

Abstract
Introduction:
Leptin resistance is believed to cause insulin resistance though the exact mechanism is not fully understood. The present study aims to investigate the temporal profile of postprandial triglyceride (PPTG) and leptin levels, and their association with each other as well as with markers of metabolic syndrome.
Materials and Methods:
Serum leptin and PPTG levels were measured longitudinally till 26 weeks in Wistar rats fed on controlled diet (group 1) and high sucrose diet (HSD) (group 2). Two additional groups fed on HSD were taken and treated with pioglitazone (group 3) and atorvastatin (group 4). Body weight, homeostasis model assessment of insulin resistance (HOMA-IR), and glucose intolerance were also measured during this period. Comparison of the groups were done and Pearson's correlation coefficient was used to ascertain the association.
Results:
Leptin levels were significantly higher in all three groups receiving HSD compared to controlled diet group from week 2 to week 26 (P < 0.01). The postprandial triglyceride area under the curve (PPTG AUCs) were significantly higher in group 2 than controls during this period (P < 0.001). Body weight, HOMA-IR and glucose AUC were found to be significantly higher in group 2 rats than controls only from week 6, 8, and 12 respectively. In HSD-fed rats, but not in control, mean serum leptin levels from 2-26 weeks as well as peak (10th week) and 26th week were strongly associated with corresponding as well as preceding PPTG levels. Leptin levels significantly predicted HOMA-IR and prediabetes in group 2.
Conclusion:
This study found significant hyperleptinemia associated with postprandial hypertriglyceridemia that predicted insulin resistance and prediabetes in high sucrose diet–fed rats.
Keywords: Glucose intolerance, high sucrose diet, HOMA-IR, hyperleptinemia, metabolic syndrome, postprandial hypertriglyceridemia
INTRODUCTION
Postprandial hypertriglyceridemia has been shown to lead to development of insulin resistance, and predict prediabetes and diabetes in diet-induced experimental models of type 2 diabetes mellitus.[1] However, development of diabetes in this model was not associated with hypoadiponectinemia.[2] Studies have reported the association of both hypo-[3,4] as well as hyper- adiponectinemia[2,5] with metabolic syndrome and insulin resistance[3,4,5] thereby suggesting that adiponectin may not be a critical determinant of insulin resistance and other factors may be more important.
It is also believed that leptin resistance and hyperleptinemia can result in insulin resistance[6,7,8,9] and could be the underlying mechanism for its development,[9,10] particularly in the obese. Several studies using experimental models have demonstrated leptin resistance and hyperleptinemia in diet-induced obesity and metabolic syndrome.[11,12,13] Leptin resistance occurs predominantly centrally at the level of the hypothalamus and is believed to work at two levels.[14] At the blood-brain barrier (BBB), this is due to the inability of leptin to cross the BBB and reach the hypothalamus in sufficient quantities to modulate leptin signaling. Further, leptin resistance also results from impaired signaling at the level of the hypothalamic neurons secondary to several dietary[15] and other factors.[14,15]
Saturated triglycerides are believed to be major modulators of leptin resistance at the level of the BBB.[16] Acute infusion of triglycerides but not fatty acids in rats have been shown to block leptin transport across the BBB.[16] Similarly, leptin resistance has also been consistently demonstrated in high fructose diet–fed rats which are known to induce hypertriglyceridemia.[17,18] Rats that were fed cream with saturated triglycerides have also been shown to induce leptin resistance both acutely at the BBB[16] as well as sub-chronically at the level of the central leptin signaling pathways.[16]
We hypothesized that the significantly higher postprandial triglyceride burden which develops very early in experimental models of diet-induced diabetes using high sucrose feeding could result in leptin resistance, hyperleptinemia and lead to the development of insulin resistance and metabolic syndrome. To test this hypothesis, levels of postprandial triglycerides as well as leptin were measured longitudinally in this study during the development of metabolic syndrome to determine their temporal profile and association with each other, as well as with the development of metabolic syndrome and prediabetes.
MATERIALS AND METHODS
This longitudinal study was conducted in eight-week-old male Wistar rats who were randomized into four groups i.e., group 1, 2, 3 and 4 (n = 24 per group). Group 1 was given a standard chow diet. The three remaining groups were given a HSD. In addition to a HSD, group 3 rats were treated with pioglitazone (10 mg/kg body-weight/day) and group 4 rats were treated with atorvastatin (20 mg/kg body-weight/day) for 26 weeks.
Body weight, fasting serum leptin, and insulin levels were measured every fortnight in all the four groups from baseline (week 0 of study, 8-week-old rats) to 26th week. Fat challenge test (to study postprandial triglyceride levels) and oral glucose tolerance test (to study glucose intolerance) were also done alternatively at fortnight intervals up to the 26th week of the study.
Fat challenge test and oral glucose test were performed after overnight fasting. Following fasting blood sampling, olive oil (2 ml/kg body-weight) was given in fat challenge test, and blood samples were collected two-hourly till eight hours for estimation of postprandial triglyceride levels. Oral glucose tolerance tests were performed with glucose (2 gm/kg body-weight) and blood samples were collected every thirty minutes till two hours.
HSD had 230 gm added sucrose and 66 gm added palm oil per kilogram of diet. Diet and water were given ad libitum unless specified. Standard environmental conditions, i.e., 20–25 °C room temperature, 50 ± 20% humidity and 12-hour light/dark cycle were maintained during the entire study.
Body weight was recorded on a dedicated electronic weighing balance. Serum leptin and insulin levels were measured by radioimmuno assay method (kit: Millipore, USA). Coefficient of variation were <5% intra-assay and <10% inter-assay.
Merck lab kits (Spain) were used for estimation of serum triglyceride. Inter-assay and intra-assay coefficients of variation were <2% for these kits. Quality control sera (BioRad, USA) were run along with each assay to ensure accuracy of test results. Blood glucose was measured by glucometer (OneTouch, LifeScan's SURESTEP, Johnson & Johnson). The glucometer was calibrated every time before each use. The estimation of some samples (n = 197) was also done through Glucose Oxidase Peroxidase (GOD-POD) method to further validate the accuracy of the glucometer. The glucose value obtained from both methods (GOD-POD method and glucometer) showed good correlation (r = 0.97).
HOMA-IR was calculated as: fasting insulin (μU/ml) x fasting glucose (mg/dl)/2430. This formula is applicable for animal models.[19]
Statistical analysis: Postprandial triglyceride/glucose area under the curves (AUC) were calculated by trapezoidal rule. Study variables between the groups were compared via generalized estimating equations followed by least significant difference pair wise comparisons. Association between the study variables were estimated by Pearson's correlation coefficient followed by linear regression. Cox regression was used (group 1 + 2) to predict incident prediabetes from leptin levels. Data were considered significantly different/associated if P values were <0.05 (SPSS 20.0).
RESULTS
Baseline parameters such as body weight, fasting serum leptin, insulin, glucose and lipids were similar in all the four study groups.
Serum leptin levels were significantly higher in all the three groups receiving HSD, viz., groups 2, 3, and 4 compared to control group 1 from week 2 to week 26. The highest increase in serum leptin levels was in group 2 followed by groups 3 and 4. The peak leptin levels in HSD–fed rats who did not receive any treatment were observed at week 10 [Figure 1a].
Figure 1.

Serum leptin levels and postprandial triglyceride responses over the period of 26 weeks
Postprandial triglyceride responses as indicated by PPTG AUC were found to be significantly higher in group 2 compared to group 1 from week 2 to week 26. PPTG AUC was also significantly higher in group 3 compared to group 1 at week 2 and 10 but was comparable thereafter at most of the time points. Similarly, PPTG AUC was significantly higher in group 4 compared to group 1 at week 2 and 6 but was comparable thereafter at all time points [Figure 1b].
Before 6 weeks, there was no significant difference in body weight between any of the study groups. Body weight was found to be significantly higher in group 2 compared to group 1 rats from week 6 (283.54 ± 21.27 vs 266.50 ± 25.42 gm, P = 0.01) and continued to be significantly higher in this group till week 26. The maximum weight gain was achieved by week 20 by the HSD–fed groups (group 2: 365.33 ± 33.72; group 3: 347.38 ± 38.89 gm; and group 4: 327.25 ± 30.61 gm) as well as the control group (339.54 ± 27.99 gm). There was a modest but non-significant increase in body weight in group 3 rats compared to group 1. No increase in body weight was seen in 4 rats compared to group 1.
Insulin resistance as indicated by HOMA-IR was significantly higher in group 2 rats compared to group 1 at week 8. It peak at week 16, and remained higher thereafter at most of the time points. Groups 3 and 4 also showed significantly higher HOMA-IR compared to group 1 from week 14 at some of the time points [Figure 2a].
Figure 2.

HOMA-IR and glucose intolerance over the period of 26 weeks
Postprandial glucose area under the curve (PPGl AUC) indicating glucose intolerance was found to be significantly higher in group 2 compared to group 1 from week 12 to week 26. Group 3 showed significantly higher PPGl AUC compared to group 1 at weeks 24 and 26. There was no significant difference in PPGl AUC between group 4 and 1 at any of the time points [Figure 2b].
Serum leptin levels showed significantly strong positive correlation with corresponding PPTG AUC at weeks 10 and 26 in group 2 only. Similarly, serum leptin levels (week 10) of group 2 showed significantly strong positive correlation with body weight (week 10) and HOMA-IR of succeeding weeks (weeks 12, 16, and 26) and PPGl AUC (week 16) [Table 1]. Also, in group 2, mean serum leptin levels from weeks 2–26 were positively associated with postprandial triglyceride burden (cumulative postprandial triglyceride burden from weeks 2–26), HOMA-IR (week 26) and PPGl AUC (week 26) [Table 1].
Table 1.
Linear regression analyses to predict leptin levels, HOMA-IR and PPGl-AUC in high sucrose diet-fed rats
| Predictor | Dependent variable | Pearson’s correlation coefficient (r) | Regression coefficient (B) | Significance (P) | 95% C. I. |
|---|---|---|---|---|---|
| PPTG-AUC wk10 | Leptin wk10 | 0.48 | 0.008 | 0.019 | 0.001-0.014 |
| PPTG-AUC wk26 | Leptin wk26 | 0.46 | 0.009 | 0.026 | 0.001-0.017 |
| Cumulative PPTG burden wk2-26 | Mean Leptin wk2-26 | 0.55 | 0.001 | 0.005 | 0.000-0.001 |
| Leptin wk10 | HOMA-IR wk12 | 0.50 | 0.064 | 0.014 | 0.015-0.114 |
| Leptin wk10 | HOMA-IR wk16 | 0.53 | 0.063 | 0.009 | 0.018-0.108 |
| Leptin wk10 | HOMA-IR wk26 | 0.60 | 0.050 | 0.003 | 0.019-0.080 |
| Leptin wk10 | PPGl-AUC wk16 | 0.43 | 2.90 | 0.035 | 0.230-5.580 |
| Mean Leptin wk2-26 | HOMA-IR wk26 | 0.51 | 0.080 | 0.015 | 0.017-0.143 |
| Mean Leptin wk2-26 | PPGl-AUC wk26 | 0.43 | 5.77 | 0.043 | 0.213-11.33 |
Confidence interval (CI), homeostasis model assessment of insulin resistance (HOMA-IR), postprandial glucose area under the curve (PPGl-AUC), postprandial triglyceride area under the curve (PPTG-AUC), week (wk)
Linear regression analyses in HSD–fed group 2 showed that serum leptin levels were significantly predicted by postprandial triglyceride parameters [Table 1]. Also, serum leptin levels at weeks 10 and 26 as well as mean leptin levels significantly predicted succeeding weeks HOMA-IR and postprandial glucose AUC in this group [Table 1].
Similarly, serum leptin levels (week 10) significantly predicted HOMA-IR of weeks 16 (r = 0.48, B = 0.14, P = 0.020, CI = 0.025 – 0.26) and week 26 (r = 0.46, B = 0.061, P = 0.033, CI = 0.005 – 0.116) in control group 1 also. Serum leptin levels in control group 1 did not show any significant association with postprandial triglyceride levels, body weight and PPGl AUC at any time point.
Cox regression analysis revealed that serum leptin levels significantly predicted the risk of prediabetes with hazard ratio (HR) of 1.47 (CI = 0.90 – 2.41, P = 0.001) [Figure 3].
Figure 3.
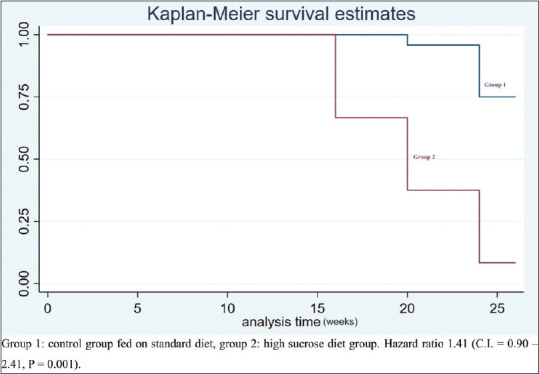
Cox regression analysis predicting prediabetes from serum leptin levels
DISCUSSION
This study found elevated postprandial triglyceride and leptin levels as early as two weeks after HSD consumption in eight-week-old male Wistar rats. Leptin levels continued to rise and peaked at 10 weeks; they were significantly higher than in rats fed standard chow diet at all time points, and remained elevated till 26 weeks of follow up. Weight gain in these rats was observed much later from week 6 with peak weight gain achieved at week 20. Insulin resistance by HOMA-IR was demonstrable only by week 8 which peaked by the week 16 of HSD feeding. By week 26, most rats in HSD–fed groups developed prediabetes.
In the HSD–fed rats, but not in control group, mean serum leptin levels from weeks 2–26 as well as peak (week 10) and week 26 were strongly associated with corresponding as well as preceding PPTG levels and burden.
These findings when taken together along with the temporal profiles of different parameters indicate that both postprandial hypertriglyceridemia and hyperleptinemia occur much before the onset of obesity, insulin resistance, and prediabetes. Clearly high leptin levels are not due to weight gain and obesity, and could be secondary to leptin resistance independent of obesity. Early hyperleptinemia before the onset of diet-induced obesity has been reported earlier[15,16,20] and believed to be secondary to leptin resistance at the level of blood-brain barrier (BBB). The strong association of leptin levels with previous as well as current PPTG levels in our study points to postprandial hypertriglyceridemia as the cause of leptin resistance. Triglycerides have been implicated in the development of central leptin resistance in earlier studies.[16] Acute infusions of saturated triglycerides in rats have been shown to cause leptin resistance by blocking its transport across the BBB.[16] Also, rats given cream in their diet failed to respond to leptin with inhibition of feeding both acutely and sub-chronically, suggesting that hypertriglyceridemia is secondary to consumption of cream induced leptin resistance.[16] These effects were believed to be due to leptin resistance at the BBB level in the acute group and secondary to more lasting effects of triglycerides on leptin signaling pathways in the central nervous system.
Hyperleptinemia can also result in postprandial hypertriglyceridemia.[21,22,23] Since elevation of PPTG and leptin levels were both observed from week 2, this possibility cannot be ruled out completely. Hepatic leptin resistance increases hepatic very low density lipoproteins (VLDLc) by promoting lipogenesis and impairing triglyceride breakdown and clearance in the liver which can cause postprandial hypertriglyceridemia.[21] However, these effects on the liver do not occur early even at 8 weeks, and have been reported in rats only after 14 weeks of high fat feeding.[23] Leptin regulation of lipid metabolism in the liver are believed to be long term effects[23] and the consequences of hepatic leptin resistance are believed to occur later in the course of diet-induced obesity.[23] In view of these observations, we believe that although possible, it is highly unlikely that it was hepatic leptin resistance that led to hyperleptinemia and hypertriglyceridemia in our study. As discussed above, it is far more likely that it is postprandial hypertriglyceridemia that is driving leptin resistance at the BBB level and hyperleptinemia.
We have previously shown that postprandial hypertriglyceridemia predicts diet-induced diabetes and insulin resistance in a diet-induced rat model,[1] and that development of metabolic syndrome in this setting is not associated with hypoadiponectinemia.[2] It would appear from the present study that the postprandial hypertriglyceridemia related diabetes risk reported by us earlier is secondary to the leptin resistance induced by them, as demonstrated in the current study.
Peak leptin levels predicted HOMA-IR of succeeding weeks in both HSD–fed rats as well as control. These findings confirm several earlier reports that leptin resistance precedes and leads to the development of insulin resistance.[6,7,8,9,10] We also demonstrated significant association of mean leptin levels from weeks 2–26 with glucose intolerance at week 26 in HSD groups only. Further, in cox regression analysis, high leptin levels predicted the development of prediabetes. Taken together, these findings further support the theory that leptin resistance could be the underlying condition that results in insulin resistance and glucose intolerance in diet-induced diabetes.
Leptin levels were not associated with body weight at most time points in the HSD groups, and all time points in those fed standard chow diet. This clearly points to a dissociation of serum leptin levels and body weight at least in rats and is contrary to the view that they primarily reflect the adipose tissue stores of the body. It further reinforces the conclusion that hyperleptinemia represents leptin resistance in the course of diet-induced obesity that is unrelated to obesity itself. Healthy men given fructose for four weeks displayed elevated serum fasting leptin levels without any change in weight confirming[18] similar observations in rats.
We used the HSD model in this study, 50% of which got converted to fructose. Hence, it is also possible that the significant postprandial hypertriglyceridemia as well as hyperleptinemia observed by us as early as the 2nd week of HSD was secondary to fructose. It is well known that fructose causes a much higher level of hypertriglyceridemia than glucose[18,24,25,26] and amounts of 30% or more of energy intake as a sugar sweetened beverage can cause postprandial hypertriglyceridemia within 24 hours.[27] This could perhaps be due to impaired triglyceride-rich lipoproteins and increased hepatic overproduction. High fructose diets have also been demonstrated to cause hepatic leptin resistance,[11,28] which could be centrally mediated at the hypothalamus[29] or peripherally mediated at the level of the liver,[29] resulting in marked hyperleptinemia.[30,31] It has been reported that diets containing 50% or more fructose rapidly induce hypertriglyceridemia within days and marked insulin resistance within four weeks.[17] However, diets containing less than 10% w/v fructose cause hypertriglyceridemia and leptin resistance early (few days to two weeks), but insulin resistance develops only after several weeks.[32] The relatively low fructose content (approximately 10%) of the high sucrose diet used in our study explains the observation of postprandial hypertriglyceridemia and hyperleptinemia which developed early at 2 weeks and peaked at 10 weeks, while insulin resistance was demonstrated much later. This pattern of fructose intake and its effects on triglycerides, leptin resistance, and insulin resistance is similar to that usually seen in humans.[18] Diet containing fructose is also known to have significant hepatic effects resulting in high VLDLc production and secretion[33,34] which also contributes to postprandial hypertriglyceridemia as well as hepatic steatosis in the long term.[29]
High sucrose diet in our study had low fat content (16%) overall, but included palm oil containing 44% palmitic acid—a saturated fatty acid. Studies have also shown that palmitic acid induces central leptin resistance in male mice in diet-induced obesity[35] and this could also have contributed to the development of leptin resistance in HSD groups.
Pretreatment with atorvastatin and pioglitazone in HSD–fed rats blunted the postprandial triglyceride responses to oral fat challenge after 6 and 10 weeks respectively. However, the leptin levels which were initially higher at 2 weeks, if they received the medications, remained similar to the HSD groups not on any medication from the week 4 onwards. The dissociation of postprandial triglyceride lowering effects of atorvastatin from that on leptin in our study is interesting and points to a leptin independent action on triglycerides. Statins in general reduce triglycerides proportionate to their low density lipoproteins (LDLc) lowering effect. However, atorvastatin is more potent in lowering triglyceride levels and complete attenuation of fructose induced increase in circulating triglycerides has been demonstrated with this agent.[36] It has been shown to prevent hepatic carbohydrate response element binding protein (CHREBP) activation secondary to protein kinase C activation,[36] which results in increased fatty acid oxidation over synthesis and storage of triglycerides, all of which result in decreased hepatic VLDLc production. The relationship between pioglitazone and leptin is far more complex. Peroxisome proliferator activated receptor gamma (PPAR-G) agonists decrease leptin levels in high fat diet–fed rats[37] while leptin itself downregulates PPAR-G receptor activity, thus creating a closely regulated system. The effect of PPAR-G agonists on postprandial hypertriglyceridemia is primarily due to increased clearance of triglycerides from the circulation into the subcutaneous adipose tissue, an effect not dependent on leptin.
The present study has few limitations. We did not measure leptin levels and leptin mRNA in cerebrospinal fluid, which would have helped to demonstrate leptin resistance at the BBB. Similarly, estimation of hepatic lipogenic and lipolytic enzymes, and hepatic fat content, as well as hepatic leptin receptor mRNA and protein levels could have helped identify hepatic leptin resistance and clarify its contribution in IR.
In conclusion, this study found significant hyperleptinemia associated with postprandial hypertriglyceridemia that predicted insulin resistance and prediabetes in HSD–fed rats. These findings help understand the pathogenic links of triglyceride-related insulin resistance. Further, high circulating leptin levels could serve as a marker to predict the risk of type 2 diabetes.
Ethical approval
Ethical approval for the study was given by Institutional Ethics Committee – Animal Research, University College of Medical Sciences. The study was conducted as per the guidelines of Ethics Committee and standard animal care was maintained throughout the study.
Financial support and sponsorship
The work was supported by the Department of Biotechnology, Ministry of Science and Technology, India (BT/PR14215/MED/30/403-2010).
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
Authors are thankful to technical staff of Department of Pharmacology and Central Animal House of the institute.
REFERRENCES
- 1.Aslam M, Aggarwal S, Sharma KK, Galav V, Madhu SV. Postprandial hypertriglyceridemia predicts development of insulin resistance glucose intolerance and type 2 diabetes. PLoS One. 2016;11:e0145730. doi: 10.1371/journal.pone.0145730. doi: 10.1371/journal.pone.0145730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aslam M, Madhu SV. Development of metabolic syndrome in high-sucrose diet fed rats is not associated with decrease in adiponectin levels. Endocrine. 2017;58:59–65. doi: 10.1007/s12020-017-1403-5. [DOI] [PubMed] [Google Scholar]
- 3.Moon HU, Ha KH, Han SJ, Kim HJ, Kim DJ. The Association of adiponectin and visceral fat with insulin resistance and β-cell dysfunction. J Korean Med Sci. 2019;34:e7. doi: 10.3346/jkms.2019.34.e7. doi: 10.3346/jkms.2019.34.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen MC, Lee CJ, Yang CF, Chen YC, Wang JH, Hsu BG. Low serum adiponectin level is associated with metabolic syndrome and is an independent marker of peripheral arterial stiffness in hypertensive patients. Diabetol Metab Syndr. 2017;9:49. doi: 10.1186/s13098-017-0247-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sumiyoshi M, Sakanaka M, Kimura Y. Chronic intake of high-fat and high-sucrose diets differentially affects glucose intolerance in mice. J Nutr. 2006;136:582–7. doi: 10.1093/jn/136.3.582. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes. 2001;50:2786–91. doi: 10.2337/diabetes.50.12.2786. [DOI] [PubMed] [Google Scholar]
- 7.Leon-Cabrera S, Solís-Lozano L, Suárez-Álvarez K, González-Chávez A, Béjar YL, Robles-Díaz G, et al. Hyperleptinemia is associated with parameters of low-grade systemic inflammation and metabolic dysfunction in obese human beings. Front Integr Neurosci. 2013;7:62. doi: 10.3389/fnint.2013.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang J-H, Lim H-S, Heo Y-R. Sasa borealis leaves extract improves insulin resistance by modulating inflammatory cytokine secretion in high fat diet-induced obese C57/BL6J mice. Nutr Res Pract. 2010;4:99–105. doi: 10.4162/nrp.2010.4.2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao S, Zhu Y, Schultz RD, Li N, He Z, Zhang Z, et al. Partial leptin reduction as an insulin sensitization and weight loss strategy. Cell Metab. 2019;30:706–19. doi: 10.1016/j.cmet.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mambiya M, Shang M, Wang Y, Li Q, Liu S, Yang L, et al. The play of genes and non-genetic factors on type 2 diabetes. Front Public Health. 2019;7:349. doi: 10.3389/fpubh.2019.00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shapiro A, Mu W, Roncal C, Cheng KY, Johnson RJ, Scarpace PJ. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am J Physiol-Regul Integr Comp Physiol. 2008;295:R1370–5. doi: 10.1152/ajpregu.00195.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scarpace PJ, Matheny M, Tümer N, Cheng KY, Zhang Y. Leptin resistance exacerbates diet-induced obesity and is associated with diminished maximal leptin signalling capacity in rats. Diabetologia. 2005;48:1075–83. doi: 10.1007/s00125-005-1763-x. [DOI] [PubMed] [Google Scholar]
- 13.Levin BE, Dunn-Meynell AA. Reduced central leptin sensitivity in rats with diet-induced obesity. Am J Physiol-Regul Integr Comp Physiol. 2002;283:R941–8. doi: 10.1152/ajpregu.00245.2002. [DOI] [PubMed] [Google Scholar]
- 14.El-Haschimi K, Pierroz DD, Hileman SM, Bjørbæk C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105:1827–32. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vasselli J, Scarpace P, Harris R, Banks W. Dietary components in the development. Adv Nutr. 2013;4:164–75. doi: 10.3945/an.112.003152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banks WA, Coon AB, Robinson SM, Moinuddin A, Shultz JM, Nakaoke R, et al. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes. 2004;53:1253–60. doi: 10.2337/diabetes.53.5.1253. [DOI] [PubMed] [Google Scholar]
- 17.Alegret M, Roglans N, Laguna JC. Fructose consumption and leptin resistance: What have we learnt from animal studies? Leptin Horm Funct Dysfunctions Clin Uses. 2011 April;:209–30. [Google Scholar]
- 18.Brkljacic J, Velickovic N, Elakovic I, Teofilovic A, Milutinovic VD, Dordevic A, et al. Fructose-enriched diet affects hepatic lipid metabolism in young male and female rats in different ways. Arch Biol Sci. 2019;71:417–24. [Google Scholar]
- 19.Cacho J, Sevillano J, De Castro J, Herrera E, Ramos MP. Validation of simple indexes to assess insulin sensitivity during pregnancy in Wistar and Sprague-Dawley rats. Am J Physiol-Endocrinol Metab. 2008;295:E1269–76. doi: 10.1152/ajpendo.90207.2008. doi: 10.1152/ajpendo.90207.2008. [DOI] [PubMed] [Google Scholar]
- 20.Izquierdo AG, Crujeiras AB, Casanueva FF, Carreira MC. Leptin, obesity, and leptin resistance: Where are we 25 years later? Nutrients. 2019;11:2704. doi: 10.3390/nu11112704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yen FT, Roitel O, Bonnard L, Notet V, Pratte D, Stenger C, et al. Lipolysis stimulated lipoprotein receptor: A novel molecular link between hyperlipidemia, weight gain, and atherosclerosis in mice. J Biol Chem. 2008;283:25650–9. doi: 10.1074/jbc.M801027200. [DOI] [PubMed] [Google Scholar]
- 22.Huynh FK, Neumann UH, Wang Y, Rodrigues B, Kieffer TJ, Covey SD. A role for hepatic leptin signaling in lipid metabolism via altered very low density lipoprotein composition and liver lipase activity in mice. Hepatology. 2013;57:543–54. doi: 10.1002/hep.26043. [DOI] [PubMed] [Google Scholar]
- 23.Guzmán-Ruiz R, Stucchi P, Ramos MP, Sevillano J, Somoza B, Fernández-Alfonso M, et al. Leptin drives fat distribution during diet induced obesity in mice. Endocrinol Nutr. 2012;59:354–61. doi: 10.1016/j.endonu.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Schaefer EJ, Gleason JA, Dansinger ML. Dietary fructose and glucose differentially affect lipid and glucose homeostasis. J Nutr. 2009;139:1257S–62S. doi: 10.3945/jn.108.098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–34. doi: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanhope KL, Griffen SC, Bair BR, Swarbrick MM, Keim NL, Havel PJ. Twenty-four-hour endocrine and metabolic profiles following consumption of high-fructose corn syrup-, sucrose-, fructose-, and glucose-sweetened beverages with meals. Am J Clin Nutr. 2008;87:1194–203. doi: 10.1093/ajcn/87.5.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teff KL, Grudziak J, Townsend RR, Dunn TN, Grant RW, Adams SH, et al. Endocrine and metabolic effects of consuming fructose- and glucose-sweetened beverages with meals in obese men and women: Influence of insulin resistance on plasma triglyceride responses. J Clin Endocrinol Metab. 2009;94:1562–9. doi: 10.1210/jc.2008-2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.C Bursac BN, Vasiljevic AD, Nestorovic NM, Velickovic NA, Milutinovic DDV, Matic GM, et al. High-fructose diet leads to visceral adiposity and hypothalamic leptin resistance in male rats – Do glucocorticoids play a role? J Nutr Biochem. 2014;25:446–55. doi: 10.1016/j.jnutbio.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, Song H, Ge Y, Ji G, Yao Z. Temporal relationship between diet-induced steatosis and onset of insulin/leptin resistance in male Wistar rats. PLoS One. 2015;10:e0117008. doi: 10.1371/journal.pone.0117008. doi: 10.1371/journal.pone. 0117008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li JM, Wang C, Hu QH, Kong LD. Fructose induced leptin dysfunction and improvement by quercetin and rutin in rats. Chin J Nat Med. 2008;6:466–73. [Google Scholar]
- 31.Lozano I, Van Der Werf R, Bietiger W, Seyfritz E, Peronet C, Pinget M, et al. High-fructose and high-fat diet-induced disorders in rats: Impact on diabetes risk, hepatic and vascular complications. Nutr Metab (Lond) 2016;13:15. doi: 10.1186/s12986-016-0074-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roglans N, Vilà L, Farré M, Alegret M, Sánchez RM, Vázquez-Carrera M, et al. Impairment of hepatic STAT 3 activation and reduction of PPARa activity in fructose-fed rats. Hepatology. 2007;45:778–88. doi: 10.1002/hep.21499. [DOI] [PubMed] [Google Scholar]
- 33.Chong T, Naples M, Federico L, Taylor D, Smith GJ, Cheung RC, et al. Effect of rosuvastatin on hepatic production of apolipoprotein B-containing lipoproteins in an animal model of insulin resistance and metabolic dyslipidemia. Atherosclerosis. 2006;185:21–31. doi: 10.1016/j.atherosclerosis.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 34.Taghibiglou C, Carpentier A, Van Iderstine SC, Chen B, Rudy D, Aiton A, et al. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster m. J Biol Chem. 2000;275:8416–25. doi: 10.1074/jbc.275.12.8416. [DOI] [PubMed] [Google Scholar]
- 35.Cheng L, Yu Y, Szabo A, Wu Y, Wang H, Camer D, et al. Palmitic acid induces central leptin resistance and impairs hepatic glucose and lipid metabolism in male mice. J Nutr Biochem. 2015;26:541–8. doi: 10.1016/j.jnutbio.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 36.Rodríguez-Calvo R, Barroso E, Serrano L, Coll T, Sánchez RM, Merlos M, et al. Atorvastatin prevents carbohydrate response element binding protein activation in the fructose-fed rat by activating protein kinase A. Hepatology. 2009;49:106–15. doi: 10.1002/hep.22570. [DOI] [PubMed] [Google Scholar]
- 37.Törüner F, Akbay E, Çakir N, Sancak B, Elbeg Ş, Taneri F, et al. Effects of PPARγ and PPAR α agonists on serum leptin levels in diet-induced obese rats. Horm Metab Res. 2004;36:226–30. doi: 10.1055/s-2004-814452. [DOI] [PubMed] [Google Scholar]