

Abstract
Background
Gaucher disease, a rare disorder, is caused by inherited deficiency of the enzyme glucocerebrosidase. It is unique among the ultra‐orphan disorders in that four treatments are currently approved by various regulatory authorities for use in routine clinical practice. Hitherto, because of the relatively few people affected worldwide, many of whom started therapy during a prolonged period when there were essentially no alternatives to imiglucerase, these treatments have not been systematically evaluated in studies such as randomized controlled trials now considered necessary to generate the highest level of clinical evidence.
Objectives
To summarize all available randomized controlled study data on the efficacy and safety of enzyme replacement therapies and substrate reduction therapy for treating Gaucher disease.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Inborn Errors of Metabolism Trials Register. Additional searches were conducted on ClinicalTrials.gov for any ongoing studies with potential interim results, and through PubMed. We also searched the reference lists of relevant articles and reviews.
Date of last search: 07 August 2014.
Selection criteria
All randomized and quasi‐randomized controlled studies (including open‐label studies and cross‐over studies) assessing enzyme replacement therapy or substrate reduction therapy, or both, in all types of Gaucher disease were included.
Data collection and analysis
Two authors independently assessed the risk of bias in the included studies, and extracted relevant data.
Main results
Of the 488 studies retrieved by the electronic searches, eight met the inclusion criteria and were analysed (300 participants). Response parameters were restricted to haemoglobin concentration, platelet count, spleen and liver volume and serum biomarkers (chitotriosidase and CCL18). Only one publication reported a 'low risk of bias' score in all parameters assessed, and all studies included were randomized.
Four studies reported the responses to enzyme replacement therapy of previously untreated individuals with type 1 Gaucher disease. Two studies investigated maintenance enzyme replacement therapy in people with stable type 1 Gaucher disease previously treated for at least two years. One study compared substrate reduction therapy, enzyme replacement therapy and a combination thereof as maintenance therapy in people with type 1 Gaucher disease previously treated with enzyme replacement therapy. One study examined substrate reduction therapy in people with chronic neuronopathic (type 3) Gaucher disease who continued to receive enzyme replacement therapy.
Treatment‐naïve participants had similar increases in haemoglobin when comparing those receiving imiglucerase or alglucerase at 60 units/kg, imiglucerase or velaglucerase alfa at 60 U/kg, taliglucerase alfa at 30 units/kg or 60 units/kg, and velaglucerase alfa at 45 units/g or 60 units/kg. For platelet count response in participants with intact spleens, a benefit for imiglucerase over velaglucerase alfa at 60 units/kg was observed, mean difference ‐79.87 (95% confidence interval ‐137.57 to ‐22.17). There were no other significant differences in platelet count response when comparing different doses of velaglucerase alfa and of taliglucerase alfa, and when comparing imiglucerase to alglucerase. Spleen and liver volume reductions were not significantly different in any enzyme replacement therapy product or dose comparison study. Although a dose effect on serum biomarkers was not seen after nine months, a significantly greater reduction with higher dose was reported after 12 months in the velaglucerase study, mean difference 16.70 (95% confidence intervaI 1.51 to 31.89). In the two enzyme replacement therapy maintenance studies comparing infusions every two weeks and every four weeks, there were no significant differences in haemoglobin concentration, platelet count, and spleen and liver volumes over a 6 to 12 month period when participants were treated with the same cumulative dose.
A total of 25 serious adverse events were reported, nearly all deemed unrelated to treatment.
There are, as yet, no randomized trials of substrate reduction therapy in treatment‐naïve patients that can be evaluated. Miglustat monotherapy appeared as effective as continued enzyme replacement therapy for maintenance of hematological, organ and biomarker responses in people with type 1 Gaucher disease previously treated with imiglucerase for at least two years. In those with neuronopathic Gaucher disease, no significant improvements in haemoglobin concentration, platelet count or organ volumes occurred when enzyme replacement therapy was augmented with miglustat.
One randomized controlled study assessing substrate reduction therapy was published immediately prior to producing the final version of this review, and this, along with a further ongoing study (expected to be published in the near future), will be assessed for eligibility in a future update of the review.
Authors' conclusions
The results reflect the limitations of analysing evidence restricted to prospective randomized controlled trials, especially when dealing with chronic rare diseases. This analysis suggests that, during the first year of treatment, different recombinant glucocerebrosidases are bio‐similar and non‐inferior in safety and efficacy for surrogate biological response parameters. Enzyme replacement therapy given at 30 to 45 units/kg body weight every two to four weeks was generally as effective as the 60 unit/kg dose for the assessed clinical outcomes. The analysis emphasise the need to determine whether it is realistic to carry out multi‐decade prospective clinical trials for rare diseases such as type 1 Gaucher disease. With large treatment effects on the classical manifestations of the disorder, therapeutic investigations in Gaucher disease mandate innovative trial designs and methodology to secure decisive data concerning long‐term efficacy and safety – with the realization that knowledge about disease‐modifying actions that are sustained are of crucial importance to people with this chronic condition.
Keywords: Humans, 1‐Deoxynojirimycin, 1‐Deoxynojirimycin/adverse effects, 1‐Deoxynojirimycin/analogs & derivatives, Enzyme Inhibitors, Enzyme Inhibitors/adverse effects, Enzyme Replacement Therapy, Enzyme Replacement Therapy/methods, Gaucher Disease, Gaucher Disease/blood, Gaucher Disease/drug therapy, Glucosylceramidase, Glucosylceramidase/therapeutic use, Hemoglobin A, Hemoglobin A/metabolism, Hepatomegaly, Hepatomegaly/drug therapy, Platelet Count, Randomized Controlled Trials as Topic, Splenomegaly, Splenomegaly/drug therapy, Substrate Specificity
Plain language summary
Treatment options for Gaucher disease
Gaucher disease, a rare disorder, is caused by inherited deficiency of the enzyme glucocerebrosidase. This defect leads to the build‐up of a fatty material called glucocerebroside in various cells in the body. Untreated individuals may suffer from anaemia, a decrease in platelet counts, massive enlargement of the liver and spleen, and damage to the bones. Two different types of treatment are available: the intravenous supplementation of the deficient protein glucocerebrosidase (enzyme replacement therapy), or the oral administration of a drug that slows down the production of the fatty material that it normally breaks down (substrate reduction therapy).
Although there are many published studies about treatment of Gaucher disease, those that are based on randomized controlled clinical studies are considered to provide the strongest evidence. Eight such studies (300 participants) were identified by systematically reviewing the medical literature. Potential risks of bias in the methods employed in the original studies and in the data reported were assessed; only one study scored 'low' in all bias domains. Six evaluated the possible beneficial effects on the disease by enzyme replacement therapy using different products, doses or frequencies of administration; two studies examined the effects of substrate reduction therapy. Owing to different study methods and data formats, the original results could not be pooled or combined in order to obtain summary measures of efficacy. Our analysis, despite being limited by the small number of studies, suggests that, at least during the first year of treatment, different enzyme replacement therapy drugs appear to have a similar safety profile and all improve some of the most obvious manifestations of the disease.
One randomized controlled study assessing substrate reduction therapy was published immediately prior to producing the final version of this review, and this, along with a further ongoing study (expected to be published in the near future), will be assessed for eligibility in a future update of the review.
Background
Description of the condition
Gaucher disease (GD) is one of the most common lysosomal disorders, affecting approximately 1 out of 75,000 births worldwide (Meikle 1999), although the reported incidence and birth prevalence of such diseases figures vary considerably. It is an inherited autosomal recessive trait, and is most prevalent amongst Ashkenazi Jews (Grabowski 1997) in whom the frequency of heterozygote carriers is estimated to be as high as 6% to 10% (Beutler 1993). Gaucher disease is classified into three major phenotypes based on clinical features, age of onset, and the degree of organ involvement (Grabowski 2008). Because all affected individuals may experience enlargement of the liver and spleen and involvement of skeletal system and bone marrow, other features are used for categorization into the three types. Type 1 (GD1) (non‐neuronopathic), the most prevalent form in Western countries, is characterized by sparing of the central nervous system (CNS) ‐ a feature that differentiates it from type 2 (GD2) (acute‐neuronopathic) and type 3 (GD3) (subacute‐neuronopathic) variants. However, intermediate phenotypes of GD are recognized, mainly between type 2 and type 3 (Conradi 1991), and the different types of GD may alternatively be considered as part of a phenotypic continuum (Cherin 2006; Sidransky 2004)
While GD is caused by an inborn defect of glycosphingolipid metabolism due to a deficiency of acid ß‐glucocerebrosidase in the lysosome (Brady 1966), the molecular link between the consequential clinico‐pathological effects of this enzyme defect and the complex multisystem disorder is not fully understood and remains the subject of intensive study, such as linking specific mutations to clinical severity (Beutler 2006). Reduced capacity to degrade exogenous and endogenous glycosphingolipids induces the accumulation of glucocerebroside within macrophages with the development of the pathognonomic 'storage' macrophages, often referred to as 'Gaucher cells' (Lee 1982). These cells are found principally in the bone marrow, spleen and liver. Prominent clinical features of GD are anaemia, bleeding (mostly attributed to thrombocytopenia), enlargement of the spleen and liver, osteopenia (Stowens 1985) and bone pain. In GD2 and GD3, the neurological manifestations are usually dominant, and neurodegenerative processes lead to tremor, parkinsonism, and abnormal eye movements. Other features of GD are osteonecrosis (which characteristically causes severe bone pain and bone crises), retarded growth (Kaplan 2006; Kauli 2000), and, occasionally, lung and even kidney disease. Given the description of GD1 as non‐neuronopathic, it is noteworthy that it has been recently shown that carriers of GD, as well as people with the disease, are at a greatly increased risk of parkinsonism (Goker‐Alpan 2004; Sidransky 2004)
Description of the intervention
Complementation of deficient enzyme activity in the lysosomal compartment of macrophages is brought about by receptor‐mediated glycoprotein uptake, via mannose receptors on the cell surface membrane; this mechanism forms the basis for enzyme replacement therapy (ERT) (Mistry 1996). Macrophage‐targeted (mannose‐terminal) human glucocerebrosidase is generally accepted as the first line therapy for GD. Recombinant enzymes, such as imiglucerase (Cerezyme®, Genzyme Corporation, Cambridge, MA) taliglucerase alfa (Elelyso®, Protalix Biotherapeutics, Carmiel, Israel) or velaglucerase alfa (VPRIV®, Shire HGT, Cambridge MA), are administered intravenously. The treatment dose usually varies from 15 units/kg to 60 units/kg body weight (Pastores 2010). Most individuals respond well, with a prompt reduction in liver and spleen volume, increased haemoglobin concentrations and platelet counts; moreover, skeletal pain and episodic symptoms originating from the involvement of bone may also resolve (Hollak 2010). These beneficial effects are usually apparent by six months of administration but, in some people, longer periods of treatment may be required for other manifestations of disease to improve. Biochemical markers of the disease in the plasma show marked reduction upon effective treatment. Usually the response to administered enzyme reaches a plateau within the first few years. Of note, the administered enzymes do not cross the blood‐brain barrier (BBB), and the neurologic manifestations seen in GD2 and GD3 are refractory to this treatment. The use of ERT is considered safe, with few hypersensitivity reactions, and rare cases of anaphylaxis (Starzyk 2007). Unwanted effects include temporary weight gain and insulin resistance (Langeveld 2008). At present, it is unclear whether prolonged survival due to therapy in affected individuals is accompanied by salutary effects or other effects on the development of complications of GD, such as multiple myeloma (Rosenbloom 2005) and Parkinson disease (Bultron 2010), which occur with increased frequency in adults.
Substrate reduction therapy (SRT) has been explored as an alternative treatment for GD. Substrate reducing agents were used to inhibit the biosynthesis of glucocerebroside (Cox 2000), thereby ameliorating the pathological accumulation of glycolipids and cognate metabolites in GD (Platt 1994;Inokuchi 1987). At present, the only approved agent in this class is miglustat (Zavesca®, Actelion Pharmaceuticals US, San Francisco, CA), which is licensed as a second‐line agent in adults who are unsuitable for or unwilling to take ERT. The drug, an iminosugar, has the advantage of being administered orally; however, it has a higher frequency of unwanted effects, such as tremor and gastrointestinal complaints, and, although comparative studies have never been performed, may be less effective for several aspects of GD than the available ERTs (Cox 2003;Kuter 2013). Unlike ERT, miglustat can partially cross the BBB (Aerts 2006). Miglustat has also been explored in people with GD3, but no clinically meaningful neurological benefit was observed (Schiffmann 2008). However, the trial was small and participants differed substantially in disease severity. Eliglustat tartrate (Genzyme, a Sanofi Corporation, Cambridge, MA, USA), a potent inhibitor of glucosylceramide synthase that is chemically unrelated to miglustat, is also orally active (McEachern 2007) and is now in the late stage of phase 3 clinical trials for the treatment of GD1 (Cox 2010; Lukina 2010) ‐ as a substrate for P‐glycoprotein, eliglustat tartrate is extruded from the central nervous system (CNS).
Why it is important to do this review
In considering clinical studies, evidence for efficacy of ERT and SRT in GD is unusually sparse. This is somewhat understandable, given that the condition is an ultra‐orphan multisystem chronic disorder with diverse clinical manifestations and severity. Although there is much anecdotal and experiential conviction that ERT for GD has long‐term benefits, in order to substantiate the effectiveness of enzyme therapy in GD, there is need for a better corpus of evidence, including clinical trials that, due to the rarity, heterogeneity and chronicity of this disorder may or may not necessarily be able to conform to accepted criteria for evidence‐based medicine. Nevertheless, a critical examination of what evidence from randomized clinical studies is now available in GD is essential as a basis for future studies. Given the exceptional commercial interest in this disorder, a robust platform for evaluating long‐term efficacy and other outcomes is desirable. The recent development of new products (miglustat, velaglucerase‐alfa, taliglucerase‐alfa, eliglustat tartrate) as alternatives to imiglucerase raises questions as to whether the multiplicity of new non‐inferior agents offers biological benefits beyond potential cost savings (a significant consideration considering the huge expense of these treatments) and redundancy in case of a supply interruption such as occurred in 2009. Here we use the methodology of the Cochrane collaboration to examine the current evidentiary status of the four approved therapies currently prescribed for more than 7000 people with GD worldwide.
Objectives
To assess the effectiveness and safety of ERTs and SRTs.
To identify knowledge gaps which may result in new areas of research.
Methods
Criteria for considering studies for this review
Types of studies
All randomized and quasi‐randomized controlled studies (including open‐label trials and cross‐over studies).
Types of participants
Individuals with GD of any age (specifically including children) and of any disease severity.
Types of interventions
ERT (of any dose) compared with SRT (of any dose)
The addition of SRT to ERT compared with ERT alone
ERT or SRT, or both (of different doses), compared with no treatment
Comparison of different ERTs (evaluated in this review)
Comparison of different SRTs
Types of outcome measures
Primary outcomes
Frequency of side effects
Haemoglobin and platelet counts
-
Hepatosplenomegaly
Spleen size (e.g. multiples of normal, tissue mL/kg body weight or mL)
Liver size (e.g. multiples of normal, tissue mL/kg body weight or mL)
Secondary outcomes
-
Change in biomarker levels
Chitotriosidase (nmol/ml/hr)
CCL18‐PARC
Angiotensin‐converting enzyme (ACE) (international units (IU)/L)
Tartrate resistant acid phosphatase (TRAP) (IU/L) levels
-
Skeletal outcomes
Frequency of bone pain
Frequency of bone crisis events (events in which severe pain is experienced due to infarction of the bones)
Bone density scores (z scores and t scoresֹׁׁ)ׁ
Search methods for identification of studies
Electronic searches
Relevant trials were identified by searching the Cochrane Cystic Fibrosis and Genetic Disorders Group's Inborn Errors of Metabolism Trials Register using the term: gauchers.
The Inborn Errors of Metabolism Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated with each new issue of The Cochrane Library), quarterly searches of MEDLINE and the prospective handsearching of one journal ‐ Journal of Inherited Metabolic Disease. Unpublished work is identified by searching through the abstract books of the Society for the Study of Inborn Errors of Metabolism conference and the SHS Inborn Error Review Series. For full details of all searching activities for the register, please see the relevant section of the Cystic Fibrosis and Genetic Disorders Group Module.
Additional searches were conducted on ClinicalTrials.gov for any ongoing studies with potential interim results, and through MEDLINE (PubMed) (Appendices).
Date of the most recent search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Inborn Errors of Metabolism Trials Register: 07 August 2014.
Searching other resources
The bibliographic references of all retrieved literature were reviewed for additional reports of studies. In addition, the authors contacted experts in the field in an attempt to retrieve any further relevant data and papers.
Data collection and analysis
Selection of studies
Two authors (ES and LD) independently assessed and discussed the papers identified by the initial searches in order to decide whether they met the inclusion criteria.
Data extraction and management
Two authors (ES and LD) independently extracted the data using standard acquisition forms, that included information about:
methods of the study (type of study, whether randomization was applied, number of participants and dropouts);
characteristics of the study population;
type of intervention;
data for assessing the risk of bias (methods of randomization, whether blinding was applied (and who was blinded), type of sequence generation, and type of allocation concealment);
outcomes and results (means, standard deviations (SD) or standard errors (SE).
Assessment of risk of bias in included studies
We assessed the included studies using the Cochrane risk of bias assessment tool, focusing on the following domains (Higgins 2011a).
Assessment of sequence generation
low: if allocation sequence is suitable to prevent selection bias (i.e. computer‐generated lists, coin tossing, shuffling cards or envelopes, random number table, etc);
high: if allocation sequence could be related to prognosis and thus introduce selection bias (i.e. date of admission, clinical judgement, participant preference, results of laboratory tests, availability of intervention, date of birth, etc);
unclear: if there is insufficient information regarding the sequence generation.
Assessment of allocation concealment
low: if investigators were shielded from predicting the assignment of participants into the intervention groups (i.e. central randomization was done by telephone or by a pharmacy, identical and sequentially numbered drug containers or sequentially numbered sealed and opaque envelopes);
high: if investigators were not shielded from predicting the assignment of participants into the intervention groups (i.e. non‐opaque envelopes, open allocation schedule, etc);
unclear: if the method of concealment is not described.
Assessment of blinding
low: if no blinding was done, but outcome was not likely to be influenced by the lack of blinding; or if blinding was ensured and was unlikely to be broken;
high: if no blinding was done, and outcome was likely to be affected by the lack of blinding; or blinding was attempted but could have been broken;
unclear: if the method of blinding is not described.
Incomplete data
We considered whether it was stated how many participants were lost to follow‐up, and if reasons for this were given; when loss to follow up did occur, we checked whether an intention‐to‐treat‐analysis was performed. According to these criteria, we defined three levels of risk of bias:
low risk: if the number or reasons (or both) for loss to follow up were mentioned, and if intention‐to‐treat‐analysis was carried out;
high risk: if number or reasons (or both) for loss to follow up were not mentioned, or intention‐to‐treat‐analysis was not carried (or both);
unclear risk: if the original study did not specify the way loss to follow‐up was handled.
Selective outcome reporting
We assessed the consistency between the outcomes originally planned to be reported during the study, and those actually reported in the published paper, by comparing the trial protocols and registries with the information in the final publication. When trial protocols were not available, the methods section of the publication were compared to the published results.
Other sources of bias
We reviewed relevant studies for other potential sources of bias, such as: deviation from study protocol; early cessation of the study; selective reporting of subgroups; or a bias due to poor delivery of the interventions.
Measures of treatment effect
We have analysed the continuous data by calculating treatment effects' mean difference (MD) and the corresponding 95% CIs. If relevant, in future updates of this review, we plan to analyse dichotomous data by calculating the odds ratio (OR) and 95% confidence intervals (CIs).
Unit of analysis issues
If cross‐over studies are included in future updates of this review, we plan to use the methods recommended by Elbourne (Elbourne 2002) to conduct a meta‐analysis combining results from such studies. Similarly, if cluster‐randomized studies are identified and included, we will check these for unit of analysis errors based on the advice given in chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
Dealing with missing data
In four of the eight included studies (de Fost 2007; Elstein 2007; Kishnani 2009; Schiffmann 2008), the data were not in the format of mean with SD or SE, or were incompletely reported. We contacted the authors of these manuscripts, and in three cases obtained further data (de Fost 2007; Kishnani 2009; Schiffmann 2008). In one instance, the authors were unable to provide original data (Elstein 2007). In this case we extracted means from the original figures and estimated SDs from the reported statistical probability values, assuming normal distribution of the variables.
In future updates of the review, if some of the numerical data are missing from further identified studies (i.e. SDs), we will contact the original investigators and request these data. If we are unable to retrieve these data, we will attempt to calculate these by utilising the available data (e.g. when a SD is not reported, calculation may be possible based on given CIs, means, and the number of participants). If this is not possible, we will attempt to impute SDs based on similar studies (i.e. interventions, time points, population).
Assessment of heterogeneity
Since a meta‐analysis was not carried out, we did not assess heterogeneity between the studies mathematically. In a visual assessment of the forest plots, no major heterogeneity between studies was noted. In future updates of this review, if we conduct a meta‐analysis, we will assess the forest plots visually, and will use the I2 statistic and the Chi2 test and its corresponding P value, as stated in chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011).
Assessment of reporting biases
As per protocol, since we identified fewer than 10 studies, we did not construct a funnel plot. Therefore, no comments regarding reporting biases are discussed in this review. If future updates of this review include 10 or more studies, we will assess potential publication bias by constructing and assessing the symmetry of a funnel plot.
Data synthesis
Time points originally considered in the analysis 0 to under 3 months, 3 months to under 6 months, 6 months to under 9 months, 9 months to under 12 months, 12 months to under 18 months, and 18 months up to 24 months. For haemoglobin, platelet counts, disease biomarkers, liver and spleen volumes, the latter two time points are evaluated in the current review. Since visceral effects and improvement in blood counts in the first 0 to 6 months of treatment add no worthwhile measure of clinical efficacy, especially in the case of biomarkers such as chitotriosidase, and because data relevant to this time frame was only reported by one group (de Fost 2007), we have chosen not to include it in the analyses. A fixed‐effect model will be used to synthesize data in future updates of this manuscript, if possible.
Subgroup analysis and investigation of heterogeneity
Different doses or regimens (or both) of therapy: up to 30 international units/kg/2 weeks; 30 to 60 international units/kg/2 weeks; and above 60 international units/kg/2 weeks
Different ERTs and SRTs
Splenectomised individuals compared with those having an intact spleen
Sensitivity analysis
As per protocol, a sensitivity analysis (excluding studies with an overall high risk of bias) was not performed, since less than 10 studies were identified. However, a sensitivity analysis was conducted excluding individuals reported in one study who patently did not have GD.
Results
Description of studies
Results of the search
A total of 488 studies and five conference abstracts were retrieved by the electronic search, and screened for eligibility. Eight studies met the inclusion criteria and were included in the analysis (Figure 1), yielding a total of 300 adults and children (males and females), of multinational origin. Studies were excluded if they were not randomized, not controlled, or if an irrelevant intervention was assessed.
1.
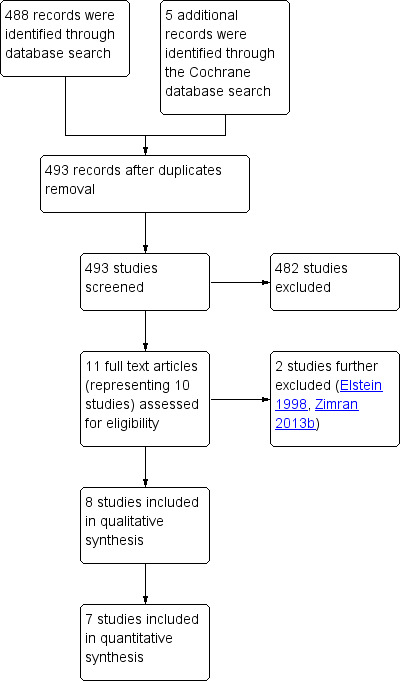
Study flow diagram.
No placebo‐controlled studies were found.
Included studies
Eight studies fulfilled the inclusion criteria for this review. Six studies (234 participants) compared different ERTs (de Fost 2007; Gonzalez 2013; Grabowski 1995; Kishnani 2009; Ben Turkia 2013; Zimran 2011b); and two studies compared the addition of SRT to ERT compared with ERT alone (66 participants) (Elstein 2007; Schiffmann 2008). No trials compared exactly the same treatments. There were a range of ERTs used (velaglucerase, imiglucerase, alglucerase or taliglucerase) which were compared to each other or the same ERT but at different doses; and one SRT used (miglustat).
Half of the included studies evaluated pre‐treated or stabilized individuals (de Fost 2007; Kishnani 2009; Schiffmann 2008; Elstein 1998). Seven studies enrolled people with GD1; one study (Schiffmann 2008) was restricted to people with GD3. Unlike the others, the primary focus of this study was on neurological rather than systemic responses.
Four studies enrolled participants who were either ERT or SRT naïve or who had an interruption of prior ERT of at least 12 months. In one of these, two participants without evidence of GD were included (Gonzalez 2013). Sub‐analyses, excluding these participants, were therefore carried out.
Of note, four of the included studies that enrolled individuals who were already receiving ERT were not primary efficacy studies, but rather were designed to test the effect of a new intervention on maintenance of previously achieved therapeutic gains.
For more details regarding the included studies, please see the Characteristics of included studies table.
Excluded studies
Two non‐randomized studies and one investigating alendronate disodium were excluded from the analysis (Elstein 1998; Wenstrup 2004; Zimran 2013b). In the Elstein study, 28 people with GD1 received a low‐dose low‐frequency imiglucerase regimen (Elstein 1998); in the Zimran study, 40 people with GD1 received different doses of velaglucerase alfa (Zimran 2013b). A further study was excluded as the intervention was not relevant (alendronate disodium) (Wenstrup 2004).
Studies awaiting classification
There is one study, published just immediately prior to producing the final version of this review, that is awaiting classification (Engage 2015). Eliglustat is the focus of this phase III randomized placebo‐controlled study of treatment‐naïve patients (Engage 2015).
Ongoing studies
There is one ongoing study (Encore 2013). This phase III randomized study compares eliglustat to a imiglucerase control arm and is expected to be published in full in the near future (Encore 2013).
Risk of bias in included studies
All studies were randomized. Four of the included studies were unblinded (de Fost 2007; Elstein 2007; Kishnani 2009; Schiffmann 2008) (Characteristics of included studies). A risk of bias table was completed for randomization, allocation concealment, blinding, incomplete outcome data, and study size (Characteristics of included studies, Figure 2). In most cases, the risk for adequate sequence generation and allocation concealment bias could not be assessed, due to insufficient details regarding the methods used. Only one publication (Grabowski 1995) reported a 'low risk of bias' score in all parameters assessed. Gonzalez was described as with high risk for other type of bias, due to the inclusion of two participants who proved not to have GD (Gonzalez 2013).
2.
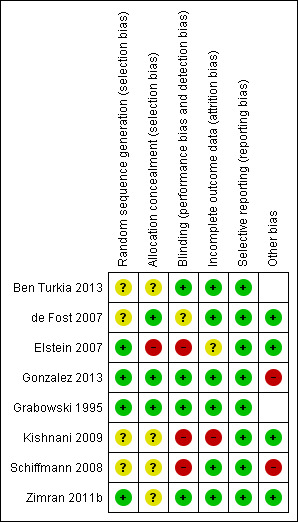
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Generation of randomization sequence
For four studies we assessed there to be a low risk of bias for the generation of the randomization sequence (Elstein 2007; Gonzalez 2013; Grabowski 1995; Zimran 2011b) and for the remaining four, an unclear risk of bias due to insufficient data (de Fost 2007; Kishnani 2009; Schiffmann 2008; Ben Turkia 2013).
Allocation concealment
Three studies described the allocation concealment in enough detail to be judged as to have a low risk of bias (de Fost 2007; Gonzalez 2013; Grabowski 1995). The risk of bias bias in four studies was considered unclear (Kishnani 2009; Schiffmann 2008; Ben Turkia 2013; Zimran 2011b), and as high in the final study (Elstein 2007).
Blinding
Three studies were considered to have a high risk for this domain (Elstein 2007; Kishnani 2009; Schiffmann 2008): in one study it was stated that participants were not blinded to treatment group (Kishnani 2009), and in two studies an oral drug was added to IV therapy (or oral drug alone) compared to IV therapy, without reporting the use of an oral placebo (Elstein 2007; Schiffmann 2008). It is therefore plausible to assume that participants were not blinded to treatment. Three studies were described as double‐blind and one stated that observers were blinded; these four were deemed to have a low risk of bias (Ben Turkia 2013; Gonzalez 2013; Grabowski 1995; Zimran 2011b). The remaining study was classified as unclear due to insufficient data (de Fost 2007).
Incomplete outcome data
Kishnani was the only study considered to have a high risk of bias for incomplete outcome reporting (Kishnani 2009). This was decided because an intention‐to‐treat analysis was carried out, despite not meeting all criteria for such analysis. Six studies were regarded as having a low risk for bias for this domain (Ben Turkia 2013; de Fost 2007; Gonzalez 2013; Grabowski 1995; Schiffmann 2008; Zimran 2011b). The final study was categorized as having an unclear risk of bias (Elstein 2007).
Selective reporting
All eight studies were assessed as having a low risk for selective reporting bias.
Other potential sources of bias
Gonzalez included participants who did not have GD and was therefore regarded as a high risk of 'other bias' (Gonzalez 2013). After careful evaluation of the Schiffmann study, it appeared that the intervention groups were not balanced in several aspects, such as sex and age of the participants (Schiffmann 2008). It was not stated in the original paper whether these differences were statistically significant.
Effects of interventions
Enzyme replacement treatment (ERT)
Six studies (234 participants) assessed this intervention (Ben Turkia 2013; de Fost 2007; Gonzalez 2013; Grabowski 1995; Kishnani 2009; Zimran 2011b).
Primary outcomes
1. Frequency of side effects
All six studies (234 participants) evaluating ERT reported on side effects.
In the Grabowski study (alglucerase versus imiglucerase), three participants (unclear from paper in which group) reported dizziness, pruritus or a skin eruption (Grabowski 1995). No discontinuation of the intervention was required.
In the Zimran study (taliglucerase alfa), 137 adverse events were reported by 23 participants (12 in the 30 units/kg and 11 in the 60 units/kg group) (Zimran 2011b). The investigators graded all adverse events as mild or moderate and transient. Nausea and headache were the most frequent. One participant in each dose group developed hypersensitivity reactions, with one of them experiencing an immediate hypersensitivity reaction and being excluded from the intention‐to‐treat efficacy analysis after pre‐dose serum IgE concentrations were found to be elevated. This participant was also subsequently determined to have a sensitivity to imiglucerase. A single participant withdrew from the study after a mast cell‐mediated hypersensitivity reaction occurred.
In the Gonzalez study, (velaglucerase alfa), no treatment‐related hypersensitivity reactions or life‐threatening serious adverse events were described (Gonzalez 2013). Fifteen of 25 participants (six in the 60 units/kg group and nine in the 45 units/kg group) experienced at least one adverse event, of which, 14 were classified as infusion‐related. Headache, nasopharyngitis, traumatic injury, arthralgia, cough, pyrexia, dizziness, nasal congestion, influenza, bone pain and prolonged activated partial thromboplastin time (aPTT) were the most common.
In the Ben Turkia study (imiglucerase versus velaglucerase alfa), three serious adverse events occurred in the participants receiving velaglucerase alfa (allergic dermatitis, severe prolonged aPTT and, in a child with no prior neurological history, a 15‐minute generalized tonic‐clonic seizure immediately after an infusion) (Ben Turkia 2013). All were deemed unrelated to treatment. Similar frequencies of drug‐related adverse events (most infusion‐related) were reported for the two ERTs (8 of 17 participants in the velaglucerase group; 6 of 17 participants in the imiglucerase group). One imiglucerase participant withdrew consent due to multiple infusion reactions.
In the Kishnani study (imiglucerase), 18 serious adverse events occurred (13 in the 33 participants in the once‐every‐two‐weeks infusion group; 5 in the 62 participants in the once‐every‐four‐weeks infusion group) (Kishnani 2009). None were graded by the investigators as infusion‐related. Fatigue, a falling haemoglobin concentration, decrease in platelet count and increasing splenomegaly, (all associated with worsening or progression of disease), were more frequent in the group infused every four weeks (Kishnani 2009).
2. Haemoglobin (Hb) concentrations and platelet counts (PLT)
All six studies (234 participants) assessing ERT evaluated the effects on Hb concentration and PLT counts.
a. Haemoglobin concentration
Four studies evaluated Hb in treatment‐naïve participants (see Characteristics of included studies), and two in participants who were actively receiving treatment (maintenance studies). Among treatment‐naïve, anaemic participants in the four relevant studies, increases in Hb concentration of 1.62 to 3.2 g/dL were reported over 6‐ to 12‐month observation periods.
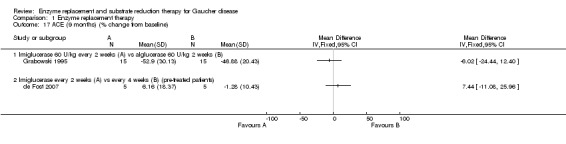
When treatment‐naïve participants were analysed, no significant difference was seen at nine months when comparing imiglucerase and alglucerase at 60 units/kg every two weeks (Analysis 1.1; Figure 3; Grabowski 1995). No significant difference was seen between imiglucerase 60 units/kg and velaglucerase alfa 60 units/kg every two weeks at nine months (Analysis 1.1; Ben Turkia 2013), even when participants with a history of splenectomy were excluded from the analysis. On comparing taliglucerase alfa at 30 units/kg to a 60 units/kg regimen (both approximately every two weeks), at nine months, there was no statistically significant difference (Analysis 1.1; Zimran 2011b).
1.1. Analysis.
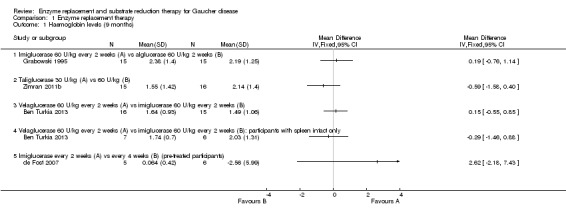
Comparison 1 Enzyme replacement therapy, Outcome 1 Haemoglobin levels (9 months).
3.
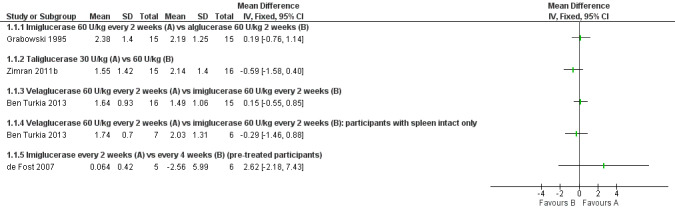
Forest plot of comparison: 1 Enzyme replacement therapy, outcome: 1.1 Haemoglobin levels (9 months).
For velaglucerase alfa, at 12 months, there was no significant difference in Hb outcome between 45 units/kg and 60 units/kg every two weeks even after exclusion of two non‐GD participants who were erroneously included (Analysis 1.2; Gonzalez 2013).
1.2. Analysis.
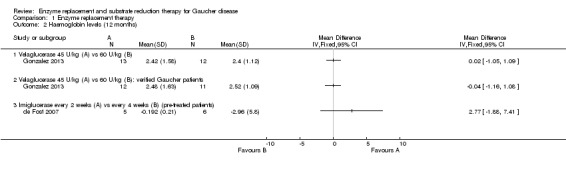
Comparison 1 Enzyme replacement therapy, Outcome 2 Haemoglobin levels (12 months).
In the two maintenance studies that could be assessed with our methodology (de Fost 2007; Kishnani 2009) ‐ in which an end‐point values analysis is reported, no statistically significant difference in Hb concentrations was observed between imiglucerase given at intervals of either two or four weeks, at the same monthly total dose, or at nine, 12 or 24 months (Analysis 1.1; Analysis 1.2, Analysis 1.3).
1.3. Analysis.
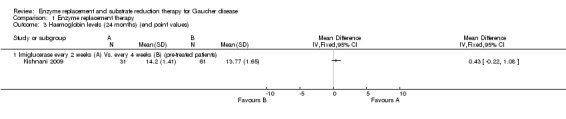
Comparison 1 Enzyme replacement therapy, Outcome 3 Haemoglobin levels (24 months) (end point values).
b. Platelet count
After a nine‐month study, there was no significant difference in PLT between participants treated with either imiglucerase 60 units/kg or alglucerase 60 units/kg every two weeks (Analysis 1.4; Figure 4; Grabowski 1995). In the nine‐month results from the comparison study of imiglucerase and velaglucerase alfa at 60 units/kg every two weeks, there was no significant difference in PLT response when all trial participants are included in the analysis (Ben Turkia 2013). However, when participants with a history of splenectomy, in whom pre‐treatment platelet counts are most often in the high normal range (Charrow 2000), are excluded from the analysis (20 of 34 enrolled participants; 10 participants from each treatment group), a significant difference was seen, in favour of imiglucerase, MD ‐79.87 (95% CI ‐137.57 to ‐22.17) (Analysis 1.4; Ben Turkia 2013). The study authors suggest that this difference may be attributable to a greater prevalence of splenic structural lesions in the velaglucerase treatment arm, a phenomenon that was not controlled in the trial and that is reported to independently affect the platelet response to ERT (Stein 2010). On comparing taliglucerase alfa at 30 units/kg to at 60 units/kg regimen (both approximately every two weeks), at nine months, there was no statistically significant difference in PLT responses (Analysis 1.4; Zimran 2011b).
1.4. Analysis.
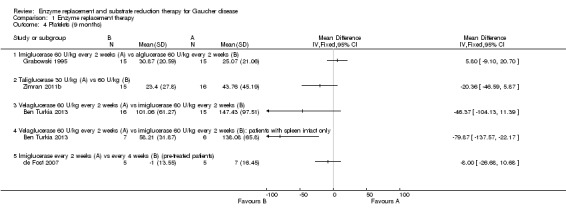
Comparison 1 Enzyme replacement therapy, Outcome 4 Platelets (9 months).
4.
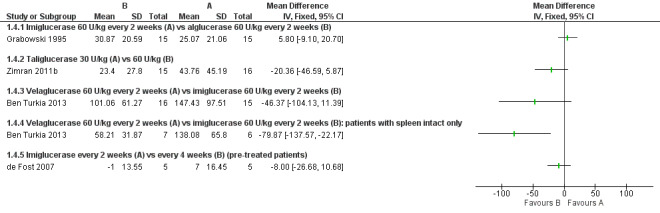
Forest plot of comparison: 1 Enzyme replacement therapy, outcome: 1.4 Platelets (9 months).
In a 12‐month assessment of PLT response to either velaglucerase alfa at 45 units/kg or 60 units/kg every two weeks, no difference was seen between the different doses. This finding remained when the two participants without GD were excluded (Analysis 1.5; Gonzalez 2013).
1.5. Analysis.
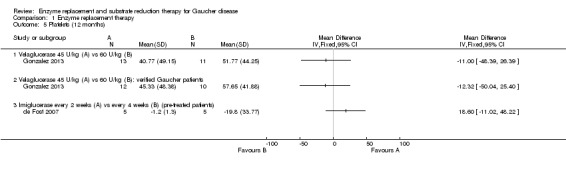
Comparison 1 Enzyme replacement therapy, Outcome 5 Platelets (12 months).
In the de Fost and Kishnani (end‐point values analysis) imiglucerase maintenance studies, no significant differences were seen between PLT response to imiglucerase infusions at intervals of either two or four weeks, at the same monthly total dose, either at nine, 12 and up to 24 months (Analysis 1.4; Analysis 1.5, Analysis 1.6; de Fost 2007; Kishnani 2009).
1.6. Analysis.

Comparison 1 Enzyme replacement therapy, Outcome 6 Platelets ‐ 24 months (end point values).
3. Hepatosplenomegaly
a. Liver volume
Six studies (234 participants) evaluated the effects of ERT on the change in liver volume. Four of these studies assessed the change in liver volume at up to nine months, and are included in the six to nine months time interval (104 participants) (Ben Turkia 2013; de Fost 2007; Grabowski 1995; Zimran 2011b). Two studies evaluated the change at 12 months (31 participants) (de Fost 2007; Gonzalez 2013), and one study at 24 months (89 participants) (Kishnani 2009). No clear difference in liver volume response was seen among ERT preparations in treatment‐naïve participants nor was there any advantage. As with the determination of Hb concentration levels, no clear benefit for higher doses of velaglucerase alfa was observed even when the analysis was restricted only to participants with verified GD (Analysis 1.7; Figure 5; Analysis 1.8). In the study that evaluated changes in liver volume after 24 months of treatment in stabilized participants, no significant difference was seen between imiglucerase infusions every two weeks to every four weeks (Analysis 1.9).
1.7. Analysis.
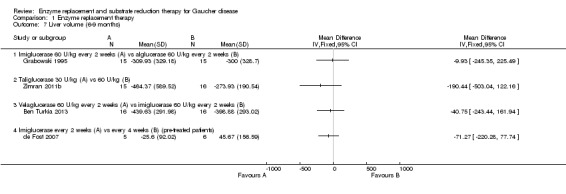
Comparison 1 Enzyme replacement therapy, Outcome 7 Liver volume (6‐9 months).
5.
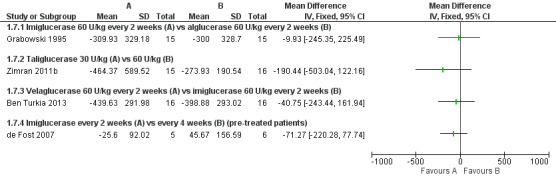
Forest plot of comparison: 1 Enzyme replacement therapy, outcome: 1.7 Liver volume (6‐9 months).
1.8. Analysis.
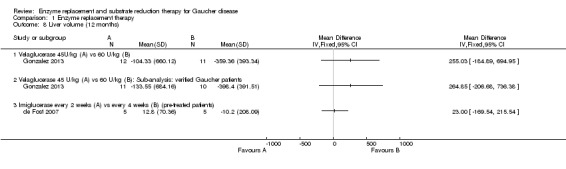
Comparison 1 Enzyme replacement therapy, Outcome 8 Liver volume (12 months).
1.9. Analysis.
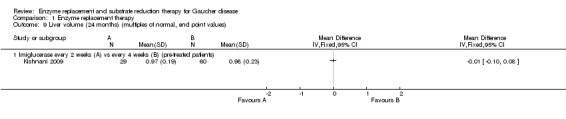
Comparison 1 Enzyme replacement therapy, Outcome 9 Liver volume (24 months) (multiples of normal, end point values).
b. Spleen volume
Four studies (82 participants) evaluated the change in spleen volume under ERT infusions in the time interval of six to nine months (Ben Turkia 2013; de Fost 2007; Grabowski 1995; Zimran 2011b).This was also assessed at 12 months (51 participants) (de Fost 2007; Gonzalez 2013), and at 24 months of treatment (64 participants) (Kishnani 2009).
In treatment‐naïve participants, when comparing imiglucerase with alglucerase, or with velaglucerase alfa, no significant difference was seen in spleen volume changes, assessed after nine months of treatment (Analysis 1.10). Of note, this result appears to be discrepant between the effects on platelet counts, suggesting a direct impact on platelet formation and survival, rather than simply spleen function. There was no significant difference in effect of ERT dose on spleen volume response in the study in which participants were treated for nine months with taliglucerase 30 units/kg/every two weeks or 60 units/kg/every two weeks (Analysis 1.10; Figure 6; Zimran 2011b). After 12 months of treatment, no significant difference in spleen volume was noted when comparing velaglucerase at 45 units/kg/every two weeks or at 60 units/kg/every two weeks (Analysis 1.11; Gonzalez 2013). In the 12‐month maintenance study, different frequencies of imiglucerase administration did not show a difference favouring one treatment arm over the other (Analysis 1.11; de Fost 2007). Even at 24 months, no significant change in spleen volume was noted between different imiglucerase administration frequencies for stabilized participants (Analysis 1.12).
1.10. Analysis.
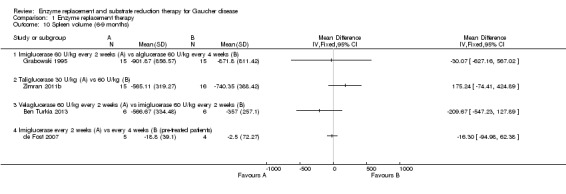
Comparison 1 Enzyme replacement therapy, Outcome 10 Spleen volume (6‐9 months).
6.
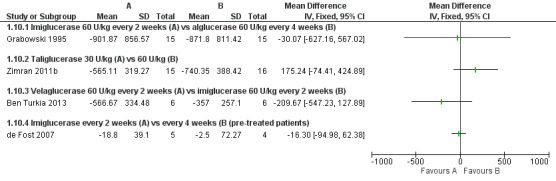
Forest plot of comparison: 1 Enzyme replacement therapy, outcome: 1.10 Spleen volume (6‐9 months).
1.11. Analysis.
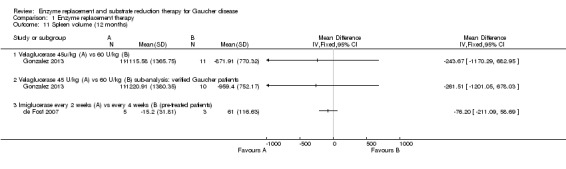
Comparison 1 Enzyme replacement therapy, Outcome 11 Spleen volume (12 months).
1.12. Analysis.
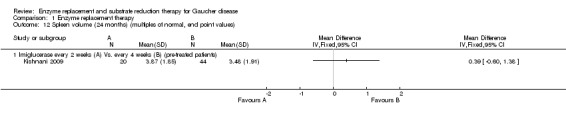
Comparison 1 Enzyme replacement therapy, Outcome 12 Spleen volume (24 months) (multiples of normal, end point values).
Secondary outcomes
1. Biomarkers (chitotriosidase, CCL 18‐PARC and ACE)
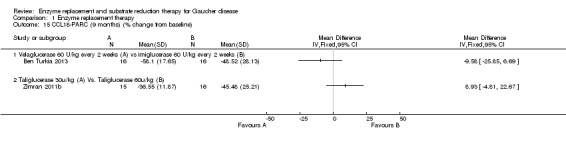
Three studies (60 participants) evaluated the change in chitotriosidase at a six to nine months time interval (Ben Turkia 2013; de Fost 2007; Zimran 2011b). The de Fost study included data both for six and nine months. Only two studies evaluated the change in chitotriosidase levels at 12 months (de Fost 2007; Gonzalez 2013), giving a total of 30 participants. Change in CCL18‐PARC levels were evaluated at nine months in two studies (63 participants) (Ben Turkia 2013; Zimran 2011b), and at 12 months in one study (24 participants) (Gonzalez 2013). After nine months, there was no significant difference in the reduction of serum chitotriosidase or CCL18‐PARC in ERT‐naïve participants treated either with velaglucerase alfa or imiglucerase at 60 units/kg/every two weeks (Ben Turkia 2013). There was also no significant dose effect for either dose of taliglucerase (Zimran 2011b) on reduction of chitotriosidase activity after nine months of treatment (Analysis 1.13; Analysis 1.15). However, after 12 months of treatment, confirmed GD participants receiving 60 units/kg of velaglucerase every two weeks had a greater per cent reduction (from baseline) in plasma chitotriosidase activity and in CCL 18‐PARC when compared with the 45 units/kg dose group, MD 22.61 (95% CI 7.65 to 37.57) and MD 16.70 (95% CI 1.51 to 31.89), respectively (Analysis 1.14; Analysis 1.16; Gonzalez 2013). Chitotriosidase and CCL 18‐PARC results were not reported in the alglucerase versus imiglucerase study because these tests were not available in 1995. However, there were no differences between the treatment arms for other serum biomarkers (acid phosphatase and angiotension converting enzyme (Grabowski 1995). In the de Fost maintenance trial, there were no significant differences in chitotriosidase activity after 12 months between participants treated with imiglucerase at two‐week or four‐week intervals (Analysis 1.14).
1.13. Analysis.
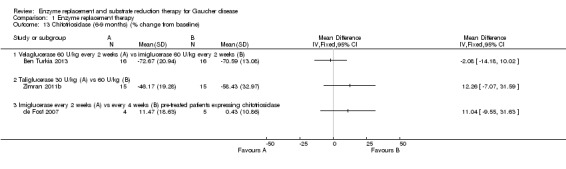
Comparison 1 Enzyme replacement therapy, Outcome 13 Chitotriosidase (6‐9 months) (% change from baseline).
1.15. Analysis.
Comparison 1 Enzyme replacement therapy, Outcome 15 CCL18‐PARC (9 months) (% change from baseline).
1.14. Analysis.
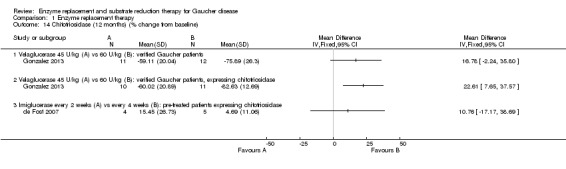
Comparison 1 Enzyme replacement therapy, Outcome 14 Chitotriosidase (12 months) (% change from baseline).
1.16. Analysis.
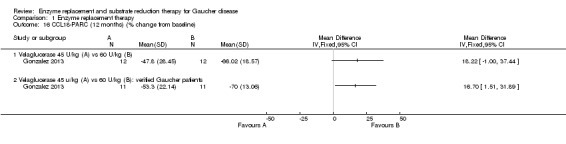
Comparison 1 Enzyme replacement therapy, Outcome 16 CCL18‐PARC (12 months) (% change from baseline).
The change in ACE levels was assessed in two studies (40 pre‐treated participants) (de Fost 2007; Grabowski 1995). No significant differences were noted when comparing imiglucerase to alglucerase, or when comparing different frequencies of imiglucerase infusions at nine months (Analysis 1.17). Only one trial reported TRAP levels (Kishnani 2009). The data available did not allow it to be included in the analysis.
1.17. Analysis.
Comparison 1 Enzyme replacement therapy, Outcome 17 ACE (9 months) (% change from baseline).
2. Skeletal outcomes
Three studies evaluated skeletal outcomes (de Fost 2007; Gonzalez 2013; Zimran 2011b). In two studies, 19 participants were evaluated (de Fost 2007; Zimran 2011b), and in the third study, no comment was made regarding the exact number of participants evaluated (Gonzalez 2013). The wide variety of reporting for this outcome, and the lack of available numerical data, allowed limited analysis. Results are therefore described as in the original studies.
In the de Fost study, a quantitative chemical shift imaging (QCSI) technique was used to assess lumbar bone marrow fat concentration, as an indicator of bone marrow involvement. In this technique, the difference between water and fat signal frequencies in magnetic resonance imaging allows the quantification of the fat signal fraction. If deterioration in disease severity occurs, an increase in Gaucher cell mass can lead to bone marrow involvement, which will cause a decrease in QCSI. No benefit was noted for participants treated with imiglucerase at two‐weekly intervals compared to the four‐weekly interval group, either at 6 or at 12 months (Analysis 1.18; Analysis 1.19). The study authors reported that two participants with low baseline QCSI enrolled in the low‐frequency group, were classified as treatment failures: in one of the participants a decrease from 29% at baseline to 24% QCSI at 12 months occurred, together with hepatomegaly, fatigue and abdominal discomfort, but without clinical bone problems; and in the other participant a decrease in QCSI from 33% to 23% at 12 months occurred, without any bone complications or other symptoms. A return to the high‐frequency regimen led to an increase in QCSI up to 27% at 18 months.
1.18. Analysis.
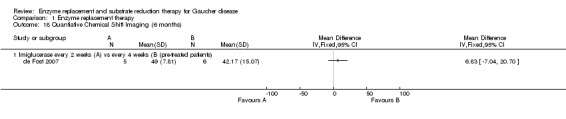
Comparison 1 Enzyme replacement therapy, Outcome 18 Quantiative Chemical Shift Imaging (6 months).
1.19. Analysis.
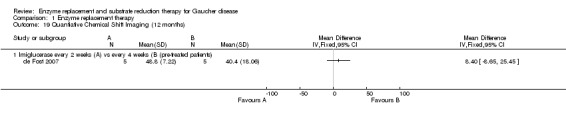
Comparison 1 Enzyme replacement therapy, Outcome 19 Quantiative Chemical Shift Imaging (12 months).
Zimran reported no deterioration in bone density in any of the treatment groups (Zimran 2011b). In the Gonzalez study, bone mineral density measurements were captured, but were not part of the declared objectives, and were not reported (Gonzalez 2013).
Substrate reducing therapy (SRT)
Two randomized controlled studies (36 GD1 and 30 GD3 participants) assessed the effect of either addition or substitution of miglustat on haemoglobin concentration, platelet count, liver and spleen volume in participants previously treated with or continuing to receive ERT (Elstein 2007; Schiffmann 2008). Elstein compared three treatment arms: miglustat; imiglucerase; and the combination of both, in pre‐treated, stabilized people with GD1 (Elstein 2007). Schiffmann evaluated ERT treatment compared with the combination of ERT and miglustat in people with GD3 over a period of 12 months, with an additional 12‐month extension phase where all participants received miglustat (not included in this review) (Schiffmann 2008). Additional clinical parameters, primarily related to neuronopathic manifestations, were reported in the latter study. There are no randomized GD clinical studies for miglustat or other SRT in treatment‐naïve patients that met the criteria for this analysis.
Primary outcomes
1. Frequency of side effects
Elstein reported no severe adverse events during the initial six month randomization phase (Elstein 2007). However, 94% of participants had gastrointestinal adverse events (diarrhoea, abdominal pain, constipation, flatulence, nausea and vomiting), 68% experienced weight decrease. Information as to how many gastrointestinal adverse events were confined to participants treated with miglustat is not included in the study report. However, these adverse events are consistent with other reports of miglustat gastrointestinal side effects attributable to off‐target inhibition of intestinal disaccharidases (Amiri 2012). A total of 62% of participants had neurological adverse events (tremor, dizziness, headache, fatigue, weakness). Electromyography abnormalities were noted in 8 of 36 enrolled participants (with no baseline measurements available), but all participants were asymptomatic. Two of the eight affected participants did not receive miglustat.
The most frequent miglustat‐associated adverse events reported by participants with GD3 enrolled in the Schiffmann study were of gastrointestinal origin ‐ diarrhoea (21 participants), abdominal pain (10 participants), vomiting (six participants) and weight loss (seven participants, up to 20% decrease) (Schiffmann 2008). Ten participants needed at least one medication for these events during the initial 13 weeks of intervention, with a decrease of those needing such medications over time. Also reported were neurological adverse events, including tremor (11 participants), convulsions (six participants), headache (six participants) and "clinical or subclinical neuropathy" in four participants with previously normal EMG recordings. Of these, three were randomized to the initial 12 months miglustat intervention and one withdrew from the study. Other adverse events included cough, pyrexia, nasopharyngitis, and an increase in serum AST and prolongation of aPTT.
2. Haemoglobin (Hb) concentrations and platelet counts (PLT)
In both studies, the available data did not allow the calculation of the mean effect change or the corresponding SDs for the haematological outcomes. Consequently, results are reported as in the original articles. In the Elstein study, Hb in clinically stable GD1 participants did not change significantly from baseline over a six‐month treatment period in the miglustat group (P = 1.01), the imiglucerase continuation group (P = 0.198), or in the miglustat‐imiglucerase group (P = 0.815) and no significant inter‐group differences occurred (Elstein 2007). Similarly, for PLT, no significant per cent changes from baseline were noted in any of the treatment arms. However, there was a statistically significant difference (20%, P = 0.035) between the imiglucerase and the miglustat arms with PLT increasing in the imiglucerase‐maintained participants and decreasing in those treated solely with miglustat.
Type 3 GD participants randomized to receive miglustat experienced a negligible change of Hb from a baseline of 12.7 g/dL (range: 10.6 to 14.9) to 12.9 g/dL (range: 11.5 to 14.5) after 12 months of treatment (n = 18). Platelet counts at baseline were 243 x 103/µL (range: 124 to 334) and after 12 months of intervention 241 x 103/µL (range: 140 to 384). In participants continued solely on imiglucerase , baseline Hb was 12.3 g/dL (range: 10.7 to 14.0) and 12.7g/dL (range: 11.1 to 13.8) after 12 months. Baseline PLT was 244 x 103/µL (range: 160 to 343) and 242 x 103/µL (range: 134 to 348) at the end point (n = 9). No P values were reported in the original manuscript for these comparisons (Schiffmann 2008).
3. Hepatosplenomegaly
Schiffmann evaluated the change in liver and spleen volumes after 12 months (no sufficient data available to be included in the analysis) (Schiffmann 2008). Elstein examined the change in liver (30 participants) and spleen volumes (22 participants) at six months (Elstein 2007). In the Elstein study, there was no significant difference on liver and spleen volume status between participants continued on imiglucerase and those in whom miglustat was substituted (Analysis 2.1; Analysis 2.2). Participants treated with a combination of imiglucerase and miglustat had a borderline significant decrease in liver volume compared to those maintained on imiglucerase alone, MD ‐8.75 (95% CI ‐15.63 to ‐1.87) (P = 0.047) (Analysis 2.1; Elstein 2007). A similar difference was not observed for spleen volume suggesting that, given the relative imprecision of quantitative spleen‐liver volumetric measurements, this might be a random observation (Analysis 2.2).
2.1. Analysis.
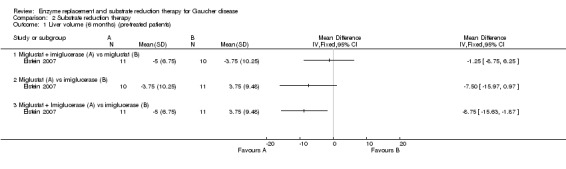
Comparison 2 Substrate reduction therapy, Outcome 1 Liver volume (6 months) (pre‐treated patients).
2.2. Analysis.
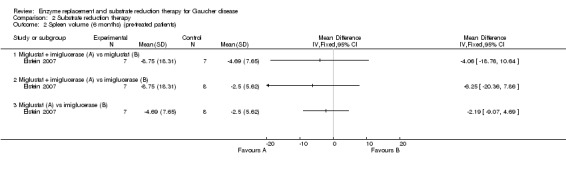
Comparison 2 Substrate reduction therapy, Outcome 2 Spleen volume (6 months) (pre‐treated patients).
In the Schiffmann study (GD3 participants), liver and spleen volumes remained essentially stable in both treatment groups at all time points (Schiffmann 2008). Participants treated with continued imiglucerase for 12 months experienced a decrease in liver volume of 0.5 L/cm3, and no change in spleen volume, while the combination of imiglucerase and miglustat resulted in decreases in liver and spleen volumes of 0.3 L/cm3 and 0.2 L/cm3, respectively.
Secondary outcomes
1. Biomarkers (chitotriosidase, CCL 18‐PARC and ACE)
The available data on biomarkers did not allow calculation of mean effect change and corresponding SDs. Results are hence reported as in the original papers. In the Schiffmann study, a decrease in chitotriosidase activity from baseline was observed in the miglustat group already at six months, while in the no miglustat group it remained unchanged up to 12 months (Schiffmann 2008). Elstein reported that all participants in each of the three treatment arms remained with stable chitotriosidase levels, although two participants experienced an increase in chitotriosidase with concurrent bone pain, which were resolved once treatment had been halted, and in one of the cases treatment with miglustat has been reinitiated afterwards (Elstein 2007).
2. Skeletal outcomes
Elstein stated that two participants who suffered from bone pain events, which disappeared once miglustat treatment was withdrawn (Elstein 2007). In one of the cases, treatment was later reinitiated. Schiffmann stated that although no cases of bone crisis events were reported during the study, one participant in the miglustat group experienced a tibial fracture after 15 months, and another participant who had history of fractures prior to enrolment in the ERT‐alone group, had two events of severe femoral fractures at 9 and 12 months (Schiffmann 2008).
Discussion
This systematic review summarizes, for the first time, all currently available randomized interventional studies assessing the effects of enzyme replacement and substrate reducing therapies in treating people with Gaucher disease (GD). The analysis allows for direct comparisons of the emerging new drugs with previously established treatment options for GD and focuses attention on some of the requirements for future research in this ultra‐orphan disorder. We also contend that the review provides an example for the strengths and limitations of the increasingly accepted methodology of evidence‐based medicine, specifically as reflected through the unusually distorted prism of rare diseases.
Several principal limitations in the conduct of this review warrant consideration. As in the case of other very rare diseases, the number of studies meeting the inclusion criteria following Cochrane methodology, and the low number of participants in each of these studies, significantly limits our ability to draw rigid conclusions. An extensive search and consultation between the authors was undertaken with every effort made to ensure that all relevant data were indeed included, and to avoid oversights or over‐interpretation of any available data, and their credibility.
Until the early 1990s, when enzyme replacement therapy (ERT) was introduced as the first specific therapy, only symptomatic drugs were available for the treatment of GD. With the support of large‐scale observational studies (Andersson 2008; Weinreb 2002) and the efforts of the International Collaborative Gaucher Group (ICGG), which established a GD registry in 1991, it was clear that enzyme infusions markedly improve the visceral and haematological manifestations of GD, with salutary changes often seen after only a few infusions. This improvement is also reflected in patients' reported quality of life, when compared with patients suffering from other chronic metabolic diseases (Damiano 1998). The clear impact reported in the first patients receiving ERT was so remarkable (Barton 1991), that no randomized controlled studies investigating the absolute effects compared with placebo were carried out. However, with such an individual history of discovery, the ability to quantify the absolute effect of ERT and to describe the actual difference between treated and untreated individuals in a solid experimental fashion is diminished. Moreover, with the emergence of new enzyme preparations, experimental studies investigating their efficacy could only compare the new drugs with the acceptable gold standard (i.e. imiglucerase), since a randomized placebo‐controlled study would no longer have been an ethical proposition. Cognizant of these issues, it is clear that this systematic review cannot focus on the relative efficacy of given treatments, but rather, on the comparison of different treatment options, attempting to address pressing questions (such as what is the optimal dosing). It is also prudent to review all the data in a systematic manner, after which, there may be a case for investigating established evidence‐based statistical methodologies.
For GD, which current unmet needs might constitute a worthy impetus for the development of new treatments? Unlike infectious and neoplastic diseases, refractoriness or resistance to imiglucerase is largely inconsequential, since neutralizing antibodies or intractable hypersensitivity reactions rarely occur (Starzyk 2007). The intravenous route of administration is problematical for some individuals with poor venous access including small children in whom prolonged use of infusaports or indwelling catheters might be disadvantageous. Nevertheless, market research indicates that globally, 80% to 90% of prescribed ERT infusions are actually received (Genzyme/Sanofi 2013 [pers comm]). The increasing global use of home infusion undoubtedly has contributed to increased compliance as well as accessibility (Hughes 2007). The astronomical expense of ERT continues to be an obstacle to access even in affluent countries and much more so in under‐developed countries where large numbers of severely affected people with GD remain untreated, despite industry programs such as Project Hope and efforts of advocacy organizations such as the European Gaucher Alliance and the US National Gaucher Foundation. Although newer competitive treatments such as miglustat, velaglucerase alfa and taliglucerase alfa have been introduced at somewhat less expensive prices than imiglucerase, treatment is still unaffordable for many individuals and societies. People with GD1 have an increased risk of developing concurrent malignancies, especially multiple myeloma, other hematological malignancies, and liver and kidney carcinomas (Rosenbloom 2013). The effect of imiglucerase treatment on this risk is currently unknown. Although substantially ameliorated by imiglucerase treatment, people with GD1, especially those with a history of splenectomy, continue to develop new episodes of skeletal avascular necrosis (Mistry 2009) and GD‐associated osteopenia or osteoporosis with increased fracture risk may respond incompletely to imiglucerase ERT, especially when treatment initiation is delayed until adulthood (Mistry 2011; Khan 2012). Since imiglucerase is a large molecule, it is excluded from the central nervous system by the blood brain barrier and ineffective for treating classical neuronopathic GD (GD2, GD3 and intermediate phenotypes). Moreover, there is, as yet, no evidence that imiglucerase prevents the development of parkinsonism and peripheral neuropathy which are now recognized as important and sometimes life‐limiting outcomes in some middle aged and elderly people with GD1 (Biegstraaten 2010; Bultron 2010; Chetrit 2013; Rosenbloom 2011). On an even more basic level, after 20 years, we cannot yet define the long‐term effects of imiglucerase therapy on health‐related quality of life or on life expectancy.
The short‐term randomized clinical studies of recently developed new treatments for GD that we have reviewed here, although vital for regulatory approval and commercialisation of these products and thus potentially relevant to some of the socio‐economic issues raised above, do not and will not address the biological and medical questions we have raised. With that in mind, what valid conclusions may be drawn from this review that is limited to data from randomized controlled studies? Information about the comparative efficacies of imiglucerase, velaglucerase alfa and taliglucerase alfa in treatment‐naïve participants is restricted to haemoglobin (Hb) concentration, platelet counts (PLT), spleen volume, liver volume and some serum biomarker responses during the initial 9 to 12 months of therapy. In that limited context, velaglucerase and taliglucerase do not appear to be inferior to imiglucerase. There is no evidence for superiority and the studies were insufficiently designed or powered to show such an effect. As regards dose effects, in the time frame of 9 to 12 months, there appeared to be little difference in Hb, PLT, spleen, liver and biomarker responses when individuals were treated with velaglucerase 45 or 60 units/kg every two weeks or taliglucerase 30 or 60 units/kg/every two weeks. However, these studies do not address the issue of even lower doses which are commonly used with imiglucerase in a number of European countries and Israel. Furthermore, in earlier dose studies with imiglucerase (de Fost 2006; Grabowski 2009), dose‐related differences in chitotriosidase response were not evident until at least 24 months of treatment and dose‐related differences in liver and spleen size were also not clearly evident at 12 months. The observed dose differences on Hb and PLT responses reported in the Grabowski study at 12 months, may be attributable to different study design (retrospective, propensity score matching, non‐linear mixed effects modelling) and much larger participant numbers (122 participants each for doses of 15, 30 and 60 units/kg/every two weeks) (Grabowski 2009). In any event, the effect of ERT dose on the long‐term clinical outcomes cited above continues to be undefined.
As regards those who appear to have an initial response and are clinically stable after at least two years of imiglucerase treatment, the randomized controlled studies (including the Kishnani study whose results were presented as composite rather than as discrete end points (Kishnani 2009)) indicate that infusion frequency can usually be reduced from every two weeks to every four weeks without adverse hematological or organ volume consequences provided that cumulative dose is maintained constant. Additionally, equal dose switches from imiglucerase to velaglucerase (Zimran 2013b; non‐randomized so not analysed above) were tolerated for 12 months without haematological or visceral changes from baseline and without unusual adverse effects. Substitution of miglustat for imiglucerase also resulted in haematological and visceral stability for 6 to 12 months but was associated with significant gastrointestinal discomfort (Elstein 2007; Schiffmann 2008). As regards all of these short‐term 'maintenance studies', it should be noted that following a 2009 to 2011 disruption in imiglucerase supply caused by a vesivirus contamination of bioreactors at the Genzyme manufacturing facility, many patients globally had their imiglucerase treatment interrupted or reduced in dose for 6 to 18 months on average. Although some individuals noted worsening fatigue and bone pain, and serum chitotriosidase activity occasionally increased in most affected by the shortage, haematological and visceral measurements generally remained unchanged (Deroma 2013; Giraldo 2011; Goldblatt 2011; Zimran 2011a). Thus, in people with GD1, 'stability' over a short observation period following a change in treatment may not necessarily be attributable to the new agent.
Limitations of our review
Although we performed an extensive literature search and consulted with study authors to ensure that all the pertinent data were included and accurately interpreted, this review is nonetheless significantly constrained. The randomized controlled studies that were eligible for inclusion in this analysis represent a tiny proportion of the total body of literature reporting therapeutic outcomes in people with GD1. Additionally, the small number of participants included in the studies we analysed, who were selected on the basis of specific eligibility criteria, may not necessarily be representative of a genotypically and phenotypically diverse GD global population. In two studies we reviewed, previously treated participants were labelled "treatment naïve" after an arbitrarily chosen 'washout' period. In fact, we do not know whether these individuals respond to new treatment in identical fashion as truly‐naïve patients. The end points selected are traditional, but insufficient, surrogates for the full spectrum of GD pathophysiology. Skeletal manifestations were not addressed and direct or even indirect (via imaging) information on tissue substrate storage and histopathology is not available. Furthermore, in the context of a chronic disease whose typical course extends for decades, trial periods of one to two years necessarily lack perspective. For these reasons, we should be very cautious in reaching conclusions about absolute or comparative effectiveness of the treatments under consideration. These limitations undoubtedly extend to other rare or ultra‐rare diseases with extended prognoses and heterogeneous phenotypes and penetrance.
Different drugs ‐ different impact on outcomes?
Taking into account the results of this systematic review, it appears that the different ERTs currently available do not, under the conditions studied, show clear differences in efficacy. Mechanistically, all ERTs help to lessen disease manifestations by replacing the deficient or defective (or both) lysosomal enzyme. Of note, however, is that the modest number of participants included in these comparisons may mean that the studies are insufficiently powered to reveal superiority of one agent over the others.
The number of studies included in this systematic review and assessing SRT in comparison with ERT is likely to be insufficient to enable the drawing of conclusions regarding the superiority or inferiority of one over the other. Eliglustat, a promising new player in the field of SRTs, currently in phase III clinical trials, is an inhibitor of N‐acylsphingosine transferase. This enzyme catalyzes the first committed step in the biosynthesis of glycosphingolipids. Therefore, it would be of particular interest to include the current phase III international trials of eliglustat in an update of this Cochrane Review. This approach may yield general data of more universal applicability and use, since the inhibition of glycosphingolipid biosynthesis has the potential to address other several glycosphingolipid disorders.
The studies reviewed here did not address the issue of switching people from one enzyme replacement therapy to another. However, other literature suggests that this can be done safely and without loss of disease control (Hollak 2012; Zimran 2013b). There is evidence from two randomised controlled studies that some people with type 1 Gaucher disease, in whom disease manifestations are controlled, may be infused with imiglucerase at four‐week rather than two‐week intervals, provided that the same cumulative imiglucerase dose is unchanged (de Fost 2007; Kishnani 2009). Similar evidence for similar studies of velaglucerase alfa and taliglucerase is not available. Given the optimal dose for ERT generally has to be determined on an individual patient basis, it is sometimes appropriate to reduce an empiric initial dose when good disease control is achieved and sustained. Clinicians occasionally elect to accomplish this by reducing the frequency of infusions without a concomitant adjustment of enzyme replacement therapy dose. At this time, there is no evidence from randomised controlled studies for this therapeutic strategy.
Dose of enzyme replacement therapy
The doses used in the eight studies included in the qualitative analysis varied from 15 units/kg up to 60 units/kg every two weeks, with only two of the studies comparing different doses head‐to‐head (Gonzalez 2013; Zimran 2011b). Deduced from the integrated comparisons provided herein, a beneficial effect for higher doses was observed on both CCL18 and chitotriosidase after 12 months of treatment. Yet, no statistically significant differences were noted between lower and higher doses in all other systemic outcomes. The same pattern of lack of superiority of higher doses remained when considering the excluded non‐randomized study data that assessed different doses (15 units/kg compared with 30 units/kg compared with 45 units/kg and 60 units/kg) of velaglucerase alfa (Zimran 2013b) (data not shown). However, several other studies have addressed long‐lasting questions about different effects of dosing, including a retrospective analysis of two cohorts from the Netherlands and Germany (de Fost 2006) and a study from the ICGG (Grabowski 2009). Both studies showed more robust effects of higher doses on biomarkers (de Fost 2006) and haematological and visceral parameters (Grabowski 2009). The clinical relevance of these effects are, however, not always clear. In addition, several cohorts followed the consequences of the restriction of imiglucerase distribution, which emerged after a viral contamination in the manufacturing site. In a Spanish cohort described by Giraldo (Giraldo 2011), a reduction of 50% in dosage to a mean dose of 34 units/kg for a six months period, did not result in a statistically significant decrease in haemoglobin, platelet count, or CCL18‐PARC activity ‐ but a significant increase in chitotriosidase activity was noted. Seven bone pain events in the group of 17 participants who underwent a dose reduction were also described in this cohort. Similar results were described in a 12‐month Italian cohort (Deroma 2013), in which the dose of ERT decreased from a median of 55.5 units/kg to 15.8 units/kg, without bone pain events.
Role of evidence‐based analyses in rare diseases
Deriving evidence of value, based on medicine in the field of rare diseases, is beset with numerous obstacles ‐ the low numbers of patients and their different geographical locations set a particular challenge for the conduct of interventional trials. This is particularly true when moving from a ground‐breaking discovery which has revolutionized therapy for such a disease (as in this case with enzyme replacement for GD) to more delicate questions aimed at optimising therapeutic interventions. Inherently, differences between more subtle protocols or specific preparations are likely to be far less robust than when a new therapeutic agent that directly targets the mechanism of disease is first introduced. These considerations inevitably mandate the need for larger numbers, in order to achieve a sufficient statistical power to reveal differences or associations with confidence.
Moving from single studies to a meta‐analysis, the high heterogeneity of such studies, often attributable to their complexity, may not allow pooled estimations, thus preventing us from fully appreciating the effects of interventions. For example, a clear distinction should be made between studies on naïve and those on pre‐treated individuals. In fact, any treatment modifies the natural course of the disease, thus different results would be predicted in untreated individuals when compared with those who receive long‐standing therapy (even if a washout period is considered). Moreover, it should be kept in mind that stable patients are less prone to experience major clinical deteriorations. Hence, when participants are stabilized before entering a study investigating a treatment switch, an underestimation of the effect of the new drug could take place.
Authors' conclusions
Implications for practice.
The limitations of the evidence summarized by this review have been discussed and should also be considered in making decisions in clinical practice.
With the recent regulatory approval of taliglucerase alfa in the USA and Israel, and with approval likely to be granted in other jurisdictions in the near future, practitioners caring for people with Gaucher disease who need treatment must choose between four agents: three recombinant glucocerebrosidase glycoproteins with minor differences in primary amino acid sequence and greater differences in carbohydrate structure (imiglucerase, velaglucerase alfa, taliglucerase alfa); and the small molecule, the oral iminosugar, miglustat. The evidence from the small number of included studies provides the clinician with little to no decisive help.
There is no randomized controlled study supporting the use of miglustat as a primary intervention for treatment‐naïve patients and this agent is not authorized as a first‐line agent by either the Food and Drug Administration (FDA) or European Medicines Agency (EMA). Two studies (with specific reference only to non‐neurological Gaucher disease manifestations) show that miglustat might be considered as a maintenance‐phase substitute for enzyme replacement therapy in individuals with good disease control, but the relatively short duration of the studies suggests that if individuals are switched from enzyme replacement therapy to miglustat (because of venous access problems or for cost containment), they should be carefully monitored over several years for evidence of disease progression (Elstein 2007; Schiffmann 2008). The studies additionally confirmed the significant burden of side effects (gastrointestinal and neurological) identified in the initial non‐randomized studies and repeatedly observed in routine clinical use. The current indication for miglustat use in those unable to tolerate, or unwilling to accept, intravenous ERT is based on a lack of evidence for any therapeutic superiority for miglustat and a substantial side effect burden.
To choose among the three approved enzyme replacement therapies, the evidence from randomized controlled studies suggests that, in treatment‐naïve patients, or after treatment interruption longer than one year, these agents are equipotent during the initial year of treatment for increasing haemoglobin concentration and platelet counts in cytopenic patients, for decreasing splenomegaly and hepatomegaly, and for achieving decreases in serum biomarkers such as chitotriosidase or CCL18‐PARC. Over the same period, there appears to be no obvious difference in the safety profile, particularly as regards infusion‐related hypersensitivity reactions. We cannot extend the impression of safety equivalence beyond the duration of the studies as the products differ in the likelihood of the development of enzyme replacement therapy antibodies (Zimran 2013b). This may have safety and efficacy implications, once a longer clinical experience is acquired. We conclude that on the basis of the randomized controlled studies we have reviewed, there is no evidence supporting the choice of one therapy over another for the treatment of naïve patients, leaving clinicians free to make recommendations to patients on the basis of non‐medical considerations such as cost.
Implications for research.
It is virtually certain that, despite the rarity of the disease, additional candidate therapies for type 1 Gaucher disease (as well as for the neuronopathic variants) will continue to emerge. Eliglustat, an oral substrate synthesis inhibition therapy (SSIT), for which two‐year results of an extended phase II study (26 participants) have been published (Lukina 2010) and five‐year results presented at the WORLD Lysosomal Disease Network Symposium (February 2013) appears to replicate the haematological, visceral, and biomarker responses described above for enzyme replacement therapies. Eliglustat is the focus of two phase III randomized controlled studies, one of which has very recently been published in full (and will be assessed for eligibility in a future update of this review) and one which will be published in full in the very near future. In one of these studies, eliglustat is compared to a placebo arm in treatment‐naïve patients ('ENGAGE'); and in the other to an imiglucerase control arm in a switch study ('ENCORE') (Encore 2013; Engage 2015). There is intense interest in pharmacological chaperone therapy (PCT) with one pilot study with ambroxol recently completed in Israel (Zimran 2013a). This therapy prevents the degradation of the defective enzyme in the endoplasmic reticulum and allows proper folding and trafficking, by acting as reversible competitive inhibitors to glucocerebrosidase (Oulaidi 2011). Pre‐clinical interest in small molecule proteostasis modulators, nonsense mutation suppression and gene modification therapy may also become clinically relevant in the future (Dietz 2010).
Is the current paradigm for evidence‐based medicine in which observational cohort studies and patient registry results are devalued relative to randomised controlled studies necessarily best adapted for comprehensively evaluating new treatments for rare, chronic conditions such as type 1 Gaucher disease, where the clinical course is protracted and disease complications are sporadic and unpredictable? When applied to chronic metabolic disorders, randomised controlled studies are rarely sustainable for more than a few years because of expense and the need for a rapid determination as to whether the candidate treatment will be a viable commercial product. A four‐arm randomised controlled study with 5000 participants to determine the "long‐term" effects of diabetes medications in people older than 30 years of age, projects an average follow up of 4.8 years (Nathan 2013); and the six‐year ORIGIN randomised controlled study (12,500 participants with pre‐ or early diabetes) is reputed to be the world’s longest for this disorder (Gerstein 2012). When sentinel clinical events may not occur for many years after starting treatment, as is often the case in people with type 1 Gaucher disease, randomised controlled studies are usually dependent on surrogate end points such as laboratory tests or organ volume measurements whose relevance to clinically important end points may be indirect and even tenuous. Thus, as shown in this report, randomised controlled studies can establish efficacy and non‐inferiority to a sufficient level to satisfy the requirements of evidence‐based medicine, but are less likely to be able to establish superiority and effectiveness (defined as the ability of an intervention to produce a clinical benefit in actual practice) necessary to comply with the demands of value‐based medicine (Black 1996; Resnick 2013). In the case of Gaucher disease, it is unlikely that any randomised controlled study will successfully define the effect of current or future treatments on important outcomes such as Parkinsonism, malignancy, quality of life and mortality. This need can be better met by strengthening existing rare disease patient registries: to include mandatory participation in order to minimize selection bias; site visits for data verification; innovative statistical analysis such as propensity scoring and case‐control matching (Cole 2011); data submitted by patients; and, regardless of sponsorship, open data access (Hollak 2011; Krumholz 2013)
What's new
| Date | Event | Description |
|---|---|---|
| 13 April 2015 | Amended | Contact details updated. |
Notes
During the final production of this Cochrane Review, two studies were completed ‐ the 'Engage' study ‐ available online, and the 'Encore' study ‐ expected to be published during 2015. These will be included in a future update of this review (Encore 2013; Engage 2015).
Acknowledgements
We thank Tracey Remmington and Kerry Dwan (Cochrane Cystic Fibrosis and Genetic Disorders Group) for helpful comments and insight. We also thank Genzyme corporation (a Sanofi company) for providing relevant data. This study was supported in part by grants from the UK Gaucher Association and by the European Working Group for Gaucher Disease (EWGGD).
Appendices
Appendix 1. Search strategy for PubMed
( ((((((((((((((randomized controlled trial[Publication Type])) OR (controlled clinical trial[Publication Type])) OR (randomized[Title/Abstract])) OR (placebo[Title/Abstract])) OR (drug therapy[MeSH Subheading])) OR (randomly[Title/Abstract])) OR (trial[Title/Abstract])) OR (groups[Title/Abstract]))) NOT (animals[MeSH Terms] NOT humans[MeSH Terms]))) AND (((gaucher[Text Word])) OR (gaucher disease[MeSH Terms]))) AND (((enzyme replac*[Text Word] OR enzyme replacement therapy[MeSH])) OR (substrate[Text Word] AND (reduc*[Text Word] OR substrate depriv*[Text Word]) OR miglustat[Text Word] OR zavesca[Text Word] OR eliglustat[Text Word] OR imiglucerase[Text Word] OR cerezyme[Text Word] OR velaglucerase[Text Word] OR VPRIV[Text Word] OR taliglucerase alfa[Text Word])))))
Data and analyses
Comparison 1. Enzyme replacement therapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Haemoglobin levels (9 months) | 4 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Imiglucerase 60 U/kg every 2 weeks (A) vs alglucerase 60 U/kg 2 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Taliglucerase 30 U/kg (A) vs 60 U/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Velaglucerase 60 U/kg every 2 weeks (A) vs imiglucerase 60 U/kg every 2 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Velaglucerase 60 U/kg every 2 weeks (A) vs imiglucerase 60 U/kg every 2 weeks (B): participants with spleen intact only | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Imiglucerase every 2 weeks (A) vs every 4 weeks (B) (pre‐treated participants) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Haemoglobin levels (12 months) | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Velaglucerase 45 U/kg (A) vs 60 U/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Velaglucerase 45 U/kg (A) vs 60 U/kg (B): verified Gaucher patients | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Imiglucerase every 2 weeks (A) vs every 4 weeks (B) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Haemoglobin levels (24 months) (end point values) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Imiglucerase every 2 weeks (A) Vs. every 4 weeks (B) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Platelets (9 months) | 4 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Imiglucerase 60 U/kg every 2 weeks (A) vs alglucerase 60 U/kg every 2 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Taliglucerase 30 U/kg (A) vs 60 U/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Velaglucerase 60 U/kg every 2 weeks (A) vs imiglucerase 60 U/kg every 2 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 Velaglucerase 60 U/kg every 2 weeks (A) vs imiglucerase 60 U/kg every 2 weeks (B): patients with spleen intact only | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 Imiglucerase every 2 weeks (A) vs every 4 weeks (B) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Platelets (12 months) | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 Velaglucerase 45 U/kg (A) vs 60 U/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Velaglucerase 45 U/kg (A) vs 60 U/kg (B): verified Gaucher patients | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Imiglucerase every 2 weeks (A) vs every 4 weeks (B) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Platelets ‐ 24 months (end point values) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6.1 Imiglucerase every 2 weeks (A) Vs. every 4 weeks (B) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Liver volume (6‐9 months) | 4 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 7.1 Imiglucerase 60 U/kg every 2 weeks (A) vs alglucerase 60 U/kg every 2 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Taliglucerase 30 U/kg (A) vs 60 U/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Velaglucerase 60 U/kg every 2 weeks (A) vs imiglucerase 60 U/kg every 2 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Imiglucerase every 2 weeks (A) vs every 4 weeks (B) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Liver volume (12 months) | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 8.1 Velaglucerase 45U/kg (A) vs 60 U/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 Velaglucerase 45 U/kg (A) vs 60 U/kg (B): Sub‐analysis: verified Gaucher patients | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 Imiglucerase every 2 weeks (A) vs every 4 weeks (B) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Liver volume (24 months) (multiples of normal, end point values) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 9.1 Imiglucerase every 2 weeks (A) vs every 4 weeks (B) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Spleen volume (6‐9 months) | 4 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 10.1 Imiglucerase 60 U/kg every 2 weeks (A) vs alglucerase 60 U/kg every 4 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 Taliglucerase 30 U/kg (A) vs 60 U/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 Velaglucerase 60 U/kg every 2 weeks (A) vs imiglucerase 60 U/kg every 2 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.4 Imiglucerase every 2 weeks (A) vs every 4 weeks (B (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Spleen volume (12 months) | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 11.1 Velaglucerase 45u/kg (A) vs 60 U/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 Velaglucerase 45 U/kg (A) vs 60 U/kg (B) sub‐analysis: verified Gaucher patients | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 Imiglucerase every 2 weeks (A) vs every 4 weeks (B (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Spleen volume (24 months) (multiples of normal, end point values) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 12.1 Imiglucerase every 2 weeks (A) Vs. every 4 weeks (B) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Chitotriosidase (6‐9 months) (% change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 13.1 Velaglucerase 60 U/kg every 2 weeks (A) vs imiglucerase 60 U/kg every 2 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 Taliglucerase 30 U/kg (A) vs 60 U/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.3 Imiglucerase every 2 weeks (A) vs every 4 weeks (B) pre‐treated patients expressing chitotriosidase | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Chitotriosidase (12 months) (% change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 14.1 Velaglucerase 45 U/kg (A) vs 60 U/kg (B): verified Gaucher patients | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 Velaglucerase 45 U/kg (A) vs 60 U/kg (B): verified Gaucher patients, expressing chitotriosidase | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.3 Imiglucerase every 2 weeks (A) vs every 4 weeks (B): pre‐treated patients expressing chitotriosidase | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 CCL18‐PARC (9 months) (% change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 15.1 Velaglucerase 60 U/kg every 2 weeks (A) vs imiglucerase 60 U/kg every 2 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 Taliglucerase 30u/kg (A) Vs. Taliglucerase 60u/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 CCL18‐PARC (12 months) (% change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 16.1 Velaglucerase 45 U/kg (A) vs 60 U/kg (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.2 Velaglucerase 45 u/kg (A) vs 60 U/kg (B): verified Gaucher patients | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17 ACE (9 months) (% change from baseline) | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 17.1 Imiglucerase 60 U/kg every 2 weeks (A) vs alglucerase 60 U/kg 2 weeks (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.2 Imiglucerase every 2 weeks (A) vs every 4 weeks (B) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18 Quantiative Chemical Shift Imaging (6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 18.1 Imiglucerase every 2 weeks (A) vs every 4 weeks (B (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 19 Quantiative Chemical Shift Imaging (12 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 19.1 Imiglucerase every 2 weeks (A) vs every 4 weeks (B (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2. Substrate reduction therapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Liver volume (6 months) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Miglustat + imiglucerase (A) vs miglustat (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Miglustat (A) vs imiglucerase (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Miglustat + Imiglucerase (A) vs imiglucerase (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Spleen volume (6 months) (pre‐treated patients) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Miglustat + imiglucerase (A) vs miglustat (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Miglustat + imiglucerase (A) vs imiglucerase (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Miglustat (A) vs imiglucerase (B) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ben Turkia 2013.
| Methods | Randomized, double‐blind, multicenter, parallel, non‐inferiority trial | |
| Participants | 34 treatment‐naïve type I Gaucher participants (including children) | |
| Interventions | Velaglucerase (N = 17) vs imiglucerase (N = 17) for 9 months | |
| Outcomes | Hb, Plt, liver and spleen volume, chitotriosidase, CCL18 | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomization method is not specified |
| Allocation concealment (selection bias) | Unclear risk | No description of the allocation process |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Described as double‐blind study |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Except where stated, all analyses were performed on an intent‐to treat basis, defined as all randomized patients who received at least one full or partial infusion" |
| Selective reporting (reporting bias) | Low risk | All data were reported (either in original paper or in supplementary tables) |
de Fost 2007.
| Methods | Randomized controlled study | |
| Participants | 11 pre‐treated, stable, Gaucher participants | |
| Interventions | Imiglucerase every 2 weeks (N = 5) vs every 4 weeks (N = 6). Participants remained on study for 12 months or until study withdrawal | |
| Outcomes | Hb, Plt, ACE, chitotriosidase, ferritin, liver enzymes, lumbar bone marrow fat content, liver ratio, spleen volume | |
| Notes | Further data acquired from original study authors | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No clear reference to the randomization method (block randomization was used, but no clear definition was given) |
| Allocation concealment (selection bias) | Low risk | "Block randomization was used to assign patients to receive infusions either once every 4 weeks (low frequency group) or to continue their original schedule (once every 1 or 2 weeks, control group)" |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No description of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data for all participants were reported |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported |
| Other bias | Low risk | |
Elstein 2007.
| Methods | Randomized, phase II, parallel group, open‐label study | |
| Participants | 36 adult, pre‐treated, stable participants with type I Gaucher disease | |
| Interventions | Miglustat (SRT, N = 12) vs imiglucerase (ERT, N = 12) vs the combination of both (N = 12). Total study period was 6 months, after which participants could enter an extension protocol | |
| Outcomes | Spleen and liver volume, quality of life, chitotriosidase, imiglucerase assay, adverse events, "routine laboratory tests" | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A minimization algorithm with a random component assigned 36 patients to treatment groups according to clinical severity, age, sex, and years on enzyme replacement therapy" |
| Allocation concealment (selection bias) | High risk | According to the above citation, participants were assigned based on clinical severity, among other parameters. Participants with severe disease received the addition of SRT, and no method was used to "protect" the allocation‐ participants knew their state of disease, and therefore knew whether they are enrolled in the intervention arm. |
| Blinding (performance bias and detection bias) All outcomes | High risk | This was an open‐label study. The interventions assessed in the paper (the addition of oral drug to existing IV therapy vs oral drug alone and the combination of an oral drug with IV therapy compared to IV therapy alone, as well as comparison of the oral drug to IV therapy), without a mention of placebo make it likely that participants, and possibly clinicians, were not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | While it was reported that 3 participants discontinued the study due to adverse events (2 in the miglustat group, 1 in the miglustat plus imiglucerase group), and 7 participants were splenectomized (with no clear description of the timing of this event), data regarding spleen volume were not reported for 3 participants in the miglustat group, 4 participants in the imiglucerase group, and for 4 in the combination arm. Some data were also missing in regards to liver volume (1 participant from the imiglucersae group, and 2 in the combination group). |
| Selective reporting (reporting bias) | Low risk | Data for most outcomes were reported, except for pharmacokinetic profile ‐ "The combination of miglustat and enzyme replacement did not affect the pharmacokinetic profile of enzyme (data not shown)." |
| Other bias | Low risk | 10 participants were enrolled in another clinical trial previously conducted by the same group |
Gonzalez 2013.
| Methods | Randomized, double‐blind, multicenter phase III parallel group study | |
| Participants | 25 treatment‐naïve anaemic type I Gaucher participants | |
| Interventions | Velaglucerase alfa 45 units/kg (N = 13) vs 60 units/kg (N = 12), study period 12 months | |
| Outcomes | Hb, Plt, liver and spleen volume, chitotriosidase, CCL18, bone mineral density (not defined as an endpoint) | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "..performed centrally using a covariate adaptive randomization algorithm.." |
| Allocation concealment (selection bias) | Low risk | "After confirmation of eligibility, participants were randomized at a ratio of 1:1 to receive velaglucerase alfa at either 45 units/kg or 60 units/kg every other week. Randomization was stratified by sex (male/female) and age (2–17 years; 18 years)" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "This was a multinational, phase 3, randomized, double‐blind..." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "All analyses were performed on an intent‐to‐treat basis, defined as all randomized patients who received at least one full or partial infusion. No patient was missing values for the primary endpoint. Where values were missing for the secondary efficacy endpoints, a pre‐specified imputation strategy was applied. After applying last observation carried forward for post‐baseline measurements, if data were still missing (including at baseline), the median value in the corresponding age group (2–17 years; 18 years) was used." |
| Selective reporting (reporting bias) | Low risk | Data are reported for all outcomes, except for bone mineral density (not an endpoint) ‐ "Data on other parameters, including bone mineral density, were collected at baseline and subsequent scheduled intervals to achieve additional, exploratory objectives, but they were not in the scope of this report." |
| Other bias | High risk | Original study included several participants who did not have Gaucher disease |
Grabowski 1995.
| Methods | Randomized, double‐blind, parallel study | |
| Participants | 30 treatment‐naïve type I Gaucher participants (including 4 children) | |
| Interventions | Imiglucerase 60 units/kg (N = 15) vs alglucerase 60 units/kg (N = 15) for 9 months | |
| Outcomes | Hb, Plt, serum acid phosphatase, ACE, liver and spleen volume, IgG antibodies | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "In each study center, patients were independently randomly assigned in blocks by age and were assigned study numbers" |
| Allocation concealment (selection bias) | Low risk | "In each study center, patients were independently randomly assigned in blocks by age and were assigned study numbers. All study personnel except the pharmacist at each institution were blinded to the allocated treatment." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "The study was a double‐blind, parallel trial with random assignment to Ceredase or Cerezyme" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study, without loss to follow up being reported |
| Selective reporting (reporting bias) | Low risk | Data for all outcomes were reported (either in original paper or in supplementary tables) |
Kishnani 2009.
| Methods | Randomized, multicenter, open‐label trial, parallel study | |
| Participants | 102 pre‐treated, stabilized, type I Gaucher participants | |
| Interventions | Imiglucerase every 2 (N = 37) weeks vs every 4 weeks (N = 65) for 24 months | |
| Outcomes | Hb, Plt, liver and spleen volume, ACE, TRAP, chitotriosidase, adverse events, bone disease progression, SF‐36 health survey | |
| Notes | Further data was acquired | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No clear description of the sequence generation process ‐ "Eligible patients were randomized 2:1 to receive imiglucerase either once every 4 weeks (Q4) or once every 2 weeks (Q2) for 24 months" |
| Allocation concealment (selection bias) | Unclear risk | No clear description of whether allocation concealment was applied |
| Blinding (performance bias and detection bias) All outcomes | High risk | "...this study was not blinded to treatment group..." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | While an intention‐to‐treat analysis was carried by the study authors, it does not answer all criteria to be regarded as such‐ "All patients who enrolled in the study and received at least one infusion were included in the primary analysis population, the Intent‐to‐Treat (ITT). The safety population was equivalent to ITT." |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported, but some were reported as figures only, with no numerical data. |
| Other bias | Low risk | |
Schiffmann 2008.
| Methods | Randomized, multicenter, open label phase II, controlled study | |
| Participants | 30 type III pre‐treated Gaucher participants | |
| Interventions | Addition of miglustat (SRT) to ERT (N = 21) vs ERT alone (N = 9) for 24 months | |
| Outcomes | Change in saccadic eye movements (horizontal and vertical) from baseline, neurological assessments (Purdue Peg Board test, Wechsler scale, Benton visual retention test, Rey auditory verbal learning test, d2 test of attention, continuous performance test, trail marking test, mental state, cranial nerves, motor skills), brain auditory‐evoked potentials), pulmonary function tests, liver and spleen volume, Hb, Plt, chitotriosidase, hexosaminidase, glucosylceramide, adverse events, tremor measurements | |
| Notes | The study had an extension phase after 12 months, in which all participants received miglustat for additional 12 months | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No clear description of the sequence generation process‐ "Patients were randomized (2:1) to receive miglustat or “no miglustat treatment” for the first 12 months" |
| Allocation concealment (selection bias) | Unclear risk | No clear description of whether allocation concealment was applied |
| Blinding (performance bias and detection bias) All outcomes | High risk | Although not discussed in the original paper, it is plausible to assume that participants were not blinded (since one treatment arm received the addition of miglustat ‐ an oral drug, while the other arm did not receive it. it is not reported whether they received an oral placebo instead) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "All efficacy evaluations for the 24‐month period were performed on all randomized patients who entered the 12‐month extension period and had at least one post baseline efficacy assessment during the 12‐month extension period." |
| Selective reporting (reporting bias) | Low risk | Data for most of the outcomes are reported in numerical manner ‐ either in the original paper or in supplementary tables. Hexoaminidase, glucosylceramide, and several eye‐movement parameters were not reported numerically (i.e. "No statistically significant between‐group differences were seen on the other secondary neurological evaluations at the end of randomized therapy or at the end of the extension phase (data not shown)." |
| Other bias | High risk | It appears that the 2 groups of participants were not balanced (it is not stated in the original study whether these differences were statistically significant or not): "There was a greater proportion of male patients in patients randomized to miglustat treatment than in those randomized to no miglustat treatment. The proportion of patients in the 2‐ to 11‐year‐old category was much greater than in the no miglustat treatment group"; "Seventeen patients (81%) in the miglustat group and all patients in the no miglustat treatment group had hepatomegaly (volume in millilitres> 25 age in years) and splenomegaly (volume in millilitres 2 age in years). Nine patients (43%) in the miglustat group and 6 patients (67%) in the no miglustat treatment group had anaemia, whereas 12 (57%) and 7 (78%) had thrombocytopenia (<150 X 10^3/ml), respectively. History of bone crisis was reported in four patients (19%) from the miglustat group and one (11%) from the no miglustat treatment group" |
Zimran 2011b.
| Methods | Randomized, double‐blind, multicenter, phase III parallel study | |
| Participants | 32 treatment‐naïve type I Gaucher participants | |
| Interventions | Taliglucerase alfa 30 units/kg (N = 16) vs 60 units/kg (N = 16) for 9 months | |
| Outcomes | Spleen and liver volume, Hb, Plt, chitotriosidase, CCL18, bone mineral density, bone marrow fat fraction | |
| Notes | van Dussen 2013 is an extension report of 8 participants from this study, focusing on bone marrow response | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patient randomization after screening eligibility was performed by computer‐generated coding." |
| Allocation concealment (selection bias) | Unclear risk | The study does not discuss the randomization method in sufficient amount of details in order to conclude regarding the sequence generation ‐ "Procedures to reduce bias (randomization, observer blinding, and inclusion of an intention to treat population who had received at least one dose and had a baseline evaluation) were incorporated" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Observers were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3 participants discontinued the study ‐ in the 30 units/kg arm, two participants (because of pregnancy and allergic reaction that occurred without receiving first full dose of treatment. The latter participant was included in the safety analysis, but not in efficacy analysis. In the 60 units/kg arm, 1 participant discontinued after the 12th infusion, due to allergic reaction, and was included in the analysis |
| Selective reporting (reporting bias) | Low risk | Data are reported for all outcomes, except for bone mineral density ‐ "There was no deterioration in bone density in any patient. Preliminary results of QCSI scans18 showed increases in fat fraction at month 9 in all 8 patients evaluated (data not shown)." |
| Other bias | Low risk | |
ACE: angiotensin converting enzyme CCL18: chemokine (C‐C motif) ligand 18 Hb: haemoglobin Plt: platelet TRAP: tartrate resistant acid phosphatase
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Elstein 1998 | Not randomized. |
| Wenstrup 2004 | Intervention not ERT or SRT, but alendronate disodium. |
| Zimran 2013b | Not randomized. |
ERT: enzyme replacement therapy SRT: substrate reduction therapy
Characteristics of studies awaiting assessment [ordered by study ID]
Engage 2015.
| Methods | RCT. Double‐blind, placebo‐controlled phase 3 trial. |
| Participants | 40 participants with untreated Gaucher disease type 1 (mean age 31.8 years; 20 males, 20 females). Participants with splenomegaly and thrombocytopenia or anemia (or both) were stratified by spleen volume. |
| Interventions | Randomized 1:1, eliglustat (50 or 100 mg twice‐daily depending on plasma levels) or placebo for 9 months. |
| Outcomes | Primary efficacy endpoint: per cent change in spleen volume (multiples of normal). Other efficacy outcomes included: haemoglobin; liver volume; platelets; disease‐related biomarkers; QoL. Safety evaluations included: spontaneously reported adverse events; laboratory evaluations; and ECGs. |
| Notes | The full paper was published just immediately prior to publication of this Cochrane Review. This RCT will be fully assessed for the next review update. |
ECG: electrocardiogram QoL: quality of life RCT: randomized controlled trial
Characteristics of ongoing studies [ordered by study ID]
Encore 2013.
| Trial name or title | ENCORE |
| Methods | RCT. Open‐label non‐inferiority, phase 3 trial. |
| Participants | 160 participants who had previously received ERT for ≥ 3 years who had met therapeutic goals. |
| Interventions | Eliglustat versus imiglucerase. |
| Outcomes | Primary efficacy endpoint: per cent of participants remaining stable after 52 weeks (using a pre‐defined composite of spleen, haemoglobin, platelet, and liver parameters). |
| Starting date | |
| Contact information | |
| Notes | Abstract only. Await full paper. |
ERT: enzyme replacement therapy RCT: randomized controlled trial
Differences between protocol and review
Several adjustments were made to the protocol when this review was carried out. To maximize the search sensitivity, the Cochrane collaboration highly sensitive search strategy was implemented, after consulting the group's Trials Search Co‐ordinator. As a consequence of the variability of data presentation in the identified studies, we considered that a meta‐analysis would be precluded. Since less than 10 RCTs were identified in type 1 Gaucher disease, we did not assess publication bias. Several changes were made to the outcomes assessed in this review ‐ tartrate‐resistant acid phosphatase activities were not consistently reported and are therefore not included in the analysis. We chose to include the levels of CCL‐18‐PARC chemokine, an increasingly used biomarker for Gaucher disease. Because of clinical irrelevancy to treatment effects, data for the zero to six months period which was available in one study, was not included in the analysis.
Skeletal outcomes were not reported in most studies, and are not included in our review. The lack of relevant data did not allow complete performance of the planned sub‐analyses. However, as stated above, in some of the cases we were able to carry out sub‐analyses of patients with intact spleens, and those with a confirmed diagnosis of Gaucher disease.
Contributions of authors
ES and TMC developed the research questions. Outcomes and subgroup analyses were discussed by all authors. writing of the first protocol draft was done by ES. The final version of the protocol was written by the help of all authors.The designing of the review was done by all authors. Data collection and analyses were done by ES and LD. Data interpretation and the writing of the manuscript were done by all authors.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Dr Elad Shemesh: none known.
Dr Laura Deroma: no direct conflicts of interest but her Institution received some equipment and consumables from pharmaceutical companies (Genzyme, Shire, Actelion). These also co‐financed several research projects.
Dr Bruno Bembi: a member of a bio‐tech university spin‐off (University of Udine) named Transactiva, operating in the field of vegetal bio reactor protein production. His institution received from pharmaceutical companies (Shire, Genzyme, Actelion) grants and co‐financing (equipments and consumables) for research projects; the author participated to meetings/advisory boards promoted by these companies. He is member of a bio‐tech university spin‐off named Transactiva, operating in the field of vegetal bio reacter protein production.
Dr Patrick Deegan: declares no bias toward any company mentioned or its products.
Prof Carla Hollak: received fees and travel reimbursements for consultancies and/or lectures in the field of lysosomal storage disorders from Genzyme, Shire, Protalix and Actelion. She has received grants for educational purposes and for a public‐private partnership TIPharma project, from Genzyme and Shire. All fees are received by the AMC Medical research BV in accordance with AMC regulations.
Dr Neal Weinreb: received research support (Genzyme/Sanofi, Shire HGT) and honoraria (Genzyme/Sanofi, Shire HGT,Pfizer) and has served on advisory boards (Genzyme/Sanofi, Shire HGT, Pfizer, Protalix Biotherapeutics) and speakers bureaus (Genzyme/Sanofi, Actelion, Pfizer).
Professor Timothy Cox: interests in this clinical and scientific field are longstanding (over 25 years) and, where possible, he endeavours to work with all companies in this ultra‐orphan field to promote competitive clinical and laboratory science for the benefit of patients with Gaucher disease; his personal rewards are relatively modest.
Edited (no change to conclusions)
References
References to studies included in this review
Ben Turkia 2013 {published data only}
- Ben Turkia H, Gonzalez DE, Barton NW, Zimran A, Kabra M, Lukina EA, et al. Velaglucerase alfa enzyme replacement therapy compared with imiglucerase in patients with Gaucher disease. American Journal of Hematology 2013;88(3):179‐84. [PUBMED: 23400823] [DOI] [PubMed] [Google Scholar]
- Ben Turkia H, Gonzalez DE, Kabra M, Lukina EA, Giraldo P, Kisinovsky I, et al. Two‐year safety and tolerability of velaglucerase alfa in patients with type 1 gaucher disease, including patients switched from imiglucerase: phase III trial HGT‐GCB‐039 and extension [abstract]. Blood 2011;118(21):Poster: 3214. [CENTRAL: 977632; CRS: 5500125000000576] [Google Scholar]
- Ben Turkia H, Gonzalez DE, Zimran A, Kabra M, Lukina EA, Giraldo P, et al. Achievement of therapeutic goals in patients with type 1 gaucher disease (GD1) on velaglucerase alfa or imiglucerase: phase III trials HGT‐GCB‐039 and extension [abstract]. Journal of Inherited Metabolic Disease 2011;34 Suppl 3:S224, Abstract no: P‐432. [CENTRAL: 977631; CRS: 5500125000000575] [Google Scholar]
- Elstein D, Ben Turkia H, Gonzalez DE, Kabra M, Lukina EA, Giraldo P, et al. Bone mineral density in adults with type 1 gaucher disease receiving velaglucerase alfa 60 U/KG every other week: 2‐year results [abstract]. Journal of Inherited Metabolic Disease 2012;35 Suppl 1:S150, Abstract no: P‐410. [CENTRAL: 977634; CRS: 5500125000000578] [Google Scholar]
- Zimran A, Kisinovsky I, Lukina EA, Elstein D, Zahrieh D, Crombez E, et al. Efficacy of long‐term velaglucerase alfa on haematological and visceral parameters in patients with type 1 gaucher disease [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S302, Abstract no: P‐728. [CENTRAL: 977639; CRS: 5500125000000583] [Google Scholar]
de Fost 2007 {published data only}
- Fost M, Aerts JM, Groener JE, Maas M, Akkerman EM, Wiersma MG, et al. Low frequency maintenance therapy with imiglucerase in adult type I Gaucher disease: a prospective randomized controlled trial. Haematologica 2007;92(2):215‐21. [PUBMED: 17296571] [DOI] [PubMed] [Google Scholar]
Elstein 2007 {published data only}
- Elstein D, Dweck A, Attias D, Hadas‐Halpern I, Zevin S, Altarescu G, et al. Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood 2007;110(7):2296‐301. [PUBMED: 17609429] [DOI] [PubMed] [Google Scholar]
- Elstein D, Heitner R, Dweck A, Attias D, Atarescu G, Zimran A. OGT 918 as substrate reduction therapy in type I gaucher disease [abstract]. 6th Meeting of the European Haematology Association; 2001 June 21‐24; Frankfurt, Germany.. 2001:Abstract no: 212. [CENTRAL: 593107; CRS: 5500100000003060]
Gonzalez 2013 {published data only}
- Elstein D, Ben Turkia H, Gonzalez DE, Kabra M, Lukina EA, Giraldo P, et al. Bone mineral density in adults with type 1 gaucher disease receiving velaglucerase alfa 60 U/KG every other week: 2‐year results [abstract]. Journal of Inherited Metabolic Disease 2012;35 Suppl 1:S150, Abstract no: P‐410. [CENTRAL: 977634; CRS: 5500125000000578] [Google Scholar]
- Gonzalez DE, Ben Dridi MF, Lukina E, Kisinovsky I, Ben Turkia H, Elstein D, et al. Clinically significant hemoglobin response observed within 3 months following treatment with velaglucerase alfa in patients with type 1 gaucher disease [abstract]. Journal of Inherited Metabolic Disease 2010;33 Suppl 1:S139, Abstract no: 442‐P. [CENTRAL: 977630; CRS: 5500125000000574] [Google Scholar]
- Gonzalez DE, Turkia HB, Lukina EA, Kisinovsky I, Dridi MF, Elstein D, et al. Enzyme replacement therapy with velaglucerase alfa in Gaucher disease: Results from a randomized, double‐blind, multinational, Phase 3 study. American Journal of Hematology 2013;88(3):166‐71. [PUBMED: 23386328] [DOI] [PubMed] [Google Scholar]
- Zimran A, Kisinovsky I, Lukina EA, Elstein D, Zahrieh D, Crombez E, et al. Efficacy of long‐term velaglucerase alfa on haematological and visceral parameters in patients with type 1 gaucher disease [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S302, Abstract no: P‐728. [CENTRAL: 977639; CRS: 5500125000000583] [Google Scholar]
Grabowski 1995 {published data only}
- Grabowski GA, Barton NW, Pastores G, Dambrosia JM, Banerjee TK, McKee MA, et al. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose‐terminated glucocerebrosidase from natural and recombinant sources. Annals of Internal Medicine 1995;122(1):33‐9. [PUBMED: 7985893] [DOI] [PubMed] [Google Scholar]
- Grabowski GA, Barton NW, Pastores G, Dambrosia JM, Banerjee TK, McKee MA, et al. Enzyme therapy in gaucher disease type 1: comparative efficacy of mannose‐terminated glucocerebrosidase from natural and recombinant sources [abstract]. Blood 1994, issue Suppl:226a. [DOI] [PubMed]
Kishnani 2009 {published data only}
- Kishnani PS, DiRocco M, Kaplan P, Mehta A, Pastores GM, Smith SE, et al. A randomized trial comparing the efficacy and safety of imiglucerase (Cerezyme) infusions every 4 weeks versus every 2 weeks in the maintenance therapy of adult patients with Gaucher disease type 1. Molecular Genetics and Metabolism 2009;96(4):164‐70. [PUBMED: 19195916] [DOI] [PubMed] [Google Scholar]
Schiffmann 2008 {published data only}
- Schiffmann R, Fitzgibbon EJ, Harris C, DeVile C, Davies EH, Abel L, et al. Randomized, controlled trial of miglustat in Gaucher's disease type 3. Annals of Neurology 2008;64(5):514‐22. [PUBMED: 19067373] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zimran 2011b {published data only}
- Zimran A, Brill‐Almon E, Chertkoff R, Petakov M, Blanco‐Favela F, Munoz ET, et al. Pivotal trial with plant cell‐expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 2011;118(22):5767‐73. [PUBMED: 21900191] [DOI] [PubMed] [Google Scholar]
- Zimran A, Gonzalez‐Rodriguez DE, Abrahamov A, Elstein D, Heitner R, Paz A, et al. A multicenter, double‐blind, randomized safety and efficacy study of two dose levels of taliglucerase alfa in pediatric patients with gaucher disease [abstract]. Blood 2012;120(21):Abstract no: 2140. [CENTRAL: 977633; CRS: 5500125000000577] [Google Scholar]
- Zimran A, Gonzalez‐Rodriguez DE, Abrahamov A, Elstein D, Paz A, Brill‐Almon E, et al. A multi‐center, double‐blind, randomized safety and efficacy study of two dose levels of taliglucerase alfa in pediatric patients with gaucher disease [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S301, Abstract no: P‐724. [CENTRAL: 977638; CRS: 5500125000000582] [Google Scholar]
- Zimran A, Mehta A, Giraldo P, Rosenbaum H, Giona F, Amato D, et al. Long‐term safety and efficacy data of taliglucerase alfa, a plant cell expressed recombinant glucocerebrosidase, in the treatment of naive gaucher disease patients [abstract]. Journal of Inherited Metabolic Disease 2012;35 Suppl 1:S11, Abstract no: O‐032. [CENTRAL: 977635; CRS: 5500125000000579] [Google Scholar]
- Dussen L, Zimran A, Akkerman EM, Aerts JM, Petakov M, Elstein D, et al. Taliglucerase alfa leads to favorable bone marrow responses in patients with type I Gaucher disease. Blood cells, Molecules & Diseases 2013;50(3):206‐11. [PUBMED: 23199589] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Elstein 1998 {published data only}
- Elstein D, Abrahamov A, Hadas‐Halpern I, Meyer A, Zimran A. Low‐dose low‐frequency imiglucerase as a starting regimen of enzyme replacement therapy for patients with type I Gaucher disease. QJM: Monthly Journal of the Association of Physicians 1998;91(7):483‐8. [PUBMED: 9797931] [DOI] [PubMed] [Google Scholar]
Wenstrup 2004 {published data only}
- Wenstrup RJ, Bailey L, Grabowski GA, Moskovitz J, Oestreich AE, Wu W, et al. Gaucher disease: alendronate disodium improves bone mineral density in adults receiving enzyme therapy. Blood 2004;104(5):1253‐7. [CENTRAL: 489109; CRS: 5500100000002632; PUBMED: 15010365] [DOI] [PubMed] [Google Scholar]
Zimran 2013b {published data only}
- Zimran A, Pastores GM, Tylki‐Szymanska A, Hughes DA, Elstein D, Mardach R, et al. Safety and efficacy of velaglucerase alfa in Gaucher disease type 1 patients previously treated with imiglucerase. American Journal of Hematology 2013;88(3):172‐8. [PUBMED: 23339116] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
Engage 2015 {published data only}
- Ben Turkia HMF, Lukina E, Amato D, Baris H, Dasouki M, Ghosn M, et al. Engage: a phase 3, randomized, double blind, placebo controlled, multi center study to investigate the efficacy and safety of eliglustat in adults with gaucher disease type 1: 9 month results [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S268, Abstract no: P‐596. [CENTRAL: 977640; CRS: 5500125000000584] [Google Scholar]
- Dasouka M, Lukina E, Ben Dridi MF, Amato D, Baris H, Ghosn M, et al. Effects of oral eliglustat on bone disease in gaucher disease type 1: results from the randomized, placebo‐controlled engage trial [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S268, Abstract no: P‐597. [CENTRAL: 977636; CRS: 5500125000000580] [Google Scholar]
- Mistry PK, Lukina E, Ben Turkia H, Amato D, Baris H, Dasouki M, et al. Effect of Oral Eliglustat on Splenomegaly in Patients With Gaucher Disease Type 1: The ENGAGE Randomized Clinical Trial. JAMA 2015;313(7):695‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
Encore 2013 {published data only}
- Cox TM, Drelichman G, Cravo R, Balwani M, Burrow T, Martins AM, et al. Encore: a multi‐national, randomized, controlled, open‐label non‐inferiority study comparing eliglustat to imiglucerase in gaucher disease type 1 (GD1) patients on enzyme replacement therapy (ERT) who have reached therapeutic goals [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S268, Abstract no: P‐598. [CENTRAL: 977637; CRS: 5500125000000581] [Google Scholar]
Additional references
Aerts 2006
- Aerts JM, Hollak CE, Boot RG, Groener JE, Maas M. Substrate reduction therapy of glycosphingolipid storage disorders. Journal of Inherited Metabolic Disease 2006;29(2‐3):449‐56. [PUBMED: 16763917] [DOI] [PubMed] [Google Scholar]
Amiri 2012
- Amiri M, Naim HY. Miglustat‐induced intestinal carbohydrate malabsorption is due to the inhibition of alpha‐glucosidases, but not beta‐galactosidases. Journal of Inherited Metabolic Disease 2012;35(6):949‐54. [PUBMED: 22976762] [DOI] [PubMed] [Google Scholar]
Andersson 2008
- Andersson H, Kaplan P, Kacena K, Yee J. Eight‐year clinical outcomes of long‐term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics 2008;122(6):1182‐90. [PUBMED: 19047232] [DOI] [PubMed] [Google Scholar]
Barton 1991
- Barton NW, Brady RO, Dambrosia JM, Bisceglie AM, Doppelt SH, Hill SC, et al. Replacement therapy for inherited enzyme deficiency‐‐macrophage‐targeted glucocerebrosidase for Gaucher's disease. New England Journal of Medicine 1991;324(21):1464‐70. [PUBMED: 2023606] [DOI] [PubMed] [Google Scholar]
Beutler 1993
- Beutler E, Nguyen NJ, Henneberger MW, Smolec JM, McPherson RA, West C, et al. Gaucher disease: gene frequencies in the Ashkenazi Jewish population. American Journal of Human Genetics 1993;52(1):85‐8. [PUBMED: 8434610] [PMC free article] [PubMed] [Google Scholar]
Beutler 2006
- Beutler E. Gaucher disease: multiple lessons from a single gene disorder. Acta Paediatrica (Oslo, Norway : 1992). Supplement 2006;95(451):103‐9. [PUBMED: 16720474] [DOI] [PubMed] [Google Scholar]
Biegstraaten 2010
- Biegstraaten M, Mengel E, Marodi L, Petakov M, Niederau C, Giraldo P, et al. Peripheral neuropathy in adult type 1 Gaucher disease: a 2‐year prospective observational study. Brain: a Journal of Neurology 2010;133(10):2909‐19. [PUBMED: 20693542] [DOI] [PubMed] [Google Scholar]
Black 1996
- Black N. Why we need observational studies to evaluate the effectiveness of health care. BMJ (Clinical research ed.) 1996;312(7040):1215‐8. [PUBMED: 8634569] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brady 1966
- Brady RO, Kanfer JN, Bradley RM, Shapiro D. Demonstration of a deficiency of glucocerebroside‐cleaving enzyme in Gaucher's disease.. The Journal of Clinical Investigation July;45(7):1112‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bultron 2010
- Bultron G, Kacena K, Pearson D, Boxer M, Yang R, Sathe S, et al. The risk of Parkinson's disease in type 1 Gaucher disease. Journal of Inherited Metabolic Disease 2010;33(2):167‐73. [PUBMED: 20177787] [DOI] [PMC free article] [PubMed] [Google Scholar]
Charrow 2000
- Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Archives of Internal Medicine 2000;160(18):2835‐43. [PUBMED: 11025794] [DOI] [PubMed] [Google Scholar]
Cherin 2006
- Cherin P, Sedel F, Mignot C, Schupbach M, Gourfinkel‐An I, Verny M, et al. Neurological manifestations of type 1 Gaucher's disease: Is a revision of disease classification needed? [Les manifestations neurologiques de la maladie de Gaucher de type 1: vers une remise en cause de la classification actuelle?]. Revue Neurologique 2006;162(11):1076‐83. [PUBMED: 17086144] [DOI] [PubMed] [Google Scholar]
Chetrit 2013
- Chetrit EB, Alcalay RN, Steiner‐Birmanns B, Altarescu G, Phillips M, Elstein D, et al. Phenotype in patients with Gaucher disease and Parkinson disease. Blood Cells, Molecules & Diseases 2013;50(3):218‐21. [PUBMED: 23265741] [DOI] [PubMed] [Google Scholar]
Cole 2011
- Cole JA, Taylor JS, Hangartner TN, Weinreb NJ, Mistry PK, Khan A. Reducing selection bias in case‐control studies from rare disease registries. Orphanet Journal of Rare Diseases 2011;6:61. [PUBMED: 21910867] [DOI] [PMC free article] [PubMed] [Google Scholar]
Conradi 1991
- Conradi N, Kyllerman M, Mansson JE, Percy AK, Svennerholm L. Late‐infantile Gaucher disease in a child with myoclonus and bulbar signs: neuropathological and neurochemical findings. Acta Neuropathologica 1991;82(2):152‐7. [PUBMED: 1718128] [DOI] [PubMed] [Google Scholar]
Cox 2000
- Cox T, Lachmann R, Hollak C, Aerts J, Weely S, Hrebicek M, et al. Novel oral treatment of Gaucher's disease with N‐butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000;355(9214):1481‐5. [PUBMED: 10801168] [DOI] [PubMed] [Google Scholar]
Cox 2003
- Cox TM, Aerts JM, Andria G, Beck M, Belmatoug N, Bembi B, et al. The role of the iminosugar N‐butyldeoxynojirimycin (miglustat) in the management of type I (non‐neuronopathic) Gaucher disease: a position statement. Journal of Inherited Metabolic Disease 2003;26(6):513‐26. [PUBMED: 14605497] [DOI] [PubMed] [Google Scholar]
Cox 2010
- Cox TM. Eliglustat tartrate, an orally active glucocerebroside synthase inhibitor for the potential treatment of Gaucher disease and other lysosomal storage diseases. Current Opinion in Investigational Drugs 2010;11(10):1169‐81. [PUBMED: 20872320] [PubMed] [Google Scholar]
Damiano 1998
- Damiano AM, Pastores GM, Ware JE Jr. The health‐related quality of life of adults with Gaucher's disease receiving enzyme replacement therapy: results from a retrospective study. Quality of Life Research : an International Journal of Quality of Life Aspects of Treatment, Care and Rehabilitation 1998;7(5):373‐86. [PUBMED: 9691718] [DOI] [PubMed] [Google Scholar]
de Fost 2006
- Fost M, Hollak CE, Groener JE, Aerts JM, Maas M, Poll LW, et al. Superior effects of high‐dose enzyme replacement therapy in type 1 Gaucher disease on bone marrow involvement and chitotriosidase levels: a 2‐center retrospective analysis. Blood 2006;108(3):830‐5. [PUBMED: 16527890] [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks J, Higgins J, Altman D. Chapter 9 Analysing data and undertaking meta‐analysis. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Deroma 2013
- Deroma L, Sechi A, Dardis A, Macor D, Liva G, Ciana G, et al. Did the temporary shortage in supply of imiglucerase have clinical consequences? Retrospective observational study on 34 italian Gaucher type I patients. JIMD reports 2013;7:117‐22. [PUBMED: 23430505] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dietz 2010
- Dietz HC. New therapeutic approaches to mendelian disorders. The New England journal of medicine 2010;363(9):852‐63. [PUBMED: 20818846] [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Genzyme/Sanofi 2013 [pers comm]
- Genzyme/Sanofi. Prescribed ERT infusions [personal communication]. Email to: Neal Weinreb 2013.
Gerstein 2012
- Gerstein HC, Bosch J, Dagenais GR, Díaz R, Jung H, Maggioni AP, et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. New England Journal of Medicine 2012;367(4):319‐28. [DOI] [PubMed] [Google Scholar]
Giraldo 2011
- Giraldo P, Irun P, Alfonso P, Dalmau J, Fernandez‐Galan MA, Figueredo A, et al. Evaluation of Spanish Gaucher disease patients after a 6‐month imiglucerase shortage. Blood Cells, Molecules & Diseases 2011;46(1):115‐8. [PUBMED: 20934891] [DOI] [PubMed] [Google Scholar]
Goker‐Alpan 2004
- Goker‐Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney‐Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. Journal of Medical Genetics 2004;41(12):937‐40. [PUBMED: 15591280] [DOI] [PMC free article] [PubMed] [Google Scholar]
Goldblatt 2011
- Goldblatt J, Fletcher JM, McGill J, Szer J, Wilson M. Enzyme replacement therapy "drug holiday": results from an unexpected shortage of an orphan drug supply in Australia. Blood cells, Molecules & Diseases 2011;46(1):107‐10. [PUBMED: 20684886] [DOI] [PubMed] [Google Scholar]
Grabowski 1997
- Grabowski GA. Gaucher disease: gene frequencies and genotype/phenotype correlations. Genetic testing 1997;1(1):5‐12. [PUBMED: 10464619] [DOI] [PubMed] [Google Scholar]
Grabowski 2008
- Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 2008;372(9645):1263‐71. [DOI] [PubMed] [Google Scholar]
Grabowski 2009
- Grabowski GA, Kacena K, Cole JA, Hollak CE, Zhang L, Yee J, et al. Dose‐response relationships for enzyme replacement therapy with imiglucerase/alglucerase in patients with Gaucher disease type 1. Genetics in Medicine: Official Journal of the American College of Medical Genetics 2009;11(2):92‐100. [PUBMED: 19265748] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hollak 2010
- Hollak CE, vom Dahl S, Aerts JM, Belmatoug N, Bembi B, Cohen Y, et al. Force majeure: therapeutic measures in response to restricted supply of imiglucerase (Cerezyme) for patients with Gaucher disease. Blood Cells, Molecules & Diseases 2010;44(1):41‐7. [PUBMED: 19804996] [DOI] [PubMed] [Google Scholar]
Hollak 2011
- Hollak CE, Aerts JM, Ayme S, Manuel J. Limitations of drug registries to evaluate orphan medicinal products for the treatment of lysosomal storage disorders. Orphanet Journal of Rare Diseases 2011;6:16. [PUBMED: 21496291] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hollak 2012
- Hollak CE. An evidence‐based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evidence 2012;7:15‐20. [PUBMED: 22654679] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hughes 2007
- Hughes DA, Mlilligan A, Mehta A. Home therapy for lysosomal storage disorders. British Journal of Nursing 2007;16(22):1386‐9. [DOI] [PubMed] [Google Scholar]
Inokuchi 1987
- Inokuchi J, Radin NS. Preparation of the active isomer of 1‐phenyl‐2‐decanoylamino‐3‐morpholino‐1‐propanol, inhibitor of murine glucocerebroside synthetase. Journal of Lipid Research 1987;28(5):565‐71. [PUBMED: 2955067] [PubMed] [Google Scholar]
Kaplan 2006
- Kaplan P, Andersson HC, Kacena KA, Yee JD. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Archives of Pediatrics & Adolescent Medicine 2006;160(6):603‐8. [PUBMED: 16754822] [DOI] [PubMed] [Google Scholar]
Kauli 2000
- Kauli R, Zaizov R, Lazar L, Pertzelan A, Laron Z, Galatzer A, et al. Delayed growth and puberty in patients with Gaucher disease type 1: natural history and effect of splenectomy and/or enzyme replacement therapy. Israel Medical Association Journal 2000;2(2):158‐63. [PUBMED: 10804944] [PubMed] [Google Scholar]
Khan 2012
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. Journal of Bone and Mineral Research : the Official Journal of the American Society for Bone and Mineral Research 2012;27(8):1839‐48. [PUBMED: 22692814] [DOI] [PubMed] [Google Scholar]
Krumholz 2013
- Krumholz HM, Ross JS, Gross CP, Emanuel EJ, Hodshon B, Ritchie JD, et al. A historic moment for open science: the yale university open data access project and medtronic. Annals of Internal Medicine 2013;158(12):910‐1. [PUBMED: 23778908] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kuter 2013
- Kuter DJ, Mehta A, Hollak CE, Giraldo P, Hughes D, Belmatoug N, et al. Miglustat therapy in type 1 Gaucher disease: clinical and safety outcomes in a multicenter retrospective cohort study. Blood Cells, Molecules & Diseases 2013;51(2):116‐24. [PUBMED: 23683771] [DOI] [PubMed] [Google Scholar]
Langeveld 2008
- Langeveld M, Fost M, Aerts JM, Sauerwein HP, Hollak CE. Overweight, insulin resistance and type II diabetes in type I Gaucher disease patients in relation to enzyme replacement therapy. Blood Cells, Molecules & Diseases 2008;40(3):428‐32. [PUBMED: 17950007] [DOI] [PubMed] [Google Scholar]
Lee 1982
- Lee RE. The pathology of Gaucher disease. Progress in Clinical and Biological Research 1982;95:177‐217. [PUBMED: 7122634] [PubMed] [Google Scholar]
Lukina 2010
- Lukina E, Watman N, Arreguin EA, Dragosky M, Iastrebner M, Rosenbaum H, et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz‐112638) treatment: 2‐year results of a phase 2 study. Blood 2010;116(20):4095‐8. [PUBMED: 20713962] [DOI] [PMC free article] [PubMed] [Google Scholar]
McEachern 2007
- McEachern KA, Fung J, Komarnitsky S, Siegel CS, Chuang WL, Hutto E, et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Molecular Genetics and Metabolism 2007;91(3):259‐67. [PUBMED: 17509920] [DOI] [PubMed] [Google Scholar]
Meikle 1999
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281(3):249‐54. [PUBMED: 9918480] [DOI] [PubMed] [Google Scholar]
Mistry 1996
- Mistry PK, Wraight EP, Cox TM. Therapeutic delivery of proteins to macrophages: implications for treatment of Gaucher's disease. Lancet 1996;348(9041):1555‐9. [PUBMED: 8950883] [DOI] [PubMed] [Google Scholar]
Mistry 2009
- Mistry PK, Deegan P, Vellodi A, Cole JA, Yeh M, Weinreb NJ. Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: effect on incidence of avascular necrosis. British Journal of Haematology 2009;147(4):561‐70. [PUBMED: 19732054] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mistry 2011
- Mistry PK, Weinreb NJ, Kaplan P, Cole JA, Gwosdow AR, Hangartner T. Osteopenia in Gaucher disease develops early in life: response to imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells, Molecules & Diseases 2011;46(1):66‐72. [PUBMED: 21112800] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nathan 2013
- Nathan DM, Buse JB, Kahn SE, Krause‐Steinrauf H, Larkin ME, Staten M, et al. Rationale and Design of the Glycemia Reduction Approaches in Diabetes: A Comparative Effectiveness Study (GRADE). Diabetes Care 2013;36(8):2254‐61. [PUBMED: 23690531] [DOI] [PMC free article] [PubMed] [Google Scholar]
Oulaidi 2011
- Oulaidi F, Front‐Deschamps S, Gallienne E, Lesellier E, Ikeda K, Asano N, et al. Second‐generation iminoxylitol‐based pharmacological chaperones for the treatment of Gaucher disease. ChemMedChem 2011;6(2):353‐61. [PUBMED: 21275057] [DOI] [PubMed] [Google Scholar]
Pastores 2010
- Pastores GM. Recombinant glucocerebrosidase (imiglucerase) as a therapy for Gaucher disease. BioDrugs: Clinical Immunotherapeutics, Biopharmaceuticals and Gene Therapy 2010;24(1):41‐7. [PUBMED: 20055531] [DOI] [PubMed] [Google Scholar]
Platt 1994
- Platt FM, Neises GR, Dwek RA, Butters TD. N‐butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. The Journal of Biological Chemistry 1994;269(11):8362‐5. [PUBMED: 8132559] [PubMed] [Google Scholar]
Resnick 2013
- Resnick D, Bozic KJ. Meta‐analysis of Trials of Recombinant Human Bone Morphogenetic Protein‐2: What Should Spine Surgeons and Their Patients Do With This Information?. Annals of Internal Medicine 2013;158(12):912‐3. [PUBMED: 23778909] [DOI] [PubMed] [Google Scholar]
Rosenbloom 2005
- Rosenbloom BE, Weinreb NJ, Zimran A, Kacena KA, Charrow J, Ward E. Gaucher disease and cancer incidence: a study from the Gaucher Registry. Blood 2005;105(12):4569‐72. [PUBMED: 15718419] [DOI] [PubMed] [Google Scholar]
Rosenbloom 2011
- Rosenbloom B, Balwani M, Bronstein JM, Kolodny E, Sathe S, Gwosdow AR, et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher Registry. Blood Cells, Molecules & Diseases 2011;46(1):95‐102. [PUBMED: 21067946] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rosenbloom 2013
- Rosenbloom BE, Weinreb NJ. Gaucher disease: a comprehensive review. Critical Reviews in Oncogenesis 2013;18(3):163‐75. [PUBMED: 23510062] [DOI] [PubMed] [Google Scholar]
Sidransky 2004
- Sidransky E. Gaucher disease: complexity in a "simple" disorder. Molecular Genetics and Metabolism 2004;83(1‐2):6‐15. [PUBMED: 15464415] [DOI] [PubMed] [Google Scholar]
Starzyk 2007
- Starzyk K, Richards S, Yee J, Smith SE, Kingma W. The long‐term international safety experience of imiglucerase therapy for Gaucher disease. Molecular Genetics and Metabolism 2007;90(2):157‐63. [PUBMED: 17079176] [DOI] [PubMed] [Google Scholar]
Stein 2010
- Stein P, Malhotra A, Haims A, Pastores GM, Mistry PK. Focal splenic lesions in type I Gaucher disease are associated with poor platelet and splenic response to macrophage‐targeted enzyme replacement therapy. Journal of Inherited Metabolic Disease 2010;33(6):769‐74. [PUBMED: 20683668] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stowens 1985
- Stowens DW, Teitelbaum SL, Kahn AJ, Barranger JA. Skeletal complications of Gaucher disease. Medicine 1985;64(5):310‐22. [PUBMED: 4033409] [DOI] [PubMed] [Google Scholar]
Weinreb 2002
- Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. American Journal of Medicine 2002;113(2):112‐9. [PUBMED: 12133749] [DOI] [PubMed] [Google Scholar]
Zimran 2011a
- Zimran A, Altarescu G, Elstein D. Nonprecipitous changes upon withdrawal from imiglucerase for Gaucher disease because of a shortage in supply. Blood Cells, Molecules & Diseases 2011;46(1):111‐4. [PUBMED: 20542712] [DOI] [PubMed] [Google Scholar]
Zimran 2013a
- Zimran A, Altarescu G, Elstein D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood cells, molecules & diseases 2013;50(2):134‐7. [PUBMED: 23085429] [DOI] [PubMed] [Google Scholar]