

Abstract
Alzheimer’s disease (AD) is the most common form of dementia affecting nearly 45 million people worldwide. However, the etiology of AD is still unclear. Accumulations of amyloid-β plaques and tau tangles, neuroinflammation, and synaptic and neuronal loss are the major neuropathological hallmarks of AD, with synaptic loss being the strongest correlating factor with memory and cognitive impairment in AD. Many of these pathological hallmarks influence each other during the onset and progression of the disease. Recent genetic evidence suggests the possibility of a causal link between altered immune pathways and synaptic dysfunction in AD. Emerging studies also suggest that immune system-mediated synaptic pruning could initiate early-stage pathogenesis of AD. This comprehensive review is toward understanding the crosstalk of neuron-microglia-astrocyte and dynamics of complement, cytokine, and chemokine systems in the regulation of synaptic function and dysfunction relevant to AD. We start with summarizing several immune pathways, involving complements, MHC-I and CX3CL1, which mediate synaptic elimination during development and in AD. We then will discuss the potential of targeting these molecules as therapeutic interventions or as biomarkers for AD.
Keywords: Alzheimer’s disease, astrocyte, complements, CX3CR1, cytokines, fractalkine, MHC-I, microglia, neurons, synaptic pruning
INTRODUCTION
Contrary to the conventional dogma, the central nervous system is not as “immune-privileged” as previously considered. Indeed, the immune and central nervous systems actively interact and share mechanisms of gene regulation, signaling, and cell communication [1]. Immune and neuronal cells interact bi-directionally through extra-synaptic communication mediated by complements, cytokines, chemokines, neuropeptides, neurotransmitters, neurotrophins, and their receptors [2, 3]. These immune system mediators are not only expressed in cells from the peripheral immune system, but they are also central nervous system (CNS)-derived. Notably, cells such as microglia, astrocytes, and neurons play important roles as immune system modulators. It is well recognized that the CNS resident immune cells (microglia and astrocytes) and immune system-mediators play essential roles in the synapse formation, neurotransmission, long-term potentiation (LTP), and synaptic pruning [4–11]. Conversely, the immune system can also affect the neuronal activity under healthy and disease conditions, including AD. Increased neuroinflammation, reflected by microglial and astroglial activation, elevated pro-inflammatory cytokine release, and early complement activation – all of these have been often observed during AD [12, 13]. Recent pre-clinical and clinical research evidences suggest that neuroinflammation is not merely the response to pathological insults; it may even precede AD pathology and drive the pathogenesis of AD [14–16]. GWAS studies have also identified various immune genes as risk factors for AD, they are TREM2, MS4A4/MS4A6E, CLU (APOJ), EPHA1, MHC II, and CR1 [17–20]. Interestingly, some of these susceptible loci/gene(s) are also involved in the regulation of synaptic function. In a recent study, TREM2 overexpression has been shown to rescue neuronal and synaptic loss [21] and EPHA1 is thought to regulate synapse formation [22]. Therefore, more studies are needed to understand if these AD risk gene(s) could contribute to the disease process exclusively via cell-autonomous manner by impacting the microglial cell function or if they also directly impair synaptic function, independent of microglial involvement. A growing body of research now suggests that the CNS resident immune cells are directly involved in synaptic pruning during postnatal brain development and AD pathogenesis. In this review, we will discuss these findings and the development of therapeutic drugs that may enhance synaptic activity and neuronal function in the AD brain.
MICROGLIA-MEDIATED, COMPLEMENT-DEPENDENT SYNAPTIC PRUNING IN THE NORMAL AND DISEASE BRAIN
The complement system is a major component of innate immunity. The complement system is responsible for recognition and lysis of invading microorganisms, clearance of apoptotic cells, and recruitment of immune cells. De novo synthesis of complement factors in the brain has been confirmed in neurons, microglia, astrocytes, and oligodendrocytes (more information about the complement system in the brain is reviewed in [23]). Surprisingly, complements in the brain had an unexpected role in the elimination of inappropriate synapses, a process named ‘synaptic pruning’, which is important for the formation of mature neuronal circuits during development (see Table 1, Fig. 1A). The mRNA of complement component 1, subcomponent q (C1q), the initiating protein of the classical complement cascade, was found to be highly upregulated in purified retinal ganglion cells (RGCs), which were exposed to astrocytes [24]. In this study, localization of C1q was also observed in vivo, in the postnatal, immature synaptic inner plexiform layer at P5. C1q and C3 (downstream complement protein) knockout (KO) mice both had significant defects in eye-specific segregation and synapse elimination, investigated by neuroanatomical and electrophysiological techniques [24]. A subsequent study by Chu et al. also showed that C1q KO mice display remarkable seizures due to failure to prune excessive excitatory synapses in the layer V pyramidal neurons during the neocortex development [25]. Schafer et al. later demonstrated that during active synaptic remodeling, it is microglia in the postnatal dorsal lateral geniculate nucleus (dLGN) of the thalamus phagocytosing RGC presynaptic terminals in a complement receptor 3 (CR3)/C3-dependent pathway [26]. The synaptic remodeling was regulated in a developmental- and neuronal activity-dependent manner and appears to be very specific to microglia since microglia are the only cell type within dLGN to express CR3 [27]. Genetic manipulations by knocking out CR3 or C3 and pharmacologically inhibiting microglial activation using minocycline both led to defects in the synaptic circuitry [26]. The astrocyte-derived factor that upregulates C1q in RGCs was also discovered to be transforming growth factor-β (TGF-β) [28]. In summary, these studies suggest a pathway engaging astrocyte-derived TGF-β, neuronal complement, and microglial phagocytosis for proper synapse elimination during development. However, C1q and C3 KO mice still display some level of synaptic pruning, suggesting the existence of a complement-independent pathway for synapse elimination.
Table 1.
Selected studies on complement-mediated synaptic pruning during development and neurodegeneration
| Material used | Molecules Investigated | inhibitors used | Summary | References |
|---|---|---|---|---|
| AD autopsy brain tissue | C1q, C3b, C3c, C3d and C4 | None | Plaques contain the complement factors. | Eikelenboom, 1982 [30] |
| AD autopsy brain tissue | C1q, C3d, C4d | None | Classical complement pathway is activated in AD autopsy brain. | McGeer, 1989 [31] |
| AD autopsy brain tissue | C1q, C4d, C5b-9 | None | Aβ binds and activates classical complement pathway. | Rogers, 1992 [32] |
| AD autopsy brain tissue | C1q, C2, C3,C4, C5, C6, C7, C8 and C9 | None | Neurons express significantly higher levels of complement mRNAs in the temporal cortex and hippocampus of AD brains compared to control brains. | Shen, 1997 [70] |
| Neurofibrillary tangle isolated from AD brain, fixed AD entorhinal cortex tissue | C1q, C4d | None | Tau protein is an antibody-independent activator of the classical complement pathway. | Shen, 2001 [33] |
| Pre-clinical AD autopsy brain tissue | C1q | None | C1q is predominantly localized in frontal cortex and hippocampus in pre-clinical AD. | Fonseca, 2004 [34] |
| Tg2576; C1q−/− | C1q | Genetic deletion of C1q | C1q deficiency in the animal model of AD decreased glial cell activation and improved neuronal integrity. | Fonseca, 2004 [35] |
| C1q KO, C3 KO, DBA/2J glaucoma mice | C1q, C3 | Genetic deletion of C1q and C3 | During development, retinal ganglion cells upregulated C1q/C3 with exposure to immature astrocytes to eliminate weaker synapses for retinogeniculate refinement. | Stevens, 2007 [24] |
| Tg2576 mice 3xTg mice | C5aR | Small molecule inhibitor of C5aR-PMX205 | Oral delivery of a C5a receptor antagonist (PMX205) for 2–3 mo. resulted in substantial reduction of fibrillar Aβ deposits, hyperphosphorylated tau and activated glia, improvement in behavioral test in two mouse models of AD. | Fonseca, 2009 [36] |
| C1q KO mice | C1q | Genetic deletion of C1q | Enhanced synaptic connectivity and epilepsy in C1q knockout mice | Chu, 2010 [25] |
| C3 KO and CR3 KO mice | C3, CR3 | Genetic deletion of C3 and CR3 | Microglia is the cell type in the CNS that phagocytose weak synapses in C3/CR3-dependent manner during development. | Schafer, 2012 [26] |
| Retinal Tgfbr2 KO mice, C1qa KO mice | C1q, TGF(3 | Genetic deletion of Tgfbr and C1q, anti–TGF-β antibody | Astrocytes secrete TGFβ, which can upregulate neuronal C1q expression associated with synaptic pruning during development. | Bialas, 2013 [28] |
| hAPP (“J20”) transgenic mice | C1q, C3 | C1q antibody-ANX-M1 | C1q-mediated excessive pruning of synapses can drive the early pathogenesis of AD. | Hong, 2016 [38] |
Fig. 1.
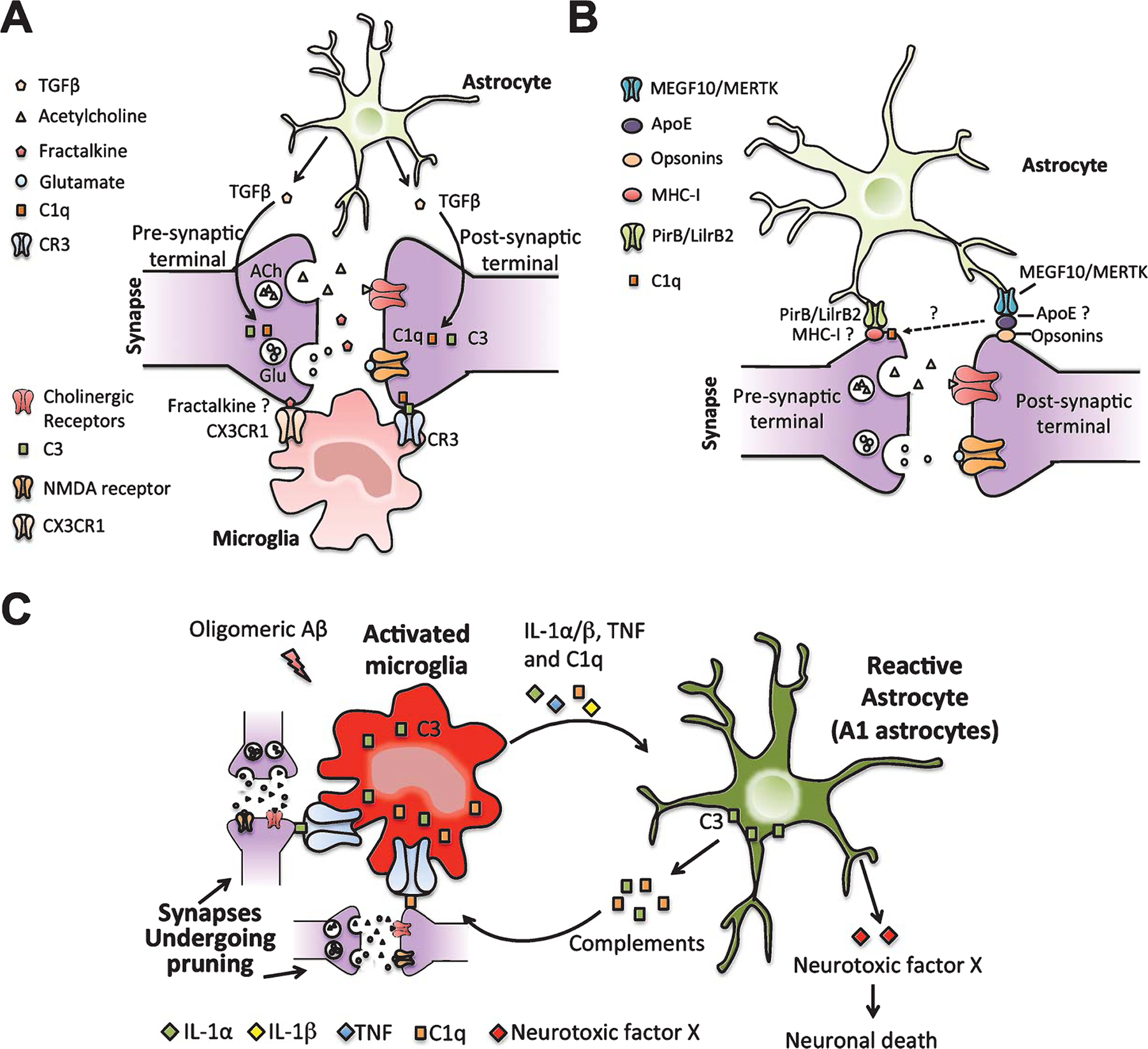
Neuron-microglia-astrocyte interaction during synaptic pruning in healthy and AD brain. A) During development and in adult, astrocytes secrete TGFβ, which upregulates neuronal C1q expression. Microglia then eliminates weak synapses in C1q/C3/CR3-dependent pathway. Fractalkine (CX3CL1) from neurons might also serve as a “find-me” signal for microglia to phagocytose weak synapses through CX3CL1/CX3CR1-dependent pathway. B) Astrocytes are also shown to phagocytose weak synapses during development through MEGF10/MERTK phagocytic receptors. ApoE mainly produced by astrocytes might help bridge opsonin-tagged synapses for phagocytosis by astrocytes. MHC-I is expressed in neurons and its receptor PirB/LilrB2 are expressed in astrocytes. MHC-I is also closely associated with C1q. These suggest astrocyte might prune synapses in an alternative pathway involving MHC-I, its receptor and C1q. C) In case of AD, increased C1q expression and synaptic loss are seen in AD animal models during early AD, even before plaques develop. Knocking out C1q rescued synaptic loss; suggesting complement-mediated over-pruning of synapses by microglia can contribute to the pathogenesis of AD. A recent study also showed that microglia activated by oligomeric Aβ can secrete several cytokines, IL-1α, TNF, and C1q. These cytokines in turn activate reactive astrocytes (A1 astrocytes). A1 astrocytes upregulate several complement genes that are involved in synaptic pruning. A1 astrocytes can also secrete an unknown neurotoxic factor X, which kills neurons. All these can contribute to the synaptic and neuronal loss in AD. It is also reported that IL-1β can upregulate MHC-I, reduce the number of synapses on the cell surface and prevent synapse stabilization.
Interestingly, complements are also involved in the pathogenesis of AD [29] (see Table 1, Fig. 1C). The amyloid-β (Aβ) plaques were shown to contain complement factors [30], and the classical complement pathway was observed to be activated in the AD patient brain [31]. In a separate study, both Aβ and tau were shown to activate the complement pathway [32, 33], especially C1q, which predominantly localized in the frontal cortex and hippocampus in pre-clinical AD [34]. Deletion of C1q in the Tg2576 and APP/PS1 models of AD suggested a detrimental role of complement activation in the AD pathogenesis, because both Tg2576;C1q−/− and APP/PS1;C1q−/− mice showed less astrocytes and microglia surrounding the plaques and increased synaptophysin at 12 and 16 months [35]. In contrast, at the pre-plaque stage (3–6 months of age), no significant changes were seen in any of the neuronal or glial markers tested [35]. In a separate study, inhibition of pro-inflammatory complement factor C5a receptor (C5aR) using a small molecule inhibitor, PMX205, reduced Aβ deposits, decreased glial activation in the hippocampus and cortex, and improved the behavioral test performance in Tg2576 mice at 12 to 15 months of age (when accumulation of Aβ plaque is rapidly developing). A significant decrease in the levels of hyperphosphorylated tau was also observed in the 3xTg mouse model of AD with a deficiency of C5aR [36]. However, how exactly the complement system contributes to the AD pathogenesis was still not clear at that time.
Synaptic loss is one of the early markers for AD and is the strongest correlating factor for the extent of dementia [37]. Considering the role of complement and microglia in the synaptic pruning during development, Hong et al. hypothesized that microglia-mediated, complement-dependent synaptic pruning might be associated to synaptic loss during in early AD [38]. Hong et al. observed increased expression of C1q in the hippocampus and frontal cortex of familial AD-mutant human Aβ protein precursor (hAPP) (“J20”) transgenic mice [39] at 1 month of age, preceding the synaptic loss [38]. An increase of C1q was also found in the hippocampus of APP/PS1 AD transgenic mice [38]. The increased C1q expression was demonstrated in microglia, suggesting that microglia as a major source of C1q in these pre-plaque brains of AD transgenic mice. Intraperitoneal infusion of compound E, a γ-secretase inhibitor, remarkably reduced soluble Aβ and C1q levels in J20 mice [38]. The significant synaptic loss observed in J20 mice was at 3 to 4 months of age, which are several months prior to the Aβ plaque deposition. Notably, knocking out C1q/C3 or neutralizing the C1q with a C1q-specific antibody ANX-M1 in non-transgenic mice injected with oligomeric Aβ blocked excessive pruning of synapses by microglia and prevented synaptic loss [9, 38, 40]. This is the first study to show the detrimental role of microglia in the over-pruning of synapses that can lead to synaptic loss in early AD, even before the plaques develop. Importantly, ANX-005, a human form of the antibody that can block C1q, is currently under Phase 1b clinical trial to be tested for safety in humans. However, the receptors of C1q and C3 on phagocytosed synapses are yet to be discovered, and the precise mechanism still needs to be defined.
ASTROCYTE-MEDIATED, MEGF10/MERTK- AND APOE-DEPENDENT SYNAPTIC PRUNING IN THE NORMAL AND DISEASED BRAIN
Astrocytes comprise one-third of the total cells in the brain and are also involved in synaptic pruning (see Table 2, Fig. 1A, B). In the developing visual system, astrocytes secrete TGF-β to increase neuronal C1q in order to tag weak synapses for elimination. However, it is also reported to directly phagocytose synapses during retinogeniculate refinement involving two phagocytic receptors, Multiple EGF-like–domains 10 (MEGF10) and MER receptor tyrosine kinase (MERTK) [41]. This process was also strongly related to neuronal activity and the phagocytic activity of astrocytes even surpassed that of microglia during several different stages of dLGN development. MEGF10 and MERTK-dependent pruning of both excitatory and inhibitory synapses by astrocytes continued in the adult CNS [41].
Table 2.
Selected studies on astrocyte-mediated synaptic pruning during development and neurodegeneration
| Material used | Molecules investigated | Inhibitors used | Summary | References |
|---|---|---|---|---|
| Megf10 KO and Mertk KO mice | MEGF10 and MERTK | Genetic deletion of Megf10 and Mertk | Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. | Chung, 2013 [41] |
| APOE KI mice | APOE2, APOE3, and APOE4 | Genetic knock-in of human APOE | APOE can facilitate or inhibit the astrocyte-mediated phagocytosis with the presence of opsonins. | Chung, 2016 [42] |
| Il1a KO, Tnf KO, C1qa KO mice, Huntington’s and Alzheimer’s diseases, and amyotrophic lateral sclerosis patient brain tissue | IL-1α, TNF, C1q, C3 | Genetic deletion of Il1a, Tnf, C1qa | Reactive microglia can secrete cytokines that induce reactive astrocytes, which in turn upregulate complement-mediated synaptic loss and also secrete neurotoxic factors to kill a subset of neurons and oligodendrocytes in multiple neurodegenerative conditions. | Liddelow, 2017 [47] |
A recent study by Chung et al. has shed light on the role of human apolipoprotein E (ApoE) in the astrocyte-mediated synapse elimination [42]. ApoE is a lipid transport protein mainly produced by astrocytes in the CNS. Humans have three common isoforms: E2, E3, and E4. APOE genotype is the strongest genetic risk factor for late-onset AD, with two copies of the E4 allele leading to an increased risk by 12-fold [43], with the E2 allele being associated with 2-fold decreased risk for AD [44], and the E3 allele being the most common form and leading to intermediate risk of AD [45]. Chung et al. obtained astrocyte-conditioned medium (ACM) from APOE2, APOE3, and APOE4 homozygous knock-in (KI) astrocytes, in which mouse Apoe gene is replaced with human APOE. Apoe knockout astrocytes were then treated with tdTomato-positive synaptosomes and different ApoE ACM. Incubation of ApoE2 ACM strongly enhanced engulfment of synaptosomes by Apoe knockout astrocytes compared to APOE3 and APOE4 ACMs, with the APOE4 ACM showing the minimum engulfment [42]. In contrast, lipidated recombinant APOE2, 3, and 4 particles did not induce differential effects on astrocyte-mediated phagocytosis, whereas adding Protein S simultaneously did. Proteins like Protein S and GAS6 are opsonins, required for phagocytic receptors like MERTK [46], suggesting that APOE can facilitate or inhibit the astrocyte-mediated phagocytosis with the presence of opsonins. In vivo experiments further demonstrated that astrocytes in APOE2 KI animals showed significantly enhanced phagocytosis of labeled RGC presynaptic terminals compared with APOE3 KI animals, whereas those from APOE4 KI animals showed decreased phagocytic capacity. The amount of C1q accumulation in the hippocampus was also APOE allele-dependent in the 9- and 18-month-old APOE KI animals [42]. Although this is the first study to link APOE allele risk to synaptic pruning dysfunction during AD, there are still some important questions that need to be answered: 1) it is not clear which cell type in the hippocampus is the major source of this increased amount of C1q, since C1q is expressed in neurons, microglial cells, and astrocytes; 2) astrocyte-dependent synaptic pruning was previously reported to be complement-independent [41]. Therefore, whether the increased amount of C1q is a result from defective synaptic pruning by astrocytes needs to be verified. Also, whether the C1q elevation in the hippocampus affects microglia-mediated, complement-dependent synaptic pruning remains elusive; 3) how exactly ApoE affects phagocytic capacity of astrocytes is not clear. The authors hypothesized ApoE may help bridge the binding of synapses to phagocytic receptors on the astrocyte surface; 4) APOE allele’s effect on the phagocytic capacity of microglia should be investigated, since microglia can express ApoE receptors as well; 5) it will be also interesting to see how ApoE, in conjunction with complements, contributes to the early stage of AD using APP or tau transgenic animal models.
A most recent study by Ben Barres’s group has shown that the secreted cytokines from reactive microglia work together to activate reactive astrocytes (A1) in vitro and in vivo (see Fig. 1C). The secreted immune mediators are interleukin 1α (IL-1α), TNF, and C1q [47]. A1 astrocytes can upregulate many classical complement cascade genes that are involved in the synaptic pruning. The A1 astrocytes formed fewer and weaker synapses and engulfed 50–70% fewer synaptosomes than the control astrocytes. A1 astrocytes also displayed decreased phagocytic capacity for synaptosomes and myelin [47]. A1-astrocyte-conditioned medium also killed RGCs, cortical neurons, embryonic spinal motor neurons, and mature, differentiated oligodendrocytes and human dopaminergic neurons [47], suggesting that an unknown neurotoxic factor(s) secreted from A1 astrocytes is/are cytotoxic in the brain. Complement component 3 (C3)-positive, A1 astrocytes were present in the post-mortem tissue from patients with AD, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and multiple sclerosis [47]. Especially in human AD, nearly 60% of astrocytes in the prefrontal cortex were positive for C3, suggesting that A1 astrocytes might be playing an important role in the disease initiation and progression of many neurodegenerative diseases, including AD. Taken together, it suggests that during AD, reactive microglia can secrete cytokines that induce reactive astrocytes, which in turn upregulate complement-mediated synaptic loss and also secrete neurotoxic factors to kill a subset of neurons and oligodentrocytes [47]. This is another important study to support glial cell-mediated over-pruning of synapses can alter synaptic function, cause imbalance in the neurotransmitter system, and may contribute to the AD pathogenesis.
MHC-I-DEPENDENT SYNAPTIC PRUNING IN THE NORMAL AND DISEASED BRAIN
Another important immune molecule, major histo-compatibility class I (MHC-I) is involved in synaptic pruning as well (see Table 3, Fig. 1B). It was also found to be expressed in neurons in the visual cortex, colocalized with postsynaptic proteins, and regulated by neuronal activity [48]. Knocking out MHC-I or its receptor PirB (paired immunoglobulin-like receptor B) in animals led to defects in eye-specific segregation and ocular dominance plasticity [49]. Especially, MHC-I molecules H2-Kb and H2-Db were closely associated with C1q at the excitatory and inhibitory LGN synapses, suggesting its role in the retinogeniculate refinement and synapse elimination [50]. Later, MHC-I molecule H2-Db was shown to be necessary and sufficient for synapse elimination in the retinogeniculate system. Since expressing just H2-Db in H2-KbDb(−/−) double knockout mice rescued defects in synapse elimination, eye-specific segregation, and long-term depression (LTD) [51]. However, the precise mechanism and the cell type mediating the MHC-I-dependent elimination of synapse is not yet defined. Since PirB and its human homolog, LilrB2, are reported to be expressed in neurons and astrocytes, but not in microglia [52], it is intriguing to see whether astrocytes are involved in the MHC-I-dependent synaptic pruning. MHC-I is highly expressed by activated microglia as well, suggesting a possible role of microglial MHC-I in the synaptic pruning.
Table 3.
Selected studies on MHC-I-dependent synaptic pruning during development and neurodegeneration
| Material used | Molecules investigated | Inhibitors used | Summary | References |
|---|---|---|---|---|
| PirB deficient mice-PirBTM mice | Mouse MHC-I receptor PirB | Genetic deletion of Pirb | Knocking out PirB in animals led to defects in eye-specific segregation and ocular dominance plasticity. | Syken, 2006 [49] |
| β2m/TAP1 double KO | MHC-I | Genetic deletion of cell surface MHC-I | MHC-I is expressed in neurons in the visual cortex, co-localized with postsynaptic proteins and regulated by neuronal activity. | Goddard, 2007 [48] |
| KbDb KO mice | MHC-I | Genetic deletion of MHC-I components | H2-Kb and H2-Db can signal through neuronal MHCI receptors to enable activity-dependent remodeling of brain circuits during developmental critical periods. | Datwani, 2009 [50] |
| PirB−/−; APP/PS1 mice | PirB, LilrB2 | Genetic deletion of PirB | LilrB2 and PirB are the receptors for Aβ oligomers and knocking out PirB in the APP/PS1 AD transgenic mice rescued not only synaptic and cognitive alterations induced in adult mice by Aβ, but also loss of plasticity during early development in visual cortex of APP/PS1 mice. | Kim, 2013 [53] |
| KbDb KO mice | MHC-I molecule H2-Kb and H2-Db | Genetic deletion of MHC-I components | Expressing just H2-Db in H2-KbDb(−/−) double knockout mice rescued defects in synapse elimination, eye-specific segregation and long-term depression (LTD). | Lee, 2014 [51] |
LilrB2/PirB were shown to be involved in Aβ-induced synaptic loss, decreased LTP, and increased LTD [53]. LilrB2 and PirB are the receptors for Aβ oligomers, and the first two extracellular immunoglobulin (Ig) domains of PirB and LilrB2 are responsible for the interaction with Aβ and neuronal cofilin signaling [53]. Knocking out PirB in the APP/PS1 AD transgenic mice rescued not only synaptic and cognitive alterations induced in adult mice by Aβ, but also loss of plasticity during early development in the visual cortex of APP/PS1 mice [53]. The authors are currently testing to see whether blocking PirB, in early stages of AD before plaques appear, can ameliorate the cognitive decline associated with synaptic loss [54].
MICROGLIA-MEDIATED, FRACTALKINE-DEPENDENT SYNAPTIC PRUNING IN THE NORMAL AND DISEASED BRAIN
CX3CL1 or Fractalkine is a unique chemokine primarily expressed in neurons and moderately by astrocytes, although at lower levels [55]. It exists in two different forms, membrane-bound form proposed as an anchoring molecule and secreted form as a chemokine to attract microglia, which exclusively expresses its receptor CX3CR1 in the CNS. Fractalkine signaling plays essential roles in mediating neuron-microglia crosstalk in the developing and mature brain, and it has been implicated in various aspects of brain physiology, including synaptic pruning (see Table 4, Fig. 1A). Fractalkine signaling is extensively reviewed in [56].
Table 4.
Selected studies on fracktalkine-dependent synaptic pruning during development and neurodegeneration
| Material used | Molecules investigated | Inhibitors used | Summary | References |
|---|---|---|---|---|
| Cx3cr1 KO mice | CX3CR1 | Genetic deletion of Cx3cr1 | Cx3cr1 KO mice had decrease in the microglial number, increase in the excitatory postsynaptic density and dendritic spine density on CA1 pyramidal neurons, accompanied by significantly increased LTD. | Paolicelli, 2011 [57] |
| Cx3cr1 KO mice | CX3CR1 | Genetic deletion of Cx3cr1 | Deficit in fractalkine signaling can cause decrease in the microglial number, defects in synaptic pruning and ultimately neurodevelopmental and neuropsychiatric disorders in mice. | Zhan, 2014 [58] |
| hAPP; Cx3cr1 KO mice | CX3CR1 | Genetic deletion of Cx3cr1 | Deficiency of fractalkine signaling worsened plaque-independent cognitive retention in hAPP-J20 mice. | Cho, 2011 [60] |
| 5xTg; Cx3cr1 KO mice | CX3CR1 | Genetic deletion of Cx3cr1 | Fractalkine deletion can prevent neuronal loss in 5xTg mice. | Fuhrmann, 2010 [62] |
| hTau; Cx3cr1 KO mice | CX3CR1 | Genetic deletion of Cx3cr1 | Deficiency of fractalkine signaling aggravates microglia-mediated hyperphosphorylation of tau and inflammatory responses in a mouse model of systematic inflammation and hTau. | Bhaskar, 2010 [14] |
| APP/PS1; Cx3cr1 KO mice | CX3CR1 | Genetic deletion of Cx3cr1 | CX3CR1 deficiency is anti-amyloidogenic in APP/PS1 transgenic mouse model of AD. | Lee, 2010 [61] |
During the first postnatal weeks, Cx3cr1 KO mice had transient decrease in the microglial number, increase in the excitatory postsynaptic density and dendritic spine density on CA1 pyramidal neurons, accompanied by significantly increased LTD [57]. This suggests that deficient fractalkine signaling in microglia can lead to defects in synaptic pruning. Another study also confirmed that a deficit in fractalkine signaling can cause decrease in the microglial number, defects in synaptic pruning, and ultimately contribute to neurodevelopmental and neuropsychiatric disorders in mice [58]. However, it remains elusive whether fractalkine serves as a chemokine to attract microglia to the synapses for phagocytosis or affects microglial proliferation or perhaps directly contributes to the pruning process by facilitating synapse recognition for pruning. Fractalkine has been shown to serve as a “find-me” signal released by neurons, which undergoes ethanol-induced apoptosis [59], suggesting that fractalkine might also serve as a “find-me” signal for synapses for phagocytosis by microglia.
Fractalkine signaling has been shown be deficient in AD brains [60], and the role of fractalkine signaling during AD pathogenesis is controversial. Deficiency of fractalkine signaling was found to aggravate microglia-mediated hyperphosphorylation of tau and inflammatory responses in a mouse model of systematic inflammation in hTau [14] mice and worsened plaque-independent cognitive retention in hAPP-J20 mice [60]. On the contrary, CX3CR1 deficiency is anti-amyloidogenic in an APP/PS1 transgenic mouse model of AD [61] and fractalkine deletion can prevent neuronal loss in 5xTg mice [62]. The contradicting roles of fractalkine signaling against Aβ versus tau pathology might be due to the requirement of pro-inflammatory/pro-phagocytic role for microglia against Aβ plaques versus antiinflammatory role of microglia against tau pathology. This is a key consideration to prevent collateral damage caused by reactive microglia when it is performing the anti-amyloidogenic function. Additional studies suggested that IL-1β is the major cytokine that links both exacerbation of tau pathology (in tau models [14, 63]) and attenuation of Aβ pathology (in APP/PS1 mouse model of AD [61]). At present, the role of fractalkine-CX3CR1 signaling axis in mediating synaptic loss during AD is still rudimentary.
Since IL-1β was one of the potential links identified in the above described studies and may regulate synaptic structure and function, in a recent study, the direct incubation of cultured neurons with IL-1β upregulated MHC-I and reduced the number of synapses on the cell surface [64]. Contrariwise, low-ering MHC-I rescued synapses from the detrimental effects of IL-1β. It is also reported that IL-1β can prevent synapse stabilization in zebrafish larvae and deficiency of IL-1β or depletion of microglia prevented the synaptic loss [64]. Additionally, IL-1β has been implicated in loss of synapses in obese mice [65]. Collectively, these data suggest a potential role of IL-1β in synaptic pruning during development and various neurological disease conditions, including AD.
SUMMARY AND CONCLUSIONS
Emerging studies now suggest that during CNS development and neurodegenerative processes, microglia, astrocyte, and neurons interact actively for synaptic pruning and regulation of neurotransmission through several different pathways. This involves immune molecules like complements, chemokines, MHC complexes and cytokines. Pharmacological inhibition using small molecule inhibitors, neutralization by antibodies, or genetic manipulation of immune molecules rescued synaptic loss and ameliorated behavioral deficits in animal models of AD. Therefore, complements/MHC-I/cytokine/chemokine systems could serve as novel, promising therapeutic targets in ameliorating AD-related synaptic dysfunction, which is the best measure for memory loss in AD. Specifically, antibody therapies or small molecule pharmaceuticals inhibiting human complements [66], IL-1α and TNF are already FDA-approved and used for other diseases, which may help accelerate testing of cytokine/complement-targeted therapeutics against AD.
The possibility of complements as biomarkers has also been revealed. The autosomal dominant form of Alzheimer’s disease (ADAD) is far less predominant than late onset Alzheimer’s disease, however, increasing evidence suggest ADAD can be a good model for studying late-onset AD. Muenchhof et al. investigated plasma protein changes at the asymptomatic and symptomatic stages of ADAD [67]. Levels of complement components C3, C5, and C6 differed significantly between non-carriers and asymptomatic mutation carriers. Similarly, the proteins that associated with the cognition or neuroimaging markers were also turned out to mainly have functions in the complement system (complement factors B and I, complement components C2, C4-A, C6, and C8 β chain, complement C1R subcomponent, C4b-binding protein β chain, and C1 inhibitor) and lipid metabolism (ApoA1, ApoM, and ApoE) [67]. A recent study by Hakobyan et al. measured five complement proteins and four activation products in the plasma samples of donors with mild cognitive impairment, AD, and controls. Clusterin, a complement analyte, was significantly elevated in AD plasma compared to control [68]. The levels of three analytes (clusterin, complement factor I, terminal complement complex) were significantly different between mild cognitive impairment patients who converted to dementia one year later compared to those who did not. Alteration in these three analytes was highly predictive of the conversion [68]. A subsequent study by the same group looked at five different complement markers in the plasma of 93 AD patients and found that plasma clusterin level showed an association with overall AD polygenic risk score, while clusterin, C1 inhibitor, and C-reactive protein levels each displayed some association with the inflammatory-specific AD polygenic risk score [69]. Taken together, these studies suggest that the plasma complement factors and associated proteins can serve as biomarkers in the disease prediction for AD.
Although the aberrant synaptic pruning by multiple cellular pathways seem to be a promising target for AD therapy and biomarker development, there are still certain important unanswered questions: 1) immune molecules (C1q, MHC-I, PirB, APOE receptor, fractalkine) involved in synaptic pruning are not specific to certain type of cells in the CNS, but rather simultaneously expressed in neurons, microglia, astrocytes. Therefore, spatial (cell-specific) and temporal (timely) targeting of these molecules without affecting other cell types remains to be investigated; 2) how different synaptic pruning pathways (microglia-mediated, C1q-dependent pathway versus Astrocyte-mediated, MEGF10/MERTK- and APOE-dependent pathway versus MHC-I-dependent pathway versus microglia-mediated, fractalkine-dependent pathway) synergistically work during development and AD remains to be investigated; 3) finally, the crosstalk among different cell types in the CNS (microglia, astrocytes, neurons) during development and AD need to be investigated in detail.
Acknowledgments:
We thank NIH/NINDS for R21NS077089 and R01NS083704 funding to KB.
Footnotes
DISCLOSURE STATEMENT
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1123r2).
REFERENCES
- [1].Kioussis D, Pachnis V (2009) Immune and nervous systems: More than just a superficial similarity? Immunity 31, 705–710. [DOI] [PubMed] [Google Scholar]
- [2].Camacho-Arroyo I, López-Griego L, Morales-Montor J (2009) The role of cytokines in the regulation of neurotransmission. Neuroimmunomodulation 16, 1–12. [DOI] [PubMed] [Google Scholar]
- [3].Selmeczy Z, Vizi ES, Csóka B, Pacher P, Haskó G (2008) Role of nonsynaptic communication in regulating the immune response. Neurochem Int 52, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Miller AH, Haroon E, Raison CL, Felger JC (2013) Cytokine targets in the brain: Impact on neurotransmitters and neurocircuits. Depress Anxiety 30, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yirmiya R, Goshen I (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25, 181–213. [DOI] [PubMed] [Google Scholar]
- [6].Baune B, Camara ML, Eyre H, Jawahar C, Anscomb H, Körner H (2012) Tumour necrosis factor - alpha mediated mechanisms of cognitive dysfunction. Transl Neurosci 3, 263–277. [Google Scholar]
- [7].Stephan AH, Barres BA, Stevens B (2012) The complement system: An unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 35, 369–389. [DOI] [PubMed] [Google Scholar]
- [8].Hong S, Dissing-Olesen L, Stevens B (2016) New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 36, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hong S, Stevens B (2016) Microglia: Phagocytosing to clear, sculpt, and eliminate. Dev Cell 38, 126–128. [DOI] [PubMed] [Google Scholar]
- [10].Chung WS, Welsh CA, Barres BA, Stevens B (2015) Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci 18, 1539–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zuchero JB, Barres BA (2015) Glia in mammalian development and disease. Development 142, 3805–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Heppner FL, Ransohoff RM, Becher B (2015) Immune attack: The role of inflammation in Alzheimer disease. Nat Rev Neurosci 16, 358–372. [DOI] [PubMed] [Google Scholar]
- [13].Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT (2010) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. [DOI] [PubMed] [Google Scholar]
- [16].Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K (2015) Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138, 1738–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bertram L, Tanzi RE (2009) Genome-wide association studies in Alzheimer’s disease. Hum Mol Genet 18, R137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Morón FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fiévet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossú P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, European Alzheimer’s Disease Initiative (EADI), Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH Jr, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nöthen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Rüther E, Schürmann B, Heun R, Kölsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Gallacher J, Hüll M, Rujescu D, Giegling I, Goate AM, Kauwe JSK, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jöckel K-H, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC; Alzheimer’s Disease Neuroimaging Initiative, van Duijn CM, Breteler MM, Ikram MA, DeStefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S; CHARGE consortium, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alpérovitch A, Lathrop M; EADI1 consortium, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Björnsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossú P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fiévet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O; European Alzheimer’s Disease Initiative Investigators, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossú P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanché H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alpérovitch A, Lathrop M, Amouyel P (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41, 1094–1099. [DOI] [PubMed] [Google Scholar]
- [21].Jiang T, Zhang YD, Chen Q, Gao Q, Zhu XC, Zhou JS, Shi JQ, Lu H, Tan L, Yu JT (2016) TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 105, 196–206. [DOI] [PubMed] [Google Scholar]
- [22].Lai KO, Ip NY (2009) Synapse development and plasticity: Roles of ephrin/Eph receptor signaling. Curr Opin Neurobiol 19, 275–283. [DOI] [PubMed] [Google Scholar]
- [23].Veerhuis R, Nielsen HM, Tenner AJ (2011) Complement in the brain. Mol Immunol 48, 1592–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Stevens B, Allen NJ, Vazquez LE, Howell GR, Christophe-rson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- [25].Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA (2010) Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A 107, 7975–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Akiyama H, McGeer PL (1990) Brain microglia constitutively express beta-2 integrins. J Neuroimmunol 30, 81–93. [DOI] [PubMed] [Google Scholar]
- [28].Bialas AR, Stevens B (2013) TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 16, 1773–1782. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [29].McGeer PL, Rogers J, McGeer EG (2016) Inflammation, antiinflammatory agents, and Alzheimer’s disease: The last 22 years. J Alzheimers Dis 54, 853–857. [DOI] [PubMed] [Google Scholar]
- [30].Eikelenboom P, Stam FC (1982) Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol 57, 239–242. [DOI] [PubMed] [Google Scholar]
- [31].McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 107, 341–346. [DOI] [PubMed] [Google Scholar]
- [32].Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P (1992) Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 89, 10016–10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shen Y, Lue L, Yang L, Roher A, Kuo Y, Strohmeyer R, Goux WJ, Lee V, Johnson GV, Webster SD, Cooper NR, Bradt B, Rogers J (2001) Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci Lett 305, 165–168. [DOI] [PubMed] [Google Scholar]
- [34].Fonseca MI, Kawas CH, Troncoso JC, Tenner AJ (2004) Neuronal localization of C1q in preclinical Alzheimer’s disease. Neurobiol Dis 15, 40–46. [DOI] [PubMed] [Google Scholar]
- [35].Fonseca MI, Zhou J, Botto M, Tenner AJ (2004) Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J Neurosci 24, 6457–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, Taylor SM, Woodruff TM, Tenner AJ (2009) Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol 183, 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Spires-Jones TL, Hyman BT (2014) The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82, 756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L (2000) High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci 20, 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Whalley K (2016) Neurodegenerative disease: Complement mediates pathological pruning. Nat Rev Neurosci 17, 336. [DOI] [PubMed] [Google Scholar]
- [41].Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, Smith SJ, Barres BA (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chung WS, Verghese PB, Chakraborty C, Joung J, Hyman BT, Ulrich JD, Holtzman DM, Barres BA (2016) Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci U S A 113, 10186–10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. [DOI] [PubMed] [Google Scholar]
- [44].Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1994) Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 7, 180–184. [DOI] [PubMed] [Google Scholar]
- [45].Liu CC, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat Rev Neurol 9, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lew ED, Oh J, Burrola PG, Lax I, Zagórska A, Través PG, Schlessinger J, Lemke G (2014) Differential TAM receptor ligand–phospholipid interactions delimit differential TAM bioactivities. Elife 3, e03385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Goddard CA, Butts DA, Shatz CJ (2007) Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci U S A 104, 6828–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Syken J, Grandpre T, Kanold PO, Shatz CJ (2006) PirB restricts ocular-dominance plasticity in visual cortex. Science 313, 1795–1800. [DOI] [PubMed] [Google Scholar]
- [50].Datwani A, McConnell MJ, Kanold PO, Micheva KD, Busse B, Shamloo M, Smith SJ, Shatz CJ (2009) Classical MHCI molecules regulate retinogeniculate refinement and limit ocular dominance plasticity. Neuron 64, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lee H, Brott BK, Kirkby LA, Adelson JD, Cheng S, Feller MB, Datwani A, Shatz CJ (2014) Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature 509, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yue J, Li W, Liang C, Chen B, Chen X, Wang L, Zang Z, Yu S, Liu S, Li S, Yang H (2016) Activation of LILRB2 signal pathway intemporal lobe epilepsy patients and in a pilocarpine induced epilepsy model. Exp Neurol 285, 51–60. [DOI] [PubMed] [Google Scholar]
- [53].Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, Hyman BT, Shatz CJ (2013) Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341, 1399–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Alzheimer Research Forum (2015) Microglia rely on mixed messages to select synapses for destruction. Available from: http://www.alzforum.org/news/conference-coverage/microglia-rely-mixed-messages-select-synapses-destruction
- [55].Hulshof S, van Haastert ES, Kuipers HF, van den Elsen PJ, De Groot CJ, van der Valk P, Ravid R, Biber K (2003) CX3CL1 and CX3CR1 expression in human brain tissue: Noninflammatory control versus multiple sclerosis. J Neuropathol Exp Neurol 62, 899–907. [DOI] [PubMed] [Google Scholar]
- [56].Paolicelli RC, Bisht K, Tremblay MÉ (2014) Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front Cell Neurosci 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. [DOI] [PubMed] [Google Scholar]
- [58].Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT (2014) Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17, 400–406. [DOI] [PubMed] [Google Scholar]
- [59].Sokolowski JD, Chabanon-Hicks CN, Han CZ, Heffron DS, Mandell JW (2014) Fractalkine is a “find-me” signal released by neurons undergoing ethanol-induced apoptosis. Front Cell Neurosci 8, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, Ransohoff RM, Gan L (2011) CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem 286, 32713–32722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT (2010) CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol 177, 2549–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J (2010) Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci 13, 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Maphis N, Xu G, Kokiko-Cochran ON, Cardona AE, Ransohoff RM, Lamb BT, Bhaskar K (2015) Loss of tau rescues inflammation-mediated neurodegeneration. Front Neurosci 9, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Alzheimer Research Forum (2015) Microglia control synapse number in multiple disease states. Available from: http://www.alzforum.org/news/conference-coverage/microglia-control-synapse-number-multiple-disease-states
- [65].Erion JR, Wosiski-Kuhn M, Dey A, Hao S, Davis CL, Pollock NK, Stranahan AM (2014) Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J Neurosci 34, 2618–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Morgan BP, Harris CL (2015) Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov 14, 857–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Muenchhoff J, Poljak A, Thalamuthu A, Gupta VB, Chatterjee P, Raftery M, Masters CL, Morris JC, Bateman RJ, Fagan AM, Martins RN, Sachdev PS (2016) Changes in the plasma proteome at asymptomatic and symptomatic stages of autosomal dominant Alzheimer’s disease. Sci Rep 6, 29078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Hakobyan S, Harding K, Aiyaz M, Hye A, Dobson R, Baird A, Liu B, Harris CL, Lovestone S, Morgan BP (2016) Complement biomarkers as predictors of disease progression in Alzheimer’s disease. J Alzheimers Dis 54, 707–716. [DOI] [PubMed] [Google Scholar]
- [69].Morgan AR, Touchard S, O’Hagan C, Sims R, Majounie E, Escott-Price V, Jones L, Williams J, Morgan BP (2017) The correlation between inflammatory biomarkers and polygenic risk score in Alzheimer’s disease. J Alzheimers Dis 56, 25–36. [DOI] [PubMed] [Google Scholar]
- [70].Shen Y, Li R, McGeer EG, McGeer PL (1997) Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res 769, 391–395. [DOI] [PubMed] [Google Scholar]