

Abstract
Alpha-1-acid glycoprotein (AGP-1) is a positive acute phase glycoprotein with uncertain functions. Serum AGP-1 (sAGP-1) is primarily derived from hepatocytes and circulates as 12 to 20 different glycoforms. We isolated a glycoform secreted from PAF-stimulated human neutrophils (nAGP-1). Its peptide sequence was identical to hepatocyte-derived sAGP-1, but nAGP-1 differed from sAGP-1 in its chromatographic behaviour, electrophoretic mobility, and pattern of glycosylation. The function of these two glycoforms also differed. sAGP-1 activated neutrophil adhesion, migration and NETosis involving MPO, PAD4, and phosphorylation of ERK in a dose-dependent fashion, while nAGP-1 was ineffective as an agonist for these events. Furthermore, sAGP-1, but not nAGP-1, inhibited LPS-stimulated NETosis. Interestingly, nAGP-1 inhibited sAGP-1-stimulated neutrophil NETosis. The discordant effect of the differentially glycosylated AGP-1 glycoforms was also observed in platelets where neither of the AGP-1 glycoforms alone stimulated aggregation of washed human platelets, but sAGP-1, and not nAGP-1, inhibited aggregation induced by Platelet-activating Factor (PAF) or ADP, but not by thrombin. These functional effects of sAGP-1 correlated with intracellular cAMP accumulation and phosphorylation of the PKA substrate Vasodilator-stimulated phosphoprotein (VASP) and reduction of Akt, ERK, and p38 phosphorylation. Thus, the sAGP-1 glycoform limits platelet reactivity while nAGP-1 glycoform also limits pro-inflammatory actions of sAGP-1. These studies identify new functions for this acute phase glycoprotein and demonstrate that the glycosylation of AGP-1 controls its effects on two critical cells of acute inflammation.
Keywords: Acute phase proteins, Inflammation, Neutrophil activation, NETosis, Platelet aggregation
Graphical Abstract
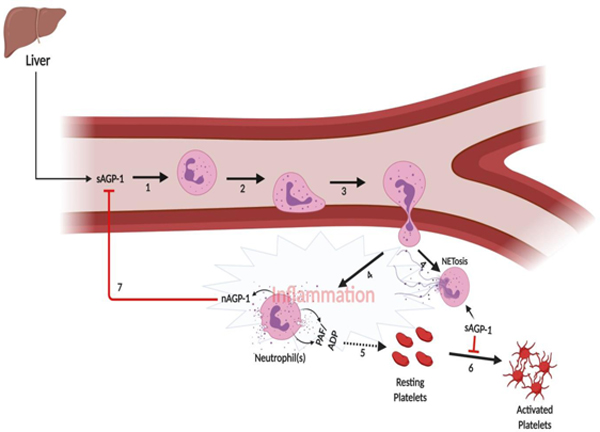
Proposed mechanism of action of sAGP-1 and nAGP-1 in an inflammatory milieu. During an inflammatory response, the level of circulating AGP-1 (sAGP-1) goes up in the serum (1). The sAGP-1 activates the first responder of the innate immune system, neutrophils, and induces adhesion and migration of the neutrophils to the site of inflammation (2 and 3). At the site of inflammation, neutrophils among other pro-inflammatory molecules like, PAF, ADP etc also secrete a different glycoform of AGP-1 (nAGP-1) (4). Parallely, some neutrophils under the influence of sAGP-1 may also undergo NETosis (4). The pro-inflammatory molecules secreted by neutrophils (PAF, ADP etc.) stimulate platelets and aggregate them (5). This event is inhibited by sAGP-1 by increasing the cAMP levels in platelets (6). Although nAGP-1 does not appear to have any effect on platelet function, the nAGP-1 secreted by neutrophils inhibits sAGP-1’s neutrophil activating function and thereby avert hyper-inflammatory reaction (7). The above illustration was created with BioRender.com
1 |. Introduction
Alpha-1-acid glycoprotein (AGP-1) is a positive acute phase glycoprotein with a carbohydrate content contributing to 45% of its total mass. Hepatic synthesis of AGP-1 increases during an inflammatory response [1, 2] and is released to the circulation (sAGP-1). Although numerous activities like immunomodulation and altering inflammatory milieu have been ascribed to AGP-1 [2, 3], its function(s) are ill defined. Other cell types including human breast epithelial cells, lymphocytes and monocytes secrete AGP-1 in response to appropriate inflammatory stimuli [3–5]. In addition, human neutrophils, secrete AGP-1 in response to platelet-activating factor (PAF), lipopolysaccharide (LPS), TNF and phorbol myristyl acetate (PMA) [6]. AGP-1 undergoes extensive glycosylation and approximately 12–20 different glycoforms of AGP-1 are present in human blood [2, 7]. This may be an underestimate, as more recent studies indicated that more than 150 isoforms of AGP-1 can occur in human plasma [8, 9].
This impressive number of glycoforms of AGP-1 is altered during both acute and chronic inflammation [5, 10–13]. There is ample evidence to support the concept that the glycosylation pattern and degree of chain branching may serve as one of the markers for specific disease conditions [3, 9, 10, 12–17]. Unraveling AGP-1 function(s), in general, remains challenging. For example, AGP-1 offers non-specific protection against various Gram-negative bacterial infection and TNF-induced lethality [18–20], but also promotes monocytes to an anti-inflammatory M2 phenotype rendering them ineffective against bacterial infections [21]. Furthermore, AGP-1 inhibits neutrophil migration in sepsis [22], contributing to infection. More recently, Higuchi et al. [23] have shown that AGP-1 is also involved in the allograft rejection after kidney transplantation.
Additionally, AGP-1 is shown to have anti-heparin effect leading to declined anti-coagulation of blood [24]. Elevated levels of AGP-1 is correlated with increased incidence of ischemic stroke and carotid plaques [25]. AGP-1 is shown to interact with plasminogen activator inhibitor type I and stabilizes its activity and is shown to induce platelet shape change and activation, with an impact on thrombosis [26, 27]. Contradicting to this, AGP-1 is shown to be a potent inhibitor of platelet aggregation, shown to have anti-thrombotic activity and offers protection against ischemia/reperfusion injury by preventing apoptosis and inflammation [20, 28–30].
Earlier studies have shown that activated TLR-4 triggers pro-thrombotic effects and that TLR-4 signaling is a potential therapeutic target [31–34]. In accordance to this, we previously observed that serum AGP-1 preferentially inhibits the TLR-4 agonist bacterial lipopolysaccharide (LPS), but not TLR-2 agonist Braun Lipoprotein (BLP) mediated inflammatory responses both in vivo and in vitro [35, 36]. The main aim of this study is to understand the effect of two different glycoforms of AGP-1 on two different cell types of innate immune system. Accordingly, we show that the hepatocyte-derived serum AGP-1 (sAGP-1) is pro-inflammatory, while the neutrophil derived glycoforms (nAGP-1) primarily display anti-inflammatory activity. As the protein sequences of two glycoforms are identical but not the glycan structure, we conclude that different glycoforms differently contributes to the inflammatory milieu and may partly be responsible for the contradictory roles of this acute-phase glycoprotein.
2 |. Materials and Methods
2.1 |. Chemicals
Cibacron F3GA agarose, DEAE-cellulose gel beads, anti-human AGP-1 antibody (A5566), anti-mouse-IgG-HRP antibody, commercial AGP-1, Foetal Bovine Serum (FBS), Lipopolysaccharide (LPS), recombinant IL-8, Micrococcal DNase, Adenosine diphosphate (ADP), Thrombin, Apyrase, Prostaglandin E1 (PGE1), Medium-199, Poly-L-Lysine, Hank’s-Balanced Salt Solution (HBSS) and RPMI media were procured from Sigma Chemicals Co., St. Louis, MO, USA. Phospho-specific antibodies to p-38, JNK, ERK, VASP; β-actin, and anti-rabbit IgG-HRP were obtained from Cell Signaling Technology, Danvers, MA, USA. NETosis marker antibodies i.e. citrulinated Histone H3, Histone H3, PAD4 and MPO antibodies were procured from Abcam, Cambridge, UK. Complete Mini EDTA-free protease inhibitor cocktail tablets were from Roche Diagnostics, Mannheim, Germany. PVDF membrane was from BioRad Laboratories, Hercules, CA, USA and Thioglycollate media was obtained from Sisco Research Laboratories, Mumbai, India. Trypsin was purchased from Promega, Madison, WI, USA. Sytox Green, Sytox Orange, Syto Green, Calcein 2 AM and molecular weight markers were procured from Invitrogen, Carlsbad, CA, USA. Collagen was from Chronolog, Havertown, PA, USA. Platelet-activating Factor was from Avanti polar Lipids, Alabaster, AL, USA. All the reagents used in the Glycomics Massspectrometry studies were procured from Sigma Aldrich Co., St. Louis, MO, USA except for PNGaseF which was from New England Biolabs, Ipswich, MA, USA.
2.2 |. Serum collection:
Blood was drawn from healthy volunteers with informed consent. Permission to draw blood was obtained from the Institutional Human Ethics Committee, University of Mysore, Mysuru (UOM No. 104 Ph.D/2015–16). Briefly, the blood was collected and coagulated blood was then centrifuged at 600 x g for 20 min at 25 °C to collect serum and stored at −20 °C till further use. All the experiments involving human blood were approved by the Institutional Human Ethics Committees of University of Mysore and University of Utah.
2.3 |. Isolation of polymorphonuclear leukocytes (Neutrophils):
Neutrophils were routinely isolated by dextran sedimentation and separated by centrifugation over Ficoll density gradient [37]. Neutrophil-rich pellets from this gradient was suspended in 1 ml of HBSS containing 0.2% human serum albumin (HBSS/A). Neutrophil isolation was also carried out by AutoMACS. Briefly, whole blood was labeled with CD15 microbeads and the neutrophils were positively selected using AutoMACS. (Miltenyi Biotech, USA)
2.4 |. Secretion of AGP-1 from isolated human neutrophils:
Ten million freshly isolated human neutrophils were stimulated or not with PAF (10−6 M), LPS (1 µg/ml), TNF (1000 U/ml) and PMA (5 µg/ml) for 60 min at 37 °C. In parallel, human neutrophils were stimulated with varied concentrations of PAF (10−4 to 10−10 M) for 60 min at 37 °C. Neutrophil supernatants derived under these conditions were concentrated and were immunoblotted against Anti-human AGP-1 monoclonal antibody with controls at both “0 min” and at “60 min”. sAGP-1 (250 ng) isolated previously in our laboratory [35] was used as the reference standard, as commercial AGP-1 many times displayed variation in electrophoretic mobility (Fig. 1B).
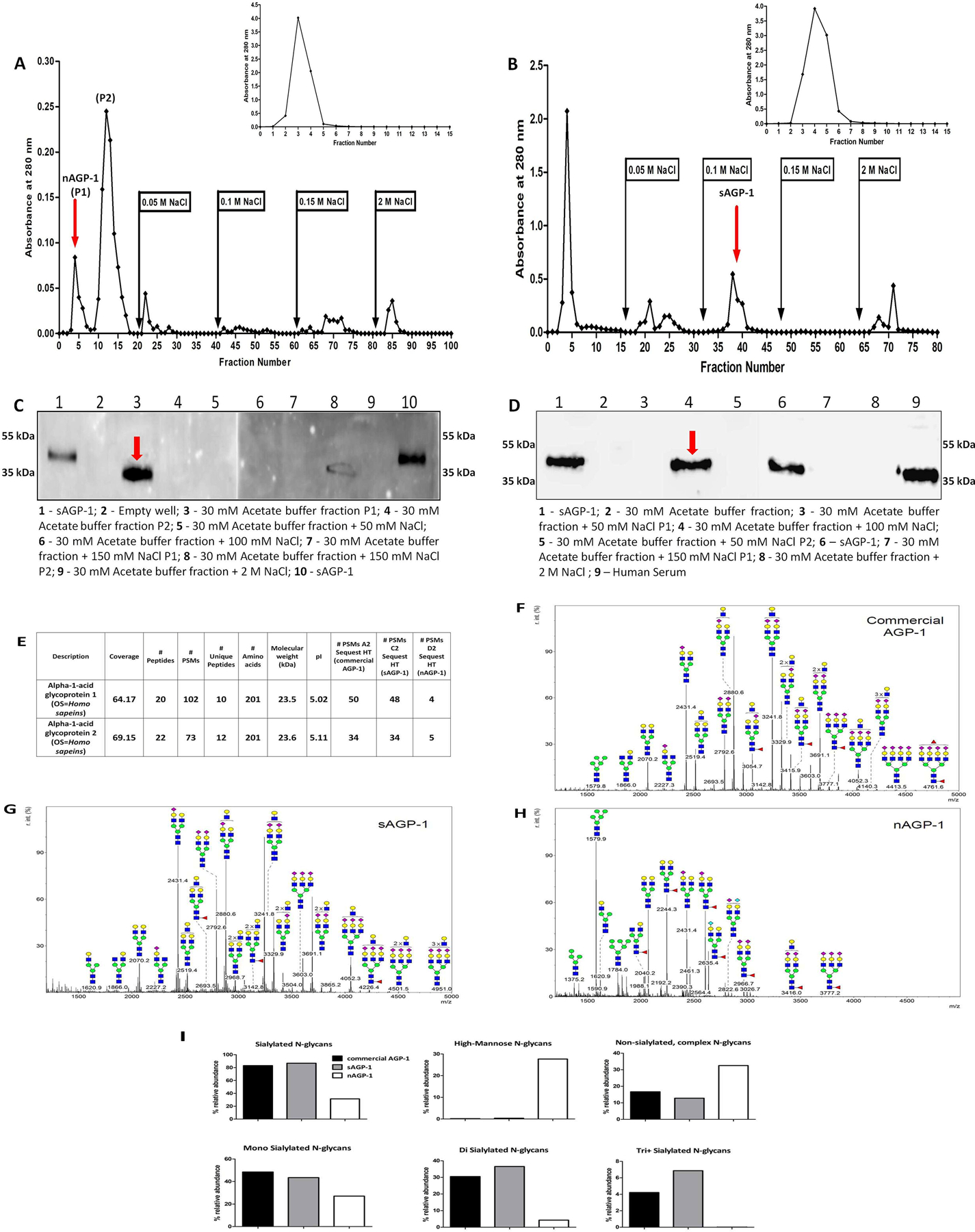
Fig. 1: Stimulated human neutrophils secrete AGP-1 that differs from sAGP-1:

(A) Freshly isolated human neutrophils secrete AGP-1 after stimulation (for 60 min). Secreted proteins were concentrated, resolved, and immunoblotted against AGP-1. Lanes represent; 1: sAGP-1 (250 ng); Lanes 2 – 7 represents nAGP-1 secreted from 2: 0 min Control neutropils; 3: 60 min Control neutrophils; 4: PAF (10−6 M) stimulated neutrophils; 5: TNF (1000U/ml) stimulated neutrophils; 6: LPS (1µg/ml) stimulated neutrophils; 7: PMA (5µg/ml) stimulated neutrophils. (B) Secretion of nAGP-1 by PAF stimulated neutrophils; Lane 1: commercial AGP-1 (250 ng); 2: 0 min Control; 3: 60 min Control; 4: empty well; 5: PAF (10−4 M); 6: PAF (10−6 M); 7: PAF (10−8 M); 8: PAF (10−10 M). nAGP-1 is indicated by red arrow.
2.5 |. Purification of AGP-1 from neutrophil supernatant
Purification of AGP-1 from PAF-stimulated neutrophil supernatants was carried out as described earlier [35]. Briefly, pooled neutrophil supernatants were loaded on to a Cibacron F3GA agarose column (10 x 1.5 cm) equilibrated with 10 mM phosphate buffer (pH 7.8). Unbound proteins containing AGP-1 were eluted at a flow rate of 1 ml/min. AGP-1 containing fractions were pooled and concentrated. The concentrate was then applied to a DEAE-cellulose column (25 x 0.5 cm) equilibrated with 30 mM acetate buffer (pH 5.0). Fractions were eluted with a sodium chloride gradient (0 to 2 M) at a flow rate of 24 ml/ hour. AGP-1 containing fractions were pooled, concentrated, and quantified for protein using Lowry’s method [38]. The endotoxin content of purified AGP-1 was assessed by Limulus amebocyte lysate (LAL) assay (Endochrome – K™, Charles River, USA) as per manufacturer’s instructions.
2.6 |. Western blotting for AGP-1
AGP-1 samples were resolved on a reducing SDS-PAGE (7.5 % acrylamide) and visualized by Western blotting. Blots were stained using appropriate primary antibody (anti-human AGP-1 monoclonal antibody; 1:10000 v/v) and secondary antibody (anti-mouse IgG HRP conjugate; 1:5000 v/v). The blots were visualized using freshly prepared ECL reagent by UV-transillumination (Uvi-Tech, Cambridge, UK).
2.7 |. Protein mass spectrometry analysis
Purified samples were resolved by SDS-PAGE, visualized using Coomassie stain; AGP-1 band was excised and destained with 30% methanol for 4 hours. Upon reduction (10 mM dithiothreitol) and alkylation (65 mM 2-chloroacetamide) of the cysteines, protein was digested overnight with sequencing grade modified trypsin. The resulting peptides were resolved on a nano-capillary reverse phase column (Acclaim PepMap C18, 2 micron, 50 cm, Thermo Scientific) using a 1% acetic acid/acetonitrile gradient at 300 nl/min and directly introduced into Q Exactive HF mass spectrometer (Thermo Scientific, USA). MS1 scans were acquired at 60 K resolution (AGC target=3e6, max IT=50ms). Data-dependent high-energy C-trap dissociation MS/MS spectra were acquired for the 20 most abundant ions (Top20) following each MS1 scan (15 K resolution; AGC target=1e5; relative CE ~28%). Proteome Discoverer (V 2.1; Thermo Scientific) software suite was used to identify the peptides by searching the HCD data against an appropriate database. Search parameters included MS1 mass tolerance of 10 ppm and fragment tolerance of 0.1 Da. False discovery rate (FDR) was determined using Fixed PSM validator and proteins/peptides with a FDR of ≤1 % were retained for further analysis.
2.8 |. Glycomics mass spectrometry analysis
Coomassie-stained AGP-1 protein bands were excised, washed with 400 µl of 50 mM AMBIC (ammonium bicarbonate) in 50% acetonitrile, dried and 200 µl of 10 mM DTT (1,4-Dithiothreitol) solution in 50mM AMBIC were added and incubated at 50º C for 30 mins. The gel was washed with 200 µl of acetonitrile and dried. The samples were incubated with 200 µl of 55 mM IAA (Iodoacetamide) in 50mM AMBIC for 30 mins at room temperature in dark. The samples were again washed with 500 µl of 50 mM AMBIC for 15 mins at room temperature followed by a wash with 200 µl of acetonitrile for 5 mins. The samples were dried prior to adding 500 µl of 50 mM AMBIC containing 10 µg of TPCK-treated trypsin and incubated overnight at 37º C. The tryptic digestion was terminated and supernatants were recovered. Further peptides were recovered by two cycles of washes with 200 µl of 50 mM AMBIC, 200 µl of 50% acetonitrile in 50 mM AMBIC and 200 µl of acetonitrile. All supernatants and washes were pooled in the same glass tube used and lyophilized. The dried peptides were resuspended in 200 µl of 50 mM AMBIC and incubated overnight with 1 µl of PNGaseF at 37º C. The enzymatic reaction was stopped by using 5% of acetic acid prior to purification of the released N-glycans over a C18 Sep-Pak (50 mg) column (Waters, USA) conditioned with 1 column volume (CV) of methanol, 1 CV of 5% of acetic acid, 1 CV of 1-propanol, and 1 CV of 5% of acetic acid. The C18 column was washed with 3 ml of 5% of acetic acid. Flow through and wash fractions were collected, pooled and lyophilized prior to permethylation. Lyophilized N-glycan samples were incubated with 1 ml of DMSO (Dimethyl Sulfoxide)-NaOH slurry solution and 500 µl of methyl iodide for 30 mins under shaking at 25 ºC. The reaction was stopped with 1 ml of MilliQ water and 1 ml of chloroform was added to purify out the permethylated N-glycans. The chloroform layer was washed 3 times with 3 ml of Milli-Q water and dried. The dried materials were re-dissolved in 200 µl of 50% methanol prior to be loaded into a conditioned (1 CV methanol, 1 CV MilliQ water, 1 CV acetonitrile and 1 CV Milli-Q Water) C18 Sep-Pak (50 mg) column. The C18 column was washed with 15% acetonitrile and then eluted with 3 ml of 50% acetonitrile. The eluted fraction was lyophilized and then redissolved in 10 µl of 75% methanol from which 1 µl was mixed with 1 µl DHB (2,5-dihydroxybenzoic acid) (5mg/ml in 50% acetonitrile with 0.1% trifluoroacetic acid) and spotted on a MALDI polished steel target plate (Bruker Daltonics, Germany). MS data was acquired on a Bruker UltraFlex II MALDI-TOF Mass Spectrometer instrument. Reflective positive mode was used and data recorded between 1000 m/z and 5000 m/z. For each MS N-glycan profiles the aggregation of 20,000 laser shots or more were considered for data extraction. Only MS signals matching an N-glycan composition were considered for further analysis. Subsequent MS post-data acquisition analysis were made using mMass [39].
2.9 |. Human neutrophil adhesion
For assessment of adhesion, the human neutrophil suspension (1x107 cells/ml) was loaded with Calcein-AM to a final concentration of 1 μM prior to incubation for 45 min at 37 °C. The labeled neutrophils (1x106 cells/well) were incubated with sAGP-1 or nAGP-1 (25, 50 and 100 µg/ml) in triplicate wells in cell culture plates (Nest Biotechnology Co. Ltd., China) pre-coated with 0.2% gelatin. Unbound neutrophils were removed by washing twice with HBSS/A and the adherent neutrophils were visualized and photographed at a magnification of 10x by fluorescent microscopy (Motic BA410 fluorescence microscope, Hong Kong; with Nikon DS-Qi2 camera, Japan) [36]. The number of cells adhered in each well was determined by counting the cells adhered in 10 randomly chosen fields using ImageJ software (ver.1.51j8) and then calculating the average number of cells adhered per field.
2.10 |. Neutrophil migration
Neutrophil migration was assessed using Transwell plates (Costar, Corning Inc., USA) with 5µm pore size inserts. 1x106 (200µl) freshly isolated neutrophils in M-199 medium were incubated in the upper chamber of the transwell and the chemoattractant, IL-8 (10 ng/ml) and/ or AGP-1 (sAGP-1 and nAGP-1) (25, 50 and 100 µg/ml) were introduced in the lower chamber. The number of neutrophils migrating to the lower chamber after incubation for 1 hour at 37 °C with 5% CO2 were counted with a hemocytometer and expressed as % neutrophil-migrated using IL-8 as positive control (100% migration).
2.11 |. Neutrophil degranulation and estimation of ROS
Neutrophil degranulation was monitored by the secretion/ production of myeloperoxidase (MPO) by stimulated neutrophils. 1x106 freshly isolated neutrophils suspended in HBSS/A were treated with varying concentrations of sAGP-1 (25, 50, and 100 µg/ml) or nAGP-1 (25, 50, and 100 µg/ml) for 1 hour at 37 °C with 5% CO2. LPS (10 µg/ml) was used as positive control. MPO activity was measured according to the method of Bradley et. al. [40]. Briefly, after the treatment, neutrophils were pelleted by centrifugation at 300 x g for 5 min at 4 °C. The supernatant were separated from the pellet. The neutrophils pellet were lysed using RIPA buffer containing protease inhibitor cocktail. Both the supernatant and the cell lysate were used as the source for MPO. Aliquots of the supernatant (250 μl) and cell lysate (50 μl)were mixed with 2.9 ml of 50 mM sodium phosphate buffer (pH 6.0) containing 167 μg/ml of o-dianisidine (SRL, Mumbai, India) and 0.0005% H2O2. The change in absorbance at 460 nm was recorded in a UV-Visible spectrophotometer (Biomate 3S, Thermo Scientific, USA).
For ROS estimation, freshly isolated neutrophils (1x106) were treated with different concentrations (25, 50, and 100 µg/ml) of sAGP-1 or nAGP-1 for 1 hour at 37 °C with 5% CO2. Again LPS (10 µg/ml) was taken as positive control. After treatment, the cells were washed and loaded with 20µM DCF-DA and incubated in dark for 15 min. The fluorescence was measured at an excitation at 488 nm and emission at 525 nm in a multi-mode plate reader (Thermo Scientific, USA) [41].
2.12 |. Assessment and quantification of Neutrophil Extracellular Traps (NETosis)
1 million freshly isolated human neutrophils were treated with LPS (10 µg/ml) in the presence / absence of AGP-1 (sAGP-1 and nAGP-1 glycoforms at 25, 50 and 100 µg/ml) before being transferred to Poly-L-Lysine coated coverslips. The neutrophils were incubated at 37 °C in 5% CO2 for 1 hour before adding Syto Green (cell-permeable) and Sytox orange (cell-impermeable) fluorescent dye mixture and visualized by fluorescent microscopy (EVOS Fluorescence microscope, Thermo Scientific, USA). The two dye images were merged using ImageJ software (ver. 1.51j8).
High-throughput NET quantification [42] was employed to quantify neutrophil NETs. For this, 24 well plates were pre-coated with poly-L-lysine before neutrophil addition followed by stimulation for 1 hour by indicated agonists at 37 °C under 5% CO2. The extracellular traps were recovered by treating the neutrophils with Micrococcal DNase and the DNA stained with cell-impermeable Sytox Green dye. These were measured with a fluorescent plate-reader with excitation at 485 nm and emission at 530 nm. Parallely, NETosis was also quantified by using NETosis assay kit (ab 235979, Abcam, Cambridge, UK). Activity of neutrophil elastase on harvested NET structures was quantified in this method according to the manufacturer’s instructions.
2.13 |. Profiling of protein markers of NETosis by western blotting
Freshly isolated human neutrophils (1x106 cells/ml) were stimulated for 1 hour with sAGP-1 (25, 50, and 100 µg/ml) and highest dose for nAGP-1 (100 µg/ml) with or without LPS (10 µg/ml). In some experiments, neutrophils were treated with a combination of sAGP-1 (50 µg/ml) and nAGP-1 (50 µg/ml). Unstimulated neutrophils served as the negative control. Lysates were prepared by using RIPA buffer containing protease inhibitor cocktail. Immunoblots were developed using specific primary and appropriate secondary antibodies for citrulinated histone H3 (R2, R8, and R17) (cit His H3), Histone H3, peptidylarginine deaminase (PAD) 4, MPO, phospho-pERK,and β-actin (1:1000 v/v).
2.14 |. Platelet aggregation using washed human platelets
Blood was drawn from healthy volunteers with informed consent. Platelet-rich plasma (PRP) was isolated from this blood using the method previously described by Zhou et al [43]. Briefly, blood was drawn into citrated tubes (1/9) and centrifuged for 15 minutes at 45 x g at 25 °C to obtain PRP. The PRP was treated with PGE1 to stop the activation of platelets. PGE1 treated PRP was centrifuged at 225 x g for 20 min at 25 °C without breaking. The resulting platelet pellet was resuspended in HEPES-Tyrode’s buffer containing 0.02 U Apyrase. Platelet aggregation was performed by using 2x108 platelets/mL in a final reaction volume of 500 μL at 37 °C with stirring at 1,200 rpm. All assays were performed using Chrono-log aggregometer and the traces were recorded using AGGRO/LINK 8 ver.1.0.1 software.
2.15 |. Signaling by stimulated platelets
Washed platelets (2x108 cells/ml) were stimulated for 5 min with thrombin (0.075 U/assay), PAF (4 µM), or ADP (500 nM) with or without sAGP-1 (50 µg/ml). Unstimulated washed platelets served as the negative control. Lysates were prepared by using perchloric acid (6 N) and reducing sample buffer (modified Laemmli buffer). Immunoblots were developed using specific primary and appropriate secondary antibodies for phospho-p38, phospho-Akt, phospho-ERK, phospho-VASP (S157) and β-actin (1:1000 v/v).
2.16 |. ELISA for platelet cAMP
cAMP levels in washed platelets were quantified after treatment with buffer, Thrombin (0.075U/assay), ADP (500 nM), or PAF (4 µM) with or without sAGP-1 using competitive ELISA kit (EMSCAMPL, Invitrogen, Austria) according to the manufacturer’s instructions.
2.17 |. Statistical analysis
All experiments are a representative of at least two or more experiments. The data are represented as mean ± SEM. Analysis between two groups were performed using Student t – Test. For analysis of more than two groups one way - analysis of variance (ANOVA) was used. All the statistical analysis was carried out using GraphPad Prism 5 software.
3 |. Results
3.1 |. Neutrophils secrete distinct AGP-1 isoforms
To isolate the extra-hepatic secretion of AGP-1, freshly isolated human neutrophils (1 x 106 cells/ml) (isolated by using both ficoll density gradient method and CD15 positive selection) were stimulated with the agonists PAF (10−6 M), TNF (1000 U/ml), PMA (5 µg/ml) or LPS (1 µg/ml) and the secreted proteins were immunoblotted against AGP-1. Unstimulated neutrophils did not secrete detectable AGP-1, while neutrophils stimulated by these agonists did, suggesting the extra-hepatic source of AGP-1 (Fig. 1A). Stimulated neutrophils, however, secreted both a 43 kDa AGP-1 corresponding to sAGP-1 and also released more slowly migrating (~60 kDa) larger form of AGP-1 which we termed nAGP-1 (Fig. 1B). The ratio of the two isoforms differed according to the inciting agonists. We sought to purify the higher molecular weight nAGP-1 isoform using conventional chromatographic techniques (Methods) and so stimulated neutrophils with PAF to obtain an AGP-1 corresponding to both sAGP-1 and nAGP-1. However, only the larger nAGP-1 was isolated by this purification protocol and was used for further experimentations. Based on immuno-detection, we found that neutrophils stimulated with lower concentrations of PAF secreted higher amount of higher molecular weight (~60 kDa) nAGP-1 than higher concentrations of PAF (Fig. 1B).
In the next series of experiments, we determined the basis for the distinct physical properties of the two AGP-1 isoforms. Unlike sAGP-1, nAGP-1 failed to bind to the DEAE-cellulose anion exchange column and so eluted in the void volume of this chromatographic separation (Figs. 2A–D), while sAGP-1 eluted at a salt concentration of 150mM. The endotoxin content of both the purified nAGP-1 and sAGP-1 was < 5 EU/mg protein, and so would not be relevant in subsequent biological analyses. The slow moving nAGP-1 (~60 kDa) before purification (Fig. 1B), now moved faster than that of the 43 kDa sAGP-1 after purification (Fig. 2C) on SDS-PAGE, probably due to the dissociation of the “uncharacterized binding factor(s)”. (See discussion)
Fig. 2: Purification and characterization of nAGP-1 in comparison with sAGP-1:
Elution profile of (A) PAF-induced Neutrophil supernatant (nAGP-1) and (B) serum derived AGP-1 (sAGP-1) on DEAE-Cellulose column. The insets represent the elution profile of nAGP-1 (A inset) and sAGP-1 (B inset) on Cibacron blue column respectively. The fraction(s) containing the respective AGP-1 is indicated by red arrow. (C) and (D) Western blotting analysis of different fractions of DEAE-cellulose column against nAGP-1 and sAGP-1 respectively (two different gels) indicated by red arrow. (E) Mass spectral analysis of commercial AGP-1, sAGP-1 and nAGP-1. The data revealed that the protein components of the two AGP-1s are identical. Glycomic analysis of (F) commercial AGP-1 and (G) sAGP-1 showed that these two preparations are similar. (H) nAGP-1 differed in glycosylation complexity and composition when compared to sAGP1.  - N-Acetyl Glucoseamine;
- N-Acetyl Glucoseamine;  - Mannose;
- Mannose;  - Galactose;
- Galactose;  - Fucose;
- Fucose;  - Sialic acid. (I) % relative abundance of sialylated, non-sialylated, high-mannose, mono- di- and tri+ - sialylated N-glycans present in commercial AGP-1, sAGP-1 and nAGP-1.
- Sialic acid. (I) % relative abundance of sialylated, non-sialylated, high-mannose, mono- di- and tri+ - sialylated N-glycans present in commercial AGP-1, sAGP-1 and nAGP-1.
3.2 |. Glycan and peptide analysis of nAGP-1 by mass spectrometry
The electrophoretic mobility of purified nAGP-1 was slower than that of sAGP-1 before purification. To understand whether this reflects size or charge differences, we subjected the two isoforms to mass spectrometry analysis. We found the sequence of the two proteins aligned and generated peptides that were identical for the entire sequence (Fig. 2E). We next examined the glycosylation, more precisely the N-linked glycans (N-glycans) of sAGP-1 and nAGP-1. sAGP-1 is mainly glycosylated with complex bi- and tri-antennary, sialylated N-glycan chains. 86% of sAGP-1 N-glycans are sialylated, with the majority of them being mono- and di-sialylated N-glycans (Figs. 2G, I). This is similar, if not identical, to commercially available, serum-derived AGP-1 (Figs. 2F, I). By contrast, nAGP-1 has different types of N-glycan. The relative abundance of complex sialylated N-glycans in nAGP-1 was significantly reduced (~31%), and these are mainly mono-sialylated N-glycans (Figs. 2H, I). High-mannose-type N-glycans are, however, much more relatively abundant among nAGP-1 N-glycans (~27%) compared to sAGP-1 and commercial AGP-1 N-glycans (<1% for both) (Figs. 2F–I). Since nAGP-1 differs from the sAGP-1 only with respect to N-glycans and not the protein core, we considered these as distinct glycoforms of AGP-1.
3.3 |. AGP-1 glycoforms differently stimulate neutrophils
AGP-1 is elaborated in response to systemic inflammation, but with incompletely defined biological effects. To determine whether either or both of the AGP-1 glycoforms contribute to the inflammatory responses, we determined whether their undefined functions include the ability to localize neutrophils to sites of inflammation. Neutrophils rapidly and avidly adhere in response to inflammatory stimuli [44, 45]. To quantify this, we labelled neutrophils with Calcein, a fluorescent vital dye and assessed adhesion to a gelatin-coated glass surface that prevents interaction with unstimulated neutrophils. We found both sAGP-1 and nAGP-1 were agonists for this event, with sAGP-1 ultimately stimulating twice the number of neutrophils to adhere than nAGP-1 (Figs. 3 A, B). At lower concentrations, sAGP-1 also proved to be a significantly more potent agonist than nAGP-1. We next examined neutrophil chemotaxis using Transwell chambers to find migration in response to AGP-1 recapitulated adhesion, with sAGP-1 being significantly more potent and more stimulatory than nAGP-1 (Fig. 3C). Thus while both glycoforms of AGP-1 are neutrophil agonists, quantitative examination showed that these glycoforms differently affect neutrophil function, with sAGP-1 acting as a more robust neutrophil chemo-attractant than nAGP-1. Following this trend, sAGP-1, but not nAGP-1, induced ROS production (Fig. 3D) and MPO secretion (Fig. 3 E, F) in a concentration – dependent manner.
Fig. 3: sAGP-1 is more potent activator of human neutrophils than nAGP-1:
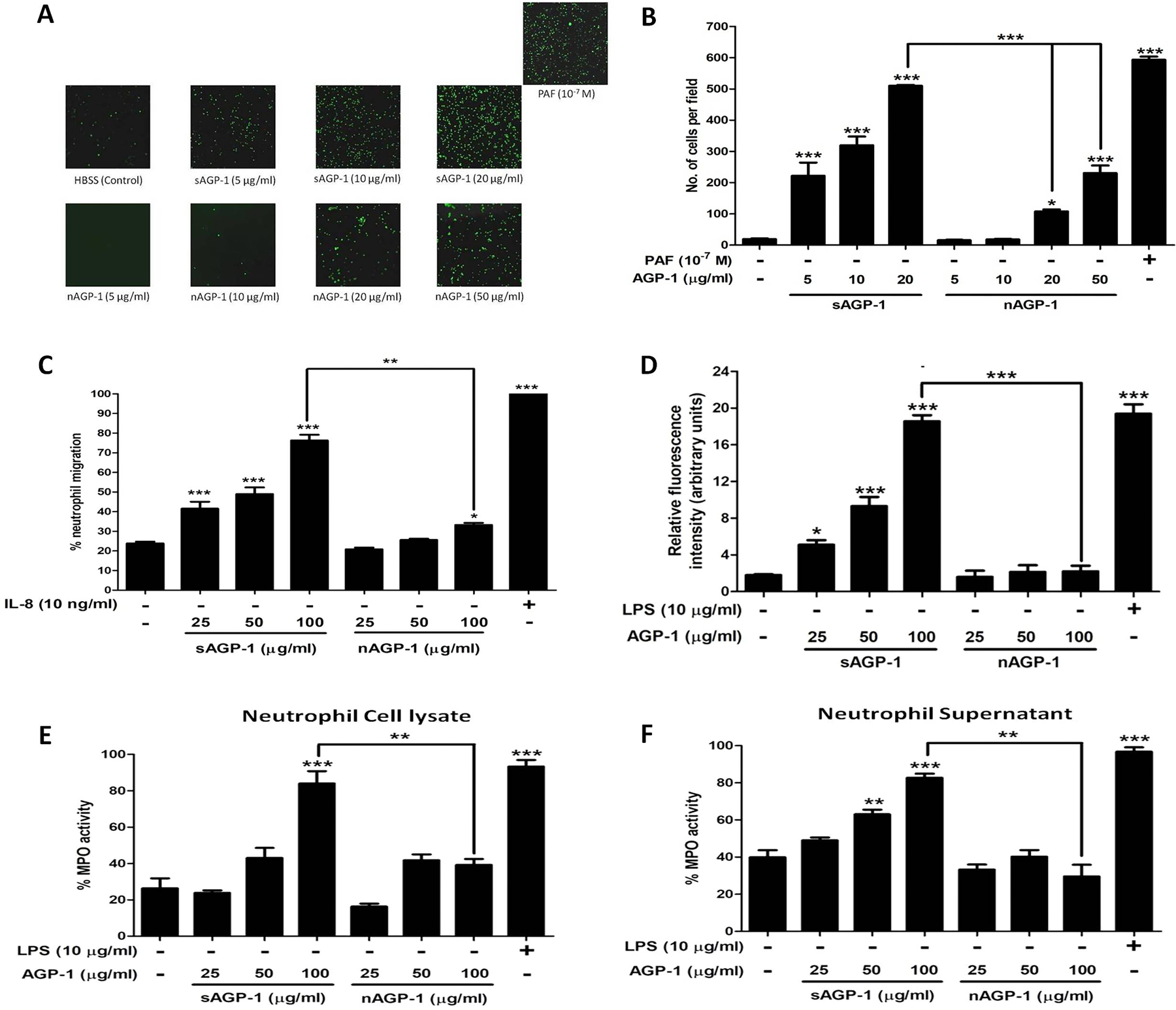
(A) Neutrophil adhesion. Neutrophils loaded with Calcein-AM in HBSS/A were treated with increasing concentrations of sAGP-1 and nAGP-1(5–50 µg/ml) separately. The adherent PMNs were visualized under fluorescent microscope at a magnification of 10X. PAF (10−7 M) was used as positive control. (B) Quantitation of neutrophils adhesion. AGP-1 induced PMN activation was quantified by counting the cells per field using ImageJ software as explained under ‘Methods‘. (C) Neutrophil migration. Chemotaxis in response to sAGP-1 and nAGP-1 (25, 50 and 100 µg/ml) was tested using Transwell plates with 5µm pore size inserts (Boyden chamber assay). IL-8 (10 ng/ml) served as the positive control. Percentage of neutrophils migrated to the lower chamber were calculated and plotted. Chemotaxis induced in response to IL-8 was considered 100%. (D) ROS production. Freshly isolated neutrophils were treated with LPS (10 µg/ml), different concentrations (25, 50, and 100 µg/ml) of sAGP-1 or nAGP-1 for 1 hour at 37 °C with 5% CO2. After treatment, the cells were washed; loaded with DCF-DA and the fluorescence was measured using fluorescence multi-mode plate reader (E, F) MPO secretion. Neutrophils were stimulated with various concentrations (25, 50, and 100 µg/ml) of sAGP-1 or nAGP-1. Both neutrophils cell lysate (E) and supernatant (F) were used to assess the MPO activity as described in the “Methods‘. LPS (10 µg/ml) served as positive control, while unstumilated neutrophils served as negative control. The data shown are mean ± SEM. ***=P <0.0001, **=P<0.001 and *=P<0.01 as determined one – way ANOVA
3.4 |. sAGP-1 induces neutrophil extracellular traps (NETosis) while nAGP-1 is inhibitory
Neutrophils extrude their DNA along with antimicrobial enzymes and peptides after encountering microbes or during inflammation that forms a net-like structure to entrap bacteria in a process termed NETosis [46]. We determined whether AGP-1s activate neutrophils to undergo NETosis to find sAGP-1, but not nAGP-1, induced this response (Fig. 4A). In fact, the highest concentration of sAGP-1 was comparable to the level of NETosis induced by the positive control LPS (Fig. 4B). The concentration-response relationship showed 50 µg/ml sAGP-1 was suboptimal, enabling us to determine the combined effect of sAGP-1 and nAGP-1 at this concentration. We found nAGP-1 at this concentration not only failed to induce NETosis, it inhibited the NETosis induced by sAGP-1 (Figs. 4 A, B inset). Interestingly, sAGP-1 and not nAGP-1 suppressed LPS-induced NETosis in a concentration-dependent fashion (Figs. 4 C, D), which is in accordance with our previous study [35]. nAGP-1, in contrast, failed to reduce LPS-induced NETosis even at 100 µg/ml, where sAGP-1 abolished the NETosis induced by LPS (Figs. 4 C, D). We also validated NETosis by measuring the neutrophil elastase bound to the extracellular DNA in a parallel experiment (Fig. 5A).
Fig. 4: sAGP-1, but not nAGP-1, induces NETosis:
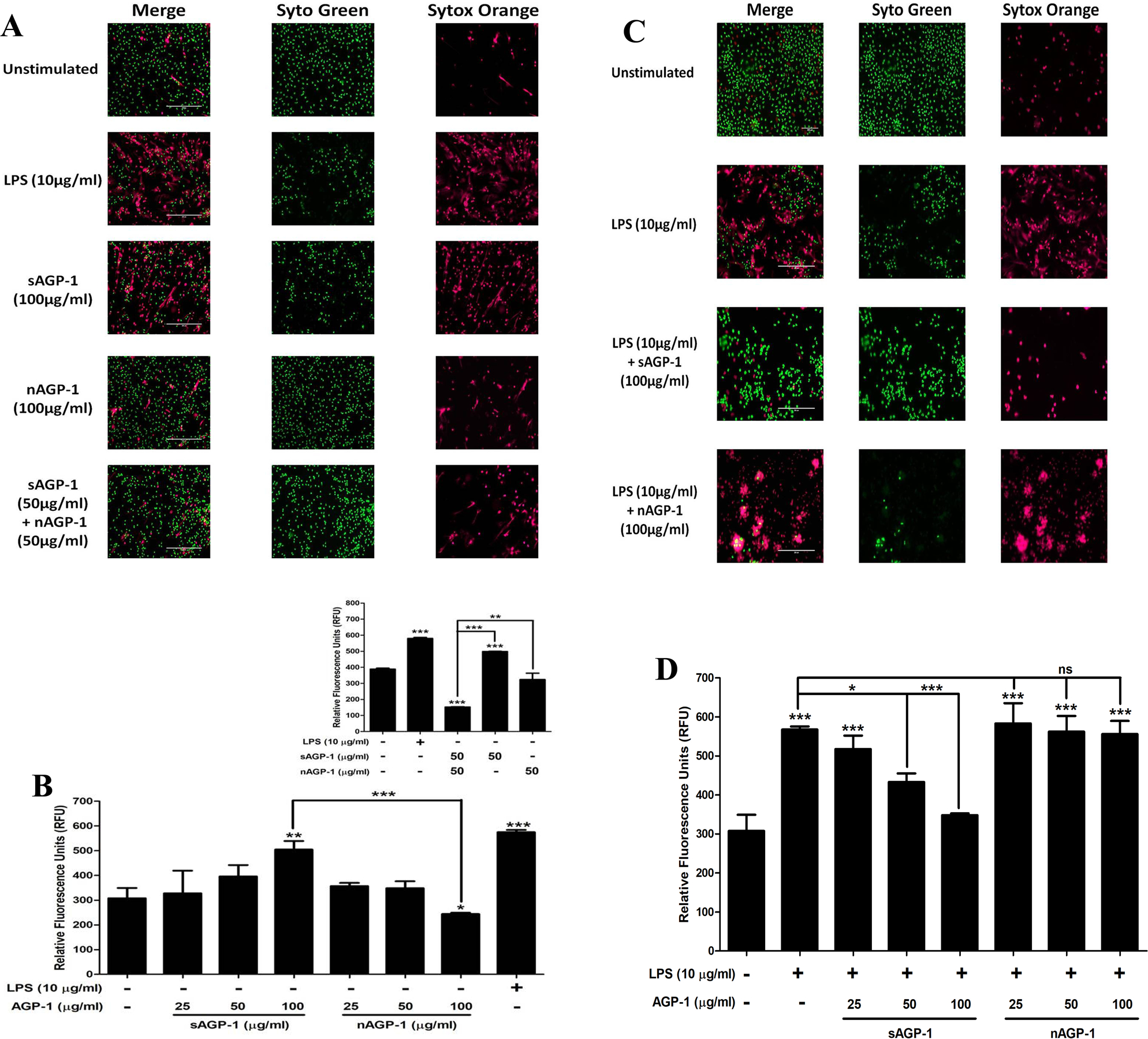
(A) NET formation. Isolated human neutrophils adhering to poly-L-Lysine coated slides were incubated with media alone, or with sAGP-1, nAGP-1 and LPS for 1 hour and were then stained with cell-permeable, Syto Green, and/ or with cell-impermeable Sytox Orange to label polymeric DNA. NETs were assessed by live cell imaging using fluorescence microscope at 20x magnification (Scale: 200µm). NET formation by LPS (red fluorescence) served as positive control. (B) Concentration-response relationships of NETosis. NETs were quantified using high-throughput method as explained in the “Methods” section. Neutrophils were stimulated with sAGP-1, nAGP-1 (25, 50 and 100 µg/ml) or LPS for 1 hour before quantifying NETs by fluorometry. In a parallel experiment, neutrophils were incubated with sAGP-1, nAGP-1 and combination of sAGP-1 and nAGP-1 (inset). LPS served as positive control. (C) Neutrophils were treated respectively with vehicle, LPS (10 µg/ml) in the presence/absence of sAGP-1 and nAGP-1 (25, 50 and 100 μg/ml). The mixture was incubated for 60 min at 37 °C and stained with Syto Green and Sytox Orange fluorescent dye mixture. The NETs were visualized under a fluorescent microscope at a magnification of 20x. sAGP-1 inhibited TLR-4 (LPS) mediated NETosis, while nAGP-1 did not have any effect on LPS – mediated effects. (D) NETosis was quantified using high-throughput method as explained under “Methods”. The data shown are mean ± SEM. ***=P <0.0001, **=P<0.001 and *=P<0.01 as determined by one – way ANOVA
Fig. 5: Profiling of sAGP-1 - induced NETosis:
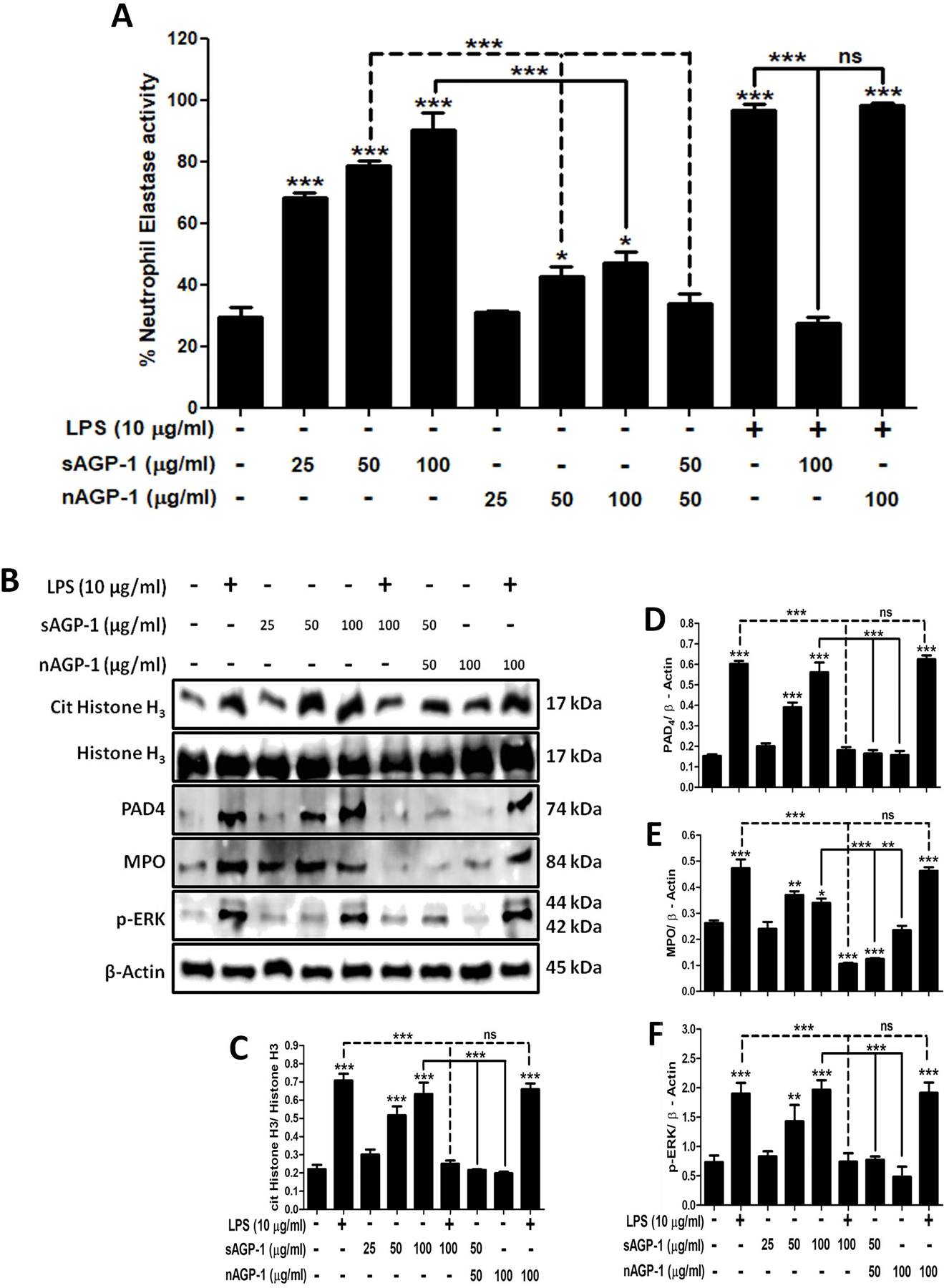
(A) Quantification of NETosis. Apart for the high-throughput method, NETosis was also quantified by using NETosis assay kit. Neutrophils were stimulated with LPS (10 µg/ml), sAGP-1 and/ or nAGP-1 (25, 50, and 100 µg/ml). In addition neutrophils were also treated with combination of LPS (10 µg/ml), sAGP-1 (100 µg/ml) and/ or nAGP-1 (100 µg/ml) and sub-maximal concentration of sAGP-1 and nAGP-1 (50 µg/ml) (B) Immunoblots for markers of NETosis. Immunoblots for the key markers of NETosis like cit His H3, PAD4, MPO and phospho-ERK was performed. sAGP-1 – induced the expression of the key markers of NETosis in a concentration-dependent manner, while it inhibited LPS – induced expression of these proteins. Although, nAGP-1 failed to induce expression of these proteins even at highest concentration (100 µg/ml), sub-maximal concentration of nAGP-1 (50 µg/ml) inhibited sAGP-1 (50 µg/ml) – induced expression of cit His H3, PAD4, MPO and phospho-ERK. (C – F) Densitometric analyses of NETosis immunoblots. Densitometric analysis of blots obtained from 3 experiments was done using ImageJ software (ver. 1.51j8). The data shown are mean ± SEM. ***p<0.0001, *p<0.01 as determined by one – way ANOVA.
3.5 |. Analysis of markers of NETosis
To determine the molecular events in play during NETosis, citrulinated Histone H3 (cit His H3), peptidylarginine deaminase 4 (PAD4), MPO and phospho-ERK were monitored by immunoblotting as these are the key markers of NETosis [47–49]. We found that, the sAGP-1 showed a concentration – dependent increase in the expression of the key markers of NETosis, while nAGP-1 failed to increase the expression of these proteins significantly (Figs. 5 B–F). Furthermore, sAGP-1 (100 µg/ml), but not nAGP-1 (100 µg/ml), inhibited the LPS – induced expression of cit His H3, PAD4, MPO and phosphorylation of ERK at the concentration tested. In accordance with the previous results (Figs. 4B inset and 5A), at sub-maximal concentration nAGP-1 (50 µg/ml) inhibited sAGP-1 (50 µg/ml) – induced expression of these proteins (Figs. 5 B–F). Moreover, since sAGP-1 induced phosphorylation of ERK and ROS production (Fig. 3D), sAGP-1 likely induces NETosis via Raf-MEK-ERK pathway.
3.6 |. sAGP-1, but not nAGP-1, suppresses platelet aggregation induced by incomplete stimuli
To understand whether AGP-1 affect platelets, we examined aggregation of washed human platelets incubated with AGP-1 alone or together with thrombocyte agonists. We found neither of the AGP-1 glycoforms stimulated platelet aggregation by themselves (Fig. 6A inset). Instead, sAGP-1 profoundly suppressed, and ultimately abolished, platelet aggregation induced by either PAF (Figs. 6A, E & F) or ADP (Figs. 6B, G & H). In contrast, nAGP-1 was without effect on stimulated platelet aggregation. Platelet aggregation in response to the single “strong” platelet agonist thrombin, however, was not affected by either of the AGP-1 glycoforms (Figs. 6C, I & J), while soluble collagen acting on non-G protein-coupled receptors was only modestly affected (Figs. 6D, K & L).
Fig. 6: sAGP-1, but not nAGP-1, inhibits stimulated platelet aggregation.
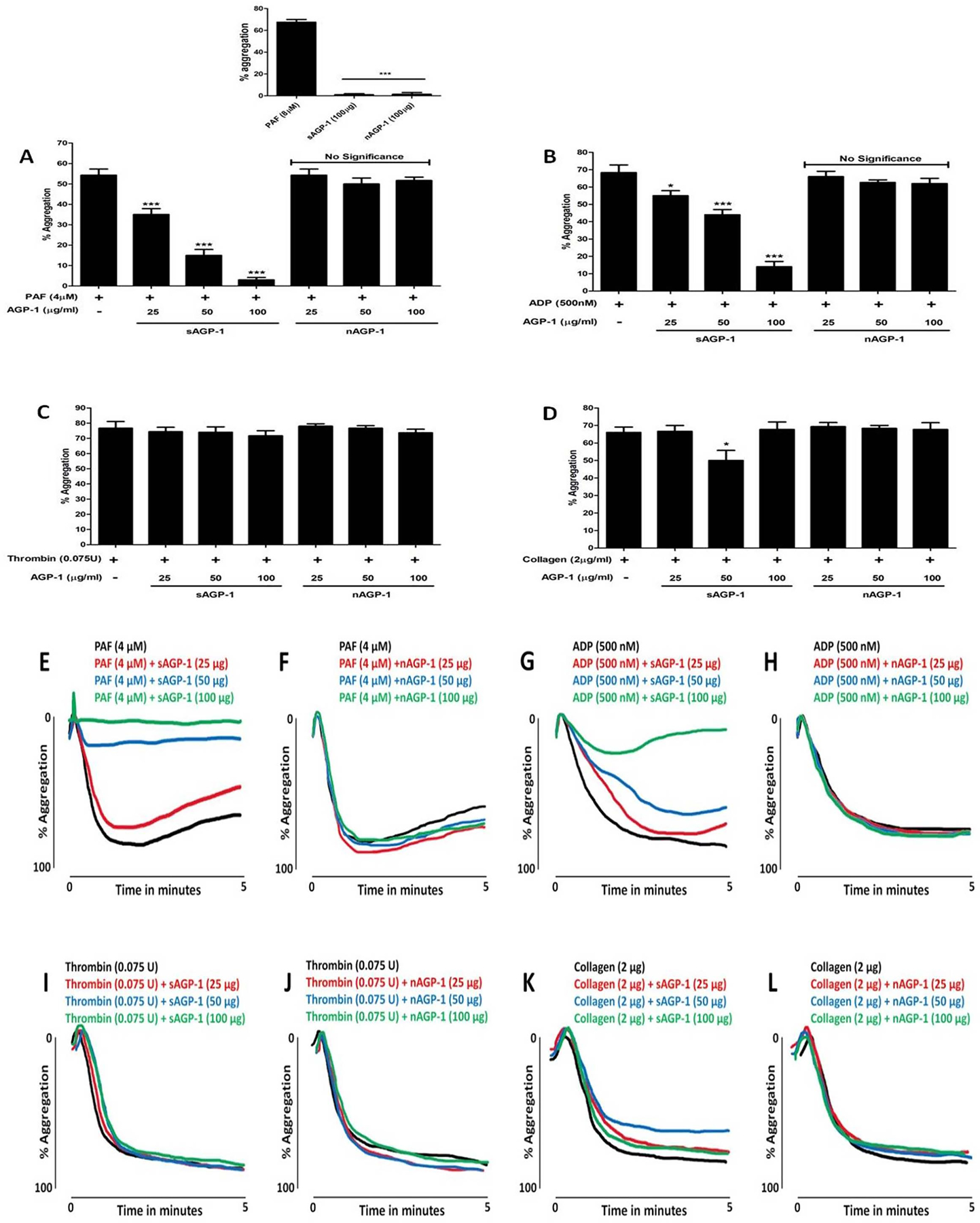
Platelet aggregation was induced by indicated amounts of PAF (A, E & F), ADP (B, G & H), thrombin (C, I & J) or collagen (D, K & L) with or without sAGP-1 and nAGP-1. (A) inset shows the effect of PAF (positive control), sAGP-1 and nAGP-1 on platelet aggregation. Each set of experiments were carried out with platelets from the same donor as well as repeated with platelets from at least three other donors. All assays were performed using Chrono-log aggregometer and the traces were recorded using AGGRO/LINK 8 ver.1.0.1 software. Representative trace of three independent experiments is shown here. The aggregation traces were merged using Wacom graphics pad. The data shown are mean ± SEM (n = 3). ***p<0.0001, *p<0.01 as determined by one – way ANOVA.
3.7 |. sAGP-1 stimulates cAMP accumulation in platelets stimulated by incomplete agonists
To understand the molecular mechanism underlying sAGP-1 inhibition of platelet aggregation, we assessed the effect of sAGP-1 on cyclic AMP (cAMP), whose levels profoundly affect platelet aggregation. We found sAGP-1 modestly, but not significantly, reduced cAMP levels in resting platelets, while the strong agonist thrombin significantly reduced cAMP abundance (Fig. 7A). sAGP-1 did not affect thrombin-suppressed cAMP, but did greatly increase cAMP levels in platelets stimulated with either PAF or ADP. We next determined whether sAGP-1 affected kinase signaling, and visualized the phosphorylation status of Akt, p38, and ERK kinases as well as the actin regulator vasodilator activated phosphoprotein (VASP) that is phosphorylated by cAMP and protein kinase A (PKA). The phosphoblot of platelets stimulated in the presence or absence of sAGP-1 showed that thrombin stimulation, like aggregation, was unaffected by this acute phase protein (Figs. 7B, C–F). These experiments also demonstrated that VASP phosphorylation was increased by sAGP-1 when cAMP levels increased (Figs. 7 A, B and F). In contrast, phosphorylation and activation of serine kinases by PAF or ADP was reduced by sAGP-1 (Figs. 7 B–F). These findings were confirmed by the results of three separate experiments that used platelets from different donors. The totality of these results shows congruence of the effect of sAGP-1 on aggregation, cAMP accumulation, kinase phosphorylation, and VASP phosphorylation across a range of agonist effectiveness.
Fig. 7: Inhibition of PAF and ADP-induced platelet aggregation by sAGP-1 is mediated by a cAMP-dependent pathway:
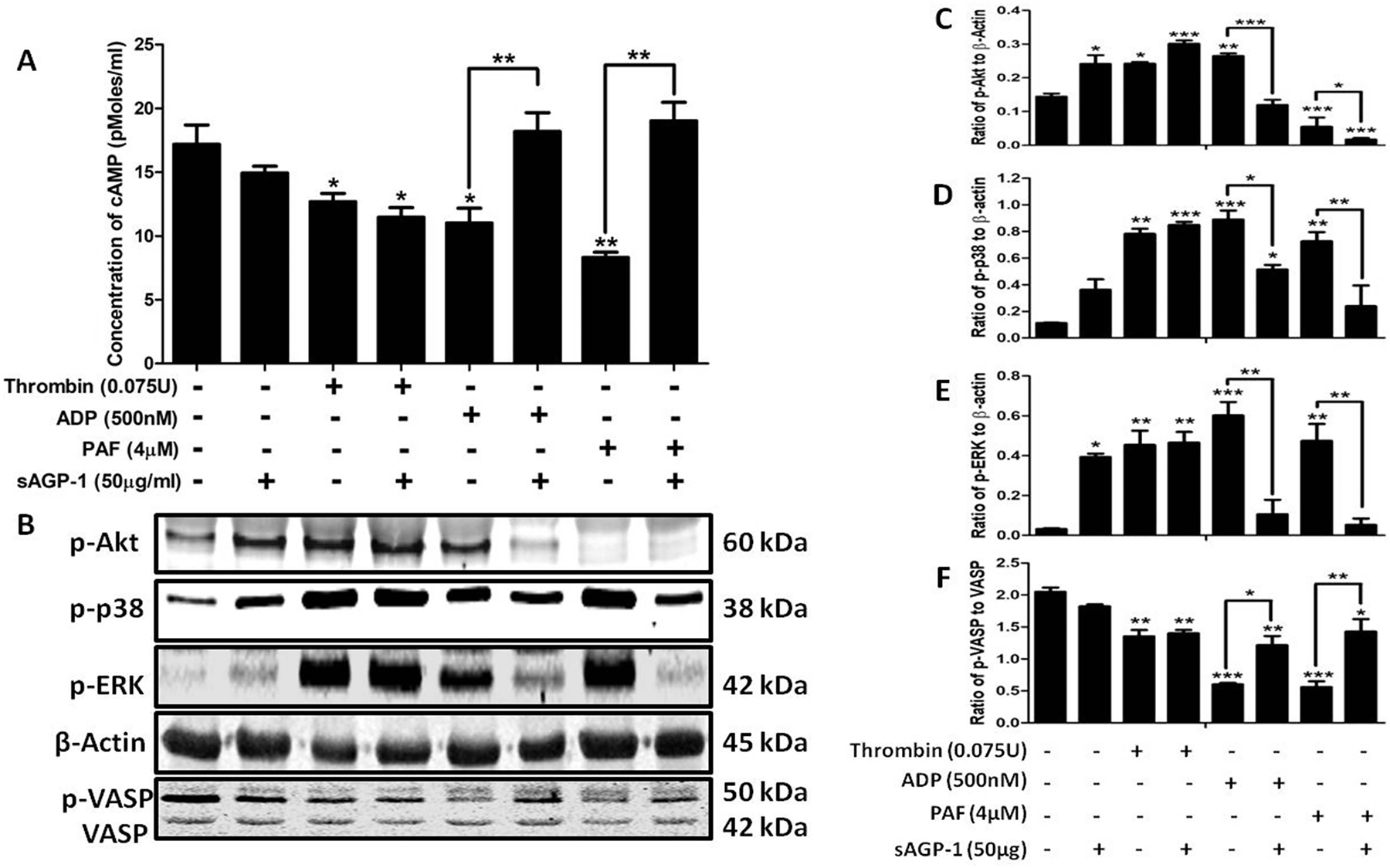
(A) Quantification of cAMP levels in plaelets. Washed platelets were stimulated with PAF, ADP or Thrombin with or without sAGP-1. After incubation, the cells were collected by centrifugation and then assayed for cAMP by ELISA. The data represents results from two different experiments. (B) Phosphokinase western blot. The phosphorylation status of the stated kinases treated, or not, with the indicated agonist as in the preceding panel was visualized by western blotting. Phosphorylation of Akt, p38, ERK, or VASP was normalized using total β-actin. (C – F) Densitometric analyses of phosphokinase immunoblots. Densitometric analysis of blots obtained from 3 experiments was done using ImageJ software (ver. 1.51j8). The data shown are mean ±SEM ***p < 0.001, **p < 0.01 and *p < 0.05 as analysed by one – way ANOVA.
4 |. Discussion
AGP-1 is an acute phase protein primarily secreted from hepatocytes, but also secreted from extra-hepatic sites [3–5]. Our results demonstrate unique biological effects of different glycoforms of the acute phase glycoprotein AGP-1, depending on its origin in either serum (sAGP-1) or from activated neutrophils (nAGP-1). AGP-1 is often considered as one of the markers of several inflammatory diseases [2, 50]. Although AGP-1 levels are elevated during inflammation, its biological function(s) are not completely understood. As there are more than 150 glycoforms of AGP-1 present in the human plasma, this molecular diversity adds to the complexity of AGP-1 biology [2, 8, 13, 14, 17]. To this end, we isolated and characterized a novel AGP-1 glycoform from PAF-stimulated human neutrophils (nAGP-1). We were focused in particular on determining the functional differences between nAGP-1 and sAGP-1, if so, does it play a role during inflammatory responses, a concept not yet examined in the field.
We determined whether an extra-hepatic source of AGP-1 (neutrophils) was identical to sAGP-1 in structure and function, or whether the plethora of glycoforms also contributes to functional diversity. We found that unstimulated human neutrophils, the first responder of the innate immune system, secreted little AGP-1, but did so in response to any of neutrophil agonists. The AGP-1 released from PAF-stimulated neutrophils consisted primarily of two immunoreactive species with distinct electrophoretic mobilities. One glycoform migrated like sAGP-1 (43 kDa) (secreted in low concentration), while the major product when PAF was the stimulus was a slower migrating glycoform (~60 kDa). There are previous reports that a higher molecular weight AGP-1 exists in the secondary granules of both human and bovine neutrophils [6, 51]. However, its structural details were not unraveled.
We found AGP-1 secretion from neutrophils in response to PAF varied with agonist concentration, where more AGP-1 was secreted with lower concentration of PAF. Although nAGP-1 (both ~60 kDa and 43 kDa glycoforms) eluted together in the void volume of Cibacron chromatography (data not shown), DEAE chromatography completely resolved both the slowly and rapidly migrating AGP-1 glycoforms. We discovered the difference in behavior between the glycoforms was the inability of the higher molecular weight (~60 kDa) nAGP-1 to bind positively charged DEAE resin but not the lower molecular weight nAGP-1 (43 kDa). The differences in behavior was not due to protein itself, as mass spectrometry evidence confirmed that the sAGP-1 retained by the DEAE column generated the same peptides after trypsin digestion as that of nAGP-1, and the same AGP-1 gene was identified by the Protein Discoverer program for both proteins. The differences between sAGP-1 and nAGP-1 were found to reside in N-glycans. Analyses revealed that the glycosylation of nAGP-1 is dramatically different from that of sAGP-1. nAGP-1 mainly expressed high-mannose, non-sialylated and mono-sialylated N-glycans, as opposed to sAGP-1, which expressed mono- and di-sialylated N-glycans. In accordance with this, nAGP-1, which only partially expresses mono-sialylated N-glycans, failed to bind to the positively charged DEAE resin.
Furthermore, higher molecular weight (~60 kDa) nAGP-1 after DEAE-purification showed faster mobility than that of the abundant 43 kDa sAGP-1 (Fig 2C). Therefore, we hypothesized the presence of uncharacterized “AGP-1 binding factor” that may account for the slower electrophoretic mobility of nAGP-1 before purification as previously hypothesized by Libert C et. al [52]. This “AGP-1 binding factor” might dissociate during the DEAE-purification step and hence the faster mobility of purified nAGP-1 (~35 kDa). Although, we performed “add-back experiment” to regain the higher molecular weight fraction (~60 kDa) of nAGP-1, these experiments were unsuccessful (data not shown). It is possible that there may be more than one binding factor(s) with low abundance. The search for this uncharacterized “AGP-1 binding factor” is still ongoing. Thus, we conclude that both the slow (~60 kDa) and rapidly migrating (~35 kDa) nAGP-1 species are glycoforms of the same nAGP-1 protein. However, the less secreted 43 kDa AGP-1, due to its similarity with that of sAGP-1, might bind to the DEAE resin like that of sAGP-1. Hence, the nAGP-1 thus purified is homogenous. Since the concentration of nAGP-1 secreted from the stimulated neutrophils is very less (~0.2–0.5µg/106 neutrophils) compared to the abundance of sAGP in serum (~0.7mg/ml), several rounds of purification for nAGP-1 were performed. The DEAE-purified nAGP-1 (~35 kDa) was pooled, characterized (for molecular weight and glycan structure) and used for the functional studies.
Our next question was whether AGP-1 glycoforms differ in their function. To address this, we first defined relevant functions for AGP-1 in the inflammatory system. Neutrophils when stimulated/ activated migrate to the site of inflammation and undergo degranulation and produce ROS. The granules of neutrophils contains many anti-microbial proteins and peptides including but not limited to MPO, matrix metalloprotesaes, elastases, cathepsins, defencins etc [53, 54]. We employed neutrophils, since activation of neutrophils and NETosis play a critical role in various inflammatory processes [55–57]. In accordance with our previous data, sAGP-1-induces concentration-dependent activation of neutrophil adhesion and migration, suggesting that the sAGP-1 induces Ca2+ influx to activate neutrophils [58, 59]. Here sAGP-1 is as effective as IL-8, but nAGP-1 is significantly less potent in activating either of these responses. In addition, sAGP-1 also induced degranulation of neutrophils (MPO secretion) and ROS production, while nGAP-1 was not an effective stimulator of neutrophils. Similarly, we found that while sAGP-1 stimulates NETosis, nAGP-1 does not. Moreover, nAGP-1 suppresses NETosis stimulated by sAGP-1, while sAGP-1, but not nAGP-1, suppresses NETosis induced by LPS. This effect might be due to direct interaction of AGP-1with LPS thereby quenching LPS’s effects as suggested by Huang et. al. [60]. To check this possibility, we performed binding studies using FITC conjugated LPS with sAGP-1. However, we could not see any significant binding of LPS to sAGP-1 (data not shown). Additionally, the carbohydrate moiety of nAGP-1 might physically prevent recognition of the carbohydrate structures displayed by sAPG-1 and LPS. NETosis is of two types; suicidal and vital. Regardless of the type, mechanism of NETosis generally follows the Raf-MEK-ERK pathway that involves ROS production by NADPH oxidase and MPO [47–49, 61, 62]. We found that sAGP-1, but not nAGP-1, induced increase in ROS production and also the expression of the MPO, PAD4 and phosphorylation of ERK in a concentration dependent fashion, while LPS-induced expression of these proteins were inhibited by sAGP-1. Hence, we hypothesize that sAGP-1 may induce NETosis via Raf-MEK-ERK pathway, a hallmark of suicidal NETosis. We conclude that plasma sAGP-1 stimulates leukocyte function, while neutrophil-derived nAGP-1 mostly does not. This difference in biological function reflects differences in post-translational modification of these proteins by glycans. Thus, stimulated neutrophils produce an anti-inflammatory glycoform of AGP-1 that counteracts the pro-inflammatory actions of circulating sAGP-1.
The variations in the effects of AGP-1 glycoforms are also extended to the responses of platelets, which play a critical role also in inflammation [63–68]. In fact, platelets are considered as immune cells, besides their role in hemostasis and blood coagulation [69–71]. While neither glycoforms alone stimulated platelet aggregation, sAGP-1 proved to be a highly effective suppressor of platelets stimulated by PAF or ADP, less so for those stimulated by soluble collagen, and was without effect on the strong agonist thrombin. Again, nAGP-1 differed in function from sAGP-1 and had no effect on stimulated platelet aggregation. The concentration-dependent inhibitory effect of sAGP-1 on platelet function induced by PAF or ADP correlated to a sharp increase in intracellular cAMP levels. In contrast, thrombin-induced aggregation and cAMP levels were not affected by sAGP-1. Phosphorylation of VASP is dependent on cAMP and the PKA pathway it controls [72, 73], and sAGP-1 upregulated phosphorylation of VASP in PAF- or ADP-stimulated platelets, but not in thrombin stimulated cells. sAGP-1 reduced PAF- and ADP-stimulated phosphorylation and activation of Akt, ERK, and p38 kinases, but again thrombin stimulation of these enzymes were unaffected by sAGP-1. These results, then, suggest that sAGP-1 inhibits platelet aggregation by weaker agonists via cAMP-PKA mediated signaling (Fig. 8); supporting the claim that AGP-1 demonstrates anti-thrombotic effects [28–30].
Fig. 8: Proposed action of sAGP-1-induced inhibition of PAF- and ADP-mediated platelet aggregation:
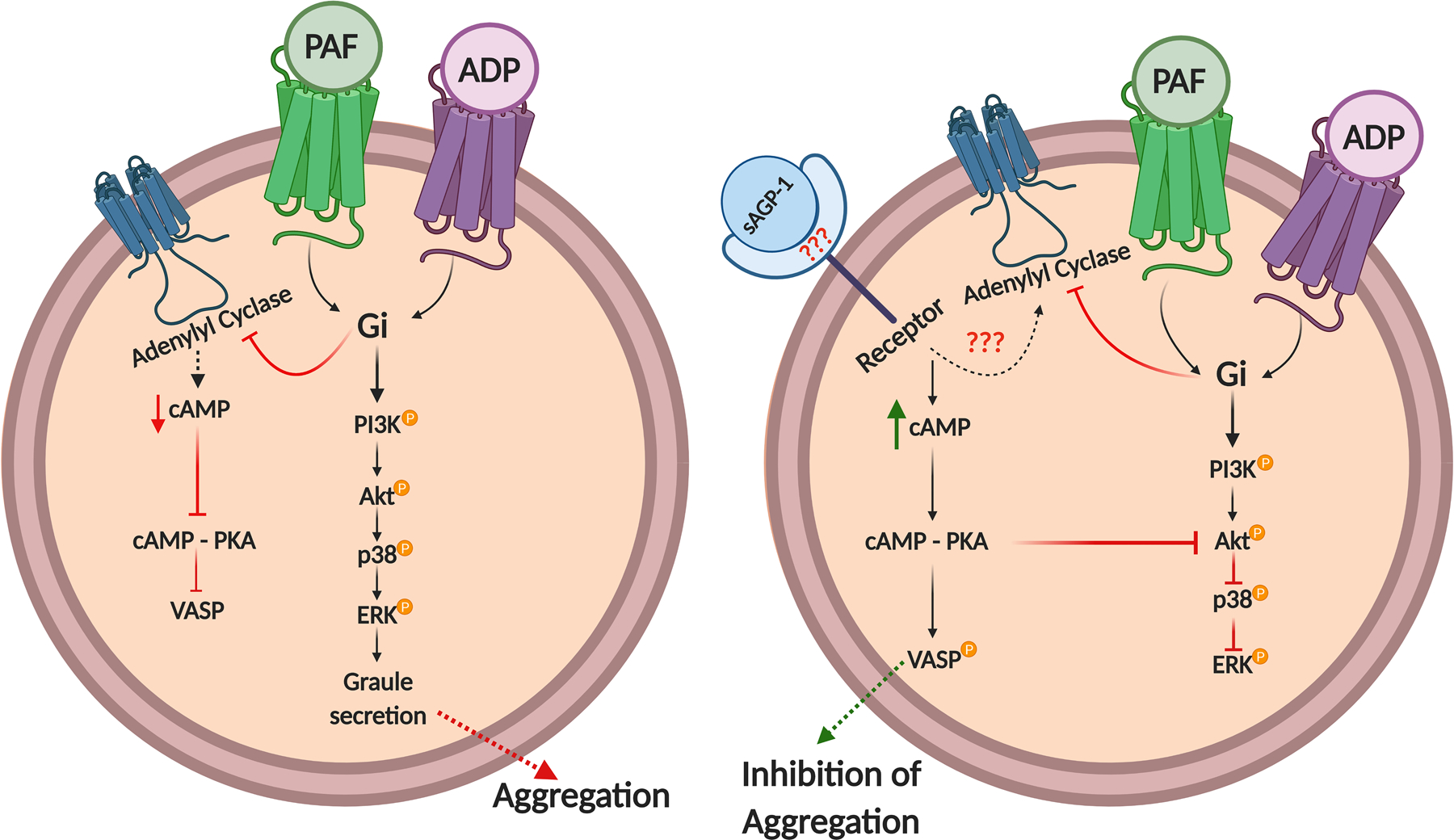
sAGP-1 up-regulates cAMP levels in PAF or ADP activated platelets, thereby activating PKA. This then activates VASP and inhibits Akt phosphorylation and activation thereby inhibiting downstream signaling and platelet aggregation. The illustration was created with BioRender.com
Previously, AGP-1 is shown to act via, a family of receptors called Sialic acid binding immunoglobulin like lectins (Siglec), Siglec – 5. Siglec-5 receptor signaling, although not well understood, demonstrates both activating as well as inhibitory inflammatory signaling in neutrophils [58, 74–76]. Moreover, we show the involvement of sAGP-1 in the cAMP pathway in platelets, which is a characteristic feature of GPCRs and not of the Siglec receptors [27, 77, 78]. Put together all these findings, we propose that sAGP-1 and nAGP-1 might act via different or related receptors and also that sAGP-1 might have different receptors on different cells.
In conclusion, our studies demonstrate that AGP-1 displays structural heterogeneity in its glycan structures associated with differences in physiological functions. Isolation and characterization of a glycoform of AGP-1 released from PAF-stimulated human neutrophils represents a non-hepatic source of extracellular AGP-1, which is less pro-inflammatory than hepatic sAGP-1 on platelets and neutrophils. The basis for these differences is the distinct carbohydrate chain composition and structure incorporated by the two cell types rather than the AGP-1 protein itself. Furthermore, our studies demonstrate that the AGP-1 glycoform released from stimulated neutrophils in the early phases of inflammation is not an effective positive acute phase protein, but instead counteracts the hepatic sAGP-1 glycoform. These findings in part explain the contradictory role of AGP-1 in various inflammatory processes. This, in turn, identifies a need for future investigations to determine functions of the multitude of other AGP-1 glycoforms and there is a need to differentially quantify AGP-1 glycoforms rather than just AGP-1 protein in health and diseases, which can be insurmountable task. The other greatest challenge lies in identifying appropriate receptors (Fig. 8) for AGP-1 glycoforms that ligate and alter intracellular signaling pathways.
Acknowledgement
The authors thank Mr. Neal Tolley and Mr. Mark Cody, at the University of Utah Molecular Medicine Program in Salt Lake City, Utah, USA for their technical assistance and Prof. B.S. Vishwanath and Mr. Rudresha G.V, Dept. of Studies in Biochemistry, University of Mysore, Mysuru, India, for graciously sharing the antibodies. The authors also thank Prof. Guy Zimmerman, University of Utah, Salt Lake City, USA and Dr. Stephen Prescott, OMRF, Oklahoma, USA for their timely advice during the course of this study. The authors thank University Grants Commission (UGC), India [for Basic Science Research fellowship – F4-1/2006(BSR)/7-366/2012(BSR) to MSS, National Fellowship for Higher Education (NFHE-ST) – F1-17/2015-16/NFST-2015-17-ST-KAR-3879/(SA-III/Website) to KVA] and Vision Group of Science & Technology (VGST), Government of Karnataka, India to the Dept. of Studies in Biochemistry, University of Mysore, Mysuru, India, and the National Center for Functional Glycomics Grant P41GM103694 (RDC and SL). This work was funded by the NHLBI (HL092746, HL126547), NICHD (HD93826), and NIA (AG048022), and supported in part by Merit Review Award Number I01 CX001696 from the United States (U.S.) Department of Veterans Affairs Clinical Sciences R&D (CSRD) Service. This material is the result of work supported with resources and the use of facilities at the George E. Wahlen VA Medical Center, in Salt Lake City, Utah. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the U.S. Government.
Abbreviation
- ADP
Adenosine diphosphate
- cAMP
cyclic Adenosine monophosphate
- DEAE-cellulose
Diethyl aminoethane-cellulose
- LPS
Lipopolysaccharide
- MPO
Myeloperoxidase
- nAGP-1
neutrophil derived AGP-1
- PAD4
Peptidylarginine deaminase 4
- PAF
Platelet-activating factor
- PKA
Protein kinase A
- sAGP-1
serum Alpha-1-acid glycoprotein
- TNF
Tumor necrosis factor
- VASP
Vasodilator stimulated phosphoprotein
Footnotes
Disclosure
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Schmid K, Mao S, Kimura A, Hayashi S, Binette JP Isolation and characterization of a serine-threonine-rich galactoglycoprotein from normal human plasma. J. Biol. Chem 1980; 255: 3221–3226. [PubMed] [Google Scholar]
- 2.Fournier T, Medjoubi-N N, Porquet D Alpha-1-acid glycoprotein. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 2000; 1482: 157–171. [DOI] [PubMed] [Google Scholar]
- 3.Hochepied T, Berger FG, Baumann H, Libert C α1-Acid glycoprotein: an acute phase protein with inflammatory and immunomodulating properties. Cytokine Growth Factor Rev 2003; 14: 25–34. [DOI] [PubMed] [Google Scholar]
- 4.Gendler SJ, Dermer GB, Silverman LM, Tökés ZA Synthesis of α1-antichymotrypsin and α1-acid glycoprotein by human breast epithelial cells. Cancer Res 1982; 42: 4567–4573. [PubMed] [Google Scholar]
- 5.Adam P, Sobek O, Táborský L, Hildebrand T, Tutterová O, Žáček P CSF and serum orosomucoid (α−1-acid glycoprotein) in patients with multiple sclerosis: a comparison among particular subgroups of MS patients. Clin. Chim. Acta 2003; 334: 107–110. [DOI] [PubMed] [Google Scholar]
- 6.Theilgaard-Mönch K, Jacobsen LC, Rasmussen T, Niemann CU, Udby L, Borup R, Gharib M, Arkwright PD, Gombart AF, Calafat J Highly glycosylated α1-acid glycoprotein is synthesized in myelocytes, stored in secondary granules, and released by activated neutrophils. J. Leukoc. Biol 2005; 78: 462–470. [DOI] [PubMed] [Google Scholar]
- 7.Ceciliani F and Pocacqua V The acute phase protein α1-acid glycoprotein: a model for altered glycosylation during diseases. Curr. Protein Pept. Sci 2007; 8: 91–108. [DOI] [PubMed] [Google Scholar]
- 8.Ongay S and Neusüβ C Isoform differentiation of intact AGP from human serum by capillary electrophoresis–mass spectrometry. Anal. Bioanal. Chem 2010; 398: 845–855. [DOI] [PubMed] [Google Scholar]
- 9.Ceciliani F and Lecchi C The Immune Functions of α1 Acid Glycoprotein. Curr. Protein Pept. Sci 2019; 20: 505–524. [DOI] [PubMed] [Google Scholar]
- 10.Moule SK, Peak M, Thompson S, Turner GA Studies of the sialylation and microheterogeneity of human serum α1-acid glycoprotein in health and disease. Clin. Chim. Acta 1987; 166: 177–185. [DOI] [PubMed] [Google Scholar]
- 11.Mackiewicz A, Marcinkowska-Pieta R, Ballou S, Mackiewicz S, Kushner I Microheterogeneity of alpha1-acid glycoprotein in the detection of intercurrent infection in systemic lupus erythematosus. Arthritis Rheum 1987; 30: 513–518. [DOI] [PubMed] [Google Scholar]
- 12.De Graaf TW, Van der Stelt M, Anbergen M, Van Dijk W Inflammation-induced expression of sialyl Lewis X-containing glycan structures on alpha 1-acid glycoprotein (orosomucoid) in human sera. J. Exp. Med 1993; 177: 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pawlowski T, Mackiewicz SH, Mackiewicz A Microheterogeneity of alpha1-acid glycoprotein in the detection of intercurrent infection in patients with rheumatoid arthritis. Arthritis Rheum 1989; 32: 347–351. [DOI] [PubMed] [Google Scholar]
- 14.Van Dijk W, Havenaar E, Brinkman-Van der Linden EC α 1-acid glycoprotein (orosomucoid): pathophysiological changes in glycosylation in relation to its function. Glycoconj. J 1995; 12: 227–233. [DOI] [PubMed] [Google Scholar]
- 15.Zhang D, Huang J, Luo D, Feng X, Liu Y Glycosylation change of alpha-1-acid glycoprotein as a serum biomarker for hepatocellular carcinoma and cirrhosis. Biomark. Med 2017; 11: 423–430. [DOI] [PubMed] [Google Scholar]
- 16.Xiao K, Su L, Yan P, Han B, Li J, Wang H, Jia Y, Li X, Xie L α−1-Acid glycoprotein as a biomarker for the early diagnosis and monitoring the prognosis of sepsis. J. Crit. Care 2015; 30: 744–751. [DOI] [PubMed] [Google Scholar]
- 17.Van Dijk W, Brinkman-Van der Linden EC, Havenaar EC Glycosylation of α1-Acid Glycoprotein (Orosomucoid) in Health and Disease. Trends Glycosci. Glycotechnol 1998; 10: 235–245. [Google Scholar]
- 18.Hochepied T, Van Molle W, Berger FG, Baumann H, Libert C Involvement of the acute phase protein α1-acid glycoprotein in nonspecific resistance to a lethal gram-negative infection. J. Biol. Chem 2000; 275: 14903–14909. [DOI] [PubMed] [Google Scholar]
- 19.Libert C, Brouckaert P, Fiers W Protection by alpha 1-acid glycoprotein against tumor necrosis factor-induced lethality. J. Exp. Med 1994; 180: 1571–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costello M, Fiedel BA, Gewurz H Inhibition of platelet aggregation by native and desialised alpha-1 acid glycoprotein. Nature 1979; 281: 677–678. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura K, Ito I, Kobayashi M, Herndon DN, Suzuki F Orosomucoid 1 drives opportunistic infections through the polarization of monocytes to the M2b phenotype. Cytokine 2015; 73: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mestriner FL, Spiller F, Laure HJ, Souto FO, Tavares-Murta BM, Rosa JC, Basile-Filho A, Ferreira SH, Greene LJ, Cunha FQ Acute-phase protein alpha-1-acid glycoprotein mediates neutrophil migration failure in sepsis by a nitric oxide-dependent mechanism. Proc. Nat. Acad. Sci. USA 2007; 104: 19595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higuchi H, Kamimura D, Jiang JJ, Atsumi T, Iwami D, Hotta K, Harada H, Takada Y, Kanno-Okada H, Hatanaka KC, Tanaka Y, Shinohara N, Murakami M Orosomucoid 1 is involved in the development of chronic allograft rejection after kidney transplantation. Int. Immunol 2020. [DOI] [PubMed] [Google Scholar]
- 24.Andersen P The antiheparin effect of α1-acid glycoprotein, evaluated by the activated partial thromboplastin time and by a factor Xa assay for heparin. Pathophysiol. Haemos. Thromb 1980; 9: 303–309. [DOI] [PubMed] [Google Scholar]
- 25.Berntsson J, Östling G, Persson M, Smith JG, Hedblad B, Engström G Orosomucoid, carotid plaque, and incidence of stroke. Stroke 2016; 47: 1858–1863. [DOI] [PubMed] [Google Scholar]
- 26.Boncela J, Papiewska I, Fijalkowska I, Walkowiak B, Cierniewski CS Acute phase protein α1-acid glycoprotein interacts with plasminogen activator inhibitor type 1 and stabilizes its inhibitory activity. J. Biol. Chem 2001; 276: 35305–35311. [DOI] [PubMed] [Google Scholar]
- 27.Gunnarsson P, Levander L, Påhlsson P, Grenegård M α1-acid glycoprotein (AGP)-induced platelet shape change involves the Rho/Rho kinase signalling pathway. Thromb. Haemost 2009; 102: 694–703. [DOI] [PubMed] [Google Scholar]
- 28.Fiedel B, Costello M, Gewurz H, Hussissian E Effects of Heparin and α1-Acid Glycoprotein on Thrombin or Activated Thrombofax Reagent-Induced Platelet Aggregation and Clot Formation. Pathophysiol. Haemos. Thromb 1983; 13: 89–95. [DOI] [PubMed] [Google Scholar]
- 29.Osikov M, Makarov E, Krivokhizhina L Effects of α 1-acid glycoprotein on hemostasis in experimental septic peritonitis. Bull. Exp. Biol. Med 2007; 144: 178–180. [DOI] [PubMed] [Google Scholar]
- 30.Daemen MA, Heemskerk VH, van’t Veer C, Denecker G, Wolfs TG, Vandenabeele P, Buurman WA Functional protection by acute phase proteins α1-acid glycoprotein and α1-antitrypsin against ischemia/reperfusion injury by preventing apoptosis and inflammation. Circulation 2000; 102: 1420–1426. [DOI] [PubMed] [Google Scholar]
- 31.Stark RJ, Aghakasiri N, Rumbaut RE Platelet-derived Toll-like receptor 4 (Tlr-4) is sufficient to promote microvascular thrombosis in endotoxemia. PloS one 2012; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rondina M, Schwertz H, Harris E, Kraemer B, Campbell R, Mackman N, Grissom C, Weyrich A, Zimmerman G The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J. Thromb. Haemost 2011; 9: 748–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jia S-J, Niu P-P, Cong J-Z, Zhang B-K, Zhao M TLR4 signaling: A potential therapeutic target in ischemic coronary artery disease. Int. Immunopharmacol 2014; 23: 54–59. [DOI] [PubMed] [Google Scholar]
- 34.Schattner M Platelet TLR4 at the crossroads of thrombosis and the innate immune response. J. Leukoc. Biol 2019; 105: 873–880. [DOI] [PubMed] [Google Scholar]
- 35.Sumanth MS, Abhilasha KV, Jacob SP, Chaithra VH, Basrur V, Willard B, McIntyre TM, Prabhu KS, Marathe GK Acute phase protein, alpha - 1- acid glycoprotein (AGP-1), has differential effects on TLR-2 and TLR-4 mediated responses. Immunobiology 2019; 224: 672–680. [DOI] [PubMed] [Google Scholar]
- 36.Lakshmikanth CL, Jacob SP, Kudva AK, Latchoumycandane C, Yashaswini PSM, Sumanth MS, Goncalves-de-Albuquerque CF, Silva AR, Singh SA, Castro-Faria-Neto HC Escherichia coli Braun Lipoprotein (BLP) exhibits endotoxemia–like pathology in Swiss albino mice. Sci. Rep 2016; 6: 34666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe J, Marathe GK, Neilsen PO, Weyrich AS, Harrison KA, Murphy RC, Zimmerman GA, McIntyre TM Endotoxins stimulate neutrophil adhesion followed by synthesis and release of platelet-activating factor in microparticles. J. Biol. Chem 2003; 278: 33161–33168. [DOI] [PubMed] [Google Scholar]
- 38.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ Protein measurement with the Folin phenol reagent. J. Biol. Chem 1951; 193: 265–275. [PubMed] [Google Scholar]
- 39.Strohalm M, Kavan D, Novak P, Volny M, Havlicek V mMass 3: a cross-platform software environment for precise analysis of mass spectrometric data. Anal. Chem 2010; 82: 4648–4651. [DOI] [PubMed] [Google Scholar]
- 40.Bradley PP, Priebat DA, Christensen RD, Rothstein G Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J. Invest. Dermatol 1982; 78: 206–209. [DOI] [PubMed] [Google Scholar]
- 41.Yoshikawa T, Furukawa Y, Murakami M, Watanabe K, Kondo M Effect of vitamin E on endotoxin-induced disseminated intravascular coagulation in rats. Thromb. Haemost 1982; 47: 235–237. [PubMed] [Google Scholar]
- 42.Carmona-Rivera C and Kaplan MJ Induction and quantification of NETosis. Curr. Protoc. Immunol 2016; 115: 14.41.01–14.41.14. [DOI] [PubMed] [Google Scholar]
- 43.Zhou L and Schmaier AH Platelet aggregation testing in platelet-rich plasma: description of procedures with the aim to develop standards in the field. Am. J. Clin. Pathol 2005; 123: 172–183. [DOI] [PubMed] [Google Scholar]
- 44.Leick M, Azcutia V, Newton G, Luscinskas FW Leukocyte recruitment in inflammation: basic concepts and new mechanistic insights based on new models and microscopic imaging technologies. Cell Tissue Res 2014; 355: 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McIntyre TM, Prescott SM, Weyrich AS, Zimmerman GA Cell-cell interactions: leukocyte-endothelial interactions. Curr. Opin. Hematol 2003; 10: 150–158. [DOI] [PubMed] [Google Scholar]
- 46.Mesa MA and Vasquez G NETosis. Autoimmune Dis 2013; 2013: 651497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, Herrmann M, Bilyy R, Müller S, Popper B PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur. J. Immunol 2016; 46: 223–229. [DOI] [PubMed] [Google Scholar]
- 48.Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol 2011; 7: 75–77. [DOI] [PubMed] [Google Scholar]
- 49.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med 2010; 207: 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Komori H, Watanabe H, Shuto T, Kodama A, Maeda H, Watanabe K, Kai H, Otagiri M, Maruyama T α1-acid glycoprotein up-regulates CD163 via TLR4/CD14 pathway: possible protection against hemolysis-induced oxidative stress. J. Biol. Chem 2012; 287: 30688–30700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rahman MM, Miranda-Ribera A, Lecchi C, Bronzo V, Sartorelli P, Franciosi F, Ceciliani F Alpha1-acid glycoprotein is contained in bovine neutrophil granules and released after activation. Vet. Immunol. Immunopathol 2008; 125: 71–81. [DOI] [PubMed] [Google Scholar]
- 52.Libert C, Hochepied T, Berger FG, Baumann H, Fiers W, Brouckaert P High-level constitutive expression of α1-acid glycoprotein and lack of protection against tumor necrosis factor-induced lethal shock in transgenic mice. Transgenic Res 1998; 7: 429–435. [DOI] [PubMed] [Google Scholar]
- 53.Borregaard N and Cowland JB Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 1997; 89: 3503–3521. [PubMed] [Google Scholar]
- 54.Lacy P Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol 2006; 2: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iba T and Levy J Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost 2018; 16: 231–241. [DOI] [PubMed] [Google Scholar]
- 56.Kapoor S, Opneja A, Nayak L The role of neutrophils in thrombosis. Thromb. Res 2018; 170: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perdomo J, Leung HH, Ahmadi Z, Yan F, Chong JJ, Passam FH, Chong BH Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun 2019; 10: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gunnarsson P, Levander L, Påhlsson P, Grenegard M The acute-phase protein α1-acid glycoprotein (AGP) induces rises in cytosolic Ca2+ in neutrophil granulocytes via sialic acid binding immunoglobulin-like lectins (Siglecs). FASEB J 2007; 21: 4059–4069. [DOI] [PubMed] [Google Scholar]
- 59.Levander L, Gunnarsson P, Grenegård M, Rydén I, Påhlsson P Effects of α1-acid Glycoprotein Fucosylation on its Ca2+ Mobilizing Capacity in Neutrophils. Scand. J. Immunol 2009; 69: 412–420. [DOI] [PubMed] [Google Scholar]
- 60.Huang JX, Azad MA, Yuriev E, Baker MA, Nation RL, Li J, Cooper MA, Velkov T Molecular characterization of lipopolysaccharide binding to human α−1-acid glycoprotein. J. Lipids 2012; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol 2007; 176: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Masuda S, Nakazawa D, Shida H, Miyoshi A, Kusunoki Y, Tomaru U, Ishizu A NETosis markers: quest for specific, objective, and quantitative markers. Clin. Chim. Acta 2016; 459: 89–93. [DOI] [PubMed] [Google Scholar]
- 63.Rondina MT and GUO L The era of thromboinflammation: platelets are dynamic sensors and effector cells during infectious diseases. Front. Immunol 2019; 10: 2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rondina MT, Weyrich AS, Zimmerman GA Platelets as cellular effectors of inflammation in vascular diseases. Circ. Res 2013; 112: 1506–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klinger MH Platelets and inflammation. Anat. Embryol 1997; 196: 1–11. [DOI] [PubMed] [Google Scholar]
- 66.Weyrich A, Lindemann S, Zimmerman G The evolving role of platelets in inflammation. J. Thromb. Haemost 2003; 1: 1897–1905. [DOI] [PubMed] [Google Scholar]
- 67.Thomas MR and Storey RF The role of platelets in inflammation. Thromb. Haemost 2015; 114: 449–458. [DOI] [PubMed] [Google Scholar]
- 68.Dewitte A, Lepreux S, Villeneuve J, Rigothier C, Combe C, Ouattara A, Ripoche J Blood platelets and sepsis pathophysiology: A new therapeutic prospect in critical ill patients? Ann. Intensive Care 2017; 7: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Semple JW, Italiano JE, Freedman J Platelets and the immune continuum. Nat. Rev. Immunol 2011; 11: 264–274. [DOI] [PubMed] [Google Scholar]
- 70.Ali RA, Wuescher LM, Worth RG Platelets: essential components of the immune system. Curr. Trends Immunol 2015; 16: 65. [PMC free article] [PubMed] [Google Scholar]
- 71.Morrell CN, Aggrey AA, Chapman LM, Modjeski KL Emerging roles for platelets as immune and inflammatory cells. Blood 2014; 123: 2759–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anton KA, Sinclair J, Ohoka A, Kajita M, Ishikawa S, Benz PM, Renne T, Balda M, Jorgensen C, Matter K PKA-regulated VASP phosphorylation promotes extrusion of transformed cells from the epithelium. J. Cell Sci 2014; 127: 3425–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y-T, Xu L-H, Lu Q, Liu K-P, Liu P-Y, Ji F, Liu X-M, Ouyang D-Y, He X-H VASP activation via the Gα13/RhoA/PKA pathway mediates cucurbitacin-B-induced actin aggregation and cofilin-actin rod formation. PLoS One 2014; 9: e93547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ali SR, Fong JJ, Carlin AF, Busch TD, Linden R, Angata T, Areschoug T, Parast M, Varki N, Murray J, Nizet V, Varki A Siglec-5 and Siglec-14 are polymorphic paired receptors that modulate neutrophil and amnion signaling responses to group B Streptococcus. J. Exp. Med 2014; 211: 1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Avril T, Freeman SD, Attrill H, Clarke RG, Crocker PR Siglec-5 (CD170) can mediate inhibitory signaling in the absence of immunoreceptor tyrosine-based inhibitory motif phosphorylation. J. Biol. Chem 2005; 280: 19843–19851. [DOI] [PubMed] [Google Scholar]
- 76.Pepin M, Mezouar S, Pegon J, Muczynski V, Adam F, Bianchini EP, Bazaa A, Proulle V, Rupin A, Paysant J, Panicot-Dubois L, Christophe O, Dubois C, Lenting P, Denis C Soluble Siglec-5 associates to PSGL-1 and displays anti-inflammatory activity. Sci. Rep 2016; 6: 37953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sadana R and Dessauer CW Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals 2009; 17: 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sassone-Corsi P The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol 2012; 4: a011148. [DOI] [PMC free article] [PubMed] [Google Scholar]