyte cells. Using the rat model of CPIP, we found that ischemia and reperfusion injury triggered the upregulation and phosphorylation of NR2B on keratinocytes, which subsequently contributed to inflammatory peripheral sensitization by activating the NF-κB and ERK pathways and increasing IL-1β production in the skin and DRG. Peripheral NR2B also mediated c-fos induction and glial activation in SDH. These findings reveal a previously unknown role of keratinocyte NR2B in peripheral and central sensitization (Fig. 9). Topical ketamine has been reported to alleviate allodynia in patients with CRPS[66]. However, as a non-specific antagonist for NMDA receptor, ketamine has many unwanted side effects. Our study therefore has clinical implications with respect to the use of selective topical NR2B antagonists for the treatment of CRPS, as topical application would be more tolerable and cause fewer adverse effects compared with systemic drug administration.
Fig. 9.
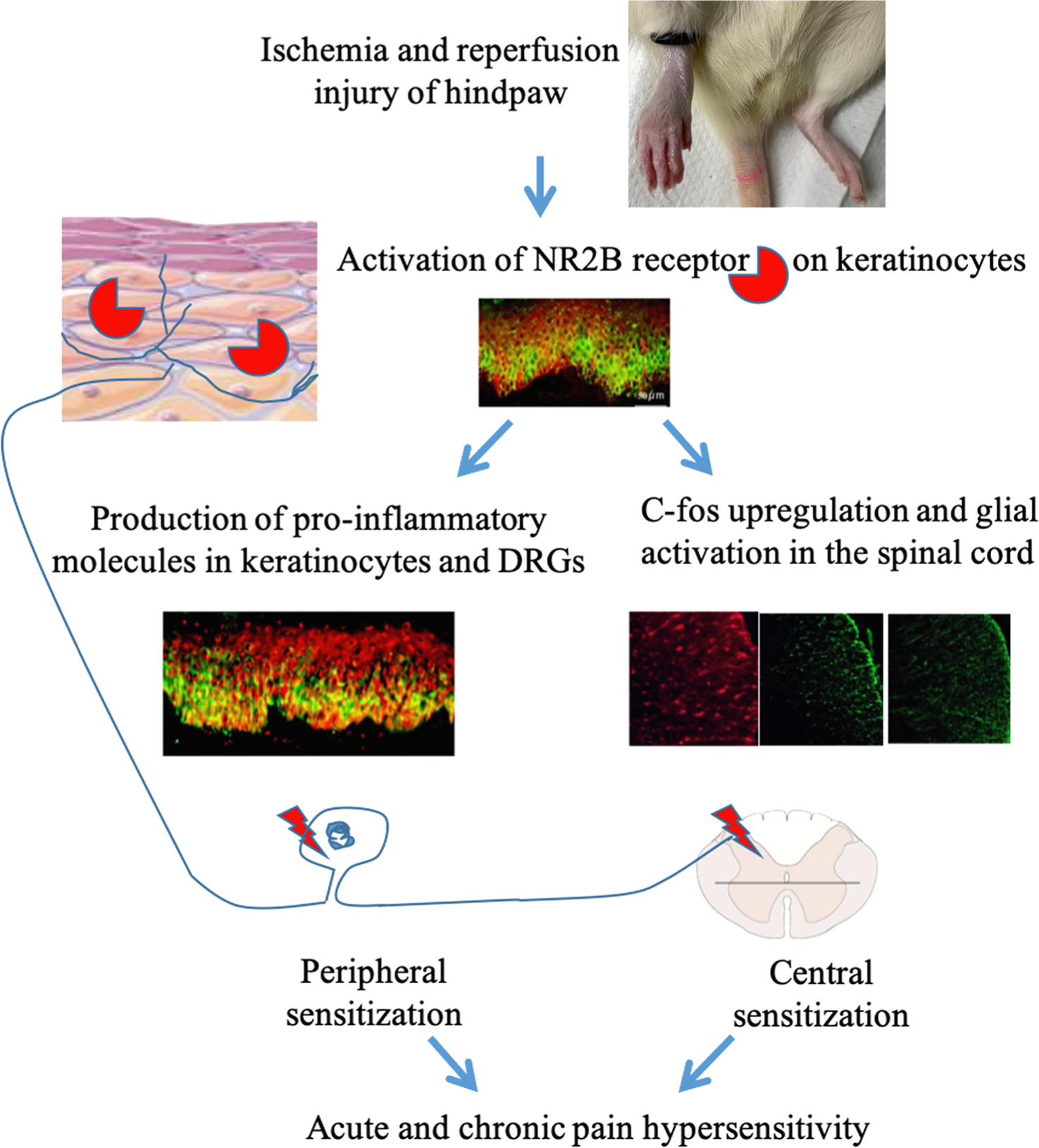
Schematic model showing the mechanistic pathway by which keratinocyte NR2B mediates peripheral and central sensitization. Ischemia and reperfusion injury of hindpaw induces activation of NR2B receptor on keratinocytes. Activation of NR2B leads to the production of pro-inflammatory molecules in keratinocytes and DRGs, which contributes to peripheral sensitization. Activation of keratinocyte NR2B also triggers central sensitization by upregulating c-fos and activating glia in the spinal cord dorsal horn. Abbreviations: NR2B, N-methyl-D-aspartate subunit 2B; DRG, dorsal root ganglion.
Supplementary Material
{kind=link}
Acknowledgements
We thank Dr. Nora Singer in Case Western Reserve University for the help on HaCaT cell culture. The work was supported by Beijing Natural Science Foundation (7182136 to LX) and Mentoring Exceptional New Translational Researchers (MENTR) grant at Cleveland Clinic Lerner Research Institute (to JX). The authors declare that they have no competing interests.
Footnotes
Declaration of Competing Interest
None.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbi.2020.02.003.
References
- Bruehl S, 2010. An update on the pathophysiology of complex regional pain syndrome. Anesthesiology 113, 713–725. [DOI] [PubMed] [Google Scholar]
- Marinus J, Moseley GL, Birklein F, Baron R, Maihofner C, Kingery WS, van Hilten JJ, 2011. Clinical features and pathophysiology of complex regional pain syndrome. Lancet Neurol. 10, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ, Xanthos DN, Francis L, Bennett GJ, 2004. Chronic post-ischemia pain (CPIP): a novel animal model of complex regional pain syndrome-type I (CRPS-I; reflex sympathetic dystrophy) produced by prolonged hindpaw ischemia and reperfusion in the rat. Pain 112, 94–105. [DOI] [PubMed] [Google Scholar]
- Ragavendran JV, Laferriere A, Khorashadi M, Coderre TJ, 2014. Pentoxifylline reduces chronic post-ischaemia pain by alleviating microvascular dysfunction. Eur. J. Pain 18, 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Liu L, Xu D, Zhang W, Zhang Y, Zhou J, Huang W, 2018. Interaction between astrocytic colony stimulating factor and its receptor on microglia mediates central sensitization and behavioral hypersensitivity in chronic post ischemic pain model. Brain Behav. Immun 68, 248–260. [DOI] [PubMed] [Google Scholar]
- David Clark J, Tawfik VL, Tajerian M, Kingery WS, 2018. Autoinflammatory and autoimmune contributions to complex regional pain syndrome. Mol Pain 14 1744806918799127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Thompson SW, 1991. The induction and maintenance of central sensitization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain 44, 293–299. [DOI] [PubMed] [Google Scholar]
- Xu J, Tang Y, Xie M, Bie B, Wu J, Yang H, Foss JF, Yang B, Rosenquist RW, Naguib M, 2016. Activation of cannabinoid receptor 2 attenuates mechanical allodynia and neuroinflammatory responses in a chronic post-ischemic pain model of complex regional pain syndrome type I in rats. Eur. J. Neurosci 44, 3046–3055. [DOI] [PubMed] [Google Scholar]
- Luo X, Tai WL, Sun L, Pan Z, Xia Z, Chung SK, Cheung CW, 2016. Crosstalk between astrocytic CXCL12 and microglial CXCR4 contributes to the development of neuropathic pain. Mol. Pain 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Q, Barr T, Gee L, Vickers J, Wymer J, Borsani E, Rodella L, Getsios S, Burdo T, Eisenberg E, Guha U, Lavker R, Kessler J, Chittur S, Fiorino D, Rice F, Albrecht P, 2011. Keratinocyte expression of calcitonin gene-related peptide beta: implications for neuropathic and inflammatory pain mechanisms. Pain 152, 2036–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birklein F, Drummond PD, Li W, Schlereth T, Albrecht N, Finch PM, Dawson LF, Clark JD, Kingery WS, 2014. Activation of cutaneous immune responses in complex regional pain syndrome. J. Pain 15, 485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo TZ, Wei T, Shi X, Li WW, Hou S, Wang L, Tsujikawa K, Rice KC, Cheng K, Clark DJ, Kingery WS, 2012. Neuropeptide deficient mice have attenuated nociceptive, vascular, and inflammatory changes in a tibia fracture model of complex regional pain syndrome. Mol. Pain 8, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WW, Guo TZ, Li XQ, Kingery WS, Clark JD, 2010. Fracture induces keratinocyte activation, proliferation, and expression of pro-nociceptive inflammatory mediators. Pain 151, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WW, Sabsovich I, Guo TZ, Zhao R, Kingery WS, Clark JD, 2009. The role of enhanced cutaneous IL-1beta signaling in a rat tibia fracture model of complex regional pain syndrome. Pain 144, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z, Sakamoto T, Tiwari V, Kim YS, Yang F, Dong X, Guler AD, Guan Y, Caterina MJ, 2015. Selective keratinocyte stimulation is sufficient to evoke nociception in mice. Pain 156, 656–665. [DOI] [PubMed] [Google Scholar]
- Fischer M, Glanz D, William T, Klapperstuck T, Wohlrab J, Marsch W, 2004. N-methyl-D-aspartate receptors influence the intracellular calcium concentration of keratinocytes. Exp. Dermatol 13, 512–519. [DOI] [PubMed] [Google Scholar]
- Nahm WK, Philpot BD, Adams MM, Badiavas EV, Zhou LH, Butmarc J, Bear MF, Falanga V, 2004. Significance of N-methyl-D-aspartate (NMDA) receptor-mediated signaling in human keratinocytes. J. Cell. Physiol 200, 309–317. [DOI] [PubMed] [Google Scholar]
- Morhenn VB, Murakami M, O’Grady T, Nordberg J, Gallo RL, 2004. Characterization of the expression and function of N-methyl-D-aspartate receptor in keratinocytes. Exp. Dermatol 13, 505–511. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R, 2010. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q, 2013. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Zhuo M, 2009. Plasticity of NMDA receptor NR2B subunit in memory and chronic pain. Mol. Brain 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo I, Sangiovanni E, Maggio R, Mattozzi C, Zava S, Corbett Y, Fumagalli M, Carlino C, Corsetto PA, Scaccabarozzi D, Calvieri S, Gismondi A, Taramelli D, Dell’Agli M, 2017. HaCaT cells as a reliable in vitro differentiation model to dissect the inflammatory/repair response of human keratinocytes. Mediators Inflamm 7435621–7435621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, Mogil JS, 2015. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci 18, 1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klafke JZ, da Silva MA, Rossato MF, de Pra SD, Rigo FK, Walker CI, Bochi GV, Moresco RN, Ferreira J, Trevisan G, 2016. Acute and chronic nociceptive phases observed in a rat hind paw ischemia/reperfusion model depend on different mechanisms. Pflugers Arch. 468, 229–241. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ, 2007. The neuropathic pain triad: neurons, immune cells and glia. Nat. Neurosci 10, 1361–1368. [DOI] [PubMed] [Google Scholar]
- Ji RR, Berta T, Nedergaard M, 2013. Glia and pain: is chronic pain a gliopathy? Pain 154 (Suppl. 1), S10–S28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Wang L, Li X, Sahbaie P, Kingery WS, Clark JD, 2011. Neuropeptides contribute to peripheral nociceptive sensitization by regulating interleukin-1beta production in keratinocytes. Anesth. Analg 113, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbauer KM, DeBerry JJ, Adelman PC, Miller RH, Hachisuka J, Lee KH, Ross SE, Koerber HR, Davis BM, Albers KM, 2015. Keratinocytes can modulate and directly initiate nociceptive responses. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Lee H, Chung MK, Park U, Yu YY, Bradshaw HB, Coulombe PA, Walker JM, Caterina MJ, 2008. Overexpressed transient receptor potential vanilloid 3 ion channels in skin keratinocytes modulate pain sensitivity via prostaglandin E2. J. Neurosci 28, 13727–13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Barr TP, Hou Q, Dib-Hajj SD, Black JA, Albrecht PJ, Petersen K, Eisenberg E, Wymer JP, Rice FL, Waxman SG, 2008. Voltage-gated sodium channel expression in rat and human epidermal keratinocytes: evidence for a role in pain. Pain 139, 90–105. [DOI] [PubMed] [Google Scholar]
- Radtke C, Vogt PM, Devor M, Kocsis JD, 2010. Keratinocytes acting on injured afferents induce extreme neuronal hyperexcitability and chronic pain. Pain 148, 94–102. [DOI] [PubMed] [Google Scholar]
- Ritter-Jones M, Najjar S, Albers KM, 2016. Keratinocytes as modulators of sensory afferent firing. Pain 157, 786–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch PM, Knudsen L, Drummond PD, 2009. Reduction of allodynia in patients with complex regional pain syndrome: a double-blind placebo-controlled trial of topical ketamine. Pain 146, 18–25. 10.1016/j.pain.2009.1005.1017. Epub 2009 Aug 1022. [DOI] [PubMed] [Google Scholar]
- Zhou S, Bonasera L, Carlton SM, 1996. Peripheral administration of NMDA, AMPA or KA results in pain behaviors in rats. NeuroReport 7, 895–900. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Shinjo K, Mizutani M, Shimada K, Ishikawa T, Menniti FS, Nagahisa A, 1997. Antinociceptive activity of CP-101,606, an NMDA receptor NR2B subunit antagonist. Br. J. Pharmacol. 122, 809–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton SM, Hargett GL, Coggeshall RE, 1995. Localization and activation of glutamate receptors in unmyelinated axons of rat glabrous skin. Neurosci. Lett 197, 25–28. [DOI] [PubMed] [Google Scholar]
- Ma QP, Hargreaves RJ, 2000. Localization of N-methyl-D-aspartate NR2B subunits on primary sensory neurons that give rise to small-caliber sciatic nerve fibers in rats. Neuroscience 101, 699–707. [DOI] [PubMed] [Google Scholar]
- Qu XX, Cai J, Li MJ, Chi YN, Liao FF, Liu FY, Wan Y, Han JS, Xing GG, 2009. Role of the spinal cord NR2B-containing NMDA receptors in the development of neuropathic pain. Exp. Neurol 215, 298–307. [DOI] [PubMed] [Google Scholar]
- Liao Y, Xu M, 2017. Efficacy and mechanism of action of etanercept in bone cancer pain. Pharmazie 72, 219–222. [DOI] [PubMed] [Google Scholar]
- Nozaki C, Vergnano AM, Filliol D, Ouagazzal AM, Le Goff A, Carvalho S, Reiss D, Gaveriaux-Ruff C, Neyton J, Paoletti P, Kieffer BL, 2011. Zinc alleviates pain through high-affinity binding to the NMDA receptor NR2A subunit. Nat. Neurosci 14, 1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Zhang K, Miao J, Zhao P, Lv M, Li J, Fu X, Luo X, Zhu P, 2018. The spinal NR2BR/ERK2 pathway as a target for the central sensitization of collagen-induced arthritis pain. PLoS One 13, e0201021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TT, Ren WH, Xiao X, Nan J, Cheng LZ, Zhang XH, Zhao ZQ, Zhang YQ, 2009. NMDA NR2A and NR2B receptors in the rostral anterior cingulate cortex contribute to pain-related aversion in male rats. Pain 146, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce S, Wyatt A, Webb JK, O’Donnell R, Mason G, Rigby M, Sirinathsinghji D, Hill RG, Rupniak NM, 1999. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology 38, 611–623. [DOI] [PubMed] [Google Scholar]
- Norcini M, Sideris A, Adler SM, Hernandez LA, Zhang J, Blanck TJ, Recio-Pinto E, 2016. NR2B expression in Rat DRG is differentially regulated following peripheral nerve injuries that lead to transient or sustained stimuli-evoked hypersensitivity. Front. Mol. Neurosci 9, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MX, Chen Y, Fu R, Liu SY, Yang QQ, Shen TB, 2016. Activation of 5-HT and NR2B contributes to visceral hypersensitivity in irritable bowel syndrome in rats. Am. J. Transl. Res 8, 5580–5590. [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Jang TJ, Moon IS, 2006. Increased expression of N-methyl-D-aspartate receptor subunit 2D in the skin of patients with fibromyalgia. J. Rheumatol 33, 785–788. [PubMed] [Google Scholar]
- Liu C, Zhang F, Liu H, Wei F, 2018. NF-κB mediated CX3CL1 activation in the dorsal root ganglion contributes to the maintenance of neuropathic pain induced in adult male Sprague Dawley rats1. Acta Cirurgica Brasileira 33, 619–628. [DOI] [PubMed] [Google Scholar]
- Arruri V, Komirishetty P, Areti A, Dungavath SKN, Kumar A, 2017. Nrf2 and NF-kappaB modulation by Plumbagin attenuates functional, behavioural and biochemical deficits in rat model of neuropathic pain. Pharmacol. Rep 69, 625–632. [DOI] [PubMed] [Google Scholar]
- Zhang A, Wang K, Ding L, Bao X, Wang X, Qiu X, Liu J, 2017. Bay11-7082 attenuates neuropathic pain via inhibition of nuclear factor-kappa B and nucleotide-binding domain-like receptor protein 3 inflammasome activation in dorsal root ganglions in a rat model of lumbar disc herniation. J. Pain Res 10, 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhu MD, Zhang X, Tian H, Zhang JH, Wu XB, Gao YJ, 2014. NFkappaB-mediated CXCL1 production in spinal cord astrocytes contributes to the maintenance of bone cancer pain in mice. J. Neuroinflamm 11, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li Z, Chen F, Liu H, Wang H, Li X, Liu X, Wang J, Zheng Z, 2017. TGF-beta1 suppresses CCL3/4 expression through the ERK signaling pathway and inhibits intervertebral disc degeneration and inflammation-related pain in a rat model. Exp. Mol. Med 49, e379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZH, Miao GS, Wang JN, Yang CX, Fu ZJ, Sun T, 2016. Resolvin D1 inhibits mechanical hypersensitivity in sciatica by modulating the expression of nuclear factor-kappaB, Phospho-extracellular signal-regulated kinase, and pro- and antiinflammatory cytokines in the spinal cord and dorsal root ganglion. Anesthesiology 124, 934–944. [DOI] [PubMed] [Google Scholar]
- Hensellek S, Brell P, Schaible HG, Brauer R, Segond von Banchet G, 2007. The cytokine TNFalpha increases the proportion of DRG neurones expressing the TRPV1 receptor via the TNFR1 receptor and ERK activation. Mol. Cell. Neurosci 36, 381–391. [DOI] [PubMed] [Google Scholar]
- Bessler H, Shavit Y, Mayburd E, Smirnov G, Beilin B, 2006. Postoperative pain, morphine consumption, and genetic polymorphism of IL-1beta and IL-1 receptor antagonist. Neurosci. Lett 404, 154–158. [DOI] [PubMed] [Google Scholar]
- Wolf G, Yirmiya R, Goshen I, Iverfeldt K, Holmlund L, Takeda K, Shavit Y, 2003. Impairment of interleukin-1 (IL-1) signaling reduces basal pain sensitivity in mice: genetic, pharmacological and developmental aspects. Pain 104, 471–480. [DOI] [PubMed] [Google Scholar]
- Hollo K, Ducza L, Hegyi Z, Docs K, Hegedus K, Bakk E, Papp I, Kis G, Meszar Z, Bardoczi Z, Antal M, 2017. Interleukin-1 receptor type 1 is overexpressed in neurons but not in glial cells within the rat superficial spinal dorsal horn in complete Freund adjuvant-induced inflammatory pain. J Neuroinflamm 14, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S, Drummond PD, 2011. Implications of a local overproduction of tumor necrosis factor-alpha in complex regional pain syndrome. Pain Med. (Malden, Mass) 12, 1784–1807. [DOI] [PubMed] [Google Scholar]
- Uceyler N, Eberle T, Rolke R, Birklein F, Sommer C, 2007. Differential expression patterns of cytokines in complex regional pain syndrome. Pain 132, 195–205. [DOI] [PubMed] [Google Scholar]
- Guo TZ, Wei T, Li WW, Li XQ, Clark JD, Kingery WS, 2014. Immobilization contributes to exaggerated neuropeptide signaling, inflammatory changes, and nociceptive sensitization after fracture in rats. J. Pain 15, 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T, Guo TZ, Li WW, Kingery WS, Clark JD, 2016. Acute versus chronic phase mechanisms in a rat model of CRPS. J. Neuroinflamm 13, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D, 2009. Cellular and molecular mechanisms of pain. Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF, 2001. Glial activation: a driving force for pathological pain. Trends Neurosci. 24, 450–455. [DOI] [PubMed] [Google Scholar]
- Ji RR, Nackley A, Huh Y, Terrando N, Maixner W, 2018. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 129, 343–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajerian M, Leu D, Yang P, Huang TT, Kingery WS, Clark JD, 2015. Differential efficacy of ketamine in the acute versus chronic stages of complex regional pain syndrome in mice. Anesthesiology 123, 1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Washiyama K, Ikeda K, 2006. Inhibition of G protein-activated inwardly rectifying K+ channels by ifenprodil. Neuropsychopharmacology 31, 516–524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.