

Abstract
Cerebral cavernous malformations (CCMs) are acquired vascular anomalies that constitute a common cause of central nervous system hemorrhage and stroke. The past two decades have seen a remarkable increase in our understanding of the pathogenesis of this vascular disease. This new knowledge spans genetic causes of sporadic and familial forms of the disease, molecular signaling changes in vascular endothelial cells that underlie the disease, unexpectedly strong environmental effects on disease pathogenesis, and drivers of disease endpoints such as hemorrhage. These novel insights are the integrated product of human clinical studies, human genetic studies, studies in mouse and zebrafish genetic models and basic molecular and cellular studies. This review addresses the genetic and molecular underpinnings of CCM disease, the mechanisms that lead to lesion hemorrhage, and emerging biomarkers and therapies for clinical treatment of CCM disease. It may also serve as an example for how focused basic and clinical investigation and emerging technologies can rapidly unravel a complex disease mechanism.
Keywords: Cerebral cavernous malformation, cavernoma, cavernous angioma, vascular malformation
Introduction
Cerebral cavernous malformations (CCMs) are a type of vascular lesion that form specifically in the central nervous system. CCMs are not a major underlying cause of cerebrovascular disease, but their investigation has been unusually fruitful and provided unexpected molecular and genetic insight into vascular development and disease. As discussed in greater detail below, multi-disciplinary studies of CCM disease performed by a group that includes clinicians, human geneticists, mouse and zebrafish biologists, cell biologists, and molecular biologists have culminated in an exceptionally rapid and deep understanding of disease pathogenesis.
This review summarizes the major developments in the CCM field over the past few decades, with the goal of highlighting recent advancements in the disease based on analysis of human genetics and murine models. Recent findings, such as the identification of PIK3CA mutations in CCM lesions, offer crucial insights into how genetics explain the highly variable penetrance in the phenotype and natural history of the disease. In addition to genetic factors, we discuss the importance of environmental contributors such as the microbiome and gut barrier in altering upstream inputs into the CCM signaling pathway. We emphasize the myriad ways in which CCM disease pathology is part of a complex network of pathways, operating at the intersection of hemodynamics, inflammation, and angiogenesis, and conclude with the current state of clinical trials and promising new avenues of therapeutics. The rapid rate of discovery in this field, and the close and productive interactions between the many different types of investigators working in it, have both brought the disease close to translational breakthroughs and provided a template for a modern approach to unraveling disease pathogenesis.
CCM Human Genetics: Mechanisms of Familial and Sporadic Disease
Heterozygous Mutation of CCM Genes: KRIT1, CCM2, or PDCD10 Causes Familial CCM
CCM disease can be broadly classified as either sporadic or familial. While sporadic CCMs typically present as solitary lesions in otherwise healthy individuals, familial CCMs typically present with a multiplicity of lesions and follow an autosomal dominant inheritance pattern1–3. Familial CCM has been shown to be caused by heterozygous loss of function (LOF) mutations in either KRIT14, 5, CCM26, 7, or PDCD108. Mutations in these genes primarily consist of nonsense, frameshift, and canonical splice site mutations. Pathogenic missense mutations have been reported in all three genes; however, they make up the minority of reported mutations. Notably, there are 4 known founder mutations in the CCM genes that account for substantial fraction of familial CCM cases. The most common of these is NM_194456.1(KRIT1):c.1363C>T(p.Gln455Ter) present in most Hispanic American cases of familial CCM5, 9. Another less frequent KRIT1 founder mutation: NM_194456.1(KRIT1):c.987C>A(p.Cys329Ter), has been found in several kindreds with Sardinian lineage10. A deletion spanning 77.6kb of CCM2 resulting in deletion of exons 2 – 10 has been founds in several Caucasian kindreds, it remains unclear whether all cases with this mutation are related or whether it has occurred independently in other families11. A splice mutation affecting CCM2: NM_031443.3(CCM2):c.30+5_30+6delinsTT, has been found in seemingly unrelated Ashkenazi Jewish probands, possibly due to an historically ancient mutation12.
Modifiers of Disease Severity
Genotype-Phenotype Correlation.
The clinical manifestations of familial CCM are highly heterogeneous. In many genetic diseases, the primary determinant of progression and severity is the causal mutation. However, this does not seem to be the case for CCM. A prime example of this is the common Hispanic founder mutation. Disease progression and severity in individuals with the Hispanic founder mutation is highly variable, despite sharing an identical pathogenic mutation13–16. The lack of correlation between clinical manifestations and mutation identity is consistent with the finding that the majority of germline mutations found in KRIT1, CCM2, and PDCD10—primarily frameshift, nonsense, and splice site mutations—result in loss of function. This suggests that the identity of mutations within the same gene does not have a significant impact on disease severity.
Though different mutations within a gene do not seem to impact disease severity, the identity of the mutated gene has been associated with several clinical characteristics. Many groups have noted an association between individuals with KRIT1 mutations and cutaneous vascular lesions16–22. Individuals with a mutation in CCM2 are more likely to be asymptomatic and have lower number of lesions compared to individuals with KRIT1 or PDCD10 mutations23. Familial CCM has been shown to be significantly more aggressive in individuals with a mutation in PDCD1024–26. The severity of CCM associated with PDCD10 mutations is attributable to the role of PDCD10 in the gut epithelium not shared with KRIT1 or CCM2 as discussed further below27.
Genetic Modifiers.
To identify genetic variants associated with disease severity, one group performed a large genetic association study of individuals with the KRIT1 founder mutation that identified several genetic polymorphisms within inflammatory and immune response genes that are associated with total lesion count, number of large lesions, and intracerebral hemorrhage28. This analysis revealed associations between clinical disease presentation and variants in several genes including: TGFBR2, CD14, IL-6R, MSR1, IGH, and TLR4. Some of these genes have been shown to have critical roles in CCM pathogenesis, highlighting the importance of further evaluating the roles of these genes, and demonstrating the power of association studies in a genetically homogenous cohort. Identification of TLR4 variants associated with disease severity are of particular interest owing to the direct role of TLR4 in propagating CCM signaling (discussed below)27. These data suggest that polymorphisms in genes other than KRIT1, CCM2, and PDCD10 may be important modifiers of CCM pathogenesis.
Radiation-Induced CCMs.
While the majority of sporadic CCMs occur in otherwise healthy individuals, numerous reports have shown that ionizing radiation is a potent inducer of CCM formation29–38. While pathologically similar to non-radiation induced sporadic CCMs, they have several distinct characteristics that hint at the underlying mechanisms driving CCM pathogenesis. One such characteristic is that individuals with radiation-induced CCMs often present with multiple lesions, in stark contrast to non-radiation induced sporadic CCMs which almost always occur as a solitary lesion. Furthermore, it was found that occurrence of multiple radiation-induced CCMs is significantly associated with younger age at time of radiation treatment29 and that the presence of multiple radiation induced CCMs may be related to higher doses of ionizing radiation37. Ionizing radiation has long been recognized as a potent source of DNA damage leading to genomic instability reviewed elsewhere39. The observation that radiation treatment may induce CCM formation—and that the multiplicity of lesions is related to radiation dose and age at radiation—support a key role for somatic mutations in CCM pathogenesis.
Role of Somatic Mutations in CCM Pathogenesis
Somatic Mutation of KRIT1, CCM2, or PDCD10 drives CCM formation.
Although germline LOF mutations in KRIT1, CCM2, or PDCD10 cause familial CCM as a clinical entity, they do not explain why CCMs present as focal lesions rather than a systemic vascular defect as might be expected if CCMs were the result of haploinsufficiency. This observation led to the hypothesis that a secondary, local event is necessary to initiate lesion formation; specifically, a somatic mutation in a CCM gene resulting in biallelic LOF. Somatic mutations resulting in biallelic LOF of KRIT1, CCM2, or PDCD10 have been reported in both familial40–42 43 and sporadic44 43 CCMs. Furthermore, laser capture microdissection and immunohistochemical staining of the CCM proteins in the lesion endothelium has established that these mutations occur in endothelial cells41, 45, 46. Together these data show that while loss of a CCM gene is dominant at the level of an individual, it is recessive on a cellular level, requiring loss of the normal allele via somatic mutation prior to lesion formation (Figure 1).
Figure 1. Somatic mutations driving CCM progression.
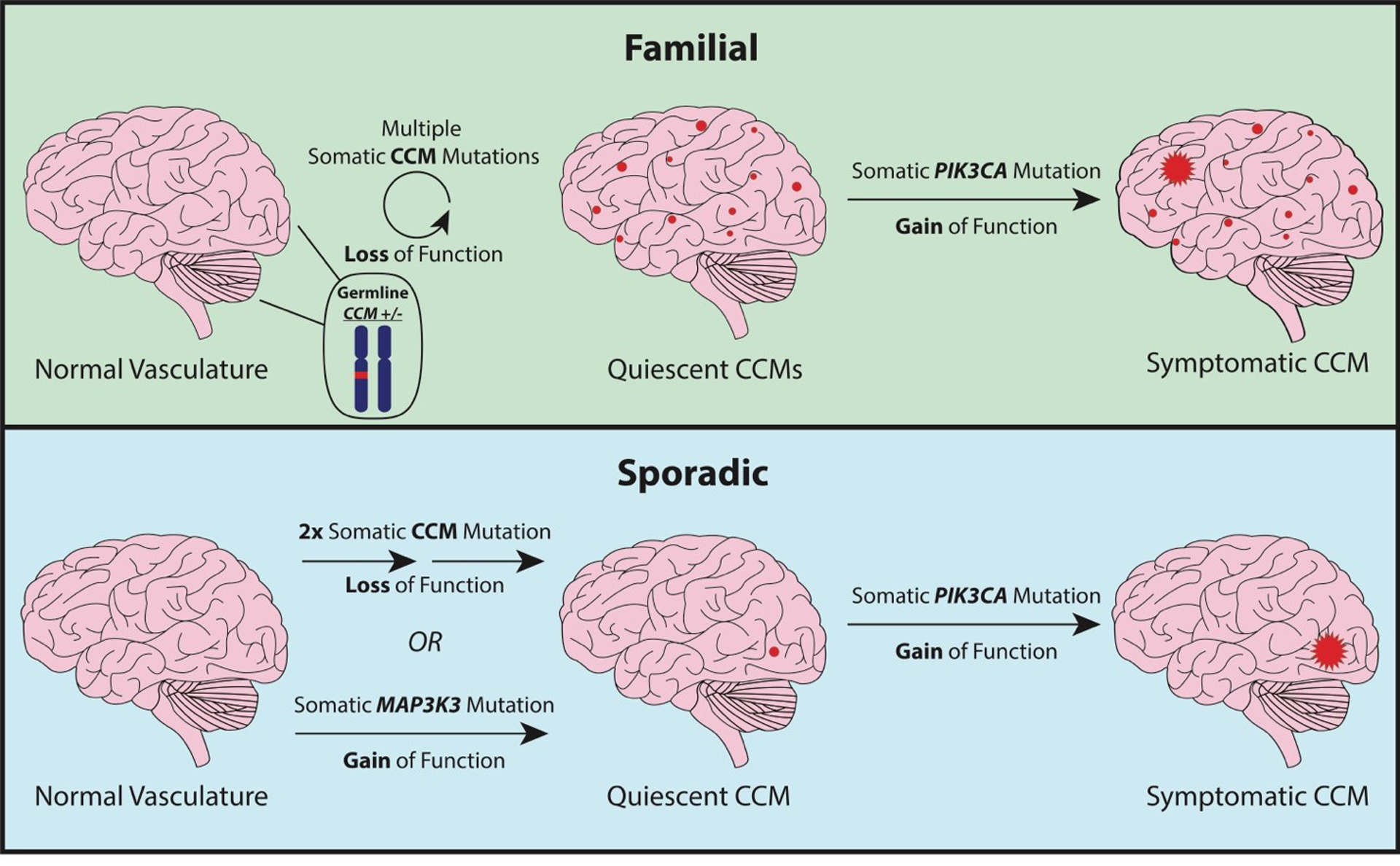
In familial CCM (top), lesion formation is initiated by a somatic mutation in a CCM gene resulting in biallelic loss of function which may occur multiple times resulting in the formation of multiple quiescent CCMs. A subset of these CCMs acquire a somatic gain of function mutation in PIK3CA which fuels lesion growth. Sporadic CCM formation (bottom) requires either two somatic CCM mutations to occur in the same cell resulting in biallelic loss of function or a single gain of function somatic mutation in MAP3K3. A CCM at this stage may or may not acquire a somatic gain of function in PIK3CA which would fuel lesion growth. The order in which the mutations occur is not yet known. This figure shows the CCM/MAP3K3 mutations as occurring first, however the PIK3CA mutation may occur first in some or all cases.
Activating PIK3CA Somatic Mutations in Familial and Sporadic CCMs.
The two-hit model of CCM pathogenesis elegantly explains why CCMs occur as focal lesions and why individuals with familial CCM have numerous lesions whereas sporadic cases almost always have a single lesion. The two-hit model has also proven true in mouse models of CCM (discussed below) where biallelic loss of Krit1, Ccm2, or Pdcd10 is required for lesion formation47, 48. One limitation of these models is that lesions only form during a brief window during early development. This observation is contradictory to what we find in human CCM disease where lesions may form throughout adulthood49, 50. This discrepancy may be explained by the recent discovery that many human CCMs harbor gain of function (GOF) mutations in the oncogene PIK3CA in addition to the previously noted LOF somatic mutations in the CCM genes. Somatic GOF mutations in PIK3CA are present in both sporadic43, 51, 52 and familial CCMs of all three genotypes43, 51, 52. PIK3CA mutations co-occur with somatic CCM gene mutations indicating that in some sporadic CCMs no less than three independent somatic mutations (two LOF somatic mutations in a CCM gene, and a GOF somatic mutation in PIK3CA) occurred during lesion development (Figure 1). Single-nucleus DNA sequencing has shown that PIK3CA and CCM gene somatic mutations are present in the same clonal population of cells suggesting that the effects of these mutations synergize in a cell autonomous manner. Consistent with the finding of activating PIK3CA somatic mutations, several previous studies have noted a link between CCM pathogenesis and PI3K signaling44, 53. Indeed, mouse models of CCM that previously only developed lesions during early development, when subjected to PIK3CA activation, develop lesions in adulthood.
Although activation of PIK3CA seems to be a common feature of resected human CCM lesions, it is unlikely that somatic activation of PIK3CA is required for lesion formation. Mouse models of CCM without PIK3CA perturbation suggest that congenital lesions may form in the absence of a PIK3CA mutation, perhaps due to a high basal level of PI3K signaling during development. Likewise, CCMs in humans may form in the absence of PIK3CA mutations during periods of elevated angiogenesis as may occur during growth or as a result of environmental factors. Furthermore, one important caveat of human CCM sequencing studies is that genetic analysis of lesions is limited to those which have been surgically resected, as asymptomatic lesions are rarely removed. This is a potent bias, selecting for aggressive lesions which are most likely to have activating mutations in PIK3CA. It is highly likely that a minority of all CCMs, but a majority of symptomatic CCMs, have activating mutations in PIK3CA.
Activating MAP3K3 Somatic Mutations in Sporadic CCMs.
As discussed above, previous work has established that familial and sporadic CCMs follow two-hit model, where somatic mutation(s) of a CCM gene initiates lesion formation. Recent work has found that many sporadic CCMs harbor a specific gain of function mutation in MAP3K3 (p.I441M), the gene that encodes MEKK351, 52. Furthermore, MAP3K3 mutations have been found to co-occur with PIK3CA mutations, however initial data suggest that CCM and MAP3K3 mutations are mutually exclusive52. This finding suggests that gain of function in MAP3K3 and loss of function in CCM genes have similar functional consequences such that either are sufficient to initiate CCM formation (Figure. 1)—distinct from mutations in PIK3CA which exacerbate lesion growth but not required for lesion formation. The equivalent effects of CCM and MAP3K3 mutations is supported by the direct role of the CCM complex in inhibiting MEKK3 activity (Figure. 2) such that increased MEKK3 activity may be achieved by either loss of the CCM complex or by gain of function in MEKK3.
Figure 2. Molecular signaling pathways involved in CCM lesion formation.
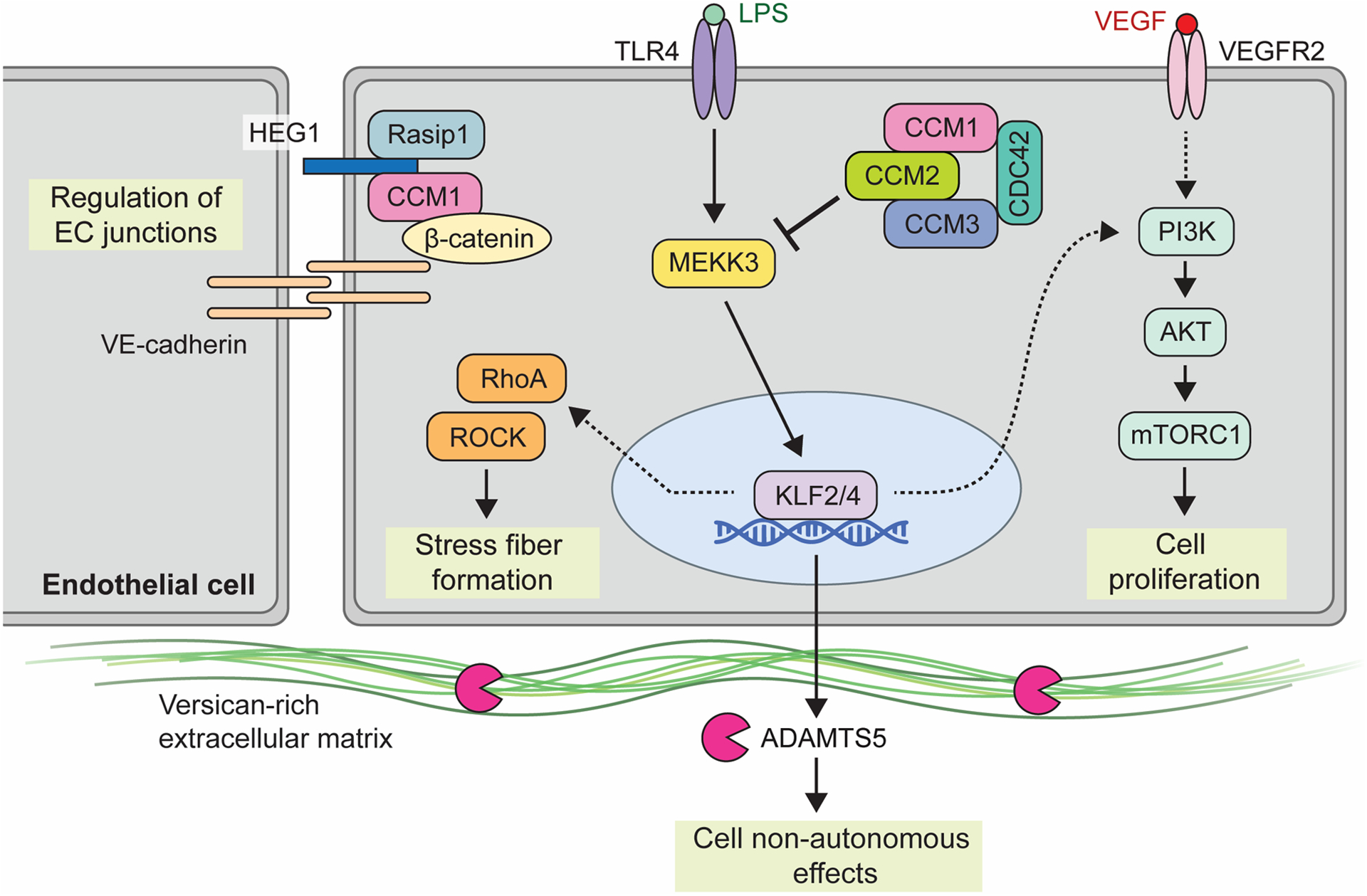
Shown is a partial summary of the various upstream inputs and downstream effectors implicated in CCM signaling. The heterotrimeric CCM complex, consisting of CCM1 (KRIT1), CCM2, and CCM3 (PDCD10), is an inhibitor of MAP3K3 Kinase, MEKK3. Inflammatory signals via TLR4 serve as the upstream input into MEKK3 signaling, as may shear forces associated with blood flow (not shown). Loss of CCM and subsequent activation of MEKK3 leads to upregulation of transcription factors KLF2 and KLF4. Downstream KLF2/4 transcriptional targets include both cell autonomous pathways through PI3K signaling and RHO/ROCK signaling, as well as cell non-autonomous effects via metalloprotease ADAMTS5 and extracellular matrix cleavage. VEGF is shown as a possible distinct input for PI3K-mTOR signaling while HEG1 has been associated with regulation of endothelial cell junctions. Of note, numerous other pathways have been found to be affected by loss of CCM function. This diagram focuses on those that are presently best understood and those that have been investigated using mouse and human genetic studies and/or are presently targeted therapeutically.
Lesion Formation and Growth.
The identification of somatic mutations in familial and sporadic CCMs raises several important questions about CCM pathogenesis. Does heterozygous loss of KRIT1, CCM2, or PDCD10 increase the rate of somatic mutations? One possible explanation for the high lesion burden is that heterozygous loss of a CCM gene, as is the case in familial CCM, causes cellular stress and/or genomic instability leading to an increase in the rate of somatic mutations. If true, one might expect a significant association between familial CCM and cancer—similar to hereditary cancer syndromes caused by mutations in DNA repair genes (e.g., Li-Fraumeni syndrome caused by mutations in p53, and Lynch syndrome caused by mutations in 4 mismatch repair genes). To date, no association between familial CCM and cancer has been noted.
Do somatically-mutated endothelial cells clonally expand similar to cancers? The initial studies identifying somatic mutations in familial and sporadic CCMs noted that the allele frequency of the somatic mutations was generally quite low, typically <10%40–42, 51. This observation may be in part explained by the presence of non-lesional cells in the sample diluting the mutant allele, however it may also suggest that CCM lesions are mosaic—consisting of both somatic mutant and wild type cells. Direct evidence of mosaicism in human lesions is limited to staining of the CCM proteins showing incomplete staining of the lesional endothelium45, 46. However, recent studies of mouse models54, 55 suggest that initially CCMs grow by clonal expansion but as they mature, they begin to incorporate wild type endothelial cells into the lesion.
CCM signaling pathways: molecular and genetic insights
Regulation of MEKK3-KLF2/4 signaling is a primary role of the CCM complex
Identification of the mutant genes responsible for CCM disease using positional cloning approaches in familial CCM disease patients revealed three non-homologous proteins with protein binding motifs, but no enzymatic or transcriptional activity. How three distinct proteins caused one clinical syndrome became clear when biochemical studies demonstrated that KRIT1, CCM2 and PDCD10 form a molecular complex56–58. However, gene expression studies did not reveal a vascular-specific pattern of these genes, and where and how this complex might be required to prevent vascular malformation remained unknown.
In vivo studies using genetic model organisms have revealed that the heterotrimeric “CCM complex” is required for cardiovascular development. Loss of ccm1 or 2 function in zebrafish results in a dilated heart phenotype associated with failure to create a lumenized aortic outflow tract59–61. Constitutive global loss in mice also results in failure to create a lumenized branchial arch artery and early embryonic lethality59, 62. These phenotypes were reproduced following endothelial-specific gene deletion63–66, suggesting that CCM function is required specifically in endothelial cells, but a molecular mechanism of action and a facile animal model with which to investigate disease pathogenesis remained elusive. This barrier was broken with the seminal demonstration that deletion of CCM genes in neonatal mice is sufficient to confer robust CCM formation63. Using this neonatal model, numerous groups have subsequently demonstrated that endothelial loss of CCM complex function is sufficient to confer lesion formation, confirming that the endothelial cell is the site of action for the CCM complex.
How does an adaptor protein complex regulate endothelial cell function? A critical clue to the molecular basis of CCM function arose prior to the identification of the disease-causing genes from studies of the MAP3K3 kinase, MEKK3. A yeast two-hybrid screen performed using MEKK3 as bait identified strong interaction with an uncharacterized protein, OSM, later identified as CCM2. This interaction was confirmed biochemically and characterized structurally67, 68, establishing that the CCM complex binds MEKK3 through the CCM2 protein that also binds KRIT1 and PDCD10. Genetic loss of function studies in the mouse demonstrated a critical role for MEKK3 in endothelial cells69 and corroborated human genetic studies that identified sporadic MEKK3 mutations in verrucous venous malformations70. Genetic studies in the developing heart defined the functional significance of this interaction, demonstrating that CCM function is required to negatively regulate MEKK3 function in vivo66. Importantly, this essential functional role was confirmed in the neonatal mouse brain, where loss of MEKK3 or either of its established downstream transcriptional effectors, KLF2 or KLF4, was found to be sufficient to prevent CCM formation following endothelial cell loss of CCM function71–73. These studies have established the MEKK3-KLF2/4 signaling pathway as a central signaling axis in CCM disease pathogenesis.
Modulators of CCM signaling: HEG1, CCM2L, STK24/25 and CDC42
In addition to the core players of the CCM complex and MEKK3-KLF2/4 signaling pathway, genetic and biochemical studies have identified a number of other proteins that appear to modulate this pathway, but for which precise roles in CCM disease remain less understood.
HEG1:
Forward genetic studies in fish demonstrated that loss of the transmembrane protein HEG1 conferred a big heart phenotype identical to that observed with loss of the CCM complex74. In mice, loss of HEG1 does not confer a developmental cardiovascular phenotype, but Heg1−/−;Ccm2+/− animals exhibit branchial arch artery defects identical to Ccm2−/− animals, consistent with action in a common pathway59. However, postnatal loss of HEG1 in endothelial cells does not confer or sensitize to CCM formation, and human genetic studies have not identified HEG1 mutations in familial CCM disease75. Biochemical studies reveal that HEG1 directly binds KRIT159, 76, and both proteins regulate endothelial junctions in cultured cells77, 78. The role of HEG1 remains incompletely understood, but present data suggest that HEG1 functions in association with the CCM complex in a manner that may be independent of MEKK3 but important for regulation of endothelial cell junctions. Biochemical and cellular studies suggest that an important mechanism for HEG1 regulation of endothelial junctions is the binding and localization of RASIP178.
CCM2L:
This is an orthologue of CCM2 with a highly homologous PTB domain by which it may also bind KRIT179–81. Unlike CCM2, CCM2L is expressed specifically in endothelial cells in a dynamic pattern during angiogenic and cardiogenic processes79. Biochemical studies reveal that CCM2L, like CCM2, binds MEKK379, 81, but whether CCM2L acts in a redundant manner with CCM2 to negatively regulate MEKK3 or as a dynamically expressed CCM2 antagonist remains controversial. Genetic interaction studies in zebrafish embryos support a redundant role with CCM2 in heart development80–82. In contrast, genetic studies in developing mice revealed no phenotype with loss of CCM2L and instead found that loss of CCM2L rescued lethal cardiovascular defects observed in Heg−/−;Ccm2+/− embryos. These findings suggested opposing roles for CCM2 and CCM2L, a conclusion also supported by biochemical studies demonstrating that CCM2L does not bind PDCD10, an essential component of the CCM complex79. Thus, CCM2L likely plays a modulatory role for CCM signaling in endothelial cells during cardiovascular growth. Whether that role is redundant with or opposing to that of CCM2 remains controversial, but there is presently no evidence that CCM2L participates in postnatal CCM lesion formation.
STK24 and STK25:
The GCKIII serine/threonine kinases STK24 (aka MST3) and STK25 associate with the PDCD10/CCM3 protein through its focal adhesion targeting (FAT) domain in numerous contexts, including the CCM complex56, 83–85. Genetic studies in zebrafish reveal that loss of both STK24 and STK25 results in the big heart phenotype characteristic of CCM function loss84. Thus, it is likely that STK24/25 function is required for CCM complex function, but this hypothesis has not yet been tested in mice and the identity of the substrate(s) of these serine-threonine kinases in this context remains unknown.
CDC42:
CDC42 was identified as an unexpected modulator of CCM function and lesion formation following postnatal endothelial-specific deletion studies in mice. Loss of endothelial CDC42 was first noted to confer capillary-venous malformations in the angiogenic neonatal retina86 and formation of lesions in the neonatal hindbrain that resembled CCMs both physically and spatially, and were associated with elevated endothelial levels of KLF487. Like CCMs, vascular lesions conferred by endothelial CDC42 loss can be reversed by loss of KLF4, and biochemical studies revealed interaction between CDC42 and the CCM complex mediated by PDCD1087. These studies suggest that CDC42 may participate in CCM disease pathogenesis, perhaps in the still undefined mechanism by which the CCM complex negatively regulates MEKK3 activity in endothelial cells.
Upstream MEKK3-KLF2/4 signaling in CCM disease
Identifying key upstream signaling inputs that drive CCM disease is a priority because the nature of these inputs is expected to shed light on both disease pathogenesis and potential CCM therapies. In vitro and in vivo studies in mice have defined two major inputs to the MEKK3-KLF2/4 signaling pathway in endothelial cells: hemodynamic shear forces and inflammatory signals. Both have been implicated in CCM disease pathogenesis, as discussed below.
The gut microbiome and brain endothelial TLR4 signaling define a CCM disease gut-brain axis.
Studies of MEKK3 signaling in cultured fibroblasts have demonstrated that it is required for innate immune inflammatory signaling in response to lipopolysaccharide (LPS) and interleukin 1b (IL1b)88. A first clue linking CCM disease to inflammatory signals in brain endothelial cells came from human genetic studies that associated polymorphisms in the genes encoding the LPS receptor TLR4 and its co-receptor CD14 with more severe familial CCM disease28. This connection remained mysterious until studies in mice serendipitously demonstrated that CCM lesion burden varied from severe to barely detectable depending upon the mouse facility the animals were housed in. Mice in a resistant colony only generated CCM lesions when they developed gram negative bacterial abscesses, suggesting that LPS derived from gram negative bacteria (GNB) drives lesion formation. This hypothesis was supported by the ability to prevent CCM lesion formation in susceptible animals with endothelial-specific loss of the LPS receptor TLR489. Susceptibility to CCM lesion formation in mice tracked with the GNB content of the gut microbiome, and treatment of susceptible animals with antibiotics or housing in germ-free conditions prevented CCM formation89, indicating that LPS from GNB that reside in the gut microbiome is an essential driver of CCM formation in the brain (Figure 3). Importantly, these studies in mice are supported by analysis of the gut microbiome in humans with and without symptomatic CCM disease. These human studies confirm the importance of LPS derived from GNB and define a permissive microbiome, characterized by more abundant Bacteroides species, in human CCM disease90. These unexpected findings have established a particularly straightforward gut-brain disease axis in which GNB in the gut microbiome provide a ligand, LPS, required to activate TLR4 receptors and downstream MEKK3-KLF2/4 signaling in brain endothelial cells. They further identify inflammatory signaling as the primary driver of CCM formation in vivo and suggest that future diagnostic and therapeutic strategies based on manipulation of the gut microbiome could be used to treat CCM disease.
Figure 3. Gut-brain axis of CCM disease.
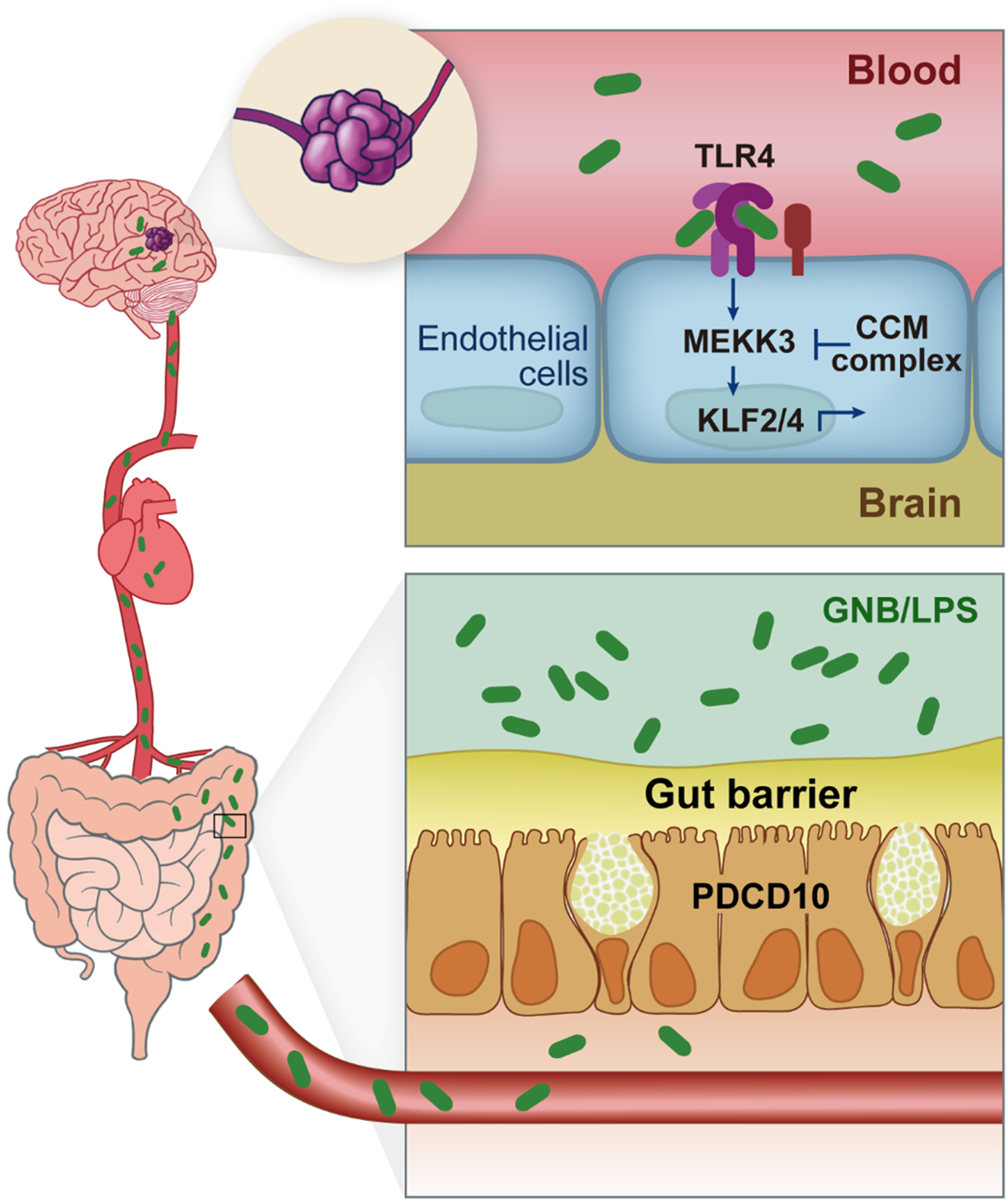
The gut microbiome and gut barrier are drivers of cavernoma formation. Gram-negative bacteria (GNB) and GNB-derived lipopolysaccharide (LPS) translocate across the mucosal and epithelial gut barrier into blood stream. LPS activates Toll-like Receptor 4 (TLR4) on brain endothelial cells and serves as an inflammatory upstream input into the MEKK3-KLF2/4 signaling pathway. PDCD10, unlike KRIT1 and CCM2, is required for the secretion of mucus by intestinal goblet cells. Germline loss of one PDCD10 allele reduces the gut barrier, thereby increasing translocation of LPS to the blood and accelerating the growth of CCM lesions in the brain. Adapted from Tang et al.27, 89
PDCD10 plays dual roles at both ends of the gut-brain CCM disease axis.
Further evidence supporting the importance of a gut-brain axis in CCM disease has come from recent studies that address the role of PDCD10/CCM3. Studies of familial CCM disease have shown that germline loss of either KRIT1 or CCM2 results in a similar disease course, with symptomatic disease most frequently arising in middle age. In contrast, familial CCM disease due to germline loss of one PDCD10 allele has been found to result in earlier and more severe disease, often presenting as stroke in childhood25. Since the three CCM proteins contribute equally to the molecular CCM complex, the basis for this difference in natural history has not been clear. The answer to this puzzle turns out to be a dual role for PDCD10 in CCM disease. Genetic deletion studies in mice demonstrate that in the brain endothelium PDCD10 is required in much the same way as KRIT1 or CCM2, i.e., as a requisite component of the CCM complex27. In the gut epithelium, however, PDCD10 is required for the secretion of mucus by goblet cells, a critical part of the gut barrier that separates the billions of bacteria from the gut wall and vasculature. This function is not shared with KRIT1 and CCM2, and is likely mediated by PDCD10 participation in the STRIPAK complex that does not contain those proteins27. Germline loss of one PDCD10 allele reduces the gut barrier, thereby enabling translocation of GNB-derived LPS to the blood and accelerating the growth of CCM lesions in the brain that form following loss of PDCD10 in brain endothelial cells. These findings highlight the importance of the gut-brain axis for CCM disease, and that of the gut barrier in preventing CCM formation over an individual’s lifetime.
The role of hemodynamic activation of MEKK3-KLF2/4 signaling in CCM disease.
The best characterized input to endothelial MEKK3-KLF2/4 signaling is hemodynamic shear force; thus it logical to consider that hemodynamic forces play a significant role in CCM disease. Studies in the developing zebrafish and mouse embryo have demonstrated that MEKK3-KLF2/4 signaling is regulated by fluid shear forces during cardiovascular development91–93. The CCM complex has been shown to regulate the MEKK3-KLF2/4 pathway in this context, and its manipulation alters cardiovascular developmental processes linked to fluid forces in the zebrafish embryo66, 94–98. These studies suggest that the CCM pathway may regulate MEKK3-KLF2/4 signaling stimulated by hemodynamic forces, but whether and to what extent pathway analysis in the developing embryo can be directly translated to CCM lesion formation in the postnatal brain remains unclear. CCM lesions in the brain first arise in post-capillary venules, a site of extremely low fluid shear. However, the beta blocker propranolol, an agent that reduces cardiac output and hemodynamic shear forces, ameliorates both zebrafish developmental phenotypes and postnatal CCM lesions conferred by CCM loss of function42. Future studies that focus on hemodynamic shear forces specifically in the venules and veins of the postnatal brain will better define the role of this upstream input in CCM pathogenesis.
Downstream MEKK3-KLF2/4 signaling effectors in CCM disease
A final signaling question remains the nature of the downstream MEKK3-KLF2/4 effectors that drive CCM lesion formation. A very large number of signaling pathways have been found to be altered in CCM lesions, and thereby implicated in CCM pathogenesis, but functional data that test causal mechanism are more scarce.
RHO/ROCK and endothelial junctions.
Studies in cultured endothelial cells and zebrafish embryos have revealed that loss of CCM function results in activation of the RHO/ROCK signaling pathway with elevated levels of phospho-myosin light chain (pMLC) and altered vascular integrity84, 99–101. These findings have stimulated therapeutic efforts aimed at normalizing this pathway in CCM-deficient cells, e.g., with the use of the ROCK inhibitor Fasudil or antagonizing microRNA inhibition of the endothelial cell junction protein VE-cadherin, both of which can reduce lesion burden in mice97, 102. Mouse genetic studies demonstrate that reducing expression of either MEKK3 or KLF2 or KLF4 blocks the increase in pMLC conferred by CCM loss71. Confusingly, endothelial loss of MEKK3 also results in increased endothelial pMLC expression68. However, preclinical studies in mice suggest that ROCK inhibition can reduce CCM lesion burden102. Thus, changes in RHO/ROCK signaling appear to be downstream of increased MEKK3-KLF2/4 signaling and participate in CCM formation, but the molecular pathway between them remains undefined.
End-MT and cell non-autonomous signals.
The rise in expression of KLF4 and other mesenchymal cell markers has led to a model in which CCM-deficient endothelial cells undergo mesenchymal transition, thereby becoming pathogenic cells creating CCM lesions72. Recent lineage tracing studies in mice have further demonstrated that as CCM lesions grow they incorporate large numbers of unlabeled, presumably wild-type, endothelial cells54, 55. These findings have generated a model in which CCM-deficient cells drive early lesion formation (perhaps through the generation of endothelial progenitor-like cells55) with subsequent cell non-autonomous effects on neighboring wild-type endothelial cells that fuel later lesion growth54. One such cell non-autonomous mechanism may be secretion of ADAMTS5, a versican-degrading metalloprotease that has been shown to be augmented in CCM-deficient endothelial cells in vivo and in vitro, with effects on CCM growth that are genetically linked to altered versican proteolysis103. Another may be reduced expression of thrombospondin 1 (TSP1), a secreted anti-angiogenic protein that suppresses CCM formation104. Future testing of the role of wild-type endothelial cells in CCM lesion formation in human lesions will be particularly valuable in understanding this disease.
PI3K-mTOR signaling is a critical downstream mechanism of CCM formation.
Very recent mouse and human genetic studies have demonstrated an unexpected downstream mechanism by which CCM loss of function and MEKK3-KLF2/4 gain of function confer lesion formation: increased PI3K-mTOR signaling51. As noted above, mouse models have identified a strong association between sites of active angiogenesis and CCM formation. Following global endothelial cell loss of CCM function, neonatal mice form lesions specifically in the hindbrain and retina, two sites of active angiogenesis after birth63. If CCM gene deletion is induced at a later timepoint when those sites are less angiogenic, CCM formation is lost105. Conversely, addition of the angiogenic growth factor VEGF-A106 or loss of the anti-angiogenic factor TSP1104 augment CCM formation. These observations suggest that loss of CCM function alone is not sufficient for lesion formation, and that other signals associated with active angiogenesis are also required. Since VEGF is a strong activator of PI3K signaling, CCMs arise in the venous system, and gain of function mutations in PIK3CA are known to drive venous and lymphatic malformations107–112 we examined the interaction between CCM loss of function and PIK3CA gain of function in vivo. In the neonatal mouse, CCM loss of function and PIK3CA gain of function drove the formation of similar venous lesions, and strong synergy was observed when both were present. In a new adult mouse model in which Cre-encoding adeno-associated virus is injected directly into the mature brain, neither alone was sufficient to create a lesion but the combination resulted in classic cavernoma formation.
As described above, bulk DNA sequencing revealed PIK3CA gain of function mutations identical to those observed in human cancer in a majority of surgically resected human CCM lesions. Single nucleus DNA sequencing further revealed that CCM mutations and PIK3CA mutations arise in the same cell, supporting an intracellular molecular interaction. Consistent with these mouse and human genetic findings, both CCM loss of function and KLF4 gain of function increase endothelial PI3K-mTOR signaling, and Rapamycin, an approved mTORC1 inhibitor, potently blocks CCM formation in both neonatal and adult mice. These new studies are consistent with prior reports that identified PI3K and mTOR signaling in CCM regulation of autophagy113 and in blinded studies of potential CCM signaling mechanisms114 and identify PI3K-mTOR signaling as a critical downstream effector mechanism in CCM disease and reveal a compound molecular and genetic pathogenesis similar to cancer in which the CCM complex plays the role of a vascular growth suppressor while PIK3CA plays the role of a vascular growth oncogene.
Other pathways implicated in CCM pathogenesis
In addition to the pathways described above, studies have implicated Notch115–117, WNT118 and TGFb signaling72, 73 in various stages of CCM disease. We have not discussed them in detail because their roles of these pathways are less investigated and their precise contributions less defined.
Future signaling directions.
There remain many outstanding questions regarding the signaling events that culminate in CCM disease. Why do CCMs arise specifically in the central nervous system? Does this reflect a specific environmental factor, e.g. the presence of perivascular versican or some other aspect of the neurovascular unit, or is it possible that they arise elsewhere but only come to clinical attention in the brain. How does the CCM complex negatively regulate MEKK3? Is MEKK3 an STK24/25 substrate? Are there still unidentified molecular players? Could CCM loss of function mutations contribute to other vascular malformations, e.g., those strongly associated with increased PI3K-mTOR signaling? Finally, are the vascular changes conferred by CCM loss of function reversible? The recent strides made in understanding CCM pathogenesis have shed great light on the mechanism of this disease and raised as many questions as they have answered.
CM signaling pathways: Mechanisms underlying hemorrhage
Why do CCM lesions bleed?
Bleeding is a major source of morbidity in CCM, yet the molecular and cellular events that underlie acute or chronic hemorrhage in this disease are unclear. CCMs are dynamic lesions that can form, enlarge, regress, or behave aggressively, producing repetitive hemorrhages49, 119, 120. Hemorrhagic strokes are a significant cause of disability in patients living with CCMs121–124 and a combination of CCM enlargement and bleeding can cause focal neurological deficits, headaches, epileptic seizures, and occasionally death123, 124. Natural history studies and MRI analysis have identified that CCM location and past hemorrhage as significant risk factors for bleeding and subsequent clinical sequelae.123, 125–127. Factors such as gender, lesion number, and size are not conclusively related to bleeding risk127, 128. Studies also estimate that symptomatic CCM hemorrhages rates are higher for familial CCMs than for sporadic CCMs123, 124, 129, 130. In addition, patients with PDCD10 mutations are more likely to present significant CCM hemorrhages earlier in life24–26. Although the annual symptomatic hemorrhage rate varies largely among different studies from 0.25 to 22.9% per patient-year,131, 132 it is thought that all CCM harbor occult bleeding123, 132 because a hemosiderin halo is the hallmark of CCMs on MRI. Here we summarize some of the potential mechanisms that may lead to bleeding in CCM (Figure 4.)
Figure 4. Mechanism of increase in TM and EPCR in CCM.
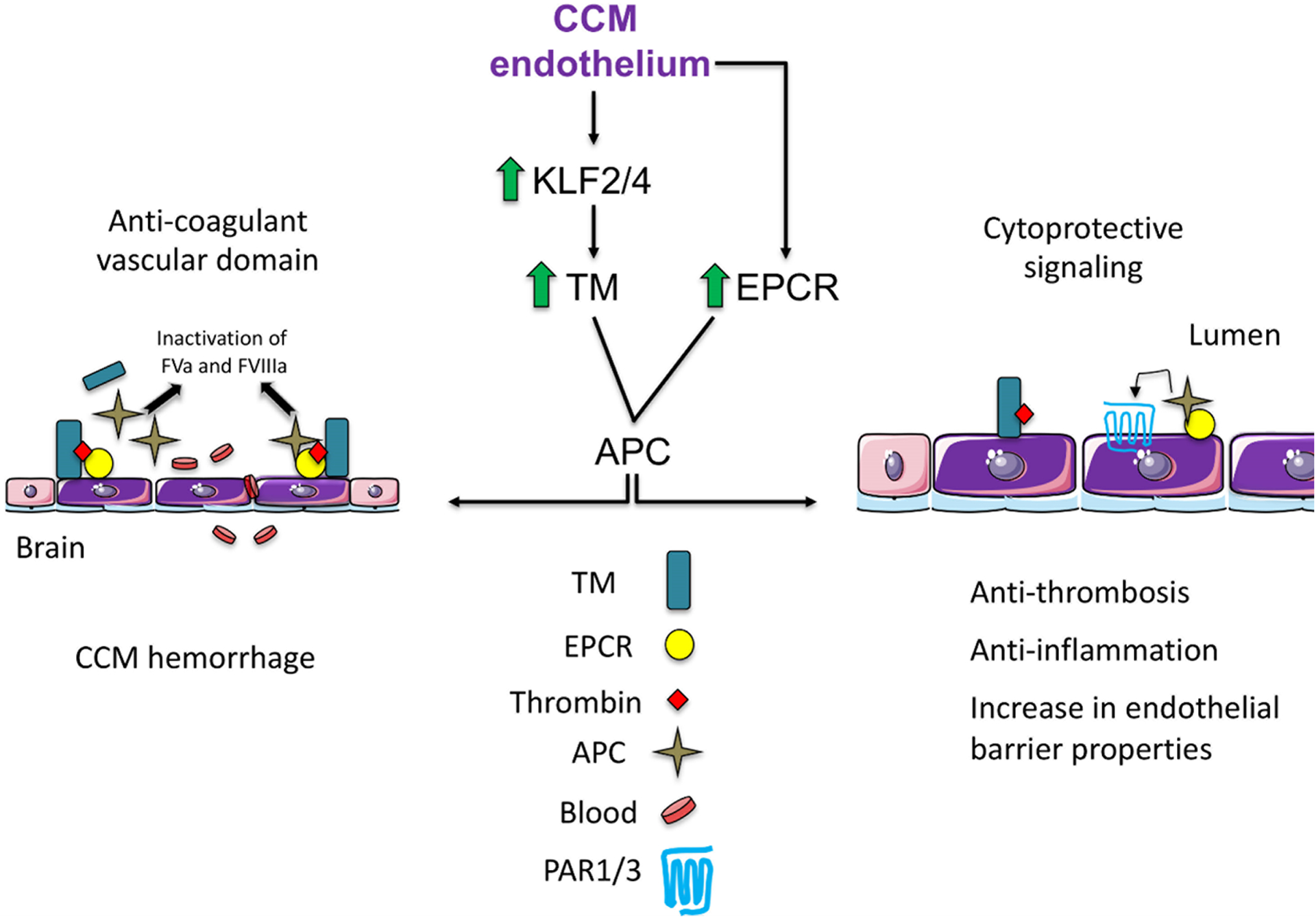
CCM endothelium is associated with locally elevated expression of anticoagulant endothelial receptors TM and EPCR. TM upregulation is due to upregulation of KLF2 and KLF4 transcription factors. Increased levels of vascular TM and EPCR result in enhanced APC and the anticoagulation cascade by inactivation of FVa and FVIIIa, thus contributing to an increase in lesion bleeding (Anti-coagulant vascular domain). Proposed cytoprotective signaling in CCMs, retention of APC to endothelial receptor EPCR allows activation of PAR1/3 and subsequent cytoprotective signaling in CCM endothelium. TM-thrombin complex reduces fibrin generation, and TM exerts anti-inflammatory properties. TM=thrombomodulin, EPCR= endothelial protein C, APC=activated protein C. Adapted from Lopez-Ramirez et al.121 and Mosnier et al.152
Loss of cell-cell junctions as a cause of CCM hemorrhage
Electron microscopic and immune histochemical analyses have documented that disruption of the endothelial barrier by disassembling of inter-brain endothelial junctions in CCMs104, 133, 134. This loss of cell-cell junctions could trigger the recurring micro-hemorrhages that cause hemosiderin deposition and the propensity for seizures119. Moreover, this mechanism is also supported by results in CCM mouse models. The expression and assembly of claudin5, ZO1, and VE-cadherin at the endothelial junctions are dramatically altered in dilated and hemorrhagic CCM lesions but normal in perilesional vessels72, 104, 105, 121. In addition, in familial CCM3 disease, there is a profound change in the tight junctions in brain endothelial cells that could affect the underlying severity of CCM3 disease134 and early-onset in brain hemorrhage26. Importantly CCM endothelium also exhibits combination an increase in cytoskeleton contractility in low flow areas98, 99, 101, 102, 127 that predispose to rupture leading to hemorrhagic stroke134, 135. Furthermore, mice harboring CCMs experience acute hemorrhage when injected with a single sub-lethal dose of LPS136, which could mimic the important effects of gut microbiome and gut barrier integrity on CCM exacerbation89 described above. Together these data suggest that CCM hemorrhages may result from unstable endothelial cell-cell contacts and that LPS-induced complete disassembly of endothelial cell-cell junctions or frank endothelial cell death can greatly exacerbate bleeding137–139.
CCMs form an anti-thrombotic vascular domain in the brain
Normal brain vasculature maintains restricted levels of thrombomodulin (TM) and endothelial protein C receptor (EPCR) compared to systemic blood vessels140, 141 suggesting that the brain is a pro-hemostatic environment. A genome-wide transcriptome analysis of the acute effects of inactivation of Krit1 or Pdcd10 in murine brain endothelial cells (BMEC) found increased levels of Thbd mRNA, which encodes the natural anticoagulant receptor, TM.104, 142 TM binds thrombin and while bound, thrombin fails to convert fibrinogen into insoluble fibrin, and instead catalyzes formation of activated protein C (APC). APC generation is enhanced by the presence of the EPCR,143, 144 the mRNA of which is also increased with loss of Krit1 or Pdcd10 in BMECs.104, 121, 142, 145 Because APC is a potent natural anticoagulant, that inactivates the coagulation factor Va and factor VIIIa, high levels of TM have been associated with a bleeding disorder146, 147 and used as a biomarker of endothelial cell dysfunction,148 these data suggested that increased TM and EPCR could create a local bleeding diathesis in CCM121.
Familial and sporadic forms of CCMs exhibited significant increases in soluble TM in plasma, suggesting that this may be a biomarker for patients at elevated risk of bleeding104. Elevated levels of TM in plasma are associated with endothelial dysfunction148, 149 and bleeding disorders.146–148. Moreover, human CCM endothelium displays increased levels of TM and EPCR mRNA and protein104. As discussed above, the increased TM can reflect an increased formation of APC in lesions which, in turn, leads to anticoagulant activity143 that facilitates bleeding. Insights gleaned from bleeding in Hemophilia increased our awareness that excessive or unbalanced APC generation can actively contribute to bleeding and that the anticoagulant activity of APC is a valid target for drug development.150, 151
APC induces cytoprotective effects on endothelial cells and neurons that are dependent on protease activated receptor (PAR) 1, PAR3, and EPCR152, 153.These cell signaling activities of APC are central to the primary mechanism of action for APC’s neuroprotective effects in a variety of acute and chronic neuropathologies.154 Strategies targeting the protein C system aimed at mitigating or preventing bleeding in CCM, therefore, need to be specific for APC anticoagulant activity while leaving APC’s neuroprotective activities unaltered (Figure 4). Several anticoagulant-specific strategies targeting the protein C pathway have been proposed for control of bleeding in hemophilia patients.150, 151 Whether these strategies can be safely adapted to CCM will have to be determined since the safety window in hemophilia and CCM is likely quite different and targeting of the acute central nervous system (CNS) bleeds versus the risk of re-bleeding in the 1 year window of increased vulnerability that follows such bleeds will be an important consideration for this.123, 132. Another consideration is that the high level of APC in CCM lesions could diminish morbidity by means of its cytoprotective effects that include protection of vascular and blood-brain-barrier integrity.144, 155
The dramatic elevation of TM and EPCR in CCM endothelium121, 145 may also serve a protective anti-thrombotic function. The low flow environment of CCM may be conducive to thrombosis, which, by obstructing outflow, could lead to lesion expansion or frank rupture. Indeed, a recent retrospective analysis suggested that CCM patients who received anti-thrombotic therapy experienced less morbidity156. In this scenario, the activation of Protein C by CCM endothelium may serve a protective role. Resistance to APC anti-coagulant activity, due to Factor V Leiden, is one of the commonest thrombophilias157 and CCM patients with this mutation may be at enhanced risk for thrombosis within CCM and at much greater likelihood to be treated with an anti-thrombotic. Indeed, pre-screening CCM patients for thrombophilia may be helpful in guiding future studies of the effect of anti-thrombotics on CCM.
The studies described above show that the potential consequences of the enhanced APC generation in CCM are manifold (Figure 4). The diverse effects of Protein C as an anticoagulant and as a cytoprotectant emphasize the need for studies to analyze these complexities and to evaluate strategies targeting the different functions of the protein C system in CCM. Studies that develop approaches to junction stabilization99, 101, 158 and the APC pathway can point to new therapies. Conversely, as the number of potentially new therapeutic interventions in CCMs grows,73, 89, 104, 117, 135 the identification of markers to identify patients with elevated risk of hemorrhage becomes imperative. Indeed, recent studies137, 159 that showed that levels of inflammatory and angiogenic molecules in peripheral blood plasma could predict CCM behavior have already begun. The work reviewed here also suggests that a thorough understanding of the hemostatic balance, and Protein C pathway in particular, can contribute to the management of CNS hemorrhage in CCM.
Translational Roadmap: Biomarkers and Trial Readiness
Biomarkers and Clinical Features of CCM disease
In CCM disease, several primary sequelae are considered strongly in clinical management, including symptomatic lesional growth and hemorrhage, and lesion burden (Figure 5).160 While the annual risk of a first symptomatic hemorrhage is quite low (0.08% per patient year), after an initial bleeding event, the risk of a subsequent bleed increases more than ten-fold.160 The clinically adjudicated definition of Cavernous Angioma with Symptomatic Hemorrhage (CASH) requires imaging evidence of bleeding or growth with directly attributable symptoms. Bleeding may be clinically silent or misdiagnosed, and other neurological symptoms such as headache or seizure can occur without an overt, new bleed.160, 161 Seizures and focal neurologic deficits can also be caused by lesion growth or by cumulative, chronic bleeding. Lesional growth may occur because of proliferation of new caverns, or as a result of expansion of one or more caverns (intralesional bleeding). Hemorrhage may also leak beyond the confines of the lesion. Lesional expansion and symptomatic hemorrhage are clinically relevant in familial disease and also in the more common sporadic lesions, where similar somatic mutations have been identified. There is a need for sensitive and specific diagnostic and prognostic biomarkers of CASH lesions.162 These biomarkers may help reveal which patients bled or may suffer a hemorrhagic stroke in the near future and therefore need more urgent or risky surgical interventions. High risk lesions may also be selectively targeted by emerging therapies. A goal of lesion stabilization may be pursued, or ultimately a more ambitious goal of lesion regression.
Figure 5. Progression of CCM Lesion Pathogenesis.
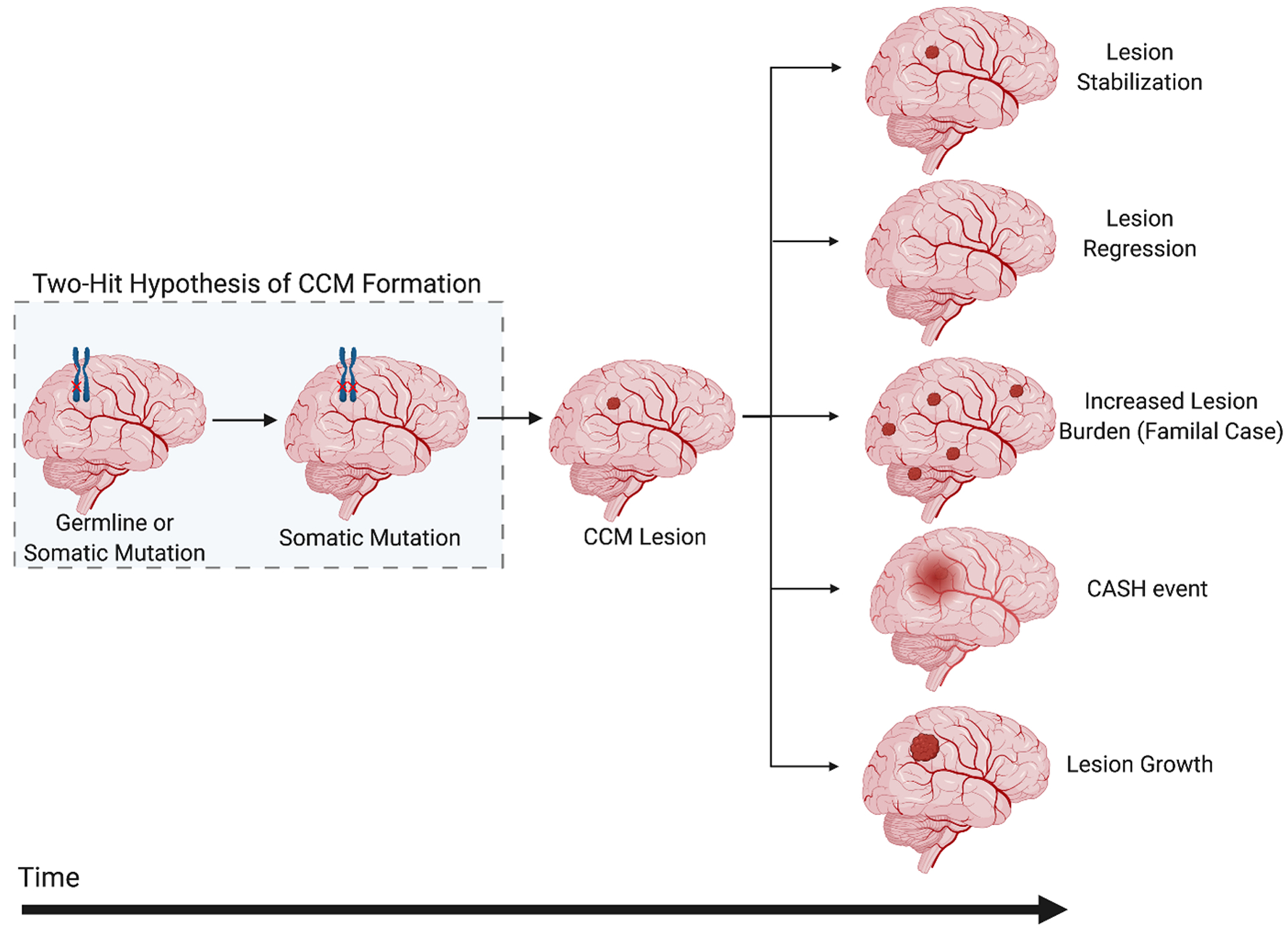
The pathogenesis of CCMs begins with an inherited or somatic mutation, followed by somatic mutations resulting in lesion genesis and growth. The natural history of CCM disease is thought to lead to five clinically relevant outcomes (1) lesion stabilization, (2) lesion regression, (3) increased lesion burden, (4) symptomatic hemorrhage (SH), and/or (5) lesional growth (shown from top to bottom). CASH, Cavernous Angioma with Symptomatic Hemorrhage.
Lesion burden is relevant in familial disease, where lesions develop stochastically during a patient’s lifetime throughout the brain and spinal cord, and in sporadic cases, where new lesions can develop in association with a pre-existing developmental venous anomaly. Lesion burden can be tracked using susceptibility magnetic resonance imaging (MRI) sequences revealing the earliest development of punctate lesions, well before any clinical manifestations. An increase in the number of CCMs increases the chances of subsequent related lesional growth and SH (Figure 5).
Mechanistic research and the transcriptome of human and mouse CCM lesions have clarified several features of CCM disease (Table 1), and disease models have been adapted to better examine various features of lesion genesis, maturation and growth, intralesional and extralesional hemorrhage.97, 136 Therapeutic targets may be better suited for one aspect or another among disease manifestations, including a potential promise of combination therapies.
Table 1.
Candidate circulating plasma and imaging biomarkers and potential CCM therapies.
| Mechanisms of disease | Plasma circulating biomarkers | Validating preclinical models | Candidate therapeutics | Lesion Pathogenesis | Development status |
|---|---|---|---|---|---|
| MAPK Signaling, PI3K-mTOR Signaling, microbiome mechanisms27, 43, 51, 52, 71, 89, 90, 103, 137 | ADAMTS4 ADAMTS5 TLR4 LPB (LPS) CD14 |
murine/human | BIX02189 (anti-MEK5) XMD17-109 (anti-ERK5) Rapamycin (mTOR inhibitor) TLR4 inhibitors |
Lesion burden Bleeding | Preclinical |
| Angiogenesis106, 137, 162, 179 | VEGF Angiopoietin 1–2 ROBO4 Thrombospondin 1 |
murine/human | Bevacizumab Semaxanib TSP1 replacement |
Lesion growth Bleeding |
Preclinical |
| Inflammatory processes, autophagy, focused immune response137, 159, 176, 178, 179, 184 | 25-OH Vitamin D CRP Endoglin IL-1β IL-10 |
murine/human | Vitamin D3 Tempol Anti-BR3 antibody |
Lesion burden Bleeding |
Preclinical |
| Coagulation domains and thrombin-endothelial interactions104, 121, 162 | Thrombomodulin | murine/human | Potential biomarker | Bleeding | Preclinical |
| Rheologic mechanisms97, 121 | Blood pressure pro-thrombotic states |
zebrafish/murine/human | Propranolol β1receptor antagonists |
Lesion growth | Preclinical Phase I-IIA |
| Endothelial-mesenchymal transition97 | TGF-β | murine | Exisulind Propranolol |
Lesion burden | Preclinical |
| Endothelial permeability (RhoA/ROCK)166, 180–182 | pMLC pMBS Leukocytes |
murine/human | Fasudil BA1049 Atorvastatin |
Lesion burden Bleeding | Preclinical Phase I-IIA |
MAPK = Mitogen Activated Protein Kinase; RhoA = Ras homolog gene family member A; ROCK = Rho-associated protein kinase; ADAMTS4 = ADAM Metallopeptidase With Thrombospondin Type 1 Motif 4; ADAMTS5 = ADAM Metallopeptidase With Thrombospondin Type 1 Motif 5; TLR4 = Toll-Like Receptor 4; LPB = Lipopolysaccharide Binding Protein; LPS = Lipopolysaccharide; CD14 = Cluster of Differentiation 14; VEGF = Vascular Endothelial Growth Factor; ROBO4 = Roundabout Homolog 4; TSP1 = Thrombospondin 1; CRP = C-Reactive Protein; IL-1β = Interleukin-1β; IL-10 = Interleukin-10; TGF-β = Transforming Growth Factor-β; pMLC = phosphorylated Myosin Light Chain; pMBS = phosphorylated Myosin Binding Subunit; MEK5 = dual specificity mitogen-activated protein kinase 5; ERK = extracellular signal-regulated kinase; BR3 = BLyS receptor 3/BAFF-R
Imaging in Preclinical Models and in CCM Patients
Imaging modalities, such as MRI and Computerized Tomography (CT), have been used clinically as standard for diagnosis of CCMs (Figure 6 A and B). In preclinical studies, MRI was originally lauded as an opportunity to study lesions at near histologic resolution at various stages of development.163, 164 However, high field murine MRI scans are expensive and time consuming, preventing high-throughput application.163 Micro-CT instruments (using paleo-CT instruments initially developed for fossil imaging) have been effectively purposed for high-throughput assessment of lesion burden and volume while complementing histology in ex-vivo pre-clinical studies in CCM mouse models.165 This diffusible iodine-based, contrast-enhanced, micro-CT protocol was originally developed to study the consequences of endothelial TLR4/CD14 signaling89 and the influence of the gut microbiome on CCMs pathogenesis.27 Later, the lesional burden, estimated using micro-CT, has been used to monitor not only the efficacy of candidate therapeutics,97, 104, 166 but also genetic manipulation in preclinical mouse models of CCM disease.103, 136
Figure 6. Imaging Biomarkers of CCM.
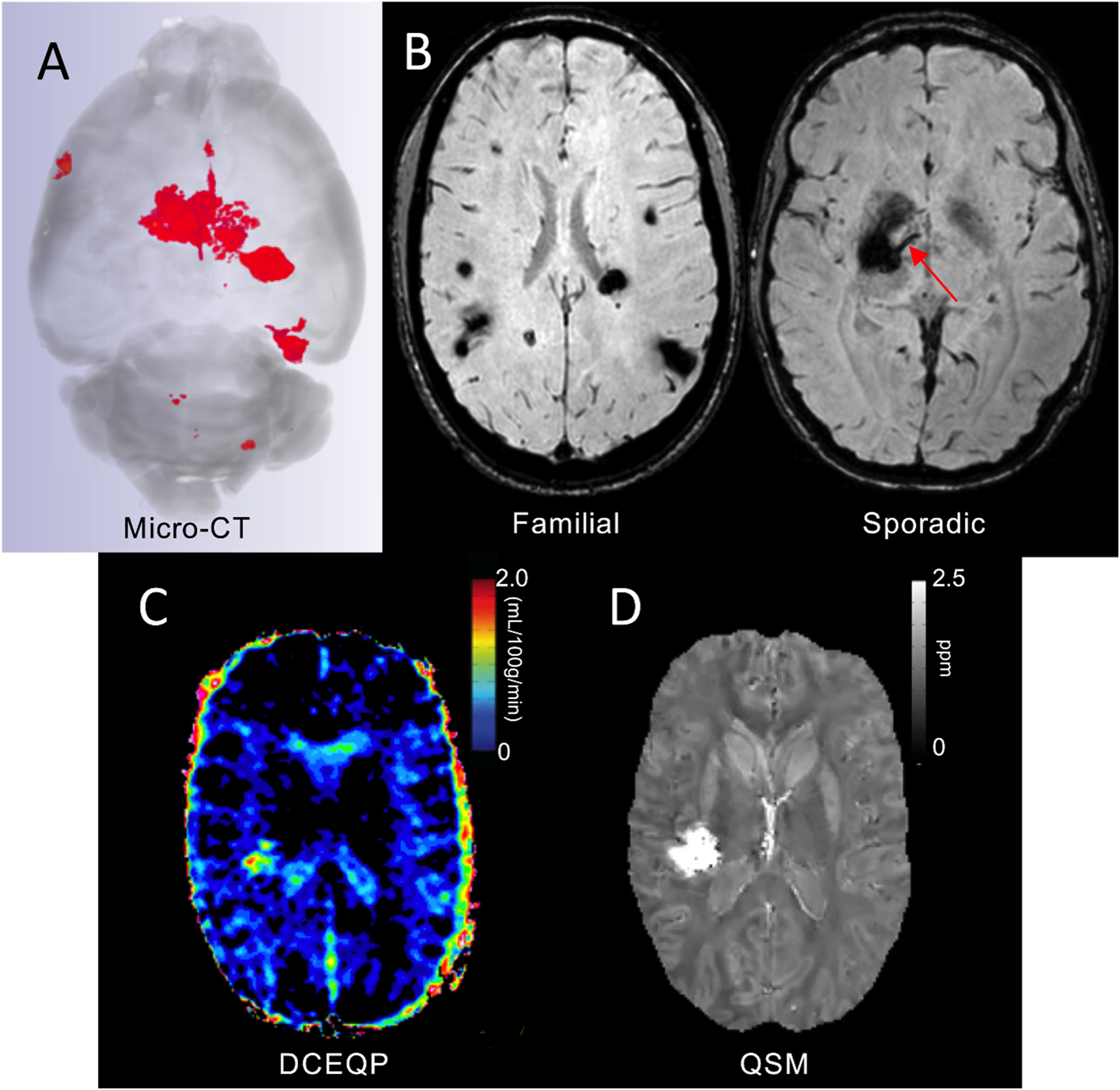
(A). Micro-CT is used to assess lesion burden, volume, and stage ex-vivo in murine brains. (B). Susceptibility weighted MRI assesses lesion burden in familial cases, and the presence of an associated developmental venous anomaly (red arrow) in sporadic cases. (C). Dynamic contrast enhanced quantitative perfusion (DCEQP) MRI assesses the vascular permeability of CCM lesions and background brain. (D). Quantitative susceptibility mapping (QSM) MRI assesses iron content in CCM lesions.
Increased vascular permeability is a hallmark of CCM disease physiopathology, mediating hemorrhage and non-heme iron accumulation,99, 101, 167, 168 In CCM patients, two in-vivo imaging biomarkers have therefore been proposed to assess lesional and background brain permeability (i.e., Dynamic Contrast Enhanced Quantitative Perfusion [DCEQP]) and iron accumulation (i.e., Quantitative Susceptibility Mapping [QSM]) (Figure 6 C and D).126, 167–171 DCEQP has been used to assess vascular properties of brain disorders, allowing estimation of perfusion, blood volume and blood-brain barrier permeability.169, 172 In fact, background brain permeability was confirmed to be greater in familial CCM disease, consistent with heterozygous germ line mutations in brain vascular bed.167 Further, longitudinal studies have reported an increased permeability within CCM lesions proper, after an SH, and in background brain parenchyma preceding new lesion formation.126 Taken together, these results support the use of DCEQP as diagnostic and monitoring biomarkers for endothelial hyperpermeability of CCMs.
QSM is a gradient echo-generated imaging modality that has been used to assess brain iron deposition in cerebrovascular disorders.173, 174 In surgically excised CCMs, QSM measurements have correlated precisely with iron concentrations measured by mass spectroscopy.170 No significant differences were observed in lesional iron deposition between sporadic and familial CCMs, nor in stable CCM lesions over time.171 The lesional QSM values increase consistently after a hemorrhagic event, suggesting that QSM can be used as a monitoring biomarker of CASH.126, 175
Circulating Plasma Biomarkers in Mice and Humans
CCMs demonstrate a complex pathophysiological milieu, involving a dynamic interplay between inflammation, angiogenesis, and the loss of endothelial barrier function.137, 159, 166, 176, 177 Preliminary studies have reported associations between the clinical course of CCM disease and systemic circulating compounds, suggesting they could act as peripheral blood biomarkers.104, 121, 137, 159, 178, 179 A weighted combination of 4 of the plasma levels of these candidates biomarkers, including sCD14, VEGF, CRP, and IL-10, was able to distinguish CASH patients with 76% sensitivity and 80% specificity thereby acting as a possible diagnostic biomarker.137, 179 In the same way, a prognostic biomarker of CASH, including the weighted plasma levels of sCD14, VEGF, IL-1B, and sROBO-4, was also able to predict an SH within the following year with 86% sensitivity and 88% specificity.137, 179 These are being optimized and validated as much needed diagnostic and prognostic biomarkers of CASH. These preliminary studies were the first insight into the development of diagnostic and prognostic biomarkers of CASH. They have since paved the way for testing new candidate biomarker molecules emerging from mechanistic studies.162
Transcriptomic identification and validation of biomarkers.
The transcriptome of lesional neuro-vascular units (NVUs) has served as a reference to validate mechanistic hypotheses related to the pathogenesis of CCMs, and also to identify candidate biomarkers;142. In humans, the transcriptome of CASH NVUs identified several DEGs related to 5 of the 6 circulating proteins composing the prognostic and diagnostic biomarkers, and therefore provided evidence that these markers are related to hemorrhagic CAs.142 In preclinical murine models, some of the mechanistic findings were then translated and validated in human patients. Lopez et al. (2019) showed increased levels of vascular Thrombomodulin (TM), an anticoagulant receptor, following an upregulation of transcription factors KLF2 and KLF4 in Ccm1−/− or Ccm3−/− BMECs and confirmed that Thbd was also dysregulated in human lesional NVUs.121 In addition, the plasma levels of TM were also increased in CCM patients, suggesting that plasma soluble TM may be used as a marker for hemorrhagic risk.121 These results provide compelling evidence that the transcriptomes can validate mechanistic discoveries, and also identify novel candidate biomarkers (Table 1).
Micro-RNAs (miRNAs).
In addition to plasma proteins, circulating micro-RNAs (miRNAs) have been suggested as possible mechanistic biomarkers. In CCM disease, 13 differently expressed (DE) miRNAs were identified in the plasma of CASH patients.179 These 13 DE miRNAs showed 16 putative gene interactions in the coding sequence (CDS) region of mRNA within the transcriptomes of human CCM NVUs. Of interest, further analyses showed that IL-10RA is the putative target of miT-185–5, one of these 13 DE miRNAs and decreased plasma levels of IL-10 have been reported in CCM subjects that experienced recent SH.179 While this is just the start of the vast utility of miRNAs clinically, further initiatives have been proposed to integrate these plasma DE miRNAs with plasma proteins to complement and/or increase the sensitivity/specificity of prognostic and diagnostic biomarkers of CASH.162 Preliminary ML simulations showed that the diagnostic biomarker of CASH was improved when plasma levels of DE miRNAs were included in the model.162 Further clinical and research utility of miRNAs may arise from their application as diagnostic and monitoring biomarkers of disease genotype.
Trial Readiness and Multisite Therapeutic Initiatives
While the development of clinically useful biomarkers as smart blood tests is important for the diagnosis, prognosis, and monitoring of the disease, current treatment of CCMs remains neurosurgical intervention and removal or ablation of a symptomatic lesion. Such intervention is associated with high cost and significant risks, particularly if the lesion involves critical brain regions.160 Neurosurgical intervention will not be necessary if a symptomatic lesion is stabilized, and its growth or hemorrhage are prevented. Therapies may also be targeted at preventing lesion development or growth, and potentially even to induce lesion regression. Therefore, pharmacotherapeutics are urgently needed in this disease, particularly given the wealth of emerging mechanistic information as described above. Several preclinical studies have already reported the effects of repurposed or novel drugs on CCM development and hemorrhage (Table 1).71, 97, 104, 166, 176, 180, 181
Potential obstacles have been identified related to accurate estimation of clinical case prevalence and the projected enrollment and event rates needed to drive clinical trials, harmonization of clinical and imaging data collection across clinical sites, and the analytic validation of risk and monitoring biomarkers that may be deployed in clinical trials. And there is a need to characterize precise event rates to monitor the impact of therapies. The CASH Trial Readiness (TR) initiative has set forth a protocol for foundational data collection and has been followed by an Exploratory Proof of Concept Trial of high dose atorvastatin as a ROCK inhibitor versus placebo.161, 182The latter trial is planned based on effect size of differences in lesional iron deposition (assessed using QSM) and lesional permeability (assessed using DCEQP).182
Another recent initiative assembles three CCM Centers of Excellence in order to optimize and validate diagnostic and prognostic biomarkers of CASH.162 As a part of this project, the aim will be to test whether individual and combined levels of candidate plasma proteins and miRNAs are associated with the diagnosis and prognosis of CASH. The project will also assess whether changes in imaging monitoring biomarkers after CASH are reflected in plasma molecules. Analyses will be done on possible confounders, such as age, hemorrhagic microangiopathy, and seizures, in the context of clinical use of these biomarkers to determine generalizability in the population.
Much mechanistic information has come to light in the past decade, to guide the therapeutic roadmap.176, 183, 184 CCM pathogenesis is a continuum from lesion development, to lesion growth, acute and chronic bleeding, with interactions including germ line mutations, somatic mutations, a permissive microbiome,90 pro-inflammatory genotypes and a defined immune response. It may be difficult to ascertain which aspect of the disease is being targeted by a drug in clinical trials, and optimal dose response considerations are required. Other determinations include how long to treat a patient, at what stage of the illness, and what therapeutic risk is justified and acceptable. This requires very focused mechanistic studies in preclinical models and rigorous translational paradigms. Biomarker driven trials offer advantages at Phase I and II stages to compare relative effects of various therapies, dose calibration and drug combinations, before committing a limited patient population to a Phase III trial with best chances at success.
Finally, there should be considerations beyond systemic pharmacotherapies. Other options may be pursued in the context of the previously articulated therapeutic goals, such as local drug therapy through an injected or endovascularly delivered viral vector, and laser or ultrasound lesion ablation. The development of multiple treatment options could aid in personalized patient treatment plans based on complexity of disease and comorbidities and calibrated risks.
Closing Remarks
The past two decades have seen a remarkable convergence of human genetic studies, genetic studies in fish and mice, and molecular and cellular insights in the field of CCM disease. These studies have shed light on the endothelial cell molecular and genetic responses to loss of CCM function, identified environmental conditions that strongly influence the cellular response and clinical disease, and, most recently, yielded updated genetic insights that support a cancer-like mechanism for the most aggressive CCM lesions. With a more sophisticated understanding of the disease mechanism in hand, the field is poised for successful translation of these findings to the creation of new, non-surgical therapies for CCM disease.
A final question that merits consideration is why so much progress has been made with CCM disease. CCM disease has benefited from the convergence of a number of scientific and medical approaches that provided synergy between multiple investigators to rapidly advance the field. Human genetics has played a major role, first defining the CCM complex through classic positional cloning studies of familial disease and then providing additional mechanistic insights through more advanced genomic sequencing techniques. Vertebrate models have also contributed important mechanistic insights, as expected, but the identification of a faithful neonatal mouse model that was adopted by a large number of investigators in the field has played a major role in moving the field forward because it enabled studies in many laboratories to be directly compared and integrated. As efforts to bring these insights to patients intensify, it will be important to remember these lessons and perform translational and clinical studies in a similarly integrated and efficient manner
Acknowledgements.
We would like to acknowledge Connie Lee and Amy Akers of Angioma Alliance for their many efforts to foster a greater scientific and clinical understanding of CCM disease. We would also like to acknowledge Dr. Jim Koenig and the NINDS for their support of the studies performed by the authors through P01NS092521 (MK, DM, IA and MG).
NONSTANDARD ABBREVIATIONS AND ACRONYMS
- CCM
cerebral cavernous malformation
- LOF
loss of function
- GOF
gain of function
- GNB
gram negative bacteria
- BMEC
murine brain endothelial cells
- CASH
cavernous angioma with symptomatic hemorrhage
- DCEQP
dynamic contrast enhanced quantitative perfusion
- QSM
quantitative susceptibility mapping
- NVU
neuro-vascular unit
Footnotes
Disclosures
None.
References Cited:
- 1.Bicknell JM, Carlow TJ, Kornfeld M, Stovring J and Turner P. Familial cavernous angiomas. Arch Neurol. 1978;35:746–9. [DOI] [PubMed] [Google Scholar]
- 2.Clark JV. Familial occurrence of cavernous angiomata of the brain. J Neurol Neurosurg Psychiatry. 1970;33:871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kidd HA and Cumings JN. Cerebral angiomata in an Icelandic family. Lancet. 1947;1:747. [DOI] [PubMed] [Google Scholar]
- 4.Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF and Tournier-Lasserve E. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189–93. [DOI] [PubMed] [Google Scholar]
- 5.Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, Touchman JW, Gallione CJ, Lee-Lin SQ, Kosofsky B, Kurth JH, Louis DN, Mettler G, Morrison L, Gil-Nagel A, Rich SS, Zabramski JM, Boguski MS, Green ED and Marchuk DA. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet. 1999;8:2325–33. [DOI] [PubMed] [Google Scholar]
- 6.Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, Verlaan D, Balogun F, Hughes L, Leedom TP, Plummer NW, Cannella M, Maglione V, Squitieri F, Johnson EW, Rouleau GA, Ptacek L and Marchuk DA. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A, Benabid AL, Comoy J, Frerebeau P, Gilbert B, Houtteville JP, Jan M, Lapierre F, Loiseau H, Menei P, Mercier P, Moreau JJ, Nivelon-Chevallier A, Parker F, Redondo AM, Scarabin JM, Tremoulet M, Zerah M, Maciazek J, Tournier-Lasserve E and Societe Francaise de N. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. 2004;74:326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, Jacquet G, Lonjon M, Moreau JJ, Neau JP, Parker F, Tremoulet M, Tournier-Lasserve E and Societe Francaise de N. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunel M, Awad IA, Finberg K, Anson JA, Steinberg GK, Batjer HH, Kopitnik TA, Morrison L, Giannotta SL, Nelson-Williams C and Lifton RP. A founder mutation as a cause of cerebral cavernous malformation in Hispanic Americans. N Engl J Med. 1996;334:946–51. [DOI] [PubMed] [Google Scholar]
- 10.Cau M, Loi M, Melis M, Congiu R, Loi A, Meloni C, Serrenti M, Addis M and Melis MA. C329X in KRIT1 is a founder mutation among CCM patients in Sardinia. Eur J Med Genet. 2009;52:344–8. [DOI] [PubMed] [Google Scholar]
- 11.Liquori CL, Berg MJ, Squitieri F, Leedom TP, Ptacek L, Johnson EW and Marchuk DA. Deletions in CCM2 are a common cause of cerebral cavernous malformations. Am J Hum Genet. 2007;80:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallione CJ, Solatycki A, Awad IA, Weber JL and Marchuk DA. A founder mutation in the Ashkenazi Jewish population affecting messenger RNA splicing of the CCM2 gene causes cerebral cavernous malformations. Genet Med. 2011;13:662–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gault J, Sain S, Hu LJ and Awad IA. Spectrum of genotype and clinical manifestations in cerebral cavernous malformations. Neurosurgery. 2006;59:1278–84; discussion 1284–5. [DOI] [PubMed] [Google Scholar]
- 14.Laurans MS, DiLuna ML, Shin D, Niazi F, Voorhees JR, Nelson-Williams C, Johnson EW, Siegel AM, Steinberg GK, Berg MJ, Scott RM, Tedeschi G, Enevoldson TP, Anson J, Rouleau GA, Ogilvy C, Awad IA, Lifton RP and Gunel M. Mutational analysis of 206 families with cavernous malformations. J Neurosurg. 2003;99:38–43. [DOI] [PubMed] [Google Scholar]
- 15.Denier C, Labauge P, Brunereau L, Cave-Riant F, Marchelli F, Arnoult M, Cecillon M, Maciazek J, Joutel A, Tournier-Lasserve E, Societe Francaise de N and Societe de Neurochirurgie de Langue F. Clinical features of cerebral cavernous malformations patients with KRIT1 mutations. Ann Neurol. 2004;55:213–20. [DOI] [PubMed] [Google Scholar]
- 16.Gianfrancesco F, Cannella M, Martino T, Maglione V, Esposito T, Innocenzi G, Vitale E, Liquori CL, Marchuk DA and Squitieri F. Highly variable penetrance in subjects affected with cavernous cerebral angiomas (CCM) carrying novel CCM1 and CCM2 mutations. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:691–5. [DOI] [PubMed] [Google Scholar]
- 17.Sirvente J, Enjolras O, Wassef M, Tournier-Lasserve E and Labauge P. Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol. 2009;23:1066–72. [DOI] [PubMed] [Google Scholar]
- 18.Musunuru K, Hillard VH and Murali R. Widespread central nervous system cavernous malformations associated with cafe-au-lait skin lesions. Case report. J Neurosurg. 2003;99:412–5. [DOI] [PubMed] [Google Scholar]
- 19.Grippaudo FR, Piane M, Amoroso M, Longo B, Penco S, Chessa L, Giubettini M and Santanelli F. Cutaneous venous malformations related to KRIT1 mutation: case report and literature review. J Mol Neurosci. 2013;51:442–5. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Liu XW, Lee N, Liu QJ, Li WN, Han T, Wei KK, Qiao S and Chi ZF. Features of a Chinese family with cerebral cavernous malformation induced by a novel CCM1 gene mutation. Chin Med J (Engl). 2013;126:3427–32. [PubMed] [Google Scholar]
- 21.Eerola I, Plate KH, Spiegel R, Boon LM, Mulliken JB and Vikkula M. KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associated with cerebral capillary malformation. Hum Mol Genet. 2000;9:1351–5. [DOI] [PubMed] [Google Scholar]
- 22.Labauge P, Enjolras O, Bonerandi JJ, Laberge S, Dandurand M, Joujoux JM and Tournier-Lasserve E. An association between autosomal dominant cerebral cavernomas and a distinctive hyperkeratotic cutaneous vascular malformation in 4 families. Ann Neurol. 1999;45:250–4. [DOI] [PubMed] [Google Scholar]
- 23.Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, Maciazek J, Vicaut E, Brunereau L, Tournier-Lasserve E and Societe Francaise de N. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006;60:550–6. [DOI] [PubMed] [Google Scholar]
- 24.Fauth C, Rostasy K, Rath M, Gizewski E, Lederer AG, Sure U, Zschocke J and Felbor U. Highly variable intrafamilial manifestations of a CCM3 mutation ranging from acute childhood cerebral haemorrhage to late-onset meningiomas. Clin Neurol Neurosurg. 2015;128:41–3. [DOI] [PubMed] [Google Scholar]
- 25.Shenkar R, Shi C, Rebeiz T, Stockton RA, McDonald DA, Mikati AG, Zhang L, Austin C, Akers AL, Gallione CJ, Rorrer A, Gunel M, Min W, De Souza JM, Lee C, Marchuk DA and Awad IA. Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet Med. 2015;17:188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riant F, Bergametti F, Fournier HD, Chapon F, Michalak-Provost S, Cecillon M, Lejeune P, Hosseini H, Choe C, Orth M, Bernreuther C, Boulday G, Denier C, Labauge P and Tournier-Lasserve E. CCM3 Mutations Are Associated with Early-Onset Cerebral Hemorrhage and Multiple Meningiomas. Mol Syndromol. 2013;4:165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang AT, Sullivan KR, Hong CC, Goddard LM, Mahadevan A, Ren A, Pardo H, Peiper A, Griffin E, Tanes C, Mattei LM, Yang J, Li L, Mericko-Ishizuka P, Shen L, Hobson N, Girard R, Lightle R, Moore T, Shenkar R, Polster SP, Roedel CJ, Li N, Zhu Q, Whitehead KJ, Zheng X, Akers A, Morrison L, Kim H, Bittinger K, Lengner CJ, Schwaninger M, Velcich A, Augenlicht L, Abdelilah-Seyfried S, Min W, Marchuk DA, Awad IA and Kahn ML. Distinct cellular roles for PDCD10 define a gut-brain axis in cerebral cavernous malformation. Sci Transl Med. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choquet H, Pawlikowska L, Nelson J, McCulloch CE, Akers A, Baca B, Khan Y, Hart B, Morrison L, Kim H and Brain Vascular Malformation Consortium S. Polymorphisms in inflammatory and immune response genes associated with cerebral cavernous malformation type 1 severity. Cerebrovasc Dis. 2014;38:433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cutsforth-Gregory JK, Lanzino G, Link MJ, Brown RD, Jr. and Flemming KD. Characterization of radiation-induced cavernous malformations and comparison with a nonradiation cavernous malformation cohort. J Neurosurg. 2015;122:1214–22. [DOI] [PubMed] [Google Scholar]
- 30.Heckl S, Aschoff A and Kunze S. Radiation-induced cavernous hemangiomas of the brain: a late effect predominantly in children. Cancer. 2002;94:3285–91. [DOI] [PubMed] [Google Scholar]
- 31.Jain R, Robertson PL, Gandhi D, Gujar SK, Muraszko KM and Gebarski S. Radiation-induced cavernomas of the brain. AJNR Am J Neuroradiol. 2005;26:1158–62. [PMC free article] [PubMed] [Google Scholar]
- 32.Burn S, Gunny R, Phipps K, Gaze M and Hayward R. Incidence of cavernoma development in children after radiotherapy for brain tumors. J Neurosurg. 2007;106:379–83. [DOI] [PubMed] [Google Scholar]
- 33.Strenger V, Sovinz P, Lackner H, Dornbusch HJ, Lingitz H, Eder HG, Moser A and Urban C. Intracerebral cavernous hemangioma after cranial irradiation in childhood. Incidence and risk factors. Strahlenther Onkol. 2008;184:276–80. [DOI] [PubMed] [Google Scholar]
- 34.Vinchon M, Leblond P, Caron S, Delestret I, Baroncini M and Coche B. Radiation-induced tumors in children irradiated for brain tumor: a longitudinal study. Childs Nerv Syst. 2011;27:445–53. [DOI] [PubMed] [Google Scholar]
- 35.Koike T, Yanagimachi N, Ishiguro H, Yabe H, Yabe M, Morimoto T, Shimizu T, Takakura H and Kato S. High incidence of radiation-induced cavernous hemangioma in long-term survivors who underwent hematopoietic stem cell transplantation with radiation therapy during childhood or adolescence. Biol Blood Marrow Transplant. 2012;18:1090–8. [DOI] [PubMed] [Google Scholar]
- 36.Martinez-Lage JF, de la Fuente I, Ros de San Pedro J, Fuster JL, Perez-Espejo MA and Herrero MT. Cavernomas in children with brain tumors: a late complication of radiotherapy. Neurocirugia (Astur). 2008;19:50–4. [DOI] [PubMed] [Google Scholar]
- 37.Novelli PM, Reigel DH, Langham Gleason P and Yunis E. Multiple cavernous angiomas after high-dose whole-brain radiation therapy. Pediatr Neurosurg. 1997;26:322–5. [DOI] [PubMed] [Google Scholar]
- 38.Baumgartner JE, Ater JL, Ha CS, Kuttesch JF, Leeds NE, Fuller GN and Wilson RJ. Pathologically proven cavernous angiomas of the brain following radiation therapy for pediatric brain tumors. Pediatr Neurosurg. 2003;39:201–7. [DOI] [PubMed] [Google Scholar]
- 39.Little JB. Radiation-induced genomic instability. Int J Radiat Biol. 1998;74:663–71. [DOI] [PubMed] [Google Scholar]
- 40.Gault J, Shenkar R, Recksiek P and Awad IA. Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke. 2005;36:872–4. [DOI] [PubMed] [Google Scholar]
- 41.Akers AL, Johnson E, Steinberg GK, Zabramski JM and Marchuk DA. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. Hum Mol Genet. 2009;18:919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gault J, Awad IA, Recksiek P, Shenkar R, Breeze R, Handler M and Kleinschmidt-DeMasters BK. Cerebral cavernous malformations: somatic mutations in vascular endothelial cells. Neurosurgery. 2009;65:138–44; discussion 144–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ren AA, Snellings DA, Su YS, Hong CC, Castro M, Tang AT, Detter MR, Hobson N, Girard R, Romanos S, Lightle R, Moore T, Shenkar R, Benavides C, Beaman MM, Mueller-Fielitz H, Chen M, Mericko P, Yang J, Sung DC, Lawton MT, Ruppert M, Schwaninger M, Korbelin J, Potente M, Awad IA, Marchuk DA and Kahn ML. PIK3CA and CCM mutations fuel cavernomas through a cancer-like mechanism. Nature. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDonald DA, Shi C, Shenkar R, Gallione CJ, Akers AL, Li S, De Castro N, Berg MJ, Corcoran DL, Awad IA and Marchuk DA. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum Mol Genet. 2014;23:4357–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pagenstecher A, Stahl S, Sure U and Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet. 2009;18:911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rath M, Pagenstecher A, Hoischen A and Felbor U. Postzygotic mosaicism in cerebral cavernous malformation. J Med Genet. 2020;57:212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDonald DA, Shenkar R, Shi C, Stockton RA, Akers AL, Kucherlapati MH, Kucherlapati R, Brainer J, Ginsberg MH, Awad IA and Marchuk DA. A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease. Hum Mol Genet. 2011;20:211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plummer NW, Gallione CJ, Srinivasan S, Zawistowski JS, Louis DN and Marchuk DA. Loss of p53 sensitizes mice with a mutation in Ccm1 (KRIT1) to development of cerebral vascular malformations. Am J Pathol. 2004;165:1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Detwiler PW, Porter RW, Zabramski JM and Spetzler RF. De novo formation of a central nervous system cavernous malformation: implications for predicting risk of hemorrhage. Case report and review of the literature. J Neurosurg. 1997;87:629–32. [DOI] [PubMed] [Google Scholar]
- 50.Brunereau L, Levy C, Laberge S, Houtteville J and Labauge P. De novo lesions in familial form of cerebral cavernous malformations: clinical and MR features in 29 non-Hispanic families. Surg Neurol. 2000;53:475–82; discussion 482–3. [DOI] [PubMed] [Google Scholar]
- 51.Hong T, Xiao X, Ren J, Cui B, Zong Y, Zou J, Kou Z, Jiang N, Meng G, Zeng G, Shan Y, Wu H, Chen Z, Liang J, Xiao X, Tang J, Wei Y, Ye M, Sun L, Li G, Hu P, Hui R, Zhang H and Wang Y. Somatic MAP3K3 and PIK3CA mutations in sporadic cerebral and spinal cord cavernous malformations. Brain. 2021. [DOI] [PubMed] [Google Scholar]
- 52.Weng J, Yang Y, Song D, Huo R, Li H, Chen Y, Nam Y, Zhou Q, Jiao Y, Fu W, Yan Z, Wang J, Xu H, Di L, Li J, Wang S, Zhao J, Wang J and Cao Y. Somatic MAP3K3 mutation defines a subclass of cerebral cavernous malformation. Am J Hum Genet. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Y, Peters C, Hallier-Neelsen M, Miller D, Pagenstecher A, Bertalanffy H and Sure U. Phosphatase and tensin homolog in cerebral cavernous malformation: a potential role in pathological angiogenesis. J Neurosurg. 2009;110:530–9. [DOI] [PubMed] [Google Scholar]
- 54.Detter MR, Snellings DA and Marchuk DA. Cerebral Cavernous Malformations Develop Through Clonal Expansion of Mutant Endothelial Cells. Circ Res. 2018;123:1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malinverno M, Maderna C, Abu Taha A, Corada M, Orsenigo F, Valentino M, Pisati F, Fusco C, Graziano P, Giannotta M, Yu QC, Zeng YA, Lampugnani MG, Magnusson PU and Dejana E. Endothelial cell clonal expansion in the development of cerebral cavernous malformations. Nat Commun. 2019;10:2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Voss K, Stahl S, Schleider E, Ullrich S, Nickel J, Mueller TD and Felbor U. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics. 2007. [DOI] [PubMed] [Google Scholar]
- 57.Hilder TL, Malone MH, Bencharit S, Colicelli J, Haystead TA, Johnson GL and Wu CC. Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res. 2007;6:4343–55. [DOI] [PubMed] [Google Scholar]
- 58.Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL and Marchuk DA. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. 2005;14:2521–31. [DOI] [PubMed] [Google Scholar]
- 59.Kleaveland B, Zheng X, Liu JJ, Blum Y, Tung JJ, Zou Z, Sweeney SM, Chen M, Guo L, Lu MM, Zhou D, Kitajewski J, Affolter M, Ginsberg MH and Kahn ML. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat Med. 2009;15:169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hogan BM, Bussmann J, Wolburg H and Schulte-Merker S. ccm1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum Mol Genet. 2008;17:2424–32. [DOI] [PubMed] [Google Scholar]
- 61.Mably JD, Chuang LP, Serluca FC, Mohideen MA, Chen JN and Fishman MC. santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development. 2006;133:3139–46. [DOI] [PubMed] [Google Scholar]
- 62.Whitehead KJ, Plummer NW, Adams JA, Marchuk DA and Li DY. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437–48. [DOI] [PubMed] [Google Scholar]
- 63.Boulday G, Blecon A, Petit N, Chareyre F, Garcia LA, Niwa-Kawakita M, Giovannini M and Tournier-Lasserve E. Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: implications for human cerebral cavernous malformations. Dis Model Mech. 2009;2:168–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He Y, Zhang H, Yu L, Gunel M, Boggon TJ, Chen H and Min W. Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci Signal. 2010;3:ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan AC, Drakos SG, Ruiz OE, Smith AC, Gibson CC, Ling J, Passi SF, Stratman AN, Sacharidou A, Revelo MP, Grossmann AH, Diakos NA, Davis GE, Metzstein MM, Whitehead KJ and Li DY. Mutations in 2 distinct genetic pathways result in cerebral cavernous malformations in mice. J Clin Invest. 2011;121:1871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou Z, Rawnsley DR, Goddard LM, Pan W, Cao XJ, Jakus Z, Zheng H, Yang J, Arthur JS, Whitehead KJ, Li D, Zhou B, Garcia BA, Zheng X and Kahn ML. The Cerebral Cavernous Malformation Pathway Controls Cardiac Development via Regulation of Endocardial MEKK3 Signaling and KLF Expression. Developmental cell. 2015;32:168–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang X, Hou Y, Deng K, Zhang Y, Wang DC and Ding J. Structural Insights into the Molecular Recognition between Cerebral Cavernous Malformation 2 and Mitogen-Activated Protein Kinase Kinase Kinase 3. Structure. 2015. [DOI] [PubMed] [Google Scholar]
- 68.Fisher OS, Deng H, Liu D, Zhang Y, Wei R, Deng Y, Zhang F, Louvi A, Turk BE, Boggon TJ and Su B. Structure and vascular function of MEKK3-cerebral cavernous malformations 2 complex. Nat Commun. 2015;6:7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deng Y, Yang J, McCarty M and Su B. MEKK3 is required for endothelium function but is not essential for tumor growth and angiogenesis. American journal of physiology Cell physiology. 2007;293:C1404–11. [DOI] [PubMed] [Google Scholar]
- 70.Couto JA, Vivero MP, Kozakewich HP, Taghinia AH, Mulliken JB, Warman ML and Greene AK. A somatic MAP3K3 mutation is associated with verrucous venous malformation. Am J Hum Genet. 2015;96:480–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou Z, Tang AT, Wong WY, Bamezai S, Goddard LM, Shenkar R, Zhou S, Yang J, Wright AC, Foley M, Arthur JS, Whitehead KJ, Awad IA, Li DY, Zheng X and Kahn ML. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature. 2016;532:122–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, Boulday G, Tournier-Lasserve E, Chapon F, Richichi C, Retta SF, Lampugnani MG and Dejana E. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature. 2013;498:492–6. [DOI] [PubMed] [Google Scholar]
- 73.Cuttano R, Rudini N, Bravi L, Corada M, Giampietro C, Papa E, Morini MF, Maddaluno L, Baeyens N, Adams RH, Jain MK, Owens GK, Schwartz M, Lampugnani MG and Dejana E. KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol Med. 2016;8:6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mably JD, Mohideen MA, Burns CG, Chen JN and Fishman MC. heart of glass regulates the concentric growth of the heart in zebrafish. Curr Biol. 2003;13:2138–47. [DOI] [PubMed] [Google Scholar]
- 75.Zheng X, Riant F, Bergametti F, Myers CD, Tang AT, Kleaveland B, Pan W, Yang J, Tournier-Lasserve E and Kahn ML. Cerebral cavernous malformations arise independent of the heart of glass receptor. Stroke. 2014;45:1505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gingras AR, Liu JJ and Ginsberg MH. Structural basis of the junctional anchorage of the cerebral cavernous malformations complex. J Cell Biol. 2012;199:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Glading AJ and Ginsberg MH. Rap1 and its effector KRIT1/CCM1 regulate beta-catenin signaling. Dis Model Mech. 2010;3:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de Kreuk BJ, Gingras AR, Knight JD, Liu JJ, Gingras AC and Ginsberg MH. Heart of glass anchors Rasip1 at endothelial cell-cell junctions to support vascular integrity. Elife. 2016;5:e11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zheng X, Xu C, Smith AO, Stratman AN, Zou Z, Kleaveland B, Yuan L, Didiku C, Sen A, Liu X, Skuli N, Zaslavsky A, Chen M, Cheng L, Davis GE and Kahn ML. Dynamic regulation of the cerebral cavernous malformation pathway controls vascular stability and growth. Developmental cell. 2012;23:342–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rosen JN, Sogah VM, Ye LY and Mably JD. ccm2-like is required for cardiovascular development as a novel component of the Heg-CCM pathway. Dev Biol. 2013;376:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cullere X, Plovie E, Bennett PM, MacRae CA and Mayadas TN. The cerebral cavernous malformation proteins CCM2L and CCM2 prevent the activation of the MAP kinase MEKK3. Proc Natl Acad Sci U S A. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choi JP, Yang X, He S, Song R, Xu ZR, Foley M, Wong JJ, Xu CR and Zheng X. CCM2L (Cerebral Cavernous Malformation 2 Like) Deletion Aggravates Cerebral Cavernous Malformation Through Map3k3-KLF Signaling Pathway. Stroke. 2021;52:1428–1436. [DOI] [PubMed] [Google Scholar]
- 83.Zhang M, Dong L, Shi Z, Jiao S, Zhang Z, Zhang W, Liu G, Chen C, Feng M, Hao Q, Wang W, Yin M, Zhao Y, Zhang L and Zhou Z. Structural mechanism of CCM3 heterodimerization with GCKIII kinases. Structure. 2013;21:680–8. [DOI] [PubMed] [Google Scholar]
- 84.Zheng X, Xu C, Di Lorenzo A, Kleaveland B, Zou Z, Seiler C, Chen M, Cheng L, Xiao J, He J, Pack MA, Sessa WC and Kahn ML. CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J Clin Invest. 2010;120:2795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kean MJ, Ceccarelli DF, Goudreault M, Sanches M, Tate S, Larsen B, Gibson LC, Derry WB, Scott IC, Pelletier L, Baillie GS, Sicheri F and Gingras AC. Structure-function analysis of core STRIPAK Proteins: a signaling complex implicated in Golgi polarization. J Biol Chem. 2011;286:25065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lavina B, Castro M, Niaudet C, Cruys B, Alvarez-Aznar A, Carmeliet P, Bentley K, Brakebusch C, Betsholtz C and Gaengel K. Defective endothelial cell migration in the absence of Cdc42 leads to capillary-venous malformations. Development. 2018;145. [DOI] [PubMed] [Google Scholar]
- 87.Castro M, Lavina B, Ando K, Alvarez-Aznar A, Abu Taha A, Brakebusch C, Dejana E, Betsholtz C and Gaengel K. CDC42 Deletion Elicits Cerebral Vascular Malformations via Increased MEKK3-Dependent KLF4 Expression. Circ Res. 2019;124:1240–1252. [DOI] [PubMed] [Google Scholar]
- 88.Huang Q, Yang J, Lin Y, Walker C, Cheng J, Liu ZG and Su B. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat Immunol. 2004;5:98–103. [DOI] [PubMed] [Google Scholar]
- 89.Tang AT, Choi JP, Kotzin JJ, Yang Y, Hong CC, Hobson N, Girard R, Zeineddine HA, Lightle R, Moore T, Cao Y, Shenkar R, Chen M, Mericko P, Yang J, Li L, Tanes C, Kobuley D, Vosa U, Whitehead KJ, Li DY, Franke L, Hart B, Schwaninger M, Henao-Mejia J, Morrison L, Kim H, Awad IA, Zheng X and Kahn ML. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature. 2017;545:305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Polster SP, Sharma A, Tanes C, Tang AT, Mericko P, Cao Y, Carrion-Penagos J, Girard R, Koskimaki J, Zhang D, Stadnik A, Romanos SG, Lyne SB, Shenkar R, Yan K, Lee C, Akers A, Morrison L, Robinson M, Zafar A, Bittinger K, Kim H, Gilbert JA, Kahn ML, Shen L and Awad IA. Permissive microbiome characterizes human subjects with a neurovascular disease cavernous angioma. Nat Commun. 2020;11:2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goddard LM, Duchemin AL, Ramalingan H, Wu B, Chen M, Bamezai S, Yang J, Li L, Morley MP, Wang T, Scherrer-Crosbie M, Frank DB, Engleka KA, Jameson SC, Morrisey EE, Carroll TJ, Zhou B, Vermot J and Kahn ML. Hemodynamic Forces Sculpt Developing Heart Valves through a KLF2-WNT9B Paracrine Signaling Axis. Developmental cell. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Steed E, Faggianelli N, Roth S, Ramspacher C, Concordet JP and Vermot J. klf2a couples mechanotransduction and zebrafish valve morphogenesis through fibronectin synthesis. Nat Commun. 2016;7:11646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pestel J, Ramadass R, Gauvrit S, Helker C, Herzog W and Stainier DY. Real-time 3D visualization of cellular rearrangements during cardiac valve formation. Development. 2016;143:2217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rodel CJ, Otten C, Donat S, Lourenco M, Fischer D, Kuropka B, Paolini A, Freund C and Abdelilah-Seyfried S. Blood Flow Suppresses Vascular Anomalies in a Zebrafish Model of Cerebral Cavernous Malformations. Circ Res. 2019;125:e43–e54. [DOI] [PubMed] [Google Scholar]
- 95.Donat S, Lourenco M, Paolini A, Otten C, Renz M and Abdelilah-Seyfried S. Heg1 and Ccm1/2 proteins control endocardial mechanosensitivity during zebrafish valvulogenesis. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Renz M, Otten C, Faurobert E, Rudolph F, Zhu Y, Boulday G, Duchene J, Mickoleit M, Dietrich AC, Ramspacher C, Steed E, Manet-Dupe S, Benz A, Hassel D, Vermot J, Huisken J, Tournier-Lasserve E, Felbor U, Sure U, Albiges-Rizo C and Abdelilah-Seyfried S. Regulation of beta1 integrin-Klf2-mediated angiogenesis by CCM proteins. Developmental cell. 2015;32:181–90. [DOI] [PubMed] [Google Scholar]
- 97.Li W, Shenkar R, Detter MR, Moore T, Benavides C, Lightle R, Girard R, Hobson N, Cao Y, Li Y, Griffin E, Gallione C, Zabramski JM, Ginsberg MH, Marchuk DA and Awad IA. Propranolol inhibits cavernous vascular malformations by beta1 adrenergic receptor antagonism in animal models. J Clin Invest. 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mleynek TM, Chan AC, Redd M, Gibson CC, Davis CT, Shi DS, Chen T, Carter KL, Ling J, Blanco R, Gerhardt H, Whitehead K and Li DY. Lack of CCM1 induces hypersprouting and impairs response to flow. Hum Mol Genet. 2014;23:6223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stockton RA, Shenkar R, Awad IA and Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. The Journal of experimental medicine. 2010;207:881–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Borikova AL, Dibble CF, Sciaky N, Welch CM, Abell AN, Bencharit S and Johnson GL. Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J Biol Chem. 2010;285:11760–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Whitehead KJ, Chan AC, Navankasattusas S, Koh W, London NR, Ling J, Mayo AH, Drakos SG, Jones CA, Zhu W, Marchuk DA, Davis GE and Li DY. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med. 2009;15:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McDonald DA, Shi C, Shenkar R, Stockton RA, Liu F, Ginsberg MH, Marchuk DA and Awad IA. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke. 2012;43:571–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hong CC, Tang AT, Detter MR, Choi JP, Wang R, Yang X, Guerrero AA, Wittig CF, Hobson N, Girard R, Lightle R, Moore T, Shenkar R, Polster SP, Goddard LM, Ren AA, Leu NA, Sterling S, Yang J, Li L, Chen M, Mericko-Ishizuka P, Dow LE, Watanabe H, Schwaninger M, Min W, Marchuk DA, Zheng X, Awad IA and Kahn ML. Cerebral cavernous malformations are driven by ADAMTS5 proteolysis of versican. The Journal of experimental medicine. 2020;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lopez-Ramirez MA, Fonseca G, Zeineddine HA, Girard R, Moore T, Pham A, Cao Y, Shenkar R, de Kreuk BJ, Lagarrigue F, Lawler J, Glass CK, Awad IA and Ginsberg MH. Thrombospondin1 (TSP1) replacement prevents cerebral cavernous malformations. The Journal of experimental medicine. 2017;214:3331–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boulday G, Rudini N, Maddaluno L, Blecon A, Arnould M, Gaudric A, Chapon F, Adams RH, Dejana E and Tournier-Lasserve E. Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. The Journal of experimental medicine. 2011;208:1835–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.DiStefano PV and Glading AJ. VEGF signalling enhances lesion burden in KRIT1 deficient mice. J Cell Mol Med. 2020;24:632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ten Broek RW, Eijkelenboom A, van der Vleuten CJM, Kamping EJ, Kets M, Verhoeven BH, Grunberg K, Schultze Kool LJ, Tops BBJ, Ligtenberg MJL and Flucke U. Comprehensive molecular and clinicopathological analysis of vascular malformations: A study of 319 cases. Genes Chromosomes Cancer. 2019;58:541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rodriguez-Laguna L, Agra N, Ibanez K, Oliva-Molina G, Gordo G, Khurana N, Hominick D, Beato M, Colmenero I, Herranz G, Torres Canizalez JM, Rodriguez Pena R, Vallespin E, Martin-Arenas R, Del Pozo A, Villaverde C, Bustamante A, Ayuso C, Lapunzina P, Lopez-Gutierrez JC, Dellinger MT and Martinez-Glez V. Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. The Journal of experimental medicine. 2019;216:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Castillo SD, Baselga E and Graupera M. PIK3CA mutations in vascular malformations. Curr Opin Hematol. 2019;26:170–178. [DOI] [PubMed] [Google Scholar]
- 110.Wetzel-Strong SE, Detter MR and Marchuk DA. The pathobiology of vascular malformations: insights from human and model organism genetics. J Pathol. 2017;241:281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Luks VL, Kamitaki N, Vivero MP, Uller W, Rab R, Bovee JV, Rialon KL, Guevara CJ, Alomari AI, Greene AK, Fishman SJ, Kozakewich HP, Maclellan RA, Mulliken JB, Rahbar R, Spencer SA, Trenor CC 3rd, Upton J, Zurakowski D, Perkins JA, Kirsh A, Bennett JT, Dobyns WB, Kurek KC, Warman ML, McCarroll SA and Murillo R Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. 2015;166:1048–54 e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Limaye N, Kangas J, Mendola A, Godfraind C, Schlogel MJ, Helaers R, Eklund L, Boon LM and Vikkula M. Somatic Activating PIK3CA Mutations Cause Venous Malformation. Am J Hum Genet. 2015;97:914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Marchi S, Corricelli M, Trapani E, Bravi L, Pittaro A, Delle Monache S, Ferroni L, Patergnani S, Missiroli S, Goitre L, Trabalzini L, Rimessi A, Giorgi C, Zavan B, Cassoni P, Dejana E, Retta SF and Pinton P. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol Med. 2015;7:1403–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Otten C, Knox J, Boulday G, Eymery M, Haniszewski M, Neuenschwander M, Radetzki S, Vogt I, Hahn K, De Luca C, Cardoso C, Hamad S, Igual Gil C, Roy P, Albiges-Rizo C, Faurobert E, von Kries JP, Campillos M, Tournier-Lasserve E, Derry WB and Abdelilah-Seyfried S. Systematic pharmacological screens uncover novel pathways involved in cerebral cavernous malformations. EMBO Mol Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schulz GB, Wieland E, Wustehube-Lausch J, Boulday G, Moll I, Tournier-Lasserve E and Fischer A. Cerebral Cavernous Malformation-1 Protein Controls DLL4-Notch3 Signaling Between the Endothelium and Pericytes. Stroke. 2015;46:1337–43. [DOI] [PubMed] [Google Scholar]
- 116.You C, Sandalcioglu IE, Dammann P, Felbor U, Sure U and Zhu Y. Loss of CCM3 impairs DLL4-Notch signalling: implication in endothelial angiogenesis and in inherited cerebral cavernous malformations. J Cell Mol Med. 2013;17:407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wustehube J, Bartol A, Liebler SS, Brutsch R, Zhu Y, Felbor U, Sure U, Augustin HG and Fischer A. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc Natl Acad Sci U S A. 2010;107:12640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bravi L, Rudini N, Cuttano R, Giampietro C, Maddaluno L, Ferrarini L, Adams RH, Corada M, Boulday G, Tournier-Lasserve E, Dejana E and Lampugnani MG. Sulindac metabolites decrease cerebrovascular malformations in CCM3-knockout mice. Proc Natl Acad Sci U S A. 2015;112:8421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Clatterbuck RE, Moriarity JL, Elmaci I, Lee RR, Breiter SN and Rigamonti D. Dynamic nature of cavernous malformations: a prospective magnetic resonance imaging study with volumetric analysis. J Neurosurg. 2000;93:981–6. [DOI] [PubMed] [Google Scholar]
- 120.Goldberg J, Jaeggi C, Schoeni D, Mordasini P, Raabe A and Bervini D. Bleeding risk of cerebral cavernous malformations in patients on beta-blocker medication: a cohort study. J Neurosurg. 2018:1–6. [DOI] [PubMed] [Google Scholar]
- 121.Lopez-Ramirez MA, Pham A, Girard R, Wyseure T, Hale P, Yamashita A, Koskimaki J, Polster S, Saadat L, Romero IA, Esmon CT, Lagarrigue F, Awad IA, Mosnier LO and Ginsberg MH. Cerebral cavernous malformations form an anticoagulant vascular domain in humans and mice. Blood. 2019;133:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lant B, Pal S, Chapman EM, Yu B, Witvliet D, Choi S, Zhao L, Albiges-Rizo C, Faurobert E and Derry WB. Interrogating the ccm-3 Gene Network. Cell Rep. 2018;24:2857–2868 e4. [DOI] [PubMed] [Google Scholar]
- 123.Al-Shahi Salman R, Berg MJ, Morrison L, Awad IA and Angioma Alliance Scientific Advisory B. Hemorrhage from cavernous malformations of the brain: definition and reporting standards. Angioma Alliance Scientific Advisory Board. Stroke. 2008;39:3222–30. [DOI] [PubMed] [Google Scholar]
- 124.Flemming KD, Kumar S, Brown RD Jr., and Lanzino G Predictors of Initial Presentation with Hemorrhage in Patients with Cavernous Malformations. World Neurosurg. 2020;133:e767–e773. [DOI] [PubMed] [Google Scholar]
- 125.Ene C, Kaul A and Kim L. Natural history of cerebral cavernous malformations. Handbook of clinical neurology. 2017;143:227–232. [DOI] [PubMed] [Google Scholar]
- 126.Girard R, Fam MD, Zeineddine HA, Tan H, Mikati AG, Shi C, Jesselson M, Shenkar R, Wu M, Cao Y, Hobson N, Larsson HBW, Christoforidis GA and Awad IA. Vascular permeability and iron deposition biomarkers in longitudinal follow-up of cerebral cavernous malformations. J Neurosurg. 2017;127:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Flemming KD and Lanzino G. Cerebral Cavernous Malformation: What a Practicing Clinician Should Know. Mayo Clin Proc. 2020;95:2005–2020. [DOI] [PubMed] [Google Scholar]
- 128.Labauge P, Brunereau L, Levy C, Laberge S and Houtteville JP. The natural history of familial cerebral cavernomas: a retrospective MRI study of 40 patients. Neuroradiology. 2000;42:327–32. [DOI] [PubMed] [Google Scholar]
- 129.Horne MA, Flemming KD, Su IC, Stapf C, Jeon JP, Li D, Maxwell SS, White P, Christianson TJ, Agid R, Cho WS, Oh CW, Wu Z, Zhang JT, Kim JE, Ter Brugge K, Willinsky R, Brown RD Jr., Murray GD, Al-Shahi Salman R and Cerebral Cavernous Malformations Individual Patient Data Meta-analysis C. Clinical course of untreated cerebral cavernous malformations: a meta-analysis of individual patient data. Lancet Neurol. 2016;15:166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Al-Shahi Salman R, Hall JM, Horne MA, Moultrie F, Josephson CB, Bhattacharya JJ, Counsell CE, Murray GD, Papanastassiou V, Ritchie V, Roberts RC, Sellar RJ, Warlow CP and Scottish Audit of Intracranial Vascular Malformations c. Untreated clinical course of cerebral cavernous malformations: a prospective, population-based cohort study. Lancet Neurol. 2012;11:217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Porter PJ, Willinsky RA, Harper W and Wallace MC. Cerebral cavernous malformations: natural history and prognosis after clinical deterioration with or without hemorrhage. J Neurosurg. 1997;87:190–7. [DOI] [PubMed] [Google Scholar]
- 132.Gross BA and Du R. Hemorrhage from cerebral cavernous malformations: a systematic pooled analysis. J Neurosurg. 2017;126:1079–1087. [DOI] [PubMed] [Google Scholar]
- 133.Zhou HJ, Qin L, Zhang H, Tang W, Ji W, He Y, Liang X, Wang Z, Yuan Q, Vortmeyer A, Toomre D, Fuh G, Yan M, Kluger MS, Wu D and Min W. Erratum: Endothelial exocytosis of angiopoietin-2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat Med. 2016;22:1502. [DOI] [PubMed] [Google Scholar]
- 134.Stamatovic SM, Sladojevic N, Keep RF and Andjelkovic AV. PDCD10 (CCM3) regulates brain endothelial barrier integrity in cerebral cavernous malformation type 3: role of CCM3-ERK1/2-cortactin cross-talk. Acta Neuropathol. 2015;130:731–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li DY and Whitehead KJ. Evaluating strategies for the treatment of cerebral cavernous malformations. Stroke. 2010;41:S92–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Detter MR, Shenkar R, Benavides CR, Neilson CA, Moore T, Lightle R, Hobson N, Shen L, Cao Y, Girard R, Zhang D, Griffin E, Gallione CJ, Awad IA and Marchuk DA. Novel Murine Models of Cerebral Cavernous Malformations. Angiogenesis. 2020;23:651–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Girard R, Zeineddine HA, Koskimaki J, Fam MD, Cao Y, Shi C, Moore T, Lightle R, Stadnik A, Chaudagar K, Polster S, Shenkar R, Duggan R, Leclerc D, Whitehead KJ, Li DY and Awad IA. Plasma Biomarkers of Inflammation and Angiogenesis Predict Cerebral Cavernous Malformation Symptomatic Hemorrhage or Lesional Growth. Circ Res. 2018;122:1716–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lopez-Ramirez MA, Fischer R, Torres-Badillo CC, Davies HA, Logan K, Pfizenmaier K, Male DK, Sharrack B and Romero IA. Role of caspases in cytokine-induced barrier breakdown in human brain endothelial cells. J Immunol. 2012;189:3130–9. [DOI] [PubMed] [Google Scholar]
- 139.Koskimaki J, Zhang D, Carrion-Penagos J, Girard R, Piedad K, Polster SP, Lyne S, Stadnik A and Awad IA. Symptomatic Brain Hemorrhages from Cavernous Angioma After Botulinum Toxin Injections, a Role of TLR/MEKK3 Mechanism? Case Report and Review of the Literature. World Neurosurg. 2020;136:7–11. [DOI] [PubMed] [Google Scholar]
- 140.Wong VL, Hofman FM, Ishii H and Fisher M. Regional distribution of thrombomodulin in human brain. Brain research. 1991;556:1–5. [DOI] [PubMed] [Google Scholar]
- 141.Laszik Z, Mitro A, Taylor FB, Jr., Ferrell G and Esmon CT. Human protein C receptor is present primarily on endothelium of large blood vessels: implications for the control of the protein C pathway. Circulation. 1997;96:3633–40. [DOI] [PubMed] [Google Scholar]
- 142.Koskimaki J, Girard R, Li Y, Saadat L, Zeineddine HA, Lightle R, Moore T, Lyne S, Avner K, Shenkar R, Cao Y, Shi C, Polster SP, Zhang D, Carrion-Penagos J, Romanos S, Fonseca G, Lopez-Ramirez MA, Chapman EM, Popiel E, Tang AT, Akers A, Faber P, Andrade J, Ginsberg M, Derry WB, Kahn ML, Marchuk DA and Awad IA. Comprehensive transcriptome analysis of cerebral cavernous malformation across multiple species and genotypes. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zlokovic BV. Antithrombotic, procoagulant, and fibrinolytic mechanisms in cerebral circulation: implications for brain injury and protection. Neurosurgical focus. 1997;2:e5. [PubMed] [Google Scholar]
- 144.Griffin JH, Zlokovic BV and Mosnier LO. Activated protein C: biased for translation. Blood. 2015;125:2898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Orsenigo F, Conze LL, Jauhiainen S, Corada M, Lazzaroni F, Malinverno M, Sundell V, Cunha SI, Brannstrom J, Globisch MA, Maderna C, Lampugnani MG, Magnusson PU and Dejana E. Mapping endothelial-cell diversity in cerebral cavernous malformations at single-cell resolution. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dargaud Y, Scoazec JY, Wielders SJ, Trzeciak C, Hackeng TM, Negrier C, Hemker HC, Lindhout T and Castoldi E. Characterization of an autosomal dominant bleeding disorder caused by a thrombomodulin mutation. Blood. 2015;125:1497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Langdown J, Luddington RJ, Huntington JA and Baglin TP. A hereditary bleeding disorder resulting from a premature stop codon in thrombomodulin (p.Cys537Stop). Blood. 2014;124:1951–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jansson JH, Boman K, Brannstrom M and Nilsson TK. High concentration of thrombomodulin in plasma is associated with hemorrhage: a prospective study in patients receiving long-term anticoagulant treatment. Circulation. 1997;96:2938–43. [DOI] [PubMed] [Google Scholar]
- 149.Kurosawa S, Stearns-Kurosawa DJ and Kinasewitz GT. Soluble thrombomodulin: a sign of bad times. Critical care medicine. 2008;36:985–7. [DOI] [PubMed] [Google Scholar]
- 150.von Drygalski A, Cramer TJ, Bhat V, Griffin JH, Gale AJ and Mosnier LO. Improved hemostasis in hemophilia mice by means of an engineered factor Va mutant. Journal of thrombosis and haemostasis : JTH. 2014;12:363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Prince R, Bologna L, Manetti M, Melchiorre D, Rosa I, Dewarrat N, Suardi S, Amini P, Fernandez JA, Burnier L, Quarroz C, Reina Caro MD, Matsumura Y, Kremer Hovinga JA, Griffin JH, Simon HU, Ibba-Manneschi L, Saller F, Calzavarini S and Angelillo-Scherrer A. Targeting anticoagulant protein S to improve hemostasis in hemophilia. Blood. 2018;131:1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mosnier LO, Zlokovic BV and Griffin JH. Cytoprotective-selective activated protein C therapy for ischaemic stroke. Thromb Haemost. 2014;112:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Soh UJ and Trejo J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through beta-arrestin and dishevelled-2 scaffolds. Proc Natl Acad Sci U S A. 2011;108:E1372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.John H Griffin BVZ, and Laurent O. Mosnier. Activated Protein C, Protease Activated Receptor 1 and Neuroprotection. Blood. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mohan Rao LV, Esmon CT and Pendurthi UR. Endothelial cell protein C receptor: a multiliganded and multifunctional receptor. Blood. 2014;124:1553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zuurbier SM, Hickman CR, Tolias CS, Rinkel LA, Leyrer R, Flemming KD, Bervini D, Lanzino G, Wityk RJ, Schneble HM, Sure U and Al-Shahi Salman R. Long-term antithrombotic therapy and risk of intracranial haemorrhage from cerebral cavernous malformations: a population-based cohort study, systematic review, and meta-analysis. Lancet Neurol. 2019;18:935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dahlbäck B Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood. 2008;112:19–27. [DOI] [PubMed] [Google Scholar]
- 158.Li J, Zhao Y, Choi J, Ting KK, Coleman P, Chen J, Cogger VC, Wan L, Shi Z, Moller T, Zheng X, Vadas MA and Gamble JR. Targeting miR-27a/VE-cadherin interactions rescues cerebral cavernous malformations in mice. PLoS biology. 2020;18:e3000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Girard R, Zeineddine HA, Fam MD, Mayampurath A, Cao Y, Shi C, Shenkar R, Polster SP, Jesselson M, Duggan R, Mikati AG, Christoforidis G, Andrade J, Whitehead KJ, Li DY and Awad IA. Plasma Biomarkers of Inflammation Reflect Seizures and Hemorrhagic Activity of Cerebral Cavernous Malformations. Translational stroke research. 2018;9:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Akers A, Al-Shahi Salman R, Awad IA, Dahlem K, Flemming K, Hart B, Kim H, Jusue-Torres I, Kondziolka D, Lee C, Morrison L, Rigamonti D, Rebeiz T, Tournier-Lasserve E, Waggoner D and Whitehead K. Synopsis of guidelines for the clinical management of cerebral cavernous malformations: consensus recommendations based on systematic literature review by the Angioma Alliance scientific advisory board clinical experts panel. Neurosurgery. 2017;80:665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Polster SP, Cao Y, Carroll T, Flemming K, Girard R, Hanley D, Hobson N, Kim H, Koenig J, Koskimaki J, Lane K, Majersik JJ, McBee N, Morrison L, Shenkar R, Stadnik A, Thompson RE, Zabramski J, Zeineddine HA and Awad IA. Trial Readiness in Cavernous Angiomas With Symptomatic Hemorrhage (CASH). Neurosurgery. 2019;84:954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Girard R, Li Y, Stadnik A, Shenkar R, Hobson N, Romanos S, Srinath A, Moore T, Lightle R, Shkoukani A, Akers A, Carroll T, Christoforidis GA, Koenig JI, Lee C, Piedad K, Greenberg SM, Kim H, Flemming KD, Ji Y and Awad IA. A Roadmap for Developing Plasma Diagnostic and Prognostic Biomarkers of Cerebral Cavernous Angioma With Symptomatic Hemorrhage (CASH). Neurosurgery. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shenkar R, Venkatasubramanian PN, Wyrwicz AM, Zhao JC, Shi C, Akers A, Marchuk DA and Awad IA. Advanced magnetic resonance imaging of cerebral cavernous malformations: part II. Imaging of lesions in murine models. Neurosurgery. 2008;63:790–7; discussion 797–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shenkar R, Venkatasubramanian PN, Zhao JC, Batjer HH, Wyrwicz AM and Awad IA. Advanced magnetic resonance imaging of cerebral cavernous malformations: part I. High-field imaging of excised human lesions. Neurosurgery. 2008;63:782–9; discussion 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Girard R, Zeineddine HA, Orsbon C, Tan H, Moore T, Hobson N, Shenkar R, Lightle R, Shi C, Fam MD, Cao Y, Shen L, Neander AI, Rorrer A, Gallione C, Tang AT, Kahn ML, Marchuk DA, Luo ZX and Awad IA. Micro-computed tomography in murine models of cerebral cavernous malformations as a paradigm for brain disease. J Neurosci Methods. 2016;271:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shenkar R, Peiper A, Pardo H, Moore T, Lightle R, Girard R, Hobson N, Polster SP, Koskimaki J, Zhang D, Lyne SB, Cao Y, Chaudagar K, Saadat L, Gallione C, Pytel P, Liao JK, Marchuk D and Awad IA. Rho Kinase Inhibition Blunts Lesion Development and Hemorrhage in Murine Models of Aggressive Pdcd10/Ccm3 Disease. Stroke. 2019;50:738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mikati AG, Khanna O, Zhang L, Girard R, Shenkar R, Guo X, Shah A, Larsson HB, Tan H, Li L, Wishnoff MS, Shi C, Christoforidis GA and Awad IA. Vascular permeability in cerebral cavernous malformations. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015;35:1632–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mikati AG, Tan H, Shenkar R, Li L, Zhang L, Guo X, Larsson HB, Shi C, Liu T, Wang Y, Shah A, Edelman RR, Christoforidis G and Awad I. Dynamic permeability and quantitative susceptibility: related imaging biomarkers in cerebral cavernous malformations. Stroke. 2014;45:598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Larsson HB, Courivaud F, Rostrup E and Hansen AE. Measurement of brain perfusion, blood volume, and blood-brain barrier permeability, using dynamic contrast-enhanced T(1)-weighted MRI at 3 tesla. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2009;62:1270–81. [DOI] [PubMed] [Google Scholar]
- 170.Tan H, Liu T, Wu Y, Thacker J, Shenkar R, Mikati AG, Shi C, Dykstra C, Wang Y, Prasad PV, Edelman RR and Awad IA. Evaluation of iron content in human cerebral cavernous malformation using quantitative susceptibility mapping. Investigative radiology. 2014;49:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tan H, Zhang L, Mikati AG, Girard R, Khanna O, Fam MD, Liu T, Wang Y, Edelman RR, Christoforidis G and Awad IA. Quantitative Susceptibility Mapping in Cerebral Cavernous Malformations: Clinical Correlations. Am J Neuroradiol. 2016;37:1209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cramer SP, Simonsen H, Frederiksen JL, Rostrup E and Larsson HB. Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. Neuroimage Clin. 2014;4:182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sun H, Klahr AC, Kate M, Gioia LC, Emery DJ, Butcher KS and Wilman AH. Quantitative Susceptibility Mapping for Following Intracranial Hemorrhage. Radiology. 2018;288:830–839. [DOI] [PubMed] [Google Scholar]
- 174.Liu T, Surapaneni K, Lou M, Cheng L, Spincemaille P and Wang Y. Cerebral microbleeds: burden assessment by using quantitative susceptibility mapping. Radiology. 2012;262:269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zeineddine HA, Girard R, Cao Y, Hobson N, Fam MD, Stadnik A, Tan H, Shen J, Chaudagar K, Shenkar R, Thompson RE, McBee N, Hanley D, Carroll T, Christoforidis GA and Awad IA. Quantitative susceptibility mapping as a monitoring biomarker in cerebral cavernous malformations with recent hemorrhage. J Magn Reson Imaging. 2018;47:1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shi C, Shenkar R, Zeineddine HA, Girard R, Fam MD, Austin C, Moore T, Lightle R, Zhang L, Wu M, Cao Y, Gunel M, Louvi A, Rorrer A, Gallione C, Marchuk DA and Awad IA. B-Cell Depletion Reduces the Maturation of Cerebral Cavernous Malformations in Murine Models. J Neuroimmune Pharmacol. 2016;11:369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zeineddine HA, Girard R, Saadat L, Shen L, Lightle R, Moore T, Cao Y, Hobson N, Shenkar R, Avner K, Chaudager K, Koskimaki J, Polster SP, Fam MD, Shi C, Lopez-Ramirez MA, Tang AT, Gallione C, Kahn ML, Ginsberg M, Marchuk DA and Awad IA. Phenotypic characterization of murine models of cerebral cavernous malformations. Lab Invest. 2019;99:319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Girard R, Khanna O, Shenkar R, Zhang L, Wu M, Jesselson M, Zeineddine HA, Gangal A, Fam MD, Gibson CC, Whitehead KJ, Li DY, Liao JK, Shi C and Awad IA. Peripheral plasma vitamin D and non-HDL cholesterol reflect the severity of cerebral cavernous malformation disease. Biomarkers in medicine. 2016;10:255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lyne SB, Girard R, Koskimäki J, Zeineddine HA, Zhang D, Cao Y, Li Y, Stadnik A, Moore T, Lightle R, Shi C, Shenkar R, Carrión-Penagos J, Polster SP, Romanos S, Akers A, Lopez-Ramirez M, Whitehead KJ, Kahn ML, Ginsberg MH, Marchuk DA and Awad IA. Biomarkers of cavernous angioma with symptomatic hemorrhage. JCI Insight. 2019;4:(12):e128577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.McKerracher L, Shenkar R, Abbinanti M, Cao Y, Peiper A, Liao JK, Lightle R, Moore T, Hobson N, Gallione C, Ruschel J, Koskimaki J, Girard R, Rosen K, Marchuk DA and Awad IA. A Brain-Targeted Orally Available ROCK2 Inhibitor Benefits Mild and Aggressive Cavernous Angioma Disease. Translational stroke research. 2020;11:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shenkar R, Shi C, Austin C, Moore T, Lightle R, Cao Y, Zhang L, Wu M, Zeineddine HA, Girard R, McDonald DA, Rorrer A, Gallione C, Pytel P, Liao JK, Marchuk DA and Awad IA. RhoA Kinase Inhibition With Fasudil Versus Simvastatin in Murine Models of Cerebral Cavernous Malformations. Stroke. 2017;48:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Polster SP, Stadnik A, Akers AL, Cao Y, Christoforidis GA, Fam MD, Flemming KD, Girard R, Hobson N, Koenig JI, Koskimaki J, Lane K, Liao JK, Lee C, Lyne SB, McBee N, Morrison L, Piedad K, Shenkar R, Sorrentino M, Thompson RE, Whitehead KJ, Zeineddine HA, Hanley DF and Awad IA. Atorvastatin Treatment of Cavernous Angiomas with Symptomatic Hemorrhage Exploratory Proof of Concept (AT CASH EPOC) Trial. Neurosurgery. 2019;85:843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.DiStefano PV, Kuebel JM, Sarelius IH and Glading AJ. KRIT1 protein depletion modifies endothelial cell behavior via increased vascular endothelial growth factor (VEGF) signaling. J Biol Chem. 2014;289:33054–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gibson CC, Zhu W, Davis CT, Bowman-Kirigin JA, Chan AC, Ling J, Walker AE, Goitre L, Delle Monache S, Retta SF, Shiu YT, Grossmann AH, Thomas KR, Donato AJ, Lesniewski LA, Whitehead KJ and Li DY. Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation. 2015;131:289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]