

Abstract
Objective
Long QT syndrome (LQTS) is an inherited primary arrhythmia syndrome associated with life-threatening ventricular arrhythmias and sudden death. This study aimed to report the clinical and genetic characteristics and outcomes of children diagnosed as having LQTS in a tertiary pediatric cardiology center in Turkey.
Methods
This was a retrospective review of pediatric patients diagnosed as having LQTS at our center from January 2011 to April 2020.
Results
A total of 145 patients (76 males) were included, with a mean age of 9.2±4.5 years and a mean weight of 35.7±18.5 kg; 38 (26.2%) were identified as having LQTS during family screening, whereas a significant proportion of patients were asymptomatic at presentation, 15 patients (10.3%) were diagnosed after previous cardiac arrest, and 26 patients (18%) had syncope. The mean Schwartz score was 4.5 points (range, 3–7.5 points). Furthermore, 107 patients (82%) were confirmed to have a pathogenic mutation for LQTS genes. All patients received beta-blockers. Implantable cardioverter-defibrillator insertion was performed in 34 patients (23.4%). Left or bilateral cardiac sympathetic denervation was performed in 9 patients (6.2%). Median follow-up time was 35.6±25.8 months. Five (3.4%) patients died during the follow-up. Statistical analyses of risk factors for major cardiac events revealed that the QTc was >500 ms and that T wave alternans, high Schwartz score, and Jervell and Lange-Nielsen syndrome were strong and significant predictors of cardiac events.
Conclusion
LQTS has a variety of clinical manifestations. Patients’ symptoms ranged between asymptomatic and sudden cardiac death (SCD). By raising the awareness of physicians regarding the disease, SCD might be prevented in the early period.
Keywords: long QT syndrome, children, genetic, treatment
Introduction
Congenital long QT syndrome (LQTS) is an inheritable arrhythmic disorder that may present with malignant arrhythmia and, occasionally, the risk of sudden death. Abnormal ventricular repolarization in these patients predisposes them to malignant cardiac arrhythmia, such as torsades de pointes and ventricular fibrillation (1). The prevalence of LQTS is believed to be as high as 1 in 2500 individuals (2). Based on mutations associated with 15 autosomal dominant genes, LQTS has been classified into 17 subtypes: LQT1–15 (3, 4). LQTS is an important disease of sudden cardiac death (SCD) in children. The risk in affected patients is not uniform because of variable penetrance and is influenced by age, gender, genotype, environmental factors, therapy, and possibly other modifier genes (5). Lifestyle modification, beta-blockers, and implantable cardioverter-defibrillator (ICD) implantation are the most important therapeutic modalities in the proper management of patients with LQTS. To establish the extent of the disease, the main focus in the management of individuals with LQTS is to identify the individuals at high risk of cardiac events (6). In this study, we review and identify the clinical characteristics, genetic profile, management strategy, and the risk factors for major cardiac events and outcomes of our local pediatric patients with LQTS.
Methods
The study included 145 patients (female-to-male ratio, 69:76) diagnosed as having congenital LQTS between January 2011 and April 2020. LQTS was diagnosed based on clinical background using the 2013 Heart Rhythm Society, European Heart Rhythm Association, and Asia Pacific Heart Rhythm Society (HRS/EHRA/APHRS) consensus guidelines (7). QT interval was measured in lead II or V5 and corrected for differences in heart rate using the Bazett’s formula. LQTS was diagnosed through careful examination of clinical and family history, including symptoms of syncope, anoxic seizures, or, occasionally, palpitations, and through targeted investigations, including electrocardiograms (ECGs), 24-hour Holter recording analysis, and exercise stress testing. Occasionally, provocative drug challenges were used to differentiate the diagnoses from other primary arrhythmia syndromes. Patients with a Schwartz score of ≥3.5 points in the absence of a secondary cause for QT prolongation were diagnosed as having LQTS. The presence of a confirmed pathogenic LQTS mutation, irrespective of the QTc, was also used to diagnose LQTS.
The collected data included demographic features, clinical presentation, family history, genetic tests, and other special cardiac investigations (i.e., routine ECG, 24-hour Holter monitoring, epinephrine challenge test, or exercise stress testing). Schwartz score (LQTS), which was calculated with the data collected from each patient, was retrospectively reviewed, and follow-up data were obtained from hospital records (using FileMaker® online database).
After detailed evaluations, patients were classified according to SCD risk stratification using the 2013 HRS/EHRA/APHRS consensus guidelines (7). We determined our treatment modality after the risk assessment. Lifestyle modifications, such as avoidance of strenuous exercise (especially swimming without super-vision) in patients with LQT1, reduction of exposure to abrupt loud noises (e.g., alarm clock or phone ringing) in patients with LQT2, and avoidance of drugs that prolong QT interval in all patients with LQTS, are routine. All patients were offered beta-blocker therapy. Device therapy (ICD) and left or bilateral cardiac sympathetic denervation (BCSD) were considered for high-risk patients.
Major cardiac events were defined as LQTS-attributable syncope, aborted cardiac arrest (ACA), appropriate ventricular fibrillation-terminating ICD shocks, and SCD. We also investigated the association of various factors, such as gender, age, QTc duration, T wave alternans, Schwartz score, genetic subtypes, and deafness, with major cardiac events.
Genetic tests
To detect the affected individuals, the genetic studies for 3 major long QT genes (KCNQ1, KCNH2, and SCN5A) were performed sequentially. Genetic testing for other relatively infrequent mutations, such as KCNJ2 (LQT7) or CACNA1C (LQT8), was performed upon specific requests. Genetic tests were offered to all patients after informed consent was obtained from their parents or guardians, according to local ethic committee recommendations. Mutations in the LQTS loci that were classified as pathogenic or likely pathogenic were considered as genotype positive in our cohort. Cascade testing was offered to first-degree relatives of patients identified as genotype positive.
Statistical analysis
The Statistical Package for the Social Sciences for Windows (SPSS) program version 22 was used for descriptive statistics. The distribution of each continuous variable was tested for normality using the Kolmogorov-Smirnov test. Demographic data are reported as mean±standard deviation, median (range), number, and percentage. Furthermore, χ2 or Fisher’s exact test was used for comparison of categorical parameters, and Mann-Whitney U test (The variables were heterogenous and few in number) was used for comparison between continuous parameters. Logistic regression was used to assess confounding variables and Kaplan-Meier analysis to assess survival analysis, with significance based on the log-rank test. P<0.05 was considered statistically significant.
Results
General observations
All demographic and clinical characteristics of patients are listed in Table 1. A total of 145 patients (76 male) were identified. The mean age at diagnosis was 9.2±4.5 years (12 days to 18 years), and the mean weight was 35.72±18.45 kg (4.1–95 kg). The mean Schwartz score was 4.5 points (range, 3–7.5 points). Notably, 12 patients (8.3%) had congenital heart defects.
Table 1.
Clinical characteristics of the study subjects
| Patients | 145 |
| Male | 76 (52) |
| Age at diagnosis (year) mean (±SD) | 9.2±4.5 |
| Weight at diagnosis (kg) mean (±SD) | 35.7±18.5 |
| Congenital cardiac disease | n (%) |
| • Atrial septal defect | 5 (3.4) |
| • Ventricular septal defect | 2 (1.4) |
| • Patent ductus arteriosus | 1 (0.7) |
| • PAPVC | 1 (0.7) |
| • Mitral valve pathology(opere) | 1 (0.7) |
| • Pulmonary stenosis | 1 (0.7) |
| • Mitral valve prolapsus | 1 (0.7) |
| Presenting symptoms | |
| • Incidental ECG findings | 71 (48.9) |
| • During medical check-ups | 30 (20.7) |
| • Family cascade screening | 38 (26.2) |
| • Evaluation before participating in competitive sports | 3 (2.1) |
| • Aborted cardiac arrest | 15 (10.3) |
| • Syncope | 26 (17.9) |
| • Palpitation | 9 (6.2) |
| • Seizure | 2 (1.4) |
| • Bradycardia | 3 (2.1) |
| • AV block | 4 (2.8) |
| • Chest pain | 15 (10.3) |
| Total Schwartz score (points) mean±SD (range) | 4.5±2.1 (3–7.5) |
| Electrocardiographic characteristics | |
| • T-wave alternans | 28 (19.3) |
| • Bradycardia | 3 (2.1) |
| • 2:1 AV block | 3 (2.1) |
| • Complete AV block | 1 (0.7) |
| • HR (bpm) mean±SD (range) | 508±27.8 ms (450–730) |
| • QTc (ms) mean±SD (range) | 80.2±20.6 (46–125) |
| Holter characteristics | |
| • T-wave alternans | 31 (21.3) |
| • Monomorphic PVC | 4 (2.8) |
| • Polymorphic PVC | 1 (0.7) |
| • Bidirectional PVC | 1 (0.7) |
| • Monomorphic VT | 1 (0.7) |
| • Polymorphic VT | 2 (1.4) |
| Medical Treatments | |
| • Propranolol | 128 (88.3) |
| • Nadolol | 7 (4.8) |
| • Propranolol+mexiletine | 7 (4.8) |
| • Propraoalol+flecainide | 3 (2.1) |
| ICD | 34 (23.4) |
| Pacemaker | 2 (1.4) |
| Left or bilateral cardiac sympathetic denervation | 9 (6.2) |
Values are expressed mean±SD or n (%)
AV - atrioventricular; bpm- beat per minute; ECG - electrocardiography; HR - heart rate; ICD - implantable cardioverter-defibrillator; QTc - corrected QT interval; PAPVC - partial anomalous pulmonary venous connection; PVC - premature ventricular contraction; SD - standard deviation; VT - ventricular tachycardia
Mode of presentation
A significant proportion of patients with LQTS (n=71; 49%) did not have clinically important presentation at diagnosis; most of them had incidental ECG findings of prolonged QTc during cardiac evaluation because of nonspecific symptoms, such as chest pain or murmur (n=30; 20.7%), during the evaluation before participating in competitive sports (n=3; 2.1%), or by family cascade screening (n=38; 26.2%). However, 15 patients (10.3%) were diagnosed after a previous cardiac arrest and successful resuscitation. At the time of diagnosis, 26 patients (18%) had syncope complaints. Syncopal episodes were found during exercise in 15 cases (57.7%) and without stress or at rest in 11 cases (42.3%).
Moreover, 3 patients presented with sinus bradycardia, 2 of whom were detected prenatally. Three patients, including 2 infants, had 2:1 atrioventricular (AV) block, and 1 patient had complete AV block at diagnosis.
The initial complaints of the patients are presented in Table 1. A total of 22 cases (15.1%) were accompanied by congenital deafness and could be classified as having Jervell and Lange-Nielsen syndrome (JLNS).
Electrocardiographic and Holter characteristics
The mean QTc of the cohort was 508±27.8 ms (range, 450–730), and the mean heart rate was 80.2±20.6 bpm (46–125). In 28 cases (19.3%), T wave alternans was observed in the ECG, and in 31 (21.4%) cases, it was observed in Holter monitoring. Detailed ECG and Holter findings of patients are presented in Table 1.
Treatment and outcome
Treatment modalities of patients with LQTS in our cohort are presented in Table 1.
Beta-blockers
All patients were offered beta-blocker therapy. Propranolol was the initial choice as the beta-blocker. There were 7 patients who received propranolol and later switched to nadolol to enhance compliance. Mexiletine was added as an adjuvant therapy for 7 patients with LQT3 who were symptomatic. Flecainide was started in addition to dideral in 3 patients with Andersen-Tawil syndrome (LQT7) with polymorphic VT.
Implantable cardioverter-defibrillator
ICD was performed in 34 patients (23.4%). The mean age at ICD implantation was 9.08±4.65 years (0.83–17.2), and the mean weight was 36.27±18.57 kg (9–95). The mean QTc was 532.5±23.6 msec. Although 19 of these (55.9%) were female, no significant difference was found in ICD implantation between genders (p=0.268). ICD implantation was performed in 13 patients for secondary protection and in 21 patients (61.8%) for primary protection. Indications for ICD implantations were presentation with resuscitated cardiac arrest (n=13; 38.2%), syncope despite beta-blocker therapy (n=9; 26.5%), a family history of sudden death (n=4; 11.8%), and patients with syncope complaints who had not received beta-blocker therapy before and who were diagnosed as having JLNS with a QTc duration of > 550 msec (n=8; 23.5%). Notably, 18 of them (53%) were transvenous, and the remainder (n=16; 47%) were epicardial. A total of 13 patients (38.2%) experienced appropriate ICD shocks despite beta-blocker treatment. T wave oversense (TWOS)-related inappropriate shock was observed in 1 patient (2.9%). Apart from this, no major complications were observed.
Left or BCSD
Initially, left cardiac sympathetic denervation (LCSD) was performed in 1 patient, but this patient was admitted owing to recurrence events after 6 months. Therefore, right cardiac sympathetic denervation was also performed in this patient. After this clinical experience, BCSD was initially performed in 8 patients. Eight of them were performed because the patients continued to experience ICD shocks despite optimal care, and the rest was because of patients’ refusal to ICD implantation. The procedure was performed at a mean age of 9.5±4.6 years (4.3–14.5). None of the patients who underwent BCSD experienced a life-threatening cardiac event after the procedure. Alteration in sweating pattern was observed in 1 patient, and he returned to normal after 6 months. No more complications were observed.
Genetic testing
The details of the genotype information of the identified LQTS genes are presented in Figure 1. Genetic testing was performed in 143 patients (98.6%), and an abnormality was identified in 117 patients (82%). Notably, 15 patients had genetic test results that were negative for known mutations, and test results are pending in 11 patients. A KCNQ1 mutation was the most commonly identified defect (n=86; 59.3%). Of these, 21 were homozygous inheritance, whereas 65 were heterozygous inheritance; 11 patients (7.6%) had LQT2 (KCNH2), whereas 7 patients (4.8%) had LQT3 (SCN5A). Moreover, 22 patients with congenital deafness (JLNS) were tested for KCNQ1 (n=21, 95.4%) and KCNE1 (n=1; 4.6%) gene mutations. There were 2 patients with LQT8 resulting from a rare pathogenic mutation in CACNA1C. LQT8 is linked to Timothy syndrome with multiple extracardiac manifestations [syndactyly, developmental delay, and congenital heart defect (ventricular septal defect)].
Figure 1.
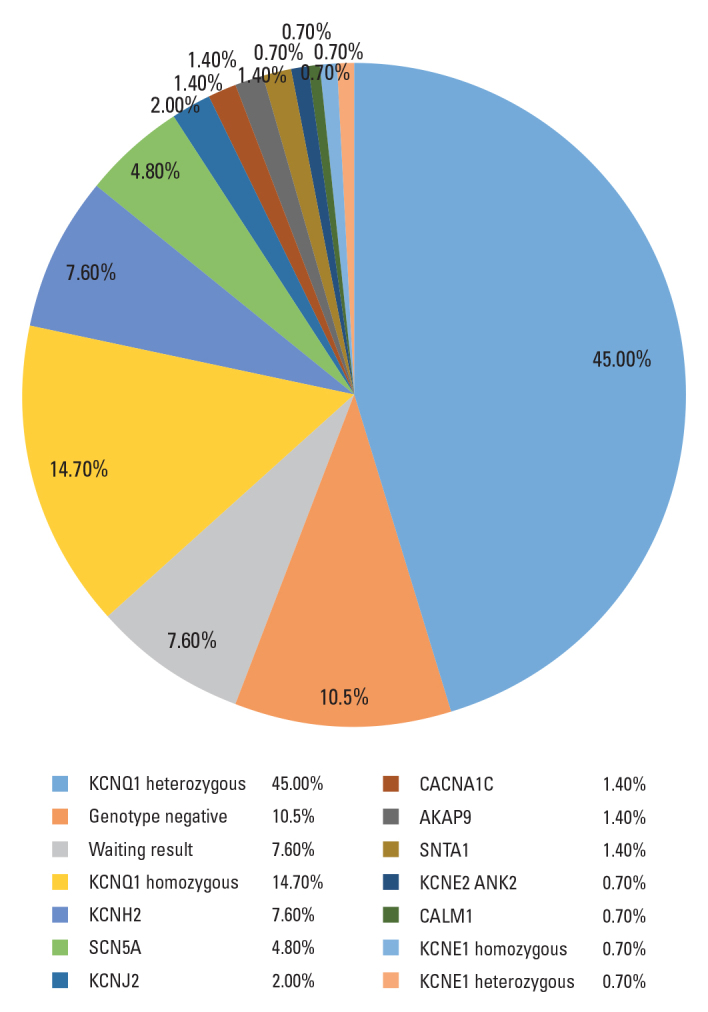
Genotypes of patients with long QT syndrome
Follow-up
The mean follow-up time was 35.6±25.8 months. During follow-up, 5 patients (3.4%) died. The mean age of exitus was 6.2±5.3 years; 3 patients were female, and 2 were male. Furthermore, 3 patients who were diagnosed after sudden cardiac arrest died shortly because of TDp. Patient with LQT1 who refused to use beta-blocker therapy died while swimming. The remaining one was followed up owing to fetal bradycardia. An epicardial pacemaker was implanted owing to sinus node dysfunction, pathological bradycardia, and long QTc interval in the postnatal follow-up of a 4.5-month-old. The patient died 29 months later. The Kaplan-Meier survival curve is depicted in Figure 2. Cardiac event-free survival was 96.0%, 94%, and 92% at 1 year, 5 years, and 10 years, respectively.
Figure 2.
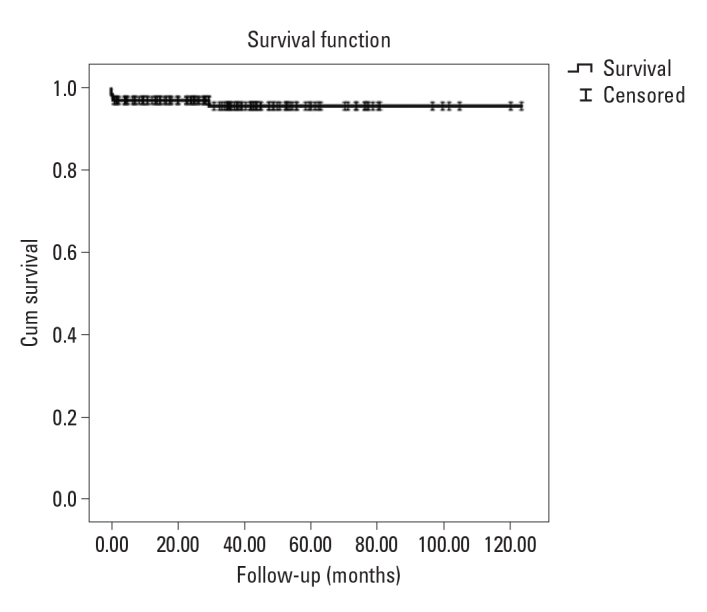
The Kaplan-Meier graph shows that the probability of survival is >90 in the average follow-up time
Risk factors affecting the development of major events
We investigated the association of various factors with major cardiac events defined as syncope, ACA, appropriate ventricular fibrillation-terminating ICD shocks, and SCD (Table 2). The QTc was significantly longer (>500 ms) in those who had experienced major cardiac events (p=0.001). The risk was 5.1 times higher in cases with QTc of >500 ms [odds ration (OR), 5.1; 95% confidence interval (CI), 2.3–11].
Table 2.
Risk factors affecting the development of major events
| Major cardiac event (−) | Major cardiac event (+) | P value | |
|---|---|---|---|
| Age (month) | 108.36±54.18 | 115.64±54.68 | 0.415 |
| Schwartz score | 4.01±0.74 | 5.1±1.2 | 0.001 |
| Gender | |||
| Female | 46 (67) | 23 (33) | 0.269 |
| Male | 54 (71) | 22 (29) | |
| Age (year) | |||
| <13 year | 77 (69) | 34 (31) | 0.778 |
| ≥13 year | 23 (68) | 11 (32) | |
| ≥13 year | |||
| Female | 14 (61) | 9 (39) | 0.271 |
| Male | 9 (82) | 2 (18) | |
| <13 year | |||
| Female | 32 (70) | 14 (30) | 0.535 |
| Male | 45 (69) | 20 (31) | |
| JNLS | |||
| (+) | 11 (50) | 11 (50) | 0.047 |
| (−) | 89 (72) | 34 (28) | |
| QTC duration (ms) | |||
| ≤500 | 65 (84) | 12 (16) | 0.001 |
| >500 | 35 (51) | 33 (49) | |
| Family history of LQTS | |||
| (+) | 70 (78 ) | 20 (22) | 0.005 |
| (−) | 30 (55) | 25 (45) | |
| Family history of SCD | |||
| (+) | 26 (68) | 12 (32) | 0.991 |
| (−) | 74 (69) | 33 (31) | |
| Holter T wave Alternans | |||
| (+) | 10 (30) | 23 (70) | 0.001 |
| (−) | 90 (80) | 22 (20) | |
Values are expressed mean±SD or n (%)
JNLS - Jervell and Lange-Nielsen syndrome; LQTS - long QT syndrome; SCD - sudden cardiac death
T wave alternans was significantly higher in patients who experienced major cardiac events (p=0.001). The presence of T wave alternans in the Holter ECG increased the probability of major cardiac events by 9 times (OR, 9; 95% CI, 3.9–21).
Notably, 22 patients with JLNS had deafness. Patients born with congenital deafness had >2.6-fold increase in the risk of major cardiac events during childhood (p=0.047; OR, 2.6; 95% CI, 1.1–6.5).
The mean Schwartz score was 5.1±1.2 in the group that had a major cardiac event, whereas it was 4.01±0.74 in the group that did not. We observed that the risk of encountering a major cardiac event was statistically higher in those with high Schwartz scores (p=0.001).
Globally, there is no statistical difference in gender and age of initial presentation. Dividing the subjects into 2 groups according to the age of initial symptom development by the age of 13 years, males predominated in the <13 category (male-to-female ratio, 20:14) and females predominated in the ≥13 category (male-to-female ratio, 2:9), without statistical significance (p=0.535, p=0.271, respectively). In terms of genetic distribution, exposure to major cardiac events was higher especially in type 1 and JLNS patients at the age of <13 years (p=0.04), whereas LQT2 was higher in patients at the age of >13 years (p=0.03). For general distribution, most patients who experienced major cardiac events had type 1 (27%). The risk of encountering a major cardiac event was significantly higher in patients who were followed up with a diagnosis of JLNS and Timothy syndrome (p=0.005).
Furthermore, a family history of sudden death in a first-degree relative was not shown to be a significant predictor of outcomes during childhood (hazard ratio, 0.82; 95% CI, 0.26–2.60; p=0.991).
Moreover, 90 patients had LQTS in a first-degree relative. We observed that an encounter with a major cardiac event was significantly lower in patients with LQTS in their family history (p=0.005). We attributed this to the fact that many asymptomatic patients were detected early, and early medical treatment was initiated, especially with family screening, owing to the increased awareness of LQTS.
Discussion
To the best of our knowledge, this is one of the largest pediatric studies that evaluated the clinical and genetic characteristics of LQTS and its outcomes in children in a single-center. This study was also the first and largest systematic study from a developing country such as Turkey.
The most noteworthy findings are as follows:
LQTS presents with a wide variety of symptoms and prognoses, ranging from no symptoms and normal life expectancy to the presence of SCD. It also presents with bradycardia and AV blocks in early infancy.
Screening of all first-degree relatives of patients with LQTS led to the identification of individuals with potentially unrecognizable disease.
Genetic analysis could be helpful in families with LQTS by providing presymptomatic diagnoses and genetic counseling.
A markedly prolonged QT interval (>500 msec), T wave alternans, high Schwartz score, and the JLNS genotype were associated with a high risk of major cardiac events.
LQTS diagnosis and presentation
LQTS is a clinical diagnosis and should be suspected in individuals on the basis of clinical presentation, family history, and ECG characteristics (6). LQTS has a variety of clinical manifestations. Patients can be detected incidentally without any complaints, or they can present with palpitations, convulsions, syncope, and even cardiac arrest. Sinus bradycardia and functional AV block are also well reported in perinatal LQTS (8, 9). Furthermore, a known family history of syncope or sudden death in a child or young adult supports the diagnosis of LQTS (6). In this study, a significant proportion of patients with LQTS (49%) did not have clinically important presentation at diagnosis; most of them had incidental ECG findings of prolonged QTc during cardiac evaluation or were identified through family cascade screening. However, 15 patients were diagnosed after ACA. There were 2 patients in our group who presented with fetal bradycardia; 3 patients, including 2 infants, had 2:1 AV block, and 1 patient had complete AV block at diagnosis. Pediatricians or gynecologists should suspect LQTS in young infants with slow heart rates (10).
LQTS genotype
Congenital LQTS results from mutations involving one of the many genes coding for ion channels that facilitate the transport of potassium, sodium, and calcium. Despite having at least 17 genetically distinct types of LQTS, at least 75% of the mutations are found in 3 genes: KCNQ1 (potassium channel), KCNH2 (potassium channel), and SCN5A (sodium channel), which cause LQT1, LQT2, and LQT3, respectively (11). LQT1, the most common subtype, affects 30% to 35% of LQTS individuals (12). Pathogenic LQTS genetic mutations were identified in 81.1% of the patients, and LQT1 was the most common type (44.8%) in our cohort. In a recent single-center study of LQTS in China and Hong Kong, LQT2 was the most common genotype (10, 13). Genetic studies could serve as a confirmative test, which will not exclude the diagnosis with negative test result in patients with probable LQTS, or it could provide additional information for the management of certain types of LQTS, such as LQT3 (14).
LQTS management
Beta-blockers are the primary treatment for LQTS and are clinically indicated in all individuals with QTc of >470 ms. They can also be useful in asymptomatic LQTS with QTc of <470 ms if there is concern for a significant family history of sudden death events or questionable history (7). It is important to know that LQT1 and LQT2 mutations are more responsive to beta-blockers than LQT3. Pharmacotherapy in LQT3 can be challenging. Beta-blockers remain the first-line drugs for treatment, and the use of a sodium channel blocker, such as mexiletine, might be considered for adjunctive therapy (7). In addition to beta-blockers, we provided mexiletine treatment in 7 of our patients diagnosed as having LQT3. Education of adult individuals and the parents of affected children, especially about beta-blocker compliance, is an important aspect of management (6).
ICDs and LCSD are therapies typically reserved for high-risk patients. High-risk patients have been defined as patients who have had cardiac events while on beta-blocker therapy, who have a history of cardiac arrest, or who have an inability to tolerate beta-blockers (i.e., severe asthma, level of evidence class I) (7). Prophylactic ICD therapy should be considered in very-high-risk patients, such as symptomatic patients with 2 or more gene mutations, including those with the JLNS variant with congenital deafness (15). In our study, ICD was performed in 34 patients (23.4%), 13 of whom had ICD implantation for secondary protection after ACA. ICD implantation comes with certain side effects. The most common ones include inappropriate shocks, lead problems, vascular occlusion, infection, psychological adjustment, and social discrimination (16, 17). TWOS-related inappropriate shock was observed in 1 patient (2.9%). Apart from this, no major complications were observed.
In the HRS guidelines, class I LCSD is recommended for patients with LQTS who are resistant to or unable to use beta-blockers, patients who refuse an ICD device, or patients who have symptoms despite ICD implantation (7). In this study, LCSD or BCSD was indicated in 8 patients who continued to experience ICD shocks despite optimal care and in 1 patient who refused to have ICD implantation. We performed LCSD in 1 patient and BCSD in 8 patients. Complications of LCSD were as follows: unilateral hand dryness, color or temperature variations between the sides of the face, and alterations in sweating patterns (18, 19). The advantage of BCSD is that it does not create asymmetric discomfort, which may be caused by LCSD (20). Alteration in sweating pattern was observed in 1 patient and returned to normal after 6 months. No more complications were observed during follow-up.
Risk factors affecting the development of major events
In a previous study, boys with LQTS experienced a significantly higher rate of fatal or near-fatal cardiac events than girls during childhood (21). Two recent studies from the International LQTS Registry that focused on adolescents (22) and adults (23) with LQTS reported that the gender-related risk reverses after childhood and that female patients maintained a higher risk than male patients throughout adolescence and during adulthood. The underlying mechanisms of this gender prevalence are incompletely defined but are believed to involve gonadal steroids (24). In accordance with the literature, in our study, encountering a major cardiac event was higher in males at the age of <13 years, whereas it was more common in females at the age of >13 years.
A baseline QTc interval of >500 ms has consistently been shown to be associated with a high risk of cardiac events (comprising syncope, ACA, or SCD) in patients with LQTS (21–23). In a study by Hobbs et al. on adolescents with LQTS, a baseline QTc duration of >530 ms was shown to be independently associated with a 2.3-fold increase in the risk of ACA or SCD compared with shorter QTc values (p=0.001) (22). Similarly, Sauer et al. (23) reported that in LQTS adults, baseline QTc durations between 500 and 549 ms were associated with a 3.3-fold increase in the risk of ACA or SCD compared with shorter QTc values and QTc >550 ms was associated with a 6.3-fold increase in risk. In our study, similar to the literature, we found that the risk of major cardiac events was 5.1 times higher in cases with a QTC duration of >500 ms (OR, 5.1; %95 CI, 2.3–11).
Furthermore, 15% of cases had JLNS, as a distinct subgroup of LQTS. This high proportion of children with JLNS among all children with LQTS can probably be explained by the fact that our institute has become a specialized center for inherited arrhythmias. As previous studies of a large number of patients with JLNS have demonstrated, these children should be considered as a high-risk group for LQTS-related symptoms (15, 25). According to the literature, we found that patients with JLNS had a 2.6-times higher risk of experiencing major cardiac events (p=0.047).
T wave alternans, a phenomenon of beat-to-beat variability in the repolarization phase of the ventricles, has been closely associated with an increased risk of ventricular tachyarrhythmic events and SCD (26). In our study, we found that patients with T wave alternans were 9 times more likely to experience major cardiac events.
Study limitations
There are some limitations in this study. The genetic tests of some patients have not been completed. The most important limitation to this study was its retrospective design. Because LQTS is defined by a set of heterogeneous clinical characteristics and genetic disorders, the number of participants with major cardiac events was not large enough to allow comparisons or relationships based on demographic features or assignment to genetic subgroups.
Conclusion
LQTS has a variety of clinical manifestations. Patient representations range from asymptomatic to SCD. By raising the awareness of physicians concerning the disease, SCD might be prevented in the early period. It is also important to screen all first-degree relatives of patients with LQTS to identify individuals with potentially unrecognizable disease. Early identification and risk assessment of LQTS could help predict the development of major cardiac events. A markedly prolonged QT interval (>500 msec), T wave alternans, high Schwartz score, and the JLNS genotype were associated with a high risk of major cardiac events.
HIGHLIGHTS .
LQTS presents a wide variety of symptoms and prognoses, ranging from no symptoms and normal life expectancy to the presence of SCD. It also presents bradycardia and AV blocks in early infancy.
Genetic analysis could be helpful in families with LQTS by providing presymptomatic diagnoses and genetic counselling.
A markedly prolonged QT interval (>500 msec), T wave alternans, high Schwartz score, and the JLN genotype were associated with a high risk of major cardiac events.
Acknowledgements
The authors would like to thank Professor Volkan Tuzcu and Mehmet Karacan for their great support in the genetic diagnostic process of LQTS patients.
Footnotes
Conflict of interest: None declared.
Peer-review: Externally peer-reviewed.
Authorship contributions: Concept – Y.E., G.T.Ş.; Design – Y.E., G.T.Ş.; Supervision – Y.E.; Fundings – None; Materials – None; Data collection and/or processing – G.T.Ş., H.C.K., E.Ö., S.Ö.; Analysis and/or interpretation – S.H., A.G.; Literature search – Y.E.; Writing – Y.E., G.T.Ş.; Critical review – Y.E., A.G.
References
- 1.Saprungruang A, Khongphatthanayothin A, Mauleekoonphairoj J, Wandee P, Kanjanauthai S, Bhuiyan ZA, et al. Genotype and clinical characteristics of congenital long QT syndrome in Thailand. Indian Pacing Electrophysiol J. 2018;18:165–71. doi: 10.1016/j.ipej.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizusawa Y, Horie M, Wilde AA. Genetic and clinical advances in congenital long QT syndrome. Circ J. 2014;78:2827–33. doi: 10.1253/circj.cj-14-0905. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5:868–77. doi: 10.1161/CIRCEP.111.962019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moss AJ. Long QT Syndrome. JAMA. 2003;289:2041–4. doi: 10.1001/jama.289.16.2041. [DOI] [PubMed] [Google Scholar]
- 6.Shah SR, Park K, Alweis R. Long QT Syndrome: A Comprehensive Review of the Literature and Current Evidence. Curr Probl Cardiol. 2019;44:92–106. doi: 10.1016/j.cpcardiol.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–63. doi: 10.1016/j.hrthm.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 8.Cuneo BF, Strasburger JF, Yu S, Horigome H, Hosono T, Kandori A, et al. In utero diagnosis of long QT syndrome by magnetocardiography. Circulation. 2013;128:2183–91. doi: 10.1161/CIRCULATIONAHA.113.004840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horigome H, Nagashima M, Sumitomo N, Yoshinaga M, Ushinohama H, Iwamoto M, et al. Clinical characteristics and genetic background of congenital long-QT syndrome diagnosed in fetal, neonatal, and infantile life: a nationwide questionnaire survey in Japan. Circ Arrhythm Electrophysiol. 2010;3:10–7. doi: 10.1161/CIRCEP.109.882159. [DOI] [PubMed] [Google Scholar]
- 10.Kwok SY, Liu AP, Chan CY, Lun KS, Fung JL, Mak CC, et al. Clinical and genetic profile of congenital long QT syndrome in Hong Kong: a 20-year experience in paediatrics. Hong Kong Med J. 2018;24:561–70. doi: 10.12809/hkmj187487. [DOI] [PubMed] [Google Scholar]
- 11.Tester DJ, Ackerman MJ. Genetics of long QT syndrome. Methodist Debakey Cardiovasc J. 2014;10:29–33. doi: 10.14797/mdcj-10-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallace E, Howard L, Liu M, O'Brien T, Ward D, Shen S, et al. Long QT Syndrome: Genetics and Future Perspective. Pediatr Cardiol. 2019;40:1419–30. doi: 10.1007/s00246-019-02151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Y, Liu W, Li C, Qiu X, Qin X, Guo B, et al. Common Genotypes of Long QT Syndrome in China and the Role of ECG Prediction. Cardiology. 2016;133:73–8. doi: 10.1159/000440608. [DOI] [PubMed] [Google Scholar]
- 14.Lee YS, Kwon BS, Kim GB, Oh SI, Bae EJ, Park SS, et al. Long QT syndrome: a Korean single center study. J Korean Med Sci. 2013;28:1454–60. doi: 10.3346/jkms.2013.28.10.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783–90. doi: 10.1161/CIRCULATIONAHA.105.592899. [DOI] [PubMed] [Google Scholar]
- 16.Gaba P, Bos JM, Cannon BC, Cha YM, Friedman PA, Asirvatham SJ, et al. Implantable cardioverter-defibrillator explantation for overdiagnosed or overtreated congenital long QT syndrome. Heart Rhythm. 2016;13:879–85. doi: 10.1016/j.hrthm.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 17.Olde Nordkamp LR, Postema PG, Knops RE, van Dijk N, Limpens J, Wilde AA, et al. Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: A systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm. 2016;13:443–54. doi: 10.1016/j.hrthm.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Ajijola OA, Lellouche N, Bourke T, Tung R, Ahn S, Mahajan A, et al. Bilateral cardiac sympathetic denervation for the management of electrical storm. J Am Coll Cardiol. 2012;59:91–2. doi: 10.1016/j.jacc.2011.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ajijola OA, Vaseghi M, Mahajan A, Shivkumar K. Bilateral cardiac sympathetic denervation: why, who and when? Expert Rev Cardiovasc Ther. 2012;10:947–9. doi: 10.1586/erc.12.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akkuş M, Seyrek Y, Kafalı HC, Ergül Y. Bilateral cardiac sympathetic denervation in children with long-QT syndrome and catecholaminergic polymorphic ventricular tachycardia. J Electrocardiol. 2020;61:32–6. doi: 10.1016/j.jelectrocard.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldenberg I, Moss AJ, Peterson DR, McNitt S, Zareba W, Andrews ML, et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation. 2008;117:2184–91. doi: 10.1161/CIRCULATIONAHA.107.701243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hobbs JB, Peterson DR, Moss AJ, McNitt S, Zareba W, Goldenberg I, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296:1249–54. doi: 10.1001/jama.296.10.1249. [DOI] [PubMed] [Google Scholar]
- 23.Sauer AJ, Moss AJ, McNitt S, Peterson DR, Zareba W, Robinson JL, et al. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329–37. doi: 10.1016/j.jacc.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 24.Li C, Hu D, Qin X, Li Y, Li P, Liu W, et al. Clinical features and management of congenital long QT syndrome: a report on 54 patients from a national registry. Heart Vessels. 2004;19:38–42. doi: 10.1007/s00380-003-0733-9. [DOI] [PubMed] [Google Scholar]
- 25.Wedekind H, Burde D, Zumhagen S, Debus V, Burkhardtsmaier G, Mönnig G, et al. QT interval prolongation and risk for cardiac events in genotyped LQTS-index children. Eur J Pediatr. 2009;168:1107–15. doi: 10.1007/s00431-008-0896-6. [DOI] [PubMed] [Google Scholar]
- 26.Gadage SN. T-wave alternans in long QT syndrome. Ann Pediatr Cardiol. 2018;11:219–21. doi: 10.4103/apc.APC_112_17. [DOI] [PMC free article] [PubMed] [Google Scholar]