

Abstract
Here we show that the capacity to manufacture IL-2 identifies constituents of the expanded CD8 T cell effector pool that display stem-like features, preferentially survive, rapidly attain memory traits, resist exhaustion, and control chronic viral challenges. The cell intrinsic synthesis of IL-2 by CD8 T cells attenuates the ability to receive IL-2-dependent STAT5 signals, thereby limiting terminal effector formation, endowing the IL-2-producing effector subset with superior protective powers. In contrast, the non-IL-2 producing effector cells respond to IL-2 signals and gain effector traits at the expense of memory formation. Despite having distinct properties during the effector phase, IL-2-producing and non-producing CD8 T cells appear to converge transcriptionally as memory matures to form populations with equal recall abilities. Therefore, the potential to produce IL-2 during the effector, but not memory stage, is a consequential feature that dictates the protective capabilities of the response.
One Sentence Summary:
IL-2 synthesis by effector CD8 T cells attenuates STAT5 signaling allowing protection against a chronic viral challenge.
Introduction
A critical step in the induction of a successful CD8 T cell response is the generation of effector and memory subsets that contribute to short-term and long-term immunological protection (1). These developmental transitions are guided by multiple factors including cytokine signals that direct cell fate decisions (2, 3). Conflicting reports have suggested roles for both autocrine and paracrine interleukin (IL)-2 in driving effector expansion and programming memory responses (4–7). CD4 T cell-derived IL-2 has been proposed as a major mechanism by which CD4 T cells help the CD8 T cell response (8–10). Nevertheless, it has also been shown that memory CD8 T cell populations capable of mounting robust recall responses can form in the absence of IL-2-producing CD4 T cells, implying that alternative sources of IL-2 can direct the CD8 T cell response (4, 5). Consequently, autocrine IL-2 was proposed to be critical for the formation of functionally competent memory CD8 T cells as the ablation of IL-2 production by CD8 T cells curtailed the secondary proliferative capacity of the resulting memory pool (5). Furthermore, the enforced expression of IL-2 by CD8 T cells has been shown to support their secondary expansion (11). Since IL-2 is typically manufactured by only a subset of the responding CD8 T cells, a key question is how the production of IL-2 by select constituents of this pool calibrates their quantitative and qualitative properties and dictates immunological protection.
We set out to analytically deconstruct the development, phenotypes, and protective powers of functionally distinct CD8 T cell subsets during the course of the anti-viral response to resolve how the production of IL-2 by CD8 T cells influences their fates and contributions to host defense. We used a system in which viable IL-2-producing and non-producing anti-viral CD8 T cells can be segregated and inspected (12). Our findings revealed that the ex vivo capacity to synthesize IL-2 identifies subsets of the expanded CD8 T cell effector pool that attain stem-like memory traits, resist exhaustion, and preferentially confer protective immunity upon secondary viral challenge. Counterintuitively, the IL-2 producing CD8 T cell population displays an attenuated ability to elaborate IL-2-dependent STAT5-signaling, which likely restricts the terminal effector properties of this subset. During the memory phase, IL-2 production is considered a hallmark trait of central-memory CD8 T cells that rapidly expand upon challenge (13–16). Nevertheless, unlike the disparities observed during the effector phase, we demonstrate that the transcriptional, phenotypic, and functional properties of IL-2-producing and non-producing CD8 T cells converge at memory. Consequently, both mature IL-2+ and IL-2− memory CD8 T cells respond equally vigorously and protect against a chronic viral challenge. Thus, there are stage specific differences in the roles of IL-2 production in establishing the properties and protective powers of the CD8 T cell pool.
Results
IL-2-producing CD8 T cells are enriched in the memory pool.
In order to deconvolute the roles of IL-2 synthesis by CD8 T cells in shaping memory T cell formation we conducted a longitudinal assessment of lymphocytic choriomeningitis virus (LCMV) epitope-specific T cells following acute infection. To accomplish this we utilized IFN- γ.Thy1.1 IL-2.GFP dual cytokine reporter mice, which accurately identify IFN-γ and IL-2 producing cells by the expression of Thy1.1 and GFP, respectively (fig. S1). By 8 days post infection, at the peak of the effector phase, a massive population of virus-specific CD8 T cells that manufactured IFN-γ upon restimulation were detected, but only a fraction of these cells co-produced IL-2 (Fig. 1A, B). Although the absolute numbers of IL-2-producing and non-producing virus-specific CD8 T cells decreased between 8 and 75 days post infection (Fig. 1B), the fraction capable of producing IL-2 became enriched as memory became established, increasing from 8–23% at day 8 to 33–44% by day 75, and 35–57% by day150 (Fig. 1C).
Fig. 1. IL-2-producing CD8 T cells are enriched as the memory pool forms.
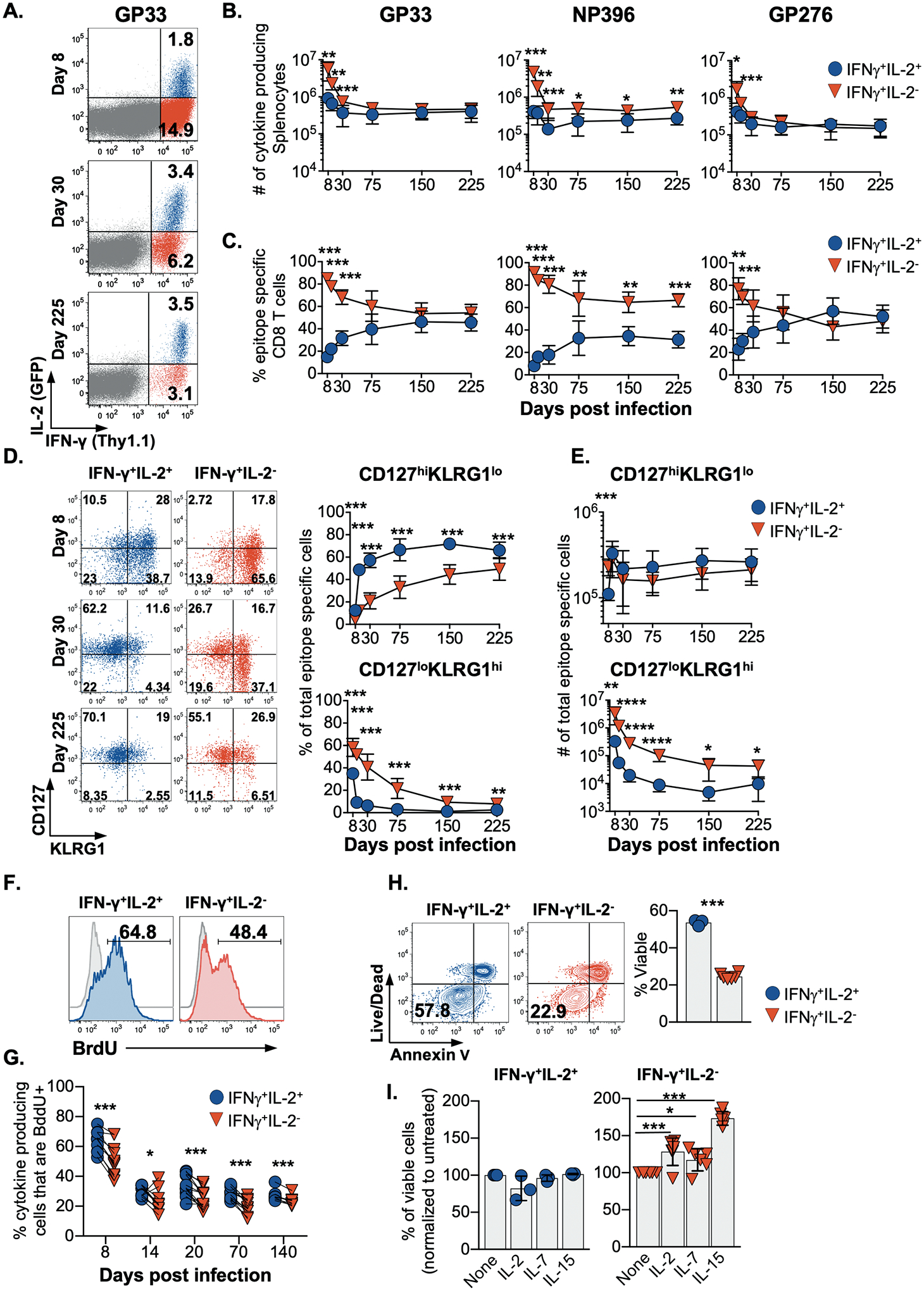
(A) Flow cytometric analysis, (B) enumeration, and (C) the proportion of splenic LCMV-specific CD8 T cells that produce IL-2 and IFN-γ following restimulation after acute infection.
Analysis of the proportion (D) and enumeration (E) of CD127hi KLRG1lo and CD127lo KLRG1hi LCMV GP33-specific IFN-γ+IL-2+ and IFN-γ+IL-2− CD8 T cells.
(F) BrdU incorporation by IFN-γ+IL-2+ and IFN-γ+IL-2− LCMV GP33-specific CD8 T cells analyzed following a 14 day BrdU pulse initiated at 8 days post-infection or (G) at the indicated times post-infection.
(H) The viability of IFN-γ+IL-2+ and IFN-γ+IL-2− LCMV GP33-specific CD8 T cells isolated at 14 days post infection and subjected to Annexin V and live/dead dye staining following culture for 18 hrs.
(I) IFN-γ+IL-2+ and IFN-γ+IL-2− LCMV-specific CD8 T cells were analyzed as in (H) with or without the addition IL-2, IL-7, or IL-15.
Flow cytometry plots show representative data; graphs show composite data depicting means ± SD. Significance was calculated using a paired two-tailed t-test except in (H) and (I) where an unpaired two-tailed test and 2-way ANOVA were used, respectively. Experiments were performed 2–4 times using a total of 6–12 mice per time point. *p<0.05, **p<0.01, ***p<0.001
To further define the connections between the capacity to produce IL-2 and the development of memory we assessed the expression of CD127 and KLRG1 (Fig. 1D and figs. S2A–S2B). Over time, the IFN-γ+IL-2+ CD8 T cells more rapidly attained a canonical CD127hiKLRG1lo memory phenotype. By contrast the IFN-γ+IL-2− subset preferentially adopted an opposing CD127loKLRG1hi phenotype, which has been ascribed to short-lived effector cells (17, 18); however, by day 225 following acute LCMV infection, all specificities of both IL-2-producing and non-producing anti-viral CD8 T cells checked were predominately CD127hiKLRG1lo. Similarly, an assessment of additional molecules that distinguish effector and memory subsets, including CD62L, CCR7, CXCR3 and CX3CR1, confirmed that by late memory time points IL-2+ and IL-2− anti-viral CD8 T cells were remarkably unified even though they did differ in the expression of CXCR3 and CX3CR1 during the effector stage (figs. S2C and S2D). Collectively, these analyses reveal that IL-2+ and IL-2− CD8 T cell subsets are more distinct during the effector stage and indicate that the IL-2+ CD8 T cells more rapidly transition to memory. Nevertheless, by late time points, both IL-2+ and IL-2− anti-viral CD8 T cells phenotypically coalesce as memory properties are cemented.
Key traits of memory T cells include self-renewal and survival; therefore, we used in vivo BrdU incorporation to probe the homeostatic proliferation of IL-2+ and IL-2− CD8 T cells. At all time points checked, the IL-2-competent CD8 T cell subsets showed slight but consistently higher levels of BrdU incorporation, demonstrating greater homeostatic turnover of this population (Figs. 1F and 1G). We next tested the survival potential of anti-viral IFN-γ+IL-2+ and IFN-γ+IL-2− CD8 T cells during the contraction phase, at 14 days post-infection. Assessment of cell viability using live/dead dye and annexin V staining following an 18 h in vitro culture showed that the IL-2+ subset was less prone to apoptosis, indicating that these cells are preferentially endowed with pro-survival memory attributes (Fig. 1H). IL-7 and IL-15, as well as IL-2, influence the survival and expansion of effector and memory CD8 T cells, so we checked whether these cytokines could preserve the IL-2+ and IL-2− CD8 T cell subsets (Fig. 1I) (19–22). By comparison with untreated control cultures, the viability of the IL-2+ cells was unaffected by the addition of these cytokines, whereas IL-2, IL-7 and IL-15 improved the survival of non-IL-2-producing CD8 T cells isolated during the early contraction phase of the response by 28.4±18.4%, 17.3±14.9% and 73.4±9.2%, respectively. Thus, at least a fraction of the non-IL-2-producing effector subset are rescuable by specific homeostatic cytokines, especially IL-15.
IL-2+ effector CD8 T cells more rapidly adopt a CD127hiKLRG1lo memory phenotype following transfer.
We tested whether the derivatives of IL-2+ or IL-2− effector CD8 T cells present at 10 days post-acute LCMV infection were capable of seeding the memory pool by taking advantage of the dual IFN-γ.Thy1.1 IL-2.GFP reporter system to procure viable IL-2-producing and non-producing LCMV NP396-specific CD8 T cells, and we traced their fates following transfer (fig. S1 and Fig. 2A). After parking the cells for 28–33 days in vivo, both IL-2+ and IL-2− donor cells were recoverable from the spleen, liver, lungs, and bone marrow (Fig. 2B), and a higher proportion of the IL-2+ donor population adopted a CD127hiKLRG1lo memory phenotype, whereas the IL-2− donor cells predominately displayed an opposing CD127loKLRG1hi phenotype (Fig. 2C). At this time point, only a minority (11.8±3.7%) of the IL-2+ donor cells retained the ability to produce IL-2 upon restimulation, and this fraction was even lower for the IL-2− donor cells (3.4±4.2%).
Fig. 2. IL-2+ effector cells more rapidly adopt a memory phenotype after transfer.
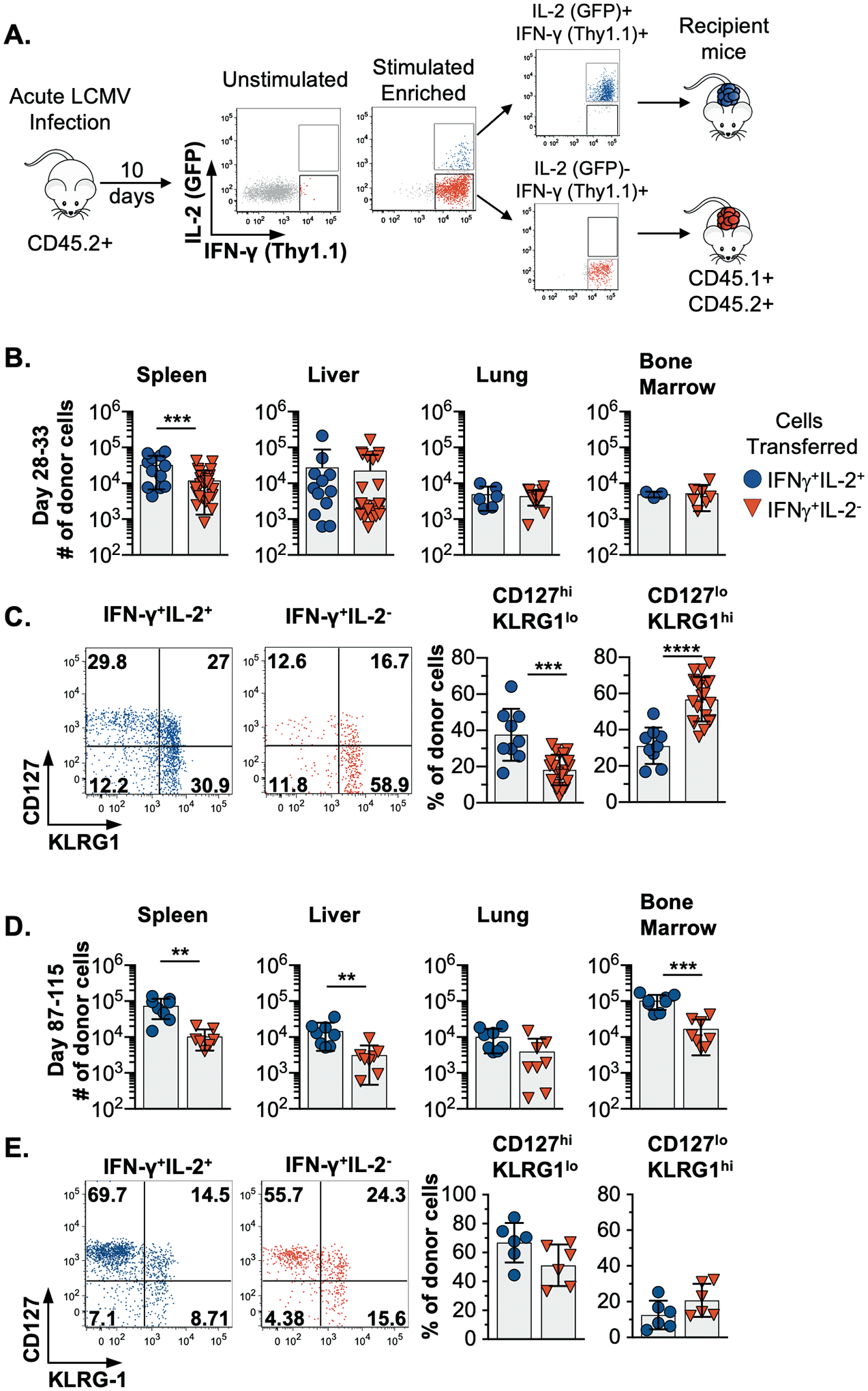
(A) Schematic of the experimental design. LCMV NP396-specific or a mix of GP33, GP276, and NP396-specific IFN-γ+IL-2+ and IFN-γ+IL-2− CD8 T cells were isolated by cell sorting at 10 days post infection and transferred into allelically marked cytokine reporter recipients. The NP396-specific only donor cell populations analyzed at 28–33 following transfer and the mix-specificity donor subsets were assessed between days 87–115 days.
(B-C) Enumeration of LCMV NP396-specific donor cells and analyses of CD127loKLRG1hi and CD127hiKLRG1lo specific donor populations at 28–33 days following transfer.
(D-E) Enumeration of the mixed specificity donor population and analyses of CD127loKLRG1hi and CD127hiKLRG1lo donor subsets at 87–115 days post transfer.
Composite or representative data are shown from 2–5 separate experiments analyzing 3–26 total mice.
Error bars show SD, and significance was determined using an unpaired two-tailed t-test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
In separate experiments, a mixture of LCMV-GP33, GP276, and NP396-specific IL-2-producing and non-producing effector cells were similarly transferred and analyzed 87–115 days later. These donor cells were present in every compartment checked (Fig. 2D), and the majority of both the IL-2-producing and non-producing donor cells were CD127hiKLRG1lo (Fig.2E). Moreover, a greater fraction of both donor cell subsets were now capable of producing IL-2 (34.7±6.0% for IL-2+ donor cells and 13.4±6.7% for IL-2− donor cells). Thus, a level of phenotypic convergence becomes apparent over time, which is consistent with the longitudinal analysis presented in Fig. 1.
The derivatives of IL-2+ effector cells and both IL-2+ and IL-2− memory subsets mount protective recall responses.
We next tested whether the capacity to produce IL-2 was associated with ability to mount protective anti-viral recall responses. LCMV NP396-specific IL-2-producing or non-producing effector or memory cells were isolated at 10 or 310–347 days, respectively, following acute infection. These donor cells were adoptively transferred and their ability to respond to a chronic viral challenge (LCMV-clone 13) assessed (Fig. 3A). Impressively, the derivatives of the IL-2+ donor effector cells mounted dramatically greater recall responses than their IL-2− counterparts (Fig. 3B). The fraction of CD8 T cells derived from the effector IL-2-producing donor cells was 39.6±13.0%, 36.7±12.0%, 54.8±14.7% and 41.7±10.7% in the blood, spleen, liver, and bone marrow, respectively. Markedly diminished responses were detected in the recipients that received non-IL-2-producing effector CD8 T cells as only 9.1±6.2%, 7.9±7.2%, 12.1±12.4% and 9.4±7.3% of CD8 T cells were donor-derived in the blood, spleen, liver and bone marrow of these cohorts, respectively. Curiously, this inferior recall capacity by the IL-2-non-producing CD8 T cells was not apparent during the memory phase as mature IL-2+ and IL-2− memory donor populations both elicited vigorous and equivalent recall responses (Fig. 3C). We also evaluated viral clearance and found that the IL-2+ effector cells as well as both the IL-2+ and IL-2− memory cells fully controlled the challenge with LCMV-clone 13, which usually establishes a chronic infection (23, 24). By contrast, the non-IL-2-producing effector cells were incapable of containing the secondary challenge, as viral titers in these recipients were identical to those present in control cohorts that did not receive donor cells (Fig. 3D and 3E). Overall, these findings show that IL-2-producing and non-producing memory CD8 T cells as well as their IL-2-producing effector counterparts are endowed with the ability to vigorously respond and combat a severe viral challenge. The IL-2 non-producing effector cells are, however, unique and cannot sufficiently amplify their response to counteract the infection.
Fig. 3. The derivatives of IL-2+ effector cells and both IL-2+ and IL-2− memory subsets mount protective recall responses.
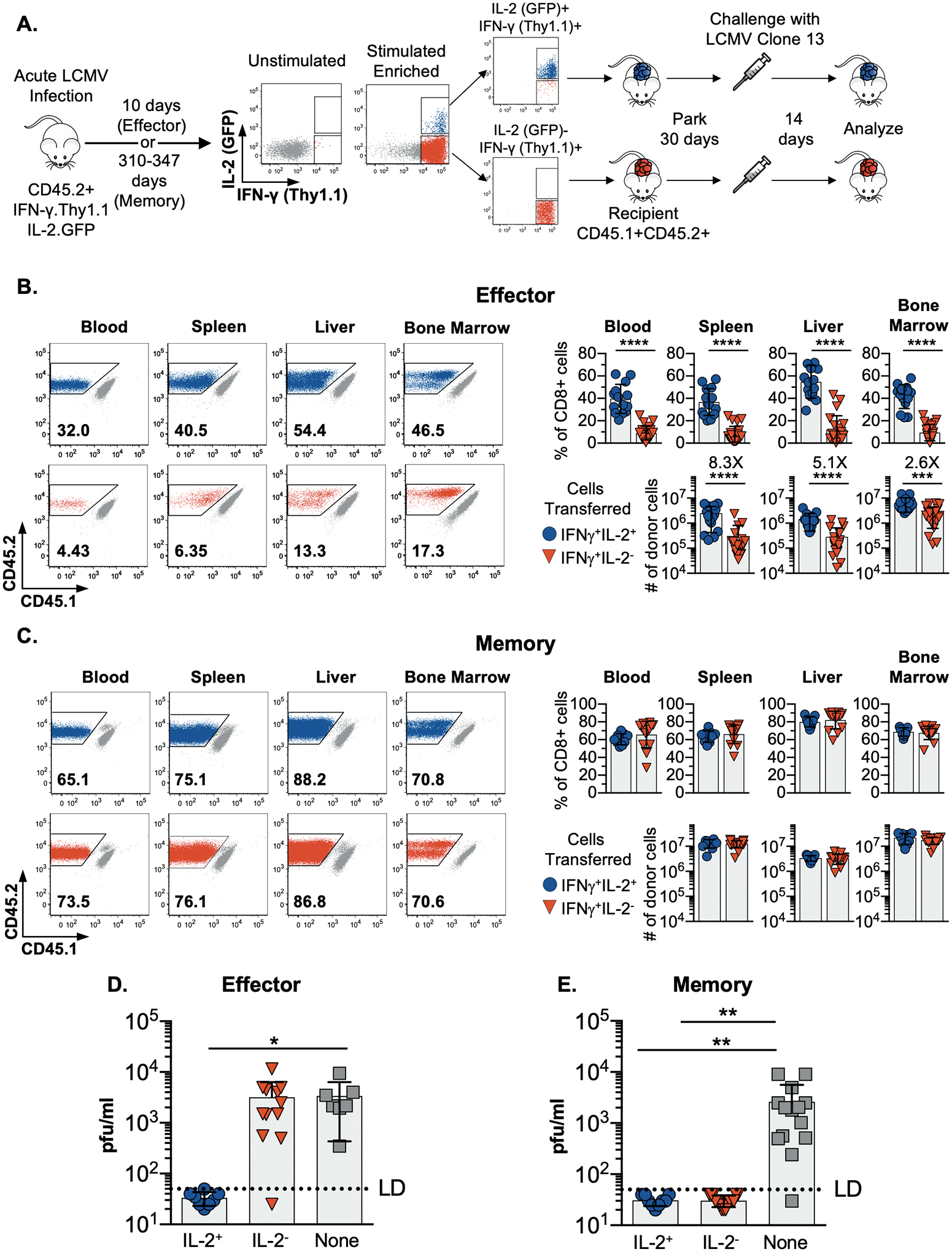
(A) Schematic of the experimental design. IFN-γ+IL-2+ and IFN-γ+IL-2− LCMV NP396-specific CD8 T cells were isolated by cell sorting either during the effector or memory phases of the response at 10 days or 310–347 days post-infection, respectively. Normalized numbers of these donor cells parked were in naïve allelically marked recipient mice for 28–30 days prior to challenge with LCMV-clone 13. Donor CD8 T cell responses were analyzed 14 days following challenge.
(B and C) Representative flow cytometry plots and composite results showing the recovery, percentage and absolute numbers of IFN-γ+IL-2+ and IFN-γ+IL-2− effector (B) and memory (C) donor cells from various sites at 14 days post challenge. Significance was calculated using unpaired two-tailed t-tests.
(D and E) Serum viral titers in challenged mice that received either effector (D) or memory (E) IFN-γ+IL-2+ or IFN-γ+IL-2− donor cells or no cells. Significance was determined using one-way ANOVA.
Error bars show SD. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
Composite or representative data are shown from 3–4 separate experiments analyzing a total of 8–22 mice per group.
IL-2+ effector as well as IL-2+ and IL-2− memory CD8 T cells resist exhaustion and rescue the endogenous response.
Evaluation of the expression of inhibitory receptors commonly upregulated by exhausted cells, including PD-1 (CD279), LAG-3 (CD223), and 2B4 (CD244) (25), showed that the failure of the IL-2− effector donor cells to contain the infection was associated with the attainment of a canonical exhausted phenotype (Fig. 4A and 4B) and that a substantial percentage of these cells co-expressed multiple inhibitory receptors (Fig. 4C). By contrast, the IL-2+ effector cells and both IL-2+ and IL-2− memory donor populations appeared refractory to exhaustion (Fig. 4A–4C upper and lower panels). Substantially fewer donor cells derived from the IL-2− effector cells produced IFN-γ, and only a fraction of these cells were capable of simultaneously synthesizing IFN-γ and TNF-α (7.9±10.7%) when compared with their IL-2+ effector donor counterparts (45.5±15.5%) (Fig. 4D–4F upper panels). By contrast, donor populations recovered from recipients that received either IL-2+ or IL-2− memory cells were highly and equally functional (Fig. 4D–4F lower panels). The apparent unique vulnerability of IL-2− effector cells to exhaustion upon secondary viral challenge was also illustrated by their relatively higher expression of granzyme B (Fig. 4G) and eomesodermin (Fig. 4H) (26).
Fig. 4. Effector CD8 T cells that do not manufacture IL-2 are selectively susceptible to exhaustion following a chronic viral challenge.
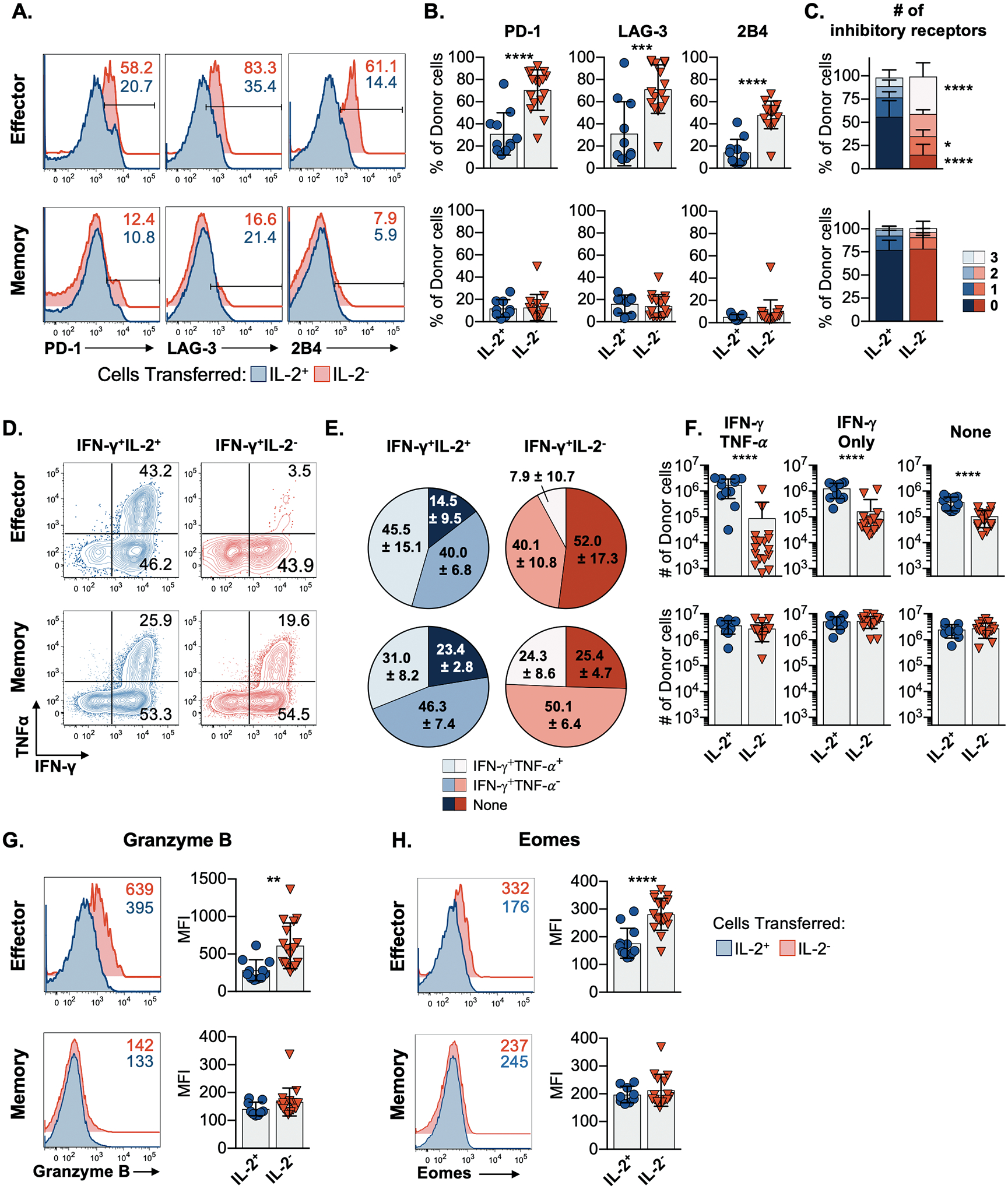
(A) Flow cytometry plots and (B) composite data showing the expression of PD-1, LAG-3, and 2B4 by donor NP396-specific IFN-γ+IL-2+ (blue) and IFN-γ+IL-2− (red) effector (top) and memory (bottom) CD8 T cells following transfer, LCMV-clone 13 challenge, and analyses as in Figure 3A.
(C) The proportion of IL-2+ and IL-2− donor effector and memory CD8 T cells which co-express 0, 1, 2, or 3 inhibitory receptors following transfer.
(D) Flow cytometry plots, (E) co-expression, and (F) numbers of IFN-γ and TNF-α producing NP396-specific cell populations derived from donor IFN-γ+IL-2+ (blue) and IFN-γ+IL-2− (red) effector (top) and memory (bottom) cells.
(G-H) Representative flow cytometry plots and bar graphs of the expression of granzyme B (G) and Eomes (H) by the indicated donor populations analyzed following transfer and challenge.
Representative or composite findings are shown from 3 experiments analyzing a total of 9–17 mice per group. Bar graphs show mean ± SD with significance calculated using an unpaired two-tailed t-test, except where a 2-way ANOVA was used (C). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
We also assessed the impact of transferring functionally distinct donor populations on the recipients’ endogenous CD8 T cell responses to the viral challenge. The robust recall responses elicited by the donor IL-2+ effector cells and both the donor IL-2+ and IL-2− memory cells curtailed the development of exhaustion by the recipients’ own CD8 T cells. In these instances, fewer of the endogenous CD8 T cells expressed any of the inhibitory receptors evaluated (figs. S3A–3C and S4), and these cells were also more functional, which was particularly evident when co-synthesis of IFN-γ and TNF-α was assessed (figs. S3D–S3I). These more functionally robust patterns were not detected in recipients that received effector non-IL-2-producing donor cells. In these recipients, the endogenous CD8 T cells succumbed to exhaustion, as they expressed elevated levels of PD-1, LAG-3, and 2B4 and were less functionally robust (figs. S3 and S4). Thus, the adoptive transfer of donor populations capable of clearing the challenge infection bolstered the overall response to virus by circumventing the exhaustion of the endogenous response.
IL-2+ and IL-2− CD8 T cells transcriptionally coalesce as the memory pool forms.
To further define the similarities and differences between IL-2+ and IL-2− effector and memory CD8 T cells, transcriptional profiling was conducted. Naïve LCMV-specific P14 TCR transgenic IFN-γ.Thy1.1 IL-2.GFP reporter CD8 T cells were transferred into allelically marked recipients and primed by acute LCMV infection. IFN-γ+IL-2+ and IFN-γ+IL-2− P14 CD8 T cells were sorted during the effector (day 9) and memory (day 308–309) phases of the response and subjected to RNA-sequencing.
Principal component analysis of the top 5000 most variable genes confirmed tight clustering of biological replicates and further showed that the transcriptional profiles of the IL-2+ and IL-2− effector populations are unique and overall distinguishable from their memory counterparts (Fig. 5A). At memory time points, the IL-2+ and IL-2− populations are more related to each other than to the IL-2+ and IL-2− subsets present during the effector phase. Hierarchical clustering analysis of the 1666 differentially expressed genes with an FDR adjusted p value <0.01 between either IL-2+ and IL-2− effector or IL-2+ and IL-2− memory cells also showed that the IL-2-producing and non-producing day 9 effector CD8 T cell subsets are distinct from each other and also from their memory counterparts (Fig. 5B).
Fig. 5. IL-2+ and IL-2− CD8 T cells transcriptionally coalesce as the memory pool forms.
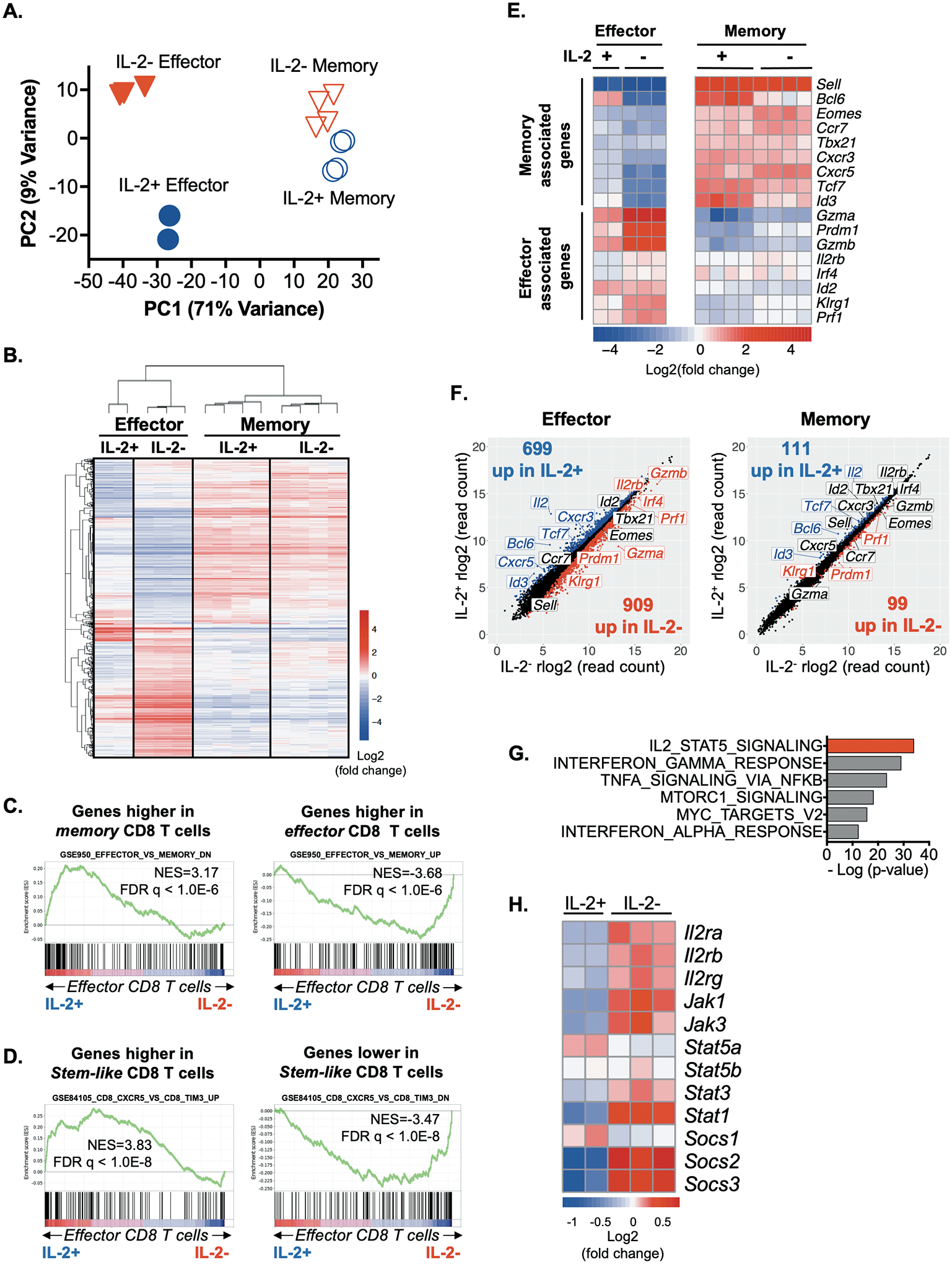
LCMV-specific P14 TCR transgenic CD8 T cells were isolated during the effector (day 9) and memory (days 308–309) phases following acute infection and subjected to RNA-sequencing.
(A) Principal component analysis of the top 5000 most variably expressed genes across all samples.
(B) Hierarchical clustering analysis of the 1666 genes differentially expressed by either IL-2+ and IL-2− effector or IL-2+ and IL-2− memory P14 CD8 T cells with an FDR adjusted p value of <0.01.
(C, D) GSEA comparing IL-2-producing and non-producing effector CD8 T cell subsets with previously published (C) effector and memory CD8 T cell signatures (GSE9650; CD8+ effector versus CD8+ memory), and (D) T cell stemness-associated signatures (GSE84105; CD8+CXCR5+ versus CD8+TIM3+).
(E) Heat map illustrating the centered log expression values of select effector- and memory-associated genes by IFN-γ+IL-2+ and IFN-γ+IL-2− cells during the effector (left) and memory (right) phases.
(F) Scatter plots show differential gene expression between IL-2+ and IL-2− CD8 T cells during the effector (left) and memory (right) phases. Select effector- and memory-associated genes are labeled. The numbers of genes statistically significantly upregulated in IL-2 producing (blue) or non-producing (red) CD8 T cells are shown.
(G) Analyses of select hallmark pathways that are differentially expressed by IL-2+ and IL-2− effector CD8 T cells.
(H) Heatmap illustrating the centered log expression values expression of select components of the JAK-STAT signaling pathway in IL-2+ and IL-2− effector CD8 T cells.
Gene set enrichment analyses (GSEA) were conducted to compare the similarities between IL-2-producing and non-producing effector cells to published effector and memory CD8 T cell transcriptomes (GSE9650_effector_vs_memory_CD8_Tcell) (27) (Fig. 5C) and to stem-like T cells (GSE84105) (28) (Fig. 5D). The IL-2+ effector subsets shared transcriptional features associated with previously characterized memory T cells and were also enriched in stemness-associated properties. Analysis of select effector and memory related genes confirmed that known memory-associated transcripts such as Bcl6, Id3, and Tcf7 are expressed at higher relative levels in IL-2-producing compared to non-producing effector cells (Fig. 5E and 5F). Conversely, effector-associated genes including Gzma, Gzmb, Prdm1, Klrg1, and Prf1 are more highly expressed by IL-2− effector cells. Thus, although the IL-2+ effector CD8 T cells do not represent a complete transcriptional homunculus of the memory population, they are enriched in the expression of memory and stemness related genes. As memory forms, the IL-2+ and IL-2− subsets transcriptionally converge because the number of genes differentially expressed (FDR adjusted p value <0.01) between IL-2+ and IL-2− CD8 T cells declines from 1608 during the effector phase to 210 at memory (Fig. 5F). This transcriptional coalescence is consistent with the results presented in Figures 1–4, and S2, showing that the phenotypic and protective properties of the IL-2+ and IL-2− populations are more similar at established memory time points.
Intrinsic IL-2 production by CD8 T cells is associated with attenuated IL-2 signaling.
To probe the differences between the IL-2-producing and non-producing CD8 T cells during the effector phase of the response, pathway analyses was conducted and revealed that these populations differentially express genes associated with IL-2-dependent STAT5-signaling (Fig. 5G). This was further confirmed by scrutinizing the levels of select transcripts of moieties within this signaling conduit (Fig. 5H). The roles of intrinsic IL-2 production and sensitivity to IL-2 signaling is intriguing since autocrine IL-2 has been proposed to be critical for the development of memory CD8 T cells with robust recall potential, but IL-2 driven STAT5 activation has been shown to promote terminal effector formation (5, 29–32). To investigate this conundrum, the levels of STAT5A and phosphorylated STAT5A/B complexes (pSTAT5) (Y694) in IL-2-producing and non-producing CD8 T cells were evaluated. Following activation effector IFN-γ+IL-2+ CD8 T cells expressed slightly higher total levels of STAT5A but displayed markedly lower levels of pSTAT5 complexes when compared with their IL-2− counterparts, implying that the ability to receive STAT5-dependent IL-2 signals is shut down in the majority of the IL-2 producing cells (Fig. 6A). To confirm that the phosphorylation of STAT5 was driven by IL-2, similar stimulations were conducted in the presence of anti-CD25 (IL-2Rα) blocking antibodies (Fig. 6B and 6C) (33). This mitigated the pSTAT5 signal in IL-2− CD8 T cells demonstrating that the observed increases in pSTAT5 levels were IL-2-dependent. Thus, intrinsic IL-2 production appears to disrupt the ability to receive IL-2 signals, which have been shown to promote terminal effector cells (29, 32).
Fig. 6. Intrinsic IL-2 production is associated with attenuated IL-2 signaling.
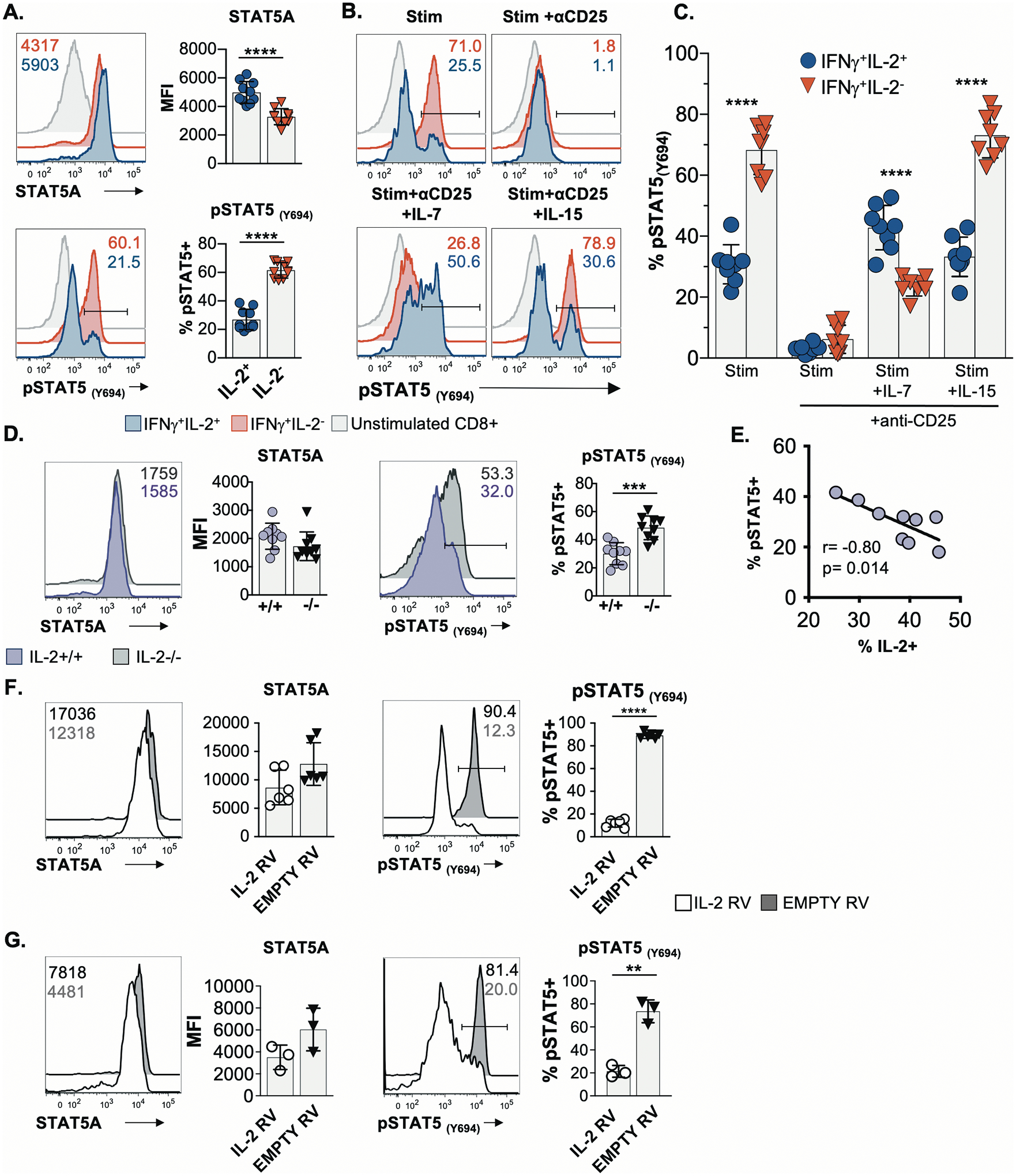
(A) Flow cytometric analyses of STAT5A and pSTAT5(Y694) levels in activated IFN-γ+IL-2+ and IFN-γ+IL-2− LCMV-specific CD8 T effector cells procured at 9 days post-infection. Representative histograms (left) and composite data (right) are shown.
(B-C) Splenic day 8 effector cells were stimulated with a cocktail of LCMV peptide epitopes for 5 hrs in the presence or absence of blocking anti-CD25 antibodies. Recombinant IL-7 or IL-15 was added to certain cultures 30 mins prior to staining for pSTAT5 levels. (B) Representative flow cytometry plots; (C) Composite data.
(D) Analyses of STAT5A and pSTAT5(Y694) levels in IL-2+/+ and IL-2−/− P14 CD8 T cells after activation, culture, and re-stimulation.
(E) Spearman rank order correlation analysis of IL-2 expression and STAT5 phosphorylation by vitro activated IL-2+/+ CD8 T cells from D.
(F-G) Analysis of STAT5A and pSTAT5(Y694) levels in P14 CD8 T cells following transduction with either a control/empty or IL-2 expressing retrovirus and transient reactivation with IL-2. The transduced populations were either kept separate (F) or co-cultured (G) prior to analyses. Gated live transduced (GFP+) cells are shown.
Data presented as mean ± SD from 3 independent experiments, except for (G) in which representative results from one of two experiments are shown. Significance calculated using unpaired two-tailed t-test (A, B, D, F, G) or a paired two-tailed t-test (C). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
Blocking CD25 allowed us to probe the sensitivity of IL-2+ and IL-2− CD8 T cells to IL-7 and IL-15, which also signal through STAT5. The IFN-γ+IL-2+ CD8 T cells preferentially phosphorylated STAT5 in response to IL-7 (Fig. 6B and 6C), which is consistent with their higher expression of CD127 (IL-7Rα) (Fig. 1D). By contrast, IL-2− CD8 T cells were more sensitive to IL-15, which promoted the survival of these cells in vitro (Fig. 1I). We also assessed the levels of pS6, an IL-2-responsive downstream target of mTOR (fig. S5A) (34, 35). Both IL-2+ and IL-2− effector CD8 T cells expressed similar levels of pS6 which were unchanged by CD25 blockade or IL-7 and IL-15 treatments, demonstrating that this pathway is intact in both populations. These results further imply that differential IL-2-driven STAT5 phosphorylation is a principal forecaster of the developmental fates of IL-2-producing and non-producing effector cells.
Modulating IL-2 synthesis by CD8 T cells alters STAT5 activation.
To ascertain how receptor expression reflected the sensitivity of IL-2+ and IL-2− effector CD8 T cell subsets to IL-2 signals we assessed the levels of CD25 (IL-2Rα), CD122 (IL-2Rβ), and CD132 (IL-2Rγ), as well as CD127 (IL-7Rα). The levels of each of these chains were similar or higher on the surface of IL-2+ CD8 T cells when compared to their non-IL-2-producing counterparts. Thus, the curtailed pSTAT5 response in the IL-2+ cells was not due to loss of receptor expression (figs. S5B and S5C), even though transcript levels were lower (Fig. 5H). The engagement of IL-2 with its receptor causes endocytosis of the complex (36) and therefore, we tested whether inhibiting this interaction affects surface expression. The blockade of CD25 resulted in a 64.2±15.1% and 24.6±7.2% increase in the levels of CD122 and CD132, respectively on the non-IL-2 producing CD8 T cells (fig. S5C), implying that these cells usually consume IL-2 and cycle the receptor from the cell surface. The increases in CD122 and CD132 levels that occurred when CD25 was blocked were overridden by IL-15 (fig. S5C), demonstrating that the IL-2− cells are responsive to IL-15, which is consistent with the survival (Fig. 1I) and signaling (Fig. 6B and 6C) findings. Inspection of CD127 levels further support the notion of cytokine-driven loss of surface receptor levels as activation in the presence of IL-7 together with CD25 blockade resulted in decreases in CD127 and CD132 levels (fig. S5C) (37).
We also conducted overexpression and knockout studies to confirm the impact of intrinsic IL-2 production on STAT5 activation. We compared wild-type (+/+) and IL-2-deficient (−/−) CD8 T cells that were differentiated in vitro using a staggered IL-2 and IL-15 culture regimen (Fig. 6D). Under these conditions both the +/+ and IL-2−/− CD8 T cells displayed similar phenotypes (fig. S6A). Following transient restimulation with antigen and IL-2, the IL-2−/− cells were more prone to pSTAT5 activation (Fig. 6D) whereas the levels of IL-2 synthesis by the +/+ cells inversely correlated with the extent of pSTAT5 (Fig. 6E). In addition, naive CD8 T cells were retrovirally transduced with either empty control or IL-2 expressing vectors. These populations were either kept segregated (Fig. 6F) or co-cultured in separate experiments (Fig. 6G), prior to assessing STAT5A and pSTAT5 levels following washing and re-exposure to exogenous IL-2. Although STAT5A levels were similar, STAT5 phosphorylation was markedly attenuated in the IL-2 transduced cells, providing further evidence that the synthesis of IL-2 can negatively impact IL-2 signaling in the producer cell. Notably, both the IL-2 transduced and control populations were phenotypically similar as assessed by the expression of STAT5A (Fig. 6F), CD122, CD132, CD27, CD43, CD62L, CD127 and KLRG1 (fig. S6B), although there was a slight diminution in CD25 levels in the IL-2 transduced cells. Collectively, these findings implicate the intrinsic manufacture of IL-2 by CD8 T cells in attenuating IL-2-dependent STAT5 signaling, which likely restricts their terminal differentiation while preserving memory potential. Conversely, non-IL-2-producing CD8 T cells are permissive to IL-2 signals which plausibly reinforces their effector phenotype at the expense of memory formation. This is consequential for immunological protection as the IL-2 producing terminal effector population mounts ineffective recall responses and fails to contain viral re-exposures.
Discussion
The manufacture of IL-2 by CD8 T cells has generally been presumed to not only provide a secreted extracellular source of the cytokine but to also deliver an autocrine signal back to the producing cell (5). In this study, however, we find that IL-2-producing CD8 T cells receive weaker STAT5-dependent IL-2 signals and more rapidly attain memory traits during the effector phase of the response. This is consistent with reports showing limited IL-2 signals favor memory development whereas stronger IL-2 signals via STAT5 drive terminal effector differentiation (3, 29, 32). Conversely, the phenotypic and transcriptional features of the non-IL-2-producing effector cells reflect the observation that these cells are more receptive to IL-2-dependent STAT5 signals. This likely contributes to their pronounced expansion, gain of effector traits, and susceptibility to exhaustion following secondary challenge, while curtailing the establishment of memory properties.
Although differences in the extent of TCR activation may contribute to discordances in IL-2-dependent STAT5 signaling in the naturally arising IL-2-producing and non-producing CD8 T cell subsets (38), the retroviral transduction analyses demonstrate that this attenuated signaling can be directly governed by the intrinsic manufacture of IL-2. By comparison with IL-2-producing effector CD8 T cells, the IL-2− cells express higher levels of Il2ra, Il2rb, and Il2rg transcripts but lower levels of these chains on the cell surface (cf. Fig. 5H vs fig. S5B, C), even though they are more prone to IL-2-driven STAT5 activation. We suspect the lower surface receptor levels reflect internalization of the receptor complex which occurs upon engagement with IL-2 (36). Notably, this interaction is prevented by blockade of CD25, which leads to an increase in surface expression on the non-IL-2-producing cells, likely reflecting a cessation of receptor uptake.
In addition to surface receptor levels, internal components including JAK1, JAK3, and suppressors of cytokine signaling (SOCS)1–3 can also impact the efficiency of STAT5 activation. The levels of Jak1, Jak3, Socs2, and Socs3 transcripts are all relatively lower in the IL-2-producing effector CD8 T cells. This fits with the attenuation of IL-2-dependent STAT5 signaling in these cells as JAK1 and 3 are upstream kinases in the signaling conduit, and SOCS2 has been demonstrate to increase IL-2 signals through the degradation of other SOCS molecules (35, 39, 40). Although SOCS3 has been shown to temper T cell activation and support memory development, IL-7 has been shown to decrease SOCS3 expression which may account for the lower levels of Socs3 transcripts in the IL-2+ population (41, 42). By contrast, Socs1 is more abundant in the IL-2 producers and has been shown to inhibit STAT5 phosphorylation (43).
An intriguing possibility is that internal STAT5-independent signaling occurs within the IL-2-producing effector cells. Recently it has been shown that the IL-2 receptor complex can assemble and engage with endogenously manufactured IL-2 within the endoplasmic reticulum (ER) and Golgi compartments of the producer cell, eliciting JAK1 and JAK3 phosphorylation (44). STAT5 is not thought to be associated with the ER and Golgi so cannot effectively participate in this signaling process (45). However, it is possible that other IL-2 signal transducers such as mTOR, which associate with the ER/Golgi complex, may be more available to receive these signals (46). We speculate that this redirected signaling may set up a scenario whereby restricted IL-2-dependent STAT5 activation limits terminal effector differentiation, whereas STAT5-independent signals may foster memory progenitor formation by the IL-2 producing cell (34, 47).
Once memory is established the ability to autonomously manufacture IL-2 becomes less tied to transcriptional and phenotypic differences and is not critical for protection against chronic LCMV challenge. IL-2 production by memory cells has been considered a hallmark feature of central-memory cells, which are thought to elicit rapid and robust recall responses (13, 15, 16). Surprisingly, we found that both IL-2-producing and non-producing memory cells mount marked and equivalent responses to the secondary viral infection. However, at the time of transfer both IL-2+ and IL-2− memory subsets displayed a CD62LhiCCR7hi central-memory phenotype and expressed closely related transcriptional profiles, by comparison with IL-2-producing and non-producing CD8 T cells analyzed at effector time points. This is consistent with the observations that as the memory pool matures the cells re-acquire certain naïve properties, attain self-renewal abilities, and maintain the capacity to reform effector subsets (48, 49).
This bifurcation between the ability of IL-2-producing and non-producing CD8 T cells to respond to IL-2 couples functional competency with fate decisions and implicates IL-2 as a critical differentiation factor. Notably, the extinguishment of IL-2-dependent STAT5 signaling by IL-2-producing CD8 T cells that develop following infection is not fully penetrant as a fraction phosphorylate STAT5 to levels comparable to that observed by the IL-2-non producers. Conversely, a sub-population of the non-IL-2 producers fail to respond to IL-2. It is plausible that these minority constituents within each functional pool account in part for some of the observed phenotypic overlap, and for the longevity and attainment of memory properties by a sub-population of non-IL-2 producing CD8 T cells. Nevertheless, the dominant trend is that differences in the ability to manufacture IL-2 during the effector phase associates with divergence in the receptiveness to IL-2 driven STAT5 signals, biased emergence of effector and memory properties, and the ability to elicit protective recall responses during the effector phase.
The relationships between IL-2 production and cell fates not only applies to CD8 T cells as studies of CD4 T cells have shown that higher extrinsic IL-2 levels as well as elevated expression of CD25 favor Th1 development whereas weaker IL-2 signals result in the formation of less terminally differentiated CXCR5+ Tfh or central memory subsets (12, 50, 51). This is further supported by the observation that the initial production of IL-2 by CD4 T cells steers their development towards Tfh formation (12). Collectively, the findings are consistent with a model in which the synthesis of IL-2 disrupts IL-2 signaling which prevents terminal differentiation and preserves memory potential. Conversely, partner subsets that do not synthesize IL-2 preferentially respond to IL-2 signals which shape their transcriptional and phenotypic properties to favor full effector formation. Although intrinsic IL-2 synthesis plays a critical role in shaping fate biases during initial stages for the response, once memory is established, highly effective recall responses are elicited independently of the cell autonomous ability to produce IL-2. These investigations were conducted using genetically engineered murine systems and evaluated responses to the natural mouse pathogen LCMV. Potential limitations of the current study include that the salient findings are not yet confirmed with human specimens and that they are restricted to analyzing CD8 T cell responses to one select pathogen.
Materials and Methods
Study Design
The purpose of this study is to ascertain the roles of intrinsic IL-2 production by virus-specific CD8 T cells in determining effector and memory T cell formation and in dictating protective immunity. Flow cytometric analyses of cellular phenotypes and cell transfer approaches were employed to determine the properties and fates of functionally distinct CD8 T cell subsets which were identified using cytokine reporter systems or intracellular cytokine staining. Most experiments were performed at least 3 times with 2–5 mice per group. The total numbers of mice used are indicated in the figure legends. Both male and female mice were used except for RNA sequencing studies and co-culture transduction experiments in which male and female mice were used, respectively. Investigators were not blinded while performing the experiments.
Mice
B6.Ifng/Thy1.1 KI (IFN-γ.Thy1.1) and B6.Cg-Tg-IL2tm1(eGFP)/Weav (IL-2.GFP) knock-in cytokine reporter mice were kindly provided by Dr. Casey Weaver (University of Alabama at Birmingham, Birmingham, AL) (12, 52). Homozygous IFN-γ.Thy1.1 IL-2.GFP double reporter mice were generated by crossing the individual IFN-γ.Thy1.1 and IL-2.GFP strains. B6.SJL-PtprcaPepcb/BoyJ mice (CD45.1) were obtained from the Jackson Laboratory (Bar Harbor, ME). CD45.1 and IFN-γ.Thy1.1 IL-2.GFP mice were crossed to produce CD45.1+CD45.2+ heterozygous reporter mice (referred to as CD45.1+CD45.2+) which were used as recipients. Homozygous IFN-γ.Thy1.1 IL-2.GFP mice expressing the P14 TCR (P14 IFN-γ.Thy1.1 IL-2.GFP) were generated by a series of crosses using P14 TCR transgenic (53) and IFN-γ.Thy1.1 IL-2.GFP mice. P14 IFN-γ.Thy1.1 IL-2.GFP chimeric mice were generated by transferring 104 purified naïve splenic P14 IFN-γ.Thy1.1 IL-2.GFP CD8 T cells into 6–8-week-old naïve CD45.1+CD45.2+ recipients. B6(Cg)-Il2tm1.1(cre)Iwsh/J (Jackson Laboratories, Bar Harbor, ME) mice in which IL-2 expression is ablated (54) were crossed to introduce the P14 TCR transgene. Comparative analyses of wild-type (+/+) and IL-2-deficient (−/−) P14 CD8 T cells were conducted. All mice were bred and/or maintained in fully accredited facilities at the University of Alabama at Birmingham. All procedures were approved by the UAB Institutional Animal Care and Use Committee in accordance with NIH guidelines.
Infections and challenges
For acute infections ~10 week old mice were infected with 2×105 pfu LCMV-Armstrong by i.p. injection. Responses in P14 IFN-γ.Thy1.1 IL-2.GFP chimeric mice were primed by infection with 104 pfu LCMV-Armstrong. For challenge studies, recipient mice were infected with 2×106 pfu LCMV-clone 13 by i.v. injection. Viral titers were quantitated by plaque assay using VERO-E6 cells (23).
Tissue Harvesting and Processing
Tissues were harvested from perfused mice. Spleens were disrupted using fine wire screens, red blood cells lysed with 0.83% (w/v) NH4Cl, and cells suspended in RPMI-1640 medium supplemented with 50μM β-mercaptoethanol, 100U/ml penicillin, 100mg/ml streptomycin and either 1% or 10% fetal calf serum (FCS). Livers and lungs were minced and digested with collagenase D (2mg/ml; Roche, Indianapolis, IN) and DNase I (0.03mg/ml; Sigma-Aldrich, St. Louis, MO) in HBSS for 30 minutes at 37°C. Digested tissue was then separated by centrifugation over a layer of Histopaque 1083 (Sigma-Aldrich) and washed with complete media. Blood was collected into 4% sodium citrate and PBMCs were isolated by centrifugation over a layer of Histopaque 1083. For bone marrow isolation, femurs were flushed with complete media and subject to red blood cell lysis.
Standard flow cytometric analyses
Cells were first treated with anti-CD16/CD32 (clone 2.4G2) antibodies (UAB Immunoreagent Core and Bio X Cell, West Lebanon, NH). For live/dead discrimination, cells were washed with PBS and subsequently treated with Live/Dead fixable Aqua dye (Invitrogen) diluted in PBS prior to antibody staining. For analysis of IL-2+ and IL-2− cells, LCMV-specific IFN-γ.Thy1.1 IL-2.GFP splenocytes were stimulated with LCMV peptide epitopes as described below and stained with anti-CD8α (53–6.7; Biolegend, San Diego, CA) and anti-Thy1.1 (OX-7; Biolegend) antibodies in addition to various combinations of antibodies specific for other surface molecules. For phenotypic analysis of stimulated, transferred, and challenged cells various combinations of the following antibodies were used: CD25 (PC61), CD132 (TUGM2), CD43 (1B11), KLRG1 (2F1), CD62L (MEL-14), PD-1 (RMPI-30), 2B4 (m2B4(B6)458.1), CXCR3 (CXCR3–173), or CX3CR1 (SA011F11), CD45.2 (104) from Biolegend and CD127 (A7R34), KLRG1 (2F1), CD122 (TM-b1), CD62L (MEL-14), CD27 (LG.7F9), CCR7 (4B12), Lag-3 (eBioC9B7W), CD45.1 (A20) from Thermo Fisher Scientific. Cells were stained in 2% BSA, 0.2% sodium azide in PBS.
To assess cytokine-induced changes in cell surface receptors, splenocytes were stimulated as described below without the addition of Brefeldin A. Anti-CD25 blocking antibodies (PC-61.5.3, 50μg/ml; Bio X Cell) were added to certain cultures either without or with the addition of IL-7 (50ng/ml) or IL-15 (50ng/ml; PeproTech, Rocky Hill, NJ) for the last 30 minutes of the activation period. Cells were then processed and stained as outlined above.
All analytical specimens were acquired using an LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR). Flow cytometry gating strategies are shown in fig. S7.
Intracellular cytokine analyses
Murine splenocytes were harvested and cultured in supplemented RPMI-1640 containing 10% FCS. Splenocytes were either left untreated or stimulated with 1μg/ml LCMV peptide epitopes (GP33, NP396, and/or GP276) in the presence of Brefeldin A (GolgiPlug; BD Biosciences, San Jose, CA) for 5 hours. Note that Brefeldin A was not added to cultures where reporter expression was used directly to distinguish cytokine producing populations. After surface staining, intracellular staining was performed after using a BD Cytofix/Cytoperm Fixation/Permeabilization kit (BD Biosciences) in conjunction with anti-IFN-γ (XMG1.2), and anti-IL-2 (JES6–5H4), and/or anti-TNFα (MP6 XT22) antibodies (55).
Eomesodermin, STAT5, and S6 staining
For Eomes analyses, surface staining was performed and cells were then processed using the eBioscience Foxp3/Transcription Factor Staining Buffer Set (eBioscience, San Diego, CA) followed by staining with anti-Eomes (Dan11mag; Thermo Fisher Scientific) antibodies.
For STAT5 analysis, splenocytes from LCMV-Armstrong infected IFN-γ.Thy1.1 IL-2.GFP mice were stimulated with GP33, NP396, and GP276 peptides (1μg/mL of each) for 5 hours without the addition of Brefeldin A. Specimens were then fixed with 0.7% paraformaldehyde in PBS and stained with anti-CD8α and anti-Thy1.1 antibodies. CD8+ Thy1.1+GFP+(IFN-γ+IL-2+) and Thy1.1+GFP− (IFN-γ+IL-2−) were then isolated by cell sorting using a FACS Aria IIIu instrument. After sorting, the cells were treated with 1.5% paraformaldehyde in PBS and permeabilized with 100% methanol for 30 minutes (56, 57). The cells were then washed in PBS 0.5% BSA and stained with anti-STAT5A (C-6; Santa Cruz Biotech) and anti-pSTAT5A (47/Stat5(pY694); BD Biosciences) antibodies in PBS 0.5% BSA.
For analysis of STAT5 and S6 phosphorylation in response to cytokines, LCMV-specific CD8+ IFN-γ.Thy1.1 IL-2.GFP cells were activated for 5 hours in the presence of LCMV-peptide epitopes without Brefeldin A. Anti-CD25 antibodies (50μg/ml; Bio X Cell) were added to specific cultures and IL-7 (50ng/ml) or IL-15 (50ng/ml) (PeproTech, Rocky Hill, NJ) added to certain samples for the last 30 minutes of the activation period. After stimulation, the cells were fixed with 1.5% paraformaldehyde in PBS for 15 minutes followed by permeabilization with 100% MeOH for 30 minutes. Specimens were then treated identically to the sorted samples, as described above, and stained at room temperature with anti-CD8α, anti-Thy1.1, anti-pSTAT5A(Y694), and anti-pS6(S235/236) (D57.2.2E; Cell Signaling Technology, Danvers MA) antibodies in PBS with 0.5% BSA. After incubation, cells were washed and suspended in PBS with 0.5% BSA for analysis.
Cell sorting and transfers
At defined times post infection splenocytes were prepared and stimulated in vitro with either NP396 peptide or a mixture of GP33, GP276, and NP396 peptides (1μg/ml each) for 5 hours. Stimulated cells were then stained with APC-conjugated anti-CD8α (53–6.7, Biolegend) and PE-conjugated anti-Thy1.1 (OX-7, Biolegend) antibodies and enriched using magnetic anti-PE microbeads (Miltenyi Biotec, San Diego, CA). Thy1.1+GFP+ (IFN-γ+IL-2+) and Thy1.1+GFP− (IFN-γ+IL-2−) CD8 T cells were then isolated using a FACS Aria IIIu cell sorter (BD Biosciences). Sorted cells were washed in PBS and equal numbers of each population were transferred into naïve CD45.1+CD45.2+ cytokine reporter recipients. For the analysis of LCMV NP396-specific CD8 T cells 4.5×105 sorted effector or 1.5 × 105 memory cells were transferred. For the analyses of mixed specificities (GP33, GP276 and NP396) 7–8.25×105 sorted effector cells were transferred. Sort gating strategies are depicted in fig. S7.
BrdU analysis
Bromodeoxyuridine (BrdU) (0.8mg/ml; Sigma-Aldrich) was administered in daily changes of drinking water beginning at days 8, 14, 20, 70, and 140 following LCMV-Armstrong infection and continued for a period of 14 days. Following the two weeks of BrdU administration splenocytes were isolated and stimulated for 5 hours in the presence of 1μg/ml LCMV GP33 peptide epitope together with Brefeldin A (GolgiPlug; BD Biosciences). Following the activation period the cells were stained with anti-CD8α antibodies, fixed and permeabilized using a cell fixation/permeabilization kit (BD Biosciences), and intracellular cytokine staining performed using anti-IFN-γ (XMG1.2), and anti-IL-2 (JES6–5H4) antibodies. The stained cells were then permeabilized overnight in PBS containing 1% paraformaldehyde and 0.05% IGEPAL (Sigma-Aldrich) at 4°C, washed with PBS, incubated for 30 minutes at 37°C in 4.2 mM MgCl2 and 50-Kunitz units/ml DNase I (Sigma-Aldrich) in PBS. Cells were then stained with anti-BrdU antibodies (3D4; BD Biosciences) in PBS with 5% FCS, 2% mouse serum, and 0.5% IGEPAL. Cells were then washed and suspended in 1% paraformaldehyde in PBS before analysis by flow cytometry (58).
Annexin V analysis
At 14 days following LCMV-Armstrong infection, splenocytes were harvested and stimulated with a pool of NP396, GP33, GP276 peptides for 5 hours (1μg/ml of each), stained with anti-CD8α Pac-Blue (53–6.7; Biolegend) and anti-Thy1.1 PE (OX-7; Biolegend) antibodies, enriched and sorted as described above. The sorted populations were then washed in complete media and incubated for 18 hours in round bottom plates either alone or in the presence of 100U/ml IL-2, 50ng/ml IL-7, or 50ng/ml IL-15 (PeproTech). After culture, cells were washed and restained with anti-CD8α and anti-Thy1.1 antibodies. The cells were then washed with PBS and stained with Live/Dead dye (Invitrogen). They were then washed with Annexin V binding buffer (10 mM HEPES, 150 mM NaCl, 2.5 mM CaCl2). APC-conjugated Annexin V (Biolegend) was added and cells incubated 15 minutes prior to further washing in Annexin V binding buffer. Stained cells were immediately analyzed by flow cytometry.
RNA preparation
Splenocytes from male P14 IFN-γ.Thy1.1 IL-2.GFP chimeric mice were harvested at days 9 and 308–309 following infection for effector and memory cells, respectively. Aliquots of splenocytes were then stimulated with the LCMV GP33 peptide epitope (1μg/mL) for 5 hours. Stimulated and control untreated splenocytes were then stained with anti-CD8α (53–6.7), anti-Thy1.1 (OX-7), anti-CD45.1 (A20), anti-CD45.2 (104) antibodies. Donor P14 CD8+ Thy1.1+ GFP+ (IFN-γ+IL-2+) and Thy1.1+ GFP− (IFN-γ+IL-2−) cells as well as control untreated donor P14 IFN-γ.Thy1.1 IL-2.GFP CD8+ T cells were then sorted directly into TRIzol-LS (Invitrogen). RNA was isolated using a Direct-zol RNA isolation kit (Zymo Research, Irvine CA). RNA samples were submitted to the UAB Heflin Center for Genomic Science (Birmingham, AL) for quality assessment, RNA sequencing and analyses.
Analyses of IL-2 deficient CD8 T cells
Naïve splenic CD8+ T cells were isolated from wild-type (+/+) and IL-2-deficient (−/−) P14 mice using the CD8a+ isolation kit (Miltenyi Biotec, San Diego, CA) supplemented with 0.003μg of biotinylated anti-CD44 antibody per million cells to ensure the procurement of naïve cells. The purified CD8 T cells were then stimulated using GP33 peptide pulsed irradiated (30 Gy) feeder cells and cultured in 24 well plates with 50U/ml IL-2 for 2 days after which the culture media was removed a replaced with media containing 50ng/ml IL-15. The following day cells were transferred to 12 well plates and fresh media with IL-15 added. After 3 days of culture with IL-15 cells were washed and then restimulated with GP33 peptide (1μg/ml) for 5 hr with IL-2 (50U/mL) added for the last 30 mins. The levels of phosphorylated STAT5 was then assessed by intracellular staining as above. pSTAT5 and IL-2 levels from the cultured specimens were separately assessed and compared for the Spearman’s rank order correlation analysis.
Retroviral transduction and analyses
The cDNA encoding murine IL-2 was excised from pCR3.1-mIL2 (Addgene, Watertown, MA) using BamHI and XhoI restriction enzymes. This fragment was then inserted by sticky end ligation, using T4 DNA ligase (Promega, Madison, WI), into the multiple cloning site, upstream of the IRES-GFP cassette of the pMSCV-IRES-GFP II plasmid (Addgene), which was previously digested with the same enzymes. The plasmids were propagated in NEB Stable competent E.coli cells (New England Biolabs, Ipswich, MA) and purified using a Maxi-Prep Kit (Qiagen, Germantown, MD). These retroviral plasmids were co-transfected together with the pCL-Eco packaging plasmid (Addgene) into 293T cells (ATCC, Manassas, VA) using Fugene HD (Promega). The parental pMSCV-IRES-GFP II plasmid vector without the mIL-2 cDNA insert was used to produce control retrovirus. Retrovirus containing supernatants were harvested between 24–72hr and used for transduction. For the transductions purified naive P14 CD8 T cells were activated using plate bound anti-CD3 (10μg/mL; clone 145–2C11; BioXCell) soluble anti-CD28 (1μg/mL; clone 37.51; ThermoFisher/eBioscience) antibodies for 20 hours. These cells were then spin-inoculated with retrovirus containing supernatants in the presence of 6μg/ml Polybrene (Sigma-Aldrich) by centrifugation at 1130g for 2 hours at 25°C and then incubated for 5hr at 37°C prior to washing. The spin-inoculation was repeated 40hr after the initial culture using fresh retroviral supernatant (59). After ~72 hours of culture cells were washed, incubated and activated with IL-2 (50U/ml) for 30 mins.
RNA sequencing
RNA-Seq was performed using 50nt paired-end reads on the Illumina HiSeq 2500 system at the UAB Heflin Center for Genomic Sciences. RNA quality was assessed using an Agilent 2100 Bioanalyzer and samples were subsequently converted to cDNA. Libraries were constructed using the TruSeq library generation kits as per the manufacturer’s instructions (Illumina, San Diego, CA). cDNA libraries were quantitated using qPCR in a Roche LightCycler 480 with the Kapa Biosystems kit for library quantitation (Kapa Biosystems, Woburn, MA) prior to cluster generation. Clusters were generated to yield approximately 725 K–825 K clusters/mm2.
The quality of the sequencing reads was verified with FastQC (version 0.11.3; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The mean Phred quality score at almost every position was found to be above 30, except in one sample, which was excluded from further analysis. Reads were aligned using Tophat 2.0.10 against the mm10 mouse reference downloaded from Illumina’s iGenome resource (https://support.illumina.com/sequencing/sequencing_software/igenome.html) (60). MultiQC was used to produce a combined QC report for sequence quality and alignment (61).
Data and Software Availability
RNA sequencing analysis generated in these studies can be found under accession number GEO137717. Data sets used for GSEA analysis of effector and memory CD8 T-cell signatures (GSE9650) (27) were downloaded from the Molecular Signatures Database (Broad Institute). Data sets used to evaluate stem-like T-cell signatures were downloaded from the NCBI GEO database (GSE84105) (28).
Flow cytometry data were analyzed using FlowJo Software. Calculations and statistical significance were determined using Excel (Microsoft) and GraphPad Prism. Graphs were made using GraphPad Prism (GraphPad Software, La Jolla, CA).
Statistical Analysis
Aligned RNA sequencing reads were quantified using the SequenceOverlap package in R Bioconductor, and differential expression analysis was performed in DESeq2 using an alpha (FDR) of 0.01.
Paired two-tailed student t-tests were used for direct analysis of IL-2 producing (IL-2+) and non-producing (IL-2−) virus-specific cells within a single sample. For analysis of independent IL-2 producing and non-producing populations, unpaired two-tailed t-tests were used. Comparisons of three or more experimental groups were determined using one-way or two-way ANOVA with Dunnett’s or Holm-Šídák multiple comparison tests. Spearman’s rank order correlation coefficient was used to assess the relationship between IL-2 synthesis and STAT5 phosphorylation. P-values are defined as *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Supplementary Material
Supplementary materials and methods.
Fig. S1. Generation and authentication of IFN-γ.Thy1.1 IL-2.GFP double cytokine reporter mice.
Fig. S2. IL-2-producing effector CD8 T cells more rapidly attain a canonical memory phenotype.
Fig. S3. IL-2+ effector as well as IL-2+ and IL-2− memory CD8 T cells rescue the endogenous response following chronic viral challenge.
Fig. S4. Endogenous CD8 T cells become exhausted in the presence of non-IL-2-producing effector cells.
Fig. S5. IL-2-producing and non-producing effector CD8 T cells are differentially sensitive to IL-2, IL-7, and IL-15.
Fig. S6. Phenotypic similarities of in vitro differentiated CD8 T cells.
Fig. S7. Flow cytometric gating strategies.
Acknowledgments:
We dedicate this report to our dear friend and colleague Rakesh Bakshi. We wish to thank Ming Du for help with resource development, Marion Spell for conducting cell sorts, Michael Crowley for assistance with RNA-seq and Troy Randall, Amy Weinmann, and Hui Hu for advice.
Funding:
This study was supported in part by awards R01 AI049360 and R01 AI156290 (to AJZ), and award UL1TR003096 (to the UAB Center for Clinical and Translational Science) from the National Institutes of Health as well as award PF-16-150-01-LIB from the American Cancer Society (to SMK).
Footnotes
Competing interests: AJZ is a paid consultant for Tentarix Biotherapeutics.
Data and materials availability:
Requests for experimental animals should be directed to Casey Weaver (cweaver@uab.edu). All other requests should be directed to Allan J. Zajac (azajac@uab.edu). RNA sequencing analysis generated in these studies can be found under accession number GEO137717.
References:
- 1.Joshi NS, Kaech SM, Effector CD8 T Cell Development: A Balancing Act between Memory Cell Potential and Terminal Differentiation. J. Immunol 180, 1309–1315 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Cox MA, Kahan SM, Zajac AJ, Anti-viral CD8 T cells and the cytokines that they love. Virology 435, 157–169 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cox MA, Harrington LE, Zajac AJ, Cytokines and the inception of CD8 T cell responses. Trends in Immunology 32, 180–186 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachmann MF, Wolint P, Walton S, Schwarz K, Oxenius A, Differential role of IL-2R signaling for CD8+ T cell responses in acute and chronic viral infections. Eur. J. Immunol 37, 1502–1512 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Feau S, Arens R, Togher S, Schoenberger SP, Autocrine IL-2 is required for secondary population expansion of CD8+ memory T cells. Nature Immunology 12, 908–913 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathieu C, Beltra J-C, Charpentier T, Bourbonnais S, Di Santo JP, Lamarre A, Decaluwe H, IL-2 and IL-15 regulate CD8+ memory T-cell differentiation but are dispensable for protective recall responses. Eur. J. Immunol 45, 3324–3338 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Williams MA, Tyznik AJ, Bevan MJ, Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature 441, 890–893 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su HC, Cousens LP, Fast LD, Slifka MK, Bungiro RD, Ahmed R, Biron CA, CD4+ and CD8+ T cell interactions in IFN-gamma and IL-4 responses to viral infections: requirements for IL-2. J. Immunol 160, 5007–5017 (1998). [PubMed] [Google Scholar]
- 9.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP, CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 421, 852–856 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Shedlock DJ, Shen H, Requirement for CD4 T Cell Help in Generating Functional CD8 T Cell Memory. Science 300, 337–339 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Redeker A, Welten SPM, Baert MRM, Vloemans SA, Tiemessen MM, Staal FJT, Arens R, The Quantity of Autocrine IL-2 Governs the Expansion Potential of CD8+ T Cells. J. Immunol 195, 4792–4801 (2015). [DOI] [PubMed] [Google Scholar]
- 12.DiToro D, Winstead CJ, Pham D, Witte S, Andargachew R, Singer JR, Wilson CG, Zindl CL, Luther RJ, Silberger DJ, Weaver BT, Kolawole EM, Martinez RJ, Turner H, Hatton RD, Moon JJ, Way SS, Evavold BD, Weaver CT, Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science 361, eaao2933 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R, Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol 4, 225–234 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A, Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Wang A, Chandran S, Shah SA, Chiu Y, Paria BC, Aghamolla T, Alvarez-Downing MM, Lee C-CR, Singh S, Li T, Dudley ME, Restifo NP, Rosenberg SA, Kammula US, The Stoichiometric Production of IL-2 and IFN-γ mRNA Defines Memory T Cells That Can Self-Renew After Adoptive Transfer in Humans. Sci. Transl. Med 4, 149ra120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin MD, Badovinac VP, Defining Memory CD8 T Cell. Front. Immunol 9 (2018), doi: 10.3389/fimmu.2018.02692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM, Inflammation Directs Memory Precursor and Short-Lived Effector CD8+ T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity 27, 281–295 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R, Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med 205, 625–640 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldrath AW, Sivakumar PV, Glaccum M, Kennedy MK, Bevan MJ, Benoist C, Mathis D, Butz EA, Cytokine Requirements for Acute and Basal Homeostatic Proliferation of NaiveI and Memory CD8+ T Cells. J Exp Med 195, 1515–1522 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell DM, Ravkov EV, Williams MA, Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J. Immunol 184, 6719–6730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rubinstein MP, Lind NA, Purton JF, Filippou P, Best JA, McGhee PA, Surh CD, Goldrath AW, IL-7 and IL-15 differentially regulate CD8+ T-cell subsets during contraction of the immune response. Blood 112, 3704–3712 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schluns KS, Williams K, Ma A, Zheng XX, Lefrançois L, Cutting Edge: Requirement for IL-15 in the Generation of Primary and Memory Antigen-Specific CD8 T Cells. J. Immunol 168, 4827–4831 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB, Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med 160, 521–540 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kahan SM, Wherry EJ, Zajac AJ, T cell exhaustion during persistent viral infections. Virology 479–480, 180–193 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DAA, Wherry EJ, Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature Immunology 10, 29–37 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, Wherry EJ, Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science 338, 1220–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R, Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue H-H, Ahmed R, Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A, Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verdeil G, Puthier D, Nguyen C, Schmitt-Verhulst A-M, Auphan-Anezin N, STAT5-Mediated Signals Sustain a TCR-Initiated Gene Expression Program toward Differentiation of CD8 T Cell Effectors. J. Immunol 176, 4834–4842 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Tripathi P, Kurtulus S, Wojciechowski S, Sholl A, Hoebe K, Morris SC, Finkelman FD, Grimes HL, Hildeman DA, STAT5 Is Critical To Maintain Effector CD8+ T Cell Responses. J. Immunol 185, 2116–2124 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R, Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity 32, 91–103 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Su EW, Moore CJ, Suriano S, Johnson CB, Songalia N, Patterson A, Neitzke DJ, Andrijauskaite K, Garrett-Mayer E, Mehrotra S, Paulos CM, Doedens AL, Goldrath AW, Li Z, Cole DJ, Rubinstein MP, IL-2Rα mediates temporal regulation of IL-2 signaling and enhances immunotherapy. Sci. Transl. Med 7, 311ra170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castro I, Yu A, Dee MJ, Malek TR, The basis of distinctive IL-2− and IL-15-dependent signaling: weak CD122-dependent signaling favors CD8+ T central-memory cell survival but not T effector-memory cell development. J. Immunol 187, 5170–5182 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross SH, Cantrell DA, Signaling and Function of Interleukin-2 in T Lymphocytes. Annual Review of Immunology 36, 411–433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duprez V, Ferrer M, Dautry-Varsat A, High-affinity interleukin 2 receptor alpha and beta chains are internalized and remain associated inside the cells after interleukin 2 endocytosis. J. Biol. Chem 267, 18639–18643 (1992). [PubMed] [Google Scholar]
- 37.Henriques CM, Rino J, Nibbs RJ, Graham GJ, Barata JT, IL-7 induces rapid clathrin-mediated internalization and JAK3-dependent degradation of IL-7Rα in T cells. Blood 115, 3269–3277 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Tkach KE, Barik D, Voisinne G, Malandro N, Hathorn MM, Cotari JW, Vogel R, Merghoub T, Wolchok J, Krichevsky O, Altan-Bonnet G, T cells translate individual, quantal activation into collective, analog cytokine responses via time-integrated feedbacks. eLife 3, e01944 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tannahill GM, Elliott J, Barry AC, Hibbert L, Cacalano NA, Johnston JA, SOCS2 Can Enhance Interleukin-2 (IL-2) and IL-3 Signaling by Accelerating SOCS3 Degradation. Mol. Cell. Biol 25, 9115–9126 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rollings CM, Sinclair LV, Brady HJM, Cantrell DA, Ross SH, Interleukin-2 shapes the cytotoxic T cell proteome and immune environment–sensing programs. Sci. Signal 11, eaap8112 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rottenberg ME, Carow B, SOCS3, a major regulator of infection and inflammation. Front. Immunol 5, 58 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pellegrini M, Calzascia T, Toe JG, Preston SP, Lin AE, Elford AR, Shahinian A, Lang PA, Lang KS, Morre M, Assouline B, Lahl K, Sparwasser T, Tedder TF, Paik J, DePinho RA, Basta S, Ohashi PS, Mak TW, IL-7 Engages Multiple Mechanisms to Overcome Chronic Viral Infection and Limit Organ Pathology. Cell 144, 601–613 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Sporri B, Kovanen PE, Sasaki A, Yoshimura A, Leonard WJ, JAB/SOCS1/SSI-1 is an interleukin-2–induced inhibitor of IL-2 signaling. Blood 97, 221–226 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Volkó J, Kenesei Á, Zhang M, Várnai P, Mocsár G, Petrus MN, Jambrovics K, Balajthy Z, Müller G, Bodnár A, Tóth K, Waldmann TA, Vámosi G, IL-2 receptors preassemble and signal in the ER/Golgi causing resistance to antiproliferative anti–IL-2Rα therapies. Proc. Natl. Acad. Sci. USA 116, 21120–21130 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeng R, Aoki Y, Yoshida M, Arai K, Watanabe S, Stat5B Shuttles Between Cytoplasm and Nucleus in a Cytokine-Dependent and -Independent Manner. J. Immunol 168, 4567–4575 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Betz C, Hall MN, Where is mTOR and what is it doing there? J. Cell Biol 203, 563–574 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Araki K, mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, Youngblood BA, Abdelsamed HA, McGuire DJ, Cohen KW, Alexe G, Nagar S, McCausland MM, Gupta S, Tata P, Haining WN, McElrath MJ, Zhang D, Hu B, Greenleaf WJ, Goronzy JJ, Mulligan MJ, Hellerstein M, Ahmed R, Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, Araki K, West EE, Ghoneim HE, Fan Y, Dogra P, Davis CW, Konieczny BT, Antia R, Cheng X, Ahmed R, Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 552, 404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Papillion A, Powell MD, Chisolm DA, Bachus H, Fuller MJ, Weinmann AS, Villarino A, O’Shea JJ, León B, Oestreich KJ, Ballesteros-Tato A, Inhibition of IL-2 responsiveness by IL-6 is required for the generation of GC-TFH cells. Science Immunology 4, eaaw7636 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pepper M, Pagán AJ, Igyártó BZ, Taylor JJ, Jenkins MK, Opposing Signals from the Bcl6 Transcription Factor and the Interleukin-2 Receptor Generate T Helper 1 Central and Effector Memory Cells. Immunity 35, 583–595 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT, Memory CD4 T cells emerge from effector T-cell progenitors. Nature 452, 356–360 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Pircher H, Bürki K, Lang R, Hengartner H, Zinkernagel RM, Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature 342, 559–561 (1989). [DOI] [PubMed] [Google Scholar]
- 54.Yamamoto M, Seki Y, Iwai K, Ko I, Martin A, Tsuji N, Miyagawa S, Love RB, Iwashima M, Ontogeny and localization of the cells produce IL-2 in healthy animals. Cytokine 61, 831–841 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fuller MJ, Khanolkar A, Tebo AE, Zajac AJ, Maintenance, loss, and resurgence of T cell responses during acute, protracted, and chronic viral infections. J. Immunol 172, 4204–4214 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Krutzik PO, Hale MB, Nolan GP, Characterization of the Murine Immunological Signaling Network with Phosphospecific Flow Cytometry. J. Immunol 175, 2366–2373 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Long M, Adler AJ, Cutting Edge: Paracrine, but Not Autocrine, IL-2 Signaling Is Sustained during Early Antiviral CD4 T Cell Response. J. Immunol 177, 4257–4261 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tebo AE, Fuller MJ, Gaddis DE, Kojima K, Rehani K, Zajac AJ, Rapid recruitment of virus-specific CD8 T cells restructures immunodominance during protective secondary responses. J. Virol 79, 12703–12713 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurachi M, Kurachi J, Chen Z, Johnson J, Khan O, Bengsch B, Stelekati E, Attanasio J, McLane LM, Tomura M, Ueha S, Wherry EJ, Optimized retroviral transduction of mouse T cells for in vivo assessment of gene function. Nat. Protoc 12, 1980–1998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL, TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ewels P, Magnusson M, Lundin S, Käller M, MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods.
Fig. S1. Generation and authentication of IFN-γ.Thy1.1 IL-2.GFP double cytokine reporter mice.
Fig. S2. IL-2-producing effector CD8 T cells more rapidly attain a canonical memory phenotype.
Fig. S3. IL-2+ effector as well as IL-2+ and IL-2− memory CD8 T cells rescue the endogenous response following chronic viral challenge.
Fig. S4. Endogenous CD8 T cells become exhausted in the presence of non-IL-2-producing effector cells.
Fig. S5. IL-2-producing and non-producing effector CD8 T cells are differentially sensitive to IL-2, IL-7, and IL-15.
Fig. S6. Phenotypic similarities of in vitro differentiated CD8 T cells.
Fig. S7. Flow cytometric gating strategies.
Data Availability Statement
RNA sequencing analysis generated in these studies can be found under accession number GEO137717. Data sets used for GSEA analysis of effector and memory CD8 T-cell signatures (GSE9650) (27) were downloaded from the Molecular Signatures Database (Broad Institute). Data sets used to evaluate stem-like T-cell signatures were downloaded from the NCBI GEO database (GSE84105) (28).
Flow cytometry data were analyzed using FlowJo Software. Calculations and statistical significance were determined using Excel (Microsoft) and GraphPad Prism. Graphs were made using GraphPad Prism (GraphPad Software, La Jolla, CA).
Requests for experimental animals should be directed to Casey Weaver (cweaver@uab.edu). All other requests should be directed to Allan J. Zajac (azajac@uab.edu). RNA sequencing analysis generated in these studies can be found under accession number GEO137717.