

Abstract
Cdc42-interacting protein 4 (CIP4), a member of the F-BAR family, which plays an important role in regulating cell membrane and actin, has been reported to interact with Cdc42 and be closely associated with tumor invadopodia formation. In this study, we found that CIP4 expression was significantly higher in human CRC tissues and correlated with the CRC infiltrating depth and metastasis, as well as the lower survival rate in patients. In cultured CRC cells, knockdown of CIP4 inhibited cell migration and invasion ability in vitro and tumor metastasis in vivo, while the overexpression of CIP4 promoted invadopodia formation and matrix degradation ability. We then identified GTP-Cdc42 as a directly interactive protein of CIP4, which was upregulated and recruited by CIP4. Furthermore, activated NF-κB signaling pathway was found in CIP4 overexpression of CRC cells contributing to invadopodia formation, while the inhibition of either CIP4 or Cdc42 led to the suppression of the NF-κB pathway and resulted in a decreased quantity of invadopodia. Our findings suggested that CIP4 targets to recruit GTP-Cdc42 and directly combines with it to accelerate invadopodia formation and function by activating NF-κB signaling pathway, thus promoting CRC infiltration and metastasis.
Keywords: CIP4, Cdc42, NF-κB, invadopodia, colorectal cancer, invasion, metastasis
Graphical Abstract
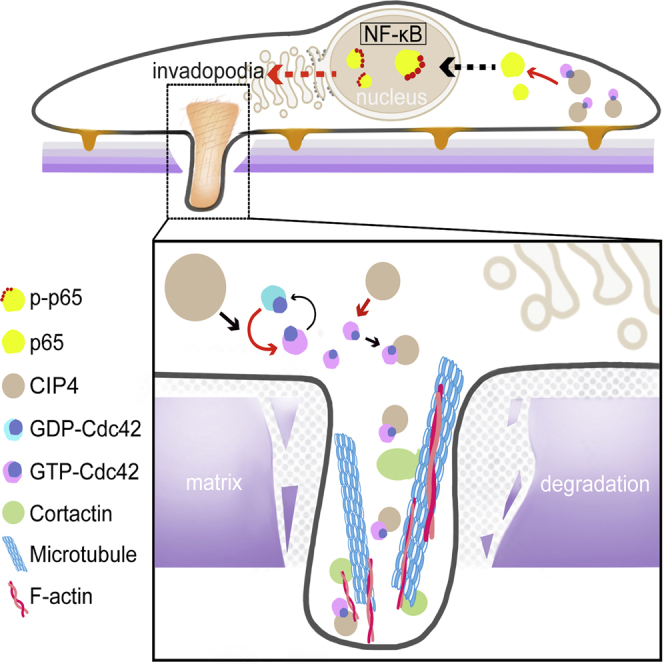
Hu and colleagues reveal the interaction between CIP4 and GTP-Cdc42 in invadopodia formation, along with the downstream NF-κB signaling pathway in response to the promotion of invasion and the metastasis of colorectal cancer, implicating a new treatment strategy that targets CIP4 against the metastasis of colorectal cancer.
Introduction
Colorectal cancer (CRC) was the third leading cancer type for new cases and deaths in 2021.1 Although surgical techniques and adjuvant therapy have advanced, the overall survival of CRC patients has not improved significantly in recent years.2 Liver and lung metastasis after radical resection and chemoradiotherapy is the most important cause of death in CRC patients.3 Metastasis is a complex biological process that needs cancer cells to form a leading edge and move forward.4 The invasion of cancer cells into surrounding tissue and the vasculature requires the chemotactic migration of cancer cells, steered by protrusive activity of the cell membrane and its attachment to the extracellular matrix (ECM).5 Recent work has uncovered a prominent actin-based cellular structure, called invadopodia, as a unique structural and functional module through which major invasive mechanisms are regulated.6, 7, 8 The co-localization of F-actin with actin-bundling protein cortactin combines with microtubules, driving the invadopodia to degrade the ECM and to facilitate distant metastasis.9
CIP4 (Cdc42-interacting protein 4) is a protein encoded by the TRIP10 gene located on human chromosome 19.10 CIP4 was first identified by using activated Cdc42 as a bait in a yeast two-hybrid screen, which contains an F-BAR domain at the N-terminal, an SH3 domain at the C-terminal, and an HR1 domain in the center.11,12 It has been reported that CIP4 plays an important role in various cellular events by regulating cell membranes and actin, such as vesicle formation, endocytosis, cytoplasmic membrane microtubule transformation, adhesion, and invadopodia formation in a variety of cells.13, 14, 15, 16, 17 Other studies have associated CIP4 with cell invasiveness and migration in different types of cancer, such as breast cancer, non-small cell lung cancer, nasopharyngeal carcinoma, and osteosarcoma , which indicates that CIP4 has a crucial part to play in tumor metastasis.18, 19, 20, 21, 22
CIP4 has been identified by the interaction with activated Cdc42. Cdc42 is a key member of the Rho family, which is well established to be central to the dynamic actin cytoskeletal assembly and rearrangement, the underpinnings of normal cell-cell adhesion, cell migration, and even transformation.23 Cdc42 is also involved in the regulation of invadopodia formation.24,25 Studies have demonstrated the podocalyxin-like 1 promotes invadopodia formation and metastasis through the activation of Rac1/Cdc42/cortactin signaling in breast cancer cells.26
Nuclear factor κB (NF-κB) is a critical cell signaling pathway that is involved in many cellular activities and can result in cancer if not appropriately regulated.27 Inappropriate activation of NF-κB leads to tumor proliferation, invasion, and metastasis.28,29 Early studies showed that Cdc42 regulates specifically in the NF-κB-dependent transcription.30,31 Therefore, in view of the special role of CIP4 and Cdc42 in invadopodia formation, along with the unknown downstream signaling pathway, whether NF-κB is in response to the activated Cdc42 interacting with CIP4 to promote CRC metastasis attracted our interest.
Our previous research found that CIP4 expression is significantly upregulated in human CRC tissues.32 In the present study, we demonstrated the expression and clinical significance of CIP4 in CRC samples, and provided evidence that CIP4 binds to GTP-Cdc42 to promote invadopodia formation and ECM degradation. Importantly, we further explored the downstream regulation mechanism of the complex of CIP4 and Cdc42 in CRC progression and metastasis, and here the NF-κB signaling pathway was confirmed to be essential.
Results
CIP4 expression in CRC tissues correlates with tumor development, invasiveness, and patient survival rate
Western blot was used to test the expression of CIP4 in 14 CRC tissues (T) and paired adjacent normal colorectal tissues (N). Our results revealed that CIP4 was upregulated in all of the 14 CRC tissues at the protein level (p < 0.0001; Figure 1A). Immunohistochemistry (IHC) staining was performed in 107 paraffin-embedded CRC tissue sections. The representative photographs showed that the expression of CIP4 was significantly elevated in human CRC tumors compared to the corresponding normal tissues. The CIP4 expression score was also higher in tumor tissues (p < 0.0001; Figure 1B). Interestingly, we found that CIP4 exhibits higher expression in the invasion front (a) than the tumor central area (b) in some CRC tissues (Figure 1C). The correlation between CIP4 expression level and CRC clinicopathological characteristics was analyzed further (Table 1). We found that the CIP4 level was closely related to tumor differentiation (p < 0.001) and invasive depth (p < 0.001).
Figure 1.

CIP4 expression in CRC tissues correlates with tumor development, invasiveness, and patient survival rate
(A) CIP4 protein expression in 14 pairs of human CRC tissues (T) and adjacent normal tissues (N) was detected by western blot. The quantification of protein levels was normalized to GAPDH. ∗∗∗p < 0.0001. (B) CIP4 protein expression in 107 paraffin-embedded normal human colorectal tissues (normal) and CRC tissues (adjacent, tumor 1, tumor 2) was detected by immunohistochemical staining (scale, 200μm, 100μm) and analyzed by scores. The error bars represent the mean ± SD; ∗∗∗p < 0.0001. (C) Comparison of CIP4 expression in the invasion front (a) and tumor central area (b) of CRC. (D) Bioinformatics analysis shows the relationship between CIP4 expression and patient survival times; p = 0.0353.
Table 1.
Clinicopathological and molecular characteristics of CIP4 expression of CRCs
| Variables | No. cases | Low | Medium | High | p |
|---|---|---|---|---|---|
| Age, y | |||||
| Younger (≤50) | 32 (29.9%) | 6 (5.6%) | 14 (13.1%) | 12 (11.2%) | 0.280 |
| Older (>50) | 75 (71.1%) | 14 (13.1%) | 44 (41.1%) | 17 (15.9%) | |
| Gender | |||||
| Male | 70 (65.4%) | 12 (11.2%) | 38 (35.5%) | 20 (18.7%) | 0.530 |
| Female | 37 (34.6%) | 8 (7.5%) | 20 (18.7%) | 9 (8.4%) | |
| Position | |||||
| Colon | 79 (73.8%) | 15 (14.0%) | 45 (42.1%) | 19 (17.8%) | 0.373 |
| Rectum | 28 (26.2%) | 5 (4.7%) | 13 (12.1%) | 10 (9.3%) | |
| Tumor size, cm (maximum diameter) | |||||
| ≤5 | 66 (61.7%) | 13 (12.1%) | 34 (31.8%) | 19 (17.8%) | 0.874 |
| >5 | 41 (38.3%) | 7 (6.5%) | 24 (22.4%) | 10 (9.3%) | |
| Tumor grade (differentiation) | |||||
| G1 | 18 (16.8%) | 10 (9.3%) | 7 (6.5%) | 1 (0.9%) | <0.001 |
| G2 | 82 (76.6%) | 10 (9.3%) | 50 (46.7%) | 22 (20.6%) | |
| G3 | 7 (6.5%) | 0 (0.0%) | 1 (0.9%) | 6 (5.6%) | |
| Invasive depth | |||||
| Submucosal | 9 (8.4%) | 5 (4.7%) | 3 (2.8%) | 1 (0.9%) | <0.001 |
| Myometrium | 16 (15.0%) | 9 (8.4%) | 4 (3.7%) | 3 (2.8%) | |
| Subserosal | 82 (76.6%) | 6 (5.6%) | 51 (47.7%) | 25 (23.4%) | |
| Mucinous component | |||||
| Absent | 23 (21.5%) | 5 (4.7%) | 9 (8.4%) | 9 (8.4%) | 0.437 |
| Present | 84(78.5%) | 15(14.0%) | 49(45.8%) | 20(18.7%) | |
To investigate whether the different levels of CIP4 expression in CRC are related to a patient’s prognosis, we performed a bioinformatic analysis of the NCBI GEO database (GEO: GSE17538). Kaplan-Meier survival analysis revealed that patients with a higher level of CIP4 expression had a worse clinical outcome (p = 0.0353, Figure 1D). These observations demonstrate that CIP4 may play an important role in CRC invasion and metastasis as well as a patient’s survival.
CIP4 promotes CRC cells migration and invasion in vitro and tumor metastasis in vivo
A high level of CIP4 expression was observed in Lovo and HT29 cells, while HCT116 and DLD1 showed a low level of CIP4 expression according to our previous research.32 To gain insight into the potential role of CIP4 in CRC invasion and metastasis, we generated Lovo-small hairpin CIP4 (shCIP4) and HT29-shCIP4 cell lines that stably downregulated CIP4, and HCT116-CIP4 and DLD1-CIP4 cell lines that stably overexpressed CIP4 (Figure 2A). Transwell migration and wound-healing assays were performed to evaluate the ability of cells tomigrate. The migration ability of Lovo-shCIP4 and HT29-shCIP4 cells was reduced compared with the control cells, while HCT116-CIP4 and DLD1-CIP4 cells showed increased migration ability compared with the control cells (p < 0.05; Figures 2B and 2C). The Matrigel-coated Boyden chamber invasion assay revealed that the knockdown of CIP4 significantly reduced the invaded cell numbers. Inversely, the overexpression of CIP4 accelerated the invasion ability of HCT116-CIP4 and DLD1-CIP4 cells (p < 0.01; Figure 2D).
Figure 2.

CIP4 promotes CRC cell migration and invasion in vitro and tumor metastasis in vivo
(A) The establishment of CIP4 stable knockdown (Lovo and HT29) and overexpression (HCT116 and DLD1) cell lines. CIP4 protein expression was detected by western blotting. (B) A wound healing assay was performed to evaluate the ability of cells to migrate (scale, 200 μm), and their migration ability was identified by wound healing percentage. The error bars represent the mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01. (C) The ability of cells to migrate in CRC cells was detected by the transwell migration assay (scale, 100 μm). The error bars represent the mean ± SD; ∗∗p < 0.01, ∗∗∗p < 0.001. (D) The invasion ability in CRC cells was detected by the Matrigel-coated Boyden chamber invasion assay (scale, 100 μm). The error bars represent the mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001. (E) The effect of CIP4 on tumor metastasis was assessed by an orthotopic xenograft CRC mouse model (scale, 100 μm). The number of liver metastatic nodules in individual mice was counted under the microscope and analyzed (∗p < 0.05, n = 3).
The effect of CIP4 on the tumor metastasis was assessed by an animal model for colon cancer metastasis. Spontaneously metastasizing colonic tumors were formed after the suture of colon cancer tumors into the cecal wall of BALB/c-nu/nu athymic mice. Eight weeks after operation, no liver metastatic nodules were found in all 5 HT29-shCIP4 groups (0 of 5), while 3 out of 5 HT29-shCtrl groups presented with liver metastasis (3 of 5). The numbers of nodules observed were 2, 3, and 5, respectively, as shown in Figure 2E. IHC staining confirmed that the tumors derived from the HT29-shCIP4 group exhibited fewer CIP4 expression levels than the tumors derived from the control cells.
CIP4 is sufficient for invadopodia formation and function in CRC cells
We evaluated the effect of CIP4 on invadopodia formation by examining the co-localization of F-actin (red) with actin-bundling protein Cortactin (green) in CRC cell lines. The suppression of CIP4 reduced the occurrence of invadopodia from 45.83% to 19.67% in Lovo cells (p < 0.001; Figure 3A) and from 26.00% to 16.67% in HT29 cells (p < 0.001, Figure S1A) and also shrank the morphology of invadopodia (Figure S1B). Meanwhile, more than 35% (HCT116) or 38% (DLD1) of cells contained invadopodia after overexpressing CIP4, which existed as bright, large puncta surrounding the nuclei, compared with 16.33% or 16.83% of control cells with smaller invadopodia, respectively (p < 0.01 and P < 0.001; Figures 3A, S1A, and S1B). In addition, we observed the effect of CIP4 on the morphology of invadopodia by scanning electron microscopy (SEM). A great quantity of elongated branching invadopodia in the ventral side of Lovo was observed. The number of invadopodia was reduced and the protrusions became shorter with the decreased expression of CIP4. The same phenomenon was found in HCT116 cells; the invadopodia was denser and more extended than control cells when CIP4 was overexpressed (Figure 3B).
Figure 3.

CIP4 is sufficient for invadopodia formation and function in CRC cells
(A) Quantification of cells with invadopodia was analyzed by immunofluorescence. The white arrowhead indicates the invadopodia. The error bars represent the mean ± SD (n = 200 cells). ∗∗p < 0.01, ∗∗∗p < 0.001. Magnification: 60×. (B) SEM showed the cell morphology in the indicated cells. Magnification: 1,000×, 2,000×, and 4,000×. (C) Matrix degradation assay was performed to analyze the invasion ability in cells. The quantification of FITC-gelatin degradation was detected by immunofluorescence. The error bars represent the mean ± SD (n = 100 cells). ∗p < 0.05, ∗∗p < 0.01. Magnification: 120×. (D) SEM showed the morphology of matrix degradation. Magnification: 150×, 2,000×.
The matrix degradation is an indispensable step in tumor metastasis,33 so we detected the ability of invadopodia to degrade matrix gels by the matrix degradation assay. The overexpression of CIP4 enlarged the cavities formed by the degradation of matrix gels by approximately 2.7-fold (HCT116, p < 0.01), while CIP4 knockdown reduced the cavities by 2.3-fold (Lovo, p < 0.05) (Figure 3C). We further observed the morphology of matrix degradation by CRC cells through SEM. As is shown in Figure 3D, the cells with high levels of CIP4 expression (Lovo-shCtrl and HCT116-CIP4) inserted the invadopodia deeply into the matrix gels and disintegrated it. On the contrary, the cells with low levels of CIP4 expression (Lovo-shCIP4 and HCT116-Ctrl) appeared to be smoother and merely adhered to the surface of the matrix gels. Based on the above data, we determined that CIP4 is necessary and sufficient to promote invadopodia formation and focal matrix degradation in CRC cells.
CIP4 promotes the expression and activation of Cdc42
Cdc42 cycles between activation with GTP and inactivation with GDP.34,35 It has been reported that the overexpression of an active form of Cdc42 is sufficient to form invadosome actin cores.36 Since CIP4 was first identified as an interacting protein of Cdc42, the regulatory mechanism between them in CRC remains to be revealed. In Figure 1A, we examined the expression levels of CIP4 in 14 pairs of CRC tissues, and we again examined the expression levels of Cdc42 in these tissues. The results showed that the protein level of Cdc42 in tumor tissue (T) was higher than that of matched normal colorectal tissue (N)—the same as CIP4 (p < 0.001; Figure 4A). As shown in Figure 4B, CIP4 and Cdc42 protein expression levels in consecutive paraffin-embedded slices of human CRC tissue were detected by IHC. We confirmed that the Cdc42 expression score was higher in tumor tissues (tumor 1, tumor 2) than normal tissues (Normal) (p < 0.01). In the meantime, we observed the consistence in expression and localization of CIP4 and Cdc42. The bioinformatic analyses indicated that there was a positive correlation between CIP4 and Cdc42 (R = 0.297, p = 0.023).
Figure 4.

CIP4 promotes the expression and activation of Cdc42
(A) Cdc42 protein expression in 14 pairs of human CRC tissues (T) and adjacent normal tissues (N) was detected by western blot. The quantification of protein levels was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). ∗∗∗p < 0.001. (B) Cdc42 expression was detected by immunohistochemistry staining in 58 paraffin-embedded normal human colorectal tissues and CRC tissues and analyzed by scores. The error bars represent the mean ± SD; ∗∗p < 0.01. Representative photographs of CIP4 and Cdc42 IHC staining (scale, 200 μm, 100 μm) of normal tissue (Normal) and CRC tissue (tumor 1, tumor 2), as indicated. Spearman’s correlation analysis showed the relationship between the CIP4 and Cdc42 expression levels in 58 human CRC tissues (r = 0.297, p = 0.023). (C) We used western blot to analyze the expression of CIP4, Cdc42, and GTP-Cdc42 in CIP4 knockdown Lovo and HT29 cells and CIP4 overexpressing HCT116 and DLD1 cells. Grayscale values were normalized to GAPDH. (D) The expression of CIP4 and Cdc42 in indicated cells was observed by immunofluorescence. Magnification: 60×.
To determine whether CIP4 regulates Cdc42 expression and activation, we detected the expression of Cdc42 and GTP-Cdc42 in Lovo and HT29 cells when CIP4 expression was blocked, and we found that the decreased protein level of Cdc42 and GTP-Cdc42 compared with the control cells. Conversely, the raised protein level of Cdc42 and GTP-Cdc42 was found in HCT116 and DLD1 cells when CIP4 expression was increased (Figure 4C). The immunofluorescence results also confirmed that the expression of Cdc42 (red) was positively correlated with CIP4 (green) (Figure 4D).
CIP4 directly interacts with activated Cdc42 to accelerate invadopodia function
Since we have demonstrated that CIP4 can promote the activation of Cdc42, whether CIP4 interacts with activated Cdc42 and participates in the process of invasion and metastasis of CRC is essential to our research. The immunofluorescence assays validated the localization of CIP4 and its partial co-localization with Cdc42 (Figure 5A). The co-localization coefficients between CIP4 and Cdc42 in Lovo cells and HT29 cells are 0.64 (n = 5) and 0.71 (n = 5); in HCT116-CIP4 and DLD1-CIP4 cells the coefficients are 0.42 (n = 5) and 0.38 (n = 5) (Figure S2A).
Figure 5.

CIP4 directly interacts with activated Cdc42 to accelerate invadopodia function
(A) Co-localization of CIP4 (green) and Cdc42 (red) in Lovo and HT29 cells was observed by immunofluorescence. Magnification: 180×. The Pearson’s correlation and overlap coefficient were shown in bar graph format. The error bars represent the mean ± SD (n = 5). (B) GST pull down experiments on CIP4 protein with mutants of activated Cdc42. GST and HIS were analyzed by western blot. (C) The localization of CIP4 and invadopdia was observed in Lovo and HT29 cells by confocal laser scanning. The white arrowhead indicates the co-localization of CIP4 and invadopodia. Magnification: 180×. (D) The localization of FLAG-tagged activated Cdc42 and invadopdia was observed in CIP4 overexpression or low-expression cells by confocal laser scanning. The white arrowhead indicates the co-localization of FLAG-tagged activated Cdc42 and invadopodia. Magnification: 180×.
Cdc42 has the ability to combine guanosine diphosphate/guanosine triphosphate (GDP/GTP). In response to extracellular stimuli, Cdc42 transitions from an inactive GDP-bound status to an active GTP-bound status and interacts with downstream effectors to propagate changes in cell behaviors. We constructed a prokaryotic vector for the mutants of activated Cdc42 (L61Cdc42), containing a Gln61-to-Leu substitution. Next, we performed the glutathione S-transferase (GST) pull-down assay to see whether CIP4 combines GTP-Cdc42 directly. Coomassie brilliant blue staining showed the clear protein bands with consistent molecular weights (GST for 26 kDa, CIP4-GST for 101 kDa, and L61Cdc42-6HIS for 27 kDa) (Figure S2C). We then detected the expression of CIP4-GST and L61Cdc42-6HIS fusion protein by western blot, which showed the combination between L61Cdc42-6HIS and CIP4-GST, while the negative control GST did not combine with L61Cdc42-6HIS (Figure 5B). According to the results, we verified that CIP4 directly interacts with GTP-Cdc42 and may form a complex to participate in the formation of invadopodia.
In the previous data, we determined that CIP4 is necessary and sufficient to promote invadopodia formation. We then validated the localization of CIP4 (green) and its partial co-localization with Cortactin (blue) and F-actin (red) in Lovo and HT29 cells by confocal laser scanning (Figure 5C). Representative photographs indicate that CIP4 not only promotes the formation of invadopodia but also assembles in the invadopodia. We further constructed a lentivirus vector FLAG- L61Cdc42 that continuously activated Cdc42 mutant L61Cdc42 and transfected it into the CIP4-overexpression or CIP4-knockdown CRC cells as well as their control cells. After performing the immunofluorescence assays to locate the invadopodia (blue for Cortactin and red for F-actin) and activated Cdc42 (green for FLAG, representing the activated Cdc42), we found that in the cells with a high expression level of CIP4 (Lovo-shCtrl, HT29-shCtrl, HCT116-CIP4, and DLD1-CIP4), the activated Cdc42 was more likely to gather in the invadopodia, while in the cells with a low expression level of CIP4 (Lovo-shCIP4, HT29-shCIP4, HCT116-Ctrl, and DLD1-Ctrl), the activated Cdc42 tended to scatter throughout the cells (Figure 5D). Therefore, we inferred that CIP4 recruits GTP-Cdc42 into the invadopodia and interacts with it, promoting the formation and function of invadopodia.
The NF-κB signaling pathway is involved in accelerating invadopodia formation regulated by CIP4 through GTP-Cdc42
GTP-Cdc42 has been reported to be able to activate the NF-κB signaling pathway.31 Since NF-κB is widely known to be a critical part of tumor invasion and metastasis,37,38 we wondered whether the invadopodia formation was accelerated by NF-κB, which was regulated by CIP4 through GTP-Cdc42. We investigated the effects of CIP4 on the key molecule of the NF-κB signaling pathway, RelA (p65), the phosphorylation status of p65 (p-p65), and the p65 in the nucleus.39,40 As is shown in Figures 6A and S4A, the downregulation of CIP4 in Lovo-shCIP4 and HT29-shCIP4 cells led to the decreased expressions of Cdc42 and GTP-Cdc42 as well as p-p65 and the p65 in the nucleus compared with the control cells. We then overexpressed the activated Cdc42 in Lovo-shCIP4 and HT29-shCIP4 cells and found the expressions of p-p65 and the p65 in the nucleus reverted. After being treated with the NF-κB activator lipopolysaccharide (LPS), the expressions of p-p65 and the p65 in the nucleus increased significantly while the expressions of CIP4, Cdc42, and GTP-Cdc42 remained low.
Figure 6.

The NF-κB signaling pathway is involved in accelerating invadopodia formation regulated by CIP4 through GTP-Cdc42
(A and B) We used western blot to analyze the expression of CIP4, Cdc42, GTP-Cdc42, p65, p-p65, and p65 in the nucleus in Lovo (the CIP4-knockdown cells were treated with LPS, 10 μg/mL for 0.5 h) and HCT116 (the CIP4-overexpressed cells were treated with ML141, 20μM for 36 h, or QNZ, 10μM for 8 h) cells, as indicated. Grayscale values were normalized to GAPDH and histone H3 in the nucleus. (C and D) The quantification of cells with invadopodia was analyzed by immunofluorescence. Lovo CIP4-knockdown cells were treated with LPS, 10 μg/mL for 0.5 h, HCT116 CIP4-overexpression cells were treated with ML141, 20μM for 36 h, or QNZ, 10μM for 8 h. The white arrowhead indicates the invadopodia. The error bars represent the mean ± SD (Lovo: p = 0.0002, HCT116: p < 0.0001, n = 200 cells). Magnification: 60×.
Correspondingly, the overexpression of CIP4 upregulated the expressions of Cdc42 and GTP-Cdc42 as well as p-p65 and the p65 in the nucleus in HCT116-CIP4 and DLD1-CIP4 cells compared with the control cells. When treated with the Cdc42 inhibitor ML141, the expressions of Cdc42 and GTP-Cdc42 decreased as did p-p65 and the p65 in the nucleus. After being treated with the NF-κB inhibitor QNZ, the expressions of p-p65 and the p65 in the nucleus was suppressed, while the expressions of CIP4, Cdc42, and GTP-Cdc42 barely changed (Figures 6B and S4B). The optimum conditions of activators and inhibitors to treat cells were examined in Figure S3. These results indicated the NF-κB was the downstream pathway regulated by CIP4 through GTP-Cdc42.
Next, we detected the influence of the expression changes of these proteins on the formation of invadopodia by immunofluorescence assays (Figures 6C, 6D, S4C, and S4D). The quantity of invadopodia was consistent with the expression level of p-p65 (p < 0.01), which revealed that the activation of the NF-κB signaling pathway was an indispensable part of invadopodia formation. Therefore, our data demonstrated that CIP4 promotes the expression and activation of Cdc42, which activates the NF-κB signaling pathway, thereby accelerating the invadopodia formation.
Discussion
CRC is one of the most common cancers worldwide.41 Although much information on the molecular basis of CRC has been provided by numerous researchers to develop all kinds of targeted therapeutic options, the clinical cure rate remains unsatisfactory.42 The main obstacle of improving the clinical treatment effect of CRC currently is metastasis, which is a complex biological process involving various molecular regulations that still need to be clearly revealed. Therefore, to elucidate the molecular mechanism of CRC infiltration and metastasis and to intervene and treat its progress are scientifically significant in improving the cure rate of malignant tumors and the survival rate of patients.
In a previous study, we found significantly higher expression of CIP4 in CRC cells compared to corresponding normal tissues, and demonstrated that PRKA kinase anchor protein 9 (AKAP-9) regulates the expression of CIP4 to promote metastasis and, consequently, the epithelial-mesenchymal transition (EMT) of CRC.32 A similar phenomenon is observed in breast cancer cells, in which CIP4 interacts with N-WASP in response to epidermal growth factor receptor (EGFR) increasing the formation of invadopodia and ECM degradation.19 While CIP4 is regarded as a suppressor of Src-induced invadopodia formation and invasiveness in MDA-MB-231 breast tumor cells.18 CIP4 also increases the expression and activity of matrix metalloproteinase-2 (MMP-2) by regulating EGFR signaling to promote metastasis in non-small cell lung cancer and nasopharyngeal carcinoma cells.20,21 The ability of CIP4 to facilitate metastasis and invasiveness is confirmed in hepatocellular carcinoma (HCC), osteosarcoma, and renal cell carcinoma as well.22,43,44 Although multiple molecular signals are involved in CIP4 function on cancer cells as reported, the specific mechanism of the interaction between CIP4 and Cdc42, which is the most well-known directly related protein to CIP4, remains unclear. Therefore, we focused on exploring the association between CIP4 and Cdc42 in invadopodia formation and ECM degradation, as well as the downstream signaling pathway in response in CRC.
Our results confirmed the high expression of CIP4 in CRC tissues, especially in the invasion front. The functional experiments in vivo and in vitro support our assumption that CIP4 plays an important role in promoting metastasis in CRC. Since the invadopodia formation is closely related to CIP4, we observed the quantity and morphology as well as the ECM degradation ability changes of invadopodia in CRC cells under different expression levels of CIP4, which indicates that CIP4 is capable of accelerating the formation and function of invadopodia. To figure out whether the interaction between CIP4 and Cdc42 contributes to this cellular structure, we verified that CIP4 can upregulate the expression and activation of Cdc42 and directly combine with the activated Cdc42. We have not been able to identify the specific binding site yet, but we believe that it may be the HR1 domain, as mentioned.45 Furthermore, we spotted an interesting phenomenon that the complex of CIP4 and GTP-Cdc42 is located on invadopodia, and GTP-Cdc42 has been proved to be recruited by CIP4 to assemble in invadopodia to maximize the impact. While further exploration of the downstream molecular mechanism is required, we noticed the wildly activated signaling pathway NF-κB in CRC, which supports tumorigenesis by enhancing the cell invasion and metastasis.46,47 As expected, the high expression level of CIP4 and activated Cdc42 results in the increased phosphorylation of RelA (p65), which controls a great amount of NF-κB activity and promotes the formation of invadopodia. While the inhibition of CIP4 and Cdc42 leads to the suppression of the NF-κB pathway ended up with decreased quantity of invadopodia. These results highlight the essential role of the NF-κB pathway in invadopodia formation and function regulation by the interaction between CIP4 and GTP-Cdc42.
Taken together, our studies reveal the molecular mechanism of CIP4 promoting CRC infiltration and metastasis. The combination with GTP-Cdc42 and the activation of the NF-κB signaling pathway associated with invadopodia formation and function are particularly important in CRC. Considering the unclarity of all of the details in the interaction, along with the recent study that identified the CIP4 phosphorylation by protein kinase A (PKA) on the modulation of cancer invasion,43 our future research will focus on the possible modification of CIP4 that leads to the interaction with Cdc42 as well as the specific mechanism of RelA (p65) phosphorylation regulating invadopodia formation and function.
Materials and methods
Clinical samples
Formalin-fixed paraffin-embedded human colorectal carcinoma tissues (n = 107) for this study were obtained from the Department of Pathology, Nanfang Hospital, Southern Medical University. Each case was confirmed a definite diagnosis of primary CRC. The fresh surgically resected CRC tissues and matched adjacent normal tissues (n = 14) were immediately frozen in liquid nitrogen till the later study. The study was approved by Nanfang Hospital, Southern Medical University, Guangzhou, China.
Cell culture and the construction of stable cell lines
CRC cell lines, including Lovo, HT29, HCT116, and DLD1 cells, were obtained from the Global Bioresource Center (American Type Culture Collection [ATCC], Manassas, VA, USA). All of the cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; GIBCO, Thermo Fisher Scientific, Waltham, MA, USA) at 37°C in a humidified atmosphere with 5% CO2. The plasmid (hU6-MCS-SV40-neomycin) carrying CIP4-repressing shRNA sequence (shCIP4, 5′-GGAGAAUAGUAAGCGUAAATT-3′) and the empty vector (shCtrl) used as control to shCIP4 were purchased from Genechem (Shanghai, China). The lentivirus vector carrying the human CIP4 sequence (CIP4) and the lentivirus containing a scrambling sequence (Ctrl) used as control to CIP4 overexpression were purchased from Genechem. The lentivirus vector carrying the FLAG-L61Cdc42 sequence, which contained a Gln61-to-Leu substitution to activated Cdc42 constantly, was purchased from Genechem. Lovo and HT29 cells were transfected with shCIP4 and shCtrl using Lipofectamine 3000 transfection reagent (Invitrogen, Thermo Fisher Scientific), referring to the specification. Stable cell lines were obtained by resistance screening with G418 (Sigma-Aldrich, St. Louis, MO, USA) at the optimal concentration for 15 days. HCT116 and DLD1 cells were transfected with CIP4, and Ctrl, Lovo-Ctrl, Lovo-shCIP4, HT29-Ctrl, HT29-shCIP4, HCT116-Ctrl, HCT116-CIP4, DLD1-Ctrl, and DLD1-CIP4 cells were transfected with FLAG-L61Cdc42, referring to the specification provided by Genechem. Stable cell lines were obtained by resistance screening with puromycin (Sigma-Aldrich) at a concentration of 5 ng/mL for 10 days. The transfection efficiency was assessed by western blot analysis.
Orthotopic xenograft CRC mouse model
Male athymic BALB/c mice 4–6 weeks old were purchased from the Central Laboratory of Animal Science at Southern Medical University. All of the animal care and experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Nanfang Hospital, Southern Medical University. The HT29-shCtrl and HT29-shCIP4 cells were prepared and suspended by fresh PBS to a concentration of 1 × 107 cells/100 μL. Eighty-microliter volumes of cells were injected into the scapular skin of each athymic BALB/c mice. The formed subcutaneous tumor tissues were planted separately into the cecal wall of athymic BALB/c mice. After 8 weeks, the orthotopic xenograft CRC masses and the livers of nude mice were surgically removed after euthanasia, fixed in formalin (neutral buffered 10%), embedded in paraffin, and prepared into 2.5-μm sections for hematoxylin and eosin (H&E) staining and IHC analysis.
Reagents and antibodies
Antibody against CIP4 (cat. no. 612556) was purchased from BD Biosciences (Franklin Lakes, NJ, USA). Antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (cat. no. 60004), Cdc42 (cat. no. 10155), cortactin (cat. no. 11381), histone H3 (cat. no. 17168), and FLAG tag (cat. no. 66008) were purchased from Proteintech (Wuhan, China). Antibodies against p65 (cat. no. 8242), p-p65 (cat. no. 3033), and the Active Cdc42 Detection Kit (cat. no. 8819) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against His tag (cat. no. 0287R) and GST tag (cat. no. 33007M) were purchased from Bioss (Beijing, China). The Cdc42 inhibitor ML141 and the NF-κB signaling inhibitor QNZ were purchased from Selleck Chemicals (Shanghai, China). The NF-κB signaling activator LPS was purchased from Sigma-Aldrich.
Western blot analysis
Total proteins from cell or tissue were separated by lysis buffer (FDbio, Hangzhou, China), and the concentration was detected by bicinchoninic acid (BCA) protein assay kits (FDbio). Proteins were separated by 10% or 12.5% SDS-PAGE gel and transferred to polyvinylidene fluoride (PVDF) membranes. After being blocked by 5% skim milk for 1 h at room temperature, the protein bands were incubated with primary antibodies at 4°C overnight. The membranes were then incubated with goat anti-mouse or anti-rabbit secondary antibody (FDbio) and detected by enhanced chemiluminescence.
IHC analysis
The IHC procedure referred to the specification of rabbit and mouse two-step immunohistochemical detection kit (ZSGB-BIO, Beijing, China). The paraffin-embedded human CRC tissue sections were incubated with primary antibodies against CIP4, Cdc42, and Ki-67 (working solution, ZSGB-BIO). Two experienced pathologists observed and scored the degree of staining in the sections independently. The staining intensity was scored as follows: 0 (no staining), 1 (light yellow), 2 (brownish yellow), and 3 (brown), and the percentage of positive staining cells was scored as follows: 1 (<10%), 2 (10%–50%), 3 (50%–70%), and 4 (>70%). Each section was comprehensively calculated by multiplying the staining intensity score and the percentage positivity score. We defined 0 as negative, 1–4 as low, 5–8 as medium, and >8 as high.
Wound-healing assay
Cells (5–10 × 105) were seeded into 6-well plates and cultured to 90% confluence. Cell monolayers were wounded by a sterile 10-μL pipette tip. The detached cells were removed by PBS. After culturing for another 48 h, the wound gaps were observed and photographed by an inverted microscope (Olympus, Tokyo, Japan).
Transwell migration and invasion assays
Cells (0.1–1 × 106) were resuspended in 200 μL serum-free medium and seeded in the 24-well transwell upper chambers (Corning, Corning, NY, USA). A total of 500 μL RPMI 1640 medium containing 10% FBS was added into the lower chambers. After culturing for 12–48 h, cells on the membrane were fixed with formalin, stained with H&E, and then calculated and photographed using the ordinary optics microscope (Olympus). Invasion assays were performed according to the same procedures with the diluted Matrigel (Corning) (RPMI 1640: Matrigel = 5:1) spread on the inside bottom of the transwell upper chambers.
Immunofluorescence
Cells were seeded on confocal disks (Nest, Wuxi, China) and cultured for 24 h, washed with PBS, fixed with 4% formaldehyde for 30 min, permeabilized with 0.25% Triton X-100 for 8 min, blocked with goat serum (ZSGB-BIO) for 45 min, and then incubated with primary antibodies at 4°C overnight. After being incubated with Alexa 488/594/355 conjugated secondary antibodies (ZSGB-BIO) or rhodamine phalloidin for 1 h and 4′,6-diamidino-2-phenylindole (DAPI) counterstaining for 10 min, the cells were observed and photographed by the Olympus confocal fluorescence microscope (FV1000).
SEM
Cells were seeded on 8-mm-diameter pre-cleaned coverslips and cultured in 24-well plates for 36 h, washed with PBS, and fixed with 2.5% glutaraldehyde at 4°C overnight. After being washed with PBS, the cells on coverslips were dehydrated by graded ethanol at 4°C, soaked in 100% acetone for 20 min, in 100% isoamyl acetate for 15 min and in propylene epoxide for 20 min at 45°C. The coverslips were put in vacuum and sprayed with metal foil. Further observations were made under SEM.
Matrix degradation assay
The confocal disks (Nest) were incubated with 200 μL of 50 μg/mL polylysine, 200 μL 0.5% glutaraldehyde, 200 μL 0.2% gelatin from pig Oregon Green 488 (Invitrogen), and 200 μL 5 mg/mL sodium borohydride, each for 15 min at room temperature and washed with PBS 3 times in between. Cells (5×103) were seeded in each disk and cultured for 48 h. Then performed the procedures of immunofluorescence.
GST pull-down
The Ctrl-GST, CIP4-GST, and L61Cdc42-6HIS prokaryotic expression vectors were constructed and purchased from Genechem. The fusion proteins were induced by isopropyl-β-d-1-thiogalactopyranoside (IPTG) in BL21 (TransGene Biotech, Beijing, China) growing in Luria-Bertani (LB) ampicillin. The optimum inducement condition for CIP4-GST is 0.5 mM of IPTG in 16°C for 20 h, and for L61Cdc42-6HIS is 0.8 mM of IPTG in 16°C for 12 h (Figure S2B). The proteins were purified by combining GST-agarose or His-agarose at 4°C overnight. The purified Ctrl-GST and CIP4-GST proteins were eluted from the GST-agarose and combined with L61Cdc42-6HIS fusion protein and His-agarose at 4°C overnight. After being washed 5 times, the remaining protein mixture combined with His-agarose was analyzed by Coomassie brilliant blue staining and western blot assays.
Statistical analysis
All of the statistical analyses were performed using SPSS version 24.0 (SPSS Statistics, Armonk, NY, USA) and presented as the mean ± standard deviation (SD). The co-localization coefficients between CIP4 and Cdc42 were analyzed using ImageJ version 2.1.0 (NIH, Bethesda, MD, USA). Graphs were plotted using Prism 7.0a (GraphPad, San Diego, CA, USA). The significance of correlation between the expression of CIP4 and histopathological factors was determined using the Pearson χ2 test. The enumeration data was analyzed using the Student’s t test or one-way ANOVA. Survival curves of CIP4 expression in CRC patients was carried out using the Kaplan-Meier method. p values of 0.05 or lower were considered statistically significant.
Data and code availability
All of the data generated or analyzed during this study are included in this article.
Acknowledgments
We thank Dr. Yanqing Ding, Dr. Li Liang, and Dr. Xin Li for providing the scientific research platform. We thank the Department of Pathology, Nanfang Hospital, Southern Medical University, China, for the clinical specimens. We thank Xia Wang, Qingcan Sun, Xiangling Lin, and Kehong Zheng for inspiring discussions and technical help. This work was supported by the National Natural Science Foundation of China (K1011482 and K117280039),the Provincial Science Foundation of Guangdong (G820281143), and Shenzhen Hospital, Southern Medical University.
Author contributions
Conceptualization, Z.L. and Z.H.; methodology, Z.H., J.Z., and Y.M.; investigation, J.Z. and Y.M.; data curation, J.Z., Y.M., and T.L.; formal analysis, J.Z., Y.M., and T.L.; software, J.Z., Y.M., and L.G.; validation, Y.Z. and X.W.; writing – original draft, J.Z. and Y.M.; writing – review & editing, J.Z. and Z.H.; visualization, J.Z.; funding acquisition, Z.L. and Z.H.; resources, Z.L. and Z.H.; supervision, Z.L.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.02.023.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2021. CA Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Eswaran J., Li D.Q., Shah A., Kumar R. Molecular pathways: targeting P21-activated kinase 1 signaling in cancer-opportunities, challenges, and limitations. Clin. Cancer Res. 2012;18:3743–3749. doi: 10.1158/1078-0432.Ccr-11-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pretzsch E., Bosch F., Neumann J., Ganschow P., Bazhin A., Guba M., Werner J., Angele M. Mechanisms of metastasis in colorectal cancer and metastatic organotropism: hematogenous versus peritoneal spread. J. Oncol. 2019;2019:7407190. doi: 10.1155/2019/7407190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paschos K.A., Majeed A.W., Bird N.C. Natural history of hepatic metastases from colorectal cancer--pathobiological pathways with clinical significance. World J. Gastroenterol. 2014;20:3719–3737. doi: 10.3748/wjg.v20.i14.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamaguchi H., Wyckoff J., Condeelis J. Cell migration in tumors. Curr. Opin. Cell Biol. 2005;17:559–564. doi: 10.1016/j.ceb.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Gimona M., Buccione R., Courtneidge S.A., Linder S. Assembly and biological role of podosomes and invadopodia. Curr. Opin. Cell Biol. 2008;20:235–241. doi: 10.1016/j.ceb.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Eddy R.J., Weidmann M.D., Sharma V.P., Condeelis J.S. Tumor cell invadopodia: invasive protrusions that orchestrate metastasis. Trends Cell Biol. 2017;27:595–607. doi: 10.1016/j.tcb.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy D.A., Courtneidge S.A. The 'ins' and 'outs' of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011;12:413–426. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowden E.T., Onikoyi E., Slack R., Myoui A., Yoneda T., Yamada K.M., Mueller S.C. Co-localization of cortactin and phosphotyrosine identifies active invadopodia in human breast cancer cells. Exp. Cell Res. 2006;312:1240–1253. doi: 10.1016/j.yexcr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Aspenstrom P. A Cdc42 target protein with homology to the non-kinase domain of FER has a potential role in regulating the actin cytoskeleton. Curr. Biol. 1997;7:479–487. doi: 10.1016/s0960-9822(06)00219-3. [DOI] [PubMed] [Google Scholar]
- 11.Liu S., Xiong X., Zhao X., Yang X., Wang H. F-BAR family proteins, emerging regulators for cell membrane dynamic changes-from structure to human diseases. J. Hematol. Oncol. 2015;8:47. doi: 10.1186/s13045-015-0144-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richnau N., Fransson A., Farsad K., Aspenstrom P. RICH-1 has a BIN/Amphiphysin/Rvsp domain responsible for binding to membrane lipids and tubulation of liposomes. Biochem. Biophys. Res. Commun. 2004;320:1034–1042. doi: 10.1016/j.bbrc.2004.05.221. [DOI] [PubMed] [Google Scholar]
- 13.Tian L., Nelson D.L., Stewart D.M. Cdc42-interacting protein 4 mediates binding of the Wiskott-Aldrich syndrome protein to microtubules. J. Biol. Chem. 2000;275:7854–7861. doi: 10.1074/jbc.275.11.7854. [DOI] [PubMed] [Google Scholar]
- 14.Saengsawang W., Taylor K.L., Lumbard D.C., Mitok K., Price A., Pietila L., Gomez T.M., Dent E.W. CIP4 coordinates with phospholipids and actin-associated proteins to localize to the protruding edge and produce actin ribs and veils. J. Cell Sci. 2013;126:2411–2423. doi: 10.1242/jcs.117473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saengsawang W., Mitok K., Viesselmann C., Pietila L., Lumbard D.C., Corey S.J., Dent E.W. The F-BAR protein CIP4 inhibits neurite formation by producing lamellipodial protrusions. Curr. Biol. 2012;22:494–501. doi: 10.1016/j.cub.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts-Galbraith R.H., Gould K.L. Setting the F-BAR: functions and regulation of the F-BAR protein family. Cell Cycle. 2010;9:4091–4097. doi: 10.4161/cc.9.20.13587. [DOI] [PubMed] [Google Scholar]
- 17.Fricke R., Gohl C., Dharmalingam E., Grevelhorster A., Zahedi B., Harden N., Kessels M., Qualmann B., Bogdan S. Drosophila Cip4/toca-1 integrates membrane trafficking and actin dynamics through WASP and SCAR/WAVE. Curr. Biol. 2009;19:1429–1437. doi: 10.1016/j.cub.2009.07.058. [DOI] [PubMed] [Google Scholar]
- 18.Hu J., Mukhopadhyay A., Truesdell P., Chander H., Mukhopadhyay U., Mak A., Craig A. Cdc42-interacting protein 4 is a Src substrate that regulates invadopodia and invasiveness of breast tumors by promoting MT1-MMP endocytosis. J. Cell Sci. 2011;124:1739–1751. doi: 10.1242/jcs.078014. [DOI] [PubMed] [Google Scholar]
- 19.Pichot C., Arvanitis C., Hartig S., Jensen S., Bechill J., Marzouk S., Yu J., Frost J., Corey S. Cdc42-interacting protein 4 promotes breast cancer cell invasion and formation of invadopodia through activation of N-WASp. Cancer Res. 2010;70:8347–8356. doi: 10.1158/0008-5472.Can-09-4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng D., Xie P., Peng L., Sun R., Luo D., Chen Q., Lv X., Wang L., Chen M., Mai H., et al. CDC42-interacting protein 4 promotes metastasis of nasopharyngeal carcinoma by mediating invadopodia formation and activating EGFR signaling. J. Exp. Clin. Cancer Res. 2017;36:21. doi: 10.1186/s13046-016-0483-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Truesdell P., Ahn J., Chander H., Meens J., Watt K., Yang X., Craig A. CIP4 promotes lung adenocarcinoma metastasis and is associated with poor prognosis. Oncogene. 2015;34:3527–3535. doi: 10.1038/onc.2014.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koshkina N., Yang G., Kleinerman E. Inhibition of Cdc42-interacting protein 4 (CIP4) impairs osteosarcoma tumor progression. Curr. Cancer Drug Targets. 2013;13:48–56. doi: 10.2174/156800913804486593. [DOI] [PubMed] [Google Scholar]
- 23.Sadok A., Marshall C.J. Rho GTPases: masters of cell migration. Small GTPases. 2014;5:e29710. doi: 10.4161/sgtp.29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakahara H., Otani T., Sasaki T., Miura Y., Takai Y., Kogo M. Involvement of Cdc42 and Rac small G proteins in invadopodia formation of RPMI7951 cells. Genes Cells. 2003;8:1019–1027. doi: 10.1111/j.1365-2443.2003.00695.x. [DOI] [PubMed] [Google Scholar]
- 25.Qadir M.I., Parveen A., Ali M. Cdc42: role in cancer management. Chem. Biol. Drug Des. 2015;86:432–439. doi: 10.1111/cbdd.12556. [DOI] [PubMed] [Google Scholar]
- 26.Lin C.W., Sun M.S., Liao M.Y., Chung C.H., Chi Y.H., Chiou L.T., Yu J., Lou K.L., Wu H.C. Podocalyxin-like 1 promotes invadopodia formation and metastasis through activation of Rac1/Cdc42/cortactin signaling in breast cancer cells. Carcinogenesis. 2014;35:2425–2435. doi: 10.1093/carcin/bgu139. [DOI] [PubMed] [Google Scholar]
- 27.Staudt L. Oncogenic activation of NF-kappaB. Cold Spring Harbor Perspect. Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu X., Yarbrough W. Negative regulation of RelA phosphorylation: emerging players and their roles in cancer. Cytokine Growth Factor Rev. 2015;26:7–13. doi: 10.1016/j.cytogfr.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 29.Thu Y., Richmond A. NF-κB inducing kinase: a key regulator in the immune system and in cancer. Cytokine Growth Factor Rev. 2010;21:213–226. doi: 10.1016/j.cytogfr.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- 31.Perona R., Montaner S., Saniger L., Sánchez-Pérez I., Bravo R., Lacal J. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- 32.Hu Z., Liu Y., Xie L., Wang X., Yang F., Chen S., Li Z. AKAP-9 promotes colorectal cancer development by regulating Cdc42 interacting protein 4. Biochim. Biophys. Acta. 2016;1862:1172–1181. doi: 10.1016/j.bbadis.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonnans C., Chou J., Werb Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peurois F., Peyroche G., Cherfils J. Small GTPase peripheral binding to membranes: molecular determinants and supramolecular organization. Biochem. Soc. Trans. 2019;47:13–22. doi: 10.1042/BST20170525. [DOI] [PubMed] [Google Scholar]
- 35.Reiner D.J., Lundquist E.A. Small GTPases. WormBook. 2018;2018:1–65. doi: 10.1895/wormbook.1.67.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Martino J., Paysan L., Gest C., Lagree V., Juin A., Saltel F., Moreau V. Cdc42 and Tks5: a minimal and universal molecular signature for functional invadosomes. Cell Adh. Migr. 2014;8:280–292. doi: 10.4161/cam.28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puar Y.R., Shanmugam M.K., Fan L., Arfuso F., Sethi G., Tergaonkar V. Evidence for the involvement of the master transcription factor NF-kappaB in cancer initiation and progression. Biomedicines. 2018;6 doi: 10.3390/biomedicines6030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaiopoulos A.G., Athanasoula K., Papavassiliou A.G. NF-kappaB in colorectal cancer. J. Mol. Med. (Berl) 2013;91:1029–1037. doi: 10.1007/s00109-013-1045-x. [DOI] [PubMed] [Google Scholar]
- 39.Viatour P., Merville M.P., Bours V., Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem. Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Sakurai H., Chiba H., Miyoshi H., Sugita T., Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 1999;274:30353–30356. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 41.Weitz J., Koch M., Debus J., Höhler T., Galle P., Büchler M. Colorectal cancer. Lancet. 2005;365:153–165. doi: 10.1016/s0140-6736(05)17706-x. [DOI] [PubMed] [Google Scholar]
- 42.Wooster A.L., Girgis L.H., Brazeale H., Anderson T.S., Wood L.M., Lowe D.B. Dendritic cell vaccine therapy for colorectal cancer. Pharmacol. Res. 2020;105374 doi: 10.1016/j.phrs.2020.105374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tonucci F., Almada E., Borini-Etichetti C., Pariani A., Hidalgo F., Rico M., Girardini J., Favre C., Goldenring J., Menacho-Marquez M., Larocca M. Identification of a CIP4 PKA phosphorylation site involved in the regulation of cancer cell invasiveness and metastasis. Cancer Lett. 2019;461:65–77. doi: 10.1016/j.canlet.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 44.Tsuji E., Tsuji Y., Fujiwara T., Ogata S., Tsukamoto K., Saku K. Splicing variant of Cdc42 interacting protein-4 disrupts beta-catenin-mediated cell-cell adhesion: expression and function in renal cell carcinoma. Biochem. Biophys. Res. Commun. 2006;339:1083–1088. doi: 10.1016/j.bbrc.2005.11.117. [DOI] [PubMed] [Google Scholar]
- 45.Kobashigawa Y., Kumeta H., Kanoh D., Inagaki F. The NMR structure of the TC10- and Cdc42-interacting domain of CIP4. J. Biomol. NMR. 2009;44:113–118. doi: 10.1007/s10858-009-9317-z. [DOI] [PubMed] [Google Scholar]
- 46.Naugler W.E., Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel M., Horgan P.G., McMillan D.C., Edwards J. NF-kappaB pathways in the development and progression of colorectal cancer. Transl. Res. 2018;197:43–56. doi: 10.1016/j.trsl.2018.02.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the data generated or analyzed during this study are included in this article.