

Abstract
Investigating the relationships between protein function and fitness provides keys for understanding biochemical mechanisms that underly evolution. Mutations with partial fitness defects can delineate the threshold of biochemical function required for viability. We utilized a previous deep mutational scan of HIV-1 protease (PR) to identify variants with 15–45 per cent defects in replication and analysed the biochemical function of eight variants (L10M, L10S, V32C, V32I, A71V, A71S, Q92I, Q92N). We purified each variant and assessed the efficiency of peptide cleavage for three cut sites (MA-CA, TF-PR, and PR-RT) as well as gel-based analyses of processing of purified Gag. The cutting activity of at least one site was perturbed relative to WT protease for all variants, consistent with cutting activity being a primary determinant of fitness effects. We examined the correlation of fitness defects with cutting activity of different sites. MA-CA showed the weakest correlation (R2 = 0.02) with fitness, suggesting relatively weak coupling with viral replication. In contrast, cutting of the TF-PR site showed the strongest correlation with fitness (R2 = 0.53). Cutting at the TF-PR site creates a new PR protein with a free N-terminus that is critical for activity. Our findings indicate that increasing the pool of active PR is rate limiting for viral replication, making this an ideal step to target with inhibitors.
Keywords: HIV-1 protease, multi-functional, selection pressure
1. Introduction
Understanding the biophysical and biochemical properties that determine the relationship of genotype with fitness has emerged as a central focus of molecular evolution (Dean and Thornton 2007; Bershtein, Serohijos, and Shakhnovich 2017; Canale et al. 2018a). A wide array of physical properties of proteins can underly phenotype and fitness, including enzyme activity (Kacser and Burns 1981; Boucher, Bolon, and Tawfik 2016) and abundance (Bershtein et al. 2013; Bhagavatula et al. 2017; Canale et al. 2018b; Socha, Chen, and Tokuriki 2019), as well as aggregation, mis-folding, or mis-localization (Geiler-Samerotte et al. 2011; Mehlhoff et al. 2020). Among these protein features, activity stands out as an important contributor because it can be directly related to fitness and many other protein features impact net activity (e.g. abundance, aggregation, mis-folding, etc.).
For enzymes, activity can be directly related to fitness using flux models (Kacser and Burns 1981). However, flux models are challenging to parameterize and have not been established for most proteins. Systematic analyses of knockouts identify genes that dramatically impact growth rate (Giaever et al. 2002). However, because knockouts are complete loss of function, they do not provide information on the impacts of reduced activity on fitness. Pioneering efforts from Kacsur and Burns demonstrated that the activity of many proteins must be dramatically reduced before growth is noticeably decreased (Kacser and Burns 1981). Importantly, this means that most essential proteins are not rate limiting for growth. The main approach taken to analyse activity–fitness relationships are based on protein perturbations. Protein activity can be manipulated by exogenous inhibitors (Kacser and Burns 1981) or engineering strategies that reduce protein abundance (Bershtein et al. 2013) or the specific activity of each protein molecule (Jiang et al. 2013). High-throughput analyses of the effects of mutations on growth rate and/or protein activity provide new approaches to investigate activity–fitness relationships (Melnikov et al. 2014; Roscoe and Bolon 2014; Mishra et al. 2016; Livesey and Marsh 2020; Mehlhoff et al. 2020).
Understanding how proteins with multiple activities contribute to fitness is important because many proteins perform ‘moonlighting’ activities (Piatigorsky and Wistow 1989; Jeffery 2003). For proteins that perform multiple activities, it can be a challenge to disentangle the impacts of each one on fitness. For example it can be difficult to determine which activities are rate limiting for growth. Here, we develop an approach building on deep mutational scanning fitness measurements to analyse function–fitness relationships in a multi-functional protein. We used previously determined high-throughput fitness measurements (Boucher et al. 2019) to identify a panel of mutations with partial growth defects and quantified the impacts of these mutations on multiple activities. We chose to analyse HIV-1 protease because it cuts at twelve different sites that each represent distinct steps in the viral life cycle.
While HIV protease has been studied extensively over the past 40 years, the relationship between cutting activity at each site and fitness remains poorly defined. A wealth of evidence demonstrates that cutting by protease is critical for viral fitness. Mutations at cut sites that prevent protease cutting result in non-infectious viral particles (Pettit et al. 1994; Wiegers et al. 1998). Mutations in protease that dramatically reduce function or inhibitors that block protease activity also result in non-infectious viral particles (Kohl et al. 1988; Meek et al. 1990; Kaplan et al. 1993). Most of the thousands of copies of matrix and capsid in an HIV virion need to be severed by protease in order to generate infections particles (Lee, Harris, and Swanstrom 2009; Muller et al. 2009). Cutting of MA-CA peptides has been used as a metric of activity to investigate how protease activity relates to fitness (Rose, Babe, and Craik 1995, 1995). This study found that the T26S mutation reduced MA-CA cutting to 25 per cent of wild-type levels and supported robust infection in cell culture, while the A28S mutation reduced cutting to about 2 per cent of wild type and resulted in non-infectious viral particles. However, a more recent kinetic model of HIV protease indicated that cutting of MA-CA is unlikely to be rate limiting for fitness (Konnyu et al. 2013) and instead highlighted that the initial cleavage of PR out of the polyprotein was more likely to be rate limiting. However, this modelling study highlighted that more experimental studies are required to appropriately parameterize rate-limiting steps for fitness.
Here, we experimentally analysed the cutting proficiency of a set of eight protease variants on the threshold of viability for multiple cut sites. We used a previously determined experimental fitness landscape of all protease point mutations (Boucher et al. 2019) to select our panel of partial loss of fitness variants. Because most mutations reduce protein function (Baase et al. 2010), we hypothesized that partial loss of fitness mutations would predominantly exhibit reduced activity. We assayed the cutting activity of our panel of protease variants for the Gag polyprotein and three peptides representing the MA-CA site, as well as sites required to free PR from the polyprotein (TF-PR, and PR-RT).
2. Results
We selected a set of eight mutations that caused partial replication defects in cell culture (Fig. 1A) based on a previously published protein fitness landscape of HIV-1 PR (Boucher et al. 2019). Mutations were selected that had lower fitness than wild-type synonyms (average − 2 standard deviations or SD) and greater fitness than null alleles (average stop + 2 SD). Within this range, eight mutations at four positions were chosen for detailed analysis (Table 1). To investigate the breadth of potential impacts of mutations on PR activity, we selected positions located in different regions. These positions included two buried locations: V32 that is close to the active site and A71 that is more distal. We also selected two positions at the interface between the protein and solvent with L10 close to the active site and Q92 further away. To distinguish if the position or the identity of the mutation has a larger role in determining functional effects, two mutations were chosen at each of these positions. We chose mutations with modest physical changes in order to reduce the likelihood of dramatically altering PR structure. Consistent with the previously observed defects in replication (Boucher et al. 2019), all the selected mutations are rarely if ever observed in drug-naïve clinical isolates (Table 1). Two of the mutations in this set (V32I and A71V) have been shown to contribute to resistance to protease drugs (Weber and Agniswamy 2009), potentially providing opportunities to further understand the mechanisms of antiviral escape.
Figure 1.
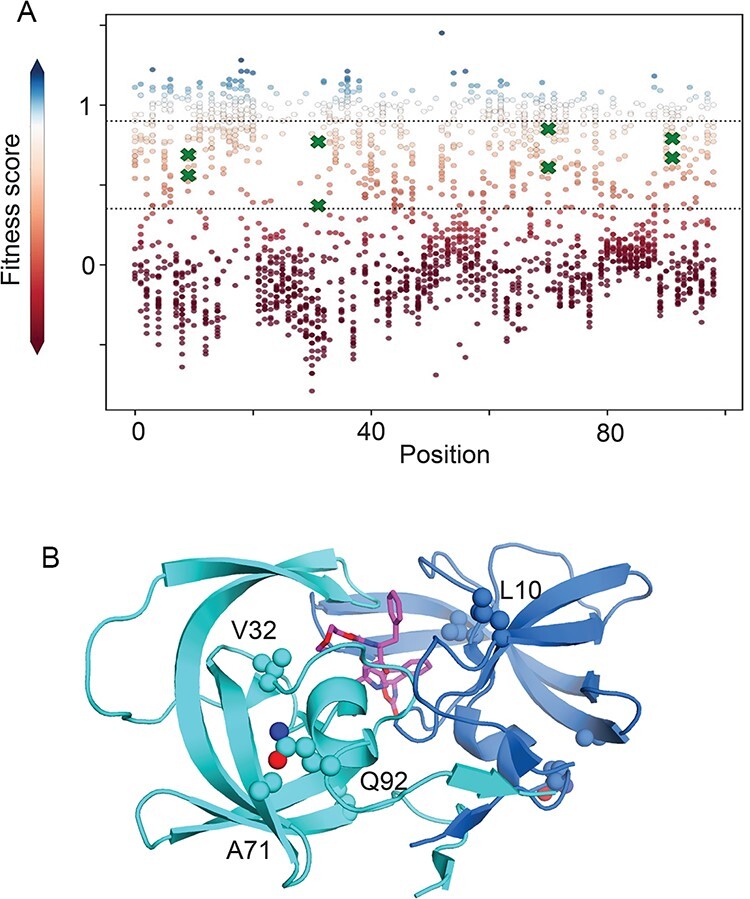
HIV-1 PR mutations selected for this study. (A) Fitness score of point mutations with partial fitness defects that were chosen for this study are indicated with green X symbols. For comparison, the fitness score for all point mutations of HIV-1 PR are indicated with circles. Fitness scores are scaled from wild type (fitness score = 1, shaded white) to null (fitness score = 0, shaded red). Dashed lines indicate the cut-offs used to identify partial fitness defects and correspond to 2 SD below the average WT synonym and 2 SD above the average stop codon. Source data for the fitness scores of all HIV-1 protease single mutants is found in Boucher et al. (2019). (B) Structural model of HIV-1 PR based on 2BB9.PDB2 (Smith et al. 2006) illustrating sites of point mutations chosen for analysis. An inhibitor that is shown in magenta is located at the PR active site.
Table 1.
Panel of PR mutants investigated.
| Mutant | FSa | Frequencyb | DRc | Codon changed |
|---|---|---|---|---|
| L10M | 0.69 | 0.0007 | CTG→ATG | |
| L10S | 0.56 | 0.00015 | TTA→TCA | |
| V32C | 0.77 | 0 | GTC→TGC | |
| V32I | 0.43 | 0.0006 | x | GTT→ATT |
| A71S | 0.61 | 0.00007 | GCG→TCG | |
| A71V | 0.85 | 0.07 | x | GCG→GTG |
| Q92I | 0.79 | 0 | CAA→ATA | |
| Q92N | 0.67 | 0.00002 | CAG→AAC |
Fitness score of mutant as determined by Boucher et al. (2019).
Frequency of mutation in drug-naïve patient samples from the Stanford data base (Rhee et al. 2003).
Association of mutation with drug resistance.
Minimal base changes required to create the amino acid change.
We quantified the enzymatic activity of each of the eight PR variants. Each variant was bacterially expressed and purified to near homogeneity (Supplementary Fig. S1). For biochemical experiments, all PR variants were generated in the HIV-1 NL4-3 sequence background. We added the Q7K mutation to all variants in order to stabilize protease to self-cleavage as in previous studies (Louis, Clore, and Gronenborn 1999).
We measured enzymatic proficiency of PR variants to three cleavage sites using peptide substrates with fluorescent probes capable of Förster Resonance Energy Transfer (FRET) similar to previous studies (Matayoshi et al. 1990). We analysed the MA-CA cut site as well as the TF-PR and PR-RT sites that must be processed in order to generate mature PR with free N- and C-termini. While we would have liked to analyse detailed kinetics of all the cutting sites in the HIV-1 proteome, optimizing peptide assays for all twelve sites is challenging and beyond the scope of this work that was focused on an initial investigation of relationships between activity and fitness in PR. Because of challenges working with the fluorescent peptides at high concentration, we were not able to determine saturation kinetics for all cut sites. We observed peptide precipitation at concentrations around 20 μM for the TF-PR and PR-RT substrates that is well below their respective Kms of 250 and 1,700 μM (Mahalingam et al. 1999). The MA-CA substrate was soluble to 40 μM, which is close to the 60 μM Km reported for wild-type PR with this substrate (Velazquez-Campoy et al. 2001). For all three substrates, we were able to estimate enzyme proficiency (kcat/Km) based on the linear increase in reaction velocity at substrate concentrations well below Km (Fig. S2). For MA-CA substrate, we could also make reasonable estimates of kcat and Km based on non-linear changes in reaction velocity at substrate concentrations close to the Km (Fig. S3).
We considered the likely biological relevance of enzyme proficiency, and kcat based on estimates of the concentration of each substrate in HIV virions (Muller et al. 2009). The Gag protein that contains most of the MA-CA sites is at a concentration of 3.6 mM in virions, which is well above the 60 μM Km for MA-CA. In contrast, the Gag-Pol protein that contains the TF-PR and PR-RT sites is at a concentration of 195 μM that is below the Kms for these sites (250 and 1,700 μM, respectively). At these estimated concentrations, the initial rate of cutting of MA-CA should be determined primarily by kcat, while cutting of TF-PR and PR-RT should be determined largely by enzyme proficiency. Of note, the concentration of uncut sites decreases as cutting proceeds over the course of viral maturation. At the later stages of viral maturation with reduced substrate concentrations, PR enzyme proficiency will play a larger role in determining reaction velocities. Because almost all MA-CA sites must be cut in order to generate infectious HIV-1 particles (Lee, Harris, and Swanstrom 2009), PR enzyme proficiency for MA-CA may be relevant to viral fitness.
As the protein purification procedure involves denaturation and refolding and PR is also subject to inactivation by autocleavage, we were concerned that different PR variants could vary in the fraction of molecules in an active conformation. To address this concern, we performed titrations with a tight-binding inhibitor (Darunavir; DRV) to estimate the concentration of active PR in preparations of each variant (e.g. Fig. S4). We noted variation in the ratio of active PR to total PR for the different variants with a range of 0.45–0.97. Because the refolding procedure was performed with purified proteins that do not include chaperones, they may not be indicative of folding in vivo. We were not able to obtain enough purified protein of one variant (L10M) to perform enzyme activity measurements, and we dropped this variant from our panel.
We compared the enzyme proficiency of each variant with its experimental fitness (Fig. 2A). Prior to these experiments, we hypothesized that mutations that caused fitness defects would generally decrease the cutting activity of PR for substrates. We observed that five of the seven variants we analysed exhibited increased enzyme proficiency relative to WT for at least one of the peptide substrates (Table 2 and Table S1). We measured enzyme proficiency for twenty-one peptide/PR variant pairs, and eight of these measurements indicated increased activity relative to WT, while thirteen decreased the observed activity (Fig. 2B). These observations indicate that mutations in PR that cause partial fitness defects are 60 per cent as likely to cause increased activity for a specific substrate compared to decreased activity. These findings are consistent with stabilizing selection (Adams 1988) on PR function where either too much or too little activity on specific cut sites is detrimental to viral propagation. As further discussed in the conclusions, multiple factors may contribute to this type of selection, including fitness dependencies on the order that sites are cut (Wiegers et al. 1998).
Figure 2.
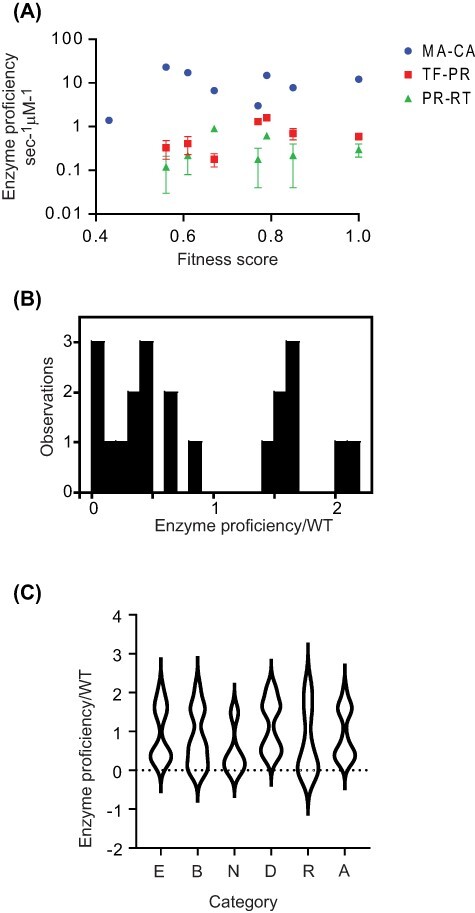
Mutations with partial fitness defects exhibit both increased and decreased enzyme activity for individual cut sites. (A) The enzyme proficiency of each variant was determined using FRET assays for three cleavage sites. Error bars indicate SD for three experimental replicates. (B) Distribution of the relative activity for all cut sites and all PR variants analysed. (C) Distribution of relative enzyme activity for different types of mutations in PR.
Table 2.
Proficiencies of mutant PR variants.
| Enzyme proficiency/WTb | ||||
|---|---|---|---|---|
| Mutant | FSa | MA-CA | TF-PR | PR-RT |
| V32I | 0.43 | 0.11 | NDc | NDc |
| L10S | 0.56 | 1.89 | 0.55 | 0.40 |
| A71S | 0.61 | 1.41 | 0.68 | 0.73 |
| Q92N | 0.67 | 0.55 | 0.30 | 3.00 |
| V32C | 0.77 | 0.25 | 2.17 | 0.60 |
| Q92I | 0.79 | 1.22 | 2.67 | 2.07 |
| A71V | 0.85 | 0.64 | 1.17 | 0.73 |
| WT | 1 | 1 | 1 | 1 |
Fitness score of mutant as determined by Boucher et al. (2019). Partial experimental replicates indicate FS RMSD of 0.1–0.15 for scores in this range.
Enzyme proficiency measured as kcat/Km for peptide substrates.
Not detected.
We examined if the structural location of mutations or association with drug resistance influenced the activity of mutant variants (Fig. 2C). Across all peptides analysed, the normalized proficiency due to mutations at buried sites (average = 0.80) was slightly lower (P > 0.05, Kruskal–Wallis) than at exposed positions (average = 0.95) despite similar impacts on fitness in both of these categories (average = 0.67 for both). Drug-resistant mutations also exhibited a slightly reduced average proficiency (0.71) compared to mutations not associated with resistance (0.92), but this also was not statistically significant (P > 0.05, Kruskal–Wallis). A far greater difference was observed between mutations grouped by distance from the active site: average activity was 0.46 for mutations closer to the active site compared to 1.16 further away (P < 0.05, Kruskal–Wallis). Mutations further away had an average fitness that was higher than those closer to the active site (0.73 compared to 0.59) that may contribute to the different impacts on activity. In addition, these findings suggest that mutations near the active site may be more likely to impact fitness by reducing activity for most cut sites, while mutations further from the active site may tend to impact fitness by altering the relative rates or the order of cutting at different sites.
We also observed a dramatic variation in observed rates for different PR variants (Table 2). For example, we observed extremely low proficiency for V32I for any of the peptide substrates, while Q92I exhibited increased proficiency relative to WT PR for all three peptide substrates. The rate of cutting of MA-CA peptide by V32I is 30-fold reduced relative to WT. To our knowledge, this is the least activity of PR that has been observed to be compatible with viral replication (Boucher et al. 2019). Of note, the V32I mutation shows strong correlations with secondary mutations in patient isolates that may alleviate its functional defects (Wu et al. 2003). The largest increase in proficiency we observed was about 2-fold for Q92I cutting PR-RT and for A71V cutting TF-PR. As modest increases in activity are often compatible with high fitness (Dean, Dykhuizen, and Hartl 1986; Dykhuizen and Dean 1990), it is plausible that the observed fitness defects are caused by perturbations to proteolysis rates at sites that we did not measure in this study. Of note, six of the seven PR variants showed a decreased proficiency for at least one of the peptide substrates we measured, consistent with the idea that reduced activity of PR variants may be a primary determinant of fitness defects.
We searched for correlations between the cutting rate of different sites and fitness effects (Fig. 3). To begin, we investigated how noise in fitness scores would impact these correlations (Fig. S5). We could not directly estimate fitness measurement variation for the set of mutants in this study because the high-throughput fitness scores were only replicated for a subset of positions (Boucher et al. 2019). Therefore, we used a bootstrapping approach to explore the correlation for randomly selected sets of mutations with similar fitness scores that were experimentally reproduced. Using this bootstrap approach we find an average R2 between fitness replicates of 0.6 with 86 per cent of sample sets having R2 > 0.4 (Fig. S5). Thus, we expect the upper bound or potential correlations with enzyme activity to be around an R2 of 0.6.
Figure 3.
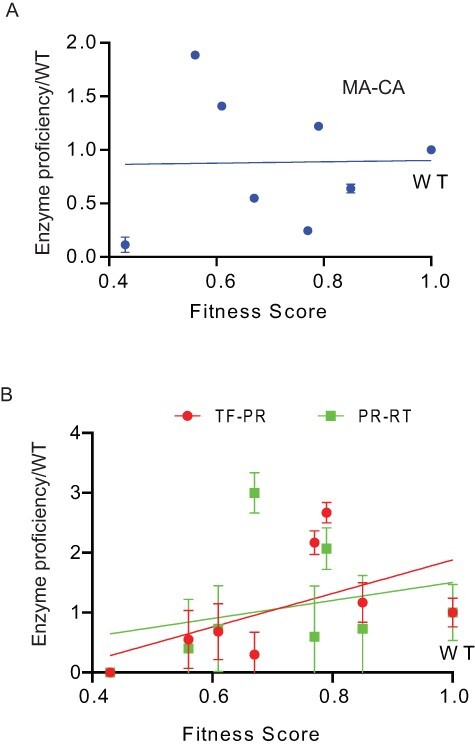
The fitness of mutations shows a stronger correlation with activity for both the TF-PR and PR-RT sites than for the MA-CA site. Plots comparing fitness with enzyme activity for the MA-CA cut site (A) as well as the TF-PR and PR-RT sites (B). Error bars indicate SDs based on three experimental replicates with errors propagated.
Recent kinetic models suggest that a wide range of PR activity for the MA-CA cut site are compatible with robust viral fitness (Konnyu et al. 2013). However, multiple biochemical analyses suggested that MA-CA cleavage would be an important determinant of viral fitness: cutting of thousands of copies of MA-CA are required for the infectivity of viral particles (Lee, Harris, and Swanstrom 2009), MA-CA is one of the fastest cut sites suggesting that it is under strong positive selection (Pettit et al. 1994), and MA-CA has been used as a standard to monitor PR activity (Rose, Babe, and Craik 1995). In experiments on our panel of PR variants, we observed almost no correlation between the proficiency of PR variants for MA-CA and fitness (Fig. 3A). We also observed very weak correlations between estimates of kcat for MA-CA cleavage and fitness score (Fig. S6). Similar to results from a kinetic model (Konnyu et al. 2013), our results indicate that a wide range of PR activity for MA-CA is compatible with efficient viral replication.
We also examined the correlation between proficiency for TF-PR and PR-RT sites (Fig. 3B). Cutting of these sites is required to free the N- and C-terminus of PR from the polyprotein. We observed a positive correlation between fitness and cutting at both TF-PR and PR-RT. The proficiency of PR variants towards TF-PR and PR-RT are better predictors of fitness effects than MA-CA. This does not indicate that these are the only sites important for fitness as cutting of other sites are also critical for HIV-1 replication. One possible reason for the correlations of TF-PR and PR-RT with fitness is that cutting of these sites leads to mature PR, which impacts total PR activity. Cleavage of PR from the HIV-1 polyprotein, especially at the N-terminus of PR (Louis, Clore, and Gronenborn 1999), dramatically increases its activity. PR variants that are slow to cut PR out of Gag-Pol will be slow to accumulate mature PR, further impacting the cutting of other sites in Gag and Gag-Pol. The impact of PR mutations on its own maturation impacts the processing of all other sites.
Freeing the N-terminus of PR has a far greater impact on PR activity than freeing the C-terminus (Louis, Clore, and Gronenborn 1999). Consistent with this property, we observe a more consistent trend between fitness and cutting of TF-PR that frees the N-terminus compared to PR-RT that frees the C-terminus. The four mutations with defects for TF-PR relative to WT also have the lowest fitness, while the three mutations that increase TF-PR activity relative to WT have the highest fitness. In addition, we find a stronger correlation between the fitness effects of PR variants (Boucher et al. 2019) and a previous report by Mahalingam et al. (1999) of cutting efficiency for TF-PR compared to CA/P2 and PR-RT (Fig. S7). These findings are consistent with the idea that the efficiency of PR maturation impacts the cutting at all other sites that in turn broadly impact viral fitness.
To investigate how each PR variant impacts the cutting of additional sites in a more native context, we monitored the cleavage of the Gag polyprotein. In viruses, Gag is generated in vast excess to Gag-Pol and, thus, its cut sites represent the most abundant sites that PR needs to process. Following previously developed procedures (Pettit et al. 2002), we monitored the conversion of Gag by WT PR into most of its constituent proteins and intermediates (Fig. 4A). Consistent with prior studies. we observe that WT PR cleaves the SP1/NC and MA-CA sites early such that the first intermediate observed is MA-CA with the unresolvable SP1 attached and NC-SP2-P6 followed by accumulation of free MA and CA (Fig. 4A and B). We were not able to observe all intermediates, including those that were too small to resolve by PAGE and those whose migration overlapped with others.
Figure 4.
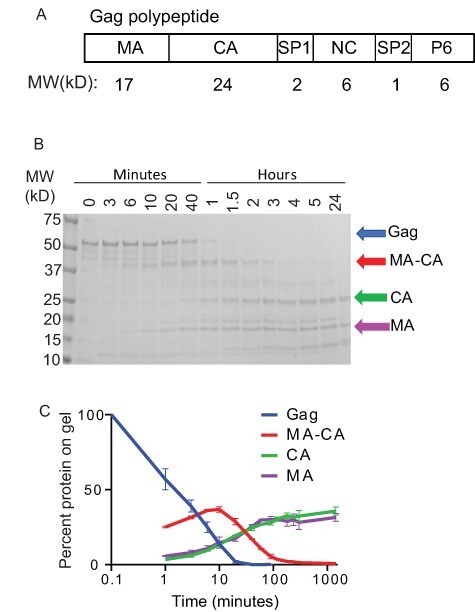
Tracking the degradation of Gag. (A) Representation of Gag illustrating each of the PR cut sites. (B) Cleavage of Gag by WT PR was followed by SDS-PAGE. (C) Quantification of each species over the course of the reaction.
The disappearance of Gag and the appearance of MA-CA were readily interpretable and we focused on these for analyses of PR variants (Fig. 5). Analysing the disappearance of Gag, we observed a range of impacts of PR mutations (Fig. 5A and B). Four variants (L10S, A71S, Q92N, and Q92I) exhibited small to modest changes in the disappearance of Gag (Fig. 5A), while V32I, V32C, and A71V showed large delays in the initial cleavage of Gag (Fig. 5B). The initial cleavage of Gag does not show a clear trend with fitness. For example, L10S had the lowest fitness score, but processed Gag faster than any other mutant variant while A71V had the highest fitness score among the mutant variants but showed a large delay in Gag cleavage. Of note, L10S has the second lowest activity for the TF-PR site in our analyses of peptide substrates, which should slow accumulation of mature protease and hinder the processing of all sites during viral maturation. The accumulation and disappearance of MA-CA also differs widely among the PR variants. The peak quantity of MA-CA (Fig. 5C) is lower than WT for three variants (L10S, Q92I, and Q92N), suggesting that the rate of formation relative to disappearance is lower for these PR variants relative to WT. In contrast, the peak quantity is dramatically higher for V32I and V32C (Fig. 5D) consistent with this intermediate being slower to process by these PR variants relative to WT. Of note, V32I and V32C also exhibited low proficiency for the MA-CA peptide (Fig. 2) consistent with their slow processing of MA-CA in the gel assay. The initial processing of Gag varies widely for PR variants that are at the threshold of replicative viability.
Figure 5.
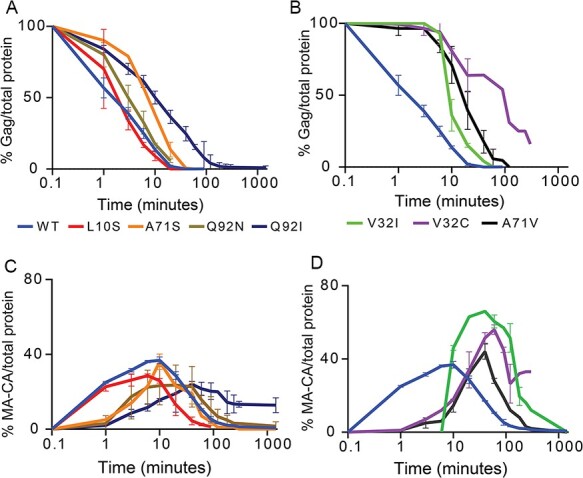
Gag processing varies widely among PR variants studied. The amount of full-length Gag (A and B) and the MA-CA intermediate (C and D) over time are shown for different PR variants.
3. Discussion
Relationships between activity and fitness are typically non-linear with large regions of activity leading to either null or WT-like fitness (Kacser and Burns 1981; Sanjuan, Moya, and Elena 2004; Wylie and Shakhnovich 2011; Bershtein et al. 2013; Jiang et al. 2013). Identifying rare partial loss of fitness mutations provides a powerful approach to investigate potential rate-limiting steps for fitness.
While our results provide insights into relationships between PR activity and fitness, there are limitations to our approach that are important to consider. For example, we assay a fraction of the PR activities and properties that contribute to fitness. We have not directly investigated stability to degradation that can impact in vivo PR function. We have not analysed the activity of PR for many of the sites in the polyprotein that are also critical for infectivity. We do not know how the unstudied sites contribute to intermediate fitness effects. Further work will be required in order to develop and assess quantitative models of how each site and the relative order of sites may impact fitness. We have assessed our results with these limitations in mind.
Mutations in PR at the threshold of viability exhibited widely varied impacts on cutting activity of different substrates. Among the PR variants and sites that we tested, we observed many instances of increased cutting activity, although we observed more instances of decreased activity. However, the magnitude of changes in activity were stronger for decreases (down to nearly undetectable levels for V32I cutting of MA-CA) compared to increases in activity (maximum of 2-fold relative to WT). All but one of the variants we analysed exhibited a decreased rate of cutting for at least one site, suggesting that decreases in activity may be a common cause of fitness defects. However, Q92I exhibited WT or better enzyme activity for the three substrates we analysed quantitatively, suggesting that hyperactive PR activity may also cause fitness defects. Alternatively, it is possible that Q92I destabilizes the accumulation of active PR in viruses so that Vmax in vivo is decreased even if kcat is increased. Further investigations of all cut sites in the proteome as well as PR abundance in virions will be required to address these issues with confidence.
Consistent with its multiple functions, there does not appear to be a single explanation or biochemical pattern for PR variants at the threshold of viability. Because HIV protease recognizes a common substrate shape (Prabu-Jeyabalan, Nalivaika, and Schiffer 2002), we conjectured that many mutations would have similar impacts on the catalytic rate of all substrates and that we might find a common threshold activity required for fitness. Instead, mutations in PR tend to vary in their impacts on different cut sites. Interestingly, cutting of MA-CA was a particularly poor predictor of intermediate fitness defects. Cutting of MA-CA is required for infectivity (Rose, Babe, and Craik 1995; Gay et al. 1998), so it is critical to cut this site. However, cutting of MA-CA does not appear to be rate limiting for viral infectivity.
Our observation that increased activity for specific cut sites was common among mutations on the threshold of viability suggests that stabilizing selection may act on protease where too much or too little activity can both be deleterious for fitness. Both the overall activity of PR (Rose, Babe, and Craik 1995) and the order of cutting of different sites (Wiegers et al. 1998) have been hypothesized to contribute to viral fitness, and both could contribute to stabilizing selection. The order of cutting can be disrupted by either increasing or decreasing the rate of an individual cut site. However, from our results, we cannot clearly distinguish the relative contributions of overall processing activity and order of cutting to the fitness of PR variants.
The cleavage of PR out of polyproteins generates mature and active PR and is a relatively strong predictor of partial fitness defects (Fig. 3). Having a free N-terminus is required for efficient PR activity (Rosé, Salto, and Craik 1993; Zybarth et al. 1994) because the structure cannot accommodate any extensions at this location. Cutting at the N-terminus of PR can control the availability of mature PR to cut all other sites (Fig. 6). This model provides a plausible explanation for the improved correlation of fitness effects with the cutting activity of TF-PR compared to MA-CA. In this model, defects in cutting MA-CA and other sites in Gag and Gag-Pol in viruses will be impacted by the amount of mature PR that is available.
Figure 6.
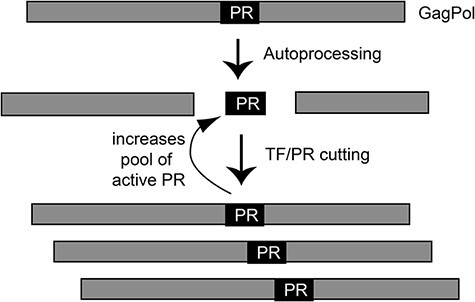
Illustration of how cutting at the TF-PR site impacts the pool of active PR available for cutting other sites. Because the total pool of active PR will impact the cutting of all other sites, the fitness impacts of cutting TF-PR may be amplified.
4. Materials and methods
4.1. Generation of HIV-1 PR variants
HIV-1 PR variants were generated as previously described (King et al. 2002; Özen et al. 2014). In brief, the protease from the HIV-1 strain, NL4-3, was constructed using codon optimization for protein expression in E. coli and was cloned into the bacterial expression vector pET11 (Novagen). The Q7K mutation was introduced to prevent autoproteolysis; the resulting construct is referred to as ‘WT HIV-1 PR’ throughout the manuscript. Additional variants of HIV-1 PR were generated by site-directed mutagenesis and confirmed by Sanger sequencing.
4.2. Protein expression and purification
The bacterial expression and purification of all HIV-1 PR variants were carried out essentially as previously described (King et al. 2002; Özen et al. 2014; Windsor and Raines 2015). Briefly, the plasmid encoding HIV-1 PR was transformed into BL-21(DE3) pRIL bacterial cells. Cells were grown at 37°C in 2xYT to an OD600 of 1, induced by the addition of 2 mM IPTG, grown for an additional hour, and harvested by centrifugation. Following lysis with a cell disruptor, HIV-1 PR was purified from inclusion bodies. The inclusion body pellet was dissolved in 50 per cent acetic acid and HIV-1 PR was further purified using a 100 ml Sephadex G-75 superfine column (Sigma Chemical) equilibrated with 50 per cent acetic acid. Fractions containing HIV-1 PR were combined and refolded by 10-fold dilution into ice cold refolding buffer (0.05 M sodium acetate, pH 5.5, 5 per cent ethylene glycol, 10 per cent glycerol, 5 mM DTT). Purified HIV-1 PR was concentrated using Amicon-10 concentrators and stored at −80°C. The total protein concentration for each variant protease dimer was measured by absorption at 280 nm using an extinction coefficient of 24,980. The percentage of active refolded HIV-1 PR for each purified variant was estimated by titration with a potent inhibitor as described below.
4.3. FRET assay to measure enzymatic activity of HIV-1 PR variants
To determine the enzymatic activity of each HIV-1 PR variant, a FRET assay was performed as previously described (Matayoshi et al. 1990). In this assay, the PR substrates consist of an eight amino acid cleavage site peptide with a fluorescent donor, 5-[(2-aminoethyl) amino] naphalene-1-sulfonic acid (EDANS), attached at the N-terminus and a quenching acceptor, 4-(4-dimethylaminophenylazo)benzoic acid (DABCYL), attached at the C-terminus. The amino acid substrate sequences are as follows (the dash denotes the site of peptide bond cleavage): MA-CA: SQNY-PIVQ; TF-PR: SFSF-PQIT; PR-RT: TLNF-PISP. The fluorescence of EDANS is low in these substrates due to quenching by DABCYL. EDANS fluorescence is increased following the cleavage of the peptides and separation from DABCYL, and thus the rate of cleavage can be measured by the increase in fluorescence emission at 492 nm over time. Due to varying degrees of solubility for each peptide, the MA-CA peptide was dissolved to a final concentration of 4–40 µM in 3–4 per cent DMSO, PR-RT was dissolved to 2–20 µM in 3–4 per cent DMSO, and TF-PR was dissolved to 1.6–15 µM in 4 per cent DMSO. We analysed the impact of DMSO concentration on PR activity and observed minimal impacts for 1–5 per cent DMSO (Supplementary Fig. S8). The peptide substrates at a range of concentrations were incubated with 20–30 nM HIV-1 PR in 50 mM sodium acetate, pH 5.5, 100 mM NaCl, in a 96-well plate. The change in fluorescence over time was measured in a PerkinElmer Envision plate reader with an excitation at 340 nm and emission at 492 nm and monitored with 200 readings over a 23-minute time course. The initial rate of cleavage was measured for a range of substrate concentrations up to the point where visible substrate precipitation began to occur (>40 μM for MA-CA, and >15 μM for TF-PR, and >20 μM for PR-RT peptides). We were unable to perform experiments at sufficiently high concentrations of the TF-PR and PR-RT substrates to determine kcat and Km independently. Instead we calculated enzyme proficiency (kcat/Km/nM of active enzyme) as the linear slope of reaction velocity versus substrate concentration. For MA-CA cleavage, we determined kcat/Km as for TF-PR and also independently estimated kcat and Km by fitting graphs of initial velocity versus substrate concentration to the Michaelis–Menten equation.
4.4. Measuring the fraction of active refolded HIV-1 PR for each variant
To determine the active enzyme concentration of each HIV-1 PR variant, the initial velocity of each variant was measured using MA-CA cutting as an assay in the presence of a varied concentration of the potent HIV-1 PR inhibitor DRV. Ten micromolars of the substrate peptide MA-CA was incubated with 10–30 nM of total HIV-1 PR protein, as calculated by absorbance at 280 nm, in the presence of 2–25 nM DRV, and the initial enzyme activity was measured using FRET as above. The effect of inhibitor concentration on HIV-1 PR activity was detected in two phases. The first one was a steep linear decrease in velocity as free protease decreased and the second was a flat phase when virtually all of the protease molecules were occupied with inhibitor (e.g. Fig. S2). The inflection point of the two linear phases represents the concentration of active protease in the assay and was calculated using GraphPad Prism 8 software. Each assay was performed in triplicate for each preparation of HIV-1 PR, and the average was taken to calculate the final active protease concentration.
4.5. Gag polyprotein cleavage assay
The pET28a plasmid containing the full-length Pr55Gag-TEV-His construct was a kind gift from the laboratory of John Flanagan. Protein expression and purification was performed as described (Bewley et al. 2017) using Co-NTA affinity chromatography. The Pr55Gag-TEV-His (Gag) protein concentration was calculated by absorption at 280 nm (extinction coefficient = 65,000 M−1cm−1).
Cleavage of Gag polyprotein by HIV-1 protease was monitored by the separation of cleavage products using SDS-PAGE (4–20 per cent gradient) (Bio-Rad) and visualized by Coomassie staining. 0.25 µM HIV-1 PR was incubated with 20–25 µM of Gag in 50 mM sodium acetate, 100 mM NaCl buffer at 25°C. Samples were taken from the reaction mixture at time points, and the cleavage reaction was quenched by adding SDS-PAGE gel running buffer containing 2 per cent SDS that was immediately boiling for 2 min. Amprenavir or DRV, potent HIV-1 protease inhibitors, were used in control reactions to indicate specificity of cleavage. The density of gel bands was quantified by an Amersham Imager 600 and analysed by the software provided by the manufacturer.
Supplementary Material
Acknowledgement
This work benefitted from useful discussions and suggestions from Mohan Somasundaran and Neha Samant.
Contributor Information
Gily Schneider-Nachum, Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, 364 Plantation St, Worcester, MA 01605, USA.
Julia Flynn, Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, 364 Plantation St, Worcester, MA 01605, USA.
David Mavor, Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, 364 Plantation St, Worcester, MA 01605, USA.
Celia A Schiffer, Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, 364 Plantation St, Worcester, MA 01605, USA.
Daniel N A Bolon, Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, 364 Plantation St, Worcester, MA 01605, USA.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
This work was supported by grant R01GM112844 from the National Institutes of Health to D.N.A.B. C.A.S. was supported by R01GM135919.
Conflict of interest
None declared.
References
- Adams M. B. (1988) ‘A Missing Link in the Evolutionary Synthesis. “Factors of Evolution: The Theory of Stabilizing Selection”. By I. I. Schmalhausen. Essay Review’, Isis, 79: 281–4. [DOI] [PubMed] [Google Scholar]
- Baase W. A. et al. (2010) ‘Lessons from the Lysozyme of Phage T4’, ProteinScience, 19: 631–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershtein S. et al. (2013) ‘Protein Quality Control Acts on Folding Intermediates to Shape the Effects of Mutations on Organismal Fitness’, MolecularCell, 49: 133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— Serohijos A. W., and Shakhnovich E. I. (2017) ‘Bridging the Physical Scales in Evolutionary Biology: From Protein Sequence Space to Fitness of Organisms and Populations’, Current Opinion in Structural Biology, 42: 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewley M. C. et al. (2017) ‘A Non-cleavable Hexahistidine Affinity Tag at the Carboxyl-terminus of the HIV-1 Pr55(Gag) Polyprotein Alters Nucleic Acid Binding Properties’, Protein Expression and Purification, 130: 137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagavatula G. et al. (2017) ‘A Massively Parallel Fluorescence Assay to Characterize the Effects of Synonymous Mutations on TP53 Expression’, MolecularCancerResearch, 15: 1301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher J. I., Bolon D. N. A., and Tawfik D. S. (2016) ‘Quantifying and Understanding the Fitness Effects of Protein Mutations: Laboratory versus Nature’, ProteinScience, 25: 1219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— et al. (2019) ‘Constrained Mutational Sampling of Amino Acids in HIV-1 Protease Evolution’, Molecular Biology and Evolution, 36: 798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs J. A. G. et al. (2004) ‘The Stoichiometry of Gag Protein in HIV-1’, Nature Structural & Molecular Biology, 11: 672–5. [DOI] [PubMed] [Google Scholar]
- Canale A. S. et al. (2018a) ‘Evolutionary Mechanisms Studied through Protein Fitness Landscapes’, Current Opinion in Structural Biology, 48: 141–8. [DOI] [PubMed] [Google Scholar]
- —— et al. (2018b) ‘Synonymous Mutations at the Beginning of the Influenza A Virus Hemagglutinin Gene Impact Experimental Fitness’, Journal of Molecular Biology, 430: 1098–115. [DOI] [PubMed] [Google Scholar]
- Dean A. M., Dykhuizen D. E., and Hartl D. L. (1986) ‘Fitness as a Function of β-Galactosidase Activity in Escherichia Coli’, Genetical Research, 48: 1–8. [DOI] [PubMed] [Google Scholar]
- —— and Thornton J. W. (2007) ‘Mechanistic Approaches to the Study of Evolution: The Functional Synthesis’, Nature Reviews Genetics, 8: 675–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykhuizen D. E., and Dean A. M. (1990) ‘Enzyme Activity and Fitness: Evolution in Solution’, Trends in Ecology & Evolution, 5: 257–62. [DOI] [PubMed] [Google Scholar]
- Gay B. et al. (1998) ‘Morphopoietic Determinants of HIV-1 Gag Particles Assembled in Baculovirus-Infected Cells’, Virology, 247: 160–9. [DOI] [PubMed] [Google Scholar]
- Geiler-Samerotte K. A. et al. (2011) ‘Misfolded Proteins Impose a Dosage-Dependent Fitness Cost and Trigger a Cytosolic Unfolded Protein Response in Yeast’, Proceedings of the National Academy of Sciences of the United States of America, 108: 680–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G. et al. (2002) ‘Functional Profiling of the Saccharomyces Cerevisiae Genome’, Nature, 418: 387–91. [DOI] [PubMed] [Google Scholar]
- Jeffery C. J. (2003) ‘Moonlighting Proteins: Old Proteins Learning New Tricks’, Trendsin Genetics, 19: 415–7. [DOI] [PubMed] [Google Scholar]
- Jiang L. et al. (2013) ‘Latent Effects of Hsp90 Mutants Revealed at Reduced Expression Levels’, PLoSGenetics, 9: e1003600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacser H., and Burns J. A. (1981) ‘The Molecular Basis of Dominance’, Genetics, 97: 639–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A. H. et al. (1993) ‘Partial Inhibition of the Human Immunodeficiency Virus Type 1 Protease Results in Aberrant Virus Assembly and the Formation of Noninfectious Particles’, Journal of Virology, 67: 4050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King N. M. et al. (2002) ‘Lack of Synergy for Inhibitors Targeting a Multi-Drug-Resistant HIV-1 Protease’, ProteinScience, 11: 418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl N. E. et al. (1988) ‘Active Human Immunodeficiency Virus Protease Is Required for Viral Infectivity’, Proceedings of the National Academy of Sciences of the United States of America, 85: 4686–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnyu B. et al. (2013) ‘Gag-Pol Processing during HIV-1 Virion Maturation: A Systems Biology Approach’, PLoSComputational Biology, 9: e1003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.-K., Harris J., and Swanstrom R. (2009) ‘A Strongly Transdominant Mutation in the Human Immunodeficiency Virus Type 1 Gag Gene Defines an Achilles Heel in the Virus Life Cycle’, Journal of Virology, 83: 8536–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey B. J., and Marsh J. A. (2020) ‘Using Deep Mutational Scanning to Benchmark Variant Effect Predictors and Identify Disease Mutations’, Molecular Systems Biology, 16: e9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis J. M., Clore G. M., and Gronenborn A. M. (1999) ‘Autoprocessing of HIV-1 Protease Is Tightly Coupled to Protein Folding’, Nature Structural Biology, 6: 868–75. [DOI] [PubMed] [Google Scholar]
- Mahalingam B. et al. (1999) ‘Structural and Kinetic Analysis of Drug Resistant Mutants of HIV-1 Protease’, European Journal of Biochemistry, 263: 238–45. [DOI] [PubMed] [Google Scholar]
- Matayoshi E. D. et al. (1990) ‘Novel Fluorogenic Substrates for Assaying Retroviral Proteases by Resonance Energy Transfer’, Science, 247: 954–8. [DOI] [PubMed] [Google Scholar]
- Meek T. D. et al. (1990) ‘Inhibition of HIV-1 Protease in Infected T-Lymphocytes by Synthetic Peptide Analogues’, Nature, 343: 90–2. [DOI] [PubMed] [Google Scholar]
- Mehlhoff J. D. et al. (2020) ‘Collateral Fitness Effects of Mutations’, Proceedings of the National Academy of Sciences of the United States of America, 117: 11597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A. et al. (2014) ‘Comprehensive Mutational Scanning of a Kinase in Vivo Reveals Substrate-Dependent Fitness Landscapes’, Nucleic AcidsResearch, 42: e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P. et al. (2016) ‘Systematic Mutant Analyses Elucidate General and Client-Specific Aspects of Hsp90 Function’, CellReports, 15: 588–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller B. et al. (2009) ‘HIV-1 Gag Processing Intermediates Trans-Dominantly Interfere with HIV-1 Infectivity’, Journal of Biological Chemistry, 284: 29692–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özen A. et al. (2014) ‘Structural Basis and Distal Effects of Gag Substrate Coevolution in Drug Resistance to HIV-1 Protease’, Proceedings of the National Academy of Sciences of the United States of America, 111: 15993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit S. C. et al. (2002) ‘Replacement of the P1 Amino Acid of Human Immunodeficiency Virus Type 1 Gag Processing Sites Can Inhibit or Enhance the Rate of Cleavage by the Viral Protease’, Journal of Virology, 76: 10226–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— et al. (1994) ‘The P2 Domain of Human Immunodeficiency Virus Type 1 Gag Regulates Sequential Proteolytic Processing and Is Required to Produce Fully Infectious Virions’, Journal of Virology, 68: 8017–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatigorsky J., and Wistow G. J. (1989) ‘Enzyme/Crystallins: Gene Sharing as an Evolutionary Strategy’, Cell, 57: 197–9. [DOI] [PubMed] [Google Scholar]
- Prabu-Jeyabalan M., Nalivaika E., and Schiffer C. A. (2002) ‘Substrate Shape Determines Specificity of Recognition for HIV-1 Protease: Analysis of Crystal Structures of Six Substrate Complexes’, Structure, 10: 369–81. [DOI] [PubMed] [Google Scholar]
- Reicin A. S. et al. (1996) ‘The Role of Gag in Human Immunodeficiency Virus Type 1 Virion Morphogenesis and Early Steps of the Viral Life Cycle’, Journal of Virology, 70: 8645–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee S. Y. et al. (2003) ‘Human immunodeficiency virus reverse transcriptase and protease sequence database’, Nucleic Acids Research, 31: 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscoe B. P., and Bolon D. N. A. (2014) ‘Systematic Exploration of Ubiquitin Sequence, E1 Activation Efficiency, and Experimental Fitness in Yeast’, Journal of Molecular Biology, 426: 2854–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose J. R., Babe L. M., and Craik C. S. (1995) ‘Defining the Level of Human Immunodeficiency Virus Type 1 (HIV-1) Protease Activity Required for HIV-1 Particle Maturation and Infectivity’, Journal of Virology, 69: 2751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosé J. R., Salto R., and Craik C. S. (1993) ‘Regulation of Autoproteolysis of the HIV-1 and HIV-2 Proteases with Engineered Amino Acid Substitutions’, Journal of Biological Chemistry, 268: 11939–45. [PubMed] [Google Scholar]
- Sanjuan R., Moya A., and Elena S. F. (2004) ‘The Distribution of Fitness Effects Caused by Single-Nucleotide Substitutions in an RNA Virus’, Proceedings of the National Academy of Sciences of the United States of America, 101: 8396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. B. et al. (2006) ‘Design, Synthesis, and Biological Evaluation of Monopyrrolinone-based HIV-1 Protease Inhibitors Possessing Augmented P2′ Side Chains’, Bioorganic & Medicinal Chemistry Letters, 16: 859–63. [DOI] [PubMed] [Google Scholar]
- Socha R. D., Chen J., and Tokuriki N. (2019) ‘The Molecular Mechanisms Underlying Hidden Phenotypic Variation among Metallo-β-Lactamases’, Journal of Molecular Biology, 431: 1172–85. [DOI] [PubMed] [Google Scholar]
- Velazquez-Campoy A. et al. (2001) ‘Catalytic Efficiency and Vitality of HIV-1 Proteases from African Viral Subtypes’, Proceedings of the National Academy of Sciences of the United States of America, 98: 6062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber I. T., and Agniswamy J. (2009) ‘HIV-1 Protease: Structural Perspectives on Drug Resistance’, Viruses, 1: 1110–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegers K. et al. (1998) ‘Sequential Steps in Human Immunodeficiency Virus Particle Maturation Revealed by Alterations of Individual Gag Polyprotein Cleavage Sites’, Journal of Virology, 72: 2846–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windsor I. W., and Raines R. T. (2015) ‘Fluorogenic Assay for Inhibitors of HIV-1 Protease with Sub-Picomolar Affinity’, Scientific Reports, 5: 11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T. D. et al. (2003) ‘Mutation Patterns and Structural Correlates in Human Immunodeficiency Virus Type 1 Protease Following Different Protease Inhibitor Treatments’, Journal of Virology, 77: 4836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie C. S., and Shakhnovich E. I. (2011) ‘A Biophysical Protein Folding Model Accounts for Most Mutational Fitness Effects in Viruses’, Proceedings of the National Academy of Sciences of the United States of America, 108: 9916–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybarth G. et al. (1994) ‘Proteolytic Activity of Novel Human Immunodeficiency Virus Type 1 Proteinase Proteins from a Precursor with a Blocking Mutation at the N Terminus of the PR Domain’, Journal of Virology, 68: 240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.