

ABSTRACT
The pharmacokinetics of ceftolozane-tazobactam (TOL-TAZ) and ceftazidime-avibactam (CEF-AVI) is influenced by renal function. Application of recommended dosages in patients with renal impairment requires the use of fractions of the full dose, as only one dosage is available for both antibiotics. The objective of this study was to evaluate the adequacy of alternative dosage regimens based on the full dose. We performed pharmacokinetic/pharmacodynamic (PK/PD) simulations of recommended and alternative dosage regimens in patients with various degrees of renal impairment by using the Pmetrics program. Alternative regimens included longer dosage interval and prolonged infusions of the full dose for both drugs. Probabilities of target attainment (PTA) were assessed considering PK/PD targets defined for cephalosporins and beta-lactamase inhibitors as well as MIC breakpoints. The risk of overexposure was also assessed. Results showed that alternative dosage regimens based on a full dose of TOL-TAZ and CEF-AVI administered every 12 or 24 h were associated with PTA similar to that of recommended dosages, especially when administered as prolonged infusion. The alternative dosage regimens were not associated with overexposure in most cases. In addition, those regimens could reduce dosing errors, drug cost, and nurse labor. Clinical investigation ovf those alternative dosage regimens would be required before implementation.
KEYWORDS: Monte Carlo simulation, beta-lactamases, cephalosporin, pharmacokinetics, population pharmacokinetics
TEXT
Ceftolozane-tazobactam (TOL-TAZ) and ceftazidime-avibactam (CEF-AVI) are combinations of a cephalosporin with a β-lactamase inhibitor that have been recently approved.
TOL-TAZ has bactericidal activity against most extended-spectrum β-lactamase (ESBL)-producing Escherichia coli, Klebsiella pneumoniae, and other Enterobacteriaceae (1, 2). The indications of TOL-TAZ are complicated intraabdominal infections (cIAI), acute pyelonephritis, complicated urinary tract infections (cUTI), and hospital-acquired pneumonia (HAP), including ventilator-associated pneumonia (VAP) (3). TOL-TAZ should be administered as a 1-h intravenous (i.v.) infusion. Both drugs are eliminated primarily by the kidneys, with about 95% and 80% of the drug dose eliminated unchanged into urine, respectively. The dosage regimen of TOL-TAZ should be adjusted in patients with renal impairment. The recommended dosage regimens of TOL-TAZ are available elsewhere (3). In patients with a creatinine clearance (ClCr) of ≤50 mL/min, the drug dose should be reduced but the dosage interval should be kept at 8 h.
The association of CEF with AVI expands ceftazidime activity on many cephalosporin- and carbapenem-resistant Enterobacteriaceae and Pseudomonas aeruginosa strains (4, 5). The indications of CEF-AVI are similar to those of TOL-TAZ: cIAI, cUTI including pyelonephritis, and HAP including VAP, as well as bacteremia associated with those infections (6). CEF-AVI is also indicated for treatment of aerobic Gram-negative bacterial infections in adults, for whom therapeutic options are limited. The dosage should be reduced in patients with renal impairment, with use of fractions of the full dose, as indicated in the drug label (6).
The recommended dosage regimens of both TOL-TAZ and CEF-AVI in patients with renal impairment raise practical issues. As only one dosage is marketed for both drugs, it is necessary to administer only a part of the vial, which is a known risk factor of errors in the administration of injectable drugs (7). As both TOL-TAZ and CEF-AVI are stable for at least 24 h at 2 to 8°C after dilution, the amount remaining might be stored for subsequent administrations (3, 6). However, this practice is prone to dose error and error in storage conditions and is not recommended. A safer practice is to discard the amount remaining in the vial after administration, but it is a waste of drug and money. Alternative dosage regimens based on the administration of the full dose may be of interest in this setting.
The objectives of the present study were to identify alternative, cost-effective dosage regimens of TOL-TAZ and CEF-AVI for administration in patients with renal impairment.
RESULTS
Pharmacokinetic/pharmacodynamic simulation results for ceftolozane-tazobactam.
Results of the different simulations for alternative dosage regimens are summarized in Table 1. For comparison, results of the simulations with recommended dosage regimens are shown in Table 2. Figure 1 illustrates the influence of dose, infusion time, and dosage interval on ceftolozane pharmacokinetic (PK) profiles in the case of patients with a ClCr of 29 mL/min. Figure 2 shows the corresponding probabilities of target attainment (PTA) as a function of ceftolozane MIC.
TABLE 1.
| ClCr (mL/min) | Infusion time | Dosing frequency | Values for drug: |
PTA |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ceftolozane |
Tazobactam |
Ceftolozane (MIC = 4 mg/L; fT>MIC ≥ 32.2%) | Tazobactam (CT = 0.25 mg/L; fT>CT ≥ 35%) | |||||||
| Cmax (mg/L) | Cmin (mg/L) | AUC0–24h (mg.h/L) | Cmax (mg/L) | Cmin (mg/L) | AUC0–24h (mg.h/L) | |||||
| 100 | 1 h | q12h | 48.0 (26.1–96.2) | 2.2 (0.2–12.3) | 348.0 (184.1–657.6) | 14.9 (7.2–30.2) | 0.0 (0.0–1.9) | 62.8 (23.9–165.4) | 98.0 | 85.7 |
| 100 | 2 h | q12h | 40.4 (22.8–71.0) | 2.5 (0.2–13.2) | 348.0 (184.1–657.5) | 10.7 (5.2–21.3) | 0.0 (0.0–2.0) | 62.8 (23.9–165.3) | 99.1 | 91.9 |
| 100 | 4 h | q12h | 30.4 (18.1–50.7) | 3.3 (0.3–15.0) | 347.8 (184.1–657.1) | 6.8 (2.9–14.6) | 0.1 (0.0–2.4) | 62.8 (23.9–165.3) | 100 | 100 |
| 100 | 6 h | q12h | 24.1 (14.1–40.0) | 4.4 (0.6–16.6) | 347.8 (184.1–656.9) | 4.9 (2.0–11.5) | 0.2 (0.0–2.9) | 62.8 (23.9–165.2) | 100 | 100 |
| 100 | 12 h | q12h | 14.4 (7.2–27.2) | 14.4 (7.2–27.2) | 344.7 (172.8–651.5) | 2.6 (0.9–6.8) | 2.6 (0.9–6.8) | 62.0 (21.7–163.1) | 100 | 100 |
| 51 | 1 h | q12h | 56.5 (32.0–110.2) | 7.5 (0.9–26.5) | 563.2 (298.0–1,064.3) | 17.8 (8.4–36.5) | 0.2 (0.0–4.5) | 98.7 (37.5–259.7) | 100 | 97.4 |
| 51 | 2 h | q12h | 50.7 (29.5–89.3) | 8.1 (1.1–27.3) | 563.2 (298.0–1,064.2) | 13.7 (6.9–26.4) | 0.3 (0.0–4.7) | 98.7 (37.5–259.6) | 100 | 98.5 |
| 51 | 4 h | q12h | 41.5 (24.8–70.5) | 9.7 (1.5–29.5) | 562.8 (297.9–1,063.6) | 9.6 (4.3–19.3) | 0.5 (0.0–5.2) | 98.6 (37.5–259.5) | 100 | 100 |
| 51 | 6 h | q12h | 34.9 (20.5–59.1) | 11.6 (2.3–31.9) | 562.8 (297.9–1,063.2) | 7.3 (3.0–16.4) | 0.7 (0.0–5.8) | 98.6 (37.5–259.4) | 100 | 100 |
| 51 | 12 h | q12h | 23.2 (11.6–43.9) | 23.2 (11.6–43.9) | 558.0 (279.7–1,054.4) | 4.1 (1.4–10.7) | 4.1 (1.4–10.7) | 97.3 (34.1–256.1) | 100 | 100 |
| 50 | 1 h | q12h | 56.8 (32.2–110.7) | 7.8 (1.0–27.1) | 571.4 (302.3–1,079.5) | 17.9 (8.5–36.6) | 0.3 (0.0–4.6) | 100.0 (38.0–263.2) | 100 | 97.4 |
| 50 | 2 h | q12h | 51.1 (29.6–89.8) | 8.4 (1.2–27.8) | 571.2 (302.2–1,079.3) | 13.8 (6.9–26.6) | 0.3 (0.0–4.8) | 100.0 (38.0–263.1) | 100 | 98.5 |
| 50 | 4 h | q12h | 41.9 (25.0–71.1) | 9.9 (1.6–30.1) | 570.8 (302.1–1,078.8) | 7.3 (2.0–17.1) | 0.5 (0.0–5.3) | 100.0 (38.0–263.0) | 100 | 100 |
| 50 | 6 h | q12h | 35.2 (20.8–59.8) | 11.9 (2.4–32.4) | 570.8 (302.1–1,078.4) | 7.3 (3.1–16.5) | 0.7 (0.0–5.9) | 100.0 (38.0–262.9) | 100 | 100 |
| 50 | 12 h | q12h | 23.6 (11.8–44.6) | 23.6 (11.8–44.6) | 565.8 (283.7–1,069.4) | 4.1 (1.4–10.8) | 4.1 (1.4–10.8) | 98.6 (34.5–259.5) | 100 | 100 |
| 30 | 1 h | q12h | 67.1 (38.3–125.2) | 15.8 (3.0–44.4) | 823.2 (435.6–1,555.2) | 20.0 (9.7–41.0) | 0.8 (0.0–7.7) | 140.8 (53.5–370.6) | 100 | 99.6 |
| 30 | 2 h | q12h | 62.2 (36.0–108.3) | 16.8 (3.4–45.8) | 823.2 (435.6–1,555.0) | 16.3 (8.3–31.5) | 1.0 (0.0–8.0) | 140.8 (53.5–370.5) | 100 | 99.9 |
| 30 | 4 h | q12h | 53.3 (31.8–90.3) | 18.9 (4.5–47.9) | 822.6 (435.3–1,554.4) | 12.1 (5.8–24.3) | 1.3 (0.0–9.0) | 140.8 (53.5–370.3) | 100 | 100 |
| 30 | 6 h | q12h | 46.7 (27.4–81.3) | 21.4 (5.8–50.1) | 822.4 (435.2–1,554.0) | 9.7 (4.2–22.0) | 1.8 (0.1–10.0) | 140.7 (53.5–370.2) | 100 | 100 |
| 30 | 12 h | q12h | 34.0 (17.0–64.2) | 34.0 (17.0–64.2) | 815.4 (408.8–1,540.9) | 5.8 (2.0–15.2) | 5.8 (2.0–15.2) | 138.9 (48.6–365.5) | 100 | 100 |
| 29 | 1 h | q24h | 55.1 (30.9–109.6) | 3.3 (0.2–16.1) | 414.3 (214.0–779.9) | 19.1 (8.4–39.5) | 0.0 (0.0–2.2) | 70.1 (25.7–183.5) | 99.5 | 88.5 |
| 29 | 2 h | q24h | 50.3 (29.2–89.6) | 3.5 (0.2–16.5) | 414.3 (214.0–779.8) | 15.5 (7.4–30.0) | 0.0 (0.0–2.3) | 70.1 (25.7–183.5) | 99.8 | 90.8 |
| 29 | 4 h | q24h | 40.8 (25.0–68.3) | 3.9 (0.3–17.3) | 414.0 (213.9–779.5) | 11.2 (5.5–21.7) | 0.1 (0.0–2.6) | 70.0 (25.7–183.4) | 99.9 | 95.6 |
| 29 | 12 h | q24h | 27.1 (15.9–45.5) | 6.6 (0.9–21.5) | 413.7 (213.8–778.8) | 5.4 (2.1–12.4) | 0.2 (0.0–3.8) | 70.0 (25.7–183.2) | 100 | 100 |
| 29 | 24 h | q24h | 17.0 (9.0–30.3) | 17.0 (9.0–30.3) | 407.1 (220.3–728.0) | 2.8 (1.1–6.9) | 2.8 (1.1–6.9) | 68.3 (26.9–165.3) | 100 | 100 |
| 15 | 1 h | q24h | 63.4 (37.1–119.8) | 10.2 (1.4–33.2) | 664.0 (343.0–1,248.4) | 21.0 (9.3–43.7) | 0.3 (0.0–5.6) | 109.0 (39.9–285.3) | 100 | 98.3 |
| 15 | 2 h | q24h | 59.6 (35.3–106.4) | 10.7 (1.5–33.9) | 663.9 (343.0–1,248.3) | 18.0 (8.7–35.2) | 0.3 (0.0–5.7) | 109.0 (39.9–285.3) | 100 | 98.7 |
| 15 | 4 h | q24h | 53.5 (32.1–90.6) | 11.6 (1.8–35.3) | 663.5 (342.8–1,248.0) | 14.2 (7.1–27.1) | 0.4 (0.0–6.1) | 109.0 (39.9–285.2) | 100 | 99.3 |
| 15 | 12 h | q24h | 39.0 (22.5–66.5) | 15.9 (3.7–40.2) | 662.8 (342.6–1,247.2) | 7.9 (3.2–17.5) | 0.9 (0.0–7.5) | 108.9 (39.9–284.9) | 100 | 100 |
| 15 | 24 h | q24h | 27.2 (14.4–48.6) | 27.2 (14.4–48.6) | 652.3 (353.0–1,166.1) | 4.4 (1.7–10.7) | 4.4 (1.7–10.7) | 106.2 (41.8–257.1) | 100 | 100 |
Concentrations and AUC0–24h at steady state were obtained from simulations on 1,000 virtual patients and are given as median (2.5 to 97.5 percentiles). Dose for all regimens is 1,000/500 mg.
Abbreviations: AUC0–24h, area under the concentration-time curve over 24 h; ClCr, creatinine clearance; Cmax, maximal concentration at the end of infusion; Cmin, concentration at steady state or plateau concentration in case of continuous infusion; q12h and q24h, administration every 12 h and 24 h, respectively; PTA, probability of target attainment.
TABLE 2.
| ClCr (mL/min) | Dose (mg) | Value for drug: |
PTA |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Ceftolozane |
Tazobactam |
||||||||
| Cmax (mg/L) | Cmin (mg/L) | AUC0–24h (mg.h/L) | Cmax (mg/L) | Cmin (mg/L) | AUC0–24h (mg.h/L) | Ceftolozane (MIC = 4 mg/L; fT>MIC ≥ 32.2%) | Tazobactam (CT = 0.25 mg/L; fT>CT ≥ 35%) | ||
| 100 | 1,000/500 | 52.2 (29.6–89.1) | 6.4 (0.7–23.4) | 511.2 (266.0–936.9) | 15.2 (7.5–27.7) | 0.3 (0.0–4.1) | 91.3 (33.8–229.5) | 99.8 | 96.7 |
| 51 | 1,000/500 | 65.7 (37.8–107.5)c | 16.5 (3.0–46.5)d | 827.3 (430.6–1,516.2)c | 18.6 (9.2–33.7)c | 1.1 (0.0–8.6) | 143.4 (53.1–360.3)c | 100 | 99.5 |
| 50 | 500/250 | 33.1 (19.0–54.1) | 8.5 (1.6–23.7) | 419.6 (218.3–768.9) | 9.4 (4.6–17.0) | 0.6 (0.0–4.4) | 72.7 (26.9–182.6) | 99.9 | 98.5 |
| 30 | 500/250 | 40.8 (23.3–66.5) | 15.4 (4.0–37.6) | 604.6 (314.6–1,107.8) | 10.8 (5.2–19.8) | 1.3 (0.1–7.5) | 102.3 (37.9–257.0) | 100 | 99.6 |
| 29 | 250/125 | 20.7 (11.8–33.9) | 8.0 (2.1–19.3) | 309.7 (161.2–567.5) | 5.5 (2.7–10.0) | 0.7 (0.0–3.9) | 52.4 (19.4–149.5) | 99.8 | 99.3 |
| 15 | 250/125 | 28.4 (16.4–46.2) | 15.4 (5.5–33.2) | 496.2 (258.2–907.6) | 6.8 (3.3–13.0) | 1.7 (0.1–6.8)d | 81.4 (30.2–204.5) | 100 | 99.8 |
Concentrations and AUC0–24h at steady state were obtained from simulations on 1,000 virtual patients and are given as median (2.5 to 97.5 percentiles). For all regimens, infusion time is 1 h and dosing frequency is q8h.
Abbreviations: AUC0–24h, area under the concentration-time curve up over 24 h; ClCr, creatinine clearance; Cmax, maximal concentration at the end of infusion; Cmin, concentration at steady state or plateau concentration in case of continuous infusion; q12h and q24h, administration every 12 h and 24 h, respectively; PTA, probability of target attainment.
Highest median Cmax and AUC0–24h for recommended dosages.
Highest median Cmin for recommended dosages.
FIG 1.

Steady-state pharmacokinetic profile of ceftolozane for three dosage regimens in patients with creatinine clearance of 29 mL/min. Simulated dosage regimens are as follows: 250 mg q8h as a 1-h infusion (top), 1,000 mg q24h as a 2-h infusion (center), 1,000 mg q24h as a 4-h infusion time (bottom). The five lines represent the 5th, 25th, 50th (median), 75th, and 95th percentiles of simulated concentrations. The gray areas are the 95% confidence intervals around percentiles.
FIG 2.
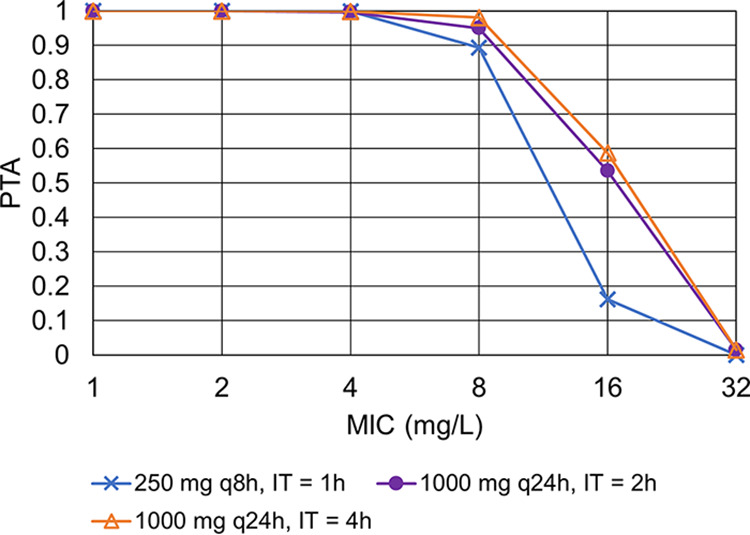
Probability of target attainment for three ceftolozane dosage regimens in patients with creatinine clearance of 29 mL/min. IT, infusion time; PTA, probability of target attainment.
In patients with ClCr between 51 to 100 mL/min, 1,000/500 mg every 12 hours (q12h) as a 1-h infusion was simulated as an alternative to q8h dosing. PTA for TOL was ≥90%. For TAZ, PTA was slightly lower than 90% (85.7%) for ClCr of 100 ml/min, but increasing the infusion time to 2 h was sufficient to achieve a PTA of ≥90%. The dosage of 1,000/500 mg q12h as a 1-h infusion was associated with acceptable PTA for both drugs with ClCr between 30 mL/min to 50 mL/min. In patients with ClCr between 15 mL/min to 29 mL/min, an alternative dosage regimen of 1,000/500 mg q24h provided interesting results. The PTA for TAZ with ClCr of 29 mL/min was slightly lower than 90% for infusion times of 1 h and 2 h. However, increasing the infusion time to 4 h was sufficient to get acceptable PTA for both agents, without overexposure.
Steady-state concentrations for alternative dosage regimens can be compared to those of the recommended dosage regimens to assess the risk of overexposure. For TOL, the recommended dosage regimen associated with the highest Cmax, Cmin, and AUC0–24h values was 1,000/500 mg q8h as a 1-h infusion in patients with a ClCr of 51 mL/min. The highest median Cmin of tazobactam was obtained with 250/125 mg q8h, in patients with a ClCr of 15 mL/min. The alternative dosage regimens for TOL-TAZ presented in Table 1 were not associated with significant overexposure. For a given dose, increasing the infusion time was associated with decreased Cmax.
Assuming that the remaining drug amount would be discarded when only a fraction had been used, simulated alternative dosage regimens of q12h administration in patients with ClCr from 51 to 100 mL/min and 30 to 50 mL/min would save one vial per day of treatment compared to the recommended dosage regimens with q8h administration. For patients with ClCr from 15 to 29 mL/min, two vials per day of treatment would be saved. So, alternative regimens could be associated with daily treatment cost savings of 33% to 66%.
Pharmacokinetic/pharmacodynamic simulation results for ceftazidime-avibactam.
Simulations results for alternative dosage regimens of CEF-AVI are summarized in Table 3. For comparison, results of the simulations with recommended dosage regimens are shown in Table 4.
TABLE 3.
| ClCr (ml/min) | Infusion time | Dosing frequency | Value for drug: |
PTA |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ceftazidime |
Avibactam |
|||||||||
| Cmax (mg/L) | Cmin (mg/L) | AUC0–24h (mg.h/L) | Cmax (mg/L) | Cmin (mg/L) | AUC0–24h (mg.h/L) | Ceftolozane (MIC = 4 mg/L; fT>MIC ≥ 32.2%) | Tazobactam (CT = 0.25 mg/L; fT>CT ≥ 35%) | |||
| 80 | 2h | q12h | 63.7 (25.1–159.7) | 4.4 (0.6–39.6) | 601.6 (257.6–1,330.3) | 10.5 (2.7–36.1) | 0.1 (0.0–3.1) | 69.6 (21.3–212.2) | 91.6 | 51.3 |
| 80 | 4h | q12h | 49.2 (20.9–104.8) | 5.7 (1.1–40.8) | 601.4 (257.5–1,329.6) | 7.0 (2.1–22.3) | 0.2 (0.0–3.5) | 69.6 (21.3–212.2) | 96.6 | 81.1 |
| 80 | 6h | q12h | 40.3 (17.6–83.4) | 7.5 (1.8–42.7) | 601.2 (257.4–1,329.3) | 5.3 (1.6–16.1) | 0.3 (0.0–3.8) | 69.6 (21.3–212.1) | 99.3 | 94.7 |
| 51 | 2h | q12h | 78.8 (31.7–206.6) | 13.1 (3.5–65.0) | 943.8 (404.1–2,081.0) | 13.2 (3.3–48.6) | 0.5 (0.1–6.2) | 111.7 (34.2–340.3) | 99.8 | 96.7 |
| 51 | 4h | q12h | 66.7 (28.0–143.3) | 15.5 (4.7–67.8) | 943.2 (403.9–2,080.6) | 9.8 (2.9–31.5) | 0.7 (0.2–6.7) | 111.6 (34.2–340.2) | 99.9 | 98.9 |
| 51 | 6h | q12h | 57.5 (24.6–119.0) | 18.9 (6.6–71.4) | 943.0 (403.7–2,080.4) | 7.8 (2.3–24.0) | 1.0 (0.3–7.8) | 111.6 (34.1–340.3) | 100 | 99.3 |
| 50 | 2h | q12h | 79.5 (32.3–208.2) | 13.6 (3.7–66.4) | 962.6 (412.1–2,121.2) | 13.3 (3.4–49.3) | 0.6 (0.1–6.4) | 114.0 (34.9–347.4) | 99.8 | 97.0 |
| 50 | 6h | q12h | 58.4 (24.9–120.7) | 19.6 (6.9–73.1) | 961.8 (411.8–2,121.1) | 8.0 (2.3–24.4) | 1.1 (0.3–8.0) | 113.9 (34.9–347.5) | 100 | 99.4 |
| 50 | 12h | q12h | 40.3 (17.9–97.0) | 40.3 (17.9–97.0) | 967.4 (431.2–2,328.1) | 4.8 (1.5–16.6) | 4.8 (1.5–16.6) | 115.0 (37.2–397.8) | 100 | 99.6 |
| 31 | 2h | q12h | 105.7 (43.3–265.3) | 33.4 (12.3–114.1) | 1,551.2 (664.7–3,399.4) | 17.1 (4.6–67.4) | 2.1 (0.6–13.8) | 188.4 (57.6–573.6) | 100 | 99.9 |
| 31 | 6h | q12h | 85.6 (35.9–175.9) | 42.1 (17.0–122.3) | 1,550.2 (663.9–3,396.1) | 11.8 (3.4–36.0) | 3.4 (1.1–16.1) | 188.2 (57.6–573.7) | 100 | 99.9 |
| 31 | 12h | q12h | 65.0 (28.8–153.1) | 65.0 (28.9–152.8) | 1,560.4 (695.4–3,674.2) | 7.9 (2.5–27.4) | 7.9 (2.5–27.4) | 190.0 (61.4–657.1) | 100 | 100 |
| 30 | 2h | q24h | 82.6 (32.6–222.5) | 7.9 (1.0–54.3) | 812.1 (357.0–1,886.0) | 15.9 (4.1–64.9) | 0.2 (0.0–5.2) | 99.1 (31.5–346.2) | 98.7 | 82.7 |
| 30 | 4h | q24h | 72.9 (29.8–161.8) | 8.8 (2.3–55.3) | 811.6 (356.8–1,884.8) | 12.6 (3.5–46.5) | 0.3 (0.0–5.5) | 99.1 (31.5–346.0) | 99.3 | 89.6 |
| 30 | 6h | q24h | 65.9 (27.8–135.4) | 9.8 (2.7–56.5) | 811.4 (356.7–1,883.8) | 10.6 (3.1–36.2) | 0.3 (0.1–6.0) | 99.1 (31.4–345.9) | 99.5 | 95.5 |
| 30 | 12h | q24h | 50.8 (22.8–104.2) | 13.8 (4.4–61.7) | 810.8 (356.4–1,881.6) | 7.1 (2.2–23.5) | 0.7 (0.2–7.0) | 99.0 (31.4–345.6) | 99.9 | 98.7 |
| 16 | 2h | q24h | 110.3 (46.4–277.2) | 29.9 (10.7–109.9) | 1,515.6 (669.4–3,472.4) | 19.7 (5.2–80.7) | 1.8 (0.5–14.1) | 191.8 (60.9–669.6) | 100 | 99.9 |
| 16 | 4h | q24h | 102.0 (44.0–223.3) | 31.9 (11.6–111.7) | 1,515.0 (669.2–3,471.8) | 17.2 (4.8–62.1) | 2.0 (0.6–14.4) | 191.8 (60.9–669.3) | 100 | 99.9 |
| 16 | 6h | q24h | 95.8 (42.2–210.7) | 34.1 (12.5–113.9) | 1,514.3 (668.8–3,470.8) | 15.3 (4.4–53.6) | 2.3 (0.7–14.9) | 191.7 (60.9–668.7) | 100 | 99.9 |
| 16 | 12h | q24h | 81.6 (37.1–173.9) | 40.5 (16.1–121.6) | 1,513.2 (668.3–3,469.4) | 11.9 (3.6–38.6) | 3.3 (1.1–16.7) | 191.5 (60.8–666.9) | 100 | 100 |
| 15 | 2h | q24h | 114.0 (48.4–283.9) | 33.7 (12.2–117.7) | 1,616.6 (714.2–3,700.1) | 20.1 (5.4–82.2) | 2.2 (0.6–15.3) | 205.0 (65.2–710.5) | 100 | 100 |
| 15 | 12h | q24h | 86.0 (38.9–183.4) | 44.6 (17.9–130.0) | 1,613.7 (712.8–3,669.6) | 12.5 (3.8–40.6) | 3.7 (1.2–18.3) | 204.7 (65.1–709.6) | 100 | 100 |
| 15 | 24h | q24h | 65.3 (29.2–137.3) | 65.1 (29.1–136.5) | 1,565.0 (704.9–3,279.6) | 8.2 (2.7–25.2) | 8.2 (2.7–25.2) | 196.7 (64.2–605.8) | 100 | 100 |
| 6 | 2h | q24h | 203.3 (87.5–461.5) | 122.1 (53.6–290.1) | 3,804.6 (1,748.8–7,942.0) | 33.0 (9.4–120.1) | 13.9 (4.6–49.6) | 526.9 (167.2–1,746.6) | 100 | 100 |
| 6 | 12h | q24h | 177.8 (80.2–374.3) | 135.8 (61.6–304.4) | 3,792.8 (1,746.9–7,910.9) | 26.3 (8.3–88.3) | 16.9 (5.5–58.1) | 525.5 (167.0–1,744.1) | 100 | 100 |
| 6 | 24h | q24h | 156.3 (64.2–314.5) | 155.3 (63.4–311.7) | 3,744.8 (1,540.4–7,519.1) | 21.2 (6.9–61.6) | 21.2 (6.9–61.4) | 508.6 (166.6–1,476.2) | 100 | 100 |
Concentrations and AUC0–24h at steady state were obtained from simulations on 1,000 virtual patients and are given as median (2.5 to 97.5 percentiles). Dose is 2,000/500 mg for all regimens.
Abbreviations: AUC0–24h, area under the concentration-time curve up over 24h; ClCr, creatinine clearance; Cmax, maximal concentration at the end of infusion; Cmin, concentration at steady state or plateau concentration in case of continuous infusion; q12h and q24h, administration every 12 h and 24 h, respectively; PTA, probability of target attainment.
TABLE 4.
| ClCr (ml/min) | Dose (mg) | Infusion time | Dosing frequency | Value for drug: |
PTA |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ceftazidime |
Avibactam |
||||||||||
| Cmax (mg/L) | Cmin (mg/L) | AUC0–24h (mg.h/L) | Cmax (mg/L) | Cmin (mg/L) | AUC0–24h (mg.h/L) | Ceftolozane (MIC = 4 mg/L; fT>MIC ≥ 32.2%) | Tazobactam (CT = 0.25 mg/L; fT>CT ≥ 35%) | ||||
| 80 | 2,000/500 | 2h | q8h | 71.9 (31.8–169.1) | 12.5 (3.9–62.8) | 914.9 (403.7–2,023.7) | 11.2 (3.3–39.2) | 0.6 (0.1–5.2) | 106.5 (34.0–322.7) | 99.4 | 97.4 |
| 51 | 2,000/500 | 2h | q8h | 95.3 (41.7–226.9)a | 29.9 (11.5–107.3) | 1,435.2 (633.2–3,165.5)a | 14.8 (4.4–53.0)a | 1.9 (0.5–11.1) | 170.8 (54.5–517.6) | 100 | 100 |
| 50 | 1,000/250 | 2h | q8h | 48.2 (21.1–115.3) | 15.4 (6.0–55.0) | 731.0 (322.9–1,613.9) | 7.5 (2.2–26.9) | 1.0 (0.3–5.8) | 87.2 (27.8–264.2) | 99.6 | 96.9 |
| 31 | 1,000/250 | 2h | q8h | 67.7 (29.4–153.8) | 32.2 (14.0–93.2) | 1,178.7 (520.8–2,550.9) | 10.2 (3.0–36.5) | 2.8 (0.9–11.7) | 144.1 (46.0–436.4) | 100 | 99.9 |
| 30 | 750/187.5 | 2h | q12h | 40.5 (16.6–101.0) | 13.2 (4.9–44.3) | 601.1 (257.6–1,317.2) | 6.5 (1.7–25.8) | 0.9 (0.3–5.4) | 73.1 (22.4–222.6) | 99.1 | 93.4 |
| 16 | 750/187.5 | 2h | q12h | 62.2 (26.1–140.3) | 34.0 (14.0–85.4) | 1,124.6 (483.0–2,435.0) | 9.6 (2.7–36.1) | 3.2 (1.0–12.2) | 141.3 (43.3–428.5) | 100 | 99.8 |
| 15 | 750/187.5 | 2h | q24h | 42.7 (18.2–106.5) | 12.6 (4.6–44.2) | 606.3 (267.8–1,387.5) | 7.6 (2.0–30.8) | 0.8 (0.2–5.7) | 76.9 (24.4–266.4) | 98.6 | 92.8 |
| 6 | 750/187.5 | 2h | q24h | 76.2 (32.8–173.1) | 45.8 (20.1–108.8)d | 1,426.7 (655.8–2,978.2) | 12.4 (3.5–45.0) | 5.2 (1.7–18.6)d | 197.6 (62.7–655.0)a | 100 | 100 |
Concentrations and AUC0–24h at steady state were obtained from simulations on 1,000 virtual patients and are given as median (2.5 to 97.5 percentiles).
Abbreviations: AUC0–24h, area under the concentration-time curve up over 24 h; ClCr, creatinine clearance; Cmax, maximal concentration at the end of infusion; Cmin, concentration at steady state or plateau concentration in case of continuous infusion; q12h and q24h, administration every 12 h and 24 h, respectively; PTA, probability of target attainment.
Highest median Cmax and AUC0–24h for recommended dosages.
Highest median Cmin for recommended dosages.
The recommended dosage regimen associated with highest median Cmax and AUC0–24h values for CEF and the highest Cmax for AVI was 2,000/500 mg q8h as a 2-h infusion in patients with ClCr of 51 mL/min. Of note, the recommended regimen associated with highest median Cmin for both drugs and the highest AUC0–24h for AVI was 750/187.5 mg q24h in patients with ClCr of 6 mL/min.
In patients with ClCr of 80 mL/min, the alternative dosage regimens using 2,000/500 mg q12h with infusion times of 2 h and 4 h were associated with a PTA of ≥90% for CEF but not for AVI. Increasing the infusion time up to 6 h was necessary to get acceptable PTA for AVI. The maximum concentrations at steady state remained lower than those of the safety references. In patients with ClCr of 51 mL/min, administration of 2,000/500 mg q12h as a 2-h infusion time was sufficient to provide a PTA of ≥90% for both CEF and AVI, without overexposure. As expected, increasing the infusion time was associated with acceptable PTA as well. In patients with ClCr from 31 mL/min to 50 mL/min, 2,000/500 mg q12h as a 2-h infusion was sufficient to provide a PTA of ≥90% for both drugs. Cmax and AUC0–24h values were very close to those of the safety references. In patients with ClCr of 30 mL/min, the administration of 2,000/500 mg q24h appeared to be acceptable in terms of PTA but only if the infusion time was extended to at least 6 h. In patients with ClCr of 16 mL/min, 2,000/500 mg q24h as a 2-h infusion was sufficient to get acceptable PTA. With that regimen, CEF Cmax and AUC0–24h values were slightly higher than those of the safety references, so extending the infusion time could be safer. In patients with a ClCr of ≥6 and ≤15 mL/min, the administration of 2,000/500 mg q24h was associated with PTA of 100% for both ceftazidime and avibactam for all tested infusion times. At the upper bound of 15 mL/min for ClCr, increasing the infusion time to 12 h was necessary to decrease Cmax toward safety reference values. In patients with ClCr as low as 6 mL/min, this regimen was associated with Cmax and AUC0–24h values much higher than those of the safety references.
The alternative dosage regimens of 2,000/500 mg q12h in patients with ClCr from 31 to 80 mL/min and 2,000/500 mg q24h in patients with ClCr from 16 to 30 mL/min could be associated with savings of 33% to 50% in daily drug cost, respectively.
DISCUSSION
Current recommended dosage regimens of TOL-TAZ and CFE/AVI are based on pharmacokinetic/pharmacodynamic (PK/PD) evaluations as well as clinical trials (3, 6, 8, 9). The recommended dosages in patients with renal impairment are sound, as they ensure PK/PD requirements of those time-dependent antibacterials. However, they require to discard a fraction of the dose, which is an issue in hospitals considering the relatively high cost of those drugs compared with that of older agents (10). Our PK/PD simulation study was designed to examine alternative dosing strategies in patients with renal impairment.
Based on our TOL-TAZ and CEF-AVI simulations, administration of a full vial every 12 h or 24 h can be associated with acceptable PTA in patients with renal impairment. Those regimens were not associated with significant overexposures except in a few subgroups of patients with very altered renal function (CEF-AVI 2,000/500 mg q24h in patients with ClCr of 6 mL/min). A simple way to optimize PTA and reduce risk of overexposure is to increase the infusion time. We showed that continuous infusion allows to achieve 100% PTA with low daily doses, but prolonging the infusion time up to 2 h, 4 h, or 6 h may be sufficient to achieve acceptable PTA in most situations.
Prolonged or continuous infusion requires sufficient stability of drugs after dilution in appropriate vehicle. For TOL-TAZ, the product is stable up to 24 h after dilution when stored at room temperature (3). For CEF-AVI, chemical and physical in-use stability after dilution has been reported for up to 24 h at 2 to 8°C, followed by up to 12 h at a temperature not exceeding 25°C (6). Therefore, increasing infusion duration up to 24 h for TOL-TAZ and 12 h for CEF-AVI is feasible.
Less-frequent administration of a full dose of TOL-TAZ and CEF-AVI could have several advantages. Dosing error associated with use of fractions of vial would be prevented. Less-frequent administration of drugs would also reduce indirect health care cost by reducing the nurse labor. Finally, this could facilitate outpatient therapy with TOL-TAZ and CEF-AVI.
Obviously, the ability to achieve a given PK/PD target for a given dosage regimen of antibiotic in patients strongly depends on the target itself. Less-frequent administration of both TOL-TAZ and CEF-AVI was associated with acceptable PTA because bactericidal activity was achieved for relatively low values of time spent above the concentration target, MIC, or threshold concentration (CT).
The choices of MIC, CT, and PK/PD targets can be discussed. We selected relatively high targets in order to ensure efficacy on a large panel of bacteria. For TOL, we used an MIC cutoff value of 4 mg/L that is relevant for P. aeruginosa, but lower values would be acceptable for Enterobacteriaceae. For example, the epidemiological cutoff value (ECOFF) of Enterobacter cloacae and E. coli is 1 mg/L, and the clinical breakpoint for Enterobacteriaceae is fixed by EUCAST at 2 mg/L. Of note, the MIC breakpoint of TOL has recently been updated, and previous studies considered a higher MIC breakpoint of 8 mg/L (11). We considered a PK/PD target defined as the time during which free concentration was above the MIC (fT > MIC) of ≥32.2% for ceftolozane, which was the median value associated with 1-log10 kill for various strains of Enterobacteriaceae (including wild types and ESBLs) and P. aeruginosa (12). While this value showed limited variability across strains and species in the murine model, the results of dosage simulations might have been different if one would consider a pathogen-specific target or a greater bactericidal effect (13). For TAZ, in vitro results with E. coli strains expressing CTX-M-15 showed that the minimal effective concentration CT varied with β-lactamase expression. It was 0.05 mg/L for strains with low and moderate expression and 0.25 mg/L for strains with the high β-lactamase expression (14). We considered the worst-case scenario by selecting CT of 0.25 mg/liter. We set the PK/PD target for TAZ as an fT>CT of ≥35%, this value being associated with net bacterial stasis in experimental studies (14). A higher target might be considered, as an fT>CT of ≥50% was required to achieve a killing effect in vitro. Overall, the target for β-lactamase inhibitors is based on limited evidence, and both the effective concentration and value of the PK/PD objectives required for clinical response are uncertain.
For CEF, an MIC cutoff value of 8 mg/L was selected, which is relevant for P. aeruginosa. Lower values could be considered for other bacteria, such as E. cloacae or E. coli, that display lower ECOFF, although the clinical MIC breakpoint value is established at 8 mg/L for all Enterobacteriaceae. For AVI, in vitro hollow-fiber experiments showed that a CT of ≤0.5 mg/L was sufficient to suppress the growth of β-lactamase-producing isolates of Enterobacteriaceae (15). However, a higher value of 1 mg/L was suggested for ceftazidime-resistant P. aeruginosa based on experiments in murine neutropenic thigh and lung infection models (16). Values of %fT>CT ranging from 0 to 21.4% in the lung model and from 14.1 to 62.5% in the thigh model were required to achieve a static effect, while an fT>CT of ≥50% was associated with 1-log10 kill of P. aeruginosa in thighs of mice. Again, we recognized that the clinical relevance of PK/PD targets defined for AVI remains uncertain.
We did not consider higher target values of fT>MIC, such as 100% fT>MIC or even 100% fT>4× MIC, that have been suggested for beta-lactams in critically ill patients (17), because those values were not consistent with targets used in drug development and labeling. Further research is necessary to determine optimal exposure of TOL-TAZ and CEF-AVI in critically ill patients.
Apart from the choice of PK/PD targets, our work has several limitations. We simulated PK parameters corresponding to only one type of infection (cIAI), so the results may not hold true for other infections such as cUTI and HAP. However, results from the original population PK studies showed that the influences of the various diseases on PK parameters are quite similar (8, 9). Indeed, cIAI appears to have the largest effect on both drug total body clearance (CL) and central volume of distribution (Vc) and might be considered a worst-case scenario in terms of drug exposure. For lung infections, the lung to plasma concentration ratio should also be taken into account in the PK/PD targets for TOL-TAZ. We did not perform simulations in other categorical groups identified for CEF-AVI (e.g., Asian patients, patients with HAP under ventilation), and those may be of interest in future studies. We did not examine exposure and PTA over the first hours of therapy, before steady state was achieved. Although both TOL-TAZ and CEF-AVI displayed a low accumulation index, close to 1 in healthy subjects (18, 19), it may be interesting to examine whether initial exposure is always optimal for those agents, for both approved and alternative dosage regimens in a dedicated study. We could not evaluate the joint target attainment of the beta-lactam and beta-lactamase inhibitor, as the joint distribution of PK parameters was not available. Finally, our findings are based on simulations and the suggested alternative regimens have not been clinically validated. However, our findings are based on updated PK/PD data and modeling and simulation approaches similar to those used in the development of both drugs.
Assessing the relevance of the dosage regimens recommended for TOL-TAZ and CEF-AVI was out of the scope of this study. However, it has been suggested that dosage reduction in patients with moderate renal impairment may result in suboptimal exposure and response (20). Dose reduction may be unnecessary, especially in case of transient acute kidney injury during the first 48 h of therapy, considering the wide therapeutic index of those beta-lactam agents (21). This cautionary statement may also apply to the alternative dosage regimens examined in our study, which have not been clinically evaluated.
Conclusions. To conclude, our PK/PD simulations performed for TOL-TAZ and CEF-AVI in patients with renal impairment showed that alternative dosage regimens based on a full dose administered every 12 or 24 h is a reasonable dosing approach, especially when administered as prolonged infusion. Such alternative dosage regimens may result in PK/PD target attainment similar to that of recommended dosage, without significant overexposure. In addition, those regimens may reduce dosing errors, drug cost, and nurse labor. However, clinical investigations of such alternative dosage regimens are necessary.
MATERIALS AND METHODS
We performed PK/PD simulations of various dosage regimens of TOL-TAZ and CEF-AVI based on previously published population PK models.
Population PK model for ceftolozane-tazobactam.
We used a published population PK model based on data from 10 different clinical studies that enrolled 376 adults, including healthy volunteers, subjects with various degrees of renal impairment, and patients with bacterial infections (cUTI and cIAI) (22). TOL-TAZ disposition was described by a linear two-compartment model. Population PK parameters followed a log-normal distribution. Values of TOL and TAZ PK parameters used in our simulations are shown in Table 5.
TABLE 5.
Ceftolozane-tazobactam and ceftazidime-avibactam pharmacokinetic parameters used in dosing simulationsa,b,c
| PK parameters | Value for drug: |
|||||||
|---|---|---|---|---|---|---|---|---|
| Ceftolozane |
Tazobactam |
Ceftazidime |
Avibactam |
|||||
| Population value | Interindividual variability (CV%) | Population value | Interindividual variability (CV%) | Population value | Interindividual variability (CV%) | Population value | Interindividual variability (CV%) | |
| CL (L/h) | (5.11 × 1.22) × (ClCr/109)0.715 | 33.0% | 18 × (ClCr/115)0.67 | 50.2% | 6.95 × (0.0103 × ClCr) × 1.16 | 11.4% | 10.2 × 1.406 × (ClCr/80)1.05 | 7.29% |
| Vc (L) | (11.4 × 1.59) × (BW/74) | 39.8% | 14.2 × 1.47 | 52.5% | 10.5 × 1.14 × (BW/70)1.01 | 31.2% | 11.1 × 1.329 × (BW/70)1.08 | 28.15% |
| Q (L/h) | 1.19 | None | 3.13 | None | 31.5 | 27.46% | 5.44 | 14.18% |
| Vp (L) | 2.88 | None | 4.29 | None | 7.57 | 17.5% | 6.91 | 13.52% |
Abbreviations: BW, body weight; CL, drug total body clearance; ClCr, creatinine clearance; CV, coefficient of variation; Vc, central volume of distribution; Vp, peripheral volume of distribution; Q, intercompartment clearance.
PK parameter values of ceftolozane-tazobactam were taken from Chandorkar et al. (22). In the original model, ceftolozane, total body clearance (CL) was influenced by ClCr and type of infection (cUTI or cIAI), while central volume of distribution (Vc) was influenced by total body weight and type of infection. For tazobactam, ClCr influenced drug CL and type of infection influenced Vc. We used parameter values defined for patients with cIAI, as tazobactam Vc had no defined value in patients with cUTI in the original publication.
PK parameter values of ceftazidime-avibactam were adapted from Li et al. (23). In the original model, ceftazidime, CL was influenced by ClCr, type of infection (cIAI or NP), and ethnicity. Vc was influenced by total body weight, type of infection (cUTI, cIAI, HAP, or acute pyelonephritis), ethnicity, and ventilation. For avibactam, CL was influenced by ClCr, end-stage renal disease, dialysis, augmented renal clearance, APACHE II score, ethnicity, and cIAI. Vc was influenced by total body weight, type of infection (cUTI, cIAI, or HAP), and ventilation. In our simulations, we used parameter values defined for non-Asian patients with cIAI, in order to be consistent with TOL-TAZ simulations. Other categorical covariates were not taken into account for sake of simplicity.
Population PK model for ceftazidime-avibactam.
We used the population PK model published by Li et al. for CEF-AVI (23). CEF and AVI concentrations were obtained in 1,975 and 2,249 adults, respectively, including patients with cIAI, cUTI, or HAP (including VAP). The final population PK model was also a two-compartment model, with log-normal distribution of random parameters. Values of CAF and AVI PK parameters used in our simulations are shown in Table 5.
Simulations of alternative dosage regimens in patients with renal impairment.
PK/PD simulations were all performed with the R package Pmetrics (24). Structural and covariate models, as well as fixed and random effects describing PK parameter distributions, were encoded within Pmetrics. Simulated values of random PK parameters were generated by sampling from appropriate Gaussian distribution of random effects, in accordance with parameter variability described in the population PK studies and equations described in Table 5. For CEF-AVI, covariance between parameters described by Li et al. was taken into account in simulations (23). Body weight values were sampled from a log-normal distribution, as follows:
where BWi is the simulated individual body weight, 74 is the typical body weight in kg, and ηBWi is the body weight random effect variable that follows a normal distribution. This model corresponds to a weight coefficient of variation of 20.7%. This weight distribution was previously used in the simulation study of TOL-TAZ by Xiao et al. and was representative of weight variability observed in clinical studies (11). The same weight distribution was assumed for simulations with CEF-AVI.
For TOL-TAZ, we simulated the approved dosage regimens provided by the manufacturer for the various stages of renal function described in the summary of product characteristics (3). We also simulated alternative dosage regimens using a full dose of 1,000/500 mg per administration. In alternative dosage regimens, we simulated infusion times of 1 h, 2 h, 4 h, 6 h, and 12 h, as well as prolonged dosing intervals of 12 h and 24 h. We also simulated continuous infusions of 12 h and 24 h. We used fixed ClCr values corresponding to the lower and upper bounds of each stage of renal impairment requiring dose adjustment as defined in the summary of product characteristics (3). Normal, moderately impaired, and severely impaired renal function were defined as ClCr values of 51 to 100 mL/min, 30 to 50 mL/min, and 15 to 29 mL/min, respectively. Simulations in patients with ClCr less than 15 mL/min and under hemodialysis were not performed.
For CEF-AVI, we simulated the approved dosage regimens for the various stages of renal function (6) as well as alternative dosage regimens using a full dose of 2,000/500 mg per administration. We simulated infusion times of 2 h, 4 h, 6 h, and 12 h associated with prolonged dosing intervals of 12 and 24 h. We also simulated continuous infusions of 12 h and 24 h. Fixed ClCr values corresponding to upper and lower bounds of renal function stages were used, as for TOL-TAZ simulations. The stages of renal impairment defined in the summary of product characteristics were 51 to 80 mL/min, 31 to 50 mL/min, 16 to 30 mL/min, and 6 to 15 mL/min (3). Simulations in patients with ClCr less than 6 mL/min and under hemodialysis were not performed.
In each simulation, 1,000 virtual patients and PK profiles were generated. Drug administration was simulated over 10 days. Drug concentrations simulated on the 10th day of therapy were analyzed in order to obtain steady-state results.
PK/PD targets and analysis of simulation results.
For TOL, the target was set at an fT>MIC of ≥32.2%, which has been associated with 1-log10 kill for Gram-negative bacilli in the neutropenic murine thigh infection model (3, 12). It has been shown that the effect of TAZ, when combined with a fixed concentration of TOL in in vitro models, correlated with the percentage of time during which the free plasma concentration of TAZ remains above a concentration threshold (%fT>CT) (14). The primary target was set at an fT>CT of ≥35%, this value being associated with net bacterial stasis of CTX-M-15-producing E. coli regardless of enzyme transcription level (11). We assumed unbound fraction of 80% and 70% for TOL and TAZ, respectively, to derive free concentrations from total concentrations (25).
For CEF, we used a target fT>MIC of ≥50%, which was associated with bactericidal effect in neutropenic mouse infection models and with microbiological eradication in patients with Gram-negative infections (26). The target for AVI was fT>CT of ≥50%, which has been associated with 1-log10 kill of P. aeruginosa resistant to CEF alone in a neutropenic mouse thigh infection model (16). We assumed unbound fractions of CEF and AVI of 90% and 92%, respectively (6).
We calculated the proportion of simulated patients who achieved the PD target (i.e., the probability of target attainment [PTA]) for a realistic range of MIC and CT values. For TOL and CEF, MICs ranging from 0.25 to 32 mg/L were considered, in agreement with MIC distribution provided by EUCAST (27). For TAZ, CT values evaluated were as follows: 0.05, 0.1, 0.25, 0.5, and 1 mg/L (14). For AVI, CT values were set at 0.5, 1, 2, and 4 mg/L (16).
We specifically examined PTA results for cutoff values of MIC and CT. The TOL MIC cutoff was 4 mg/L, which is the TOL-TAZ epidemiological cutoff value (ECOFF) as well as the clinical breakpoint determined by EUCAST for P. aeruginosa (28). The TAZ cutoff value of CT was 0.25 mg/L, corresponding to high level CTX-M-15 producer E. coli in vitro (14). The CEF cutoff value was 8 mg/L, which is P. aeruginosa ECOFF value for CEF alone and also the EUCAST clinical breakpoint for CEF-AVI for Enterobacteriaceae and P. aeruginosa. For AVI, a CT cutoff of 1 mg/L was chosen, because this value best correlated with efficacy on P. aeruginosa resistant to CEF alone in the neutropenic mouse thigh infection model (16). A PTA of ≥90% was considered acceptable in all simulations.
In addition to PD target attainment, we evaluated the risk of overexposure associated with alternative dosage regimens by computing maximal concentration at the end of infusion (Cmax), trough concentration at steady-state or plateau concentration in case of continuous infusion (both denoted as Cmin), and area under the concentration-time curve over 24 h (AUC0–24h). The median and 2.5th and 97.5th percentiles of those quantities were compared between recommended and alternative dosage regimens. The highest Cmax, Cmin, and AUC0–24h values obtained with recommended regimens irrespective of renal function were considered safety upper limits.
Data availability.
All simulated data are available upon request.
ACKNOWLEDGMENTS
This study was carried out as part of our routine work, which is funded by Hospices Civils de Lyon and the University of Lyon.
The authors have no conflicts of interest that are related with this study.
REFERENCES
- 1.Zhanel GG, Chung P, Adam H, Zelenitsky S, Denisuik A, Schweizer F, Lagace-Wiens PR, Rubinstein E, Gin AS, Walkty A, Hoban DJ, Lynch JP, 3rd, Karlowsky JA. 2014. Ceftolozane/tazobactam: a novel cephalosporin/beta-lactamase inhibitor combination with activity against multidrug-resistant gram-negative bacilli. Drugs 74:31–51. 10.1007/s40265-013-0168-2. [DOI] [PubMed] [Google Scholar]
- 2.van Duin D, Bonomo RA. 2016. Ceftazidime/avibactam and ceftolozane/tazobactam: second-generation beta-lactam/beta-lactamase inhibitor combinations. Clin Infect Dis 63:234–241. 10.1093/cid/ciw243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.European Medicines Agency. Zerbaxa summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/zerbaxa-epar-product-information_en.pdf. European Medicines Agency, Amsterdam, The Netherlands. [Google Scholar]
- 4.Zasowski EJ, Rybak JM, Rybak MJ. 2015. The beta-lactams strike back: ceftazidime-avibactam. Pharmacotherapy 35:755–770. 10.1002/phar.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shirley M. 2018. Ceftazidime-avibactam: a review in the treatment of serious Gram-negative bacterial infections. Drugs 78:675–692. 10.1007/s40265-018-0902-x. [DOI] [PubMed] [Google Scholar]
- 6.European Medicines Agency. Zavicefta summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/zavicefta-epar-product-information_en.pdf. European Medicines Agency, Amsterdam, The Netherlands. [Google Scholar]
- 7.Grissinger M. 2010. Reducing errors with injectable medications: unlabeled syringes are surprisingly common. P T 35:428–451. [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao AJ, Miller BW, Huntington JA, Nicolau DP. 2016. Ceftolozane/tazobactam pharmacokinetic/pharmacodynamic-derived dose justification for phase 3 studies in patients with nosocomial pneumonia. J Clin Pharmacol 56:56–66. 10.1002/jcph.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das S, Li J, Riccobene T, Carrothers TJ, Newell P, Melnick D, Critchley IA, Stone GG, Nichols WW. 2019. Dose selection and validation for ceftazidime-avibactam in adults with complicated intra-abdominal infections, complicated urinary tract infections, and nosocomial pneumonia. Antimicrob Agents Chemother 63. 10.1128/AAC.02187-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.National Institute for Health Care Excellence. 2017. Antimicrobial prescribing: ceftazidime/avibactam. Evidence review on ceftazidime/avibactam. https://www.nice.org.uk/advice/es16/resources/evidence-review-pdf-4662945757.
- 11.Xiao AJ, Caro L, Popejoy MW, Huntington JA, Kullar R. 2017. PK/PD target attainment with ceftolozane/tazobactam using Monte Carlo simulation in patients with various degrees of renal function, including augmented renal clearance and end-stage renal disease. Infect Dis Ther 6:137–148. 10.1007/s40121-016-0143-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Craig WA, Andes DR. 2013. In vivo activities of ceftolozane, a new cephalosporin, with and without tazobactam against Pseudomonas aeruginosa and Enterobacteriaceae, including strains with extended-spectrum beta-lactamases, in the thighs of neutropenic mice. Antimicrob Agents Chemother 57:1577–1582. 10.1128/AAC.01590-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lepak AJ, Reda A, Marchillo K, Van Hecker J, Craig WA, Andes D. 2014. Impact of MIC range for Pseudomonas aeruginosa and Streptococcus pneumoniae on the ceftolozane in vivo pharmacokinetic/pharmacodynamic target. Antimicrob Agents Chemother 58:6311–6314. 10.1128/AAC.03572-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.VanScoy B, Mendes RE, Nicasio AM, Castanheira M, Bulik CC, Okusanya OO, Bhavnani SM, Forrest A, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2013. Pharmacokinetics-pharmacodynamics of tazobactam in combination with ceftolozane in an in vitro infection model. Antimicrob Agents Chemother 57:2809–2814. 10.1128/AAC.02513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coleman K, Levasseur P, Girard AM, Borgonovi M, Miossec C, Merdjan H, Drusano G, Shlaes D, Nichols WW. 2014. Activities of ceftazidime and avibactam against beta-lactamase-producing Enterobacteriaceae in a hollow-fiber pharmacodynamic model. Antimicrob Agents Chemother 58:3366–3372. 10.1128/AAC.00080-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berkhout J, Melchers MJ, van Mil AC, Seyedmousavi S, Lagarde CM, Schuck VJ, Nichols WW, Mouton JW. 2016. Pharmacodynamics of ceftazidime and avibactam in neutropenic mice with thigh or lung infection. Antimicrob Agents Chemother 60:368–375. 10.1128/AAC.01269-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guilhaumou R, Benaboud S, Bennis Y, Dahyot-Fizelier C, Dailly E, Gandia P, Goutelle S, Lefeuvre S, Mongardon N, Roger C, Scala-Bertola J, Lemaitre F, Garnier M. 2019. Optimization of the treatment with beta-lactam antibiotics in critically ill patients-guidelines from the French Society of Pharmacology and Therapeutics (Societe Francaise de Pharmacologie et Therapeutique-SFPT) and the French Society of Anaesthesia and Intensive Care Medicine (Societe Francaise d'Anesthesie et Reanimation-SFAR). Crit Care 23:104. 10.1186/s13054-019-2378-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller B, Hershberger E, Benziger D, Trinh M, Friedland I. 2012. Pharmacokinetics and safety of intravenous ceftolozane-tazobactam in healthy adult subjects following single and multiple ascending doses. Antimicrob Agents Chemother 56:3086–3091. 10.1128/AAC.06349-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merdjan H, Rangaraju M, Tarral A. 2015. Safety and pharmacokinetics of single and multiple ascending doses of avibactam alone and in combination with ceftazidime in healthy male volunteers: results of two randomized, placebo-controlled studies. Clin Drug Invest 35:307–317. 10.1007/s40261-015-0283-9. [DOI] [PubMed] [Google Scholar]
- 20.Bidell MR, Lodise TP. 2018. Suboptimal clinical response rates with newer antibiotics among patients with moderate renal impairment: review of the literature and potential pharmacokinetic and pharmacodynamic considerations for observed findings. Pharmacotherapy 38:1205–1215. 10.1002/phar.2184. [DOI] [PubMed] [Google Scholar]
- 21.Crass RL, Rodvold KA, Mueller BA, Pai MP. 2019. Renal dosing of antibiotics: are we jumping the gun? Clin Infect Dis 68:1596–1602. 10.1093/cid/ciy790. [DOI] [PubMed] [Google Scholar]
- 22.Chandorkar G, Xiao A, Mouksassi MS, Hershberger E, Krishna G. 2015. Population pharmacokinetics of ceftolozane/tazobactam in healthy volunteers, subjects with varying degrees of renal function and patients with bacterial infections. J Clin Pharmacol 55:230–239. 10.1002/jcph.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Lovern M, Green ML, Chiu J, Zhou D, Comisar C, Xiong Y, Hing J, MacPherson M, Wright JG, Riccobene T, Carrothers TJ, Das S. 2019. Ceftazidime-avibactam population pharmacokinetic modeling and pharmacodynamic target attainment across adult indications and patient subgroups. Clin Transl Sci 12:151–163. 10.1111/cts.12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. 2012. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 34:467–476. 10.1097/FTD.0b013e31825c4ba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wooley M, Miller B, Krishna G, Hershberger E, Chandorkar G. 2014. Impact of renal function on the pharmacokinetics and safety of ceftolozane-tazobactam. Antimicrob Agents Chemother 58:2249–2255. 10.1128/AAC.02151-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nichols WW, Newell P, Critchley IA, Riccobene T, Das S. 2018. Avibactam pharmacokinetic/pharmacodynamic targets. Antimicrob Agents Chemother 62. 10.1128/AAC.02446-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.EUCAST. Antimicrobial wild type distributions of microorganisms. https://mic.eucast.org/.
- 28.EUCAST. Clinical breakpoints - breakpoints and guidance. https://www.eucast.org/clinical_breakpoints/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All simulated data are available upon request.