

Abstract
Background
Benralizumab is a humanised, anti-interleukin-5 receptor α monoclonal antibody with anti-eosinophilic activity. Lack of fucose (afucosylation) increases its affinity to CD16a and significantly enhances antibody-dependent cell-mediated cytotoxicity by natural killer (NK) cells. Although benralizumab proved clinically efficacious in clinical trials for patients with severe asthma and hypereosinophilic syndrome, in-depth characterisation of its anti-eosinophilic mechanisms of action remains elusive.
Methods
Here, we further investigated the mechanisms involved in benralizumab's anti-eosinophilic activities by employing relevant primary human autologous cell co-cultures and real-time-lapse imaging combined with flow cytometry.
Results
In the presence of NK cells, benralizumab induced potent eosinophil apoptosis as demonstrated by the upstream induction of Caspase-3/7 and upregulation of cytochrome c. In addition, we uncovered a previously unrecognised mechanism whereby benralizumab can induce eosinophil phagocytosis/efferocytosis by macrophages, a process called antibody-dependent cellular phagocytosis. Using live cell imaging, we unravelled the stepwise processes leading to eosinophil apoptosis and uptake by activated macrophages. Through careful observations of cellular co-culture assays, we identified a novel role for macrophage-derived tumour necrosis factor (TNF) to further enhance benralizumab-mediated eosinophil apoptosis through activation of TNF receptor 1 on eosinophils. TNF-induced eosinophil apoptosis was associated with cytochrome c upregulation, mitochondrial membrane depolarisation and increased Caspase-3/7 activity. Moreover, activated NK cells were found to amplify this axis through the secretion of interferon-γ, subsequently driving TNF expression by macrophages.
Conclusions
Our data provide deeper insights into the timely appearance of events leading to benralizumab-induced eosinophil apoptosis and suggest that additional mechanisms may contribute to the potent anti-eosinophilic activity of benralizumab in vivo. Importantly, afucosylation of benralizumab strongly enhanced its potency for all mechanisms investigated.
Short abstract
New insights explaining the potent anti-eosinophilic activity of benralizumab, an anti-IL-5Rα afucosylated monoclonal antibody with enhanced depleting potency in patients with severe eosinophilic inflammation https://bit.ly/3yUnsnn
Introduction
Accumulation of tissue eosinophils in chronic inflammatory diseases can result in tissue damage through the release of cytotoxic products contained in their granules [1, 2]. The indispensable role of interleukin-5 (IL-5) in eosinophil development, differentiation and activation [3, 4] prompted the development of antibody-based therapies disrupting the IL-5/IL-5 receptor (IL-5R) axis for the reduction of eosinophil numbers and their activation in afflicted tissues [5]. Indeed, two monoclonal antibodies targeting IL-5, mepolizumab (Nucala) and reslizumab (Cinqair), and one targeting IL-5Rα, benralizumab (Fasenra), are now approved therapies for the treatment of severe asthma with an eosinophilic phenotype, and also eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome) in the case of mepolizumab. While all three antibodies have been shown to reduce eosinophil counts in tissues of patients to various degrees [6, 7], they differ in their mechanism of action. Mepolizumab and reslizumab bind to and inhibit IL-5, whereas benralizumab triggers antibody-dependent cell-mediated cytotoxicity (ADCC) via binding to IL-5Rα on eosinophils. We have previously reported that afucosylation of the oligosaccharide chain in the CH2 region of benralizumab's Fc domain resulted in enhanced Fcγ receptor IIIa (CD16a) binding and subsequent ADCC-mediated eosinophil apoptosis in vitro in the presence of natural killer (NK) cells [8]. Due to its high potency [8], fast onset of action [9] and pronounced eosinophil depletion in patients [10], further investigations into mechanisms that may contribute to benralizumab's anti-eosinophilic activity are important.
In order to do so, we a developed live cell imaging approach combined with flow cytometry and biochemical analysis using primary human eosinophils and effector cells in vitro, rather than performing in vivo studies in mice, due to the lack of benralizumab's cross-reactivity to murine IL-5Rα and differences in Fc receptor biology between mouse and human. Here, we demonstrate that benralizumab induces potent Caspase-3/7 activation and cytochrome c upregulation on eosinophils in the presence of NK cells, further substantiating its pro-apoptotic activity. In addition, we show for the first time that benralizumab mediates eosinophil phagocytosis/efferocytosis by macrophages, a process known as antibody-dependent cellular phagocytosis (ADCP), and stimulates tumour necrosis factor (TNF)-dependent macrophage cytotoxicity. Since TNF has previously been reported to induce eosinophil apoptosis via TNF receptor 1 (TNFR1) [11], we investigated benralizumab-induced TNF secretion by macrophages. We were able to demonstrate enhanced expression of TNF by activated macrophages and increased expression of TNFR1 on apoptotic eosinophils. Blockade of TNF/TNFR1 resulted in reduced ADCP of eosinophils and TNF/TNFR1-mediated eosinophil apoptosis by macrophages. The presence of NK cells further enhanced TNFR1-mediated apoptosis of eosinophils through the release of interferon-γ (IFN-γ). Importantly, the observed anti-eosinophilic activities of benralizumab were significantly enhanced in comparison with the parent fucosylated isoform of benralizumab. Taken together, our data provide additional explanations for benralizumab's potent anti-eosinophilic activity in patients.
Methods
A complete description of the methods is provided in the supplementary material.
Study approval
All healthy donors were anonymously enrolled in the AstraZeneca Research Specimen Collection Program. These volunteers provided written informed consent prior to enrolment. An independent ethics committee approved the establishment of the AstraZeneca Research Specimen Collection Program and use of specimens for research purposes. This study was performed in accordance with the Declaration of Helsinki, International Council for Harmonisation/Good Clinical Practice guidelines, the applicable regulatory requirements and AstraZeneca policy on bioethics.
Results
Previously, we demonstrated benralizumab-mediated eosinophil apoptosis (half-maximal effective concentration (EC50) 0.9 pmol·L−1) through ADCC [8] by quantifying annexin V-expressing eosinophils in the presence of NK cells. To further explore this initial observation, we combined flow cytometry with live cell imaging to investigate autologous human eosinophil/NK cell co-culture assays.
Benralizumab mediates caspase-dependent eosinophil apoptosis through NK cytotoxicity
Human eosinophils were isolated from peripheral blood of healthy donors, treated with antibodies, stained with far-red lipid membrane dye and mixed with autologous phycoerythrin–Texas Red-labelled NK cells in a 1:3 ratio (eosinophil:NK cell) (supplementary figure S1a). At 6 h, cell apoptosis was detected in CD66b+ Siglec-8+ eosinophils by flow cytometry with blue cationic nucleic acid dye PO-PRO-1, which permeates plasma membranes at apoptotic cascade initiation. As expected, in the presence of NK cells, benralizumab significantly enhanced eosinophil death compared with fucosylated anti-IL-5Rα antibody, as evidenced by an increased percentage of PO-PRO-1+ CD66b+ Siglec-8+ cells (figure 1a, supplementary figure S1b and supplementary videos S1 and S2). Benralizumab alone had no effect on eosinophil death in the absence of NK cells (supplementary figure S1c). Benralizumab-induced PO-PRO-1+ eosinophils were also positive for Caspase-3/7 activity and cytochrome c, which increased concomitantly (figure 1b and c). Consistent with this observation, live imaging of caspase activity confirmed the incorporation of CellEvent Caspase-3/7 reagent prior to detection of PO-PRO-1 dye in dying eosinophils (figure 1d and supplementary video S3).
FIGURE 1.

Benralizumab induces caspase-dependent eosinophil (Eos) apoptosis by natural killer (NK) cells. Primary human NK cells and eosinophils isolated from healthy donors were labelled individually. NK cells were loaded with LysoTracker Green to selectively label NK lytic granules. Eosinophils were pre-treated with 10 nM of the indicated antibodies, then mixed with NK cells for 6 h and analysed. a–c) Eosinophil cell death, evaluated by PO-PRO-1 blue dye for detection of a) apoptotic and dead cells (n=7), b) caspase activation assessed by CellEvent Caspase-3/7 (Casp3/7) reagent (n=3), and c) cytochrome c (n=3), represented as percentage of total CD66b+ Siglec-8+ eosinophils. Data are mean±sem. d) Representative frames selected from live imaging data, with 10 min intervals, displaying caspase activation in eosinophils with CellEvent Caspase-3/7 reagent, indicated in white. Confocal microscopy was used for live imaging for 90 min. Images were acquired every 5 min. Scale bar: 2 µm (n=4). Two-way ANOVA was employed with Tukey's multiple comparisons. **: p<0.01; ***: p<0.001; ****: p<0.0001, benralizumab versus controls (anti-interleukin (IL)-5 or parent fucosylated anti-IL-5 receptor α (PF anti-IL-5Rα)). Statistical values are presented in supplementary table S2.
Benralizumab binds NK cells and eosinophils, bringing them in close contact and facilitating NK cell-mediated eosinophil apoptosis [12]. These direct NK cell–eosinophil interactions can trigger the release of specialised secretory lysosomes carrying lytic products, such as perforin and granzyme B, that are required for NK-mediated cell killing [13]. Once activated, NK cells upregulate CD137, a costimulatory receptor for CD16 [14], and lysosome-associated membrane protein (LAMP-1)/CD107a [15], an indicator of NK cell degranulation. By monitoring the expression of these markers during ADCC assays, we observed significantly increased CD137 surface expression on CD56+ CD94+ NK cells compared with the parent fucosylated anti-IL-5Rα antibody. This response was associated with increased LAMP-1 density and production of perforin and granzyme B in activated NK cells in the presence of brefeldin A (figure 2a and b). While in the presence of benralizumab a higher percentage of activated NK cells displayed increased expression of CD137 and LAMP-1 (33%), a lower proportion stained positive for granzyme B and perforin (14%) (figure 2a and b). A plausible explanation for the reduced internal stock of NK granules after 6 h stimulation is the early degranulation of NK cells upon establishment of the immune synapse with eosinophils (figure 2c and supplementary videos S3 and S4).
FIGURE 2.

Natural killer (NK) cell cytotoxicity mediates benralizumab-induced eosinophil (Eos) apoptosis. Primary eosinophils and NK cells were prepared as described in figure 1. a, b) Changes in NK activation (CD137 and CD107a) and NK cytolytic (granzyme B (GranzB) and perforin) markers: a) assessed by flow cytometry and b) represented as percentage of total CD56+ CD94+ NK cells (n=4). Data are mean±sem. c) Stepwise stages of NK-mediated eosinophil death induced by benralizumab. Cells were live imaged by confocal microscopy for 90 min. The images are representative frames acquired approximately every 2 min. 1) NK immunological synapse is established between a NK cell and an eosinophil. 2) NK lytic granules are docked at the cellular interface of the eosinophil initiating cell death as PO-PRO-1 enters the cell. 3) NK detaches from the eosinophil after delivery of NK lytic granules. 4) The eosinophil undergoes apoptosis as indicated by chromatin condensation, membrane blebbing and nuclear collapse. Scale bar: 2 µm. Two-way ANOVA was employed with Tukey's multiple comparisons. *: p<0.05; **: p<0.01, benralizumab versus parent fucosylated anti-interleukin-5 receptor α (PF anti-IL-5Rα). Statistical values are presented in supplementary table S2.
To decipher the cellular interactions leading to eosinophil apoptosis induced by benralizumab, we conducted real-time live imaging of NK cell–eosinophil interactions every 2 min for 2 h using confocal microscopy. Eosinophils and NK cells were co-incubated at a ratio of 1:3 in the presence of benralizumab and PO-PRO-1 dye for 2 h prior to image acquisition. Our image analyses confirmed that cell stimulation with benralizumab initiated the killing process through the formation of NK cell–eosinophil immune synapses (figure 2c-1), which triggered granule polarisation (the migration of lytic granules to the NK cellular surface) (figure 2c-2). Subsequently, these granules fused with the eosinophil membrane, delivering cytotoxic mediators into the eosinophil's cytosol (figure 2c-3), leading to cell membrane blebbing and nuclear fragmentation (figure 2c-4), the hallmarks of apoptotic death. Although granule polarisation occurred rapidly, initiation of eosinophil death, marked by PO-PRO-1 entry, was not visualised until ∼20 min later and increased over time (supplementary videos S3 and S4).
Taken together, our data argue for apoptosis as the main mechanism of NK cell-mediated eosinophil death in the presence of benralizumab.
Benralizumab upregulates eosinophil uptake by macrophages
Given that macrophages express CD16a, mediate potent Fcγ receptor-mediated phagocytosis [16] and induce cell death through secretory cytotoxic mechanisms [17, 18], we questioned whether benralizumab can induce eosinophil death through macrophage activation. By combining real-time imaging and flow cytometry, we developed a new assay to quantify eosinophil uptake by macrophages. Human blood CD16+ monocyte-derived macrophages were co-cultured with autologous eosinophils treated with antibodies at an optimised ratio of 1:5 in serum-free media supplemented with 50 ng·mL−1 macrophage colony-stimulating factor and imaged every 5 min for 6 h. Under these conditions, we observed individual macrophages were often able to engulf either living (phagocytosis) (figure 3a) or apoptotic eosinophils (efferocytosis) (figure 3b). Total count of eosinophil death was evaluated as the sum of engulfed alive (phagocytosis) or dead PO-PRO-1+ eosinophils (efferocytosis) after 6 h stimulation.
FIGURE 3.
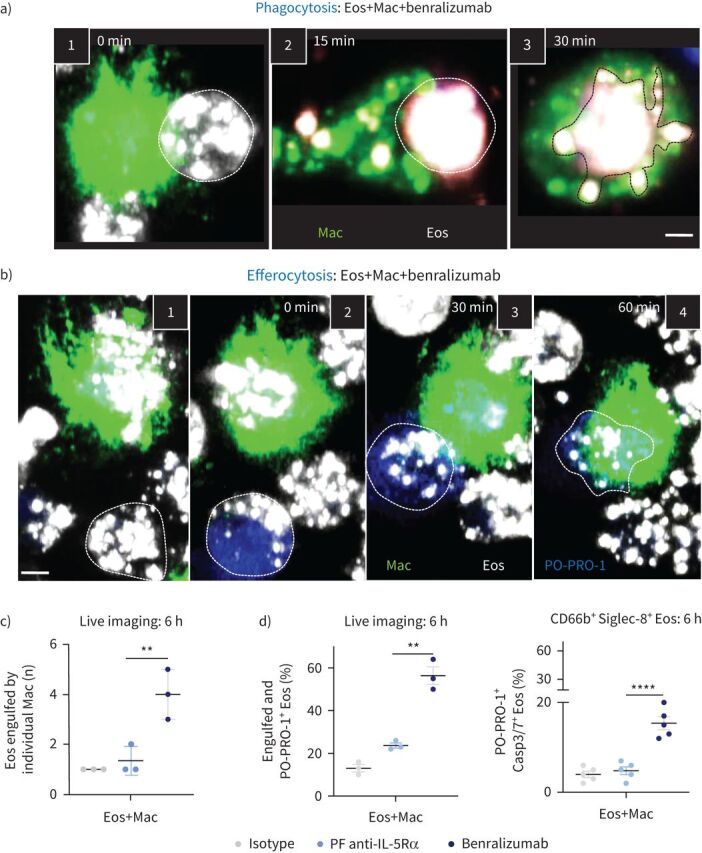
Documenting the steps of benralizumab-mediated eosinophil (Eos) depletion by macrophage (Mac) phagocytosis or efferocytosis. Human primary natural killer (NK) cells and eosinophils isolated from healthy donors were labelled individually. Macrophages were stained with green lipid membrane dye. Eosinophils were pre-treated with 10 nM of benralizumab then labelled with red lipid membrane dye. Red-labelled eosinophils were co-incubated with green-labelled NK cells in the presence of PO-PRO-1 for detection of cell death. Conjugates were live cell imaged by confocal microscopy for 90 min. a) Stepwise stages of macrophage-mediated eosinophil phagocytosis targeted by benralizumab. 1) Macrophage–eosinophil immune synapse is established. 2) Formation of the phagocytic cup. 3) Eosinophil internalisation by macrophages. b) Stepwise stages of macrophage-mediated eosinophil efferocytosis. 1, 2) Eosinophil undergoing apoptosis prior to 3) uptake and 4) engulfment by macrophages. c) Quantification of eosinophils engulfed by individual macrophages post-treatment with the indicated antibodies via live imaging (n=5). d) Left: quantification of eosinophil death (macrophage-engulfed and PO-PRO-1+ eosinophils) by live imaging, as percentage of total far-red-labelled eosinophils (n=3), in the presence of macrophages; right: changes in caspase activation assessed by flow cytometry using CellEvent Caspase-3/7 (Casp3/7) reagent and represented as percentage of total CD66b+ Siglec-8+ eosinophils (n=3). a, b) Images are frames acquired approximately every 5 min, then representative frames were selected with 15 or 30 min intervals for illlustration. Scale bar: 2 µm. c, d) Data are mean±sem. Two-way ANOVA with Tukey's and Šídák's multiple comparisons. **: p<0.01; ****: p<0.0001, benralizumab versus parent fucosylated anti-interleukin-5 receptor α (PF anti-IL-5Rα). Statistical values are presented in supplementary table S2.
To further document the cellular events involved in benralizumab-mediated phagocytosis and efferocytosis, we closely examined the acquired live cell image sequences upon benralizumab treatment. When an activated macrophage and living eosinophil came in contact, a phagocytic synapse was established (figure 3a-1), followed by changes in the macrophage membrane, giving rise to pseudopods that surrounded the living eosinophils. At the site of contact, a phagocytic cup (depression on the membrane) formed (figure 3a-2), initiating the engulfment of eosinophils. Within 30 min, the membranes closed at the distal end, creating new phagosomes [19, 20] required for eosinophil degradation (figure 3a-3 and supplementary videos S6 and S7). In addition to the engulfment of live eosinophils, detailed analysis of image sequences (supplementary videos S6 and S7) unexpectedly revealed eosinophil death prior to their physical interaction with macrophages as evidenced by the incorporation of PO-PRO-1 dye (figure 3b-2 and d, right panel). After recognition of dead eosinophils, an efferocytic synapse was formed (figure 3b-3), leading to the engulfment and subsequent degradation of PO-PRO-1+ eosinophils by activated macrophages (figure 3b-4).
With benralizumab, individual macrophages were able to engulf up to four eosinophils when compared with the parent fucosylated control (figure 3c), resulting in uptake of 55% of all eosinophils (figure 3d, left). Under these conditions, about two-thirds of eosinophils were taken up by phagocytosis (living eosinophils), while one-third underwent efferocytosis (dead eosinophils). Dead CD66b+ Siglec-8+ eosinophils were determined by Caspase-3/7 activation and PO-PRO-1 staining (figure 3d, right). The occurrence of eosinophil death prior to macrophage engagement suggested the presence of an additional secretory mechanism that may trigger eosinophil death [17, 18].
NK cells enhanced benralizumab-mediated eosinophil death by macrophages
When NK cells were added to the cell mix (1:5:15, macrophages:eosinophils:NK cells), we observed an enhancement in eosinophil uptake, live or dead, by activated macrophages in response to benralizumab using real-time imaging (figure 4a). Although the total number of activated macrophages engulfing eosinophils remained unchanged in the presence of NK cells (supplementary figure S2), the count of eosinophils engulfed per individual macrophage was greatly enhanced, from four up to eight eosinophils per macrophage, with the addition of benralizumab (figure 4a and supplementary video S8). Quantification of total eosinophil death (all engulfed PO-PRO-1+ and live eosinophils) showed a 12% increase in the percentage of dying eosinophils in the presence of NK cells and benralizumab (figure 4b), resulting in 67% total eosinophil death (figure 4b). 34.5% CD66b+ Siglec-8+ PO-PRO-1+ eosinophils exhibited a concomitant upregulation of Caspase-3/7 activity (figure 4c and supplementary video S9). Furthermore, image analysis highlighted the frequent, but transient, formation of immune synapses between NK cells and macrophages, typically ranging from nine to 14 synapses per macrophage (supplementary video S10 and figure 4d). NK cells were observed to engage shortly with activated macrophages, to then detach without being phagocyted. These observations prompted us to question whether NK–macrophage synapses have a stimulatory effect on macrophage behaviour. Therefore, we characterised benralizumab-mediated macrophage activation and the enhancement of macrophage phagocytic and cytotoxic responses by NK cells. We tracked cell surface regulation of the opsonic Fcγ receptor I (CD64) on CD163+ cells, as an indicator of macrophage activation during benralizumab-mediated eosinophil phagocytosis/efferocytosis [21], and observed upregulation of enhanced CD64 expression on CD163+ macrophages with benralizumab (figure 5a and b). CD64 upregulation was accompanied by enhanced expression of the scavenger receptor CD163 (isotype mean fluorescence intensity (MFI): 433; benralizumab MFI: 3998) and of CD107a (LAMP-1), suggesting degradation of uptaken eosinophils in macrophage phagolysosomes (figure 5a and b). In contrast, the inhibitory “don't-eat-me” signal, signal regulatory protein α (SIRPα) [22], was decreased on activated macrophages (isotype MFI: 11 066; benralizumab MFI: 2497) (figure 5a and b). These findings suggest that benralizumab can induce macrophage phagocytosis/efferocytosis through CD163, CD64 and CD107a upregulation and concomitant SIRPα downregulation, as well as mediate NK cell-mediated macrophage activation when compared with its parent fucosylated anti-IL-5Rα control (figure 5a and b).
FIGURE 4.
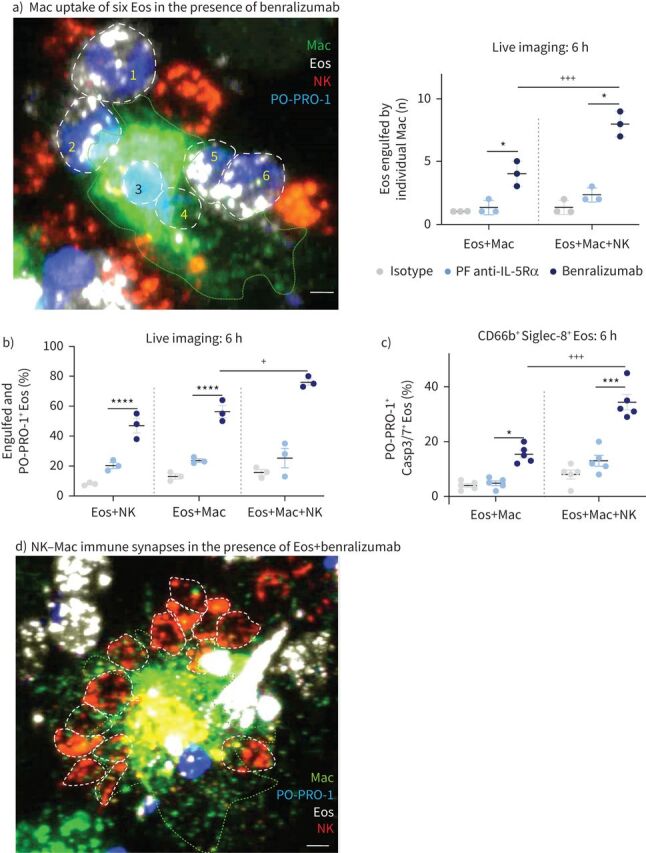
Natural killer (NK) cells enhance benralizumab-induced eosinophil (Eos) depletion by macrophages (Mac). Human macrophages were derived from peripheral blood monocytes after 7 days stimulation with 50 ng·mL−1 macrophage colony-stimulating factor. Primary NK cells and eosinophils were isolated from autologous healthy donors, and each cell type was labelled individually. Eosinophils were pre-treated with 10 nM of indicated antibodies, then labelled with far-red lipid membrane dye and co-incubated with green-labelled macrophages and/or red-labelled NK cells in the presence of PO-PRO-1. Cells were mixed for 6 h and analysed by flow cytometry or confocal microscopy for live cell imaging. a) Left: representative image depicting increased phagocytic activity of macrophages (engulfment of six eosinophils) in the presence of NK cells upon benralizumab treatment (interacting eosinophils are delineated by white dashed lines); right: quantification of eosinophils engulfed by individual macrophages post-treatment with the indicated antibodies via live imaging (n=5). Scale bar: 2 µm. b) Quantification of eosinophil death (macrophage-engulfed and PO-PRO-1+ eosinophils) by live imaging, as percentage of total far-red-labelled eosinophils (n=3), in the presence of macrophages and/or NK cells. c) Changes in caspase activation assessed by flow cytometry using CellEvent Caspase-3/7 (Casp3/7) reagent and represented as percentage of total CD66b+ Siglec-8+ eosinophils (n=3). d) Representative image of significant immune synapses between NK cells and macrophages. A representative highly activated macrophage is delineated by the green dashed line. Scale bar: 2 µm. a–c) Data are mean±sem. Two-way ANOVA with Tukey's multiple comparisons. *: p<0.05; ***: p<0.001; ****: p<0.0001, benralizumab versus parent fucosylated anti-interleukin-5 receptor α (PF anti-IL-5Rα). +: p<0.05; +++: p<0.001, Eos+Mac+NK versus Eos+Mac. Statistical values are presented in supplementary table S2.
FIGURE 5.

Natural killer (NK) cells enhance benralizumab-mediated macrophage (Mac) phagocytosis and cytotoxicity. Human macrophages were derived from peripheral blood monocytes after 7 days stimulation with 50 ng·mL−1 macrophage colony-stimulating factor. Primary NK cells and eosinophils (Eos) were isolated from autologous healthy donors. Eosinophils were pre-treated with 10 nM of indicated antibodies. Cells were mixed for 6 h and analysed by flow cytometry. Supernatants were collected for quantification of tumour necrosis factor (TNF) by ELISA. a, b) Changes in macrophage markers involved in target recognition/internalisation (CD163, CD64 and signal regulatory protein α/β (SIRPα/β)) and phagolysosome activation (CD107a), represented as percentage of total CD163+ CD11b+ macrophages (n=5). c) TNF amounts quantified by ELISA post-treatment by benralizumab in the presence and absence of macrophages and NK cells (n=4). d) Quantification of TNF expression in macrophages in the presence and absence of NK cells represented as percentage of total CD163+ CD11b+ macrophages (n=4). b, c, d) Data are mean±sem. Two-way ANOVA test with Tukey's multiple comparisons. *: p<0.05; **: p<0.01; ***: p<0.001, benralizumab versus controls (isotype or parent fucosylated anti-interleukin-5 receptor α (PF anti-IL-5Rα)). +: p<0.05; ++: p<0.01, Eos+Mac+NK versus Eos+Mac. Statistical values are presented in supplementary table S2.
Benralizumab induces TNFR1-mediated eosinophil apoptosis by macrophage-derived TNF
We next investigated whether benralizumab can induce the expression of macrophage-derived TNF and promote TNFR1-mediated eosinophil apoptosis. We first evaluated soluble TNF in the supernatants of human macrophages cultured alone or with benralizumab-treated eosinophils in the presence or absence of NK cells. While a small but significant increase in TNF quantities was detected in the supernatants of macrophages mixed with benralizumab-treated eosinophils, a robust upregulation was induced by adding NK cells to the cell mix. In contrast, TNF amounts were barely detectable in the absence of eosinophils (figure 5c). Consistently, we demonstrated an upregulation of activated macrophages (CD11b+ CD163high) expressing TNF, whose frequency further increased upon the addition of NK cells (figure 5d). In addition, we identified a significant upregulation of TNFR1 on CD66b+ Siglec-8+ eosinophils co-cultured with macrophages, which was further enhanced by adding NK cells to the mix (figure 6a). Taken together, our data highlight the requirement of target cells for benralizumab-induced TNF production by macrophages that was associated with elevated TNFR1 expression on eosinophils, and also confirm the stimulatory effect of NK cells on TNF and TNFR1 expression. To validate whether benralizumab's anti-eosinophilic activities are relevant in severe eosinophilic asthma patients, we confirmed the expression of TNFR1 and IL-5Rα on eosinophils, CD16 on NK cells and macrophages, as well as benralizumab-mediated eosinophil depletion in vitro (unpublished data).
FIGURE 6.
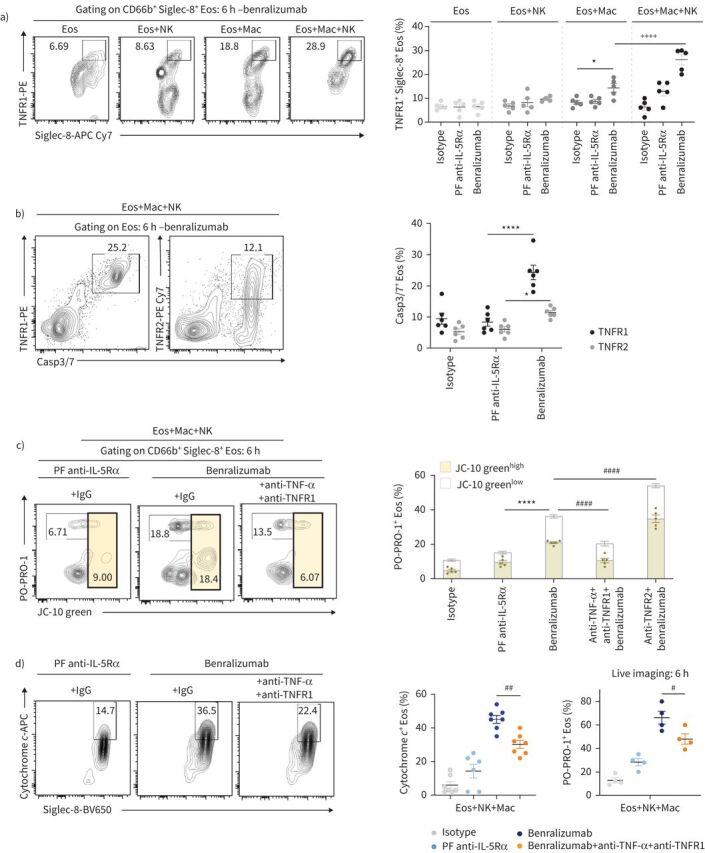
Benralizumab promotes eosinophil (Eos) apoptosis by macrophage (Mac) cytotoxicity through the tumour necrosis factor (TNF)/TNF receptor 1 (TNFR1) pathway. Human eosinophils were pre-treated with 10 nM of indicated antibodies, then labelled and co-incubated with macrophages and natural killer (NK) cells in the presence of PO-PRO-1 for 6 h. a) Quantification of TNFR1-induced expression in eosinophils in the presence and absence of macrophages and NK cells, represented as percentage of total CD66b+ Siglec-8+ cells (n=4). b) TNFR1 expression was compared with TNF receptor 2 (TNFR2) in apoptotic Caspase-3/7+ (Casp3/7) eosinophils in the presence and absence of macrophages and NK cells, represented as percentage of total CD66b+ Siglec-8+ cells (n=3). c, d) Reduction of apoptotic eosinophils, as assessed by c) PO-PRO-1 and JC-10 green mitochondrial membrane potential assay and d) cytochrome c, after inhibition of TNF and TNFR1 in the presence of indicated antibodies, macrophages and NK cells (n=4). Data are mean±sem. Two-way ANOVA with Tukey's multiple comparisons. *: p<0.05; ****: p<0.0001, benralizumab versus controls (isotype or parent fucosylated anti-interleukin-5 receptor α (PF anti-IL-5Rα)). ++++: p<0.0001, Eos+Mac+NK versus Eos+Mac. #: p<0.05; ##: p<0.01; ####: p<0.0001, blocking antibodies+benralizumab versus benralizumab. Statistical values are presented in supplementary table S2.
Since TNF was reported to also bind TNF receptor 2 (TNFR2), whose activation has been linked to pro-survival pathways in human eosinophils [23], we evaluated its expression on eosinophils by flow cytometry upon benralizumab treatment for 6 h in the presence of macrophages and NK cells. Although the majority of Caspase-3/7-expressing eosinophils expressed TNFR1 (25%), only half of them (12.5%) upregulated TNFR2 expression (figure 6b), associating caspase-dependent eosinophil death to TNFR1 upregulation. Although TNFR2 expression on apoptotic eosinophils suggests activation of a pro-survival signal, it seemed counter-balanced in favour of eosinophil apoptosis.
To prevent TNF signalling through TNFR2 (pro-survival) and to simultaneously ensure successful blockade of TNFR1-mediated eosinophil apoptosis, a combination of anti-TNF-α and anti-TNFR1 blocking antibodies was applied to the cell mix of macrophages, NK cells and antibody-treated eosinophils. Under these condition, early apoptosis was evaluated by the depolarisation of mitochondrial membrane potential, assessed by the increase in JC-10 green staining in CD66b+ Siglec-8+ eosinophils. Late apoptosis was indicated by the increased incorporation of PO-PRO-1 dye in dying eosinophils and the concomitant decrease of JC-10 green, which diffuses to other cell compartments after mitochondrial depolarisation. As expected, we observed a mix of early apoptosis (JC-10 greenhigh and PO-PRO-1+ eosinophils) as well as late apoptosis (PO-PRO-1+ and JC-10 greenlow cells) when cells were treated with benralizumab (figure 6c). While blocking of TNFR2 alone impaired eosinophil survival (figure 6c), inhibition of TNF/TNFR1 resulted in a partial but significant reduction of apoptotic eosinophil death as evidenced by the decreased incorporation of PO-PRO-1 and JC-10 green (figure 6c), as well as the downregulation of cytochrome c in CD66b+ Siglec-8+ eosinophils (figure 6d, left panel). Consistent with our findings, live cell imaging analysis confirmed the reduced eosinophil apoptosis in response to TNF/TNFR1 blockade, as indicated by the decreased PO-PRO-1 labelling in eosinophils (figure 6d, right panel). Our data suggest the presence of a macrophage TNF/eosinophil TNFR1 axis that when activated through the formation of macrophage–benralizumab–eosinophil complexes, results in eosinophil apoptosis.
TNF-dependent macrophage cytotoxicity can be triggered by NK-derived IFN-γ in the presence of benralizumab-stimulated eosinophils
Macrophage activation is triggered by several NK-derived mediators, including the most potent activating cytokine IFN-γ [14, 17, 24]. Therefore, we quantified intracellular IFN-γ expression in NK cells co-cultured with benralizumab-stimulated eosinophils. Although IFN-γ expression was upregulated in CD56+ NK cells in the presence of benralizumab-treated eosinophils, its levels were not changed by adding macrophages to the cell mix, suggesting that NK cell activation by benralizumab-treated eosinophils occurred upstream of macrophage activation under our experimental conditions (figure 7a).
FIGURE 7.

Natural killer (NK)-derived interferon-γ (IFN-γ) stimulates tumour necrosis factor (TNF)-dependent macrophage cytotoxicity in the presence of benralizumab-stimulated eosinophils (Eos). a) Quantification of IFN-γ expression in NK cells upon benralizumab treatment, represented as percentage of total CD56+ CD94+ cells (n=4). b) Quantification of TNF expression in macrophages in the presence of eosinophils after stimulation with 10 ng·mL−1 IFN-γ, represented as percentage of total CD163+ CD11b+ macrophages (n=4). c) Quantification of the induced expression of TNFR1 on eosinophils in the presence of macrophages after stimulation with 10 ng·mL−1 IFN-γ, represented as percentage of total CD66b+ Siglec-8+ cells (n=4). Data are mean±sem. Two-way ANOVA with Tukey's multiple comparisons. ***: p<0.001, benralizumab versus parent fucosylated anti-interleukin-5 receptor α (PF anti-IL-5Rα). +++: p<0.001; ++++: p<0.0001, 10 ng·mL−1 IFN-γ+Eos+Mac versus Eos+Mac. Statistical values are presented in supplementary table S2.
Given that short-term treatment with IFN-γ can enhance both ADCC and ADCP by macrophages [17], we investigated the direct effect of IFN-γ on benralizumab-mediated macrophage responses in the presence of eosinophils and/or NK cells. Stimulation with IFN-γ induced significant TNF expression in activated CD163high macrophages, co-cultured with benralizumab-stimulated eosinophils, comparable with those observed in the presence of NK cells (figure 7b). Under these conditions, TNFR1 expression was also induced in CD66b+ Siglec-8+ eosinophils, reaching comparable levels to NK cells co-cultured with macrophages (figure 7c). Taken together, our data confirmed the stimulatory effect of NK-derived IFN-γ upstream of TNFR1-mediated eosinophil apoptosis by macrophage-derived TNF.
Lastly, we investigated whether complement activation could also contribute to benralizumab-mediated eosinophil depletion in vitro. Benralizumab did not induce complement-dependent cytotoxicity of eosinophils, most likely due to the low copy number of IL-5Rα on the surface of eosinophils (supplementary figure S3).
Discussion
Although eosinophils have been associated with host defence against certain infections [25, 26], their elevated peripheral or tissue levels are linked to chronic inflammatory diseases, notably severe asthma, eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome), hypereosinophilic syndrome and many more [2, 3, 5]. The intricate dependence of eosinophils on IL-5 for their survival and maturation has encouraged the development of several immunotherapies directed against the IL-5 pathway [4], including the IL-5Rα-targeting eosinophil-depleting antibody benralizumab [4, 7, 10]. Elucidating benralizumab's mechanism of action has been challenging due to its lack of cross-reactivity to murine IL-5Rα and differences in immunoglobulin Fc receptor biology between human and mouse. However, by employing relevant primary human autologous cell co-cultures and real-time-lapse imaging combined with flow cytometry, we discovered new mechanisms that may contribute to benralizumab's potent anti-eosinophilic activity in vivo (figure 8). For the first time we “visualised” the stepwise processes deployed by benralizumab's anti-eosinophilic activity and also demonstrated that α-fucosylation of benralizumab was central to all these activities, as evidenced by its enhanced potency in comparison with its parent fucosylated anti-IL-5Rα control.
FIGURE 8.

Proposed mechanisms for benralizumab-induced eosinophil (Eos) depletion through antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP) and tumour necrosis factor-α (TNF-α)-dependent macrophage cytotoxicity. 1) In the ADCC process, benralizumab induced eosinophil apoptosis by activated natural killer (NK) cells. 2) Also, benralizumab promoted ADCP by macrophages to deplete eosinophils either by phagocytosis or efferocytosis. 3) Activated NK cells release stimulatory factors such as interferon-γ (IFN-γ) to induce macrophage cytotoxicity through TNF-α that can trigger TNF receptor 1 (TNFR1)-dependent eosinophil apoptosis; these apoptotic cells are removed by macrophage efferocytosis. Lastly, complement-dependent cytotoxicity (CDC) seems not to be involved in benralizumab-mediated eosinophil depletion. IL-5Rα: interleukin-5 receptor α.
In NK-mediated ADCC assays, we previously documented NK cytotoxicity mediating eosinophil apoptosis induced by benralizumab. Here, we further substantiate our initial finding with the upregulation of cytochrome c and caspase activity in benralizumab-targeted eosinophils, as well as associated morphological changes in eosinophils, starting with the formation of an eosinophil/NK cell synapse and ultimately resulting in eosinophil death. This synapse may induce NK cytotoxicity, where stimulator signals can bypass the role of inhibitor receptors expressed on NK cells, namely killer immunoglobulin-like receptors (KIRs) [27]. Such mechanisms may explain the potency of benralizumab-mediated eosinophil depletion observed in good responders despite the increased KIR expression on NK cells in severe eosinophilic asthmatic subjects [28].
In addition, our data highlight new mechanisms whereby benralizumab can induce eosinophil death by macrophages through two distinct processes involving either the direct uptake of eosinophils by macrophage phagocytosis/efferocytosis (ADCP) or the induction of TNFR1-mediated eosinophil apoptosis via macrophage-derived TNF. The latter being further enhanced by NK cell-derived IFN-γ. Although TNF/TNFR1 has previously been reported to enhance eosinophil survival through NF-κB activation [11, 29] via the JNK/AP-1 pathway [30], inhibition of this pathway was demonstrated to unmask the ability of TNF to induce eosinophil apoptosis [31]. Here, we demonstrated that increased TNFR1 expression was associated with upregulation of several hallmarks of cell death by apoptosis, particularly cytochrome c, membrane depolarisation and Caspase-3/7 activity under our conditions, thus arguing for the direct contribution of TNFR1 in eosinophil apoptosis, seemingly via NF-kB inhibition or activation of pro-apoptotic pathways. To validate whether benralizumab's anti-eosinophilic activities are relevant in severe eosinophilic asthma patients, we confirmed the expression of TNFR1 and IL-5Rα on eosinophils, CD16 on NK cells and macrophages, as well as benralizumab-mediated eosinophil depletion in vitro (unpublished data). In association with elevated levels of granzyme B and IFN-γ, a complete eosinophil depletion by benralizumab was demonstrated in sputum of patients with severe prednisone-dependent asthma [28, 32]; this argues for the involvement of the aforementioned effector mechanisms in the airways of severe asthma patients.
Although TNF is considered a trophic factor delaying eosinophil apoptosis in asthma [29, 33], here we showed that induced expression of TNF through the immune synapse, directed by benralizumab between macrophages and eosinophils, enhanced TNFR1-mediated eosinophil pro-apoptotic signals in vitro. While TNF-blocking immunotherapies such as golimumab failed to demonstrate a favourable risk–benefit profile in severe asthma and exhibited a limited effect reserved to a small subgroup [34], benralizumab appears to deploy several mechanisms for depleting eosinophils in severe asthma, including the newly identified TNF/TNFR1-mediated eosinophil apoptosis through benralizumab-directed immune synapses, thus extending its potent anti-eosinophilic activities in the clinic. Further investigations are warranted to elucidate the downstream signalling of the TNF/TNFR1 pathway promoting eosinophil apoptosis in response to benralizumab treatment. In addition, ADCP of eosinophils by macrophages may represent an important mechanism for the “silent” removal of eosinophils from tissues, as evidenced in vitro by real-time imaging (supplementary videos S6–S10). Our findings are aligned with the clinical observation that benralizumab-mediated eosinophil depletion coincided with reduced free eosinophilic granules, such as eosinophil-derived neurotoxin and eosinophil cationic protein, in serum of severe asthma patients [35]. However, in a recent study in patients with hypereosinophilic syndrome, Kuang et al. [36] described symptoms that included fever, chills, headache, nausea and fatigue, starting ∼6 h after the first dose of benralizumab. These self-limited episodes did not recur with subsequent doses. Interestingly, pre-treatment numbers of eosinophils and NK cells and the activation status of NK cells were not associated with the development of post-treatment reactions, indicating that tissue localisation and the participation of other effector mechanisms, like those described in the current article, may govern the appearance of such transient reactions in patients.
The extent to which benralizumab deploys the described mechanisms (figure 8) in patient tissues remains unknown, and is most likely dependent on the degree of inflammation, type of tissue and presence of effector cells. In addition to eosinophils, a recent paper reported benralizumab-mediated depletion of new target cells, namely IL-5Rα-expressing type 2 innate lymphoid cells, a major source of IL-5 [28]. Other studies identified the presence of autoimmune complexes in uncontrolled severe asthmatic subjects that may contribute to an airway pro-inflammatory environment resulting in enhanced IFN-γ and TNF levels [37, 38], thus allowing us to speculate that autoimmune-dependent mechanisms as well as infections may facilitate benralizumab-mediated eosinophil depletion in severe eosinophilic asthma.
We cannot rule out the possibility that additional effector cells expressing CD16, such as neutrophils and dendritic cells, may contribute to benralizumab's anti-eosinophilic activity in tissues [39, 40]. Also, genetic polymorphisms in the gene encoding CD16 may modulate its activity [41]. These questions require further investigations. Because afucosylated monoclonal antibodies, such as benralizumab, represent an emerging class of powerful therapeutics [42], understanding their effector functions is highly relevant beyond benralizumab itself, with implications for other cell-depleting antibodies in the clinic.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material. ERJ-04306-2020.Supplement (313.1KB, pdf)
Supplementary video S1. Videos S1, S2 and S3 are related to figure 1. Real-time videos of benralizumab inducing caspase-dependent eosinophil apoptosis through ADCC. Primary human NK cells and eosinophils were isolated from healthy donors and green-labelled individually. NK cells were loaded with green LysoTracker. To live image benralizumab-mediated ADCC, red-labelled eosinophils were pretreated with 1 μg·mL−1 of parent fucosylated anti-IL-5Rα (video S1) or benralizumab (video S2), then mixed with green-labelled NK cells and PO-PRO-1 for 4 h. Afterwards, mixed cells were live imaged for 2 h with confocal microscopy. Images were acquired every 2 or 5 min. ERJ-04306-2020.Video_S1 (15.1MB, mov)
Supplementary video S2. ERJ-04306-2020.Video_S2 (16.2MB, mov)
Supplementary video S3. Real-time video illustratingcaspase activation. CellEvent Caspase-3/7 reagent was added prior to live cell acquisition. Caspase-positive cells are depicted in white. ERJ-04306-2020.Video_S3 (7.4MB, mp4)
Supplementary video S4. Videos S4 and S5 are related to figure 2. Real-time videos of benralizumab-mediatied eosinophil ADCC by NK cells. Video S4 highlights the biological check points in assessing NK cell (green) killing of eosinophils (red), starting with the immunological synapse, release of NK-green granules into red-esosinophilic cytosol and ending with eosinophil membrane blebbing and loss of cell integrity, also depicted in video S5. Green-labelled NK lytic granules converged at the immunological synapse and docked at the plasma membrane of red-labelled eosinophils that underwent apoptosis as evidenced by the incorporations of PO-PRO-1 blue nuclear dye. Supplementary video S4. ERJ-04306-2020.Video_S4 (9.2MB, avi)
Supplementary video S5. ERJ-04306-2020.Video_S5 (1.5MB, mp4)
Supplementary video S6. Videos S6 and S7 are related to figure 3. Real-time videos of benralizumab-mediated eosinophil apoptosis via ADCP. To live image benralizumab-mediated ADCP (videos S6 and S7), primary monocytes were differentiated into macrophages for 6 days. Then, eosinophils were isolated from the autologous healthy donors and labelled individually. After pretreatment with 1 μg·mL−1 of benralizumab, far-red-labelled eosinophils (white colour) were mixed with green-labelled macrophages and blue nuclear dye PO-PRO-1. Afterwards, mixed cells were live imaged for 6 h with confocal microscopy. Images were acquired every 5 min. ERJ-04306-2020.Video_S6 (6.4MB, mp4)
Supplementary video S7. ERJ-04306-2020.Video_S7 (2.1MB, mp4)
Supplementary video S8. Videos S8, S9 and S10 are related to figure 4. Real-time videos highlighting NK enhancement of benralizumab-mediated macrophage phagocytosis/efferocytosis, and NK–macrophage immune synapses. Real-time ideo S8 illustrates the effect of NK (red) on macrophage (green) phagocytic behavior induced by benralizumab. ERJ-04306-2020.Video_S8 (2.4MB, mp4)
Supplementary video S9. Real-time Video S9 demonstrates that NK cells were interacting actively with phagocytic macrophages (green) upon benralizumab treatment. Eosinophils are labelled in white and nuclear dye PO-PRO-1 in blue. ERJ-04306-2020.Video_S9 (4.7MB, mp4)
Supplementary video S10. Real-time video S10 demonstrates that a white-labelled eosinophil incorporating blue dye, a hallmark of apoposis, was cleared by an active green-labeled macrophage (efferocytosis) that was also able to engulf another red-labelled NK cell. ERJ-04306-2020.Video_S10 (23.4MB, mp4)
Shareable PDF
Acknowledgements
We thank Richard N. Hanna (AstraZeneca, Gaithersburg, MD, USA) for his assistance in imaging and John Tillinghast (AstraZeneca, Gaithersburg, MD, USA) for reviewing the statistical testing.
Footnotes
Author contributions: R. Dagher coordinated the project, contributed to the study concept, established the study design, was responsible for data acquisition/analysis/interpretation and drafted the manuscript. V. Kumar performed cytokine measurements/analysis, flow cytometry acquisition/analysis for the TNFR1-mediated apoptosis and helped draft the manuscript. A.M. Copenhaver performed the caspase assay and flow cytometry acquisition/analysis for the ADCC process. M. Ghaedi provided scientific support. J. Boyd helped acquire live images for the ADCC and ADCP processes. S. Gallagher conducted the CDC assay. P. Newbold and A.A. Humbles provided guidance for this study. R. Kolbeck was responsible for the study concept and supervision, obtaining funding, and drafting the manuscript.
Conflict of interest: R. Dagher is employed by and shareholder of AstraZeneca.
Conflict of interest: V. Kumar is employed by and shareholder of AstraZeneca.
Conflict of interest: A.M. Copenhaver is a former employee and shareholder of AstraZeneca, and is employed by and shareholder of Takeda Pharmaceuticals.
Conflict of interest: S. Gallagher is a former employee of AstraZeneca.
Conflict of interest: M. Ghaedi is employed by and shareholder of AstraZeneca.
Conflict of interest: J. Boyd is employed by and shareholder of AstraZeneca.
Conflict of interest: P. Newbold is employed by and shareholder of AstraZeneca.
Conflict of interest: A.A. Humbles is an owner of AstraZeneca shares.
Conflict of interest: R. Kolbeck is an owner of AstraZeneca shares.
Support statement: This work was completely supported by AstraZeneca.
References
- 1.Persson C, Uller L. Theirs but to die and do: primary lysis of eosinophils and free eosinophil granules in asthma. Am J Respir Crit Care Med 2014; 189: 628–633. doi: 10.1164/rccm.201311-2069OE [DOI] [PubMed] [Google Scholar]
- 2.Wilson SJ, Rigden HM, Ward JA, et al. . The relationship between eosinophilia and airway remodelling in mild asthma. Clin Exp Allergy J 2013; 43: 1342–1350. doi: 10.1111/cea.12156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rådinger M, Lötvall J. Eosinophil progenitors in allergy and asthma – do they matter? Pharmacol Ther 2009; 121: 174–184. doi: 10.1016/j.pharmthera.2008.10.008 [DOI] [PubMed] [Google Scholar]
- 4.Matucci A, Maggi E, Vultaggio A. Eosinophils, the IL-5/IL-5Rα axis, and the biologic effects of benralizumab in severe asthma. Respir Med 2019; 160: 105819. doi: 10.1016/j.rmed.2019.105819 [DOI] [PubMed] [Google Scholar]
- 5.Cafone J, Ruffner MA, Spergel JM. The role of eosinophils in immunotherapy. Curr Allergy Asthma Rep 2020; 20: 1. doi: 10.1007/s11882-020-0895-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eger KA, Bel EH. The emergence of new biologics for severe asthma. Curr Opin Pharmacol 2019; 46: 108–115. doi: 10.1016/j.coph.2019.05.005 [DOI] [PubMed] [Google Scholar]
- 7.Jackson DJ, Korn S, Mathur SK, et al. . Safety of eosinophil-depleting therapy for severe, eosinophilic asthma: focus on benralizumab. Drug Saf 2020; 43: 409–425. doi: 10.1007/s40264-020-00926-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolbeck R, Kozhich A, Koike M, et al. . MEDI-563, a humanized anti-IL-5 receptor alpha mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J Allergy Clin Immunol 2010; 125: 1344–1353. doi: 10.1016/j.jaci.2010.04.004 [DOI] [PubMed] [Google Scholar]
- 9.Busse WW, Katial R, Gossage D, et al. . Safety profile, pharmacokinetics, and biologic activity of MEDI-563, an anti-IL-5 receptor alpha antibody, in a phase I study of subjects with mild asthma. J Allergy Clin Immunol 2010; 125: 1237–1244. doi: 10.1016/j.jaci.2010.04.005 [DOI] [PubMed] [Google Scholar]
- 10.Laviolette M, Gossage DL, Gauvreau G, et al. . Effects of benralizumab on airway eosinophils in asthmatic patients with sputum eosinophilia. J Allergy Clin Immunol 2013; 132: 1086–1096. doi: 10.1016/j.jaci.2013.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 2015; 15: 362–374. doi: 10.1038/nri3834 [DOI] [PubMed] [Google Scholar]
- 12.Wu Y, Li JJ, Kim HJ, et al. . A neutralizing antibody assay based on a reporter of antibody-dependent cell-mediated cytotoxicity. AAPS J 2015; 17: 1417–1426. doi: 10.1208/s12248-015-9798-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mace EM, Dongre P, Hsu H-T, et al. . Cell biological steps and checkpoints in accessing NK cell cytotoxicity. Immunol Cell Biol 2014; 92: 245–255. doi: 10.1038/icb.2013.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin GHY, Chai V, Lee V, et al. . TTI-621 (SIRPαFc), a CD47-blocking cancer immunotherapeutic, triggers phagocytosis of lymphoma cells by multiple polarized macrophage subsets. PLoS One 2017; 12: e0187262. doi: 10.1371/journal.pone.0187262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krzewski K, Gil-Krzewska A, Nguyen V, et al. . LAMP1/CD107a is required for efficient perforin delivery to lytic granules and NK-cell cytotoxicity. Blood 2013; 121: 4672–4683. doi: 10.1182/blood-2012-08-453738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs 2015; 7: 303–310. doi: 10.1080/19420862.2015.1011450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi Y, Fan X, Deng H, et al. . Trastuzumab triggers phagocytic killing of high HER2 cancer cells in vitro and in vivo by interaction with Fcγ receptors on macrophages. J Immunol 2015; 194: 4379–4386. [DOI] [PubMed] [Google Scholar]
- 18.Yeap WH, Wong KL, Shimasaki N, et al. . CD16 is indispensable for antibody-dependent cellular cytotoxicity by human monocytes. Sci Rep 2016; 6: 34310. doi: 10.1038/srep34310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon S. Phagocytosis: an immunobiologic process. Immunity 2016; 44: 463–475. doi: 10.1016/j.immuni.2016.02.026 [DOI] [PubMed] [Google Scholar]
- 20.Russell DG, Vanderven BC, Glennie S, et al. . The macrophage marches on its phagosome: dynamic assays of phagosome function. Nat Rev Immunol 2009; 9: 594–600. doi: 10.1038/nri2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ostrowski PP, Grinstein S, Freeman SA. Diffusion barriers, mechanical forces, and the biophysics of phagocytosis. Dev Cell 2016; 38: 135–146. doi: 10.1016/j.devcel.2016.06.023 [DOI] [PubMed] [Google Scholar]
- 22.Weiskopf K, Ring AM, Schnorr PJ, et al. . Improving macrophage responses to therapeutic antibodies by molecular engineering of SIRPα variants. Oncoimmunology 2013; 2: e25773. doi: 10.4161/onci.25773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Temkin V, Levi-Schaffer F. Mechanism of tumour necrosis factor alpha mediated eosinophil survival. Cytokine 2001; 15: 20–26. doi: 10.1006/cyto.2001.0890 [DOI] [PubMed] [Google Scholar]
- 24.Van Schie RC, Verstraten RG, Van de Winkel JG, et al. . Effect of recombinant IFN-gamma (rIFN-gamma) on the mechanism of human macrophage IgG FcRI-mediated cytotoxicity. rIFN-gamma decreases inhibition by cytophilic human IgG and changes the cytolytic mechanism. J Immunol 1992; 148: 169–176. [PubMed] [Google Scholar]
- 25.Ravin KA, Loy M. The eosinophil in infection. Clin Rev Allergy Immunol 2016; 50: 214–227. doi: 10.1007/s12016-015-8525-4 [DOI] [PubMed] [Google Scholar]
- 26.Akuthota P, Wang H, Weller PF. Eosinophils as antigen-presenting cells in allergic upper airway disease. Curr Opin Allergy Clin Immunol 2010; 10: 14–19. doi: 10.1097/ACI.0b013e328334f693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raulet DH, Vance RE. Self-tolerance of natural killer cells. Nat Rev Immunol 2006; 6: 520–531. doi: 10.1038/nri1863 [DOI] [PubMed] [Google Scholar]
- 28.Poznanski SM, Mukherjee M, Zhao N, et al. . Asthma exacerbations on benralizumab are largely non-eosinophilic. Allergy 2021; 76: 375–379. doi: 10.1111/all.14514 [DOI] [PubMed] [Google Scholar]
- 29.Gordy C, Liang J, Pua H, et al. . c-FLIP protects eosinophils from TNF-α-mediated cell death in vivo. PLoS One 2014; 9: e107724. doi: 10.1371/journal.pone.0107724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kankaanranta H, Ilmarinen P, Zhang X, et al. . Tumour necrosis factor-α regulates human eosinophil apoptosis via ligation of TNF-receptor 1 and balance between NF-κB and AP-1. PLoS One 2014; 9: e90298. doi: 10.1371/journal.pone.0090298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujihara S, Ward C, Dransfield I, et al. . Inhibition of nuclear factor-kappaB activation un-masks the ability of TNF-alpha to induce human eosinophil apoptosis. Eur J Immunol 2002; 32: 457–466. doi: [DOI] [PubMed] [Google Scholar]
- 32.Sehmi R, Lim HF, Mukherjee M, et al. . Benralizumab attenuates airway eosinophilia in prednisone-dependent asthma. J Allergy Clin Immunol 2018; 141: 1529–1532. doi: 10.1016/j.jaci.2018.01.008 [DOI] [PubMed] [Google Scholar]
- 33.Kankaanranta H, Lindsay MA, Giembycz MA, et al. . Delayed eosinophil apoptosis in asthma. J Allergy Clin Immunol 2000; 106: 77–83. doi: 10.1067/mai.2000.107038 [DOI] [PubMed] [Google Scholar]
- 34.Wenzel SE, Barnes PJ, Bleecker ER, et al. . A randomized, double-blind, placebo-controlled study of tumor necrosis factor-alpha blockade in severe persistent asthma. Am J Respir Crit Care Med 2009; 179: 549–558. doi: 10.1164/rccm.200809-1512OC [DOI] [PubMed] [Google Scholar]
- 35.Pham T-H, Damera G, Newbold P, et al. . Reductions in eosinophil biomarkers by benralizumab in patients with asthma. Respir Med 2016; 111: 21–29. doi: 10.1016/j.rmed.2016.01.003 [DOI] [PubMed] [Google Scholar]
- 36.Kuang FL, Legrand F, Makiya M, et al. . Benralizumab for PDGFRA-negative hypereosinophilic syndrome. N Engl J Med 2019; 380: 1336–1346. doi: 10.1056/NEJMoa1812185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mukherjee M, Forero DF, Tran S, et al. . Suboptimal treatment response to anti-IL-5 monoclonal antibodies in severe eosinophilic asthmatics with airway autoimmune phenomena. Eur Respir J 2020; 56: 2000117. doi: 10.1183/13993003.00117-2020 [DOI] [PubMed] [Google Scholar]
- 38.Ueki S, Mukherjee M, Nair P. Luminal eosinophil cell death as a biomarker for loss of asthma control? Chest 2020; 157: 1680–1681. doi: 10.1016/j.chest.2020.01.021 [DOI] [PubMed] [Google Scholar]
- 39.Golay J, Valgardsdottir R, Musaraj G, et al. . Human neutrophils express low levels of FcγRIIIA, which plays a role in PMN activation. Blood 2019; 133: 1395–1405. doi: 10.1182/blood-2018-07-864538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmitz M, Zhao S, Deuse Y, et al. . Tumoricidal potential of native blood dendritic cells: direct tumor cell killing and activation of NK cell-mediated cytotoxicity. J Immunol 2005; 174: 4127–4134. [DOI] [PubMed] [Google Scholar]
- 41.Dall'Ozzo S, Tartas S, Paintaud G, et al. . Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res 2004; 64: 4664–4669. doi: 10.1158/0008-5472.CAN-03-2862 [DOI] [PubMed] [Google Scholar]
- 42.Garber K. No added sugar: antibody makers find an upside to “no fucose”. Nat Biotechnol 2018; 36: 1025–1027. doi: 10.1038/nbt1118-1025 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material. ERJ-04306-2020.Supplement (313.1KB, pdf)
Supplementary video S1. Videos S1, S2 and S3 are related to figure 1. Real-time videos of benralizumab inducing caspase-dependent eosinophil apoptosis through ADCC. Primary human NK cells and eosinophils were isolated from healthy donors and green-labelled individually. NK cells were loaded with green LysoTracker. To live image benralizumab-mediated ADCC, red-labelled eosinophils were pretreated with 1 μg·mL−1 of parent fucosylated anti-IL-5Rα (video S1) or benralizumab (video S2), then mixed with green-labelled NK cells and PO-PRO-1 for 4 h. Afterwards, mixed cells were live imaged for 2 h with confocal microscopy. Images were acquired every 2 or 5 min. ERJ-04306-2020.Video_S1 (15.1MB, mov)
Supplementary video S2. ERJ-04306-2020.Video_S2 (16.2MB, mov)
Supplementary video S3. Real-time video illustratingcaspase activation. CellEvent Caspase-3/7 reagent was added prior to live cell acquisition. Caspase-positive cells are depicted in white. ERJ-04306-2020.Video_S3 (7.4MB, mp4)
Supplementary video S4. Videos S4 and S5 are related to figure 2. Real-time videos of benralizumab-mediatied eosinophil ADCC by NK cells. Video S4 highlights the biological check points in assessing NK cell (green) killing of eosinophils (red), starting with the immunological synapse, release of NK-green granules into red-esosinophilic cytosol and ending with eosinophil membrane blebbing and loss of cell integrity, also depicted in video S5. Green-labelled NK lytic granules converged at the immunological synapse and docked at the plasma membrane of red-labelled eosinophils that underwent apoptosis as evidenced by the incorporations of PO-PRO-1 blue nuclear dye. Supplementary video S4. ERJ-04306-2020.Video_S4 (9.2MB, avi)
Supplementary video S5. ERJ-04306-2020.Video_S5 (1.5MB, mp4)
Supplementary video S6. Videos S6 and S7 are related to figure 3. Real-time videos of benralizumab-mediated eosinophil apoptosis via ADCP. To live image benralizumab-mediated ADCP (videos S6 and S7), primary monocytes were differentiated into macrophages for 6 days. Then, eosinophils were isolated from the autologous healthy donors and labelled individually. After pretreatment with 1 μg·mL−1 of benralizumab, far-red-labelled eosinophils (white colour) were mixed with green-labelled macrophages and blue nuclear dye PO-PRO-1. Afterwards, mixed cells were live imaged for 6 h with confocal microscopy. Images were acquired every 5 min. ERJ-04306-2020.Video_S6 (6.4MB, mp4)
Supplementary video S7. ERJ-04306-2020.Video_S7 (2.1MB, mp4)
Supplementary video S8. Videos S8, S9 and S10 are related to figure 4. Real-time videos highlighting NK enhancement of benralizumab-mediated macrophage phagocytosis/efferocytosis, and NK–macrophage immune synapses. Real-time ideo S8 illustrates the effect of NK (red) on macrophage (green) phagocytic behavior induced by benralizumab. ERJ-04306-2020.Video_S8 (2.4MB, mp4)
Supplementary video S9. Real-time Video S9 demonstrates that NK cells were interacting actively with phagocytic macrophages (green) upon benralizumab treatment. Eosinophils are labelled in white and nuclear dye PO-PRO-1 in blue. ERJ-04306-2020.Video_S9 (4.7MB, mp4)
Supplementary video S10. Real-time video S10 demonstrates that a white-labelled eosinophil incorporating blue dye, a hallmark of apoposis, was cleared by an active green-labeled macrophage (efferocytosis) that was also able to engulf another red-labelled NK cell. ERJ-04306-2020.Video_S10 (23.4MB, mp4)
This one-page PDF can be shared freely online.
Shareable PDF ERJ-04306-2020.Shareable (287.6KB, pdf)