

Abstract
Early detection and intervention are key principles in clinical medicine and psychiatry. In this issue of Neuron, Cabungcal et al. (2014) demonstrate that prophylactic treatment with antioxidants in adolescence prevent adult deficits in a rat model relevant to schizophrenia.
Schizophrenia (SZ) is a heterogeneous clinical syndrome with typical onset in late adolescence and early adulthood. SZ is typically characterized by psychotic experiences such as hallucinations and delusions, as well as deficits in emotion, motivation, social and cognitive functions. The disease course and outcome is frequently chronic and devastating. Thus, this severe psychiatric condition has been a major subject of study in psychiatry and neuroscience. Although the onset of SZ occurs, in most cases, after puberty, many lines of evidence indicate that the primary risks for the disease emerge during earlier neurodevelopment (Insel, 2010).
To overcome the high social burden of this difficult condition, two contrasting approaches are currently utilized. Given that SZ is highly heritable, large-scale genetic and genomic studies have taken place, and over 100 genetic risk loci for the disease have been identified (Schizophrenia Working Group of the Psychiatric Genomics, 2014). This genetic information can be a promising lead to build pathophysiological hypotheses that are to be tested in the near future. In contrast to such a bottom-up approach focused on molecular etiology, is a “top-down” strategy, in which detailed and multifaceted phenotypic analyses are undertaken with patients, ideally in a longitudinal study design. Such analyses range from clinical measures such as symptoms and cognition to magnetic resonance imaging (MRI) measures of brain chemistry and molecular profiling with specimens obtained from patients via biopsy. These two approaches are complementary and mutually beneficial.
Involvement of oxidative stress in the pathophysiology in SZ has been suggested for over 80 years. Top-down approaches that include cutting-edge technology have recently shed light on this classic hypothesis (Emiliani et al., 2014). For example, neuronal cells from SZ patients, such as induced pluripotent stem cell (iPSC)-derived neural progenitors and olfactory immature neuronal cells directly obtained from nasal biopsy are useful to address the cellular predisposition to the disease. Discovery-based approaches, such as microarray gene expression and quantitative proteomic mass spectrometry analyses applied to iPSC-derived neural progenitor cells identified abnormal gene expression and protein levels related to cytoskeletal remodeling and oxidative stress in SZ (Brennand et al., 2014). Genome-wide profiling of multiple histone methylations in olfactory neuronal cells also implied cellular susceptibility to oxidative stress in SZ (Kano et al., 2013). A recent meta-analysis of the studies on oxidative stress in blood samples from SZ patients compared with those from normal controls, considering the effect of clinical status and antipsychotic treatment, has also suggested a role for oxidative stress and its potential implication as a biomarker in SZ (Flatow et al., 2013). The next question is whether such alterations associated with oxidative stress indeed occur in the brain in the disease condition. Thus, direct assessment of this question by brain imaging, in particular magnetic resonance spectroscopy, theoretically allows us to quantitatively measure the levels of a major antioxidant glutathione in regions of interest inside the brain. Note that, although we are optimistic about this strategy, the methodology still has room for further improvement, and thus far, no consistent conclusion has been obtained in regard to the levels of brain glutathione in SZ, compared with normal controls (Emiliani et al., 2014).
There may be alternatives paths to move this research strategy forward. In this issue of Neuron, the collaborative team of Kim Do and Patricio O’Donnell (Cabungcal et al., 2014) show data from a rat model with a neonatal ventral hippocampal lesion. This model shows altered prefrontal maturation, including in inhibitory interneurons during adolescence and displays SZ-relevant electrophysiological, histological, and behavioral abnormalities after puberty. Thus, this model is a useful tool to test possible developmental pathophysiological scenarios, in particular to explore mechanisms involved in delayed emergence of anomalies driven by developmental alterations. The collaborative team assessed whether oxidative stress during presymptomatic stages causes adult anomalies (Cabungcal et al., 2014): this model confirmed excess oxidative stress in the prefrontal cortex in adulthood, and further narrowed down the cells under stress to be parvalbumin (PV)-positive interneurons. Juvenile and adolescent treatment with the antioxidant N-acetyl cysteine (NAC) prevented the reduction of prefrontal PV-interneuron activity observed in this model, as well as electrophysiological and behavioral deficits relevant to SZ. Furthermore, adolescent treatment with the glutathione peroxidase mimic ebselen also reversed behavioral deficits in this animal model. This study has particular significance in translational medicine, as the effects of these antioxidants in the rodent model were assessed by mismatch negativity (a component of the event-related potential to an odd stimulus in a sequence of stimuli) and prepulse inhibition (an indicator of sensorimotor gating), both of which are known to be abnormal in many patients with SZ. Therefore the authors suggest that presymptomatic oxidative stress yields abnormal adult brain function in a developmentally compromised brain, highlighting redox modulation as a potential target for early intervention. This conclusion may not be limited to this specific model, as excess oxidative stress in a developmental rodent model useful for SZ has also been reported in a transgenic animal model in which Disc1, a susceptibility factor for major mental illnesses with neurodevelopmental functions, is perturbed (Johnson et al., 2013).
The present study also raises new scientific questions. First, it is still unclear how a neonatal hippocampal lesion results in excess oxidative stress in PV-interneurons. This specific question is a part of more general working hypothesis that presymptomatic oxidative stress during adolescence may be a common pathophysiology downstream of multiple etiopathologies of SZ. In addition to the hippocampal lesion and Disc1 models (Cabungcal et al., 2014; Johnson et al., 2013), it would be meaningful to re-address and examine the levels of juvenile oxidative stress in multiple animal models that have been classically studied in different contexts as possible models for SZ. Then, we may be able to determine mechanisms of how diverse disease etiopathologies can converge into common pathways. Second, it is very important to examine whether and how excess oxidative stress affects deficits in neural circuitry in the prefrontal cortex relevant to SZ. The outstanding aims include the influences of oxidative stress on the NMDA-type glutamate receptor as well as characteristics of PV-interneurons and dendritic spines in the pyramidal neurons (Emiliani et al., 2014; Steullet et al., 2014).
In addition to its preclinical significance, the present study provides high potential for translation. It is known that early intervention is associated with better clinical outcomes in persons with SZ, raising the hope that treatment during the prodromal phase of illness could prevent the development of a psychotic disorder and thus reduce risk of chronic symptoms and disability (Perkins et al., 2005). Understanding the mechanism by which duration of untreated psychosis influences prognosis may improve treatment strategies. The significance of “early detection and early intervention” has also been well appreciated in other medical conditions, such as cancers and cardiovascular conditions. Supported by this clinical approach, systematic longitudinal studies with subjects carrying familial high-risk as well as a broader group of people with more clinically defined at-risk mental status have taken place all over the world (Keshavan et al., 2011). These studies, including the North American Prodrome Longitudinal Study (NAPLS), have aimed to define markers that predict transition of such at-risk status (the prodromal stages) to full onset of the disease. Of great interest to these considerations, recent results from the second phase of NAPLS have suggested that inflammation, oxidative stress, and dysregulation of hypothalamic-pituitary axes may be prominently changed in the prodromal stages of psychosis (Perkins et al., 2014; Walker et al., 2013). Thus, the present observations from the hippocampal lesion rat model are reasonably compatible with those from the prodromal subjects. The translational utility of this model (and possibly, of other models that display similar adolescent changes in stress cascades before SZ-relevant behavioral deficits, such as the Disc1 transgenic model as described above) is therefore highly underscored.
Based on the above observations, we discuss future perspectives of “early detection and early intervention” by utilizing both preclinical models and clinical studies in parallel. First, we need to clinically validate the effectiveness of antioxidants and redox regulators, such as NAC, in prodromal subjects and early stages of SZ. An outstanding question is to determine whether antioxidants and redox regulators are most effective by themselves or administered together with lower doses of neuroleptics. Because antioxidants may have less adverse effects compared with those of neuroleptics, avoiding metabolic changes and extrapyramidal signs, a cost-benefit balance of efficacy and side effects may determine the optimal treatment strategy. Preclinical models may also be able to provide useful information to this question. Second, if we add antioxidants and redox regulators into the treatment strategy, it is important to establish markers for treatment response in biological pathways of oxidative stress. We expect that such molecular markers correlate well with clinical indicators, possibly including prepulse inhibition, mismatch negativity, and working memory. Interconnected efforts between preclinical and clinical studies will efficiently answer to this question. Third, it is an interesting question whether oxidative stress may be augmented prior to relapses and whether treatment with antioxidants and redox regulators during remission phases is effective in “preventing” such deteriorating relapses. Lastly, intervention strategies may not be limited to medications. For example, a different research group, utilizing the same hippocampal lesion rat model that was examined for the antioxidant trials, found that adolescent cognitive training could prevent the adult cognitive control impairment (Lee et al., 2012): the early intervention by cognitive training normalized physiological function of the brain, enhancing cognition-associated synchrony of neural oscillations, a measure of brain function that indexed cognitive ability. Clinically, research to improve cognitive deficits by training is actively taking place, based on principles of brain plasticity (Keshavan et al., 2014; Subramaniam et al., 2012). These efforts include new strategies for at-risk subjects in adolescence and young adulthood to psychosis, such as computer-associated or mobile phone-associated applications for training.
By enhancing translational research infrastructure in which both preclinical and clinical studies are meaningfully connected, we are now coming to a promising stage in the exploration of prophylactic intervention to major mental illnesses. Combating adolescent oxidative stress for adult psychosis and schizophrenia is one of the most promising routes to establish prophylactic psychiatry, hopefully in near future.
Figure 1. Utility of preclinical animal models of neurodevelopmental etiopathology in the translational study of schizophrenia.
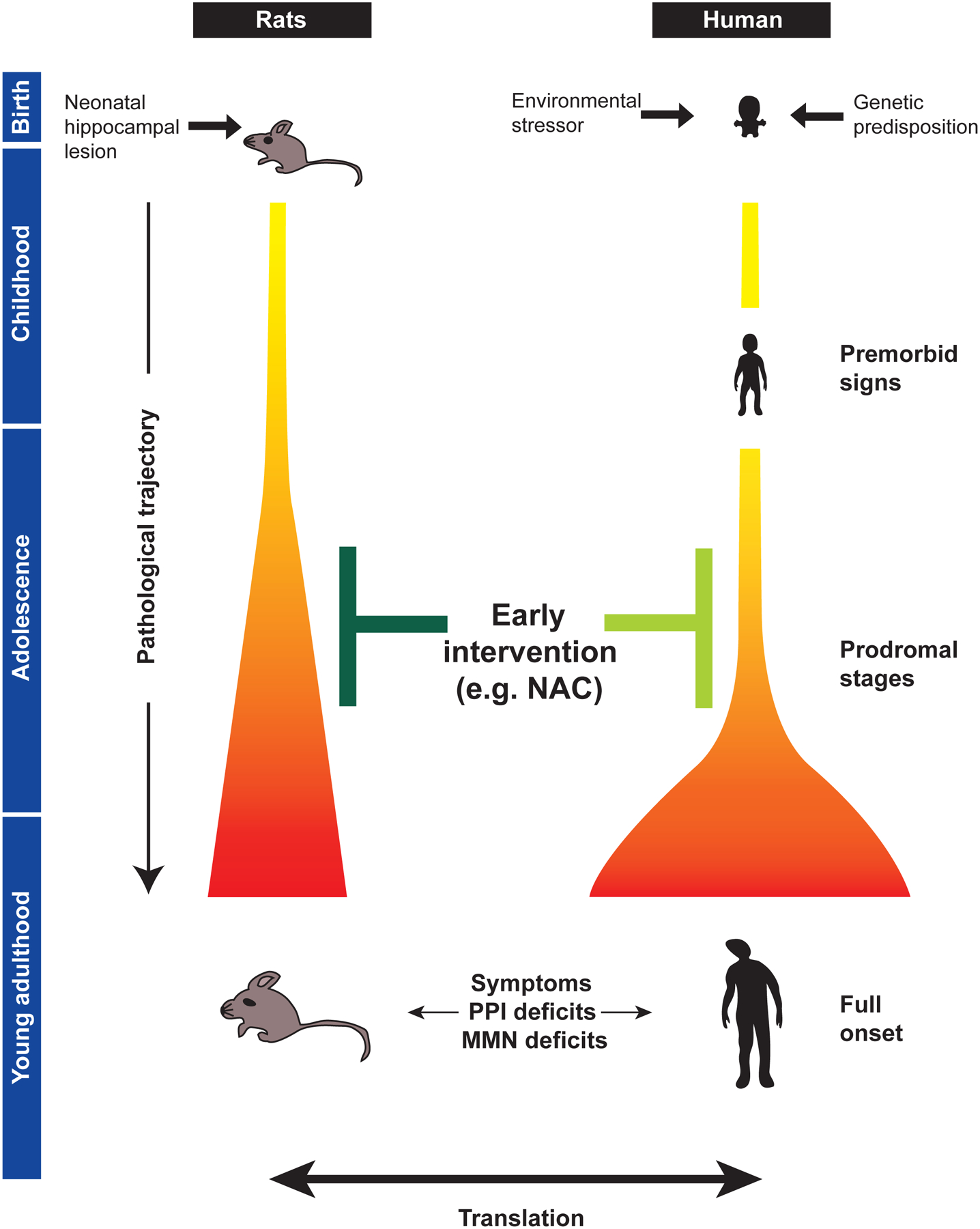
Animal models can be utilized to address biological questions on the pathological trajectory to full onset of schizophrenia after puberty. The trajectory from the primary risk beginning in earlier neurodevelopment to prodromal stages and full onset is illustrated in the gradation from yellow to orange and red. These models are potentially useful for developing presymptomatic, prophylactic intervention during adolescence, including those with the antioxidant N-acetyl cysteine (NAC) (depicted in dark green). Therapeutic concepts initially obtained from preclinical models need to be tested in clinical trials in the near future (depicted in light green). PPI, prepulse inhibition; MMN, mismatch negativity.
REFERENCES
- Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, Beaumont KG, Kim HJ, Topol A, Ladran I, et al. (2014). Mol Psychiatry. [DOI] [PMC free article] [PubMed]
- Cabungcal JH, Counotte DS, Lewis E, Tejeda HA, Piantadosi P, Pollock C, Calhoon GG, Sullivan E, Presgraves E, Kil J et al. (2014). Neuron 83, this issue, 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emiliani FE, Sedlak TW, and Sawa A (2014). Curr Opin Psychiatry 27, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatow J, Buckley P, and Miller BJ (2013). Biol Psychiatry 74, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR (2010). Nature 468, 187–193. [DOI] [PubMed] [Google Scholar]
- Johnson AW, Jaaro-Peled H, Shahani N, Sedlak TW, Zoubovsky S, Burruss D, Emiliani F, Sawa A, and Gallagher M (2013). Proc Natl Acad Sci U S A 110, 12462–12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano S, Colantuoni C, Han F, Zhou Z, Yuan Q, Wilson A, Takayanagi Y, Lee Y, Rapoport J, Eaton W, et al. (2013). Mol Psychiatry 18, 740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshavan MS, DeLisi LE, and Seidman LJ (2011). Schizophr Res 126, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshavan MS, Vinogradov S, Rumsey J, Sherrill J, Wagner, and Wagner A. (2014). Am J Psychiatry 171, 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Dvorak D, Kao HY, Duffy AM, Scharfman HE, and Fenton AA (2012). Neuron 75, 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DO, Gu H, Boteva K, and Lieberman JA (2005). Am J Psychiatry 162, 1785–1804. [DOI] [PubMed] [Google Scholar]
- Perkins DO, Jeffries CD, Addington J, Bearden CE, Cadenhead KS, Cannon TD, Cornblatt BA, Mathalon DH, McGlashan TH, Seidman LJ, et al. (2014). Schizophr Bull.
- Schizophrenia Working Group of the Psychiatric Genomics, C. (2014). Nature 511, 421–427.25056061 [Google Scholar]
- Steullet P, Cabungcal JH, Monin A, Dwir D, O’Donnell P, Cuenod M, and Do KQ (2014). Schizophr Res. [DOI] [PMC free article] [PubMed]
- Subramaniam K, Luks TL, Fisher M, Simpson GV, Nagarajan S, and Vinogradov S (2012). Neuron 73, 842–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EF, Trotman HD, Pearce BD, Addington J, Cadenhead KS, Cornblatt BA, Heinssen R, Mathalon DH, Perkins DO, Seidman LJ, et al. (2013). Biol Psychiatry 74, 410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]