

Abstract
Raynaud’s phenomenon can be either primary (idiopathic) or secondary to underlying disease including systemic sclerosis. Primary Raynaud’s phenomenon is very common, affecting approximately 3%–5% of the general population. Although much rarer, systemic sclerosis–related Raynaud’s phenomenon can be particularly severe, progressing to digital ulceration in approximately 50% of patients. Raynaud’s phenomenon can have a major impact on quality of life. This review has a focus on the systemic sclerosis–related Raynaud’s phenomenon (which is the most researched form of Raynaud’s phenomenon and probably the most challenging to treat) and on recent advances. Epidemiology (including transition from ‘isolated’ to systemic sclerosis–related Raynaud’s phenomenon), pathogenesis, diagnosis and assessment are discussed, followed by the treatment of both ‘uncomplicated’ and ‘complicated’ Raynaud’s phenomena (i.e. Raynaud’s phenomenon which has progressed to digital ulceration and/or critical ischaemia). Finally, some of the major challenges for the next 5–10 years are highlighted.
Keywords: Systemic sclerosis, Raynaud’s phenomenon, diagnosis, assessment, management
Introduction
Raynaud’s phenomenon (RP) is defined as episodic colour change of the extremities (usually best seen in the fingers), in response to cold exposure or to emotional stress. Typically, the fingers turn white, then blue and then red – representing vasospasm (white), then followed by deoxygenation (blue) and then reperfusion hyperaemia (red), respectively. It is generally accepted that for a patient to be considered as having RP, at least a biphasic colour change should occur.
The vast majority of people with RP have primary (idiopathic) RP, which does not progress to tissue damage. However, RP can also be secondary to a large number of different diseases/conditions1,2 including: connective tissue diseases (especially systemic sclerosis (SSc)), hand-arm vibration syndrome (vibration white finger), extrinsic vascular compression (including cervical rib), other large vessel causes, intravascular diseases including those associated with increased viscosity and certain drugs (including beta-blockers, some chemotherapeutic agents and stimulants (e.g. methylphenidate), recently reviewed by Khouri et al. 3 ).
Why is RP important to clinicians and scientists with an interest in SSc? First, RP is the commonest presenting feature of SSc, providing a window of opportunity for early diagnosis. Although only a small minority of patients with RP will have SSc, early diagnosis for this minority is crucial because this will improve outcome by allowing early detection and treatment of, for example, internal organ complications. Second, RP in patients with SSc is often very severe with a major impact on quality of life, even (as demonstrated in a recent study) in early disease and during the summer months. 4 Treatment of SSc-related RP can be particularly challenging, but progress is being made.
This review will discuss epidemiology, pathogenesis (albeit briefly), diagnosis and treatment, and finally, some of the main challenges for the next 5–10 years. Three short clinical scenarios are presented to give context. SSc-related RP can progress to/become complicated by digital ulceration and/or critical ischaemia, and the treatment section will be divided into ‘uncomplicated’ and ‘complicated’ RP. The aim of this review is to describe best practice and also to highlight some of the key recent findings and advances across all aspects of RP in the last 2–3 years.
Epidemiology and ‘transition’ from primary to SSc-related RP
Although reports of incidence and prevalence of RP vary widely, it is generally accepted that 3%–5% of the general population have RP. 5 Prevalence is influenced by geographic/climatic variation and also by how RP is defined. Women are more commonly affected than men. Among patients with SSc, approximately 95% have RP. 6
Given that the estimated prevalence of RP is approximately 5%, 5 and of SSc is 0.025%, 7 approximately 1 in 200 individuals with RP will have SSc (0.5%). This seems at odds with the estimate of approximately 3 transitions per 100 patient years of ‘isolated’ RP to connective tissue disease–related RP. 8 This discrepancy most likely reflects selection bias in reported series, that is, those referred for further assessment are unlikely to be typical of patients with RP in the general population.
Recent findings/points to highlight
Ingegnoli et al. 9 recently undertook a systematic review and meta-analysis which included 4051 patients with primary RP and 657 with suspected secondary RP (as defined by antinuclear antibody (ANA) positivity and/or capillary abnormalities), and concluded that the mean incidence rate of transition from primary RP to a connective tissue disease was 2.65/100 patient years (95% confidence interval (CI) = 0.44–5.73) and to SSc was 0.93/100 patient years (95% CI = 0.27–2.13). The transition rate was much higher in patients with suspected secondary RP: 11.01/100 patient years (95% CI = 0.11–22.12) to connective tissue disease and 7.10/100 patient years (95% CI = 1.02–13.19) to SSc. The combination of positive ANA and abnormal nailfold capillaries conferred particularly high risk: risk ratio for development of connective tissue disease was 16.96 and for SSc was 40.45. It is worth highlighting that in the 4051 patients with primary RP, average age at onset was 34.1 years, confirming how patients included were not representative of patients with primary RP in the general population, most of them having a younger age of onset.
Regarding triggering/aggravating factors for RP, a recent systematic review suggested that beta-blockers differ in their tendency to exacerbate vasoconstriction and that those with intrinsic sympathomimetic activity can most likely be safely used in patients with RP. 10
Pathogenesis
The pathogenesis of RP is not fully understood, although much more is known about the underlying molecular and cellular mechanisms than 20 years ago. Pathogenesis is reviewed more fully elsewhere.11–13 A key point is that pathogenesis will depend upon whether RP is primary or secondary and if secondary, then to what? Primary RP is considered purely vasospastic, whereas SSc-related RP is also associated with structural vascular change, at both small vessel (including capillary) and digital artery level, explaining its severity.
Of all the secondary types of RP, SSc-related RP has been most studied. Many different mechanisms contribute. 11 The key problem is an imbalance between vasoconstriction and vasodilation in favour of vasoconstriction, with a defect in thermoregulation. 12 Intravascular factors also contribute, including increased platelet activation and oxidant stress. Markers of endothelial activation/damage have been reported to be elevated especially in patients with SSc-related RP, or in those at high risk of its development, 14 suggesting that endothelial injury is likely to play a key role early on in pathogenesis.
Recent findings/points to highlight
Despite a recognised familial component to primary RP, its genetics have been little studied. A recent study of 4276 individuals from the TwinsUK database (15.0% of whom had RP as judged by questionnaire) reported that RP was associated with a polymorphism in the NOS1 gene. 15 NOS1 encodes neuronal nitric oxide synthase which could be implicated in vasoreactivity.
Scenarios
1. A 53-year-old woman was referred for assessment of RP since the age of 16 years. Her feet were also affected. Otherwise, she kept well and she was on no drug treatment. On examination, there were no abnormalities. Full blood count was normal, immunological testing was essentially negative and nailfold capillaroscopy was normal (Figure 1). Thermography16–18 is also shown in Figure 1. Treatment recommendations were ‘general measures’ and to commence a calcium channel blocker if symptoms became more troublesome.
Figure 1.

Nailfold capillaroscopy (left panel) is normal. Thermography showed that although the fingertips were cool at a room temperature of 23°C (middle panel), at 30°C (right panel), there were no significant persistent temperature gradients (as defined by a fingertip more than 1°C cooler than the dorsum of hand) along any of the fingers. This pattern is consistent with primary RP.16,17
2. A 35-year-old man was referred with a 10-year history of RP (evidenced by a photograph taken on his mobile phone), worse over the last 2–3 years. His feet were affected, but to a lesser extent. His general health was very good. He was on no drug treatment. ANA was negative. Capillaroscopy was normal in most nailfolds, but abnormal in the left index finger (Figure 2, which also shows thermography). A calcium channel blocker was recommended alongside general measures and he is being kept under review.
Figure 2.

Capillaroscopy (left index finger) shows an enlarged capillary and haemorrhages. Thermography shows cool fingertips at a room temperature of 23°C (middle panel), but not persisting at 30°C.
Diagnosis: very early SSc21–23 on the basis of the objective evidence of RP and abnormal capillaroscopy.
3. A 36-year-old woman was referred with a 2-year history of RP and a recent ulcer of the tip of the right middle finger, which healed after 2 months. For 4 months, she had some finger puffiness. For 1 year, she had had heartburn and was on omeprazole. On examination, she had digital pitting of her right index and middle fingers and puffiness of the index and middle fingers bilaterally. Anticentromere antibody was positive. Nailfold capillaroscopy (Figure 3, which also shows thermography) showed typical abnormalities of an SSc-spectrum disorder.18,24,25 Nifedipine (sustained release) was recommended in the first instance, with a view to adding in sildenafil if a calcium channel was insufficient to control symptoms or not tolerated.
Figure 3.

Nailfold capillaroscopy was very abnormal with dilated loops, avascularity, distortion of the normal nailfold architecture and haemorrhages. Thermographic testing showed that at a room temperature of 30°C, there were persisting temperature gradients (fingertips >1°C cooler than dorsum of hand) along several of the fingers. This pattern is consistent with SSc.16,17
Diagnosis: limited cutaneous SSc on the basis of RP, puffy fingers, digital pitting, positive anticentromere antibody and abnormal capillaroscopy.26,27
Diagnosis and assessment of RP
As stated above, the presence of RP is determined on the basis of the history. An interesting development in the last few years has been the capturing of RP attacks by patients on their mobile phones (Figure 4), providing objectivity to the diagnosis.
Figure 4.
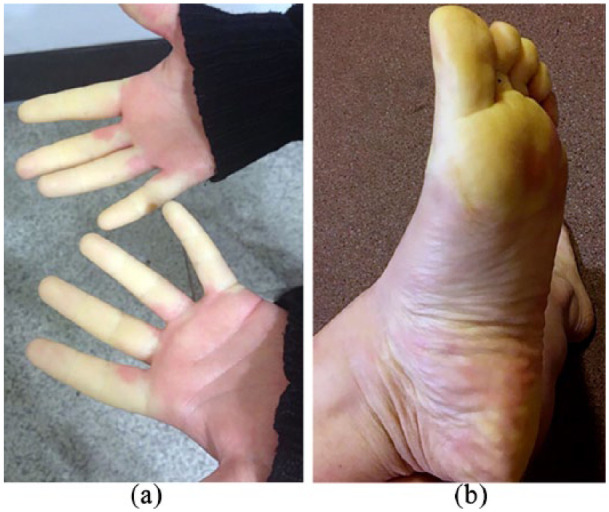
Mobile phone photographs of (a) hands and (b) foot (same patient) showing the ‘white’ phase of RP.
However, it is important to stress that RP is not a diagnosis in itself – it is a symptom complex requiring a diagnosis. So, the first question is ‘Why does this person have RP?’ Rheumatologists are referred patients with RP because among those reaching secondary care, a significant proportion will have connective tissue disease. However, patients may have other underlying diseases, for example, haematological disorders.1,2 The minimal assessment in the patient presenting with RP comprises the following:
A detailed history, including occupational and drug history.
A physical examination, especially of the hands, looking for signs of connective tissue disease (as noted in Scenario 3). Sclerodactyly (scleroderma of the fingers), puffy fingers (Figure 5), digital pitting and telangiectases all point towards a diagnosis of SSc.
A full blood count and testing for ANA. These should be normal/negative in primary RP.19,20 Although a normal erythrocyte sedimentation rate (ESR) is also reassuring, the criteria for primary RP proposed by Maverakis et al. 20 do not insist on this.
Nailfold capillaroscopy. The nailfold capillaries should be normal in primary RP,19,20 and conversely, most patients with SSc will have abnormal nailfold capillaries. It is now well established that abnormal nailfold capillaries, which are included in the 2013 classification criteria for SSc,26,27 are an independent risk factor for SSc 28 and so the early capillaroscopic findings in Scenario 2 are worrying. In a recent study involving 347 patients, a ‘scleroderma pattern’ on capillaroscopy had a positive predictive value of 84% and a negative predictive value of 90%. 29 In some specialist centres, thermography is also available: this helps differentiate between primary and SSc-related RP.16–18,30,31 The thermography pattern in Scenario 3 (persisting cold fingertips even in a warm environment of 30°C) is highly suggestive of an underlying structural vascular abnormality.
Figure 5.
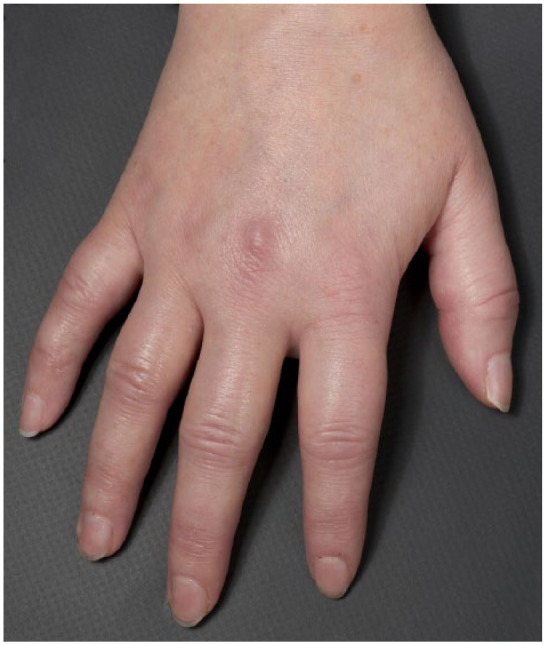
Puffy fingers – a ‘red flag’ for SSc in the patient presenting with RP.
Recent findings/points to highlight
For the clinician
The main point is the increasing international interest in nailfold capillaroscopy, resulting in large part from the inclusion of abnormal nailfold capillaries in the 2013 classification criteria for SSc.26,27 Although the gold standard for nailfold capillaroscopy is high magnification videocapillaroscopy, 25 it seems unlikely that all general rheumatologists seeing patients with RP will purchase a videomicroscope. For this reason, there is increasing interest in lower magnification systems such as the dermatoscope and USB microscope. The dermatoscope compares favourably to nailfold videocapillaroscopy: although as might be expected it is less sensitive in identifying abnormality, it is more specific.32,33 An ideal solution would be for specialist centres to have access to nailfold videocapillaroscopy, but for all rheumatologists to have at least a low-cost capillaroscopy system, for example, a dermatoscope or USB microscope.
For the clinical researcher
Although outwith the scope of this review, there is increasing interest in developing and validating reliable outcome measures, which are sensitive to change, for use in both early proof-of-concept and later phase trials of RP. These include both patient-reported outcome measures34,35 and non-invasive imaging modalities including thermography, laser Doppler methods including laser speckle contrast imaging,36,37 and dynamic Doppler ultrasound of the digital artery. 38
Management
The first step in management is to establish why the patient has RP. An underlying cause may be amenable to specific intervention (e.g. avoidance of vibratory equipment and treatment of paraproteinaemia) and/or dictate RP severity (the treatment of a patient with primary RP is very different from the treatment of a patient with severe SSc-related RP).
Having established the diagnosis and removed any remediable factors, the same general principles then apply in all patients with RP. Treatment of ‘uncomplicated’ RP will first be discussed and then treatment of ‘complicated’ RP (RP which has progressed to digital ulceration and/or critical digital ischaemia) with a focus on SSc-related RP. Treatment options for RP have recently been comprehensively reviewed by Hinze and Wigley. 39 It should be emphasised that the evidence base for the treatment of RP39,40 is relatively weak, and few of the treatments advocated in guidelines41–43 are licenced for RP. This reflects at least, in part, the challenges of clinical trials in RP 44 including (1) the effect of environmental temperature/seasonality on RP, meaning that trials have to be squeezed into the winter months (during which temperature can still fluctuate substantially); (b) the lack of objective outcome measures of RP, although progress is being made; 36 and (c) the well-recognised placebo effect in patients with RP, well demonstrated during a recent clinical trial of selexipag. 45
Treatment of uncomplicated RP
General measures
After removal of any aggravating factors, the first step is to avoid cold exposure whenever possible. The importance of dressing warmly cannot be overemphasised. Patient education is therefore an important aspect of management, and information leaflets produced by patient organisations are very helpful. Patients should be advised to stop smoking. In patients with SSc, there is evidence that smoking is associated with increased severity of digital vasculopathy. 46
Many patients, especially those with primary RP as in Scenario 1, will respond to ‘general’ (conservative) measures alone. ‘Non-drug’ approaches to treating RP have been comprehensively reviewed by Kwakkenbos and Thombs. 47
Drug treatment
Those patients who do not respond to general measures will require drug treatment, and this is the case for the majority of patients with SSc. 48 An approach to drug treatment is summarised in Figure 6, which gives an update of the UK Scleroderma Study Group consensus best pathway for Raynaud’s phenomenon. 41 The different groups of drug used (oral therapies summarised in Table 1) will be discussed in turn.
Figure 6.

Modification of the UK Scleroderma Study Group best practice recommendations on the management of Raynaud’s phenomenon. 41 Phosphodiesterase inhibition has been ‘moved up’ the original pathway to be positioned along with other oral vasodilator therapies.
ACE: angiotensin-converting enzyme; ARB: angiotensin receptor blocker: CCB: calcium channel blockers; PDE5: phosphodiesterase type 5; SSRI: selective serotonin reuptake inhibitor.
Table 1.
Examples of oral drugs most widely used in the treatment of complicated and uncomplicated RP.
| Drug or group of drugs | Main mechanism of action/other points to highlight | Examples | Usual dose range |
|---|---|---|---|
| Calcium channel blockers | Act on smooth muscle to cause vasodilation | Nifedipine (sustained release) | 10 mg twice daily to 40 mg twice daily |
| Amlodipine | 5 mg once daily to 10 mg once daily | ||
| Phosphodiesterase type 5 inhibitors | Inhibit degradation of cyclic guanosine monophosphate (and therefore increase nitric oxide effect) | Sildenafil | 20 mg/25 mg three times daily to 50 mg three times daily |
| Tadalafil | 20 mg alternate days to 20 mg once daily | ||
| Angiotensin II receptor blockers | Block action of angiotensin II on vascular smooth muscle | Losartan | 25 mg once daily to 100 mg once daily |
| Alpha-adrenergic blockers | Block vasoconstriction | Prazosin | 500 µg twice daily to 2 mg twice daily |
| Selective serotonin reuptake inhibitors | Block uptake of serotonin, a vasoconstrictor | Fluoxetine | 20 mg once daily |
| Endothelin-1 receptor antagonists | Block action of endothelin-1 on smooth muscle cells. For prevention of recurrent digital ulcers in patients with SSc | Bosentan | 62.5 mg twice daily increasing to 125 mg twice daily |
RP: Raynaud’s phenomenon; SSc: systemic sclerosis.
Calcium channel blockers
These are the widely considered first-line treatment, yet the evidence base for their use is disappointing. Recent Cochrane reviews have examined calcium channel blockers in patients with primary 49 and both primary and secondary RP. 50 The Cochrane review of primary RP alone 49 included seven randomised controlled trials (RCTs) representing 296 patients and concluded that calcium channel blockers were minimally effective: compared to placebo, calcium channel blockers reduced weekly RP attack frequency by 1.72 (95% CI = 0.60–2.84). The more recent review by Rirash et al., 50 which included 38 RCTs representing 982 patients (nine RCTs of primary RP, five of secondary RP and the others a mixture of patients with either primary or secondary RP), suggested that calcium channel blockers reduced weekly attack frequency by 6.07 (95% CI = −6.53, −5.61), although the reduction in attack frequency was only 2.93 per week if the trial with the largest reduction in attack frequency was excluded. The evidence was judged of low-to-moderate quality. 50 Subgroup analysis suggested that dihydropyridine calcium channel blockers may be more effective in primary than in secondary RP. 50
A standard approach is to commence a low-dose of a sustained release preparation and then gradually up-titrate the dose: a cautious approach is often better tolerated and less likely to result in discontinuation of treatment because of adverse effects. The commonest side-effects are headache, dizziness, nausea, palpitations and ankle oedema, with Rirash et al. 50 also commenting on the absence of serious side-effects.
Phosphodiesterase type 5 inhibitors
Phosphodiesterase type 5 (PDE5) inhibitors are being increasingly prescribed for RP, at least in SSc-related RP. They inhibit the degradation of cyclic guanosine monophosphate (cGMP), thus augmenting the action of nitric oxide (NO), a powerful vasodilator. NO relaxes smooth muscle by stimulating soluble guanylate cyclase, increasing cGMP.
Although earlier RCTs gave conflicting results, more recent RCTs have suggested benefit,51–54 and a meta-analysis published in 2013 55 concluded that PDE5 inhibitors have ‘significant but moderate efficacy in secondary RP’. Of the 244 patients included in the meta-analysis, only 8 had primary RP and most of 236 with connective tissue disease–related RP had SSc. 55 The key findings of this meta-analysis (six RCTs) were that the mean Raynaud’s Condition Score decreased by −0.46 (95% CI = −0.74, −0.17) (p = 0.002) and the daily frequency and duration of RP attacks also decreased significantly. 55 Subsequent to this meta-analysis, an RCT in 29 patients with connective tissue disease–related RP reported that udenafil 100 mg/day had similar efficacy to amlodipine 10 mg/day 54 in terms of reducing the frequency of RP attacks. All these studies were of short duration (treatment period 6 weeks or less). There is very little evidence base for PDE5 inhibitors in primary RP, indicating the need for well-designed RCTs for this indication.
PDE5 inhibitors can be prescribed together with a calcium channel blocker for additive effect, or substituted in patients experiencing no benefit from a calcium channel blocker or in whom a calcium channel blocker is not tolerated.
Other oral therapies
Other oral vasodilators are frequently used in the treatment of RP, but there is very little evidence base for this approach.56–59 Angiotensin II receptor antagonists are favoured by some clinicians, but there has been only one controlled trial (which included 25 patients with primary RP and 27 with SSc-related RP) and this was an open-label trial − 12-week treatment with losartan 50 mg/day reduced frequency and severity of RP attacks more than nifedipine 40 mg/day: 60 the benefit was more noticeable in the subgroup of patients with primary RP. Alpha-blockers, oral nitrates, and angiotensin-converting enzyme inhibitors are sometimes prescribed. Disappointingly, a recent RCT of the oral IP prostacyclin receptor agonist selexipag showed no reduction in SSc-related RP attacks. 45
A major limiting factor of all oral vasodilators is their propensity to cause vasodilatory side-effects. The recent European League Against Rheumatism (EULAR)-revised recommendations 42 suggest that the selective serotonin reuptake inhibitor – fluoxetine – ‘might be considered in the treatment of SSc-RP’. Fluoxetine is not associated with the same vasodilatory side-effects as other oral therapies for RP. The evidence to support its use comes from one open-label cross-over study comparing fluoxetine 20 mg daily to nifedipine 40 mg daily: 61 frequency and severity of RP attacks reduced on fluoxetine.
Figure 6 includes reference to antiplatelet therapies. SSc is associated with platelet activation: 11 antiplatelet agents could therefore improve microcirculatory blood flow by reducing ‘stickiness’. However, at present, there is no good evidence base for this approach, and given the complexity of clinical trials in RP, it seems unlikely that this will be obtained in the short term.
Topical vasodilators
None are licenced for RP. When topical glyceryl trinitrate (GTN) is prescribed as a systemic vasodilator, it is often poorly tolerated. 62 Although a multicentre, placebo-controlled trial which included 69 patients with primary RP and 150 patients with secondary RP (of whom 131 had SSc) demonstrated benefit from a novel gel formulation of GTN, MQX-503, 63 there are currently no approved topical treatments for RP. The aim of the study by Chung et al. was to examine ‘as required’ prevention of attacks: the gel was applied to the fingers (therefore primarily for a local effect) immediately before or within 5 min of onset of a Raynaud’s attack over a 4-week period. 63 This approach (which deserves further research) might therefore be optimally used in combination with a ‘baseline’ oral vasodilator. A recent small study in 10 patients 64 with secondary RP examined the use of topical 10% nifedipine and 5% sildenafil (one hand treated with nifedipine and one with sildenafil), pointing to a renewed interest in topical therapies.
Intravenous prostanoid therapy
The use of intravenous (IV) prostanoids tends to be mainly in patients with complicated RP, or a history of such, in the context of SSc-spectrum disorders. However, practice varies between countries, and iloprost may also be prescribed in severe uncomplicated SSc-related RP. 42
Recent findings/points to highlight
Probably, the main clinical advance has been the increasing use of PDE5 inhibitors as clinicians have become more familiar with their use in RP and with the fall in cost with patent expiry. Otherwise (and perhaps disappointingly), the focus of most recent meta-analyses has been to emphasise the lack of evidence base for many of our treatments, although a recent systematic review and meta-analysis of topical nitrates (which included seven studies and 346 patients 65 ) reported ‘significant efficacy’ in both primary and secondary RP. One of the studies included was from 1951, highlighting how the potential for topical treatments has long been recognised and that this approach deserves further research.
Treatment of complicated RP (digital ulceration and critical ischaemia)
Primary RP by definition is not associated with tissue damage. Therefore, a patient in whom RP has progressed to digital ulceration (as in Scenario 3) or critical ischaemia always has an underlying disease or condition which may require specific treatment. For example, an ischaemic finger in a patient with known vasculitis is likely to require corticosteroid and/or immunosuppressant therapy.
Here, only SSc-related ‘complicated RP’ will be considered. In other words, digital ulceration and critical ischaemia in the patient known to have SSc will be considered. The principles of management of digital ulceration and critical ischaemia are very similar and so both are considered together. Figure 7 shows an update of the UK Scleroderma Study Group consensus best pathway for digital ulceration. 41
Figure 7.

Modification of the UK Scleroderma Study Group best practice recommendations on the management of SSc-related digital ulceration. 41
PDE5: phosphodiesterase type 5.
Establishing the diagnosis and treating any remediable cause
This might seem a surprising first step, given that this is a discussion of SSc-related digital ulceration and critical ischaemia. However, it should never be forgotten that although complicated RP in the patient with SSc usually relates to a non-inflammatory angiopathy, other causes can contribute, including large vessel disease, a concomitant vasculitis (very unusual in the context of SSc) or a coagulopathy. 66 Large vessel disease is the most likely ‘additional’ cause and should always be considered in the patient with SSc who develops (especially unilateral) worsening of digital ulceration and/or critical ischaemia.
Key principles of management therefore include the following:
Early assessment of all patients with SSc and digital ulceration and/or critical ischaemia. Patient education is all important here – patients must be encouraged to seek urgent medical advice in the event of a digital ulcer or permanent finger discolouration. Prompt medical intervention may (a) prevent an ulcer enlarging or deepening, and becoming infected (with the risk of underlying bone infection) and (a) potentially save the digit(s) in the case of critical ischaemia.
Feel for the peripheral pulses. If absent, then in the case of critical ischaemia, this is a vascular surgical emergency. The patient will require urgent assessment with arterial Doppler in the first instance and possibly with angiography.
Vasoactive therapies for SSc-related digital ulceration and critical ischaemia
These therapies are additional to those discussed above under ‘uncomplicated RP’ and target the three major pathways thought to be key players in SSc pathogenesis: 67
Supplementation of the L-arginine/NO pathway;
Supplementation of the prostacyclin pathway;
Inhibition of endothelin-1 (ET-1).
PDE5 inhibitors
Although the SEDUCE study, 68 which compared 12-week treatment with sildenafil 20 mg three times daily to placebo, did not reach its primary endpoint (time to healing), there was some benefit from sildenafil in terms of a greater healing rate compared to placebo at week 8 (p = 0.01) and week 12 (p = 0.03).
Prostanoid therapy
A Cochrane review from 2000 indicated that IV prostanoids reduced frequency and severity of RP attacks and healed digital ulcers.69,70 Prescribing practice varies internationally: IV iloprost has been most studied and is most widely used, and in varying regimes, 71 but is not available in the United States, IV epoprostenol is another option. 72 Cruz et al. 73 have recently reviewed the use of IV epoprostenol. A major disadvantage of IV prostanoids is the need for hospitalisation, as well as systemic vasodilatory side-effects. Bellando-Randone et al. 71 recently reported that patients with early (oedematous) SSc may be more likely than patients with established disease to experience adverse effects including painful finger swelling with IV iloprost: this may be relevant when discussing with patients the advantages and disadvantages of trying a further course of treatment.
RCTs examining oral prostanoids have overall been disappointing. The most recent was of oral treprostinil: in a 20-week placebo-controlled study (parallel group design) in 147 patients, the primary endpoint of change in net digital ulcer burden was not reached, but patients receiving active treatment 74 experienced small (statistically insignificant) reduction in net ulcer burden (−0.43 ulcers) compared to placebo (−0.10 ulcers). Patient receiving treprostinil in an open-label extension also demonstrated a small reduction in net ulcer burden. In 51 patients in whom treprostinil was withdrawn after the open-label extension and for whom follow-up data were available (retrospective study design), 75 digital ulcer burden significantly increased after discontinuing treprostinil, suggesting that further studies of oral treprostinil should be considered.
ET-1 receptor antagonists
Bosentan is licenced for the prevention of recurrent SSc-related digital ulcers, having been shown in two placebo-controlled RCTs to reduce the number of new ulcers (but with no effect on ulcer healing).76,77 Disappointingly, macitentan, another dual ET-1 receptor antagonist, was not shown to confer benefit in either the DUAL-1 or DUAL-2 studies (of 289 and 265 patients, respectively). 78
Other drugs with effects on blood flow and/or coagulation
Many clinicians prescribe antiplatelet agents, as discussed above. For the patient with progressive critical ischaemia, there may be a case for short-term anticoagulation. Statins could potentially confer benefit on SSc-related digital vasculopathy via several different mechanisms,79,80 but further research is required before these can be generally recommended.
Other drug treatments – analgesics and antibiotics
Digital ulceration and critical ischaemia can be excruciatingly painful – adequate analgesia, often with opiates in the short term, is a key aspect of management. If there is any clinical suspicion of infection, then an antibiotic should be prescribed. Bone infection requires prolonged antibiotic therapy, and should always be suspected if an ulcer is chronic, and tender on palpation. Plain radiographs often do not demonstrate bone infection (and are often difficult to interpret when contractures are present), and if there is a high index of suspicion, magnetic resonance (MR) scanning should be performed.
Surgical management, botulinum toxin injections and fat grafting
Surgery
It is very difficult to establish an evidence base for surgery in a rare, heterogeneous disease, 81 but anecdotally, patients who do not respond to drug treatment may benefit from surgery. 82 Surgical debridement is helpful when there is necrotic tissue to be removed. In recent years, many case series have been published describing experience with digital sympathectomy, and this should be considered in refractory ulceration.82–85 However, this is a highly specialised procedure which should be performed only by an experienced surgeon, and the role of a skilled multi-disciplinary team is all important. 85 If all other approaches fail, amputation may (rarely) be required.
Botulinum toxin
Also attracting attention in recent years are botulinum toxin injections of the fingers86–89 with a recent small series of three patients suggesting benefit from botulinum toxin A in the toes. 90 Botulinum toxin A is thought to exert its action through blocking the sympathetic innervations. 91 However, a recent placebo-controlled RCT in 40 patients with SSc (botulinum toxin A was injected into one hand and sterile saline into the other) failed to confirm benefit as assessed with laser Doppler imaging, although there was some improvement in secondary endpoints. 92 Further RCTs are required to determine whether or not this is a treatment approach which should be more widely advocated. A recent study of 45 patients with SSc-related RP (randomised to treatment versus no-treatment, but not placebo-controlled) reported benefit from botulinum toxin B injections. 93
Fat grafting
A relatively new approach to treatment of SSc-related digital ulceration is autologous fat grafting/local injection of adipose-derived stromal vascular fraction94–96 with recent open-label follow-up data from 12 patients suggesting sustained benefit for 22–30 months. 97 Results of further studies are eagerly awaited.
Recent findings/points to highlight
As with ‘uncomplicated’ RP, the main recent advance has been the increasing use of PDE5 inhibitors. The use of bosentan is now well established. Although topical therapies still have a ‘long way to go’ before they are likely to be widely used, it is encouraging that these are again attracting interest. 65 Experience in procedural treatments – not only surgery but also botulinum toxin injections and ‘fat grafting’ – is increasing and it is encouraging to see that patients are being recruited into RCTs. It should not be forgotten that a key aspect of management is skilled nursing care, including wound cleansing and appropriate choice of dressings. 98
Main challenges for next 5–10 years
The ultimate aim is for patients with RP to be attack free, for progression to digital ulceration or critical ischaemia to be prevented in patients with SSc and (if patients with SSc do nonetheless develop ulcers) for ulcers to be treated early and effectively. Although progress has been made in the last 10 years, treatments remain sub-optimal. The specific challenges are as follows:
To continue to elucidate the pathogenesis of RP in order to develop new therapeutic targets. And, although some of these targets have already been identified, we need to find more effective ways to approach these, free from systemic side-effects (see below).
To improve our outcome measures for both early phase and later phase studies. Progress here will facilitate clinical trials of both primary and secondary RP.
To improve our infrastructure for investigator-led and industry-led clinical trials, with the aim of increasing ‘throughput’ of Phase 2 proof-of-concept (laboratory-based) studies in order to select the most appropriate candidates for Phase 3 trials. This is especially important for SSc-related RCTs, when the pool of patients eligible for clinical trials is finite and we need to choose wisely which drugs to put to the test in later phase trials.
To develop ‘local’ therapies for both RP and digital ulceration. Patients with cold extremities and/or digital ulceration need treatments which will improve the circulation to their fingers and toes, as opposed to treatments which cause systemic vasodilation and which are therefore often poorly tolerated. It seems counterintuitive to admit a patient to hospital for systemic vasodilation with IV iloprost when the problem is, for example, at the tip of a finger.
To prevent the structural digital vasculopathy of SSc. For clinicians with an interest in SSc, this is perhaps the greatest challenge of all. This will involve not only further elucidation of pathogenesis but also creating the infrastructure to ensure very early diagnosis of all patients with SSc (i.e. screening of patients with RP, including with capillaroscopy). This would then pave the way for most patients to have the opportunity of taking part in RCTs and observational studies of drugs with vascular remodelling potential.
Conclusion
The assessment and treatment of patients with RP, both primary and SSc-related, have progressed in the last 5 years. Increasing availability of nailfold capillaroscopy is facilitating early diagnosis in those patients with SSc. Our therapeutic options have increased with wider use of PDE5 inhibitors, and algorithms in different countries should ensure that patients are escalated along clinical pathways according to clinical need. However, many patients still experience a large burden of morbidity from RP and digital ulceration, and we must now focus on developing new therapies and putting these to the test in clinical trials.
Acknowledgments
The author is grateful to Dr Graham Dinsdale for his help with the images.
Footnotes
Declaration of conflicting interests: A.L.H. received research funding from Actelion.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Ariane L Herrick  https://orcid.org/0000-0003-4941-7926
https://orcid.org/0000-0003-4941-7926
References
- 1. Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Eng J Med 2016; 375: 556–565. [DOI] [PubMed] [Google Scholar]
- 2. Herrick AL. The pathogenesis, diagnosis and treatment of Raynaud phenomenon. Nat Rev Rheumatol 2012; 8(8): 469–479. [DOI] [PubMed] [Google Scholar]
- 3. Khouri C, Blaise S, Carpentier P, et al. Drug-induced Raynaud’s phenomenon: beyond beta-adrenoceptor blockers. Br J Clin Pharmacol 2016; 82(1): 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sandqvist G, Wollmer P, Scheja A, et al. Raynaud’s phenomenon and its impact on activities in daily life during one year of follow-up in early systemic sclerosis. Scand J Rheumatol 2018; 47(3): 206–209. [DOI] [PubMed] [Google Scholar]
- 5. Maundrell A, Proudman SM. Epidemiology of Raynaud’s phenomenon. In: Wigley FM, Herrick AL, Flavahan NA. (eds) Raynaud’s phenomenon: a guide to pathogenesis and treatment. New York: Springer, 2015, pp. 21–35. [Google Scholar]
- 6. Meier FM, Frommer KW, Dinser R, et al. Update on the profile of the EUSTAR cohort: an analysis of the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis 2012; 71(8): 1355–1360. [DOI] [PubMed] [Google Scholar]
- 7. Mayes MD, Lacey JV, Beebe-Dimmer J, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum 2003; 48: 2246–2255. [DOI] [PubMed] [Google Scholar]
- 8. Spencer-Green G. Outcomes in primary Raynaud phenomenon. Arch Intern Med 1998; 158(6): 595–600. [DOI] [PubMed] [Google Scholar]
- 9. Ingegnoli F, Ughi N, Crotti C, et al. Outcomes, rates and predictors of transition of isolated Raynaud’s phenomenon: a systematic review and meta-analysis. Swiss Med Wkly 2017; 147: w14506. [DOI] [PubMed] [Google Scholar]
- 10. Khouri C, Jouve T, Blaise S, et al. Peripheral vasoconstriction induced by beta-adrenoceptor blockers: a systematic review and a network meta-analysis. Br J Clin Pharmacol 2016; 82(2): 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herrick AL. Pathogenesis of Raynaud’s phenomenon. Rheumatology (Oxford) 2005; 44(5): 587–596. [DOI] [PubMed] [Google Scholar]
- 12. Flavahan NA. A vascular mechanistic approach to understanding Raynaud phenomenon. Nat Rev Rheumatol 2015; 11(3): 146–158. [DOI] [PubMed] [Google Scholar]
- 13. Flavahan NA. Pathophysiological regulation of the cutaneous vascular system in Raynaud’s phenomenon. In: Wigley FM, Herrick AL, Flavahan NA. (eds) Raynaud’s phenomenon: a guide to pathogenesis and treatment. New York: Springer, 2015, pp. 57–79. [Google Scholar]
- 14. Gualtierotti R, Ingegnoli F, Griffini S, et al. Detection of early endothelial damage in patients with Raynaud’s phenomenon. Microvasc Res 2017; 113: 22–28. [DOI] [PubMed] [Google Scholar]
- 15. Munir S, Freidin MB, Brain S, et al. Association of Raynaud’s phenomenon with a polymorphism in the NOS1 gene. PLoS ONE 2018; 13(4): e0196279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clark S, Hollis S, Campbell F, et al. The ‘distal-dorsal difference’ as a possible predictor of secondary Raynaud’s phenomenon. J Rheumatol 1999; 26: 1125–1128. [PubMed] [Google Scholar]
- 17. Anderson ME, Moore TL, Lunt M, et al. The ‘distal-dorsal difference’: a thermographic parameter by which to differentiate between primary and secondary Raynaud’s phenomenon. Rheumatology (Oxford) 2007; 46(3): 533–538. [DOI] [PubMed] [Google Scholar]
- 18. Herrick AL, Murray A. The role of capillaroscopy and thermography in the assessment and management of Raynaud’s phenomenon. Autoimmun Rev 2018; 17(5): 465–472. [DOI] [PubMed] [Google Scholar]
- 19. LeRoy EC, Medsger TA., Jr. Raynaud’s phenomenon: a proposal for classification. Clin Exp Rheumatol 1992; 10(5): 485–488. [PubMed] [Google Scholar]
- 20. Maverakis E, Patel F, Kronenberg DG, et al. International consensus criteria for the diagnosis of Raynaud’s phenomenon. J Autoimmun 2014; 48–49: 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. LeRoy EC, Medsger TA., Jr. Criteria for the classification of early systemic sclerosis. J Rheumatol 2001; 28(7): 1573–1576. [PubMed] [Google Scholar]
- 22. Matucci-Cerinic M, Allanore Y, Czirják L, et al. The challenge of early systemic sclerosis for the EULAR Scleroderma Trial and Research Group (EUSTAR) community – it is time to cut the Gordian knot and develop a prevention or rescue strategy. Ann Rheum Dis 2009; 68: 1377–1380. [DOI] [PubMed] [Google Scholar]
- 23. Avouac J, Fransen J, Walker UA, et al. Preliminary criteria for the very early diagnosis of systemic sclerosis: results of a Delphi Consensus Study from EULAR Scleroderma Trials and Research Group. Ann Rheum Dis 2011; 70(3): 476–481. [DOI] [PubMed] [Google Scholar]
- 24. Maricq HR, LeRoy EC. Patterns of finger capillary abnormalities in connective tissue disease by ‘widefield’ microscopy. Arthritis Rheum 1973; 16(5): 619–628. [DOI] [PubMed] [Google Scholar]
- 25. Cutolo M, Sulli A, Smith V. How to perform and interpret capillaroscopy. Best Pract Res Clin Rheumatol 2013; 27(2): 237–248. [DOI] [PubMed] [Google Scholar]
- 26. Van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification criteria for systemic sclerosis: an American College of Rheumatology/European League against rheumatism collaborative initiative. Arthritis Rheum 2013; 65(11): 2737–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification criteria for systemic sclerosis: an American College of Rheumatology/European League against rheumatism collaborative initiative. Ann Rheum Dis 2013; 72: 1747–1755. [DOI] [PubMed] [Google Scholar]
- 28. Koenig M, Joyal F, Fritzler MJ, et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: a twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum 2008; 58(12): 3902–3912. [DOI] [PubMed] [Google Scholar]
- 29. Bissell LA, Abignano G, Emery P, et al. Absence of scleroderma pattern at nail fold capillaroscopy valuable in the exclusion of scleroderma in unselected patients with Raynaud’s phenomenon. BMC Musculoskelet Disord 2016; 17(1): 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O’Reilly D, Taylor L, El-Hadidy K, et al. Measurement of cold challenge responses in primary Raynaud’s phenomenon and Raynaud’s phenomenon associated with systemic sclerosis. Ann Rheum Dis 1992; 51(11): 1193–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pauling JD, Flower V, Shipley JA, et al. Influence of the cold challenge on the discriminatory capacity of the digital distal-dorsal difference in the thermographic assessment of Raynaud’s phenomenon. Microvasc Res 2011; 82(3): 364–368. [DOI] [PubMed] [Google Scholar]
- 32. Hughes M, Moore T, O’Leary N, et al. A study comparing videocapillaroscopy and dermoscopy in the assessment of nailfold capillaries in patients with systemic sclerosis-spectrum disorders. Rheumatology (Oxford) 2015; 54(8): 1435–1442. [DOI] [PubMed] [Google Scholar]
- 33. Dinsdale G, Peytrignet S, Moore T, et al. The assessment of nailfold capillaries: comparison of dermoscopy and nailfold videocapillaroscopy. Rheumatology (Oxford) 2018; 57: 1115–1116. [DOI] [PubMed] [Google Scholar]
- 34. Pauling JD, Frech TM, Hughes M, et al. Patient-reported outcome instruments for assessing Raynaud’s phenomenon in systemic sclerosis: a SCTC Vascular Working Group report. J Scleroderma Relat Disord 2018; 3: 249–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pauling JD, Domsic RT, Saketkoo LA, et al. Multinational qualitative research study exploring the patient experience of Raynaud’s phenomenon in systemic sclerosis. Arthritis Care Res (Hoboken) 2018; 70: 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilkinson JD, Leggett SA, Marjanovic EJ, et al. A multicenter study of the validity and reliability of responses to hand cold challenge as measured by laser speckle contrast imaging and thermography: outcome measures for systemic sclerosis-related Raynaud’s phenomenon. Arthritis Rheumatol 2018; 70: 903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cutolo M, Vanhaecke A, Ruaro B, et al. Is laser speckle contrast analysis (LASCA) the new kid on the block in systemic sclerosis? A systematic literature review and pilot study to evaluate reliability of LASCA to measure peripheral blood perfusion in scleroderma patients. Autoimmun Rev 2018; 17: 775–780. [DOI] [PubMed] [Google Scholar]
- 38. Toprak U, Ozbalkan Z, Erdugan M, et al. Follow-up of treatment response with dynamic Doppler ultrasound in Raynaud phenomenon. AJR Am J Roentgenol 2017; 209(6): W388–W394. [DOI] [PubMed] [Google Scholar]
- 39. Hinze AM, Wigley FM. Pharmacotherapy options in the management of Raynaud’s phenomenon. Curr Treat Options in Rheum 2018; 4: 235–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Herrick AL. Evidence based management of Raynaud’s phenomenon. Ther Adv Musculoskelet Dis 2017; 9(12): 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hughes M, Ong VH, Anderson ME, et al. Consensus best practice pathway of the UK Scleroderma Study Group: digital vasculopathy in systemic sclerosis. Rheumatology (Oxford) 2015; 54(11): 2015–2024. [DOI] [PubMed] [Google Scholar]
- 42. Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis 2017; 76: 1327–1339. [DOI] [PubMed] [Google Scholar]
- 43. Belch J, Carlizza A, Carpentier PH, et al. ESVM guidelines – the diagnosis and management of Raynaud’s phenomenon. Vasa 2017; 46(6): 413–423. [DOI] [PubMed] [Google Scholar]
- 44. Wilkinson J. Design and reporting of randomised controlled trials for Raynaud’s phenomenon. In: Wigley FM, Herrick AL, Flavahan NA. (eds) Raynaud’s phenomenon: a guide to pathogenesis and treatment. New York: Springer, 2015, pp. 287–297. [Google Scholar]
- 45. Denton CP, Hachulla E, Riemekasten G, et al. Efficacy and safety of selexipag in adults with Raynaud’s phenomenon secondary to systemic sclerosis: a randomized, placebo-controlled, phase II study. Arthritis Rheumatol 2017; 69(12): 2370–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harrison BJ, Silman AJ, Hider SL, et al. Cigarette smoking as a significant risk factor for digital vascular diseases in patients with systemic sclerosis. Arthritis Rheum 2002; 46: 3312–3316. [DOI] [PubMed] [Google Scholar]
- 47. Kwakkenbos L, Thombs BD. Non-drug approaches to treating Raynaud’s phenomenon. In: Wigley FM, Herrick AL, Flavahan NA. (eds) Raynaud’s phenomenon: a guide to pathogenesis and treatment. New York: Springer, 2015, pp. 299–313. [Google Scholar]
- 48. Moinzadeh P, Riemekasten G, Siegert E, et al. Vasoactive therapy in systemic sclerosis: real-life therapeutic practice in more than 3000 patients. J Rheumatol 2016; 43(1): 66–74. [DOI] [PubMed] [Google Scholar]
- 49. Ennis H, Hughes M, Anderson ME, et al. Calcium channel blockers for primary Raynaud’s phenomenon. Cochrane Database Syst Rev 2016; 2: CD002069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rirash F, Tingey PC, Harding SE, et al. Calcium channel blockers for primary and secondary Raynaud’s phenomenon. Cochrane Database Syst Rev 2017; 12: CD000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shenoy PD, Kumar S, Jha LK, et al. Efficacy of tadalafil in secondary Raynaud’s phenomenon resistant to vasodilator therapy: a double-blind randomized cross-over trial. Rheumatology (Oxford) 2010; 49(12): 2420–2428. [DOI] [PubMed] [Google Scholar]
- 52. Herrick AL, Van den Hoogen F, Gabrielli A, et al. Modified-release sildenafil reduces Raynaud’s phenomenon attack frequency in limited cutaneous systemic sclerosis. Arthritis Rheum 2011; 63(3): 775–782. [DOI] [PubMed] [Google Scholar]
- 53. Caglayan E, Axmann S, Hellmich M, et al. Vardenafil for the treatment of Raynaud phenomenon: a randomized, double-blind, placebo-controlled crossover study. Arch Intern Med 2012; 172(15): 1182–1184. [DOI] [PubMed] [Google Scholar]
- 54. Lee EY, Park JK, Lee W, et al. Head-to-head comparison of udenafil vs amlodipine in the treatment of secondary Raynaud’s phenomenon: a double-blind, randomized, cross-over study. Rheumatology (Oxford) 2014; 53(4): 658–664. [DOI] [PubMed] [Google Scholar]
- 55. Roustit M, Blaise S, Allanore Y, et al. Phosphodiesterase-5 inhibitors for the treatment of secondary Raynaud’s phenomenon: systematic review and meta-analysis of randomised trials. Ann Rheum Dis 2013; 72(10): 1696–1699. [DOI] [PubMed] [Google Scholar]
- 56. Herrick AL. Raynaud’s phenomenon (secondary). BMJ Clin Evid 2008; 2008: 1125. [PMC free article] [PubMed] [Google Scholar]
- 57. Huisstede BM, Hoogvliet P, Paulis WD, et al. Effectiveness of interventions for secondary Raynaud’s phenomenon: a systematic review. Arch Phys Med Rehabil 2011; 92(7): 1166–1180. [DOI] [PubMed] [Google Scholar]
- 58. Stewart M, Morling JR. Oral vasodilators for primary Raynaud’s phenomenon. Cochrane Database Syst Rev 2012; (7): CD006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Garcia de la Pena Lefebvre P, Nishishinya MB, Pereda CA, et al. Efficacy of Raynaud’s phenomenon and digital ulcer pharmacological treatment in systemic sclerosis patients: a systematic literature review. Rheumatol Int 2015; 35(9): 1447–1459. [DOI] [PubMed] [Google Scholar]
- 60. Dziadzio M, Denton CP, Smith R, et al. Losartan therapy for Raynaud’s phenomenon and scleroderma: clinical and biochemical findings in a fifteen-week, randomized, parallel-group, controlled trial. Arthritis Rheum 1999; 42(12): 2646–2655. [DOI] [PubMed] [Google Scholar]
- 61. Coleiro B, Marshall SE, Denton CP, et al. Treatment of Raynaud’s phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology (Oxford) 2001; 40(9): 1038–1043. [DOI] [PubMed] [Google Scholar]
- 62. Teh L-S, Manning J, Moore T, et al. Sustained-release transdermal glyceryl trinitrate patches as a treatment for primary and secondary Raynaud’s phenomenon. Br J Rheumatol 1995; 34(7): 636–641. [DOI] [PubMed] [Google Scholar]
- 63. Chung L, Shapiro L, Fiorentino D, et al. MQX-503, a novel formulation of nitroglycerin, improves the severity of Raynaud’s phenomenon. Arthritis Rheum 2009; 60(3): 870–877. [DOI] [PubMed] [Google Scholar]
- 64. Wortsman X, Del Barrio-Diaz P, Meza-Romero R, et al. Nifedipine cream versus sildenafil cream for patients with secondary Raynaud phenomenon: a randomized, double-blind, controlled pilot study. J Am Acad Dermatol 2018; 78(1): 189–190. [DOI] [PubMed] [Google Scholar]
- 65. Curtiss P, Schwager Z, Cobos G, et al. A systematic review and meta-analysis of the effects of topical nitrates in the treatment of primary and secondary Raynaud’s phenomenon. J Am Acad Dermatol 2018; 78(6): 1110–1118.e3. [DOI] [PubMed] [Google Scholar]
- 66. Sharp C, Akram Q, Hughes M, et al. Differential diagnosis of critical digital ischemia in systemic sclerosis: report of five cases and review of the literature. Semin Arthritis Rheum 2016; 46(2): 209–216. [DOI] [PubMed] [Google Scholar]
- 67. Herrick AL. Therapeutic implications from the pathogenesis of Raynaud’s phenomenon. Expert Rev Clin Immunol 2017; 13(7): 723–735. [DOI] [PubMed] [Google Scholar]
- 68. Hachulla E, Hatron P-Y, Carpentier P, et al. Efficacy of sildenafil on ischaemic digital ulcer healing in systemic sclerosis: the placebo controlled SEDUCE study. Ann Rheum Dis 2016; 75(6): 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wigley FM, Wise RA, Seibold JR, et al. Intravenous iloprost infusion in patients with Raynaud phenomenon secondary to systemic sclerosis – a multicenter, placebo-controlled, double-blind study. Ann Intern Med 1994; 120(3): 199–206. [DOI] [PubMed] [Google Scholar]
- 70. Pope J, Fenlon D, Thompson A, et al. Iloprost and cisaprost for Raynaud’s phenomenon in progressive systemic sclerosis. Cochrane Database Syst Rev 2000; (2): CD000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bellando-Randone S, Bruni C, Lepri G, et al. The safety of iloprost in systemic sclerosis in a real-life experience. Clin Rheumatol 2018; 37(5): 1249–1255. [DOI] [PubMed] [Google Scholar]
- 72. Law ST, Farber HW, Simms RW. Use of intravenous epoprostenol as a treatment for the digital vasculopathy associated with the scleroderma spectrum of diseases. J Scleroderma Related Disorders 2017; 2: 208–212. [Google Scholar]
- 73. Cruz JE, Ward A, Anthony S, et al. Evidence for the use of epoprostenol to treat Raynaud’s phenomenon with or without digital ulcers. Ann Pharmacotherapy 2016; 50: 1060–1067. [DOI] [PubMed] [Google Scholar]
- 74. Seibold JR, Wigley FM, Schiopu E, et al. Digital ulcers in SSc treated with oral treprostinil: a randomized, double-blind, placebo-controlled study with open-label follow-up. J Scleroderma Relat Disord 2017; 2: 42–49. [Google Scholar]
- 75. Shah AA, Schiopu E, Chatterjee S, et al. The recurrence of digital ulcers in patients with systemic sclerosis after discontinuation of oral treprostinil. J Rheumatol 2016; 43(9): 1665–1671. [DOI] [PubMed] [Google Scholar]
- 76. Korn JH, Mayes M, Matucci-Cerinic M, et al. Digital ulcers in systemic sclerosis – prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum 2004; 50(12): 3985–3993. [DOI] [PubMed] [Google Scholar]
- 77. Matucci-Cerinic M, Denton CP, Furst DE, et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis 2011; 70(1): 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Khanna D, Denton CP, Merkel PA, et al. Effect of macitentan on the development of new ischemic digital ulcers in patients with systemic sclerosis. JAMA 2016; 315(18): 1975–1988. [DOI] [PubMed] [Google Scholar]
- 79. Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35(9): 1801–1808. [PubMed] [Google Scholar]
- 80. Ladak K, Pope JE. A review of the effects of statins in systemic sclerosis. Semin Arthritis Rheum 2016; 45(6): 698–705. [DOI] [PubMed] [Google Scholar]
- 81. Herrick A, Muir L. Raynaud’s phenomenon (secondary). BMJ Clin Evid 2014; 2014: 1125. PMCID: PMC4200538. [PMC free article] [PubMed] [Google Scholar]
- 82. Muir L. Surgical management. In: Wigley FM, Herrick AL, Flavahan NA. (eds) Raynaud’s phenomenon: a guide to pathogenesis and treatment. New York: Springer, 2015, pp. 361–372. [Google Scholar]
- 83. Merritt WH. Role and rationale for extended periarterial sympathectomy in the management of severe Raynaud syndrome: techniques and results. Hand Clin 2015; 31(1): 101–120. [DOI] [PubMed] [Google Scholar]
- 84. Momeni A, Sorice SC, Valenzuela A, et al. Surgical treatment of systemic sclerosis – is it justified to offer peripheral sympathectomy earlier in the disease process? Microsurgery 2015; 35(6): 441–446. [DOI] [PubMed] [Google Scholar]
- 85. Chiou G, Crowe C, Suarez P, et al. Digital sympathectomy in patients with scleroderma: an overview of the practice and referral patterns and perceptions of rheumatologists. Ann Plastic Surg 2015; 75: 637–643. [DOI] [PubMed] [Google Scholar]
- 86. Neumeister MW. The role of botulinum toxin in vasospastic disorders of the hand. Hand Clin 2015; 31(1): 23–37. [DOI] [PubMed] [Google Scholar]
- 87. Uppal L, Dhaliwal K, Butler PE. A prospective study of the use of botulinun toxin injections in the treatment of Raynaud’s syndrome associated with scleroderma. J Hand Surg 2014; 39E: 876–880. [DOI] [PubMed] [Google Scholar]
- 88. Motegi S, Yamada K, Toki S, et al. Beneficial effect of botulinum toxin A on Raynaud’s phenomenon in Japanese patients with systemic sclerosis: a prospective, case series study. J Dermatol 2016; 43(1): 56–62. [DOI] [PubMed] [Google Scholar]
- 89. Segreto F, Marangi GF, Cerbone V, et al. The role of botulinum toxin A in the treatment of Raynaud phenomenon. Ann Plast Surg 2016; 77(3): 318–323. [DOI] [PubMed] [Google Scholar]
- 90. Dhaliwal K, Griffin M, Denton CP, et al. The novel use of botulinum toxin A for the treatment of Raynaud’s phenomenon in the toes. BMJ Case Rep. Epub ahead of print 9 March 2018. DOI: 10.1136/bcr-2017-219348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhou Y, Liu Y, Hao Y, et al. The mechanism of botulinum A on Raynaud syndrome. Drug Des Devel Ther 2018; 12: 1905–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bello RJ, Cooney CM, Melamed E, et al. The therapeutic efficacy of botulinum toxin in treating scleroderma-associated Raynaud’s phenomenon: a randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol 2017; 69(8): 1661–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Motegi SI, Uehara A, Yamada K, et al. Efficacy of botulinum toxin B injection for Raynaud’s phenomenon and digital ulcers in patients with systemic sclerosis. Acta Derm Venereol 2017; 97(7): 843–850. [DOI] [PubMed] [Google Scholar]
- 94. Bank J, Fuller SM, Henry GI, et al. Fat grafting to the hand in patients with Raynaud phenomenon: a novel therapeutic modality. Plast Reconstr Surg 2014; 133(5): 1109–1118. [DOI] [PubMed] [Google Scholar]
- 95. Guillaume-Jugnot P, Daumas A, Magalon J, et al. State of the art – autologous fat graft and adipose tissue-derived stromal vascular fraction injection for hand therapy in systemic sclerosis patients. Curr Res Transl Med 2016; 64(1): 35–42. [DOI] [PubMed] [Google Scholar]
- 96. Del Papa N, Zaccara E, Di Luca G, et al. Adipose-derived cell transplantation in systemic sclerosis: state of the art and future perspectives. J Scleroderma Relat Disord 2017; 2: 33–41. [Google Scholar]
- 97. Daumas A, Magalon J, Jouve E, et al. Long-term follow-up after autologous adipose-derived stromal vascular fraction injection into fingers in systemic sclerosis patients. Curr Res Transl Med 2017; 65(1): 40–43. [DOI] [PubMed] [Google Scholar]
- 98. Lebedoff N, Frech TM, Shanmugam VK, et al. Review of local wound management for scleroderma-associated digital ulcers. J Scleroderma Relat Disord 2018; 3:66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]