

Abstract
The prognosis of patients with microvascular lesions remains poor because vascular remodeling eventually obliterates the lumen. Here we have focused our efforts on vessel dysfunction in two different organs, the lung and brain. Despite tremendous progress in understanding the importance of blood vessel integrity, gaps remain in our knowledge of the underlying molecular factors contributing to vessel injury, including microvascular lesions. Most of the ongoing research on these lesions have focused on oxidative stress but have not found major molecular targets for the discovery of new treatment or early diagnosis. Herein, we have focused on elucidating the molecular mechanism(s) based on two new emerging molecules NRF1 and ID3, and how they may contribute to microvascular lesions in the lung and brain. Redox sensitive transcriptional activation of target genes depends on not only NRF1, but the recruitment of co-activators such as ID3 to the target gene promoter. Our review highlights the fact that targeting NRF1 and ID3 could be a promising therapeutic approach as they are major players in influencing cell growth, cell repair, senescence, and apoptotic cell death which contribute to vascular lesions. Knowledge about the molecular biology of these processes will be relevant for future therapeutic approaches to not only PAH but cerebral angiopathy and other vascular disorders. Therapies targeting transcription regulators NRF1 or ID3 have the potential for vascular disease-modification because they will address the root causes such as genomic instability and epigenetic changes in vascular lesions. We hope that our findings will serve as a stimulus for further research towards an effective treatment of microvascular lesions.
INTRODUCTION
Microvascular lesions are characterized by an imbalance in cell death and growth resulting in vessel remodeling and luminal narrowing (1). These types of lesions occur in small vessels common to the brain, kidney, and lung. Consequences of these lesions include ischemia, hypertension, myocardial infarction, and stroke (2). Although these lesions are found in several organs in humans, we have focused our efforts on hypertension-related microvascular lesions found in both the lung and brain. These types of small vessel lesions show similar morphological changes, including early loss of vascular cells, luminal narrowing, vessel wall thickening, and inflammation. In the lung, proliferative and obliterative vascular disease from these lesions contribute to pulmonary arterial hypertension (PAH) by increasing mean pulmonary arterial pressure leading to heart failure. Brain vascular lesions are found in cases of cerebral angiopathy particularly in patients with cerebral amyloid angiopathy (CAA) resulting in microbleeds and microinfarcts. Brain and pulmonary vascular lesions are lethal with high morbidity and no available treatments except surgical interventions. Mechanisms of these diseases are not fully understood, and this demands new approaches to understand the molecular basis for these lesions for the discovery of new molecular targets.
Recently, using ChIP-seq and RNA-seq coupled with dynamic machine learning approaches, we have uncovered two transcription regulatory proteins: nuclear respiratory factor-1 (NRF1) and inhibitor of DNA binding and differentiation protein 3 (ID3) that affect vascular cell dysfunction (3). NRF1 is a pioneer transcription factor that increases chromatin opening activity by binding to the DNA in “closed” heterochromatin triggering relaxation of the chromatin landscape to provide accessibility to non-pioneer transcription regulators, including ID3 (4). These transcription regulatory proteins bind to the promoters and enhancers of several thousand genes across the genomic landscape. How these redox-sensitive transcription regulatory molecules such as NRF1 and ID3 contribute to vessel dysfunction remains to be understood. NRF1 and ID3 are activated in response to environmental stress as well as oxidative stress and they regulate hundreds of genes involved in cell injury, cell growth, cell repair, apoptosis, and vessel remodeling. Herein, we have briefly discussed the origin and development of microvascular lesions in both the lung and brain, environmental and molecular risk factors in this disease; and the emerging role of NRF1 and ID3 in microvascular lesion development.
MICROVASCULAR LESIONS
The microcirculation consisting of arterioles, capillaries, and venules are small vessels that deliver oxygenated blood directly to organs and remove carbon dioxide from the tissue. Arterioles control the local blood flow into the organ via vasoconstriction. Capillaries form a network of tiny branching vessels between the arterioles and venules essential for the exchange of oxygen and carbon dioxide in the tissue surrounding the capillary. Brain and lung tissues consist of numerous capillaries because they are metabolically active or specialize in the absorption of compounds in the blood. Microvascular lesions are found in the brain and lung because they are highly vascularized organs. In this section, lung microvascular lesions will be discussed in the context of PAH, whereas the brain vascular lesion will be related to cerebral angiopathy. Both lesions show similar morphological changes, including early loss of vascular cells, luminal narrowing, vessel wall thickening, and inflammation. We have briefly discussed the lesions here.
PAH:
The pathogenesis of PAH starts in the walls of the small pulmonary artery and arterioles. Changes to the vessel intima include endothelial injury, endothelial cell proliferation, invasion of the intima by fibroblast-like cells, fibrosis and obstruction of the lumen by plexiform lesions. Plexiform lesions are a hallmark of severe PAH that form glomeruloid-like vascular structures originating from the pulmonary arteries. These lesions disproportionally affect women (F:M≈ 2.8:1) (5–7). This results in high pulmonary arterial pressure and symptoms like fatigue and shortness of breath, but the major cause of increased morbidity and mortality is from right ventricular (RV) dysfunction. PAH has an estimated prevalence ranging from 10 to 52 cases per million (8). Idiopathic pulmonary arterial hypertension (IPAH) patients with end-stage occlusive vasculopathy have a grim prognosis for survival of 5–7 years even after treatment. RV hypertrophy is implicated in the poorer survival of male IPAH patients (9). Despite tremendous progress in understanding IPAH, current therapy (epoprostenol and derivatives, endothelin receptor antagonists, and phosphodiesterase type 5 inhibitors) has become a significant clinical barrier for treating patients with end-stage IPAH. Although these drugs allow clinical, functional, and hemodynamic improvements, the prognosis of IPAH patients remains challenging as a crucial aspect of severe PAH is the obliterative growth of vascular lesions consisting of endothelial and smooth muscle cells with an anti-apoptotic and pro-proliferative phenotype.
CAA:
Cerebral amyloid angiopathy (CAA) is a type of small vessel disease in which amyloid-beta (Aβ) protein deposits in the walls of small arteries and capillaries in the leptomeninges and cerebral cortex. CAA is a major cause of lobar intracerebral hemorrhage (ICH) and cognitive impairment in the elderly (10). The prevalence of CAA in cognitively normal elderly is estimated to be 5% to 7% (11). Progressive accumulation of Aβ in the cerebral blood vessels results in brain vascular lesions with several phenotype changes, including loss of smooth muscle cells, luminal narrowing, and vessel wall thickening (12). Leptomeningeal arteries in CAA frequently show double barrel-like changes of the wall and obliterative intimal changes with or without amyloid deposition (13). Other changes to leptomeningeal vessels include dilations with disruptions of the internal elastic lamina and medial degeneration, fibrous thickening, and onion skin-like changes. Complex pulmonary arterial lesions share similar features with CAA-related lesions, such as onion-skin lesions and dilation lesions (14). Changes observed in cortical vessels of CAA patients include microaneurysms and fibrinoid necrosis of vessels (15). Occlusion of capillaries by Aβ in CAA have been reported to disturb cerebral blood flow and contribute to ischemia in both the animal model and the human brain (16). No effective prevention or treatment strategies currently exist for CAA. Immunotherapy using the anti-human APOE antibody (HAE-4) in mice was reported to reduce amyloid pathology while protecting cerebrovascular function (17). In the following sections, we have discussed the effect of sex, age, and molecular and environmental factors in the pathogenesis of microvascular brain and lung lesions.
Sex-dependent risk of microvascular lesions:
Sex is a risk factor for both PAH and CAA. The influence of sex and APOE4 genotype was shown to be associated with CAA patients. A study by Shinohara et al. reported that histological severity scores for CAA were higher in males than females when carrying one or more alleles of APOE4 (18). However, the association of CAA in experimental models of CAA and AD show an opposite trend. In Tg-SwDI mice, a CAA model, aging female mice were reported to have more cerebral microbleeds and cognitive impairment compared to males (19). Sexual dimorphism in cytokine levels was observed in the TgSwDI CAA murine model, as female mice had a significantly lower level of anti-inflammatory cytokines IL-1α, IL-2, IL-9, and IFN-γ (19). A potential explanation for the cognitive deficits observed in female mice with CAA was from exposure to higher inflammation from the lack of anti-inflammatory cytokines. And in EFAD transgenic mice carrying human ApoE alleles with familial AD genes (FAD), there was a 3 to10-fold increase in female cases of cerebral microbleeds and of CAA compared to males (20). Evidence from both experimental and population studies show that sex influences the risk of CAA, particularly in human males with the APOE4 genotype.
Epidemiological studies have shown that female sex is a risk factor for IPAH, with women up to four times as likely to present with severe PAH than men (21). The contribution of estrogen to the sex-bias in PAH remains controversial. Experimental studies in the SuHx model of PAH show that reducing endogenous E2 by ovariectomy or aromatase inhibitor treatment decreased vessel-obliterating lesions in female rats (22). However, exogenous E2 has been shown to reduce pulmonary vascular remodeling in monocrotaline- and hypoxia-induced PH in male rats and in ovariectomized female mice (23). Evidence from earlier studies have implicated estrogen in the sex bias. Clinical studies of pulmonary hypertension and vessel lumen-obliterating lesions report associations with exposure to oral contraceptives (24) (25). Experimental studies show that chronic E2 exposure results in hypoxic pulmonary hypertension in ovariectomized female rats (26–28). The influence of E2 and its metabolites on vessel remodeling are apparent from their reported mitogenic effects on both pulmonary arterial endothelial and pulmonary arterial smooth muscle cells (29). Consistent with sex differences in PAH, female patients have a 2.8-fold higher number of lumen-obliterating lesions than their male counterparts. The influence of estrogens on vascular remodeling may partly explain why women are more susceptible to pulmonary vascular lesions (30). Estrogen production may not be solely responsible for the differences in vascular lesions seen in males and females. Genetic studies on the impact of sex-chromosomes on pulmonary hypertension suggest that other mechanisms are possible. For example, complete loss of function or alteration of several X chromosome genes linked to pulmonary hypertension (HDAC6, IL3RA1, ACE2, MAOA, MAOB, NOX1, XIAP, ELK1, XIST, MIR424) worsens hypertension initiated by chronic hypoxia, monocrotaline, bleomycin, and chronic schistosomiasis infection (31–41).
Age as a risk factor of microvascular lesions:
The most significant risk factor for the development of CAA is old age (42). In murine models, older CAA Tg2576 mice developed signs of stroke faster and exhibited an increased number of cerebral microhemorrhages compared to control (43). The prevalence of moderate to severe CAA is reported to be 2.3% between 65 to 74 yrs, 8% for ages 75 to 84 yrs, and 12.1 % in patients over 85 yrs (44). There is a higher prevalence of CAA among patients with dementia. CAA pathology was found in 60% of patients with dementia compared to less than 40% in those without dementia (45). The contribution of aging to the increase of cerebral arterial hypertension is likely to impact the formation of microbleeds in patients with CAA. A majority of studies focused on capillary density in the brains of aging animals and people, report a reduction of vascular density leading to increased cerebral arterial hypertension (46). Hypertension and CAA are major risk factors for intracerebral hemorrhages.
Emerging studies are now showing older PAH patients being diagnosed with high mortality. The mean age of patients enrolled in the US NIH PAH registry was 36 years at the time of diagnosis (47). However, the age of IPAH patients has increased with reports in Germany of the mean age of 65 years with new IPAH diagnosis (48), and from the Swedish registry, the mean age at diagnosis was 69 years (49). Older patients (>65 years) diagnosed with IPAH had a significantly higher mortality risk than younger patients (18–65 years) (50). Age was a dependent factor for PAH in patients diagnosed with scleroderma (a disease associated with PAH). A 52% increased risk of PAH was demonstrated for every 10 years of age in patients from the scleroderma registry study (51).
Molecular risk factors of microvascular lesions:
Apolipoprotein E (APOE) plays a significant role in cholesterol transport in the bloodstream (52). The major alleles of the APOE gene include E2, E3, and E4. Among these three alleles, E4 has a lower transport affinity and binding capacity for lipids resulting in higher blood cholesterol levels (53,54). APOE4 is a significant genetic risk factor for CAA. Population studies have reported that CAA severity and CAA-related vasculitis are significantly associated with carriers of the E4 allele (55). In a meta-analysis, APOE4 was associated with vasculopathic changes of severe CAA (56). While only the E4 allele is associated with the presence of CAA, both E4 and E2 are associated with lobar intracerebral hemorrhages commonly found in CAA (57). Experimental studies of PAH have also reported on the effect of APOE gene knockout. Evidence shows that APOE deficiency contributes to pulmonary hypertension in the mouse APOE KO model (58–60). Male APOE KO mice receiving a high-fat Western diet developed severe PAH associated with insulin resistance and lower plasma adiponectin levels than female mice (61). Furthermore, these studies suggest an essential role for systemic APOE in maintaining lung homeostasis by preventing the pathological lung and vascular remodeling changes associated with hypercholesterolemia induced inflammation and oxidative stress (62).
BMPR2 mutations are the most studied mutation underlying hereditary PAH. BMP receptor type 2 (BMPR2) serves as a central gatekeeper in the balance of transforming growth factor-beta (TGFβ)/bone morphogenetic protein (BMP)-signaling essential for tissue formation and homeostasis (63). BMPR2 is expressed in pulmonary artery-endothelial cells, -smooth muscle cells, and adventitial fibroblasts (64). Heritable PAH is associated with autosomal dominant mutations in BMPR2, and women with this mutation are 2.5 times more likely to develop PAH (65). The BMPR2 mutation remains strongly associated with PAH pathogenesis, with an estimated 80% of familial and 20% of idiopathic PAH patients carrying a heterozygous BMPR2 mutation (66). The SMAD family, SMAD9 and SMAD1, represent genetic risk factors for PAH. SMADs are members of the bone morphogenetic protein (BMP) pathway. SMADs 1 and 9, were determined to have variants that specifically reduced signaling activity in the BMP pathway in a PAH cohort (67). CAV1 frameshift mutations were found to be associated in rare cases of familial PAH where no TGF beta mutation was identified in PAH patients following whole-exome sequencing (68). KCNK3 homozygous mutations have contributed to aggressive forms of hereditary PAH where the disease afflicts patients at young adult age (69). EIF2AK4 is a relatively new molecular risk factor identified in the past decade in PAH. A homozygous mutation of EIF2AK4 results in a population group highly prone to developing a severe form of familial PAH (70).
Environmental risk factors of microvascular lesions:
Air pollution is implicated as a risk factor for vascular disease. Air pollutants lead to ROS production, oxidative tissue damage, and inflammation that trigger pathways involved in vascular lesion formation (71). However, there is limited data on the contribution to cerebral angiopathy. A population-based study examining the association between air pollutants and a higher risk of stroke amongst the CAA population found that ozone exposure was associated with a higher incidence of lobar ICH in this population (72). Furthermore, several population studies investigating the effect of ambient fine particulate matter exposure report the increased incidence of ICH in chronically exposed populations (71). Smoking is a well-known risk factor for ischemic stroke. A prospective cohort study among 22,022 US male physicians found an increased risk for ICH associated with cerebral microbleeds (73). In clinical patients with a history of smoking and idiopathic intraventricular hemorrhages, CAA was at a much higher risk in these patients due to ependymal vessel ruptures (73). Diesel exhaust (DE) is a significant source of particulate matter, a primary air pollutant found in urban air. Mice exposed to DE particulate matter combined with hypoxia-induced PAH resulted in increased levels of endothelial cell apoptosis (74). Exposure of mice to PM2.5 exacerbates heart failure-induced PAH (75). Several population studies studying the association between air pollution and PAH incidence suggested air pollutant exposure is a risk factor for PAH severity (76–78).
Drug-induced PAH is a significant environmental risk from exposure to diet pills, amphetamines, and other stimulants. The weight loss drug, aminorex, caused PAH in 70% patients exposed (79). The diet drug fenfluramine was also associated with PAH. A cohort study examining the effects of fenfluramine-associated PAH found that patients exposed to fenfluramine had the same median survival and hemodynamic features as patients with idiopathic PAH, suggesting that fenfluramine exposure is a potent trigger for PAH (80). Case reports and retrospective epidemiologic studies have shown that methamphetamine is a likely risk factor for PAH (81–83). Drugs such as cocaine, opioids/morphine, and methamphetamine are often injected into the bloodstream, allowing for a more significant risk for toxic insults and endothelial cell injury to the pulmonary vascular system. Intravenous drug use in HIV infection has been postulated to create a “second hit” vascular event that injures the pulmonary arterial endothelium (84).
Environmental exposure to chlorinated solvents have been associated with PAH (85). Epidemiological studies show strong evidence that chronic exposure to polychlorinated biphenyls (PCBs) is associated with lung toxicity (86) and hypertension (87). High levels of PCBs have been reported in human lung tissue (88). Inhalation exposure to vapor-phase PCBs was demonstrated to be even more critical than ingestion under some circumstances (89). Prenatal exposures to PCBs are associated with decreased lung function in 20-year-old offspring (86). Furthermore, population-based studies have provided evidence that PCBs damage the vasculature with reports of increased risk for cardiovascular disease (90–93). In-vivo animal studies have shown that PCBs produce placental vascular lesions and trophoblastic lesions (94). We reported that exposure to PCB153 [1ng/ml] at a concentration observed in human serum [0.60 – 1.63 ng/ml] (95), altered pulmonary endothelial as well as smooth muscle cell phenotypes (95). PCB153 effects on endothelial and smooth muscle cells were more pronounced than E2 regarding vasculosphere formation and angiogenesis. Based on these pieces of evidence, we postulate that exposure to PCB pollutants may contribute to vascular lesion susceptibility.
EMERGING ROLE OF NRF-1 AND ID3 IN MICROVASCULAR LESIONS
Despite tremendous progress in understanding the importance of blood vessel integrity, gaps remain in our knowledge of the underlying molecular factors contributing to vessel injury (96), including microvascular lesions. Herein, we have focused on elucidating the molecular mechanism(s) based on two new emerging molecules NRF1 and ID3, and how they may contribute to microvascular lesions in the lung and brain.
Nuclear Respiratory Factor 1 and vascular dysfunction:
NRF1 (nuclear respiratory factor-1) also known as alpha-palindrome-binding protein is a transcription factor that binds as a homodimer to the DNA sequence motif YGCGCAYGCGCR, where Y and R indicate a pyrimidine (T/C) or purine (A/G) nucleotide. Our discussion on transcription factor NRF1 is not to be confused with ‘nuclear factor (erythroid-derived 2)-like 1’, which has an official symbol of NFE2L1. The human gene symbol for nuclear respiratory factor 1 is NRF1. Confusion between NRF1 and Nrf1 (NFE2Ll) started in the early 1990s (97). A shown in figure 1, NRF1 is made up of 503 amino acids consisting of a DNA-binding region (109–305), dimerization region (1–78), transcriptional activation region (88–116), and a nuclear localization signaling motif (88–116) (98,99). NRF1 is a substrate of AKT kinase, and activation of AKT controls translocation of NRF1 to the nucleus (100). We have confirmed that NRF1 is a direct substrate of AKT. The serine residues 97, 108, and 116 are the major sites in NRF1 that are phosphorylated by AKT (101). Upon phosphorylation NRF1 is translocated into the nucleus.
Figure 1.
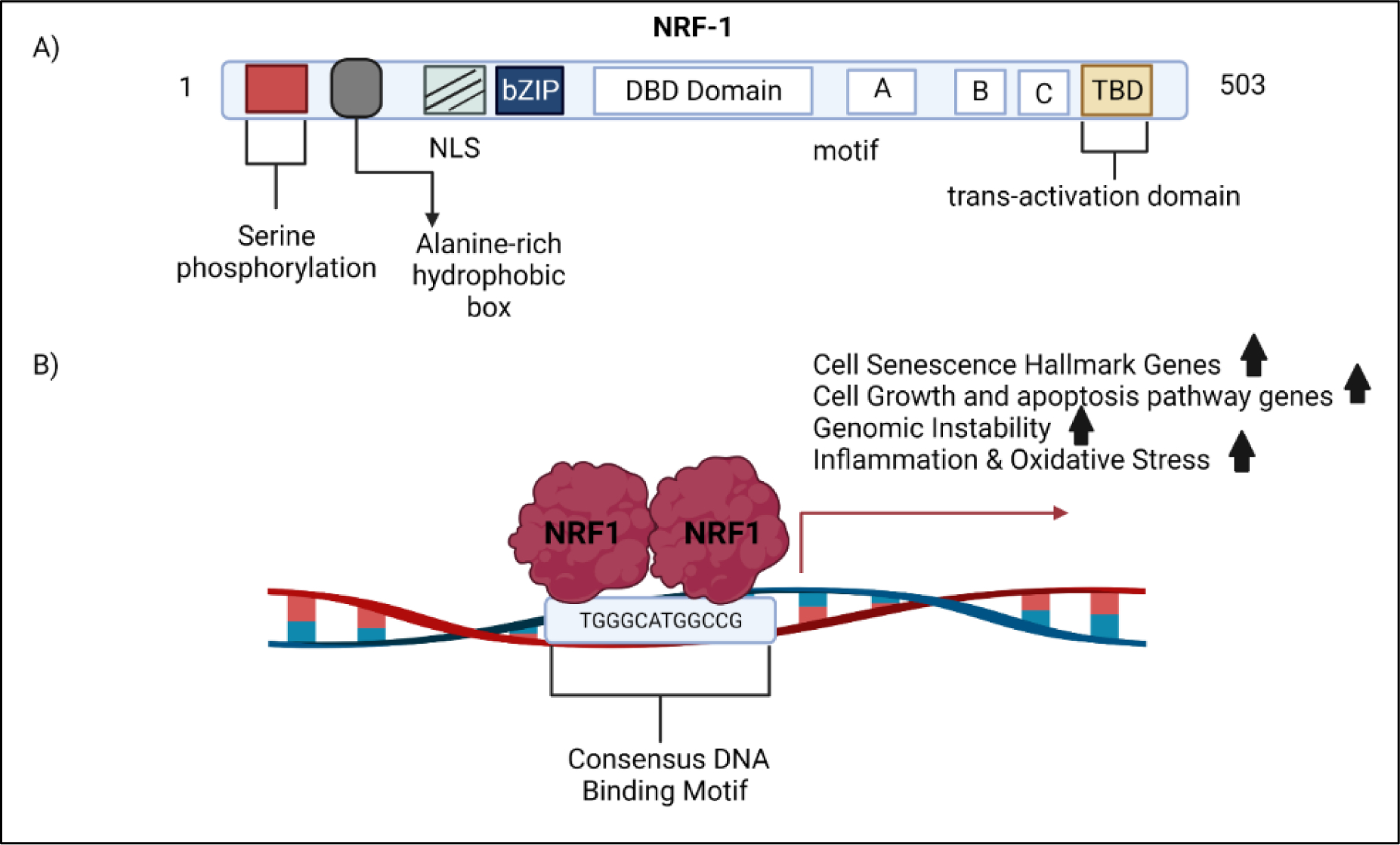
NRF1 activation of target genes. A) The structural domains of the NRF1 protein. B) Scheme illustrates NRF1 binding to its consensus DNA binding motif. This motif is found in the promoter of genes involved in cell senescence, cell growth, apoptosis, genomic instability, and oxidative stress. NRF1 may influence the transcriptional activity of gene networks involved in the vascular hallmarks of vessel dysfunction through its DNA binding motif.
Phosphorylation of the N-terminal serine residues increases NRF1 binding to its DNA motif in target gene promoters. Target genes under control of NRF1 include nuclear encoded genes for mitochondrial respiratory complex subunits, enzymes for heme synthesis, and regulatory factors involved in the replication and transcription of mtDNA essential for mitochondrial biogenesis and respiratory function (102). NRF1 regulates cytochrome c oxidase (COX) subunits in cortical neurons (103)and is a target of the mitochondrial biogenesis regulator PGC-1 (104). In addition, NRF1 controls genes that go beyond mitochondria. Recently, Satoh et al. reported 2470 NRF1 target genes that go beyond mitochondrial processes, including RNA metabolism, splicing, cell cycle, DNA damage repair, protein translation initiation, and ubiquitin-mediated protein degradation. Our studies have also established that overexpression of NRF1 produces cancer stem cells stem via cell reprogramming (105).
NRF1 is one of the least studied transcription factor proteins in vascular disease. Increasing evidence indicates mitochondrial dysfunction occurs in endothelial cells in pulmonary hypertension (106)and in CAA (107). Emerging NRF1 targets found in neurodegenerative disease-related genes, such as Parkin and Pink1 have expanded the role of NRF1 in pathways that impact vessel remodeling. Both Pink1 and Parkin genes are important in mitophagy, but little is known about the influence of mitophagy on the balance of proliferation and apoptosis in pulmonary vascular remodeling in PAH. Excessive cell proliferation is a key event leading to pulmonary vascular remodeling, and new evidence suggests that hypoxia induced Pink1/Parkin expression contributes to proliferation of pulmonary arterial smooth muscle cells and inhibits apoptosis (108). Our laboratory has shown that NRF1 overexpression increases both Pink1 and Parkin expression (109). Therefore, we postulate that oxidative stress from hypoxic conditions may signal NRF1 increased expression of the Pink1/Parkin pathway to influence growth of pulmonary vascular lesions. Mitochondrial respiratory function is influenced by NRF1 as it was shown to rank first in promoter binding sites in complex I genes, and in complex II-V genes (110). Therefore, we also looked at potential NRF1 pathways involved in mitochondrial dysfunction observed in PAH and pulmonary vascular remodeling. For example, an increase in mitochondrial ROS from a loss in Complex I and a shift of ATP production from oxidative phosphorylation to aerobic glycolysis has been shown in pulmonary hypertensive smooth muscle cells (111). Since a glycolytic shift may underlie the resistance to apoptosis and increased vascular cell proliferation observed in vascular lesions, alterations in NRF1 mediated Complex I gene transcription may influence metabolism. Several genes involved in the glycolytic pathway, such as PFKB2, PGAM1, PGKM5, and ALDOA, from ChIP-seq studies of NRF1 suggest its role in reprogramming metabolic processes in addition to directly impacting mitochondrial function (102).
Beyond NRF1 contributions to mitochondrial function, there are also growth pathways implicated in pulmonary hypertension that may be affected by this transcription factor. For example, loss of BMPR2 signaling and increased signaling via TGF-β and its downstream mediator SMAD has been proposed to drive lung vascular remodeling (112). A decrease in SMAD3 protein was observed in PAH patients and PAH experimental models. Furthermore, silencing SMAD3 increased proliferation and migration in human pulmonary arterial endothelial and smooth muscle cells by inhibiting the tumor suppressor p15INK4b. The anti-apoptotic phenotype seen in pulmonary vascular lesions may be associated with defects in this pathway. Our previous study showed aberrant NRF1 activity in regulating pathways involved with TGF-β1 and p15INK4b in glioblastoma patients (113). Since NRF1 was shown to bind to SMAD protein and repress TGF-β induced growth arrest (112), the potential exists for NRF1 to interfere with the tumor suppressive effects of the TGF-β pathway allowing for the uncontrolled growth of vascular lesions.
We have shown that NRF1 responds to oxidative stress upon exposure to estrogenic chemicals and mediates processes such as cell growth and stemness (114,115). Sporadic PAH is a multifactorial and complex disease that depends on the activation of many genes. Recent studies have reported that the NRF1 binding motif is present in at least 50% of known breast cancer susceptibility genes indicating regulation over epithelial-mesenchymal transition (EMT), stemness, cell apoptosis, cell cycle regulation, chromosomal integrity, and DNA damage and repair (116). Pathways that contribute to growth of cancer may also impact vascular remodeling. We have reported NRF1 motif-enriched WNT signaling pathway genes—WNT7B, DLV1, DLV2, GSK3B to be strongly associated with HER2-amplified breast cancer (117). Wnt/β-catenin signaling is essential in vascular remodeling (118). Wnt associated genetic signatures were reported across multiple cell types in PAH patients (119). And abberant expression of target genes in the Wnt/β-catenin pathway have been reported in end stage IPAH patients. NRF1 activation of major PAH related pathways including Wnt/β-catenin are potential transcriptional targets in pulmonary vascular remodeling that promote the growth of vascular lesions.
Genomic alterations from mutations to chromosome rearrangements may contribute to the quasi-malignant vascular lesions observed in PAH patients. In supporting this concept, chromosomal abnormalities and increased DNA damage have been observed in vessel-lumen obliterating lesions from PAH patients (120). Telomere dysfunction can generate chromosomal instability commonly found in human cancer. Overexpression of the enzyme telomerase reverse transcriptase has been shown to be associated with PA-SMC growth in PH and supports a role for telomerase in growth of vascular lesions (121). NRF1 has been reported to regulate transcription of human telomeres (122). Whether there is a link between NRF1 mediated genomic instability via dysregulated telomere transcription is not known. Epigenetic mechanisms such as DNA methylation and histone post-translational modifications (PTMs) have been known to regulate chromatin structure important to pulmonary hypertension. Plexiform lesions have been shown to undergo epigenetic instability (123), and this is one pathway by which NRF1 may contribute to the formation of vascular lesions via cell reprogramming. NRF1 is a pioneer transcription factor that increases chromatin opening activity during mouse embryonic stem cell development (124). NRF1 pioneer activity implies that it initially binds to the DNA in “closed” heterochromatin triggering relaxation of the chromatin landscape to provide accessibility to non-pioneer transcription factors. Hence, NRF1 may be a driver of vessel remodeling via epigenetic programming by opening the chromatin structure allowing for DNA methylation, miRNAs-dependent gene regulation, and acetylation/deacetylation which are known to be associated with PAH (125).
Chemokines and chemokine receptors have a significant impact on initiating and progressing neointimal formation. Chemotaxis of leukocytes to the vessel wall is regulated by chemokines and may contribute to inflammation and subsequent vascular injury resulting in remodeling of the vessel wall. Abnormal proliferation and migration of pulmonary artery ECs (PAECs) contribute to angiogenesis in the plexiform lesion (126). Endothelial cell migration is essential to angiogenesis and can be impacted by chemokines. NRF1 transcriptional activation was shown to control pathological angiogenesis via activation of its target gene chemokine receptor 4 (CXCR4) under hypoxic conditions (127). We have shown that overexpression of NRF1 activates CXCR4 (109). Since CXCR4 expression is increased in rats exposed to chronic hypoxia and pulmonary arterioles of PAH patients (128), NRF1 mediated CXCR4 activation in pulmonary endothelial cells may contribute to the formation of plexiform lesions through signaling disordered angiogenesis.
The previously mentioned pathways describe a potential role for NRF1 in the development of pulmonary vascular remodeling. We will now discuss the potential impact of NRF1 in CAA which eventually leads to the degeneration of cerebral vessels causing microbleeds and microinfarcts. The protective effects of E2 from oxidative stress in cerebral vessels has been shown to be mediated by NRF1 through transcriptional activation of nuclear gene-encoded mitochondrial genes in vivo (129). As mentioned previously, NRF1 has promoter binding sites in mitochondrial respiratory complex genes involved in the electron transport chain (ETC). Damage to the ETC and oxidative phosphorylation due to Aβ in cerebral endothelial cell mitochondria has been shown to cause excessive ROS (130). A decrease in NRF1 activity in CAA patients, may contribute to disruption of mitochondrial ETC efficiency in cerebral endothelial cells that may increase susceptibility to injury from ROS when exposed to elevated levels of Aβ. Because mitochondrial dysfunction accompanies vascular disease in CAA, we did a preliminary study on the transcriptional activity of NRF1 in CAA patients. Our preliminary data from CAA patients indicates that NRF1 activity for the brain endothelial gene signature was downregulated (131). The implication of a reduction of NRF1 transcriptional activity in CAA patients may be on mitochondrial function and susceptibility to vascular injury from oxidative stress. Excessive ROS production from amyloid beta exposure in transgenic CAA models implicate the role of mitochondria and NADPH oxidase in cerebrovascular dysfunction and CAA-related microhemorrhage (132). CAA pathology has been shown to be exacerbated by knockdown of mitochondrial superoxide dismutase 2 (SOD2)—which increases mitochondria-derived ROS (133). Since SOD2 is a target gene of NRF1 (134), a reduction in NRF1 mediated SOD2 may increase mitochondrial ROS in cerebral vessels exposed to amyloid beta. Inflammation from chronic oxidative stress is a known contributor to vascular injury. NRF1 mediated regulation of miR-378a was reported to reduce vascular remodeling by preventing vascular smooth muscle cell proliferation, migration and inflammation (135). A reduction in NRF1 activity in CAA may impact the NRF1-miR-378 axis exacerbating vessel remodeling and lesion formation from elevated levels of oxidative stress and inflammation. Besides exposure to amyloid beta, APOE4 is a significant risk factor for CAA, but the molecular mechanism is not known. The NRF1 DNA binding motif was reported in APOE4 (136) on exon 4 and may potentially affect the expression of multiple genes in its vicinity responsible for increased risk in CAA. In conclusion, NRF1 pathways have the potential to alter the balance of cell growth and death in vessels contributing to pulmonary and cerebral vascular lesions. In the next section, we will discuss the role of the inhibitor of DNA binding and differentiation protein 3 (ID3), a transcription regulator in vascular dysfunction.
Inhibitor of DNA binding and differentiation protein 3 and vascular dysfunction:
Inhibitor of DNA binding and differentiation protein 3 (ID3) is a transcription regulator that belongs to the HLH class of proteins. ID3 gene knockout studies have demonstrated that it is required for proper angiogenesis in the neuroectoderm during mouse development (137). Single cell RNA sequencing studies of mouse capillary endothelium have shown endothelial cells dealing with vascular tone and blood pressure exhibit high ID3 expression (138). The transcription of the ID3 gene is redox sensitive in vascular smooth muscle cells(139). And we have shown that estrogen induced oxidative stress increases the phosphorylation and expression of ID3 in endothelial cells (140). ID3 protein-protein interactions occur via the HLH-motif in which ID3 dimerizes with basic HLH transcription factors including the E proteins (i.e. E12/E47, E2–2, HEB) encoded by the TCF3, TCF4, and TCF12 gene, respectively (141). ID3 serves a critical role in promoting angiogenesis during development, tumor growth, and wound repair by functioning as dominant-negative regulator of bHLH transcription factors in the vascular endothelium (142). These ID3 interactions regulate the transcriptional activity of E proteins that bind to the E-box DNA binding motif (CANNTG) in the promoter of target genes. Serine phosphorylation affects ID3 dimerization partner specificity in vitro (143). Phosphorylation of ID3 inhibits E-protein mediated transcription of p21Cip1 resulting in the proliferation of vascular smooth muscle cells (144). We have shown that exposure to E2 and PCB153 induced ID3 tyrosine phosphorylation in brain endothelial cells (145,146). Our results showed novel ID3 tyrosine phosphorylation sites confirmed by MALDI-TOF MS/MS spectra data. These sites included Tyr-11 and Tyr-48 in peptides from ID3. Tyr-48 is positioned in the HLH motif that is essential for ID3 protein-protein interactions, however, the function of this phosphorylation is not known at this time. Dominant negative regulation by ID3 interactions with E-proteins in the expression of p21 and p16 are not the only regulatory function of ID3. The proteins E12/E47 suppress embryonic genes OCT4, SOX2, and NANOG leading to cell differentiation. We have shown that overexpression of ID3 in endothelial cells increased OCT4 and SOX2 expression and the acquisition of the vascular stem cell signature: CD133+ VEGFR3+ CD34+ (147). In conclusion, ID3 is a redox sensitive transcription regulator that impacts the activation and inactivation of E-protein regulated genes important in differentiation, senescence, growth, and self-renewal which directly impact vascular remodeling in the pathogenesis of vascular lesions.
A molecular risk factor for PAH that we have been investigating is ID3, which is part of a class of ID proteins that are expressed by embryonic and somatic stem cells; and contribute to stemness by enhancing proliferation and inhibiting differentiation. ID3 is upregulated in the pulmonary vasculature following prolonged exposure to hypoxia specifically in VSMCs found in the small muscularized vessels distal to terminal bronchioles (148). We have previously demonstrated that exposure to E2 and estrogenic PCB153 increased ID3 phosphorylation and ID3 protein stability; and both estrogenic compounds increased vasculogenesis and proliferation that depended on oxidant sensitive ID3 signaling (145–147). Determination of ID3 expression in vascular lesions from a human cardiovascular tissue array indicated that ID3 protein expression was significantly higher in benign and malignant vascular lesions compared to normal tissues (149). Moreover, we have shown increased expression of ID3 in vivo plexiform lesions induced by the SuHx PAH model in rodents (147). Collective epidemiological and research studies indicate that clonal hematopoiesis (CH) is a newly recognized risk factor for vascular disease. Clonal hematopoiesis is a phenomenon in which somatic mutations of several genes in blood cell progenitors contribute to the clonal expansion of distinct sub-populations of abnormal blood cells.
We postulate that ID3 contributes to clonal hematopoiesis in PAH via generating pulmonary vascular stem cells. There is evidence that ID3 mediates genome stability through recruiting MDC1 to DNA damage sites (150). ID3 has been shown to modulate the expression of genes essential to maintain genome integrity during cell division (151), to induce centrosome amplification (152), and promote chromosomal instability (153). More specifically, ID3 expression has been correlated with accumulation of excess centrosomes (154) that is a key step in genetic instability. Thus, cells undergoing cell division upon exposure to oxidative stress coupled with ID3 activation may be susceptible to mitotic abnormalities that lead to genetic instability in obliterative vascular lesions. Genetic instability usually associated with pathological disorders and referring to a range of genetic alterations from mutations to chromosome rearrangements may contribute to the quasi-malignant vascular lesions observed in PAH patients. In support of this concept, chromosomal abnormalities and increased DNA damage have been observed in vessel-lumen obliterating lesions from PAH patients (155) and we have shown a positive correlation between ID3 expression and a marker of oxidative DNA damage (8-OHdG) in benign and malignant vascular tissues (145). Our RNA-seq data (unpublished) showed that ID3 regulates many chromosomal instability-regulating genes including PCNA, TOP2A, ATAD2, CKAP5, RAD21, TTK, ECT2, CDC6, MCM10, and CEP55. Therefore, ID3 may act as a molecular risk factor under oxidative stress by producing genetic instability in vascular stem cells via these chromosomal instability genes. Recent studies have also shown that clonal hematopoiesis with JAK2V617F contributed to the development of PAH with ALK1 upregulation (156). There are numerous clinical and experimental data of vessel stem cells in the blood and the lungs of various forms of PAH (157). Obliterating vascular lesions found in end-stage PAH are multicellular and just like solid tumors contain stem cells. Multipotent CD44+ stem cells have been shown in obliterative vascular lesions (158) and are able to differentiate into pericytes and smooth muscle cells (SMC) (159). We have previously reported ID3 expression in the obliterative lesions from the SuHx model of PAH (149). Recent studies suggest that endothelial-to-mesenchymal transition (EndMT) is necessary for the development of obliterative vascular lesion “initiating” cells, which we postulate are susceptible to genetic and epigenetic instability by estrogenic stress and progressively form in vivo obliterative vascular lesions (160–162). Our preliminary observations indicate that ID3 overexpression in combination with E2 or PCB153 stress generate obliterative vascular lesion “initiating” cells. This concept originates from the similarity between the generation of neoplastic stem cells and induced pluripotent stem cells. The generation of these mesenchymal-like obliterative vascular lesion “initiating cells” also known as lesion “stem cells” requires the overexpression of the same embryonic transcription factors (OCT4, NANOG, SOX2) because they are master regulators of pluripotency. Evidence in support of these embryonic transcription factors in IPAH and obliterative vascular lesions come from studies that show OCT4 to be upregulated in cells isolated from patients with IPAH (163) while the enrichment of OCT4, NANOG, and SOX2 have been observed in both benign and malignant vascular tumors (164). We have shown that overexpression of ID3 in endothelial cells increased OCT4 and SOX2 expression and the acquisition of the vascular stem cell signature: CD133+ VEGFR3+ CD34+ (147). Therefore, chromosomal and epigenetic instability driven by ID3 in obliterative vascular lesion “initiating” cells are proposed to contribute to the progression of the vessel-lumen obliterating lesion in PAH.
The previous section primarily described a role for ID3 in PAH. We will now discuss the potential impact of ID3 in CAA which eventually leads to the degeneration of cerebral vessels. Id proteins have been gaining attention in recent articles about amyloid-associated brain disease (165). Amyloid-beta has been shown to increase expression of the ID family of proteins. In CAA vascular injury from amyloid-beta protein accumulation results in double-barrel hemorrhagic lesions or ischemic lesions derived from cells that compose the blood-brain barrier. ID3 is known to be important in the development of the brain vasculature. Id1/Id3 double KO mice display vascular malformations in the forebrain and the absence of sprouting of blood vessels in the neuroectoderm (166). Because ID proteins expression has been associated with amyloid-associated brain disease, we did a preliminary study on the transcriptional activity of ID3 in CAA patients. Our preliminary data from CAA patients indicated that ID3 activity for the brain endothelial gene signature was upregulated (131). We have stably overexpressed ID3 in human cerebral microvascular endothelial cells (hCMEC/D3) and exposed cells to oxidative stress conditions. ID3 overexpressing endothelial cells showed increased neovascularization, cell migration, and spheroid growth in human cerebral microvascular endothelial cells in-vitro (146). Exposure of cerebral microvascular endothelial cells to vascular toxicant PCB153 induced reactive oxygen species (ROS) producing a high tube branched neovascular phenotype dependent on ID3 (145,167,168). In addition, we have shown that exosomal ID3 derived from endothelial cells helped NRF1-induced cancer stem cells cross the blood-brain barrier (BBB) (169). Several phenotype changes occur in CAA including loss of smooth muscle cells, luminal narrowing, and vessel wall thickening. In the Tg2576 AD model mouse model, there are reports of amyloid beta induced angiogenesis and hypervascularization that underlie breakdown of the BBB that is linked to CAA (170). Increase in BBB disruption and leakiness were reported to be influenced by angiogenesis, resulting in the redistribution of tight junctions that maintain the barrier. Disrupted TGFβ1-Smad3 signaling in endothelial cells increases vascular sprouting, branching and proliferation, leading to vascular dysplasia and cerebral hemorrhage in vivo (171). We have observed abnormal vascular sprouting in hCMEC/D3 brain endothelial cells overexpressing ID3. Therefore, it is plausible that the increase in ID3 in CAA patients may contribute to brain vascular lesions such as microbleeds in response to amyloid beta toxicity. Moreover, ID3 has been shown as a biomarker for stroke (172), but whether it is associated with stroke from CAA is not known. To summarize the contributions of ID3 and NRF1 to lung and brain microvascular disease, we have proposed pathways in Figures 3 and 4. In figure 3, both NRF1 and ID3 are illustrated to transcriptionally activate gene networks involved in the hallmarks of cerebral angiopathy including extracellular matrix degradation, endothelial senescence, cerebral amyloid inflammation, pathological angiogenesis, and endothelial cell degeneration. In figure 4, NRF1 and ID3 are proposed to transcriptionally activate gene networks involved in the hallmarks of PAH in response to oxidative stressor.
Figure 3.
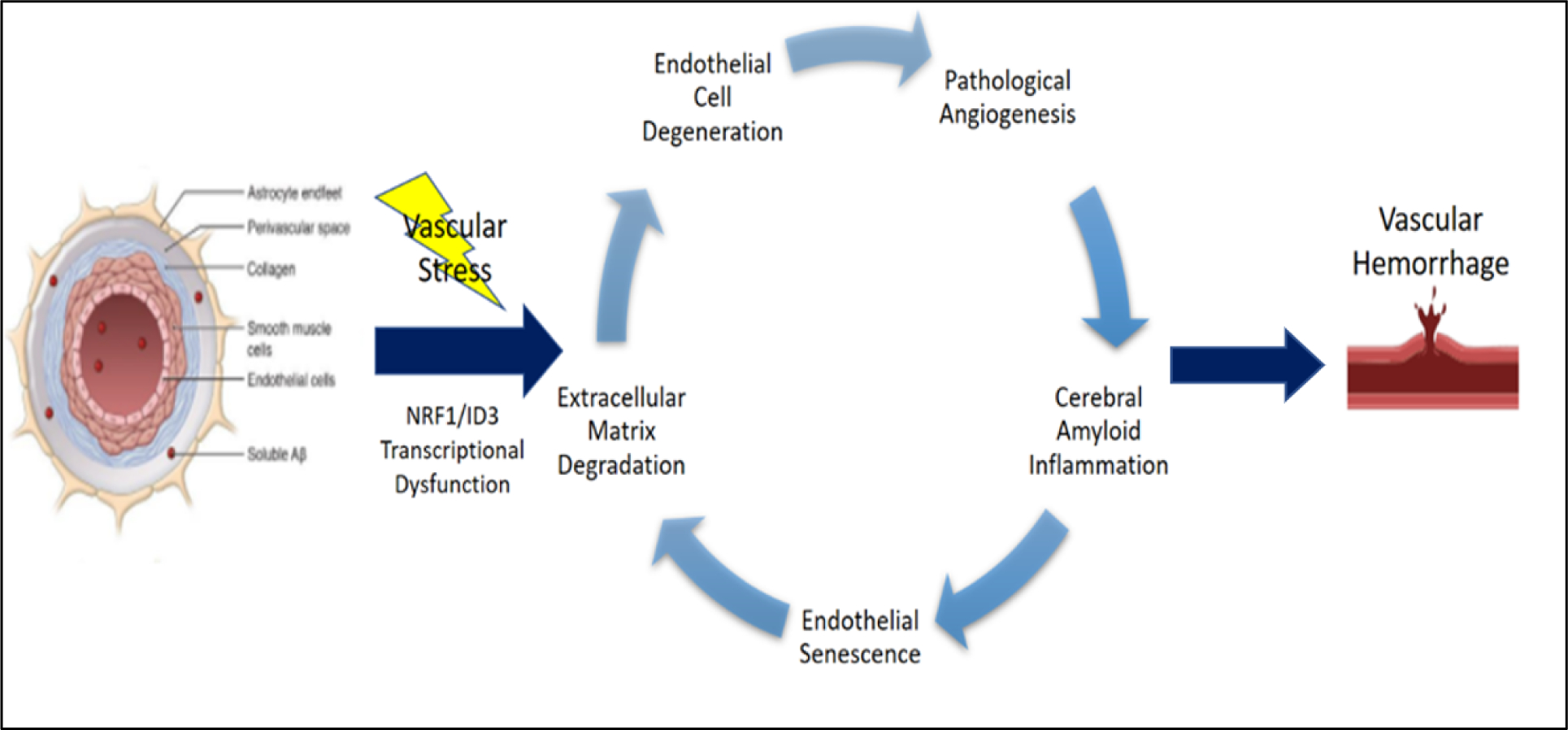
NRF1/ID3 pathway of brain microvascular lesions in CAA. Illustration of oxidative stress induced activation of redox sensitive transcription regulators: NRF1 and ID3. The proposed scheme implicates NRF1/ID3 transcriptional regulation of genes involved in vascular hallmarks of cerebral angiopathy. Aberrant regulation of hallmark genes involved in extracellular matrix degradation, endothelial senescence, cerebral amyloid inflammation, pathological angiogenesis, and endothelial cell degeneration are implicated in vascular lesions including microbleeds and microinfarcts.
Figure 4:
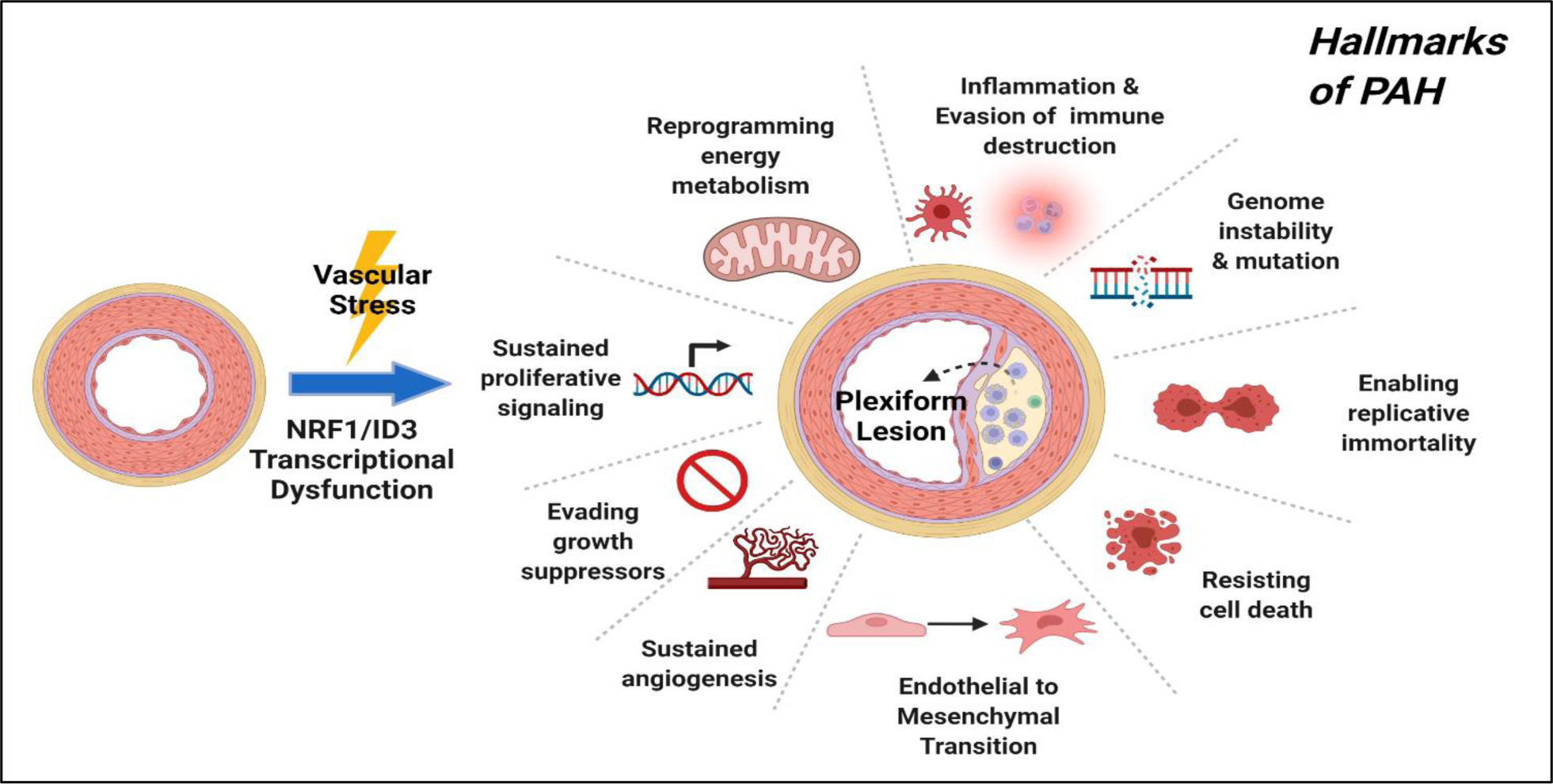
NRF1/ID3 pathway of pulmonary microvascular lesions in PAH. Illustration of oxidative stress induced activation of redox sensitive transcription regulators: NRF1 and ID3. The proposed scheme implicates NRF1/ID3 transcriptional regulation of genes involved in the formation of the plexiform lesion. Abnormal activation of gene networks involved in hallmarks of PAH that include EndMT, resistance to apoptosis, DNA damage, cell growth, angiogenesis and metabolism may influence the development of plexiform lesions from exposure to oxidative stress.
Joint Contribution of NRF1 and ID3 in vascular dysfunction:
As previously discussed, NRF1 and ID3 are redox sensitive transcription regulators that influence pathways implicated in vascular dysfunction and formation of vascular lesions. NRF1 is a pioneer transcription factor responsible for relaxing the chromatin landscape that provides accessibility to non-pioneer transcription regulators, including ID3. To understand the interaction between NRF1 and ID3 we used Encyclopedia of DNA Elements (ENCODE) ChIP-seq of these two proteins and analyzed interaction between their target genes. Furthermore, we also carried out a functional pathway analysis to understand their biological significance. NRF1 and ID3 bound DNA gene targets were almost more than 12,000. Using these targets, we conducted a Venn diagram analysis and uncovered 5,546 genes (35.9 %) that were common targets for both NRF1 and ID3 (Figure 5). To further understand the implication of these genes in cellular processes related to vascular dysfunction, we conducted a functional analysis of these genes using the Biological Networks Gene Ontology tool (BiNGO) (173). We also performed a hypergeometric Benjamini & Hochberg False Discovery Rate (FDR) test to determine NRF1 or ID3 target genes associated with various pathways involved in vascular dysfunction. After determining the set of genes involved in each GO Ontology pathway in BiNGO, we utilized the STRING database (174) to determine protein to protein interaction networks for the set of genes in each vascular dysfunction pathway (Tables 1–6). Our analysis revealed the most significant pathways of the following vascular hallmark pathways: cell apoptosis, cell assembly, oxidative stress, inflammation, genomic instability, cell repair, and cell cycle regulation. Apoptosis is a vital component of maintaining the health of the microvascular system, acting as a protection mechanism that eliminates old, useless, and damaged cells. In CAA, an overabundance of apoptosis in response to amyloid beta-induced stress contributes to the excessive loss of endothelial cells leading to vascular degeneration (175). In PAH, hyperproliferative apoptosis-resistant endothelial cells form into microvascular lesions that result in vascular remodeling of the pulmonary vasculature (176). In Table 1, we show NRF1 and ID3 regulated common genes that were significantly enriched in pathways of apoptosis (p-value = 0.012), regulation of cell death (p-value = 3.11E-04), regulation of apoptosis (p-value = 3.95E-04), and regulation of programmed cell death (p-value = 4.97E-04) amongst others.
Figure 5.
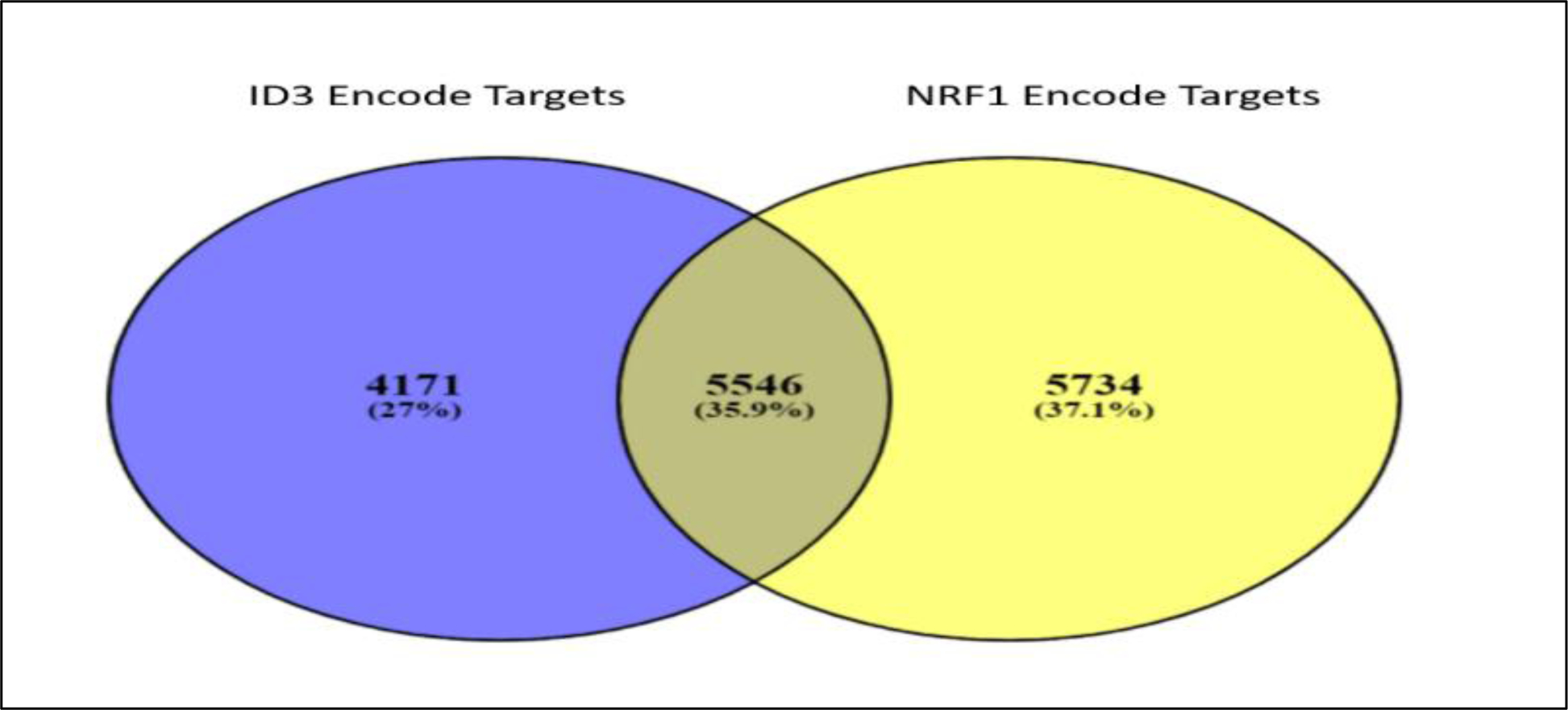
Venn Diagram comparing NRF1 and ID3 target genes. NRF1 (in yellow) and ID3 (in blue) bound DNA gene targets were obtained using the Encyclopedia of DNA Elements (ENCODE) ChIP-seq of NRF1 and ID3 (ENCSR005NMT: Homo sapiens K562 stably expressing ID3 cell line). Between both proteins, a total of 5,546 genes were common targets for both NRF1 and ID3.
Table 1:
Apoptosis enriched pathways for NRF1 and ID3 target genes
| GO ID | GO Description | p-value | Genes |
|---|---|---|---|
| 10941 | Regulation of cell death | 3.11E-04 | ATF1,ATF2, PGAP2, ITSN1, RBPJ, PREX1, PPP2R1A, CHEK2, AKT2, FTH1, DPF2, IL12B, SOX9, PRKACA, SOX4, IER3, PRKCI, PRKCE, DAPK2, DAPK3, SLC11A2, PRKCA, SCRIB, MIF, ACTN4, RUNX3, BTC, TIAM1, RRAGA, ARC, CFDP1, DCUN1D3, NAIF1, XPA, TWIST2, GRIN2A, PRDX5, PPP3R1, APH1B, NUAK2, CHST11, TERT, BLOC1S2, PRDX1, PLCG2, TP53BP2, ALX4, HMOX1,SFN, APOE, FADD, ATG5, STAT5A, BARD1, WNT10B, STAT5B, JUN, XRCC4, XRCC5, XRCC2, BRAF, PRDX6, PAFAH2, FOSL1, PPT1, CD27, ID3, ATM, TP73, BTG2, BTG1, CSRNP3, HIP1, CITED2, FAF1, AATF, SPPL3, BBC3, HERPUD1, ROBO1, IKBKB, ING4, SHH, ING2, PPP3CC, PPP2R4, MECOM, AIFM2, SMPD1, RAC1, JAK2, IKBKE, HRAS, HTATIP2, CTSB, IL10, VAV3, DUSP1, RIPK2, SPHK2, ANXA4, ANXA5, NME2, NME3, DYNLL1, TERF1, SIRT1, DUSP6, RHOA, VAV2, NME1, NME6, PAX7, ARHGEF3, TAX1BP1, G2E3, GAS1, PPARG, ARHGEF2, TLR4, CD44, CSTB, GSTP1, RPL11, HTT, NOL3, ASCL1, EGFR, RELA, FGD4, PPP2CB, MNT, WRN, BCLAF1, PLAGL1, RPS3, PRAME, APBB1, PAK3, MBD4, CD74, WWOX, MAP3K1, CDKN2C, STAT1, CD70, MLH1, HIPK3, HIPK2, WDR11, VEGFA, SKI, TFAP4, NMNAT1, GPAM, STK17A, STK17B, CTNNB1, MTCH1, NDUFA13, PDCD7, PDCD6, PDCD5, IRS2, CLU, PARK2, IGF1R, PYCARD, MAEA, CDH1, ARHGDIA, TNFRSF8, PIM3, PIM2, PHLDA1, IL6R, MAP3K5, SH3GLB1, KCNH8, OSR1, F2R, LIG4, MSH6, ERN1, ADAM17, MSH2, SMO, TMX1, MADD, PHIP, DAP3, FAIM, VCP, CUL5, GLO1, CUL3, HPCA, CUL2, PSEN2, CUL1, PHB, FOXO3, STK4, FOXO1, RTN4, HSP90B1, STK3, SOCS2, NEUROD1, SOCS3, BAG4, BCL2L11, IRAK1, PSAP, PMAIP1, NAE1, NLRP1, SKIL, BCL2L14, MAP2K6, MCL1, TXNDC5, HSPA9, PRNP, SMAD4, TGFB1, SMAD3, HSPA5, UBE2B, CBX4, API5, VDR, TGFB3, TNFRSF9, TNFRSF10B, TNFRSF10A, IGF2R, IL2, ESR2, TNFRSF10D, KLHL20, NFKBIA, NR4A2, DAP, GCLC, IL6, SON, BCL6, IL7, BCL3, JMY, NF1, MAP3K10, CYCS, LHX4, MAP3K11, FGFR1, YWHAE, AVEN, GSK3B, DYRK2, B4GALT1, YWHAB, TNFAIP3, ZC3H8, HSPB1, SPN, POLB, CASP9, SART1, SIN3A, RB1CC1, CASP3, NUP62, MYO18A, IFT57, CTNNBL1, ARHGEF11, ARHGEF12, CAMK1D, DCC, ARHGEF18, FOXL2, PPP2R5C, NGF, MMP9, YWHAZ, TGFBR1, ADAMTS20, DNM2, ALDH1A3, TRAF7, CREB1, RRM2B, TRAF4, DAD1, LCK, TRAF3K, DNAJC5k RASA1, CAT, TRAF5, CRH, TMBIM6, BIRC6, DTL, SOS2, BIRC2, CEBPB, HDAC1, TM2D1, DDX20, KATNB1, NOD1, RNF7, CLN8, THBS1, UACA, DNAJB6, MAPK1, VHL, BAK1, EYA1, SOD2, USP28, PML, DHODH, P2RX7, PC, P2RX4, ERCC3, RPS6KB1, APC, IL2RB, ERCC2, ERCC6 |
| 42981 | Regulation of apoptosis | 3.95E-04 | ATF1, ATF2, PGAP2, ITSN1, RBPJ, PREX1, PPP2R1A, CHEK2, AKT2, DPF2, IL12B, SOX9, PRKACA, SOX4, IER3, PRKCI, PRKCE, DAPK2, DAPK3, SLC11A2, PRKCA, SCRIB, MIF, ACTN4, RUNX3, BTC, TIAM1, ARC, CFDP1, DCUN1D3, NAIF1, XPA, TWIST2, GRIN2A, PRDX5, PPP3R1, APH1B, NUAK2, CHST11, TERT, BLOC1S2, PRDX1, TP53BP2, ALX4, HMOX1, SFN, APOE, FADD, ATG5, STAT5A, BARD1, WNT10B, STAT5B, JUN, XRCC4, XRCC5, XRCC2, BRAF, PRDX6, PAFAH2, FOSL1, PPT1, CD27, ID3, ATM, TP73, BTG2, BTG1, CSRNP3, HIP1, CITED2, FAF1, AATF, SPPL3, BBC3, HERPUD1, ROBO1, IKBKB, ING4, SHH, ING2, PPP3CC, PPP2R4, AIFM2, SMPD1, RAC1, JAK2, IKBKE, HRAS, HTATIP2, CTSB, IL10, VAV3, DUSP1, RIPK2, SPHK2, ANXA4, ANXA5, NME2, NME3, DYNLL1, TERF1, SIRT1, DUSP6, RHOA, VAV2, NME1, NME6, PAX7, ARHGEF3, TAX1BP1, G2E3, GAS1, PPARG, ARHGEF2, TLR4, CD44, CSTB, GSTP1, RPL11, HTT, NOL3, ASCL1, EGFR, RELA, FGD4, PPP2CB, MNT, WRN, BCLAF1, PLAGL1, RPS3, PRAME, APBB1, PAK3, MBD4, CD74, WWOX, MAP3K1, CDKN2C, STAT1, CD70, MLH1, HIPK3, HIPK2, WDR11, VEGFA, SKI, TFAP4, NMNAT1, GPAM, STK17A, STK17B, CTNNB1, MTCH1, NDUFA13, PDCD7, PDCD6, PDCD5, IRS2, CLU, PARK2, IGF1R, PYCARD, MAEA, CDH1, ARHGDIA, TNFRSF8, PIM3, PIM2, PHLDA1, IL6R, MAP3K5, SH3GLB1, KCNH8, OSR1, F2R, LIG4, MSH6, ERN1, ADAM17, MSH2, SMO, TMX1, MADD, PHIP, DAP3, FAIM, VCP, CUL5, GLO1, CUL3, HPCA, CUL2, PSEN2, CUL1, PHB, FOXO3, STK4, FOXO1, RTN4, HSP90B1, STK3, SOCS2, NEUROD1, SOCS3, BAG4, BCL2L11, IRAK1, PSAP, PMAIP1, NAE1, NLRP1, SKIL, BCL2L14, MAP2K6, MCL1, TXNDC5, HSPA9, PRNP, TGFB1, SMAD3, HSPA5, UBE2B, CBX4, API5, VDR, TGFB3, TNFRSF9, TNFRSF10B, TNFRSF10A, IGF2R, IL2, ESR2, TNFRSF10D, KLHL20, NFKBIA, NR4A2, DAP, GCLC, IL6, SON, BCL6, IL7, BCL3, JMY, NF1, MAP3K10, CYCS, LHX4, MAP3K11, FGFR1, YWHAE, AVEN, GSK3B, DYRK2, B4GALT1, YWHAB, TNFAIP3, ZC3H8, HSPB1, SPN, POLB, CASP9, SART1, SIN3A, RB1CC1, CASP3, NUP62, MYO18A, IFT57, CTNNBL1, ARHGEF11, ARHGEF12, CAMK1D, DCC, ARHGEF18, FOXL2, PPP2R5C, NGF, MMP9, YWHAZ, TGFBR1, ADAMTS20, DNM2, ALDH1A3, TRAF7, CREB1, RRM2B, TRAF4, DAD1, LCK, TRAF3, DNAJC5, RASA1, CAT, TRAF5, CRH, TMBIM6, BIRC6, DTL, SOS2, BIRC2, CEBPB, HDAC1, TM2D1, DDX20, NOD1, RNF7, CLN8, THBS1, UACA, DNAJB6, MAPK1, VHL, BAK1, EYA1, SOD2, USP28, PML, DHODH, P2RX7, PC, P2RX4, ERCC3, RPS6KB1, APC, IL2RB, ERCC2, ERCC6 |
| 43067 | Regulation of programmed cell death | 4.97E-04 | ATF1, ATF2, PGAP2, ITSN1, RBPJ, PREX1, PPP2R1A, CHEK2, AKT2, DPF2, IL12B, SOX9, PRKACA, SOX4, IER3, PRKCI, PRKCE, DAPK2, DAPK3, SLC11A2, PRKCA, SCRIB, MIF, ACTN4, RUNX3, BTC, TIAM1, ARC, CFDP1, DCUN1D3, NAIF1, XPA, TWIST2, GRIN2A, PRDX5, PPP3R1, APH1B, NUAK2, CHST11, TERT, BLOC1S2, PRDX1, TP53BP2, ALX4, HMOX1, SFN, APOE, FADD, ATG5, STAT5A, BARD1, WNT10B, STAT5B, JUN, XRCC4, XRCC5, XRCC2, BRAF, PRDX6, PAFAH2, FOSL1, PPT1, CD27, ID3, ATM, TP73, BTG2, BTG1, CSRNP3, HIP1, CITED2, FAF1, AATF, SPPL3, BBC3, HERPUD1, ROBO1, IKBKB, ING4, SHH, ING2, PPP3CC, PPP2R4, AIFM2, SMPD1, RAC1, JAK2, IKBKE, HRAS, HTATIP2, CTSB, IL10, VAV3, DUSP1, RIPK2, SPHK2, ANXA4, ANXA5, NME2, NME3, DYNLL1, TERF1, SIRT1, DUSP6, RHOA, VAV2, NME1, NME6, PAX7, ARHGEF3, TAX1BP1, G2E3, GAS1, PPARG, ARHGEF2, TLR4, CD44, CSTB, GSTP1, RPL11, HTT, NOL3, ASCL1, EGFR, RELA, FGD4, PPP2CB, MNT, WRN, BCLAF1, PLAGL1, RPS3, PRAME, APBB1, PAK3, MBD4, CD74, WWOX, MAP3K1, CDKN2C, STAT1, CD70, MLH1, HIPK3, HIPK2, WDR11, VEGFA, SKI, TFAP4, NMNAT1, GPAM, STK17A, STK17B, CTNNB1, MTCH1, NDUFA13, PDCD7, PDCD6, PDCD5, IRS2, CLU, PARK2, IGF1R, PYCARD, MAEA, CDH1, ARHGDIA, TNFRSF8, PIM3, PIM2, PHLDA1, IL6R, MAP3K5, SH3GLB1, KCNH8, OSR1, F2R, LIG4, MSH6, ERN1, ADAM17, MSH2, SMO, TMX1, MADD, PHIP, DAP3, FAIM, VCP, CUL5, GLO1, CUL3, HPCA, CUL2, PSEN2, CUL1, PHB, FOXO3, STK4, FOXO1, RTN4, HSP90B1, STK3, SOCS2, NEUROD1, SOCS3, BAG4, BCL2L11, IRAK1, PSAP, PMAIP1, NAE1, NLRP1, SKIL, BCL2L14, MAP2K6, MCL1, TXNDC5, HSPA9, PRNP, TGFB1, SMAD3, HSPA5, UBE2B, CBX4, API5, VDR, TGFB3, TNFRSF9, TNFRSF10B, TNFRSF10A, IGF2R, IL2, ESR2, TNFRSF10D, KLHL20, NFKBIA, NR4A2, DAP, GCLC, IL6, SON, BCL6, IL7, BCL3, JMY, NF1, MAP3K10, CYCS, LHX4, MAP3K11, FGFR1, YWHAE, AVEN, GSK3B, DYRK2, B4GALT1, YWHAB, TNFAIP3, ZC3H8, HSPB1, SPN, POLB, CASP9, SART1, SIN3A, RB1CC1, CASP3, NUP62, MYO18A, IFT57, CTNNBL1, ARHGEF11, ARHGEF12, CAMK1D, DCC, ARHGEF18, FOXL2, PPP2R5C, NGF, MMP9, YWHAZ, TGFBR1, ADAMTS20, DNM2, ALDH1A3, TRAF7, CREB1, RRM2B, TRAF4, DAD1, LCK, TRAF3, DNAJC5, RASA1, CAT, TRAF5, CRH, TMBIM6, BIRC6, DTL, SOS2, BIRC2, CEBPB, HDAC1, TM2D1, DDX20, NOD1, RNF7, CLN8, THBS1, UACA, DNAJB6, MAPK1, VHL, BAK1, EYA1, SOD2, USP28, PML, DHODH, P2RX7, PC, P2RX4, ERCC3, RPS6KB1, APC, IL2RB, ERCC2, ERCC6 |
| 6915 | Apoptosis | 0.012 | TRAF3IP2, ITSN1, CCAR1, PREX1, GJA1, PEG10, RASSF5, ZC3H12A, RASSF6, DPF2, CHAC1, IER3, UNC5A, UNC5B, PRKCB, DAPK2, DAPK3, SLC11A2, SCRIB, JMJD6, MFSD10, TIAM1, RRAGA, RRAGC, DDIT4, TRIB3, EPHA2, NAIF1, FXR1, RHOT2, PPP3R1, SGPL1, APH1B, RNF216, TP53BP2, SFN, FADD, FIS1, JUN, CADM1, GADD45B, GADD45A, EIF2AK1, SIAH2, SIAH1, GADD45G, AIMP2, PPT1, CD27, TNFSF9, TP73, CSRNP1, HIP1, FASTKD1, SHB, AATF, BBC3, ROBO1, ING4, PPP3CC, MECOM, AIFM2, TCTN3, RAC1, HRAS, VAV3, DFFB, RIPK2, NME3, DYNLL1, TERF1, VAV2, RHOB, SLTM, NME6, ARHGEF3, TAX1BP1, FASTKD5, PPARG, ARHGEF2, SGK1, ARF6, HTT, FGD4, PAK1, RPS3, PAK3, PAK2, WWOX, EGLN3, MAP3K1, STAT1, OSM, SULF1, BLCAP, HIPK3, HIPK2, PDCL3, RYBP, STK17A, STK17B, TAOK2, MTCH1, NDUFA13, RTKN, PDCD7, PDCD6, CSE1L, PDCD5, BUB1B, AHR, CLU, PYCARD, EP300, PIM2, PHLDA1, MAP3K5, SH3GLB1, SEMA6A, MAGI3, F2R, THOC1, DNAJB13, LIG4, UBE4B, ERN1, ELMO2, RAF1, DAP3, RTN3, VCP, PSEN2, CUL1, FOXO3, RTN4, TIMM50, FAM188A, BAG4, BCL2L11, BAG2, PSAP, PMAIP1, NLRP1, BCL2L14, MCL1, SMAD3, API5, TNFRSF10B, TNFRSF10A, TNFRSF10D, GULP1, NFKBIA, GGCT, DAP, IL6, CYCS, MDM4, CLPTM1L, YWHAE, AVEN, DYRK2, YWHAB, APIP, TNFAIP3, ZC3H8, ECE1, DBI, ATP2A1, DDX41, CASP9, OPA1, BCL7C, CASP3, IFT57, DNM1L, ARHGEF11, KLF11, ARHGEF12, PDCD6IP, PEG3, DCC, AXIN1, ARHGEF18, FOXL2, GLRX2, TNFRSF1B, NGF, TNFRSF1A, DUSP22, TRAF7, ESPL1, DAD1, LCK, C1D, LUC7L3, DNASE2, BIRC6, SOS2, TNFRSF21, SLC25A6, RNF34, LRP1, TM2D1, NOD1, THBS1, PPM1F, DRAM1, UBQLN1, PTK2B, BAK1, BUB1, CDK11A, SOD2, CDK11B, PML, PDCD4, PTPN6 |
| 45767 | Regulation of anti-apoptosis | 0.015 | PRKAA1, BTG2, RTKN, LRP1, DUSP1, CAV1, PIK3R2, SIRT1, RTN4, IGF1R, SLC9A1, SMAD7, IL6, OPA1, STAR, RASA1, SERBP1, PTK2B, HMOX1, CTNNB1, ERC1, IL6ST, IL6R |
| 8629 | Induction of apoptosis by intracellular signals | 0.024 | DYRK2, CUL5, CUL3, CUL2, XPA, CUL1, RNF7, UACA, SART1, CHEK2, CASP3, HMOX1, SFN, PRKACA, JAK2, IKBKE, MBD4, PRKCA, PPP2R5C, MLH1, USP28, PML, HIPK2, MSH6, BCL3, ATM, ERCC6, TP73 |
| 45768 | Positive regulation of anti-apoptosis | 0.038 | PRKAA1, BTG2, LRP1, DUSP1, CAV1, SIRT1, IGF1R, SLC9A1, SMAD7, IL6, OPA1, STAR, RASA1, PTK2B, HMOX1, CTNNB1, ERC1, IL6ST, IL6R |
Table 6:
Cell cycle enriched pathways for NRF1 and ID3 target genes
| GO ID | GO Description | p-value | Genes |
|---|---|---|---|
| 22402 | cell cycle process | 0.00000003 | CCNK, GFI1, CCNF, SMC3, SMC2, PSMD8, PSMD9, PSMD6, PSMD7, MYC, PSMD2, CDC27, STMN1, RCC1, PSMD1, IL12B, BTRC, TIPIN, DDX11, OVOL1, UHMK1, EML4, DYNC1LI1, CDC34, KIF20B, SLBP, MACF1, CDCA2, SEH1L, CDCA3, ANAPC16, RPN2, RPN1, NOLC1, NCAPH, DSN1, RACGAP1, WBP2NL, BARD1, GADD45A, XRCC2, PLK1, CDC7, OSGIN2, CDC6, ZWINT, ANLN, PPP5C, STAG1, ID4, HBP1, ATM, TP73, CDKN1C, SUV39H2, MEI1, NCAPG2, CETN3, KIF11, ING4, NIPBL, NUF2, HRAS, CEP55, PBRM1, NUDC, KIF22, TERF1, MLF1, SIRT2, CIT, PSMA5, RAD51B, PSMA6, NCOR1, TUBB2A, PSMA1, KIF2A, INCENP, PSMA2, CKS2, GAS1, MAPRE3, ARHGEF2, CEP63, PAFAH1B1, CDC123, AKAP8, NEDD9, CENPA, EGFR, PSMB7, PSMB4, PSMB3, PLAGL1, ASZ1, RAD54L, APBB1, MBD3, LMLN, MPP1, DYNC1H1, CGREF1, CENPV, CDKN2C, CSNK1A1, USP9X, NDE1, RPA1, MRE11A, MLH1, MPP6, HINFP, MAD2L2, NEDD1, MIS18A, PSMC4, SGSM3, CCNG2, CENPJ, PSMC1, TACC3, CTNNB1, NCAPD3, TRIP13, SPC24, MAD2L1, CDKN3, SPC25, CEP120, CASC5, BUB1B, STK11, PTTG1, SESN3, CDH1, SESN1, SESN2, NUSAP1, CEP250, OIP5, KNTC1, NBN, PIM2, MAP2K1, CEP135, FBXW7, VASH1, CGRRF1, CDC25B, MSH6, ERN1, ADAM17, CLIP1, ILF3, MSH2, FANCD2, ZNF318, ANAPC4, ANAPC5, ARL8A, MAD1L1, ANAPC1, KMT2E, KHDRBS1, CUL5, TNKS, CUL3, CUL2, CUL1, UBR2, PDS5B, PKMYT1, PDS5A, NPAT, SYCE2, CCNB1, CLTCL1, E4F1, FAM83D, SKIL, MYH10, MAP2K6, KLHDC3, PRNP, TGFB1, UBE2I, SMAD3, UBE2B, VDR, BOD1, FANCA, NDC80, FBXO31, KIF18A, CDK6, JMY, MDM2, MDM4, CNTROB, MAP3K11, EIF4G2, PINX1, CEP164, PKD1, HK2, PPP1R9B, SPN, PPP6C, SART1, CYP26B1, SGOL1, EXO1, SIN3A, WDR6, POLK, RBM7, SUN2, DYNLT1, HAUS3, SMC1A, CKAP5, DNM2, CCNA2, LATS1, ASPM, PSRC1, LATS2, ESPL1, PAPD7, PAPD5, MNS1, BCAT1, RGS14, DCTN2, RNF8, KATNB1, PPM1D, THBS1, AURKB, FZR1, KATNA1, BUB3, FSD1, BUB1, EIF4E, CDK11A, PPP2R3B, CDK11B, PML, RAD52, RSPH1, WEE1, RAD51, RPS6KB1, APC, RAN |
| 87 | M phase of mitotic cell cycle | 0.00000079342 | CCNK, CCNF, CASC5, BUB1B, SMC3, SMC2, PTTG1, CDH1, CDC27, NUSAP1, OIP5, KNTC1, MAP2K1, TIPIN, DDX11, CDC25B, EML4, DYNC1LI1, CLIP1, ANAPC4, ANAPC5, KIF20B, ARL8A, MAD1L1, ANAPC1, CDCA2, SEH1L, CDCA3, ANAPC16, CUL3, NOLC1, PDS5B, PKMYT1, PDS5A, NCAPH, DSN1, CCNB1, CLTCL1, E4F1, FAM83D, UBE2I, BOD1, PLK1, CDC6, ZWINT, NDC80, FBXO31, ANLN, PPP5C, STAG1, KIF18A, NCAPG2, CETN3, PINX1, CEP164, KIF11, HK2, NIPBL, SGOL1, NUF2, POLK, CEP55, PBRM1, NUDC, DYNLT1, HAUS3, KIF22, SMC1A, TERF1, CKAP5, SIRT2, CIT, CCNA2, LATS1, ASPM, PSRC1, LATS2, TUBB2A, ESPL1, KIF2A, PAPD7, INCENP, PAPD5, MAPRE3, ARHGEF2, CEP63, PAFAH1B1, RGS14, DCTN2, AKAP8, RNF8, KATNB1, NEDD9, AURKB, FZR1, KATNA1, BUB3, FSD1, BUB1, LMLN, MPP1, CENPV, CDK11A, CSNK1A1, USP9X, NDE1, CDK11B, MPP6, MAD2L2, NEDD1, WEE1, MIS18A, APC, CCNG2, NCAPD3, SPC24, RAN, MAD2L1, SPC25 |
| 51726 | Regulation of cell cycle | 0.00000000007 | MDC1, CCNK, CCNF, SMC3, UBE2L3, CLP1, MYC, CHEK2, CDK5RAP1, RCC1, IL12B, FBXO6, PRKACA, BTRC, JUNB, TSPYL2, TIPIN, CSNK2A1, SCRIB, MIF, RUNX3, GTPBP4, THAP1, OVOL2, UHMK1, BCR, BTC, DYNC1LI1, THAP5, CDC37, SIK1, KIF20B, AURKAIP1, SLBP, MACF1, DCUN1D3, XPC, BRCC3, BRIP1, TP53BP2, SFN, CEP76, HEXIM1, PLK4, STAT5A, BARD1, STAT5B, CDT1, CREBBP, JUN, GADD45B, GADD45A, INTS3, INSR, PLK1, LIF, CDC7, NR2F2, CDC6, ZWINT, SMARCA4, GADD45G, FOSL1, ANLN, PKIA, ID2, ID3, HBP1, ATM, CDK13, BAP1, TP73, ATR, CDKN1C, DIRAS3, BTG3, CITED2, AATF, ING4, UBL5, MECOM, UIMC1, HRAS, WNT9A, TERF1, MLF1, SIRT2, RHOB, RNF167, NME6, CKS2, GAS1, CEP63, CDC123, EGFR, MNT, RPS15A, PLAGL1, TMEM67, APBB1, MPP1, CGREF1, CDKN2C, OSM, MRE11A, GTF2H1, GPR3, WDR11, HINFP, MAD2L2, TFAP4, SGSM3, CCNG2, RFWD3, TACC3, SIPA1, NEK11, MAD2L1, CDKN3, CCNT2, CCNT1, GMNN, CASC5, BUB1B, STK11, CCND3, CCND2, MAEA, SESN3, CDH1, SESN1, SESN2, NUSAP1, PIM3, KNTC1, NBN, MAP2K1, FBXW7, AIP, VASH1, CGRRF1, GEM, ERN1, ADAM17, MSH2, MADD, TIPRL, PHIP, SRSF5, MAD1L1, KMT2E, CUL9, KHDRBS1, CUL5, CUL3, CUL2, CUL1, PKMYT1, CCNB1, TPR, E4F1, NAE1, CLSPN, SKIL, MAP2K6, ZBTB17, PRNP, TGFB1, SMAD3, UBE2B, VDR, ZZEF1, FBXO31, PTPRC, CDK6, BCL6, JMY, MDM2, PIN1, EIF4G2, PKD1, PPP1R9B, SPN, SART1, CASP3, WDR6, PPP2R5C, SMC1A, NGF, CCNA2, LATS1, PSRC1, LATS2, ESPL1, SNURF, DTL, UHRF2, HDAC1, ROCK2, RNF4, THBS1, FZR1, CDC45, BUB3, BUB1, EIF4E, PPP2R3B, USP28, PML, TICRR, ERCC3, RPS6KB1, APC, ERCC2, PDCD4, PTPN3 |
Both NRF1 and ID3 are involved in the assembly of proteins involved in various pathways associated with vascular cell senescence and the pathogenesis of age-related vascular diseases (177, 178). Therefore, we carried out protein-protein interaction of common target genes shared by NRF and ID3 involved in various cellular pathways which are shown in Figure 6. A significant interaction was observed between DYNLL1, DNM1L, NRF1, FIS1, CYCS, SOD2, PPARG, FOXO3, SMAD3, EP300, and ID3 which were associated with apoptosis associated proteins (Fig 6A). Further analysis of the cellular macromolecular complex assembly pathway showed significant interaction between SMAD2, SMAD3, EP300, ID3, NRF1, CAT, HMOX1, and SOD2 (Fig 6B). DNA Repair Pathway interacting genes were PAX5, ID3, ELAVL1, SIRT1, and NRF1 (Fig 6C). Cellular response to stress pathway showed significant interaction between ID3, KLF2, EP300, BCL6, FOXO3, SIRT1, NFE2L2, CAT, SOD2, NRF1, EIF2S1, and PPP1R15B (Fig 6D).
Figure 6.
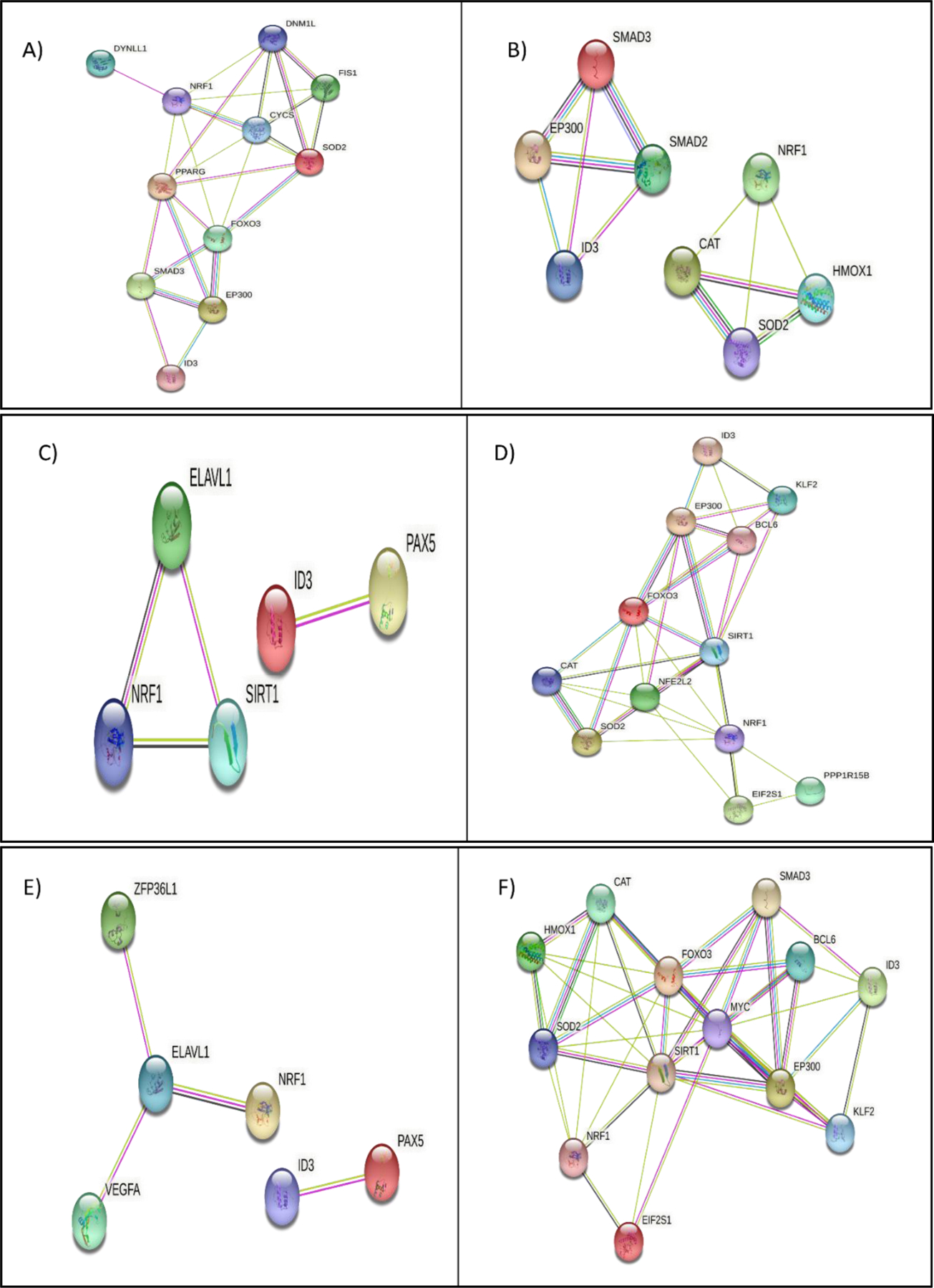
Molecular pathway analysis of NRF1/ID3 joint gene targets. A) String Interaction of 11 NRF1 and ID3 shared ENCODE target genes (DYNLL1, DNM1L, NRF1, FIS1, CYCS, SOD2, PPARG, FOXO3, SMAD3, EP300, ID3) directly connected network derived from the BINGO apoptosis pathway (p-value = 0.01 in Hypergeometric Benjamini & Hochberg False Discovery Rate (FDR) test. B) String Interaction of 8 NRF1 and ID3 shared ENCODE target genes (SMAD2, SMAD3, EP300, ID3, NRF1, CAT, HMOX1, SOD2) directly connected network derived from the BINGO cellular macromolecular complex assembly pathway p-value = 0.000004 in FDR test. C) String Interaction of 5 NRF1 and ID3 shared ENCODE target genes (PAX5, ID3, ELAVL1, SIRT1, NRF1) directly connected network derived from the BINGO DNA Repair Pathway p-value =0.000000007 in FDR test. D) String Interaction of 12 NRF1 and ID3 shared ENCODE target genes (ID3, KLF2, EP300, BCL6, FOXO3, SIRT1, NFE2L2, CAT, SOD2, NRF1, EIF2S1, PPP1R15B) directly connected network derived from the BINGO cellular response to stress pathway (p-value = 0.0000000001 in FDR test. E) String Interaction of 6 NRF1 and ID3 shared ENCODE target genes (ZFP36L1, ELAVL1, VEGFA, NRF1, ID3, PAX5) directly connected network derived from the BINGO regulation of RNA stability pathway (p-value = 0.01 in FDR test. F) String Interaction of 13 NRF1 and ID3 shared ENCODE target genes (CAT, HMOX1, FOXO3, SOD2, NRF1, EIF2S1, SIRT1, MYC, SMAD3, BCL6, EP300, ID3, KLF2) directly connected network derived from the BINGO regulation of cell cycle pathway (p-value = 0.00000000007 in FDR test. Network nodes represent proteins. Edges represent protein-protein interactions. Interactions in blue represent known interactions from curated databases and interactions in magenta represent experimentally determined interactions. Green interactions represent predicted interactions of gene neighborhoods. Red interactions represent interactions involved in gene fusions. Blue interactions represent gene co-occurrence. Yellow interactions represent text mining determined protein interaction. Black interactions represent co-expression. Light blue interactions represent protein homology. Disconnected nodes showed no protein-protein interaction.
In Table 2, we show NRF1 and ID3 regulated shared genes that were significantly enriched ontology pathways of cell assembly, such as cellular component assembly (p-value = 2.59E-09), cellular macromolecular complex assembly (p-value = 4.69E-06), and chromatin assembly or disassembly (p-value = 0.00002). Genomic instability is a central pathobiological presence featured in PAH along with increased levels of DNA damage in PAH lung vascular cells; this contributes to the pathogenic apoptosis-resistant and proliferating characteristics of microvascular lesions in the disease (179). Our lab previously showed that DNA damage/repair genes are highly enriched with NRF1 motifs following bioinformatics analysis (98). These were the reasons we carried out interactions between NRF1 and ID3 target genes involved in genomic instability. In Table 3, we show NRF1 and ID3 regulated genes that were significantly enriched pathway involved in genomic instability such as DNA Repair (p-value = 0.000000008), regulation of RNA stability (p-value = 0.010), regulation of mRNA stability (p-value= 0.019), and RNA stabilization (p-value = 0.024). Oxidative stress results in the degradation of the vascular endothelium in CAA and aberrant behavior of endothelial to mesenchymal transition of endothelial and vascular smooth muscle cells in PAH. NRF1 responds to oxidative stress upon exposure to estrogenic chemicals and mediates cell growth and stemness (105,180). ID3 is a redox-sensitive transcriptional regulator activated in response to oxidative stress and regulates genes involved in cell growth, repair, and apoptosis. Therefore, identification of NRF1 and ID3 interacting genes of oxidative stress pathway was carried out. In Table 4, we show that NRF1 and ID3 regulated shared genes significantly associated with pathways of oxidative stress, such as cellular response to stress (p-value = 0.0000000001) and regulation of cellular response to stress (p-value = 0.025). Cellular repair is a crucial component to maintaining the vascular endothelium utilizing circulating endothelial progenitor cells to repair damage to the vascular lumen. Endothelial progenitor cells (EPCs) incorporate at sites of endothelial damage in CAA and PAH, where repair of endothelial injury is required following vascular insults (181). EPC contribution to tumor neovascularization results in vessel growth in an angiogenesis-defective, Id1 +/− Id3−/− mouse models and donor-derived vascular cells (182). In Table 5, we show that NRF1 and ID3 genes that were significantly associated with pathways of cellular repair/genomic stability such as DNA Repair (p-value = 0.000000008), nucleotide-excision repair (p-value = 0.039448), and double-strand break repair (p-value = 0.00007). Lastly, we examined the interactions between NRF1 and ID3 regulated genes involved in cell cycle regulation. NRF1 can modulate cell cycle progression in response to estrogenic stress, as shown in our lab (183). The ID family of proteins in which ID3 controls normal and abnormal cell cycle progression (184–186). In Table 6, we show that NRF1 and ID3 target genes that were significantly associated with the cell cycle (p-value = 0.00000003), M phase of mitotic cell cycle (p-value = 0.0000007), and regulation of cell cycle (p-value = 0.00000000007).
Table 2:
Cellular assembly enriched pathways for NRF1 and ID3 target genes
| GO ID | GO Description | p-value | Genes |
|---|---|---|---|
| 22607 | cellular component assembly | 2.59E-09 | ATF1, CYFIP1, ZFYVE9, TESK2, EPRS, PREX1, PSMD9, GJA1, CLP1, TSPYL2, MBNL1, TSPYL1, DICER1, PRKAB1, ATG13, ATG12, DDX39B, PARD3, CLNS1A, MAP1B, TBPL1, NUP98, ASF1A, TSPYL4, SKAP2, TSPYL5, NUP205, MTMR2, SHMT1, PDGFA, THY1, TERT, NUP85, PRKAR2A, WBP2NL, HMOX1, ATG5, CREBBP, CADM1, RAB3IP, H3F3A, BRAF, SNUPN, PPP5C, VANGL2, EIF6, STRN, FXYD5, FERMT3, CEP57, ACHE, HIST2H2AB, SRP54, MED16, CLINT1, TUBA1C, MED14, CHAF1A, MED13, NDC1, KCND2, ANXA5, ATPIF1, TERF1, TAF6L, SLC7A6, SLC7A7, NCOR1, TUBB2A, CEP63, PAFAH1B1, KCNA2, ATL2, CENPA, FGD4, TMEM67, AP2S1, STX4, STX2, SRSF10, SRSF12, CENPV, BRF2, SF3A2, MBD2, GTF2H1, GNMT, SKI, TTLL3, VMA21, CENPJ, TAOK2, GTF2B, CASC5, PARK2, CDH2, CDH1, SMARCAD1, KPNA3, GTF2I, MAGI1, MAP2K1, PIAS1, KAT6A, FSCN1, SNRPE, SRSF5, SNRPF, FKBP4, SNRPC, SNRNP200, SRSF9, SNRPB, CD151, TNKS, CUL3, MAZ, ARL2, ITPR3, CORO1A, GTF2E2, ACAT1, HSP90B1, SLC9A3R1, ATXN2, DVL1, SKIL, MDN1, FANCA, FANCC, ARPC4, FBXO31, MDM2, MDM4, TUBGCP4, MAP3K11, CELF1, ITGB3, ARRB1, SART1, OPA1, NCK2, HSP90AA1, APOA1, HAUS3, TNFRSF1A, LCP1, DCTPP1, BIRC2, GTF2A1, PRKAA2, NUFIP1, GTF3C4, NOD1, HSD17B10, HDAC7, SRR, NFS1, PARD6B, POLR2A, BAHD1, POLR2E, E2F3, PCBD1, PCBD2, WASF1, WASF2, WASF3, POLR2L, SH2B1, BAIAP2, PML, P2RX7, P2RX4, RAD51, APC, CTH, IL2RB, APOC1, PFKP, EIF3A, CCNH, CCT6B, RPS14, SNRPD2, PPP2R1A, TRIM24, LONP1, CCNL1, KIFAP3, PDK1, EIF2A, COG4, CIRBP, MIF, ACTN4, MYO7A, BCS1L, GNPAT, SOAT1, KCTD13, CD2BP2, KCNQ5, EZR, STXBP3, PGS1, APOM, SPAST, RACGAP1, HIST1H3F, TSPAN4, HIST1H3H, ASL, APOE, FADD, HIST1H3B, DECR1, HIST1H3E, SRPK2, BBS1, HIST1H4K, INSR, PILRB, PTK2, SNAP91, CCDC88A, ANLN, HIST1H4A, HIST1H4G, HIST1H4H, GRB2, NDUFAF1, HIST1H4J, HIST1H4C, HIST1H4E, BAP1, AHCTF1, CD40, HIP1, C2CD3, RSF1, IKBKAP, IDE, PCM1, DNER, RAC2, RAC1, BBS5, HRAS, CTSD, RAB8A, ATPAF2, VAV3, MTSS1, VAV2, BBS10, NAPB, NEDD9, TUBD1, CTNNA1, TCEB2, EPS15, CBR4, CD74, MAP3K1, GCH1, MTSS1L, RANBP9, SORBS1, MLH1, PEX13, MPP6, PEX14, FKBP1A, TFAP4, CAPZA1, PCDHB16, CAPZA2, CTNNB1, APBA1, DPAGT1, TTF1, IGF1R, HIST1H2AE, EP300, NRCAM, KNTC1, PTRF, PRND, SBF2, HIST1H2AC, HIST1H2AB, TMOD1, HIST1H2AL, HIST1H2AI, HIST1H2AH, HIST1H2AK, ACOT13, CYBA, OXA1L, PRPF6, HIST1H2BH, PPIH, HIST1H2BG, GEMIN7, HIST1H2BD, HIST1H2BC, VCP, COX18, DDX1, HPCA, CCNB1, IRAK1, CALD1, H1FX, MYH11, COX10, SMAD2, PRNP, SMAD4, TGFB1, SMAD3, UBE2B, HSPA4, CAV1, HMGA1, TBCA, ARHGAP26, SMAD7, TSGA10, H1F0, TAF4B, LSM14A, PICALM, YWHAB, HP1BP3, HMGB2, CBY1, USP39, HK2, SNAPC5, RB1CC1, HIST1H1D, HIST1H1E, HIST1H1B, HIST1H1C, H2AFY, NCBP1, PDPK1, NCBP2, H2AFV, SHROOM1, DNM3, NDUFS7, NDUFS5, CAT, ATG4C, PLEC, HIST1H2BN, HIST1H2BJ, HIST1H2BL, HIST1H2BK, DDX23, DDX20, NRXN2, ADD1, CRYZ, TGS1, BMS1, PTK2B, PDSS2, STOM, VHL, RRM1, RRM2, TAF11, NAP1L1, SOD2, TICRR, NAP1L4, ERCC3, ERCC2, PMP22, CD9, AQP11, TAF4 |
| 34622 | cellular macromolecular complex assembly | 4.69E-06 | ATF1, ZFYVE9, CCT6B, RPS14, PREX1, PSMD9, SNRPD2, CLP1, HIST1H2AE, EP300, CCNL1, HIST1H2AC, HIST1H2AB, EIF2A, TSPYL2, HIST1H2AL, MBNL1, TSPYL1, HIST1H2AI, HIST1H2AH, COG4, HIST1H2AK, CIRBP, CYBA, DICER1, BCS1L, PIAS1, OXA1L, PRPF6, DDX39B, CLNS1A, KAT6A, CD2BP2, HIST1H2BH, SNRPE, SRSF5, NUP98, HIST1H2BG, SNRPF, GEMIN7, FKBP4, SNRPC, SNRNP200, HIST1H2BD, ASF1A, TSPYL4, SRSF9, HIST1H2BC, SNRPB, TSPYL5, NUP205, COX18, DDX1, HPCA, ARL2, ATXN2, DVL1, HIST1H3F, HIST1H3H, H1FX, MYH11, HIST1H3B, COX10, HIST1H3E, SRPK2, SMAD2, SMAD4, TGFB1, SMAD3, HIST1H4K, HSPA4, H3F3A, PILRB, ARPC4, TBCA, PTK2, SNAP91, SNUPN, FBXO31, ANLN, HIST1H4A, H1F0, EIF6, HIST1H4G, HIST1H4H, HIST1H4J, HIST1H4C, LSM14A, HIST1H4E, PICALM, AHCTF1, HIP1, CELF1, HP1BP3, HIST2H2AB, HMGB2, SRP54, RSF1, USP39, CLINT1, TUBA1C, SART1, CHAF1A, HIST1H1D, NCK2, HIST1H1E, RAC1, HIST1H1B, HIST1H1C, ATPAF2, NDC1, HSP90AA1, H2AFY, NCBP1, NCBP2, H2AFV, TUBB2A, NDUFS7, NDUFS5, HIST1H2BN, NUFIP1, HIST1H2BJ, HIST1H2BL, HIST1H2BK, DDX23, DDX20, TUBD1, CENPA, TGS1, BMS1, AP2S1, PTK2B, WASF1, EPS15, SRSF10, WASF3, SRSF12, CENPV, SF3A2, MBD2, NAP1L1, MLH1, PEX13, TICRR, NAP1L4, FKBP1A, VMA21, CENPJ, EIF3A |
| 6333 | chromatin assembly or disassembly | 0.00002 | SUV39H2, CHD9, SUV39H1, CHD7, HP1BP3, HIST2H2AB, CHD6, HMGB2, RSF1, ARID4B, CHAF1A, KAT5, HIST1H2AE, HIST1H1D, HIST1H1E, KAT8, CCNL1, HIST1H2AC, HIST1H1B, HIST1H2AB, HIST1H1C, TSPYL2, HIST1H2AL, SMARCC1, TSPYL1, HIST1H2AI, H2AFY, HIST1H2AH, HIST1H2AK, MSL3, H2AFV, BAZ1B, CDYL2, KAT6A, HIST1H2BH, HIST1H2BG, HIST1H2BD, ASF1A, TSPYL4, HIST1H2BC, TSPYL5, HIST1H2BN, HIST1H2BJ, HIST1H2BL, HIST1H2BK, CENPA, BAHD1, HIST1H3F, HIST1H3H, H1FX, MTA2, HIST1H3B, HIST1H3E, SMARCE1, CBX8, CBX7, CENPV, CBX6, HIST1H4K, CBX4, H3F3A, HMGA1, NAP1L1, NAP1L4, HIST1H4A, H1F0, HIST1H4G, HIST1H4H, HIST1H4J, HIST1H4C, MPHOSPH8, HIST1H4E |
Table 3:
Genomic instability enriched pathways for NRF1 and ID3 target genes
| GO ID | GO Description | p-value | Genes |
|---|---|---|---|
| 6281 | DNA Repair | 0.000000008 | MDC1, DCLRE1C, MPG, CCNH, MUM1, NUDT1, SMC6, SMC3, EME1, PTTG1, KAT5, EME2, CDH1, FBXO6, EPC2, NBN, FBXO18, USP3, NSMCE2, RECQL, REV1, LIG4, SETMAR, WRNIP1, CSNK1D, ESCO2, RBX1, MSH6, NEIL2, MSH2, POLG2, FANCD2, UBE2V2, DNA2, NEIL1, ASF1A, VCP, XPA, XPC, BRCC3, RAD51AP1, BRIP1, NTHL1, TP53BP1, CLSPN, FANCI, BARD1, UVRAG, XRCC6, UBE2B, XRCC4, FANCL, XRCC5, GADD45A, INTS3, XRCC2, XRCC3, FANCA, FANCC, UBE2A, FANCE, FANCF, GADD45G, TDP2, TDP1, JMY, UBE2N, ATM, RAD18, TP73, ATR, BTG2, SMG1, FEN1, OGG1, CEP164, HMGB2, POLB, CHAF1A, EXO1, UIMC1, REV3L, POLL, POLK, POLH, POLG, PARP3, RBM14, PARP4, RFC4, RFC1, RFC2, RAD23A, KIF22, SMC1A, SENP2, BAZ1B, SIRT1, DDB2, RNF168, RAD51B, KIN, SFPQ, RRM2B, PAPD7, CRY2, CRY1, DTL, FAN1, USP10, PNKP, RNF8, TYMS, FZR1, WRN, SLX4, POLD2, PMS2, RAD54L, ZSWIM7, PMS1, MUTYH, MBD4, EYA1, EYA3, RPA1, APEX2, MRE11A, RPA2, GTF2H1, MLH1, USP28, APTX, GTF2H5, TICRR, HINFP, RAD52, EEPD1, XAB2, RAD51, ERCC3, POLE2, RFWD3, RPA3, APLF, ERCC2, ERCC6, TRIP13 |
| 43487 | Regulation of RNA stability | 0.010 | ATF1, ATF2, BRF2, DHX9, CIRBP, HNRNPU, TNFRSF1B, ELAVL1, ZFP36L2, VEGFA, ZFP36L1, SYNCRIP, SERBP1, MAPKAPK2, HNRNPD, PABPC1, TARDBP, PAIP1 |
| 43488 | Regulation of mRNA stability | 0.019 | ATF1, ATF2, BRF2, DHX9, CIRBP, HNRNPU, ELAVL1, ZFP36L2, VEGFA, ZFP36L1, SYNCRIP, SERBP1, MAPKAPK2, HNRNPD, PABPC1, TARDBP, PAIP1 |
| 43489 | RNA stabilization | 0.024 | ATF1, ATF2, DHX9, CIRBP, HNRNPU, ELAVL1, VEGFA, SYNCRIP, MAPKAPK2, HNRNPD, PABPC1, TARDBP, PAIP1 |
Table 4:
Oxidative stress enriched pathways for NRF1 and ID3 target genes
| GO ID | GO Description | p-value | Genes |
|---|---|---|---|
| 33554 | Cellular response to stress | 0.0000000001 | MDC1, MAPKBP1, PGAP2, CLPB, CCNH, NUDT1, SMC6, SMC3, KAT5, CHEK2, LONP1, FBXO6, EPC2, EIF2A, TIPIN, NSMCE2, SLC11A2, RECQL, ATG10, REV1, SETMAR, CSNK1D, ESCO2, ATG13, MAPK8IP3, ATG12, NEIL2, MAP1B, DNA2, NEIL1, TRIB1, ATF6, ASF1A, ATF4, DHX9, EPAS1, XPA, XPC, BRCC3, PRDX5, BRIP1, NUAK2, NTHL1, PRDX1, HMOX1, SCAP, SFN, APOE, TP53BP1, ATG5, BARD1, TRAP1, JUN, XRCC6, XRCC4, XRCC5, GADD45A, EIF2AK1, INTS3, XRCC2, PLK1, XRCC3, EIF2AK4, PRDX6, GADD45G, TDP2, TDP1, ATM, RAD18, TP73, ATR, BTG2, SMG1, FEN1, MRPS11, OGG1, CTSV, AATF, HERPUD1, ING4, NIPBL, CHAF1A, UIMC1, REV3L, JAK2, IKBKE, CTSD, MAP4K1, SREBF1, MRPS26, PARP3, PARP4, DBNL, MINK1, KIF22, BAZ1B, SIRT1, DDB2, RNF168, RAD51B, KIN, MRPS9, CEP63, MDFI, CHST3, SP100, MRPS35, FAM129A, DERL1, TYMS, FGD4, WRN, PAK1, SLX4, RPS3, PMS2, RAD54L, STX3, ZSWIM7, APBB1, MAP4K5, PMS1, MAP4K3, MBD4, MAP3K1, ATAD5, OSM, RPA1, APEX2, MRE11A, RPA2, GTF2H1, MLH1, APTX, GTF2H5, HIPK2, HINFP, XAB2, TFAP4, NEDD4, RFWD3, RPA3, PKN1, TRIP13, SIPA1, NEK11, DCLRE1C, MPG, MUM1, FGF1, EME1, PTTG1, EME2, CDH1, SESN1, EP300, NBN, FADS1, MAP3K4, MAP3K5, MAP2K4, FBXO18, USP3, LIG4, WRNIP1, RBX1, MSH6, ERN1, MSH2, SMO, POLG2, FANCD2, TIPRL, UBE2V2, TNIK, VCP, INSIG2, INSIG1, FBXO45, FOXO3, RAD51AP1, UBR5, NAE1, CLSPN, MAP2K6, FANCI, UVRAG, GCKR, EIF2B3, HSPA5, UBE2B, FANCL, CAV1, FANCA, FANCC, UBE2A, FANCE, COQ7, MT3, EIF2S1, FBXO31, FANCF, DAP, BCL6, BCL3, JMY, UBE2N, MAP3K10, MAP3K13, FGF12, MAP3K11, MAP3K12, NFE2L2, GSK3B, BMPR2, DYRK2, CEP164, HMGB2, ATP2A1, POLB, DUSP10, EXO1, RB1CC1, CASP3, POLL, POLK, POLH, POLG, RBM14, RFC4, RFC1, GPX4, SH2D3A, RFC2, ARNT, RAD23A, GLRX2, PPP2R5C, SMC1A, SENP2, ETV5, CCNA2, SFPQ, RRM2B, PAPD7, CAT, CRY2, CRY1, ATG4C, DTL, FAN1, PPP1R15B, USP10, PNKP, RNF8, THBS1, UACA, FZR1, POLD2, MAPK1, RBBP7, MUTYH, EYA1, EYA3, SOD2, USP28, PML, KLF2, TICRR, RAD52, EEPD1, RAD51, ERCC3, APC, POLE2, APLF, ERCC2, STUB1, CCM2, ERCC6 |
| 80135 | Regulation of cellular response to stress | 0.025 | MECOM, AKT2, UIMC1, MAP3K4, POLH, MAP3K5, NDEL1, MAP4K1, DBNL, RIPK2, AXIN1, MIF, SIRT1, MAPK8IP3, RNF168, ZEB2, NCOR1, TRAF4, UBE2V2, UBE2V1, ZNF675, TLR4, MDFI, CD44, RNF8, NOD1, TNFRSF11A, BRCC3, FOXO1, RTN4, FGD4, PAK1, PRDX1, RPS3, PTK2B, MAP4K5, CD74, GCKR, MAP3K1, EYA1, EIF2AK1, EYA3, SEMA4C, HIPK3, MTOR, HIPK2, FGF19, TAOK2, PDCD4, MDM2, UBE2N, MAP3K10, CD27, ATM, PKN1, ERCC6, MAP3K11, TP73, ATR |
Table 5:
Cell repair & genome stability enriched pathways for NRF1 and ID3 target genes
| GO ID | GO Description | p-value | Genes |
|---|---|---|---|
| 6281 | DNA Repair | 0.000000008 | MDC1, DCLRE1C, MPG, CCNH, MUM1, NUDT1, SMC6, SMC3, EME1, PTTG1, KAT5, EME2, CDH1, FBXO6, EPC2, NBN, FBXO18, USP3, NSMCE2, RECQL, REV1, LIG4, SETMAR, WRNIP1, CSNK1D, ESCO2, RBX1, MSH6, NEIL2, MSH2, POLG2, FANCD2, UBE2V2, DNA2, NEIL1, ASF1A, VCP, XPA, XPC, BRCC3, RAD51AP1, BRIP1, NTHL1, TP53BP1, CLSPN, FANCI, BARD1, UVRAG, XRCC6, UBE2B, XRCC4, FANCL, XRCC5, GADD45A, INTS3, XRCC2, XRCC3, FANCA, FANCC, UBE2A, FANCE, FANCF, GADD45G, TDP2, TDP1, JMY, UBE2N, ATM, RAD18, TP73, ATR, BTG2, SMG1, FEN1, OGG1, CEP164, HMGB2, POLB, CHAF1A, EXO1, UIMC1, REV3L, POLL, POLK, POLH, POLG, PARP3, RBM14, PARP4, RFC4, RFC1, RFC2, RAD23A, KIF22, SMC1A, SENP2, BAZ1B, SIRT1, DDB2, RNF168, RAD51B, KIN, SFPQ, RRM2B, PAPD7, CRY2, CRY1, DTL, FAN1, USP10, PNKP, RNF8, TYMS, FZR1, WRN, SLX4, POLD2, PMS2, RAD54L, ZSWIM7, PMS1, MUTYH, MBD4, EYA1, EYA3, RPA1, APEX2, MRE11A, RPA2, GTF2H1, MLH1, USP28, APTX, GTF2H5, TICRR, HINFP, RAD52, EEPD1, XAB2, RAD51, ERCC3, POLE2, RFWD3, RPA3, APLF, ERCC2, ERCC6, TRIP13 |
| 6289 | nucleotide-excision repair | 0.039448 | CCNH, PNKP, OGG1, XPA, XPC, SLX4, NTHL1, POLD2, POLL, RFC4, RFC1, RFC2, RPA1, LIG4, RPA2, FANCC, RAD23A, GTF2H1, GTF2H5, DDB2, XAB2, NEIL2, ERCC3, POLE2, RPA3, ERCC2, ERCC6, NEIL1, FAN1 |
| 6302 | Double-strand break repair | 0.00007 | VCP, FEN1, DCLRE1C, RNF8, BRCC3, RAD51AP1, BRIP1, SLX4, KAT5, UIMC1, RAD54L, NBN, POLK, XRCC6, EYA1, XRCC4, XRCC5, EYA3, XRCC2, RECQL, RPA1, MRE11A, LIG4, MLH1, BAZ1B, APTX, RNF168, RAD52, RAD51, MSH2, PAPD7, TDP2, TDP1, APLF, UBE2V2, UBE2N, TRIP13, FAN1 |
CONCLUSION
There is a critical need to identify and address molecular factors that are driving microvascular lesions to help in the development of disease-modifying treatments to slow or stop the progression of pulmonary and cerebrovascular disease. Redox sensitive transcriptional activation of target genes depends on not only NRF1, but the recruitment of co-activators such as ID3 to the target gene promoter. Our review highlights the fact that targeting NRF1 and ID3 could be a promising therapeutic approach as they are major players in influencing cell growth, cell repair, senescence, and resistance to apoptotic cell death which contribute to vascular lesions. The failure of clinical trials testing new drugs in PAH patients may be because of epigenetic modifications in the endothelial and smooth muscle cells by NRF1 and/or ID3 redox signaling. Knowledge about the molecular biology of these processes will be relevant for future therapeutic approaches to not only PAH but amyloid related angiopathy and other vascular disorders. Therapies targeting transcription regulators NRF1 and/or ID3 have the potential for vascular disease-modification because they will address the root causes such as genomic instability and epigenetic changes in vascular lesions. We hope that our findings will serve as a stimulus for further research towards an effective treatment of microvascular lesions.
Figure 2.
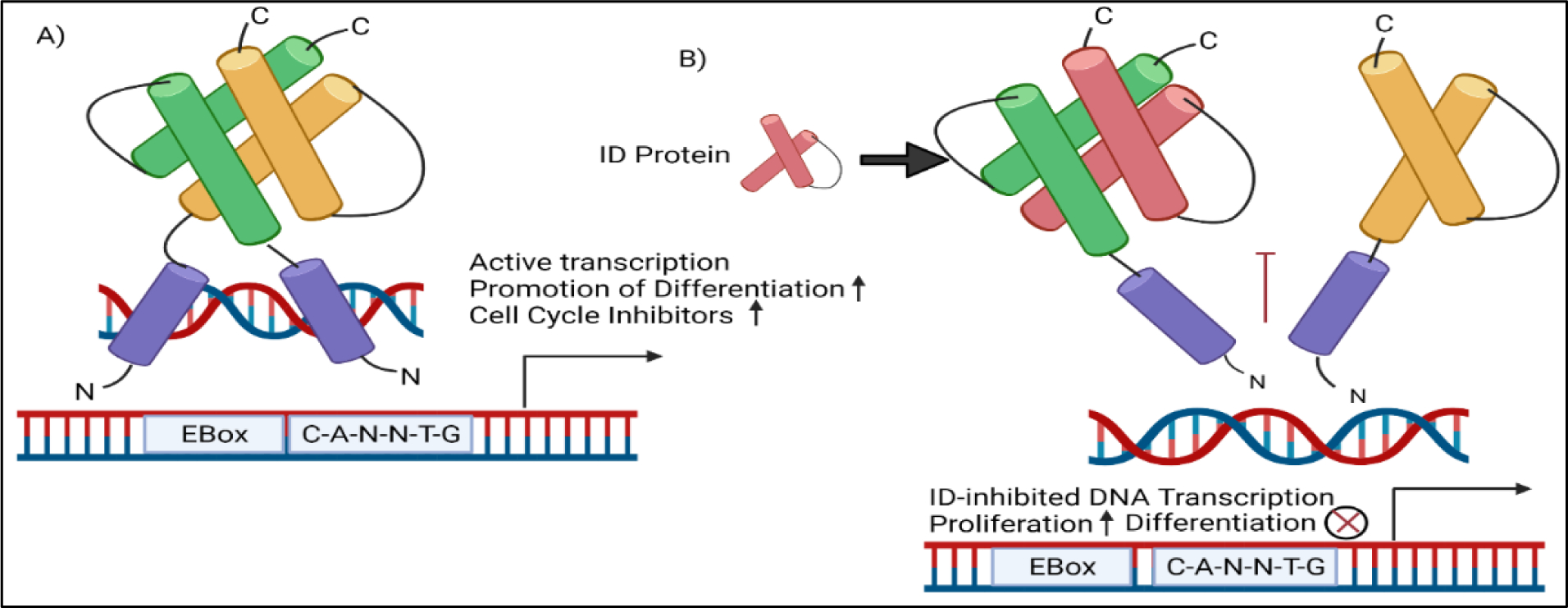
ID3 dominant negative regulation of cell proliferation and differentiation (A) bHLH proteins such as E-proteins (in green and yellow) bind to E-box DNA motifs to activate transcription of cell cycle inhibitor p21 which inhibits cell cycle progression. (B) ID3 (in red) binds to E-proteins where it negatively regulates p21. The inhibition of p21 allows cell cycle progression. ID3 can also positively regulate the expression of embryonic genes Oct4 and Sox2 by binding to E-proteins and prevent differentiation.
Highlights.
Knowledge of how emerging transcription regulators NRF1 and ID3 may contribute to microvascular lesions.
Bioinformatic analysis of the interaction between NRF1 and ID3.
Potential of NRF1 and ID3 redox sensitive transcription regulators as therapeutic targets of vascular remodeling
Acknowledgements:
This research is supported in part by the NIH R15 Award (1R15HL145652–01). We are thankful to the FIU Biomolecular Science Institute providing the license to use BioRender in the design of our illustrations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Benditt EP, Benditt JM. Evidence for a monoclonal origin of human atherosclerotic plaques. Proceedings of the National Academy of Sciences of the United States of America. 1973;70(6):1753–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boehm M, Nabel EG. The cell cycle and cardiovascular diseases. Progress in Cell Cycle Research. 2003. Jan 1;5:19–30. [PubMed] [Google Scholar]
- 3.Ramos J, Felty Q, Roy D. Integrated chip-seq and RNA-seq data analysis coupled with bioinformatics approaches to investigate regulatory landscape of transcription modulators in breast cancer cells. In: Methods in Molecular Biology. 2020. [DOI] [PubMed] [Google Scholar]
- 4.Mayran A, Drouin J. Pioneer transcription factors shape the epigenetic landscape. The Journal of Biological Chemistry [Internet]. 2018. Sep 7 [cited 2022 Jan 29];293(36):13795. Available from: /pmc/articles/PMC6130937/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cool CD, Groshong SD, Oakey J, Voelkel NF. Pulmonary hypertension: cellular and molecular mechanisms. Chest. 2005;128(6 Suppl):565S–571S. [DOI] [PubMed] [Google Scholar]
- 6.Tu L, Dewachter L, Gore B, Fadel E, Dartevelle P, Simonneau G, et al. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. American journal of respiratory cell and molecular biology. 2011. Aug 1;45(2):311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. The American Journal of Pathology. 1994. Feb 1;144(2):275–85. [PMC free article] [PubMed] [Google Scholar]
- 8.Hoeper MM, Gibbs JSR. The changing landscape of pulmonary arterial hypertension and implications for patient care. European Respiratory Review [Internet]. 2014. Dec 1 [cited 2022 Jan 29];23(134):450–7. Available from: https://err.ersjournals.com/content/23/134/450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawat SM, Al-Namnani N, Agerstrand C, Rosenzweig EB, Rowan C, Barst RJ, et al. Determinants of Right Ventricular Ejection Fraction in Pulmonary Arterial Hypertension. CHEST. 2009. Mar 1;135(3):752–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.JJ W, Y M, J N. Cerebral amyloid angiopathy-related inflammation: current status and future implications. Chinese medical journal. 2021. Feb 23;134(6):646–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jäkel L, de Kort AM, Klijn CJM, Schreuder FHBM, Vserbeek MM. Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimer’s & dementia : the journal of the Alzheimer’s Association [Internet]. 2021. [cited 2022 Jan 29]; Available from: https://pubmed.ncbi.nlm.nih.gov/34057813/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeze WM, Bacskai BJ, Frosch MP, Jacobs HIL, Backes WH, Greenberg SM, et al. Blood-Brain Barrier Leakage and Microvascular Lesions in Cerebral Amyloid Angiopathy. Stroke. 2019;50(2):328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamada M, Itoh Y, Otomo E, Hayakawa M, Miyatake T. Subarachnoid haemorrhage in the elderly: a necropsy study of the association with cerebral amyloid angiopathy. Journal of neurology, neurosurgery, and psychiatry [Internet]. 1993. [cited 2022 Jan 29];56(5):543–7. Available from: https://pubmed.ncbi.nlm.nih.gov/8505648/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. European Respiratory Journal [Internet]. 2019. Jan 1 [cited 2022 Jan 29];53(1). Available from: https://erj.ersjournals.com/content/53/1/1801887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li XQ, Su DF, Chen HS, Fang Q. Clinical Neuropathological Analysis of 10 Cases of Cerebral Amyloid Angiopathy-Related Cerebral Lobar Hemorrhage. Journal of Korean Neurosurgical Society [Internet]. 2015. Jul 1 [cited 2021 Nov 16];58(1):30. Available from: /pmc/articles/PMC4534736/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.M H, K LM, CAF von A, M O, DR T. Capillary cerebral amyloid angiopathy in Alzheimer’s disease: association with allocortical/hippocampal microinfarcts and cognitive decline. Acta neuropathologica. 2018. May 1;135(5):681–94. [DOI] [PubMed] [Google Scholar]
- 17.M X, H J, JR S, ER G, C W, M G, et al. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Science translational medicine. 2021. Feb 17;13(581). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shinohara M, Murray ME, Frank RD, Shinohara M, DeTure M, Yamazaki Y, et al. Impact of sex and APOE4 on cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathologica. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maniskas ME, Mack AF, Morales-Scheihing D, Finger C, Zhu L, Paulter R, et al. Sex differences in a murine model of Cerebral Amyloid Angiopathy. Brain, Behavior, & Immunity - Health. 2021. Jul 1;14:100260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finch CE, Shams S. ApoE and Sex Bias in Cerebrovascular Aging of Men and Mice. Trends in neurosciences [Internet]. 2016. Sep 1 [cited 2022 Jan 29];39(9):625. Available from: /pmc/articles/PMC5040339/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, et al. Pulmonary Arterial Hypertension: Baseline Characteristics From the REVEAL Registry. Chest. 2010. Feb 1;137(2):376–87. [DOI] [PubMed] [Google Scholar]
- 22.Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. American journal of respiratory and critical care medicine [Internet]. 2014. Aug 15 [cited 2022 Jan 29];190(4):456–67. Available from: https://pubmed.ncbi.nlm.nih.gov/24956156/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kostyunina DS, McLoughlin P. Sex Dimorphism in Pulmonary Hypertension: The Role of the Sex Chromosomes. Antioxidants [Internet]. 2021. May 1 [cited 2022 Jan 29];10(5). Available from: /pmc/articles/PMC8156365/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleiger RE, Boxer M, Ingham RE, Harrison DC. Pulmonary hypertension in patients using oral contraceptives. A report of six cases. Chest. 1976;69(2):143–7. [DOI] [PubMed] [Google Scholar]
- 25.Irey NS, Norris HJ. Intimal vascular lesions associated with female reproductive steroids. undefined. 1973;29(3):178–81. [PubMed] [Google Scholar]
- 26.Kovaleva YO, Artemyeva MM, Medvedev OS, Medvedeva NA. [Chronic administration of estrogen receptors antagonist reduces degree of hypoxia-induced pulmonary hypertension caused by chronic injections of estrogen in ovariectomised female Wistar rats]. Eksperimental’naia i Klinicheskaia Farmakologiia. 2013. Jan 1;76(7):15–8. [PubMed] [Google Scholar]
- 27.Artem’eva MM, Kovaleva YO, Medvedev OS, Medvedeva NA. Effect of Chronic Administration of Estradiol on Responsiveness of Isolated Systemic and Pulmonary Blood Vessels from Ovariectomized Wistar Rats with Hypoxic Pulmonary Hypertension. Bulletin of experimental biology and medicine. 2015. Aug 1;159(4):427–30. [DOI] [PubMed] [Google Scholar]
- 28.Kovaleva YO, Artem’eva MM, Medvedev OS, Medvedeva NA. [Chronic administration of estradiol to ovariectomized female Wistar rats causes development of hypoxic pulmonary hypertension]. Eksperimental’naia i Klinicheskaia Farmakologiia. 2013. Jan 1;76(5):7–9. [PubMed] [Google Scholar]
- 29.Sun Y, Sangam S, Guo Q, Wang J, Tang H, Black SM, et al. Sex Differences, Estrogen Metabolism and Signaling in the Development of Pulmonary Arterial Hypertension. Frontiers in Cardiovascular Medicine. 2021. Sep 10;0:1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV., et al. Modern age pathology of pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2012. Aug 1;186(3):261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kostyunina DS, McLoughlin P. Sex Dimorphism in Pulmonary Hypertension: The Role of the Sex Chromosomes. Antioxidants 2021, Vol 10, Page 779. 2021. May 14;10(5):779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O B, S C, R P, I T, A B, F P, et al. HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension. Scientific reports. 2017. Dec 1;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA, et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nature Medicine 2012 19:1. 2012. Dec 23;19(1):74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.BB G, MM M-K, H E-H, S P, L Z, A Z, et al. Schistosomiasis-induced experimental pulmonary hypertension: role of interleukin-13 signaling. The American journal of pathology. 2010;177(3):1549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.H S, J Z, C W, PP J, M X, X S, et al. MDM2-Mediated Ubiquitination of Angiotensin-Converting Enzyme 2 Contributes to the Development of Pulmonary Arterial Hypertension. Circulation. 2020;142(12):1190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun X-Q, Peters EL, Schalij I, Axelsen JB, Andersen S, Kurakula K, et al. Increased MAO-A Activity Promotes Progression of Pulmonary Arterial Hypertension. https://doi.org/101165/rcmb2020-0105OC. 2021. Mar 1;64(3):331–43. [DOI] [PubMed] [Google Scholar]
- 37.JF S, W X, KB S. Co-segregation of Norrie disease and idiopathic pulmonary hypertension in a family with a microdeletion of the NDP region at Xp11.3-p11.4. Journal of medical genetics. 2010. Nov;47(11):786–90. [DOI] [PubMed] [Google Scholar]
- 38.IA G, S S, DN M, JH A, DS de J, J S, et al. Endothelial Nox1 oxidase assembly in human pulmonary arterial hypertension; driver of Gremlin1-mediated proliferation. Clinical science (London, England : 1979). 2017. Aug 1;131(15):2019–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.A S, OA O, RJ T, J J, ZZ S, SN K, et al. Decreased OLA1 (Obg-Like ATPase-1) Expression Drives Ubiquitin-Proteasome Pathways to Downregulate Mitochondrial SOD2 (Superoxide Dismutase) in Persistent Pulmonary Hypertension of the Newborn. Hypertension (Dallas, Tex : 1979). 2019. Oct 1;74(4):957–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.M P, D P, R T, C B, J P, S B, et al. A novel p38 mitogen-activated protein kinase/Elk-1 transcription factor-dependent molecular mechanism underlying abnormal endothelial cell proliferation in plexogenic pulmonary arterial hypertension. The Journal of biological chemistry. 2013. Sep 6;288(36):25701–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.S Q, DN P, M P, P D, B G, SA P. Sex differences in the proliferation of pulmonary artery endothelial cells: implications for plexiform arteriopathy. Journal of cell science. 2020. May 1;133(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.SM G, JP V. Diagnosis of cerebral amyloid angiopathy. Sensitivity and specificity of cortical biopsy. Stroke. 1997;28(7):1418–22. [DOI] [PubMed] [Google Scholar]
- 43.Shih AY, Hyacinth I, Hartmann DA, Van Veluw SJ. Rodent models of cerebral microinfarct and microhemorrhage. Stroke. 2018;49(3):803–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenberg SM, Vonsattel JPG. Diagnosis of Cerebral Amyloid Angiopathy. Stroke [Internet]. 1997. [cited 2022 Jan 29];28(7):1418–22. Available from: 10.1161/01.STR.28.7.1418 [DOI] [PubMed] [Google Scholar]
- 45.Keage HAD, Carare RO, Friedland RP, Ince PG, Love S, Nicoll JA, et al. Population studies of sporadic cerebral amyloid angiopathy and dementia: a systematic review. BMC neurology [Internet]. 2009. Jan 13 [cited 2022 Jan 29];9. Available from: https://pubmed.ncbi.nlm.nih.gov/19144113/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ambrose CT. Angiogenesis, aging, and alzheimer’s disease. American Scientist. 2016. Mar 1;104(2):82–5. [Google Scholar]
- 47.Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A national prospective study. Annals of internal medicine. 1987;107(2):216–23. [DOI] [PubMed] [Google Scholar]
- 48.Hoeper MM, Huscher D, Pittrow D. Incidence and prevalence of pulmonary arterial hypertension in Germany. International journal of cardiology. 2016. Jan 15;203:612–3. [DOI] [PubMed] [Google Scholar]
- 49.Rådegran G, Kjellström B, Ekmehag B, Larsen F, Rundqvist B, Blomquist SB, et al. Characteristics and survival of adult Swedish PAH and CTEPH patients 2000–2014. Scandinavian cardiovascular journal : SCJ. 2016. Jul 3;50(4):243–50. [DOI] [PubMed] [Google Scholar]
- 50.Hoeper MM, Huscher D, Ghofrani HA, Delcroix M, Distler O, Schweiger C, et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. International journal of cardiology. 2013. Sep 30;168(2):871–80. [DOI] [PubMed] [Google Scholar]
- 51.Schachna L, Wigley FM, Chang B, White B, Wise RA, Gelber AC. Age and risk of pulmonary arterial hypertension in scleroderma. Chest. 2003;124(6):2098–104. [DOI] [PubMed] [Google Scholar]
- 52.Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein E-deficient mouse: A decade of progress. Vol. 24, Arteriosclerosis, Thrombosis, and Vascular Biology. Arterioscler Thromb Vasc Biol; 2004. p. 1006–14. [DOI] [PubMed] [Google Scholar]
- 53.Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal Domain Stability Mediates Apolipoprotein E Aggregation into Neurotoxic Fibrils. Journal of Molecular Biology. 2006. Sep 1;361(5):932–44. [DOI] [PubMed] [Google Scholar]
- 54.Vance JE. Dysregulation of cholesterol balance in the brain: Contribution to neurodegenerative diseases. Vol. 5, DMM Disease Models and Mechanisms. Dis Model Mech; 2012. p. 746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Renard D, Tatu L, Collombier L, Wacongne A, Ayrignac X, Charif M, et al. Cerebral Amyloid Angiopathy and Cerebral Amyloid Angiopathy-Related Inflammation: Comparison of Hemorrhagic and DWI MRI Features. Journal of Alzheimer’s Disease. 2018. Jan 1;64(4):1113–21. [DOI] [PubMed] [Google Scholar]
- 56.Rannikmäe K, Kalaria RN, Greenberg SM, Chui HC, Schmitt FA, Samarasekera N, et al. APOE associations with severe CAA-associated vasculopathic changes – collaborative meta-analysis. Journal of neurology, neurosurgery, and psychiatry. 2014;85(3):300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.A B, A S, CD A, B K, JM J, H S, et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Annals of neurology. 2010. Dec;68(6):934–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Renshall L, Arnold N, West L, Braithwaite A, Pickworth J, Walker R, et al. Selective improvement of pulmonary arterial hypertension with a dual ETA/ETB receptors antagonist in the apolipoprotein E−/− model of PAH and atherosclerosis: https://doi.org/101177/2045893217752328. 2018. Jan 9;8(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hansmann G, Wagner RA, Schellong S, Perez VA de J, Urashima T, Wang L, et al. Pulmonary Arterial Hypertension Is Linked to Insulin Resistance and Reversed by Peroxisome Proliferator–Activated Receptor-γ Activation. Circulation. 2007. Mar 13;115(10):1275–84. [DOI] [PubMed] [Google Scholar]
- 60.Lawrie A, Hameed AG, Chamberlain J, Arnold N, Kennerley A, Hopkinson K, et al. Paigen Diet–Fed Apolipoprotein E Knockout Mice Develop Severe Pulmonary Hypertension in an Interleukin-1–Dependent Manner. The American Journal of Pathology. 2011. Oct 1;179(4):1693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansmann G, Wagner RA, Schellong S, Perez VA de J, Urashima T, Wang L, et al. Pulmonary Arterial Hypertension Is Linked to Insulin Resistance and Reversed by Peroxisome Proliferator–Activated Receptor-γ Activation. Circulation. 2007. Mar 13;115(10):1275–84. [DOI] [PubMed] [Google Scholar]
- 62.Yao X, Gordon EM, Figueroa DM, Barochia AV, Levine SJ. Emerging Roles of Apolipoprotein E and Apolipoprotein A-I in the Pathogenesis and Treatment of Lung Disease. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hiepen C, Jatzlau J, Hildebrandt S, Kampfrath B, Goktas M, Murgai A, et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLOS Biology. 2019;17(12):e3000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tatius B, Wasityastuti W, Astarini FD, Nugrahaningsih DAA. Significance of BMPR2 mutations in pulmonary arterial hypertension. Respiratory Investigation. 2021. Jul 1;59(4):397–407. [DOI] [PubMed] [Google Scholar]
- 65.Paulin R, Michelakis ED. The Estrogen Puzzle in Pulmonary Arterial Hypertension. Circulation. 2012. Aug 21;126(9):1016–9. [DOI] [PubMed] [Google Scholar]
- 66.Andruska A, Spiekerkoetter E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and Beyond: Contributions to Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2018. Sep 1;19(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nasim MdT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Human Mutation. 2011. Dec 1;32(12):1385–9. [DOI] [PubMed] [Google Scholar]
- 68.Austin ED, Ma L, LeDuc C, Rosenzweig EB, Borczuk A, John A Phillips I, et al. Whole Exome Sequencing to Identify a Novel Gene (Caveolin-1) Associated With Human Pulmonary Arterial Hypertension. Circulation: Cardiovascular Genetics. 2012. Jun;5(3):336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tejedor PN, Castaño JT, Doza JP, Lajara PA, Trujillo GG, Meseguer ML, et al. An homozygous mutation in KCNK3 is associated with an aggressive form of hereditary pulmonary arterial hypertension. Clinical Genetics. 2017. Mar 1;91(3):453–7. [DOI] [PubMed] [Google Scholar]
- 70.Tenorio J, Navas P, Barrios E, Fernández L, Nevado J, Quezada CA, et al. A founder EIF2AK4 mutation causes an aggressive form of pulmonary arterial hypertension in Iberian Gypsies. Clinical Genetics. 2015. Dec 1;88(6):579–83. [DOI] [PubMed] [Google Scholar]
- 71.X R, J Z, RD B, S R. Effect of Particulate Matter Air Pollution on Cardiovascular Oxidative Stress Pathways. Antioxidants & redox signaling. 2018. Mar 20;28(9):797–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.EH W, E M, A F, P K, A W, A C, et al. Ambient Pollutants and Spontaneous Intracerebral Hemorrhage in Greater Boston. Stroke. 2018. Nov;49(11):2764–6. [DOI] [PubMed] [Google Scholar]
- 73.T K, CS K, K B, ES S, JE B, JM G. Smoking and the risk of hemorrhagic stroke in men. Stroke. 2003. May 1;34(5):1151–5. [DOI] [PubMed] [Google Scholar]
- 74.Liu J, Ye X, Ji D, Zhou X, Qiu C, Liu W, et al. Diesel exhaust inhalation exposure induces pulmonary arterial hypertension in mice. Environmental Pollution. 2018. Jun 1;237:747–55. [DOI] [PubMed] [Google Scholar]
- 75.Humbert M, Deng Z, Simonneau G, Barst RJ, Sitbon O, Wolf M, et al. BMPR2 germline mutations in pulmonary hypertension associated with fenfluramine derivatives. European Respiratory Journal [Internet]. 2002. Sep 1 [cited 2021 Sep 16];20(3):518–23. Available from: https://erj.ersjournals.com/content/20/3/518 [DOI] [PubMed] [Google Scholar]
- 76.CP A, EA H, SM K, JHM A, M B, ED M, et al. Ambient air pollution and pulmonary vascular volume on computed tomography: the MESA Air Pollution and Lung cohort studies. The European respiratory journal. 2019. Jun 1;53(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johannson KA, Vittinghoff E, Lee K, Balmes JR, Ji W, Kaplan GG, et al. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. European Respiratory Journal. 2014. Apr 1;43(4):1124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.KA J, E V, J M, PJ W, EM N, JR B, et al. Air Pollution Exposure Is Associated With Lower Lung Function, but Not Changes in Lung Function, in Patients With Idiopathic Pulmonary Fibrosis. Chest. 2018. Jul 1;154(1):119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Montani D, Seferian A, Savale L, Simonneau G, Humbert M. Drug-induced pulmonary arterial hypertension: a recent outbreak. European Respiratory Review. 2013. Sep 1;22(129):244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.R S, M H, B S, X J, A Y, J LP, et al. Pulmonary arterial hypertension associated with fenfluramine exposure: report of 109 cases. The European respiratory journal. 2008. Feb;31(2):343–8. [DOI] [PubMed] [Google Scholar]
- 81.G S, MA G, I A, D C, C D, A G, et al. Updated clinical classification of pulmonary hypertension. Journal of the American College of Cardiology. 2013. Dec 24;62(25 Suppl). [DOI] [PubMed] [Google Scholar]
- 82.Reddy PKV, Ng TMH, Oh EE, Moady G, Elkayam U. Clinical Characteristics and Management of Methamphetamine- Associated Cardiomyopathy: State- of- the- Art Review. Journal of the American Heart Association. 2020. Jun 2;9(11):16704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.KM C, RN C, LJ R. Is methamphetamine use associated with idiopathic pulmonary arterial hypertension? Chest. 2006;130(6):1657–63. [DOI] [PubMed] [Google Scholar]
- 84.George MP, Champion HC, Gladwin MT, Norris KA, Morris A. Injection Drug Use as a “Second Hit” in the Pathogenesis of HIV-associated Pulmonary Hypertension. American Journal of Respiratory and Critical Care Medicine. 2012. Jun 1;185(11):1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Montani D, Lau EM, Descatha A, Jaïs X, Savale L, Andujar P, et al. Occupational exposure to organic solvents: a risk factor for pulmonary veno-occlusive disease. Eur Respir J. 2015;46:1721–31. [DOI] [PubMed] [Google Scholar]
- 86.Hansen S, Strøm M, Olsen SF, Dahl R, Hoffmann HJ, Granström C, et al. Prenatal exposure to persistent organic pollutants and offspring allergic sensitization and lung function at 20 years of age. Clinical & Experimental Allergy. 2016. Feb 1;46(2):329–36. [DOI] [PubMed] [Google Scholar]
- 87.Kreiss K, Zack MM, Jones BT, Kimbrough RD, Needham LL, Smrek AL. Association of Blood Pressure and Polychlorinated Biphenyl Levels. JAMA. 1981. Jun 26;245(24):2505–9. [PubMed] [Google Scholar]
- 88.Rallis GN, Boumba VA, Sakkas VA, Fragkouli K, Siozios G, Albanis TA, et al. Residues of selected polychlorinated biphenyls (PCB) and organochlorine pesticides (OCP) in postmortem lungs from Epirus, northwestern Greece. Journal of toxicology and environmental health Part A. 2014. Jul 18;77(13):767–75. [DOI] [PubMed] [Google Scholar]
- 89.Carpenter DO. Exposure to and health effects of volatile PCBs. Reviews on environmental health. 2015. May 1;30(2):81–92. [DOI] [PubMed] [Google Scholar]
- 90.Gustavsson P, Hogstedt C. A cohort study of Swedish capacitor manufacturing workers exposed to polychlorinated biphenyls (PCBs). American Journal of Industrial Medicine. 1997. Sep 1;32(3):234–9. [DOI] [PubMed] [Google Scholar]
- 91.Goncharov A, Haase RF, Santiago-Rivera A, Morse G, McCaffrey RJ, Rej R, et al. High serum PCBs are associated with elevation of serum lipids and cardiovascular disease in a Native American population. Environmental Research. 2008. Feb 1;106(2):226–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sergeev A V, Carpenter DO. Hospitalization rates for coronary heart disease in relation to residence near areas contaminated with persistent organic pollutants and other pollutants. Environmental Health Perspectives. 2005. Jun;113(6):756–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tokunaga S, Kataoka K. A longitudinal analysis on the association of serum lipids and lipoproteins concentrations with blood polychlorinated biphenyls level in chronic “Yusho” patients. Fukuoka igaku zasshi = Hukuoka acta medica. 2003;94(5):110–7. [PubMed] [Google Scholar]
- 94.Bäcklin BM, Persson E, Jones CJP, Dantzer V. Polychlorinated biphenyl (PCB) exposure produces placental vascular and trophoblastic lesions in the mink (Mustela vison): a light and electron microscopic study. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 1998;106(8):785–99. [DOI] [PubMed] [Google Scholar]
- 95.Charlier CJ, Albert AI, Zhang L, Dubois NG, Plomteux GJ. Polychlorinated biphenyls contamination in women with breast cancer. Clinica Chimica Acta. 2004. Sep 1;347(1–2):177–81. [DOI] [PubMed] [Google Scholar]
- 96.Zhang X, Bishawi M, Zhang G, Prasad V, Salmon E, Breithaupt JJ, et al. Modeling early stage atherosclerosis in a primary human vascular microphysiological system. Nature Communications. 2020. Dec 1;11(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roy D, Tamuli R. NRF1 (nuclear respiratory factor 1) Recommended Citation. 2008; [Google Scholar]
- 98.Ramos J, Das J, Felty Q, Yoo C, Poppiti R, Murrell D, et al. NRF1 motif sequence-enriched genes involved in ER/PR −ve HER2 +ve breast cancer signaling pathways. Breast cancer research and treatment. 2018. Nov 1;172(2):469–85. [DOI] [PubMed] [Google Scholar]
- 99.Das JK, Felty Q, Poppiti R, Jackson RM, Roy D. Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis. Cells. 2018. Nov 27;7(12):234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Piantadosi CA, Suliman HB. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. The Journal of biological chemistry. 2006. Jan 6;281(1):324–33. [DOI] [PubMed] [Google Scholar]
- 101.Das J, Felty Q, Poppiti R, Jackson R, Roy D. Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis. Cells. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Satoh JI, Kawana N, Yamamoto Y. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regulation and Systems Biology [Internet]. 2013. Nov 4 [cited 2022 Jan 29];7(7):139. Available from: /pmc/articles/PMC3825669/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dhar SS, Ongwijitwat S, Wong-Riley MTT. Nuclear Respiratory Factor 1 Regulates All Ten Nuclear-encoded Subunits of Cytochrome c Oxidase in Neurons. The Journal of biological chemistry [Internet]. 2008. Feb 8 [cited 2022 Jan 29];283(6):3120. Available from: /pmc/articles/PMC2669777/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell [Internet]. 1999. Jul 9 [cited 2022 Jan 29];98(1):115–24. Available from: https://pubmed.ncbi.nlm.nih.gov/10412986/ [DOI] [PubMed] [Google Scholar]
- 105.Das J, Felty Q, Poppiti R, Jackson R, Roy D. Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis. Cells [Internet]. 2018. Nov 27 [cited 2021 Nov 17];7(12):234. Available from: https://pubmed.ncbi.nlm.nih.gov/30486409/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yu Q, Chan SY. Mitochondrial and Metabolic Drivers of Pulmonary Vascular Endothelial Dysfunction in Pulmonary Hypertension. Advances in experimental medicine and biology [Internet]. 2017. [cited 2022 Jan 29];967:373. Available from: /pmc/articles/PMC5810356/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parodi-Rullán R, Sone JY, Fossati S, Albensi B. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Journal of Alzheimer’s disease : JAD [Internet]. 2019. [cited 2022 Jan 29];72(4):1019–39. Available from: https://pubmed.ncbi.nlm.nih.gov/31306129/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Linqing L, Yuhan Q, Erfei L, Yong Q, Dong W, Chengchun T, et al. Hypoxia-induced PINK1/Parkin-mediated mitophagy promotes pulmonary vascular remodeling. Biochemical and Biophysical Research Communications. 2021. Jan 1;534:568–75. [DOI] [PubMed] [Google Scholar]
- 109.Das JK, Roy D. Abstract 3312: Transcriptional regulation of chemokine receptor 4 (CXCR4) by nuclear respiratory factor 1 (NRF1) controls estrogen-induced malignant transformation of breast epithelial cells to breast cancer stem cells. Cancer Research [Internet]. 2016. Jul 15 [cited 2022 Jan 29];76(14 Supplement):3312–3312. Available from: https://cancerres.aacrjournals.org/content/76/14_Supplement/3312 [Google Scholar]
- 110.van der Lee R, Szklarczyk R, Smeitink J, Smeets HJM, Huynen MA, Vogel R. Transcriptome analysis of complex I-deficient patients reveals distinct expression programs for subunits and assembly factors of the oxidative phosphorylation system. BMC Genomics [Internet]. 2015. Sep 15 [cited 2022 Jan 29];16(1):1–13. Available from: 10.1186/s12864-015-1883-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rafikov R, Sun X, Rafikova O, Louise Meadows M, Desai AA, Khalpey Z, et al. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biology [Internet]. 2015. Dec 1 [cited 2022 Jan 29];6:278. Available from: /pmc/articles/PMC4556771/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zabini D, Granton E, Hu Y, Miranda MZ, Weichelt U, Bonnet SB, et al. Loss of SMAD3 Promotes Vascular Remodeling in Pulmonary Arterial Hypertension via MRTF Disinhibition. American journal of respiratory and critical care medicine [Internet]. 2018. Jan 15 [cited 2022 Jan 29];197(2):244–60. Available from: https://pubmed.ncbi.nlm.nih.gov/29095649/ [DOI] [PubMed] [Google Scholar]
- 113.Bhawe K, Felty Q, Yoo C, Ehtesham NZ, Hasnain SE, Singh VP, et al. Nuclear Respiratory Factor 1 (NRF1) Transcriptional Activity-Driven Gene Signature Association with Severity of Astrocytoma and Poor Prognosis of Glioblastoma. Molecular neurobiology [Internet]. 2020. Sep 1 [cited 2022 Jan 29];57(9):3827–45. Available from: https://pubmed.ncbi.nlm.nih.gov/32594352/ [DOI] [PubMed] [Google Scholar]
- 114.Felty Q, Xiong WC, Sun D, Sarkar S, Singh KP, Parkash J, et al. Estrogen-induced mitochondrial reactive oxygen species as signal-transducing messengers. Biochemistry. 2005. May 1;44(18):6900–9. [DOI] [PubMed] [Google Scholar]
- 115.Das J, Felty Q, Poppiti R, Jackson R, Roy D. Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis. Cells. 2018. Nov 27;7(12):234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhou Y, Xu Z, Quan D, Zhang F, Zhang H, Xiao T, et al. Nuclear respiratory factor 1 promotes spheroid survival and mesenchymal transition in mammary epithelial cells. Oncogene 2018. 37:47. 2018 Jul 11;37(47):6152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ramos J, Das J, Felty Q, Yoo C, Poppiti R, Murrell D, et al. NRF1 motif sequence-enriched genes involved in ER/PR −ve HER2 +ve breast cancer signaling pathways. Breast Cancer Research and Treatment 2018 172:2 [Internet]. 2018. Aug 20 [cited 2022 Jan 29];172(2):469–85. Available from: 10.1007/s10549-018-4905-9 [DOI] [PubMed] [Google Scholar]
- 118.Goodwin AM, D’Amore PA. Wnt signaling in the vasculature. Angiogenesis [Internet]. 2002. [cited 2022 Jan 29];5(1–2):1–9. Available from: https://pubmed.ncbi.nlm.nih.gov/12549854/ [DOI] [PubMed] [Google Scholar]
- 119.West JD, Austin ED, Gaskill C, Marriott S, Baskir R, Bilousova G, et al. Stem Cell Physiology and Pathophysiology: Identification of a common Wnt-associated genetic signature across multiple cell types in pulmonary arterial hypertension. American Journal of Physiology - Cell Physiology [Internet]. 2014. Sep 1 [cited 2022 Jan 29];307(5):C415. Available from: /pmc/articles/PMC4154073/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Federici C, Drake KM, Rigelsky CM, McNelly LN, Meade SL, Comhair SAA, et al. Increased mutagen sensitivity and DNA damage in pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine. 2015. Jul 15;192(2):219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mouraret N, Houssaïni A, Abid S, Quarck R, Marcos E, Parpaleix A, et al. Role for telomerase in pulmonary hypertension. Circulation [Internet]. 2015. [cited 2022 Jan 29];131(8):742–55. Available from: 10.1161/CIRCULATIONAHA.114.013258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Diman A, Boros J, Poulain F, Rodriguez J, Purnelle M, Episkopou H, et al. Nuclear respiratory factor 1 and endurance exercise promote human telomere transcription. Science Advances [Internet]. 2016. [cited 2022 Jan 29];2(7). Available from: 10.1126/sciadv.1600031 [DOI] [PMC free article] [PubMed]
- 123.Doran AC, Lehtinen AB, Meller N, Lipinski MJ, Slayton RP, Oldham SN, et al. Id3 Is a novel atheroprotective factor containing a functionally significant single-nucleotide polymorphism associated with intima-media thickness in humans. Circulation Research. 2010. Apr;106(7):1303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhou Y, Xu Z, Quan D, Zhang F, Zhang H, Xiao T, et al. Nuclear respiratory factor 1 promotes spheroid survival and mesenchymal transition in mammary epithelial cells. Oncogene 2018. 37:47. 2018 Jul 11;37(47):6152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Luna RCP, de Oliveira Y, Lisboa JVC, Chaves TR, de Araújo TAM, de Sousa EE, et al. Insights on the epigenetic mechanisms underlying pulmonary arterial hypertension. Brazilian Journal of Medical and Biological Research [Internet]. 2018. [cited 2022 Jan 29];51(12). Available from: /pmc/articles/PMC6207290/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Howell K, Preston RJ, McLoughlin P. Chronic hypoxia causes angiogenesis in addition to remodelling in the adult rat pulmonary circulation. The Journal of Physiology [Internet]. 2003. Feb 15 [cited 2022 Jan 29];547(Pt 1):133. Available from: /pmc/articles/PMC2342608/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen P, Cai X, Yang Y, Chen Z, Qiu J, Yu N, et al. Nuclear respiratory factor-1 (NRF-1) regulates transcription of the CXC receptor 4 (CXCR4) in the rat retina. Investigative Ophthalmology and Visual Science. 2017; [DOI] [PubMed] [Google Scholar]
- 128.Wei L, Zhang B, Cao W, Xing H, Yu X, Zhu D. Inhibition of CXCL12/CXCR4 suppresses pulmonary arterial smooth muscle cell proliferation and cell cycle progression via PI3K/Akt pathway under hypoxia. Journal of Receptors and Signal Transduction. 2015. Jul 4;35(4):329–39. [DOI] [PubMed] [Google Scholar]
- 129.Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Molecular Pharmacology. 2005; [DOI] [PubMed] [Google Scholar]
- 130.Parodi-Rullán R, Sone JY, Fossati S, Albensi B. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Journal of Alzheimer’s disease : JAD [Internet]. 2019. [cited 2022 Jan 29];72(4):1019–39. Available from: https://pubmed.ncbi.nlm.nih.gov/31306129/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Perez C, Felty Q, Roy D, Yoo C, Deoraj A. Abstract 4373: Molecular Contributions of NRF1 and ID3 to Vascular Toxicity in Cerebral Amyloid Angiopathy. Toxicological Sciences. 2022. Mar;(The Toxicologist). [Google Scholar]
- 132.Han BH, Zhou ML, Johnson AW, Singh I, Liao F, Vellimana AK, et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2015. Feb 24 [cited 2022 Jan 29];112(8):E881–90. Available from: /pmc/articles/PMC4345564/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, et al. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. The Journal of neuroscience : the official journal of the Society for Neuroscience [Internet]. 2006. [cited 2022 Jan 29];26(19):5167–79. Available from: https://pubmed.ncbi.nlm.nih.gov/16687508/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang GM, Deng MT, Lei ZH, Wan YJ, Nie HT, Wang ZY, et al. Effects of NRF1 on steroidogenesis and apoptosis in goat luteinized granulosa cells. Reproduction [Internet]. 2017. Aug 1 [cited 2022 Jan 29];154(2):111–22. Available from: https://rep.bioscientifica.com/view/journals/rep/154/2/REP-16-0583.xml [DOI] [PubMed] [Google Scholar]
- 135.Chong H, Wei Z, Namuhan, Sun G, Zheng S, Zhu X, et al. The PGC-1α/NRF1/miR-378a axis protects vascular smooth muscle cells from FFA-induced proliferation, migration and inflammation in atherosclerosis. Atherosclerosis [Internet]. 2020. Mar 1 [cited 2022 Jan 29];297:136–45. Available from: https://pubmed.ncbi.nlm.nih.gov/32120345/ [DOI] [PubMed] [Google Scholar]
- 136.Anne UB, Urfer R. Identification of a nuclear respiratory factor 1 recognition motif in the apolipoprotein e variant APOE4 linked to Alzheimer’s disease. Scientific Reports. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature 1999 401:6754 [Internet]. 1999. Oct 14 [cited 2022 Jan 29];401(6754):670–7. Available from: https://www.nature.com/articles/44334 [DOI] [PubMed] [Google Scholar]
- 138.Brulois K, Rajaraman A, Szade A, Nordling S, Bogoslowski A, Dermadi D, et al. A molecular map of murine lymph node blood vascular endothelium at single cell resolution. Nature Communications [Internet]. 2020. Dec 1 [cited 2021 May 26];11(1):1–15. Available from: 10.1038/s41467-020-17291-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.NICKENIG G, BAUDLER S, MÜLLER C, WERNER C, WERNER N, WELZEL H, et al. Redox-sensitive vascular smooth muscle cell proliferation is mediated by GKLF and Id3 in vitro and in vivo. FASEB journal : official publication of the Federation of American Societies for Experimental Biology [Internet]. 2002. Jul [cited 2022 Jan 29];16(9):1077–86. Available from: https://pubmed.ncbi.nlm.nih.gov/12087069/ [DOI] [PubMed] [Google Scholar]
- 140.Felty Q, Porther N. Estrogen-induced redox sensitive Id3 signaling controls the growth of vascular cells. Atherosclerosis [Internet]. 2008. May [cited 2021 Jun 15];198(1):12–21. Available from: https://pubmed.ncbi.nlm.nih.gov/18281048/ [DOI] [PubMed] [Google Scholar]
- 141.Wang LH, Baker NE. E-proteins and ID-proteins: Helix-loop-helix partners in development and disease. Developmental cell [Internet]. 2015. Nov 9 [cited 2022 Jan 29];35(3):269. Available from: /pmc/articles/PMC4684411/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee D, Shenoy S, Nigatu Y, Plotkin M. Id proteins regulate capillary repair and perivascular cell proliferation following ischemia-reperfusion injury. PLoS ONE [Internet]. 2014. Feb 7 [cited 2021 May 31];9(2):88417. Available from: /pmc/articles/PMC3917915/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.KF M, N S, G H, D B, T T, K K, et al. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes & development. 1995. Apr 15;9(8):935–44. [DOI] [PubMed] [Google Scholar]
- 144.Forrest ST, Taylor AM, Sarembock IJ, Perlegas D, McNamara CA. Phosphorylation Regulates Id3 Function in Vascular Smooth Muscle Cells. Circulation research. 2004. Sep 17;95(6):557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Das JK, Felty Q. PCB153-induced overexpression of ID3 contributes to the development of microvascular lesions. PloS one [Internet]. 2014. Aug 4 [cited 2022 Jan 29];9(8). Available from: https://pubmed.ncbi.nlm.nih.gov/25090023/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Das JK, Felty Q. Microvascular lesions by estrogen-induced ID3: its implications in cerebral and cardiorenal vascular disease. Journal of molecular neuroscience : MN [Internet]. 2015. Mar 1 [cited 2022 Jan 29];55(3):618–31. Available from: https://pubmed.ncbi.nlm.nih.gov/25129100/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Das JK, Voelkel NF, Felty Q. ID3 contributes to the acquisition of molecular stem cell-like signature in microvascular endothelial cells: Its implication for understanding microvascular diseases. Microvascular Research [Internet]. 2015. Mar 1 [cited 2018 Oct 8];98:126–38. Available from: https://www.sciencedirect.com/science/article/pii/S0026286215000072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lowery JW, Frump AL, Anderson L, DiCarlo GE, Jones MT, de Caestecker MP. ID family protein expression and regulation in hypoxic pulmonary hypertension. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology [Internet]. 2010. Dec [cited 2022 Jan 29];299(6):R1463. Available from: /pmc/articles/PMC3007180/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Das JK, Felty Q. PCB153-induced overexpression of ID3 contributes to the development of microvascular lesions. PLoS ONE. 2014. Aug 4;9(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee JH, Park SJ, Hariharasudhan G, Kim MJ, Jung SM, Jeong SY, et al. ID3 regulates the MDC1-mediated DNA damage response in order to maintain genome stability. Nature communications [Internet]. 2017. Dec 1 [cited 2022 Jan 29];8(1). Available from: https://pubmed.ncbi.nlm.nih.gov/29026069/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ji Y, Pos Z, Rao M, Klebanoff CA, Yu Z, Sukumar M, et al. Repression of the DNA-binding inhibitor Id3 by Blimp-1 limits the formation of memory CD8+ T cells. Nature Immunology [Internet]. 2011. [cited 2022 Jan 29];12(12):1230–7. Available from: https://www.academia.edu/13527781/Repression_of_the_DNA_binding_inhibitor_Id3_by_Blimp_1_limits_the_formation_of_memory_CD8_T_cells [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hasskarl J, Duensing S, Manuel E, Münger K. The helix-loop-helix protein ID1 localizes to centrosomes and rapidly induces abnormal centrosome numbers. Oncogene [Internet]. 2004. Mar 11 [cited 2022 Jan 29];23(10):1930–8. Available from: https://pubmed.ncbi.nlm.nih.gov/14755252/ [DOI] [PubMed] [Google Scholar]
- 153.Wang X, Di K, Zhang X, Han HY, Wong YC, Leung SCL, et al. Id-1 promotes chromosomal instability through modification of APC/C activity during mitosis in response to microtubule disruption. Oncogene 2008 27:32 [Internet]. 2008. Mar 31 [cited 2022 Jan 29];27(32):4456–66. Available from: https://www.nature.com/articles/onc200887 [DOI] [PubMed] [Google Scholar]
- 154.Manthey C, Mern DS, Gutmann A, Zielinski AJ, Herz C, Lassmann S, et al. Elevated endogenous expression of the dominant negative basic helix-loop-helix protein ID1 correlates with significant centrosome abnormalities in human tumor cells. BMC cell biology [Internet]. 2010. Jan 14 [cited 2022 Jan 29];11. Available from: https://pubmed.ncbi.nlm.nih.gov/20070914/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Federici C, Drake KM, Rigelsky CM, McNelly LN, Meade SL, Comhair SAA, et al. Increased Mutagen Sensitivity and DNA Damage in Pulmonary Arterial Hypertension. American journal of respiratory and critical care medicine [Internet]. 2015. Jul 15 [cited 2022 Jan 29];192(2):219–28. Available from: https://pubmed.ncbi.nlm.nih.gov/25918951/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kimishima Y, Misaka T, Yokokawa T, Wada K, Ueda K, Sugimoto K, et al. Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension with ALK1 upregulation in lung neutrophils. Nature Communications [Internet]. 2021. Dec 1 [cited 2022 Jan 29];12(1). Available from: /pmc/articles/PMC8548396/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Felty Q, Sakao S, Voelkel NF. Pulmonary Arterial Hypertension: A Stem Cell Hypothesis. 2015. [cited 2022 Jan 29];289–306. Available from: 10.1007/978-3-319-16232-4_16 [DOI] [Google Scholar]
- 158.Ohta-Ogo K, Hao H, Ishibashi-Ueda H, Hirota S, Nakamura K, Ohe T, et al. CD44 expression in plexiform lesions of idiopathic pulmonary arterial hypertension. Pathology international [Internet]. 2012. Apr [cited 2022 Jan 29];62(4):219–25. Available from: https://pubmed.ncbi.nlm.nih.gov/22449225/ [DOI] [PubMed] [Google Scholar]
- 159.Klein D, Weißhardt P, Kleff V, Jastrow H, Jakob HG, Ergün S. Vascular wall-resident CD44+ multipotent stem cells give rise to pericytes and smooth muscle cells and contribute to new vessel maturation. PloS one [Internet]. 2011. [cited 2022 Jan 29];6(5). Available from: https://pubmed.ncbi.nlm.nih.gov/21637782/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Abu El-Asrar AM, de Hertogh G, van den Eynde K, Alam K, van Raemdonck K, Opdenakker G, et al. Myofibroblasts in proliferative diabetic retinopathy can originate from infiltrating fibrocytes and through endothelial-to-mesenchymal transition (EndoMT). Experimental eye research [Internet]. 2015. Mar 1 [cited 2022 Jan 29];132:179–89. Available from: https://pubmed.ncbi.nlm.nih.gov/25637870/ [DOI] [PubMed] [Google Scholar]
- 161.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. The Journal of clinical investigation [Internet]. 2015. Dec 1 [cited 2022 Jan 29];125(12):4514–28. Available from: https://pubmed.ncbi.nlm.nih.gov/26517696/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature [Internet]. 2013. [cited 2022 Jan 29];498(7455):492–6. Available from: https://pubmed.ncbi.nlm.nih.gov/23748444/ [DOI] [PubMed] [Google Scholar]
- 163.Firth AL, Yao W, Remillard C v., Ogawa A, Yuan JXJ. Upregulation of Oct-4 isoforms in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. American journal of physiology Lung cellular and molecular physiology [Internet]. 2010. Apr [cited 2022 Jan 29];298(4). Available from: https://pubmed.ncbi.nlm.nih.gov/20139178/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Amaya CN, Bryan BA. Enrichment of the embryonic stem cell reprogramming factors Oct4, Nanog, Myc, and Sox2 in benign and malignant vascular tumors. BMC Clinical Pathology [Internet]. 2015. Sep 26 [cited 2022 Jan 29];15(1):1–8. Available from: 10.1186/s12907-015-0018-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Das JK, Doke M, Deoraj A, Felty Q, Roy D. Abstract 1128: Exosomal ID3 is pro-metastatic through guiding NRF1-induced breast cancer stem cells across the blood-brain-barrier. In: Cancer Research. American Association for Cancer Research (AACR); 2018. p. 1128–1128. [Google Scholar]
- 166.Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999. Oct 14;401(6754):670–7. [DOI] [PubMed] [Google Scholar]
- 167.Doke M, Felty QH. Abstract 3274: VEGF—receptor antagonist, Sugen 5416, sensitizes pulmonary endothelial stem-like cells to estrogens: A microvascular model for the progression of lung cancer. In American Association for Cancer Research (AACR); 2016. p. 3274–3274. [Google Scholar]
- 168.Doke M, Das J, Felty Q. Abstract 5052: ID3 mediated vascular reprogramming of PCB exposed endothelial cells and its potential contribution to lung tumorigenicity. In 2018. [Google Scholar]
- 169.Das JK, Doke M, Deoraj A, Felty Q, Roy D. Abstract 1128: Exosomal ID3 is pro-metastatic through guiding NRF1-induced breast cancer stem cells across the blood-brain-barrier. In: Cancer Research. American Association for Cancer Research (AACR); 2018. p. 1128–1128. [Google Scholar]
- 170.Biron KE, Dickstein DL, Gopaul R, Jefferies WA. Amyloid Triggers Extensive Cerebral Angiogenesis Causing Blood Brain Barrier Permeability and Hypervascularity in Alzheimer’s Disease. PLoS ONE [Internet]. 2011. [cited 2022 Jan 29];6(8):23789. Available from: /pmc/articles/PMC3166122/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Arnold TD, Niaudet C, Pang MF, Siegenthaler J, Gaengel K, Jung B, et al. Excessive vascular sprouting underlies cerebral hemorrhage in mice lacking αVβ8-TGFβ signaling in the brain. Development (Cambridge) [Internet]. 2014. Dec 1 [cited 2022 Jan 29];141(23):4489–99. Available from: /pmc/articles/PMC4302931/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Theofilatos K, Korfiati A, Mavroudi S, Cowperthwaite MC, Shpak M. Discovery of stroke-related blood biomarkers from gene expression network models. BMC Medical Genomics [Internet]. 2019. Aug 7 [cited 2022 Jan 29];12(1):1–15. Available from: 10.1186/s12920-019-0566-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005. Aug 15;21(16):3448–9. [DOI] [PubMed] [Google Scholar]
- 174.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research. 2019. Jan 8;47(D1):D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hsu MJ, Hsu CY, Chen BC, Chen MC, Ou G, Lin CH. Apoptosis signal-regulating kinase 1 in amyloid beta peptide-induced cerebral endothelial cell apoptosis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007. May 23;27(21):5719–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Masri FA, Xu W, Comhair SAA, Asosingh K, Koo M, Vasanji A, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2007. Sep;293(3). [DOI] [PubMed] [Google Scholar]
- 177.Quan Y, Xin Y, Tian G, Zhou J, Liu X. Mitochondrial ROS-Modulated mtDNA: A Potential Target for Cardiac Aging. Oxidative Medicine and Cellular Longevity [Internet]. 2020. [cited 2022 Jan 29];2020. Available from: /pmc/articles/PMC7139858/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gilles Freyssenet D, Berthon P. Mitochondrial Biogenesis in Skeletal Muscle in Response to Endurance Exercises Article in Archives of Physiology and Biochemistry. 1996; [DOI] [PubMed] [Google Scholar]
- 179.Sharma S, Aldred MA. DNA Damage and Repair in Pulmonary Arterial Hypertension. Genes. 2020. Oct 1;11(10):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Felty Q, Xiong WC, Sun D, Sarkar S, Singh KP, Parkash J, et al. Estrogen-induced mitochondrial reactive oxygen species as signal-transducing messengers. Biochemistry. 2005. May 1;44(18):6900–9. [DOI] [PubMed] [Google Scholar]
- 181.Jickling G, Salam A, Mohammad A, Hussain MS, Scozzafava J, Nasser AM, et al. Circulating endothelial progenitor cells and age-related white matter changes. Stroke. 2009. Oct;40(10):3191–6. [DOI] [PubMed] [Google Scholar]
- 182.Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999. Oct 14;401(6754):670–7. [DOI] [PubMed] [Google Scholar]
- 183.Das J, Felty Q, Poppiti R, Jackson R, Roy D. Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis. Cells. 2018. Nov 27;7(12):234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Forrest ST, Barringhaus KG, Perlegas D, Hammarskjold ML, McNamara CA. Intron retention generates a novel Id3 isoform that inhibits vascular lesion formation. Journal of Biological Chemistry. 2004. Jul 30;279(31):32897–903. [DOI] [PubMed] [Google Scholar]
- 185.Zebedee Z, Hara E. Id proteins in cell cycle control and cellular senescence. Oncogene. 2001. Dec;20(58):8317–25. [DOI] [PubMed] [Google Scholar]
- 186.Hasskarl J, Duensing S, Manuel E, Münger K. The helix–loop–helix protein ID1 localizes to centrosomes and rapidly induces abnormal centrosome numbers. Oncogene 2004 23:10. 2004. Feb 2;23(10):1930–8. [DOI] [PubMed] [Google Scholar]