

Abstract
Background
Chemotherapy resistance is a primary reason of ovarian cancer therapy failure; hence it is important to investigate the underlying mechanisms of chemotherapy resistance and develop novel potential therapeutic targets.
Methods
RNA sequencing of cisplatin‐resistant and ‐sensitive (chemoresistant and chemosensitive, respectively) ovarian cancer organoids was performed, followed by detection of the expression level of fibrillin‐1 (FBN1) in organoids and clinical specimens of ovarian cancer. Subsequently, glucose metabolism, angiogenesis, and chemosensitivity were analyzed in structural glycoprotein FBN1‐knockout cisplatin‐resistant ovarian cancer organoids and cell lines. To gain insights into the specific functions and mechanisms of action of FBN1 in ovarian cancer, immunoprecipitation, silver nitrate staining, mass spectrometry, immunofluorescence, Western blotting, and Fӧrster resonance energy transfer‐fluorescence lifetime imaging analyses were performed, followed by in vivo assays using vertebrate model systems of nude mice and zebrafish.
Results
FBN1 expression was significantly enhanced in cisplatin‐resistant ovarian cancer organoids and tissues, indicating that FBN1 might be a key factor in chemoresistance of ovarian cancer. We also discovered that FBN1 sustained the energy stress and induced angiogenesis in vitro and in vivo, which promoted the cisplatin‐resistance of ovarian cancer. Knockout of FBN1 combined with treatment of the antiangiogenic drug apatinib improved the cisplatin‐sensitivity of ovarian cancer cells. Mechanistically, FBN1 mediated the phosphorylation of vascular endothelial growth factor receptor 2 (VEGFR2) at the Tyr1054 residue, which activated its downstream focal adhesion kinase (FAK)/protein kinase B (PKB or AKT) pathway, induced the phosphorylation of signal transducer and activator of transcription 2 (STAT2) at the tyrosine residue 690 (Tyr690), promoted the nuclear translocation of STAT2, and ultimately altered the expression of genes associated with STAT2‐mediated angiogenesis and glycolysis.
Conclusions
The FBN1/VEGFR2/STAT2 signaling axis may induce chemoresistance of ovarian cancer cells by participating in the process of glycolysis and angiogenesis. The present study suggested a novel FBN1‐targeted therapy and/or combination of FBN1 inhibition and antiangiogenic drug for treating ovarian cancer.
Keywords: angiogenesis, chemoresistance, Fibrillin‐1, glycolysis, organoid, ovarian cancer, STAT2, VEGFR
Abbreviations
- ECAR
Extracellular acidification rate
- EGFP
Enhanced Green Fluorescent Protein
- EMT
Epithelial‐to‐mesenchymal transition
- FBN1
Fibrillin‐1
- FRET‐FLIM
Fӧrster resonance energy transfer‐fluorescence lifetime imaging
- HUVEC
Human umbilical vein endothelial cells
- IC50
half maximal inhibitory concentration
- IHC
Immunohistochemistry
- IP
Immunoprecipitation
- ISV
Intersegmental vessels
- KO
Knockout
- MS
Mass spectrometry
- OCR
Oxygen consumption rate
- OS
Overall survival
- PET/CT
Positron emission tomography/computed tomography imaging
- PFS
Progression‐free survival
- qRT‐PCR
Quantitative real time PCR
- STAT2
Signal transducer and activator of transcription 2
- SUV
Standard uptake value
- TGF‐β
Transforming growth factor beta
- VEGFR2
Vascular endothelial growth factor receptor 2
1. BACKGROUND
Ovarian cancer is the fifth leading cause of female cancer‐induced death, with the majority of cases diagnosed at an advanced stage of metastatic disease when therapeutic options are limited [1]. The overall mortality has plateaued over the last two decades but remains high worldwide, making ovarian cancer one of the deadliest gynecological malignancies [2, 3]. Worldwide, an estimated 313,959 new ovarian cancer cases and 207,252 resulting deaths occurred in 2020 [4, 5]. The majority of ovarian cancers are epithelial in origin and preferentially treated by cytoreductive surgery followed by cytotoxic platinum and taxane chemotherapy [6]. However, around two‐thirds of patients with ovarian cancer develop resistance to these drugs, ultimately resulting in disease relapse [7]. Moreover, after episodes of recurrent disease, patients often develop chemoresistance, eventually leading to low 5‐year survival rates of 25%‐35% [8, 9]; this highlights the urgent requirement for exploring the chemoresistance of ovarian cancer and developing new potential therapeutic targets to improve treatment outcomes [10].
Fibrillin‐1 (FBN1) belongs to the fibrillin family, which includes FBN1, FBN2, and FBN3, and contributes to the diagnosis of genetically determined diseases such as Marfan syndrome [11] and congenital contractural arachnodactyly [12]. As reported previously, FBN1 protein was a large extracellular matrix glycoprotein that formed thread‐like microfibril filaments, which provided structural support for tissues, formed elastic fibers in the skin and vessels, regulated the availability of the transforming growth factor beta (TGF‐β) protein [13, 14, 15, 16, 17], and modulated the microenvironment [18, 19, 20, 21]. Mutations in the FBN1 gene are known to cause autosomal dominant disorders, Marfan syndrome, and other related diseases of connective tissues, collectively known as type‐1 fibrillinopathies [22, 23]. Recent data suggested that FBN1 was markedly overexpressed in testicular germ cell tumors and contributed to in situ tumor development [22]. Guo et al. [18] reported that methylation modification of FBN1 promoter was a distinctive molecular marker in colorectal cancer, indicating an effective means for its screening. Overexpression of FBN1 was indicative of early recurrence of ovarian cancer patients who were initially platinum‐sensitive [24]. However, the role of FBN1 in ovarian cancer and the underlying molecular mechanisms remain to be established.
In our present study, we established cisplatin‐resistant and ‐sensitive three‐dimensional (3D) organoids of ovarian cancer for investigating chemoresistance. By using ovarian cancer models of organoids, tissues, and cell lines, we examined the expression level of FBN1 and its role in cisplatin‐resistance. Finally, the mechanism of FBN1‐induced chemoresistance was elucidated.
2. MATERIALS AND METHODS
2.1. Clinical data of ovarian cancer patients
After signing informed consent, high‐grade serous ovarian carcinoma samples were gained from patients with epithelial ovarian cancer at Xinhua Hospital, Shanghai Jiaotong University School of Medicine (Shanghai, China) between January 2008 and December 2017. The samples were confirmed by 3 pathologists. Patients with other tumors were excluded. Overall survival (OS) was assessed as the interval time from surgery to death of any reasons or the most recent follow‐up. Progression‐free survival (PFS) was measured from the surgery to disease relapse or progression. PFS shorter than 6 months was considered as chemoresistant; otherwise, it was chemosensitive.
2.2. Organoid formation and culture
The organoids were generated and cultured as follows. Firstly, fresh high‐grade serous ovarian cancer tissues were promptly brought into cell culture room after surgery, and then suspended in Advanced Dulbecco's Modified Eagle's Medium (DMEM)/F12 (10‐092‐CVRC, Corning, Corning, NY, USA) supplemented with 1% penicillin‐streptomycin (15140122, Gibco, Carlsbad, CA, USA). Tumor tissues were carefully sliced into 2‐3 mm pieces and digested at 37°C for 1 h with type IV collagenase (C9407, Sigma‐Aldrich, St. Louis, MO, USA). Subsequently, 70‐μm filters (352350, Corning) were used to filter above samples. Cell pellets were produced by centrifuging for 5 min at 1000 revolutions per minute, washed for 3 times with red blood and then well mixed with growth factor‐reduced Matrigel (CB‐40230C, Corning). When the Matrigel was solidified, 500 μL advanced DMEM/F12 medium containing specific growth factors was added by the modified method of Willert et al. [25] with 10 mmol/L nicotinamide (N0636, Sigma, St. Louis, MO, USA) added. Organoids derived from ovarian cancer patients were cultured at 37°C with 5% CO2. The culture medium was changed every 2 or 3 days.
In our study, chemosensitive organoids were generated from cisplatin‐sensitive primary ovarian cancer tissues. The concentration gradient increasing method was applied to construct the chemoresistant organoids based on the corresponding cisplatin‐sensitive ovarian cancer organoids.
2.3. Generation of FBN1‐knockout (KO) organoids and cell lines
With the aid of CRISPR/Cas9 gene editing system, we successfully built two FBN1‐KO chemoresistant ovarian cancer organoids and OVCA433 cells [American Type Culture Collection (ATCC), Rockville, MD, USA], designated as FBN1‐KO1 and FBN1‐KO2 as described earlier [26, 27]. Sequencing was performed to verify the knockout effect. After cells and organoids were infected with lentivirus packaging system for 72 h, puromycin (2 μg/mL) was used for selection of the stable transgenic models for 3 days. Western blotting was finally conducted to confirm the deficiency of FBN1 protein.
2.4. Plasmid construction and viral infection
For generating stable cell lines, lentiviruses (System Biosciences, San Francisco, CA, USA) harboring scrambled shRNA (shNC) or specific shRNAs against signal transducer and activator of transcription 2 (shSTAT2) were transduced into chemoresistant ovarian cancer organoids and OVCA433 cells. The sequences of shSTAT2 were as follows: 5’‐CCACAUAGCUGAUCUGAAAUU‐3’. Lentiviruses were generated by HEK293T cells with recombinant vectors and pPACK Packaging Plasmid Mix (System Biosciences). shRNA vectors were produced by cloning short hairpin RNA fragments into pSIH‐H1‐Puro (SBI) and overexpression vectors were generated by inserting amplified gene fragments into pCDH (System Biosciences).
2.5. Establishment of chemoresistant ovarian cancer cells and cell culture
The concentration gradient increasing method was used to generate cisplatin‐resistant OVCA433 ovarian cancer cells (OVCA433‐CisR). OVCA433 and OVCA433‐CisR cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum (FBS) (Gibco) at 37°C in 5% CO2.
2.6. Colony formation assay
OVCA433‐CisR cells (1,500 cells/dish) were seeded onto 3.5 cm plates and cultured under 5% CO2 at 37°C for 24 h. The cells were treated with or without drugs at 37°C for 7 days. In this study, 2.5 μg/mL cisplatin (S1166, Selleck, Houston, TX, USA), 20 μmol/L apatinib (B2303, APExBIO, Houston, TX, USA), and 2.5 mmol/L glycolysis inhibitor 2‐deoxy‐d‐glucose (2‐DG, ab142242, Abcam, Cambs, Cambridge, UK) were used. Subsequently, cells were rinsed with phosphate‐buffered saline (PBS, 21‐031‐CVC, Corning) three times, and then stained with 1 mL crystal violet solution for 10 min at 20°C. After a PBS wash, cell colonies were counted.
Organoids were harvested and dissociated into single cells. Subsequently, cells were washed with PBS, resuspended at 2 × 106 cells/mL in Basement Membrane Extract (3533‐010‐02, Amyjet, Wuhan, Hubei, China), seeded onto standard 96‐well cell culture plates in triplicate and cultured for 7 days. Finally, the number of colonies formed in each well was assessed using an Olympus microscope (Shinjuku‐ku, Tokyo, Japan).
2.7. Tube formation assay
Tube formation assay was utilized for angiogenesis examination in vitro with Matrigel (BD Biosciences, Franklin, NJ, USA) following the manufacturer's protocol.
Shortly, 24‐well plate coated Matrigel was prepared in advance, and then 2 × 104 human umbilical vein endothelial cells (HUVECs, ATCC) were inoculated in the plate with 200 μL DMEM medium. After 6 h, tube formation of HUVECs was observed and calculated with an Olympus microscope.
2.8. Immunoprecipitation (IP)
IP was conducted via the Pierce Crosslink Immunoprecipitation kit (26148, Thermo Fisher Scientific, Waltham, MA, USA) in keeping with the manufacturer's protocol. Simply put, ovarian cancer organoids were collected and lysed by IP lysis solution. Then, 1 mg lysates of the organoids were used for this study, and 10 μg anti‐Flag antibody was covalently crosslinked with agarose A/G beads via crosslinker disuccinimidyl suberate. Finally, sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) was performed to test the protein expression and association.
2.9. Mass Spectrometry (MS)
Sediments from organoids or cells were lysed in IP buffer containing ethylenediamine tetraacetic acid and underwent IP. Agarose beads were eluted with FBN1 peptide. After SDS‐PAGE and silver‐staining, MS was conducted as previously described [28]. Subsequently, MS data were acquired from Data Dependent Analysis mode and analyzed with the PLGS 2.4 software (Waters, Shanghai, China). Finally, with the aid of MASCOT search engine, the resulting peak list was searched in the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/genome/).
2.10. Fӧrster resonance energy transfer‐fluorescence lifetime imaging (FRET‐FLIM)
FRET‐FLIM experiments were conducted as described earlier [29]. In short, acceptor proteins (fused to red fluorescent protein) and donor proteins (fused to green fluorescent protein) were expressed from vectors MV3‐C‐OFPSpark (RG80654‐ACR, Sino Biological, Beijing, China) and pCMV3‐C‐GFPSpark (CV026, Sino Biological), separately. Using a Leica TCS SMD FLCS confocal microscope (Weztlar, Frankfurt, Germany), FRET‐FLIM imaging was operated. The experiment was repeated three times.
2.11. Western blotting
Protein extracts from organoids or cell lines were separated by 8%‐12% SDS‐PAGE and electrotransferred to 0.2 μm nitrocellulose membranes. After blocking for 1 h in bovine serum albumin, membranes were incubated with the corresponding primary antibodies (Supplementary Table S1) at 4°C overnight, followed by treatment of secondary antibodies.
2.12. High‐throughput RNA sequencing (RNA‐seq)
The RNA‐seq assay of 6 pairs of human ovarian cancer organoids was operated as previously described [29]. In short, TRIzol reagent (Takara, Osaka, Japan) was used to gain total RNA, following by treatment of VAHTS mRNA Capture Beads (Vazyme, Nanjing, Jiangsu, China) to enrich polyAþ RNA. Next, using VAHTS mRNA‐seq v2 Library Prep Kit (Vazyme), the RNA library was prepared. The genes expressed with P < 0.05 and fold changes > 2 between 2 groups were identified as differentially expressed genes (DEGs). Gene ontology and Kyoto Encyclopedia of Genes and Genomes of DEGs were utilized to assess the association network and molecular biological function. Gene Set Enrichment Analysis (GSEA) was performed with the GSEA V4.0.3 software (Broad Institute, Cambridge, MA, USA; https://www.gseamsigdb.org/gsea/index.jsp).
2.13. Whole‐body 18F‐fluorodeoxyglucos (18F‐FDG) positron emission tomography/computed tomography (PET/CT)
Whole‐body 18F‐FDG PET/CT was conducted as previously described [29]. Patients were fasted for at least 8 h prior to PET/CT technology to generate low level of glucose metabolism (at least 4 h for nude mice). Standard uptake value (SUV) was used to measure metabolic level, and maximum of SUV (SUVmax) was calculated in lesions of adnexal carcinomas. A total of 100 patients with ovarian cancer were tested in this experiment.
2.14. Prokaryotic expression of vascular endothelial growth factor receptor 2 (VEGFR2) D1‐4/glutathione S‐transferase (GST) fusion protein in Escherichia coli (E. coli)
A prokaryotic expression vector for the expression of VEGFR2 D1‐4/GST fusion protein was constructed. The first, second, third, and fourth extracellular domains of human VEGFR2 gene fragment were synthesized and separately subcloned into pGEX4T‐1 vector downstream of the GST fragment, an E. coli expression vector, to construct the recombinant plasmids pGEX4T‐VEGFR D1‐4. The plasmid was transformed into E. coli BL21 (DE3) pLysS and induced to express fusion proteins VEGFR2 D1‐4/GST with isopropyl‐β‐D‐thiogalactoside (IPTG, Takara). The expressed protein was purified by washing with urea and detected by SDS‐PAGE and Western blotting.
2.15. Immunohistochemistry (IHC) assay
Tumor tissues from 261 epithelial ovarian cancer patients were collected for microarrays. IHC slides were analyzed by three independent pathologists who were blinded to the patients’ clinical data. A consensus was reached based on the average value of immunoreactive score (IRS). Paraffin‐embedded epithelial ovarian cancer tissues in Xinhua Hospital were sliced into 3 μm thick sections. Before immunostaining, the specimens were dewaxed using xylene and rehydrated using decreasing concentration of ethanol. After PBS washing three times, the tissues were treated with 0.01 mol/L citrate buffer (C9999, Merck, Darmstadt, Germany) to retrieve antigen, followed by bovine serum albumin (SW3015, Solarbio, Beijing, China) blocking for 30 min at 20°C. Subsequently, tissues were incubated at 4°C overnight with primary antibodies against FBN1 (ab231094, Abcam), p‐VEGFR2 (Tyr1054) (441046, Thermo Fisher Scientific) and p‐STAT2 (Tyr690) (ab53132, Abcam) and subsequently with peroxidase‐labeled anti‐rabbit (or anti‐mouse) secondary antibodies for 1 h at 37°C. IRS was determined as multiplicity of staining intensity and percentage of positively stained cells. The quartile of the multiplicity score was set as the boundary between low and high expression. The staining intensity was classified as negative (0), weak (light yellow, 1), moderate (brownish, 2), and strong (brown, 3); the proportion of positive cells was scored as 0 to 5% (0), 5% to 25% (1), 26% to 50% (2), 51% to 75% (3), and 76% to 100% (4).
2.16. Immunofluorescence assay
In this study, 4% paraformaldehyde (Sangon Biotech, Shanghai, China) was used to fix the specimens including organoids, cells, and tumor tissues for 15 min, and 0.3% Triton X‐100 (Sangon Biotech) was applied for permeation. After blocking with 5% goat serum (Life Technologies, New York, NY, USA) for 1 h at 20°C, the specimens were washed with PBS and incubated with corresponding primary antibodies (Supplementary Table S1) overnight at 4°C, followed by corresponding secondary antibodies [either FITC‐ or Texas red‐conjugated donkey F(ab)2 fragments against mouse IgG or rabbit IgG (Jackson ImmunoResearch Laboratory, West Grove, PA, USA)] and 4’,6‐diamidino‐2‐phenylindole (Life Technologies). Finally, the staining effect was observed and imaged by Leica SP5 confocal fluorescence microscope.
2.17. Quantitative real‐time PCR (qRT‐PCR)
TRIzol reagent was used for RNA extraction, and miScript Reverse Transcription Kit (Qiagen, Frankfurt, Hesse‐Darmstadt, Germany) was utilized for reverse transcription. Primer sequences are displayed in Supplementary Table S2. RNA quantification was measured by QuantiTect Probe RT‐PCR kit (Qiagen) following the condition: an initial denaturation at 95°C for 30 s, followed by 40 cycles of 95°C for 5 s, 55°C for 30 s and 72°C for 30 s.
2.18. Glycolysis assay
Glycolysis was assessed through glucose metabolism associated kits such as Glucose Uptake Colorimetric Assay kit (BioVision, Palo Alto, CA, USA), ATP Assay kit (Sigma), Lactate Colorimetric Assay kit (BioVision), and Amplite Colorimetric NADPH Assay kit (AAT Bioquest, Sunnyvale, CA, USA) following the manufacturers' protocols.
2.19. Cell viability assay
Viability inhibition of organoids and cells were assessed by Cell Counting Kit‐8 (CCK‐8) (Dojindo Laboratories, Bunkyo, Tokyo, Japan). An aliquot of 5 × 103 cancer cells was planted onto 96‐well plates per well. To determine the half maximal inhibitory concentration (IC50) of cisplatin, cells were treated with different concentrations of cisplatin with or without apatinib/2‐DG treatment. To determine the IC50 of apatinib, cells were treated with various concentrations of apatinib with or without cisplatin. After 48 h, cells were carefully treated with 10 μL CCK‐8 solution, which was diluted in DMEM medium supplemented with 10% FBS at 37°C for 2 h. Finally, optical density (OD) was measured at 450 nm by a microplate reader (Thermo Fisher Scientific). All experiments were conducted in triplicate. Half maximal inhibitory concentration (IC50) was calculated as previously described [30].
2.20. Subcellular fractionation
According to the manufacturer's instructions, we performed nuclear and cytosolic fractionation assay in cisplatin‐resistant ovarian cancer cells with the nuclear and cytoplasmic extraction Kit (P0028, Beyotime, Beijing, China). Briefly, chemoresistant ovarian cancer OVCA433 cells were obtained by cell scraper and then treated with cytoplasmic protein extraction reagent. After centrifuging for 5 min at 12,000 ×g at 4°C, cytoplasmic protein was produced in the supernatant. Subsequently, cell precipitation was treated with nuclear protein extraction reagent and centrifuged for 10 min at 12,000 ×g. Finally, the supernatant was collected as cellular nuclear protein.
2.21. Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR)
The ECAR and OCR assays were performed as described previously [29]. For ECAR assay, 25 μL each of 10 mmol/L glucose, 1 mmol/L oligomycin, and 100 mmol/L 2‐DG were used. For OCR analysis, 1 mmol/L oligomycin, 1 mmol/L uncoupler carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (FCCP), and 1 mmol/L rotenone were used. OCR and ECAR values were calculated after normalization to cell number and plotted as mean ± standard deviation (SD).
2.22. Zebrafish angiogenesis study
Zebrafish individuals were fed with or without cisplatin (0.2 mmol/L) and/or apatinib (40 μmol/L) for 24 h. To evaluate blood vessel formation, in vivo, fertilized one‐cell fli1a‐enhanced green fluorescent protein (EGFP) transgenic line embryos were injected with 4 ng FBN1 morpholino plasmid. At 24 h post‐fertilization, embryos were dechlorinated and anesthetized with 0.016% MS‐222 (tricaine methanesulfonate; Sigma). Each zebrafish embryo was mounted with 3% methylcellulose on a depression slide for fluorescence microscopy.
The effect of FBN1 morpholino and the VEGFA expression were determined by qRT‐PCR. The zebrafish embryos were divided into 5 groups (15 embryos per group): negative control with or without cisplatin treatment, FBN1 morpholino with or without cisplatin treatment, and FBN1 morpholino with combination treatment of cisplatin and apatinib.
2.23. Animal tumor model and in vivo imaging
Experimental procedures and animal care were performed following the Guidelines for Animal Experiments and approved by the Ethics Committee at Xinhua Hospital. Cisplatin‐resistant ovarian cancer organoids were harvested and dissociated into single cells. Subsequently, cells were washed with PBS. Tumor models were established by a subcutaneous injection of cells (∼5 × 106) into 25 4‐week‐old female BALB/c nude mice (Department of Laboratory Animals, Xinhua Hospital). The nude mice were divided into 5 groups (5 mice per group): negative control with or without 3 mg/kg cisplatin treatment, FBN1 knockout with or without 3 mg/kg cisplatin treatment, and FBN1 knockout with combination treatment of 3 mg/kg cisplatin and 50 mg/kg apatinib. Tumor volumes were measured 3 times a week and calculated using the following formula: V (volume) = L (length) × W (width)2. Once an average tumor volume of 100 mm3 was reached, mice were intraperitoneally administered with drugs every 3 days. Mice were ultimately euthanized with carbon dioxide (CO2) when they were in a state of obvious cachexia or the diameter of tumor ulcer was greater than 0.5 cm. All tumors were dissected and weighed. Intraperitoneal tumors of nude mice were quantified using a luminescence imaging system.
2.24. Enzyme‐linked immunosorbent assay (ELISA) assay
Human VEGFA concentration was tested by ELISA kit (ab119566, Abcam) according to the manufacturer's instructions. The OD value was measured at 450 nm using a microplate reader (Sunrise, TECAN, Männedorf, Switzerland).
2.25. Databases and online survival analysis platform
We examined the expression of FBN1, FBN2, and FBN3 in ovarian cancer in the Oncomine website (https://www.oncomine.org/resource/login.html) and analyzed the survival using the Gene Expression Omnibus (GEO) database via an online survival analysis platform (http://kmplot.com/analysis) [31]. The data of FBN1 in patients with gastric, breast, lung, and bladder cancers were also obtained from the GEO database.
2.26. Statistical analysis
Data were analyzed by GraphPad Prism (Version 8.0, GraphPad Software, San Diego, CA, USA) and are presented as means ± SD. Paired t‐test or one‐way analysis of variance (ANOVA) was performed to analyze the significant difference between experimental group (FBN1 knockout/STAT2 knockdown cells or organoids) and the control (cells or organoids without gene knockout or knockdown). Kaplan‐Meier method with log‐rank analysis was utilized to estimate survival. P < 0.05 was considered significant. SPSS software (Version 24.0, IBM, Chicago, IL, USA) was used for statistical analysis.
3. RESULTS
3.1. FBN1 was significantly highly expressed in cisplatin‐resistant ovarian cancer organoids and tissues
To probe into the crucial molecular factors of chemoresistance in ovarian cancer and investigate its potential underlying mechanism, we generated 10 pairs of cisplatin‐sensitive and ‐resistant ovarian cancer organoids, which were confirmed by CCK‐8 cell viability assay after treatment of cisplatin (Figure 1A‐B). Subsequent high‐throughput sequencing technology was used to discover the genes associated with chemoresistance in 6 cisplatin‐sensitive and 6 ‐resistant ovarian cancer organoids. Finally, we identified 2808 DEGs, including 1532 up‐regulated genes and 1276 down‐regulated genes (Supplementary Figure S1A‐B). Gene ontology assay displayed that metabolic process and angiogenesis were associated with the cisplatin‐resistance. Interestingly, GSEA analysis indicated that FBN family was particularly significantly enriched in cisplatin‐resistant ovarian cancer organoids (Figure 1C, Supplementary Table S3). To detect the clinical significance of FBN family members in ovarian cancer, we performed survival analysis using the public GEO database and Xinhua hospital dataset. We found that only high expression of FBN1 was significantly associated with OS and PFS in ovarian cancer samples from both GEO database and Xinhua Hospital (Supplementary Figure S1C‐F). Additionally, high FBN1 expression was negatively associated with OS in patients with other tumor types (Supplementary Figure S1G). Immunofluorescence and RT‐PCR assays further verified that FBN1 was highly expressed in cisplatin‐resistant ovarian cancer organoids (Figure 1D‐E). Subsequently, we surveyed whether the expression of the FBN1 protein was upregulated in cisplatin‐resistant tissues. An independent patient cohort (132 cisplatin‐resistant and 129 cisplatin‐sensitive) was enrolled to perform IHC assays. Clinical data of the patients with ovarian cancer are summarized in Supplementary Table S4. The expression of FBN1 protein was drastically upregulated in cisplatin‐resistant ovarian cancer tissues compared with that in cisplatin‐sensitive specimens (Figure 1F), and FBN1 expression was elevated in 80.3% cisplatin‐resistant tumor samples, compared to 52.7% cisplatin‐sensitive ovarian cancer tissues (Figure 1G). Consistently, the H‐score of FBN1 and FBN1 mRNA level in cisplatin‐resistant ovarian cancer tissues was significantly higher than those in cisplatin‐sensitive samples (Figure 1H‐I), which was confirmed by immunofluorescence assay (Figure 1J). These findings suggest that high expression of FBN1 contributes to chemoresistance and poor outcome of patients with ovarian cancer.
FIGURE 1.
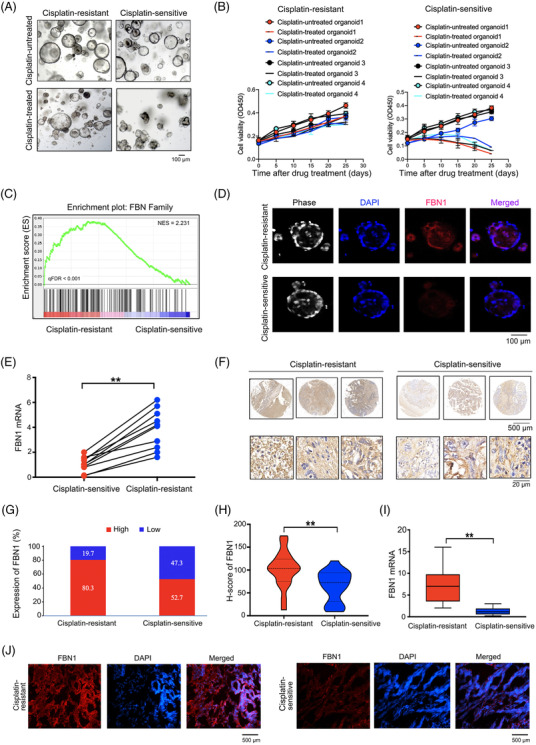
FBN1 expression level is associated with cisplatin‐resistance in patients with ovarian cancer. (A) Images of cisplatin‐resistant and ‐sensitive organoids derived from ovarian cancer patients with or without 5 μg/L cisplatin treatment for 21 days. (B) Cell viability assay of organoids treated with 5 μg/L cisplatin in different time intervals. The data of 4 pairs of ovarian cancer organoids are shown. Data are presented as mean ±SD of triplicate measurements repeated three times. (C) GSEA analysis was performed using 6 cisplatin‐resistant and 6 ‐sensitive organoids derived from ovarian cancer patients. (D) Immunofluorescence assay was performed to detect the association between FBN1 and cisplatin‐sensitive ovarian cancer organoids. (E) FBN1 mRNA expression in 10 pairs of cisplatin‐resistant and ‐sensitive organoids of ovarian cancer. (F) Representative images of immunohistochemistry staining of FBN1 in 132 cisplatin‐resistant and 129 ‐sensitive ovarian cancer tissues. (G) High‐low expression ratio of FBN1 in cisplatin‐resistant and ‐sensitive ovarian cancer tissues. (H) H‐score of FBN1 in cisplatin‐resistant and ‐sensitive ovarian cancer tissues. (I) qRT‐PCR analysis of FBN1 mRNA expression in 45 pairs of cisplatin‐resistant and ‐sensitive ovarian cancer tissues. (J) Immunofluorescence of FBN1 in cisplatin‐resistant and ‐sensitive ovarian cancer tissues. Red signals, FBN1; blue signals, DAPI. **, P < 0.01. Abbreviations: FBN1, fibrillin‐1; SD, standard deviation; GSEA, Gene Set Enrichment Analysis; qRT‐PCR, quantitative real‐time PCR; DAPI, 2‐(4‐Amidinophenyl)‐6‐indolecarbamidine dihydrochloride
3.2. FBN1 promoted cisplatin‐resistance in ovarian cancer by regulating glycolysis and angiogenesis
To further illustrate the function of FBN1 on chemoresistance of ovarian cancer, we established cisplatin‐resistant ovarian cancer cell lines by increasing cisplatin concentration gradient (OVCA433‐CisR, Supplementary Figure S2A). By performing Western blotting assay, we found that FBN1 was elevated in OVCA433‐CisR cells (Supplementary Figure S2B). Subsequently, we constructed two FBN1‐KO ovarian cancer organoids and cell lines (CR‐organoid/FBN1‐KO1/2 and OVCA433‐CisR/FBN1‐KO1/2) and the controls (CR‐organoid/NC and OVCA433‐CisR/NC) (Figure 2A). By performing CCK8 and colony formation assays, we found that FBN1 KO significantly increased cisplatin sensitivity (Supplementary Figure S2C‐H). Next, on the basis of RNA‐seq, the expression of key regulatory genes enriched in glycolysis and angiogenesis was shown to be significantly decreased in CR‐organoid/FBN1‐KO1 as compared to that in the control CR‐organoid (Figure 2B, Supplementary Table S5). MS experiments validated that FBN1 deficiency significantly affected genes and pathways involved in glycolysis and angiogenesis in ovarian cancer organoids (Figure 2C‐D) along with a decrease in metabolites of glycolysis (Figure 2E). The expression of genes involved in glycolysis and angiogenesis, such as fructose‐bisphosphate aldolase A (ALDOA), glucose transporter 1 (GLUT1), vascular endothelial growth factor A (VEGFA), and angiopoietin‐1 (Ang‐1), was significantly decreased, as determined by RT‐PCR. Notably, levels of key genes of the FAK/AKT1 pathway were attenuated in FBN1‐KO ovarian cancer organoids and cells (Figure 2F).
FIGURE 2.
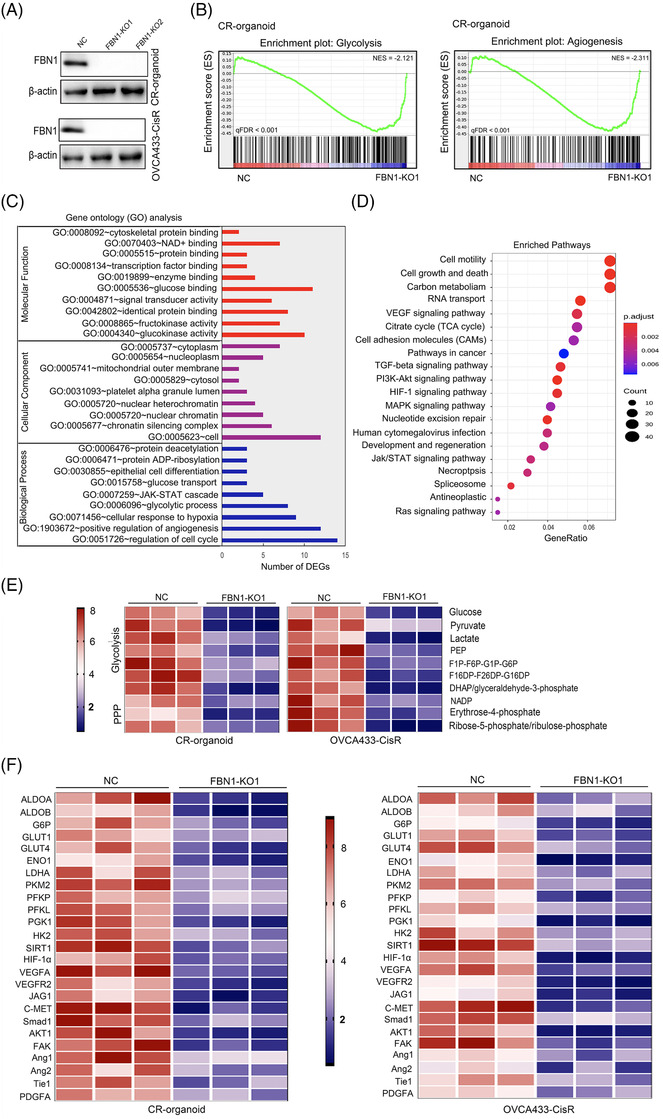
FBN1 knockout decreases glycolysis and angiogenesis. (A) FBN1‐knockout cisplatin‐resistant ovarian cancer organoids and cell lines were verified by Western blotting. (B) GSEA analysis was performed using FBN1‐knockout and control cisplatin‐resistant ovarian cancer organoids (CR‐organoid/FBN1‐KO1 and NC). The signature was defined by genes showing significant expression changes. (C) Gene ontology of mass spectrum analysis in CR‐organoid/FBN1‐KO1 and CR‐organoid/NC. (D) Pathway examination of mass spectrum analysis in CR‐organoid/FBN1‐KO1 and CR‐organoid/NC. (E) Metabolites in pathways of glucose metabolism. (F) mRNA levels of genes altered by FBN1 knockout in cisplatin‐resistant ovarian cancer organoids and cell lines. Abbreviations: FBN1, fibrillin‐1; GSEA, Gene Set Enrichment Analysis; CR, cisplatin‐resistant; KO, knockout; NC, negative control
Subsequently, we applied cellular glycolysis assays to determine the influence of FBN1 on glycolysis in chemoresistant ovarian cancer. Glycolytic phenotypes of ovarian cancer organoids and cells were markedly decreased by FBN1 KO (Figure 3A–C). To further ascertain the effects of FBN1 on angiogenesis, the tube formation assay was performed using HUVECs treated with cellular supernatant from OVCA433‐CisR/FBN1‐KO1/2 and control cells. Tube formation of HUVECs in FBN1‐KO groups was significantly inhibited (Figure 3D), supporting important roles of FBN1 in glycolysis and angiogenesis in ovarian cancer. Apatinib, a tyrosine kinase inhibitor, targets VEGFR2 and inhibits glycolysis to exert anti‐tumor effects [32]. Here, we demonstrated that the combination of FBN1‐knockout and apatinib increased cisplatin sensitivity more significantly, compared with FBN1‐knockout or apatinib treatment alone (Figure 3E‐G). Moreover, our study showed that the glycolysis inhibitor 2‐DG enhanced cisplatin sensitivity in cisplatin‐resistant ovarian cancer organoids and cells, as determined by CCK‐8 and colony formation (Figure 3H‐J). Our collective results indicate that FBN1 deficiency induces anticancer effects through inhibition of glycolysis and angiogenesis in ovarian cancer organoids and cells.
FIGURE 3.
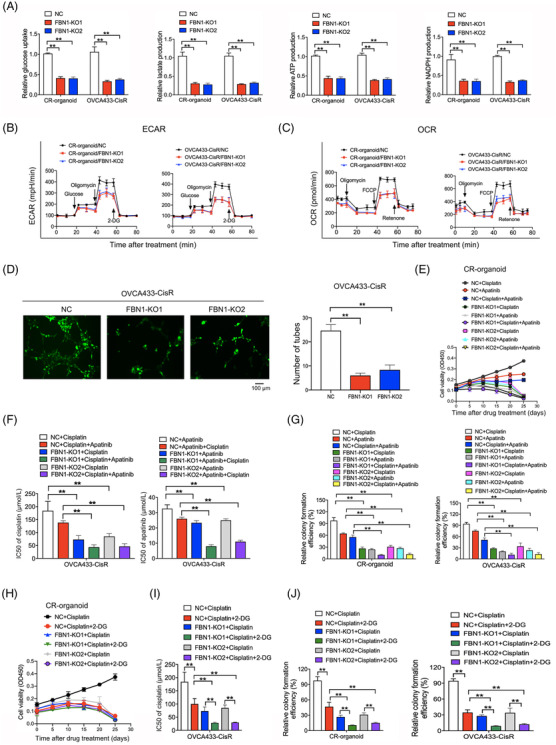
FBN1 knockout inhibits glycolysis and angiogenesis, leading to increased cisplatin sensitivity in cisplatin‐resistant ovarian cancer organoids and cells. (A) Glucose uptake, lactate, ATP and NADPH production in cisplatin‐resistant ovarian cancer organoids and cell line. Data are presented as mean ± SD of triplicate measurements repeated three times. (B‐C) ECAR (B) and OCR (C) in cisplatin‐resistant ovarian cancer organoids and cells. (D) Effect of FBN knockout on HUVEC tube formation. HUVEC cells were treated with supernatant obtained from OVCA433‐CisR/FBN1‐KO1, OVCA433‐CisR/FBN1‐KO2, or the corresponding control cells. (E) Cell viability assay of organoids treated with 5 μg/L cisplatin and/or 20 μmol/L apatinib in different time intervals. (F) IC50 values of cisplatin for FBN1‐knockout and control ovarian cancer cells treated with different concentrations of cisplatin for 48 h with or without 20 μmol/L apatinib; IC50 values of apatinib for the cells treated with various concentrations of apatinib for 48 h with or without 2.5 μg/mL cisplatin. (G) Relative colony formation efficiency of cisplatin‐resistant ovarian cancer organoids and cells treated without drugs or with 2.5 μg/mL cisplatin and/or 20 μmol/L apatinib for 7 days. (H) Cell viability assay of organoids treated with 5 μg/L cisplatin alone or in combination with 2.5 mmol/L 2‐DG in different time intervals. (I) IC50 values of cisplatin for ovarian cancer cells treated with different concentrations of cisplatin with or without 5 mmol/L 2‐DG for 48 h. (J) Relative colony formation efficiency of cisplatin‐resistant ovarian cancer organoids and cells treated with 2.5 μg/mL cisplatin alone or in combination with 2.5 mmol/L 2‐DG for 7 days. **, P < 0.01. Abbreviations: FBN1, fibrillin‐1; SD, standard deviation; CR: cisplatin‐resistant; KO, knockout; NC, negative control; ECAR, extracellular acidification rate; OCR, oxygen consumption rate; HUVECs, human umbilical vein endothelial cells. IC50, half maximal inhibitory concentration; 2‐DG, 2‐deoxy‐D‐glucose
3.3. FBN1 regulated the VEGFR2/STAT2 pathway to affect glycolysis and angiogenesis in chemoresistant ovarian cancer organoids and cells
To identify the interacting proteins of FBN1 in chemoresistant ovarian cancer, IP and MS analyses were conducted in cisplatin‐resistant organoids with high FBN1 expression. Finally, 53 proteins were detected (Supplementary Table S6). Interestingly, VEGFR2, a key regulator of angiogenesis, was identified as a molecule that binds to the FBN1 protein, which was confirmed by the following co‐IP assay (Figure 4A) and FRET‐FLIM (Figure 4B‐C). Hence, VEGFR2 could be considered as a molecule that directly interacts with FBN1. Tyrosine 1054 residue (Tyr1054) of VEGFR2 was identified as a phosphorylation site by MS, which was further verified by Western blotting and immunofluorescence experiments in cisplatin‐resistant ovarian cancer organoids and cells (Figure 4D‐E). On the basis of the abovementioned findings, downstream molecules of the VEGFR2‐mediated signaling pathway were assessed by Western blotting; the results revealed a significant reduction in the expression of proteins involved in the FAK/AKT1 pathway (Figure 4D) and that VEGFR2 overexpression activated AKT1 again (Figure 4F).
FIGURE 4.
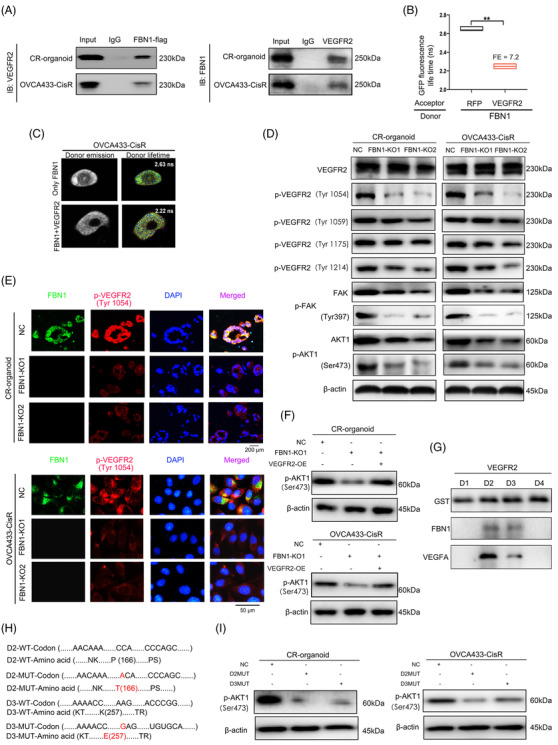
FBN1 combines directly with VEGFR2 in ovarian cancer cells. (A) Co‐IP assay of interactions between FBN1 and VEGFR2 proteins in cisplatin‐resistant ovarian cancer organoids and OVCA433‐CisR cells. (B‐C) Interactions between FBN1 and VEGFR2 confirmed by FRET‐FLIM upon transient co‐expression in OVCA433‐CisR cells. **, P < 0.01. (D) Western blotting assay for determining the relationship between FBN1 and VEGFR2, p‐VEGFR2, downstream molecules of VEGFR2‐mediated signaling in ovarian cancer cells. (E) Immunofluorescence assay for determining the relationship between FBN1 and p‐VEGFR2 (Tyr1054) in cisplatin‐resistant ovarian cancer organoids and cell lines. Green signals, FBN1; red signals, p‐VEGFR2 (Tyr1054); blue signals, DAPI. (F) Western blotting assay of p‐AKT1 (Ser473) in cisplatin‐resistant ovarian cancer organoids and OVCA433‐CisR cells. (G) Co‐IP assay demonstrated an interaction between VEGFR2 domains 2 & 3 and FBN1 protein. Binding of VEGFA and the extracellular domains 2 & 3) of VEGFR2 was used as the positive control. (H) Mutant codons and amino acids of D2 and D3 of VEGFR2. (I) Western blotting assay of p‐AKT1 (Ser473) in cisplatin‐resistant ovarian cancer organoids and OVCA433‐CisR cells. Abbreviations: FBN1, fibrillin‐1; CR, cisplatin‐resistant; KO, knockout; NC, negative control; SDS‐PAGE, sodium dodecyl sulfate‐polyacrylamide gel electrophoresis; AKT, protein kinase B; Co‐IP, co‐Immunoprecipitation; VEGFR2, vascular endothelial growth factor receptor 2; DAPI, 2‐(4‐amidinophenyl)‐6‐indolecarbamidine dihydrochloride; FRET‐FLIM, Fӧrster resonance energy transfer‐fluorescence lifetime imaging. FE, FRET efficiency
VEGFR2 has 7 IgG‐like extracellular domains (D1‐7) [33], and the domains D1‐4 of VEGFR2 are crucial for ligand receptor interaction [34, 35]. A subsequent Co‐IP assay showed an interaction between the VEGFR2 domain and the FBN1 protein, and domains D2 and D3 were verified to be the binding sites (Figure 4G). Further analysis indicated that Proline 166 (P166) of D2 and Lysine 257 (K257) of D3 were the precise mutant sites (Figure 4H), and AKT1 could not be activated if these sites were mutated (Figure 4I).
By performing RNA‐seq of CR‐organoid/FBN1‐KO1 and the control, we observed that the expression of several genes changed significantly, but the STAT family ranked first among the decreased transcription factors (Figure 5A, Supplementary Table S7). RT‐PCR assay further proved the reduction in STAT expression, especially STAT2, in CR‐organoid/FBN1‐KO1/2 and OVCA433‐CisR/FBN1‐KO1/2 as compared to their controls (Figure 5B). Interestingly, we also found that FBN1 KO contributed to significantly less protein aggregation of STAT2 and p‐STAT2 (Tyr690) in the nucleus, but minimal effect on their levels were observed in the cytoplasm (Figure 5C), which was consistent with immunofluorescence results (Figure 5D). To further investigate the factors underlying STAT2 downregulation upon FBN1 KO, we overexpressed VEGFR2 in FBN1‐KO cisplatin‐resistant ovarian cancer organoids and cells and examined the effects on STAT2 and p‐STAT2 (Tyr690). Interestingly, the protein levels of STAT2 and p‐STAT2 (Tyr690) were effectively rescued under the condition of VEGFR2 overexpression (Figure 5E). Next, shRNA vectors of STAT2 were transfected into VEGFR2‐overexpressing organoids and cells. The results of RT‐PCR assay indicated that the expression levels of genes involved in glycolysis and vascular formation pathways, such as ALDOA, Hexokinase 2 (HK2), FAK, and AKT, were enhanced by VEGFR overexpression but attenuated upon STAT2 silencing (Figure 5F). Moreover, VEGFR2 overexpression partially rescued changes in tube formation (Figure 5G), cisplatin sensitivity (Supplementary Figure S3A‐C), and glycolytic level (Supplementary Figure S3D‐F) induced by FBN1 KO, which was effectively reversed following STAT2 knockdown (Supplementary Figure S3).
FIGURE 5.
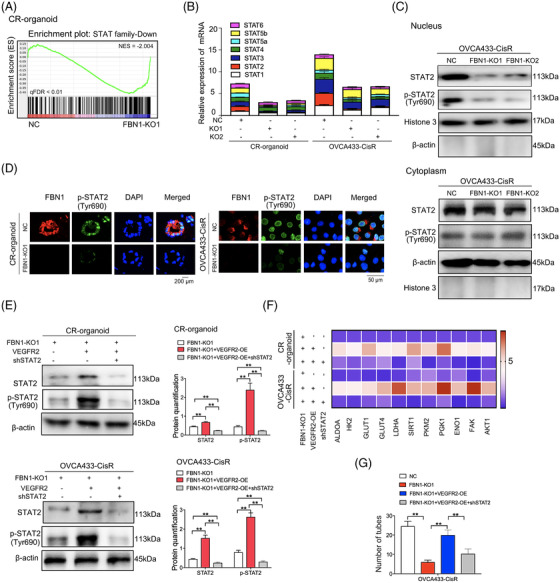
STAT2 is the downstream target molecule of the FBN1/VEGFR2 signaling axis. (A) GSEA analysis was performed using FBN1‐knockout and control cisplatin‐resistant ovarian cancer organoids (CR‐organoids/FBN1‐KO1 and CR‐organoids/NC). The signature was defined by genes showing significant expression changes. (B) Effects of FBN1 knockout on mRNA levels of STAT family members. (C) FBN1 knockout greatly altered the distribution and expression of STAT2 and p‐STAT2 (Tyr690) in cytoplasm and nucleus of OVCA433‐CisR ovarian cancer cells. (D) Immunofluorescence assay to determine the association between p‐STAT2 (Tyr690) and FBN1 in cisplatin‐resistant ovarian cancer organoids and cells. (E) Western blotting assay to detect expression of STAT2 and p‐STAT2 (Tyr690) in cisplatin‐resistant ovarian cancer organoids and OVCA433‐CisR cells. The protein quantification was analyzed by Image J software. (F) mRNA expression of glycolysis and angiogenesis‐associated genes in FBN1‐knockout and control cisplatin‐resistant ovarian cancer organoids and OVCA433‐CisR cells. (G) Effect of FBN1 knockout, VEGFR2 overexpression, and STAT2 knockdown on HUVEC tube formation. HUVEC cells were treated with supernatant obtained from groups of NC, FBN1 KO‐1, FBN1 KO‐1+VEGFR2 OE, and FBN1 KO‐1+VEGFR2 OE+shSTAT2 in OVCA433‐CisR cells. Abbreviations: FBN1, fibrillin‐1; CR, cisplatin‐resistant; KO, knockout; NC, negative control; OE, overexpression; STAT, signal transducer and activator of transcription; VEGFR2, vascular endothelial growth factor receptor 2
Based on the above results, we propose that FBN1 KO leads to sensitivity to cisplatin in ovarian cancer by restraining glycolysis and angiogenesis through modulating VEGFR2/STAT2 axis.
3.4. FBN1 KO suppressed tumor progression and enhanced cisplatin sensitivity in ovarian cancer in vivo
In vivo experiment was subsequently performed to evaluate the effect of FBN1 KO on chemosensitivity. Ovarian cancer CR‐organoids harboring either FBN1 KO or the control were subcutaneously injected into 4‐week‐old female nude mice. The results demonstrated that FBN1 depletion led to a decrease in speed of tumor growth and overall tumor weight either with or without cisplatin treatment; FBN1 KO combined with apatinib and cisplatin further enhanced drug sensitivity compared with cisplatin treatment alone (Figure 6A‐C). Then we performed PET‐CT, FBN1 KO greatly restrained glucose uptake of 18F‐FDG in above mouse models of ovarian cancer, resulting in lower SUVmax (Figure 6D‐E). For vascular formation test in vivo, we established FBN1 morpholino zebrafish development models and finally discovered that FBN1 morpholino suppressed angiogenesis with or without cisplatin treatment (Figure 6F‐G). Moreover, combination of FBN1 morpholino and apatinib restrained vascular formation more significantly. We found that VEGFA mRNA was decreased significantly by FBN1 morpholino (Figure 6H). Our further study demonstrated that FBN1 KO inhibited the expression of vascular endothelial specific marker CD31 (Figure 6I). Subsequent RT‐PCR analysis of in vivo tumors with or without cisplatin treatment showed that levels of glycolysis‐ and angiogenesis‐associated genes, such as ALDOA, HK2, and AKT1, were significantly reduced (Figure 6J‐K). Immunofluorescence assay of in vivo tumors subjected to cisplatin therapy confirmed positive associations among FBN1, p‐VEGFR2 (Tyr1054), and p‐STAT2 (Tyr690) (Supplementary Figure S4). Thus, we demonstrate that FBN1 KO inhibits glycolysis and angiogenesis and promotes cisplatin sensitivity in vivo.
FIGURE 6.
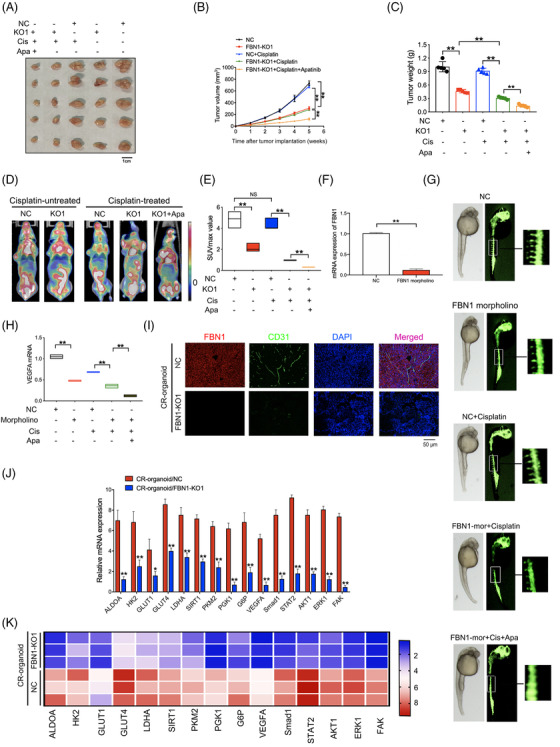
FBN1 knockout inhibits progression of ovarian cancer and sensitizes response to cisplatin in vivo. (A) Images of tumors generated by FBN1‐knockout and control cisplatin‐resistant ovarian cancer organoids with or without cisplatin and/or apatinib. (B) Growth curves of xenograft tumors in mice. (C) Average tumor weights in nude mice. (D) Representative images of PET‐CT used for detection of glucose uptake. Each group contained 5 mice. (E) Average SUVmax values of nude mice bearing tumors. (F) Effect of FBN1 morpholino in zebrafish model was tested by qRT‐PCR. (G) Zebrafish model treated with or without cisplatin (0.2 mmol/L) and/or apatinib (40 μmol/L). (H) VEGFA mRNA in zebrafish models. (I) Immunofluorescence of FBN1 and specific angiogenesis marker CD31 in the xenograft tumors of FBN1 knockout and control groups with cisplatin treatment. (J) qRT‐PCR analysis of the indicated genes in FBN1‐knockout group and the control in nude mouse tumor tissues without drug treatment. Error bars, 95% CIs. *, P < 0.05, **, P < 0.01. (K) Heatmap showing that FBN1‐affected genes are involved in glycolysis and angiogenesis with cisplatin treatment. Abbreviations: FBN1, fibrillin‐1; CR, cisplatin‐resistant; KO, knockout; NC, negative control; OE, overexpression; VEGFA, vascular endothelial growth factor A; qRT‐PCR, quantitative real‐time PCR; CD31, platelet endothelial cell adhesion molecule‐1
3.5. Expression of FBN1, VEGFR2, and STAT2 in human ovarian cancer tissues
Via IHC staining, we confirmed high expression of p‐VEGFR2 (Tyr1054) and p‐STAT2 (Tyr690) in ovarian cancer tissues with high FBN1 expression (Figure 7A). ELISA of ovarian cancer showed that FBN1 expression level was associated with VEGFA concentration (Figure 7B). PET/CT assay was performed to detect tumor dissemination in ovarian cancer patients by measuring glucose uptake. The results demonstrated a lower SUVmax in patients with low FBN1 expression than that in patients with high FBN1 expression (Figure 7C‐D). Interestingly, high SUVmax values were closely related to ovarian cancer patients’ poorer OS (Figure 7E). Application of RT‐PCR to examine the genes involved in glycolysis and angiogenesis revealed that levels of ALDOA, HK2, GLUT4, FAK1, and AKT1 were markedly attenuated in ovarian cancer tissues with low FBN1 expression (Figure 7F). The immunofluorescence assay showed associations among FBN1, p‐VEGFR2 (Tyr1054), and p‐STAT2 (Tyr690) (Figure 7G), consistent with data from ovarian cancer cell lines and mice. Thus, our collective data indicate the participation of the FBN1/VEGFR2/STAT2 signaling pathway in chemoresistance of ovarian cancer (Figure 7H).
FIGURE 7.
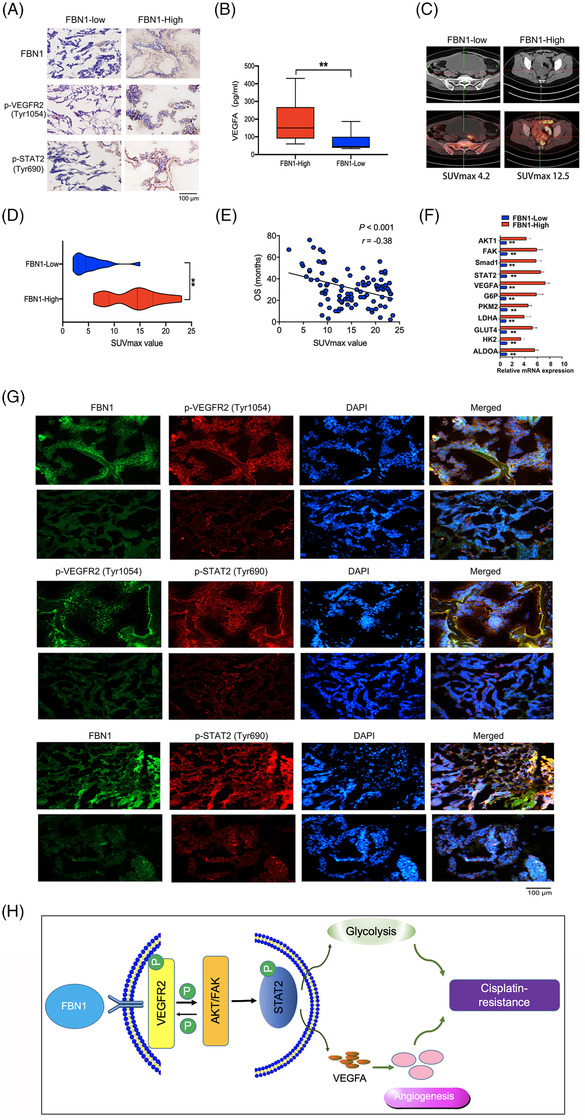
Immunohistochemical staining and immunofluorescence of FBN1, p‐VEGFR2, and p‐STAT2. (A) Representative images of immunohistochemistry of FBN1, p‐VEGFR2 (Tyr1054), and p‐STAT2 (Tyr690) in ovarian cancer tissues. (B) Human serum VEGFA concentration in 50 pairs of ovarian cancer patients measured by ELISA kit. (C‐D) Association between SUVmax of PET/CT technology and FBN1 expression in the lesions of adnexal carcinomas from 100 ovarian cancer patients. (E) The relationship between SUVmax of PET/CT image and overall survival of 100 ovarian cancer patients. (F) mRNA expression levels of genes associated with glycolysis and angiogenesis assessed via qRT‐PCR in 45 pairs of ovarian cancer samples with high or low FBN1 expression. (G) Representative images of immunofluorescence of FBN1, p‐VEGFR2 (Tyr1054), and p‐STAT2 (Tyr641) in ovarian cancer patients’ tissues. Green signals, FBN1 or p‐VEGFR2 (Tyr1054); red signals, p‐VEGFR2 (Tyr1054) or p‐STAT2 (Tyr641); blue signals, DAPI. (H) Schematic model on the proposed role of the FBN1/VEGFR2/STAT2 signaling axis in modulating glycolysis, angiogenesis, and cisplatin sensitivity. Abbreviations: FBN1, fibrillin‐1; ELISA, enzyme‐linked immunosorbent assay; SUVmax, maximum of standardized uptake value; PET‐CT, positron emission tomography‐computed tomography; VEGFA, vascular endothelial growth factor A; STAT2, signal transducer and activator of transcription 2; VEGFR2, vascular endothelial growth factor receptor 2; DAPI, 2‐(4‐amidinophenyl)‐6‐indolecarbamidine dihydrochloride
4. DISCUSSION
The present study clarified a new molecular mechanism of cisplatin‐resistance of ovarian cancer by using models of organoids and cells in vitro and in vivo. Briefly, initial RNA‐seq revealed that FBN1 was highly expressed in cisplatin‐resistant organoids of ovarian cancer; subsequently, FBN1 was verified to be able to regulate glycolysis and angiogenesis through the VEGFR2/STAT2 pathway, which reduces sensitivity to cisplatin.
As is well known, conventional tumor study models such as cells and xenografts were not able to well recapitulate the characteristic and heterogeneity of human cancer, thereby impeding the transformation of scientific research results from bench to bedside. In vitro 3D cultured patient‐derived organoids were generated from self‐organizing stem cells, which have more advantages of imitating human cancer biological processes in vivo, especially in drug‐sensitivity of numerous human cancers [30]. We established cisplatin‐resistant and ‐sensitive ovarian cancer organoids, and used the models to explore the mechanisms of ovarian cancer chemoresistance by in vitro and in vivo experiments. Finally, we identified FBN1 as a key target for ovarian cancer chemotherapy.
Angiogenesis was considered to be a crucial determinant of tumor dissemination and metastasis [36, 37], and antiangiogenic treatments have shown efficacy for epithelial ovarian cancer [38]. Antiangiogenic agents, including anti‐VEGF antibodies and multireceptor tyrosine kinase inhibitors (TKIs, such as VEGFR inhibitors), present an attractive therapeutic option for ovarian cancer [39]. Apatinib is a small‐molecule TKI that selectively binds and inhibits VEGFR2 with encouraging antitumor activity and tolerable toxicity against several malignant tumors, including cisplatin‐resistant ovarian cancer [38, 40‐42]. Moreover, a recent study discovered that apatinib regulated glucose metabolism and suppressed the viability and proliferation of ovarian cancer cells in vitro and in vivo [32], which was verified by our experiments using models of cisplatin‐resistant ovarian cancer organoids. Interestingly, we proved that FBN1 KO in combination with apatinib therapy enhanced the chemosensitivity of ovarian cancer, which implied that FBN1 induced chemoresistance by not only VEGFR2‐mediated pathway but also other tumor‐associated pathways probably.
FBN1, produced by fibroblasts, is an important structural factor of the extracellular matrix, which provides great support to connective tissues [43, 44]. As reported earlier, FBN1 mediated stretching of ligaments, skin, and blood vessels; supported rigid tissues, including muscle, nerve, and lens of the eyes; and regulated TGF‐β bioavailability [45], which induced epithelial‐to‐mesenchymal transition (EMT) in cancers of epithelial origin, thereby stimulating molecular and phenotypical changes that resulted in prometastatic characteristics [46, 47]. Moreover, the activation of TGF‐β is reported to enhance invasion of ovarian cancer [48]. Therefore, the association between FBN1 and TGF‐β in ovarian cancer requires further investigation.
Accumulating studies have shown that FBN1 was highly expressed in some tumor types, such as mammary gland cancer [46] and papillary thyroid carcinoma [49], and participated in tumor progression [49]. Moreover, FBN1 promoted both ovarian tumorigenesis and metastasis in vivo and may serve as a biomarker for predicting recurrence of platinum‐sensitive ovarian cancer [24]. However, the specific roles and mechanisms of action of FBN1 in cisplatin sensitivity of cancers remain to be established. Our experiments have shown that FBN1 KO enhanced cisplatin sensitivity by inhibiting glycolysis and angiogenesis. Moreover, besides high‐grade serous carcinomas, we also analyzed the expression of FBN1 in clear‐cell carcinoma of ovarian cancer and discovered that FBN1 was highly expressed in both tumor types though there was no significant difference between them (data not shown).
Aerobic glycolysis, known as Warburg effect, is an anomalous phenomenon of tumor cell metabolism, which is associated with progression of multiple human cancers [50] and resistance to chemotherapy [51, 52]. Now, glycolysis has been proved to be associated with ovarian cancer therapy. For example, anexelekto inhibition enhanced the sensitivity of ovarian cancer cells to cisplatin via decreasing glycolysis [53]. Moreover, several enzymes in the glycolytic pathway have been identified as promising therapeutic targets for anticancer intervention [54, 55, 56]. In the present study, we demonstrated that many glycolytic enzymes were greatly inhibited by FBN1 KO directly or indirectly, which led to a significant increase in cisplatin sensitivity.
Our further experiments showed that FBN1 regulated glycolysis and angiogenesis by the VEGFR2/STAT2 signaling axis. In the present study, STAT2 silencing induced a marked reduction in glycolysis and angiogenesis, which in turn promoted cisplatin sensitivity in ovarian cancer.
Our present study has some limitations. For example, the mechanism of STAT2‐induced glycolysis and vascular formation was not sufficiently clear and need further study; other possible pathways of FBN1 mediated in glycolysis regulation need to be further studied.
5. CONCLUSIONS
Taken together, the FBN1/VEGFR2/STAT2 signaling pathway may promote chemoresistance of ovarian cancer by modulating glycolysis and angiogenesis, and FBN1 may be a novel therapy target in ovarian cancer.
DECLARATIONS
AUTHOR CONTRIBUTIONS
ZLW, HSW, HZS, JC and WC designed the research. LZ, ZLW, JC and WC performed the in vitro experiments. HZS and WC conducted the in vivo experiments. MDX, YW and JMH analyzed the data. XZ, HZS, YHL and JW wrote the manuscript. All authors read and approved the final version of the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The specimens of patients with ovarian cancer were conducted with permission from the Institutional Research Ethics Committee of Xinhua Hospital, Shanghai Jiaotong University School of Medical (Shanghai, China). All animal experiments were performed following the Guidelines for Animal Experiments and approved by the Ethics Committee at Xinhua Hospital.
CONSENT FOR PUBLICATION
All authors agreed on the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supporting information
Supporting information
ACKNOWLEDGEMENTS
We are indebted to all the authors and our colleagues for the fruitful suggestions and discussions. This study was funded by Project of the Shanghai Municipal Health Commission (No. 20194Y0039 to HZ.S) and Natural Science Foundation of China (No. 81872117 and 81502235 to ZL.W).
Wang Z, Chen W, Zuo L, Xu M, Wu Y, Huang J, et al. The Fibrillin‐1/VEGFR2/STAT2 signaling axis promotes chemoresistance via modulating glycolysis and angiogenesis in ovarian cancer organoids and cells. Cancer Commun. 2022;42:245–265. 10.1002/cac2.12274
Ziliang Wang, Wei Chen, Ling Zuo contributed equally to this work.
Contributor Information
Ziliang Wang, Email: wangziliang@xinhuamed.com.cn.
Jing Chen, Email: cj1275@126.com.
Husheng Wang, Email: wanghusheng@xinhuamed.com.cn.
Huizhen Sun, Email: sunhuizhen@xinhuamed.com.cn.
DATA AVAILABILITY STATEMENT
This study includes no data deposited in external repositories.
REFERENCES
- 1. Katiyi A, Zorea J, Halstuch A, Elkabets M, Karabchevsky A. Surface roughness‐induced absorption acts as an ovarian cancer cells growth sensor‐monitor. Biosens Bioelectron. 2020;161:112240. [DOI] [PubMed] [Google Scholar]
- 2. Xiao Y, Yu Y, Jiang P, Li Y, Wang C, Zhang R. The PI3K/mTOR dual inhibitor GSK458 potently impedes ovarian cancer tumorigenesis and metastasis. Cell Oncol (Dordr). 2020. [DOI] [PubMed] [Google Scholar]
- 3. Lan CY, Wang Y, Xiong Y, Li JD, Shen JX, Li YF et al. Apatinib combined with oral etoposide in patients with platinum‐resistant or platinum‐refractory ovarian cancer (AEROC): a phase 2, single‐arm, prospective study. Lancet Oncol. 2018;19:1239‐46. [DOI] [PubMed] [Google Scholar]
- 4. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021. [DOI] [PubMed] [Google Scholar]
- 5. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: a time‐trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond). 2021;41:1037‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moufarrij S, Dandapani M, Arthofer E, Gomez S, Srivastava A, Lopez‐Acevedo M et al. Epigenetic therapy for ovarian cancer: promise and progress. Clin Epigenetics. 2019;11:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vaughan S, Coward JI, Bast RC, Jr., Berchuck A, Berek JS, Brenton JD et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11:719‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng W, Dean DC, Hornicek FJ, Shi H, Duan Z. Exosomes promote pre‐metastatic niche formation in ovarian cancer. Mol Cancer. 2019;18:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Z, Liu H, Xu C. Cellular Senescence in the Treatment of Ovarian Cancer. Int J Gynecol Cancer. 2018;28:895‐902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo J, Li X, Zhang W, Chen Y, Zhu S, Chen L et al. HSP60‐regulated Mitochondrial Proteostasis and Protein Translation Promote Tumor Growth of Ovarian Cancer. Sci Rep. 2019;9:12628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Du Q, Zhang D, Zhuang Y, Xia Q, Wen T, Jia H. The Molecular Genetics of Marfan Syndrome. Int J Med Sci. 2021;18:2752‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maslen C, Babcock D, Raghunath M, Steinmann B. A rare branch‐point mutation is associated with missplicing of fibrillin‐2 in a large family with congenital contractural arachnodactyly. Am J Hum Genet. 1997;60:1389‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang D, Zhao D, Chen X. MiR‐133b inhibits proliferation and invasion of gastric cancer cells by up‐regulating FBN1 expression. Cancer Biomark. 2017;19:425‐36. [DOI] [PubMed] [Google Scholar]
- 14. Davis MR, Andersson R, Severin J, de Hoon M, Bertin N, Baillie JK et al. Transcriptional profiling of the human fibrillin/LTBP gene family, key regulators of mesenchymal cell functions. Mol Genet Metab. 2014;112:73‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zilberberg L, Todorovic V, Dabovic B, Horiguchi M, Courousse T, Sakai LY et al. Specificity of latent TGF‐beta binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J Cell Physiol. 2012;227:3828‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Massam‐Wu T, Chiu M, Choudhury R, Chaudhry SS, Baldwin AK, McGovern A et al. Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF beta. J Cell Sci. 2010;123:3006‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R et al. Latent transforming growth factor beta‐binding protein 1 interacts with fibrillin and is a microfibril‐associated protein. JBC. 2003;278:2750‐7. [DOI] [PubMed] [Google Scholar]
- 18. Guo Q, Song Y, Zhang H, Wu X, Xia P, Dang C. Detection of hypermethylated fibrillin‐1 in the stool samples of colorectal cancer patients. Med Oncol. 2013;30:695. [DOI] [PubMed] [Google Scholar]
- 19. Dong J, Bu J, Du W, Li Y, Jia Y, Li J et al. A new novel mutation in FBN1 causes autosomal dominant Marfan syndrome in a Chinese family. Mol Vis. 2012;18:81‐6. [PMC free article] [PubMed] [Google Scholar]
- 20. Sengle G, Tsutsui K, Keene DR, Tufa SF, Carlson EJ, Charbonneau NL et al. Microenvironmental regulation by fibrillin‐1. PLoS Genet. 2012;8:e1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goldstein C, Liaw P, Jimenez SA, Buchberg AM, Siracusa LD. Of mice and Marfan: genetic linkage analyses of the fibrillin genes, Fbn1 and Fbn2, in the mouse genome. Mamm Genome. 1994;5:696‐700. [DOI] [PubMed] [Google Scholar]
- 22. Cierna Z, Mego M, Jurisica I, Machalekova K, Chovanec M, Miskovska V et al. Fibrillin‐1 (FBN‐1) a new marker of germ cell neoplasia in situ. BMC Cancer. 2016;16:597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Whiteman P, Handford PA. Defective secretion of recombinant fragments of fibrillin‐1: implications of protein misfolding for the pathogenesis of Marfan syndrome and related disorders. Hum Mol Genet. 2003;12:727‐37. [DOI] [PubMed] [Google Scholar]
- 24. Zhang W, Ota T, Shridhar V, Chien J, Wu B, Kuang R. Network‐based survival analysis reveals subnetwork signatures for predicting outcomes of ovarian cancer treatment. PLoS Comput Biol. 2013;9:e1002975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T et al. Wnt proteins are lipid‐modified and can act as stem cell growth factors. Nature. 2003;423:448‐52. [DOI] [PubMed] [Google Scholar]
- 26. Wang Y, Chen X, Xie J, Zhou S, Huang Y, Li YP et al. RNA Helicase A Is an Important Host Factor Involved in Dengue Virus Replication. J Virol. 2019;93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao H, Lin Y, He J, Zhou S, Liang M, Huang C et al. Role of heparan sulfate in the Zika virus entry, replication, and cell death. Virology. 2019;529:91‐100. [DOI] [PubMed] [Google Scholar]
- 28. Jin BF, He K, Wang HX, Wang J, Zhou T, Lan Y et al. Proteomic analysis of ubiquitin‐proteasome effects: insight into the function of eukaryotic initiation factor 5A. Oncogene. 2003;22:4819‐30. [DOI] [PubMed] [Google Scholar]
- 29. Cao D, Qi Z, Pang Y, Li H, Xie H, Wu J et al. Retinoic Acid‐Related Orphan Receptor C Regulates Proliferation, Glycolysis, and Chemoresistance via the PD‐L1/ITGB6/STAT3 Signaling Axis in Bladder Cancer. Cancer Res. 2019;79:2604‐18. [DOI] [PubMed] [Google Scholar]
- 30. Sun H, Wang H, Wang X, Aoki Y, Wang X, Yang Y et al. Aurora‐A/SOX8/FOXK1 signaling axis promotes chemoresistance via suppression of cell senescence and induction of glucose metabolism in ovarian cancer organoids and cells. Theranostics. 2020;10:6928‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gyorffy B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput Struct Biotechnol J. 2021;19:4101‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen L, Cheng X, Tu W, Qi Z, Li H, Liu F et al. Apatinib inhibits glycolysis by suppressing the VEGFR2/AKT1/SOX5/GLUT4 signaling pathway in ovarian cancer cells. Cell Oncol (Dordr). 2019;42:679‐90. [DOI] [PubMed] [Google Scholar]
- 33. Ruch C, Skiniotis G, Steinmetz MO, Walz T, Ballmer‐Hofer K. Structure of a VEGF‐VEGF receptor complex determined by electron microscopy. Nat Struct Mol Biol. 2007;14:249‐50. [DOI] [PubMed] [Google Scholar]
- 34. Zhang S, Gao X, Fu W, Li S, Yue L. Immunoglobulin‐like domain 4‐mediated ligand‐independent dimerization triggers VEGFR‐2 activation in HUVECs and VEGFR2‐positive breast cancer cells. Breast Cancer Res Treat. 2017;163:423‐34. [DOI] [PubMed] [Google Scholar]
- 35. Guo XF, Zhang YY, Kang J, Dou QH, Zhu XF. A bispecific decoy receptor VEGFR‐EGFR/Fc binding EGF‐like ligands and VEGF shows potent antitumor efficacy. J Drug Target. 2021:1‐11. [DOI] [PubMed] [Google Scholar]
- 36. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182‐6. [DOI] [PubMed] [Google Scholar]
- 37. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐74. [DOI] [PubMed] [Google Scholar]
- 38. Miao M, Deng G, Luo S, Zhou J, Chen L, Yang J et al. A phase II study of apatinib in patients with recurrent epithelial ovarian cancer. Gynecol Oncol. 2018;148:286‐90. [DOI] [PubMed] [Google Scholar]
- 39. Pujade‐Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G et al. Bevacizumab combined with chemotherapy for platinum‐resistant recurrent ovarian cancer: The AURELIA open‐label randomized phase III trial. J Clin Oncol. 2014;32:1302‐8. [DOI] [PubMed] [Google Scholar]
- 40. Li J, Qin S, Xu J, Xiong J, Wu C, Bai Y et al. Randomized, Double‐Blind, Placebo‐Controlled Phase III Trial of Apatinib in Patients With Chemotherapy‐Refractory Advanced or Metastatic Adenocarcinoma of the Stomach or Gastroesophageal Junction. J Clin Oncol. 2016;34:1448‐54. [DOI] [PubMed] [Google Scholar]
- 41. Hu X, Cao J, Hu W, Wu C, Pan Y, Cai L et al. Multicenter phase II study of apatinib in non‐triple‐negative metastatic breast cancer. BMC Cancer. 2014;14:820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang L, Liang L, Yang T, Qiao Y, Xia Y, Liu L et al. A pilot clinical study of apatinib plus irinotecan in patients with recurrent high‐grade glioma: Clinical Trial/Experimental Study. Medicine (Baltimore). 2017;96:e9053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sabatier L, Djokic J, Hubmacher D, Dzafik D, Nelea V, Reinhardt DP. Heparin/heparan sulfate controls fibrillin‐1, ‐2 and ‐3 self‐interactions in microfibril assembly. FEBS Lett. 2014;588:2890‐7. [DOI] [PubMed] [Google Scholar]
- 44. Summers KM, Raza S, van Nimwegen E, Freeman TC, Hume DA. Co‐expression of FBN1 with mesenchyme‐specific genes in mouse cell lines: implications for phenotypic variability in Marfan syndrome. Eur J Hum Genet. 2010;18:1209‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B et al. Dysregulation of TGF‐beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407‐11. [DOI] [PubMed] [Google Scholar]
- 46. Lien HC, Lee YH, Juang YL, Lu YT. Fibrillin‐1, a novel TGF‐beta‐induced factor, is preferentially expressed in metaplastic carcinoma with spindle sarcomatous metaplasia. Pathology. 2019;51:375‐83. [DOI] [PubMed] [Google Scholar]
- 47. Wang L, Ai M, Nie M, Zhao L, Deng G, Hu S et al. EHF promotes colorectal carcinoma progression by activating TGF‐beta1 transcription and canonical TGF‐beta signalling. Cancer Sci. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Basu M, Bhattacharya R, Ray U, Mukhopadhyay S, Chatterjee U, Roy SS. Invasion of ovarian cancer cells is induced byPITX2‐mediated activation of TGF‐beta and Activin‐A. Mol Cancer. 2015;14:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma X, Wei J, Zhang L, Deng D, Liu L, Mei X et al. miR‐486‐5p inhibits cell growth of papillary thyroid carcinoma by targeting fibrillin‐1. Biomed Pharmacother. 2016;80:220‐6. [DOI] [PubMed] [Google Scholar]
- 50. Yang J, Ren B, Yang G, Wang H, Chen G, You L et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. 2020;77:305‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bhattacharya B, Mohd Omar MF, Soong R. The Warburg effect and drug resistance. Br J Pharmacol. 2016;173:970‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Morandi A, Indraccolo S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim Biophys Acta Rev Cancer. 2017;1868:1‐6. [DOI] [PubMed] [Google Scholar]
- 53. Tian M, Chen XS, Li LY, Wu HZ, Zeng D, Wang XL et al. Inhibition of AXL enhances chemosensitivity of human ovarian cancer cells to cisplatin via decreasing glycolysis. Acta Pharmacol Sin. 2021;42:1180‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mondal S, Roy D, Sarkar Bhattacharya S, Jin L, Jung D, Zhang S et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int J Cancer. 2019;144:178‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xintaropoulou C, Ward C, Wise A, Queckborner S, Turnbull A, Michie CO et al. Expression of glycolytic enzymes in ovarian cancers and evaluation of the glycolytic pathway as a strategy for ovarian cancer treatment. BMC Cancer. 2018;18:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Baig MH, Adil M, Khan R, Dhadi S, Ahmad K, Rabbani G et al. Enzyme targeting strategies for prevention and treatment of cancer: Implications for cancer therapy. Semin Cancer Biol. 2019;56:1‐11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
This study includes no data deposited in external repositories.