

SUMMARY
Contact inhibition of cell proliferation regulates tissue size and prevents uncontrolled cell expansion. When cell density increases, contact inhibition can force proliferating cells into quiescence. Here we show that the variable memory of local cell density experienced by a mother cell controls the levels of the cyclin-dependent kinase (CDK) activator cyclin D1 and inhibitor p27 in newborn daughters, which direct cells to proliferation or quiescence. Much of this regulation can be explained by rapid suppression of ERK activity by high cell density in mothers, which leads to lower cyclin D1 and higher p27 levels in daughters. Strikingly, cell density and mitogen signals compete by shifting the ratio of cyclin D1/p27 levels below or above a single sharp threshold that controls the proliferation decision. Thus, the history of competing cell density and mitogen signals experienced by mothers is funneled into a precise activator-inhibitor balance that decides the fate of daughter cells.
In brief
Using live single-cell microscopy, Fan and Meyer show that the decision of newborn daughter cells to proliferate or become quiescent is controlled by the memory of local cell density inherited from mother cells. This memory is mediated by an ultrasensitive activator-inhibitor balance between cyclin D1 and p27.
Graphical Abstract
INTRODUCTION
Contact inhibition of cell proliferation is a main mechanism by which epithelial and many mesenchymal cells restrict cell proliferation and tissue size (McClatchey and Yap, 2012). Suppression of proliferation at high cell density is clinically relevant because loss of contact inhibition is a major mechanism driving cancer progression (Hanahan and Weinberg, 2011; McClatchey and Yap, 2012; Yu et al., 2015). When cell density increases during normal cell proliferation, inhibitory signals are generated by formation of increased junctional contacts between cells as well as changes in mechanical forces (Aragona et al., 2013; Kim et al., 2011; McClatchey and Yap, 2012; Puliafito et al., 2012; Streichan et al., 2014; Zhao et al., 2007). Because there are multiple pathways mediating contact inhibition in addition to cell-cell contacts per se, an overall strength of contact inhibition is often determined by measuring cell density (Aoki et al., 2013; Aragona et al., 2013; Zhao et al., 2007), and we use this measure of cell density throughout this study. At the end of a signal integration process that involves mitogen signaling, cell density, and other inputs, proliferating cells exit to quiescence when newborn cells fail to activate the core drivers of cell cycle entry: cyclin D-cyclin-dependent kinase (CDK) 4/6 and cyclin E-CDK2 (Malumbres and Barbacid, 2001; Morgan, 1997; Sherr, 1993).
The decision whether newborn cells activate CDK4/6 and CDK2-type kinases is regulated by multiple mechanisms, including expression of D-type cyclins and the CDK inhibitors p27KIP and p21CIP (Malumbres and Barbacid, 2001; Morgan, 1997). Previous studies have shown that CDK2 activity and cell cycle exit in newborn cells are regulated in a bimodal fashion by mitogen and DNA damage signals from the previous cell cycle, and upregulation of p21 has been found to be a rate-limiting step inhibiting activation of CDK2 (Arora et al., 2017; Barr et al., 2017; Hitomi and Stacey, 1999; Min et al., 2020; Spencer et al., 2013; Yang et al., 2017). In these studies, cells were typically plated sparsely to facilitate single-cell tracking, which likely reduced the relevance of p27 because p27 levels typically increase with higher cell density (Polyak et al., 1994; St Croix et al., 1998). Nevertheless, unlike p21, whose deletion has only small effects on mouse development (Brugarolas et al., 1995; Deng et al., 1995), p27 deletion in mice results in increased cell proliferation, multiorgan hyperplasia, and larger body size (Fero et al., 1996; Kiyokawa et al., 1996; Nakayama et al., 1996), highlighting the importance of p27 in restricting cell proliferation during normal development.
The respective roles of cyclin D, p21, and p27 in triggering cell density-mediated cell cycle exit in newborn cells were difficult to establish using bulk cell analysis because of a need to pharmacologically arrest and synchronize cells in mitosis. The goal of our study was to monitor and perturb the endogenous levels of cyclin D, p21, and p27 in asynchronously dividing cells that experience different local cell densities and mitogen signals without stress perturbations. Specifically, we sought to address the following open questions: (1) when during the cell cycle cells sense local cell density; (2) whether cell density-mediated changes in cyclin D, p21, and p27 expression are rate limiting in regulating the decision of a newborn cell to exit the cell cycle; and (3) how cell density and mitogen signaling compete to decide whether newborn cells exit to quiescence or keep proliferating.
We investigated cell density-regulated cell cycle exit by developing a fluorescence microscopy approach that tracks local cell density and signaling activities in mother and newborn daughter cells over time. In the tracked newborn cells, we also measured their cell cycle status as well as expression levels of cyclin D, p21, and p27. We find that, rather than the local cell density experienced by newborn cells, it is the memory of local cell density experienced by mother cells that directs newborn cells to quiescence. We show that high local cell density rapidly suppresses ERK activity and that low ERK activity in mother cells mediates an increase in p27 and decrease in cyclin D1 levels in newborn cells. Strikingly, we find that a small decrease in the nuclear expression ratio of cyclin D1 over p27 below a sharp threshold triggers exit to quiescence in a highly ultrasensitive and predictable manner. We go on to show that cell density and mitogen signals compete in mother cells to cause opposing shifts in the cyclin D1/27 ratio, which positions each newborn cell above or below this single threshold and directs cells toward proliferation or quiescence, respectively. Our study supports a model where each newborn cell decides between quiescence and proliferation based on a precise activator-inhibitor balance that is controlled by local cell density and mitogen signals experienced by its mother cell.
RESULTS
Local cell density in mother cells predicts whether newborn cells exit to quiescence
We first examined when during the cell cycle cells sense local cell density. To this end, we developed an assay to monitor local cell density changes over time in single cells. We transduced a fluorescently tagged nuclear histone (H2B-mTurquoise) into non-transformed human retinal pigment epithelial cells (RPE-1 hTERT). The nuclear marker allowed us to computationally monitor chromosome segregation during anaphase of mitosis, track the movement of cells, and measure changes in local cell density in thousands of cells. As a metric for the dynamically changing local cell density, we measured the fraction of total area within a fixed distance from each nucleus that is occupied by neighboring nuclei (Figure 1A; hereafter referred to as “local density”). This metric of local density is closely correlated with other metrics of cell density (Figures S1A and S1B).
Figure 1. Local cell density in mother cells predicts whether newborn cells exit to quiescence.
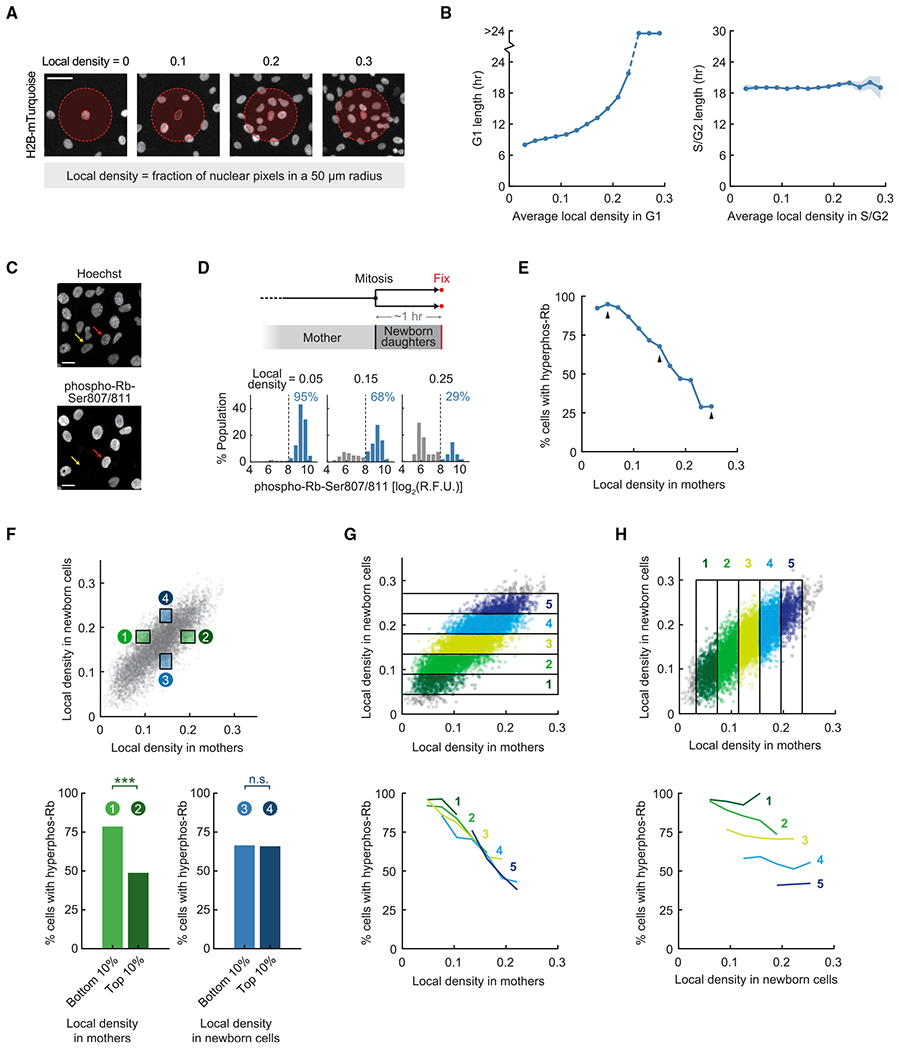
(A) Measuring local cell density in RPE-1 hTERT cells expressing histone H2B-mTurquoise. Scale bar, 50 μm.
(B) Asynchronously cycling cells expressing H2B-mTurquoise and the mCherry-tagged APC/C degron were imaged. Cells were binned by average local density in G1 or S/G2 phase. Line plots are population medians in each bin; shaded error bars are 95% confidence intervals (n ≥ 50 cells per bin, n ≥ 9,379 cells total). Data are representative of 2 independent experiments.
(C) Immunofluorescence of phospho-Rb-Ser807/811. Yellow arrow, cell with hypophosphorylated Rb; red arrow, cell with hyperphosphorylated Rb. Scale bar, 20 μm.
(D) Top: schematic of the experimental setup. Cells expressing H2B-mTurquoise were plated at different densities, imaged, and fixed at the end of the experiment. Newborn cells were gated in silico for analysis. Bottom: sample histograms of phospho-Rb-Ser807/811 in newborn cells with different histories of local density in mothers. Percentages indicate cells that had hyperphosphorylated Rb (high phospho-Rb-Ser807/811). R.F.U., relative fluorescence unit.
(E) Newborn cells were binned by local density experienced by their mothers (n ≥ 50 cells per bin, n = 8,574 cells total). Arrows indicate data points represented by sample histograms in (D). Logistic regression slope, −19.1; likelihood ratio test, p = 3 × 10−276. Cross-validation (10-fold) was performed: slope, −19.1 ± 0.1 (median ± s.t.d.); area under the curve (AUC) for the receiver operating characteristic (ROC) curve, 0.74 ± 0.01. Data are representative of 4 independent experiments.
(F) Top: green boxes gate for cells with similar local density in newborn cells (50–70 percentile) but different local density in mothers (top and bottom 10%, excluding 1% extreme outliers); blue boxes gate for the inverse. Local density in newborn cells is averaged during 0 h to 1–1.5 h after mitosis (from mitosis until cells were fixed). Bottom left: Fisher’s exact-test, p = 1.95 × 10−8 (n = 168 cells). Bottom right: Fisher’s exact test, p = 1 (n = 170 cells). Data are representative of 4 independent experiments.
(G) Top: gating for cells with similar local density in newborn cells but varying history of local density in mothers. Bottom: correlation between Rb hyperphosphorylation in newborn cells and local density in mothers after controlling for local density in newborn cells (n ≥ 20 cells per bin). Logistic regression slope, −18.0 ± 1.8 (median ± s.t.d. for 5 bins of local density in newborn cells). Cross-validation (10-fold) was performed on each bin: slope, −18.2 ± 1.8; AUC-ROC, 0.63 ± 0.02 (n = 5). Data are representative of 4 independent experiments.
(H) Same experiment as in (G) but gating for cells with similar local density in mothers but varying history of local density in newborn cells. Bottom: weak correlation between Rb hyperphosphorylation in newborn cells and local density in newborn cells after controlling for local density in mothers (n ≥ 20 cells per bin). Logistic regression slope, −3.7 ± 3.7 (median ± s.t.d. for 5 bins of mother density). Cross-validation (10-fold) was performed on each bin: slope, −3.6 ± 3.8;AUC-ROC, 0.53 ± 0.04 (n = 5). Data are representative of 4 independent experiments.
See also Figures S1 and S2. The following p value convention is used throughout: n.s., p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001.
We used this single-cell analysis to compare the behavior of different cells in the same cell sheet and observed considerable cell-to-cell heterogeneity in local density within cell sheets plated at the same density (Figure S1C). To investigate when cells sense local cell density during the cell cycle, we stably transduced cells with a degron-based APC/C activity reporter, which is actively degraded during G1 phase and accumulates during S/G2 phase (Sakaue-Sawano et al., 2008). We measured the length of G1 and S/G2 phases of the cell cycle in each cell (Figure S1D) and correlated them with average local cell density measured in G1 and S/G2 phases, respectively. We found that the length of G1 phase increased as local density increased, whereas the length of S/G2 phase was independent of local density (Figure 1B). This result suggests that local cell density controls proliferation primarily by regulating G1 length and exit to quiescence.
A key event in G1 regulation is hyperphosphorylation of the retinoblastoma protein (Rb) at approximately 15 sites by CDK4/6 and CDK2 activity (Rubin, 2013). Hyperphosphorylation inactivates Rb and relieves Rb-mediated repression of the E2F transcription factors, which induce target genes required for DNA replication (Bertoli et al., 2013; Malumbres and Barbacid, 2001; Nevins, 2001). Hyperphosphorylation of Rb can be used as a marker for proliferation because newborn cells with hyperphosphorylated Rb after mitosis keep proliferating, whereas cells with hypophosphorylated Rb remain temporarily in G1 or exit to quiescence (Chung et al., 2019; Moser et al., 2018; Spencer et al., 2013; Yang et al., 2017). Thus, we next focused on regulation of Rb hyperphosphorylation in G1 by local cell density. Previous studies using low-density cell cultures have shown that Rb hyperphosphorylation and the proliferation-quiescence decision in G1 phase are regulated by mitogen and DNA damage signals from the previous cell cycle (Arora et al., 2017; Barr et al., 2017; Hitomi and Stacey, 1999; Min et al., 2020; Spencer et al., 2013; Yang et al., 2017). Our goal was not only to understand how cell density regulates Rb hyperphosphorylation but also whether Rb hyperphosphorylation in a newborn cell is regulated similarly by the memory of local cell density experienced by its mother cell.
To measure Rb hyperphosphorylation in individual newborn cells, we performed quantitative single-cell immunofluorescence, where cells are fixed and stained using a previously characterized antibody recognizing phospho-Rb-Ser807/811 (Chung et al., 2019; Spencer et al., 2013; Yang et al., 2017). The nuclear signal intensity of this antibody is bimodal (Figures 1C and 1D, bottom panel), where cells in the high peak have most of their Rb protein hyperphosphorylated and inactive (Chung et al., 2019). In this way, whether a cell is in the hyperphosphorylated or hypophosphorylated Rb peak serves as a proxy of whether cells will continue to proliferate or exit to quiescence.
We combined live-cell microscopy with fixed-cell immunofluorescence at the end of the experiment to measure Rb hyperphosphorylation. This retrospective time-lapse analysis allowed us to match the Rb phosphorylation status of each newborn daughter cell with the local cell density its mother experienced before mitosis. Specifically, we tracked and fixed asynchronously cycling cells, gated for G1-phase daughter cells that recently underwent mitosis (1–1.5 h before fixation) (Figure 1D, top panel), and averaged local density over a 3-h period before mitosis. This single-cell analysis showed that a decreasing percentage of newborn cells had hyperphosphorylated Rb as the local density in the corresponding mother cell increased (Figures 1D and 1E), a result we confirmed using logistic regression (Figures S1E–S1G; slope, −19.1). The large negative slope derived from logistic regression demonstrates that the variable local cell density for each mother cell is a strong predictor of whether its newborn daughters will have hyperphosphorylated Rb after mitosis. Control experiments showed a similar correlation (1) when we used different metrics of local cell density (Figure S2A); (2) when we used another non-transformed cell line, human breast epithelial MCF10A cells (Figure S2B); and (3) when we used a live-cell reporter that primarily measures CDK2 activity in G1 as a readout of the proliferative status of newborn cells (Figures S2C–S2E; Barr et al., 2017; Chung et al., 2019; Hahn et al., 2009; Schwarz et al., 2018; Spencer et al., 2013; Yang et al., 2020).
Although the negative correlation shows that local cell density in mother cells is highly predictive of the newborn fate, we also tested whether local density in newborn cells contributes to regulation of Rb hyperphosphorylation. To deconvolve the effects of local density in mothers versus newborn cells on Rb hyperphosphorylation, we controlled for one factor while analyzing the effect of the other. We first gated for cells that had the same local density in newborn cells but experienced higher or lower local density in mother cells (Figure 1F, green boxes; Figure S2F). When selecting newborn cells that have the same local density, variation in the local density in mothers remained a strong predictor of Rb hyperphosphorylation in newborn cells (Figure 1F, bottom left panel). In contrast, when selecting mother cells that have the same local density, variation in the local density in newborn cells was not a strong predictor of Rb hyperphosphorylation in newborn cells (Figure 1F, blue boxes, bottom right panel).
In an additional control, we expanded the same analysis over a much wider range of local cell density in mother and newborn cells (Figure 1G). We found that Rb hyperphosphorylation remained correlated with the local density in mothers after gating for the same local density in newborn cells (logistic regression slope, −18.0 ± 1.8), with plots from different gates collapsing onto the same curve. In contrast, there was a substantially weaker correlation between Rb hyperphosphorylation and local density in newborn cells when controlling for mother density (Figure 1H; logistic regression slope, −3.7 ± 3.7). The same result could also be shown by a logistic regression model that separated the effects of mother density and daughter density as predictors (Figure S2G). Finally, we found that pairs of sister cells were more likely to share the same fate than randomly chosen pairs of cells (95.3% versus 75.1%). We conclude that it is primarily the history of local cell density experienced by mothers rather than density experienced by newborn cells that closely predicts the likelihood of newborn cells to lose Rb hyperphosphorylation, inactivate CDK2, and exit to quiescence.
High cell density in mother cells directs newborn cells to quiescence
We next tested whether the prediction of Rb hyperphosphorylation in newborn cells from the local cell density in mothers reflects a causal link. We plated cells over a broad range of cell density (Figure 2A, top schematic) and re-plated the same cells a day later at the lowest density, after which cells were live imaged and fixed. In this analysis, we selected newborn cells that underwent mitosis after re-plating using automated image analysis. These newborn cells were born into similar low-density environments (Figure S3A), whereas their mothers experienced a wide range of local density (Figure 2A, bottom gray line plot). This analysis allows one to test whether there is a memory of different cell densities experienced by mother cells that is passed down to their daughters that are now experiencing the same cell density. Indeed, newborn cells whose mothers experienced higher cell density were proportionally less likely to have hyperphosphorylated Rb (Figure 2A, blue bars).
Figure 2. High cell density in mother cells directs newborn cells to quiescence.
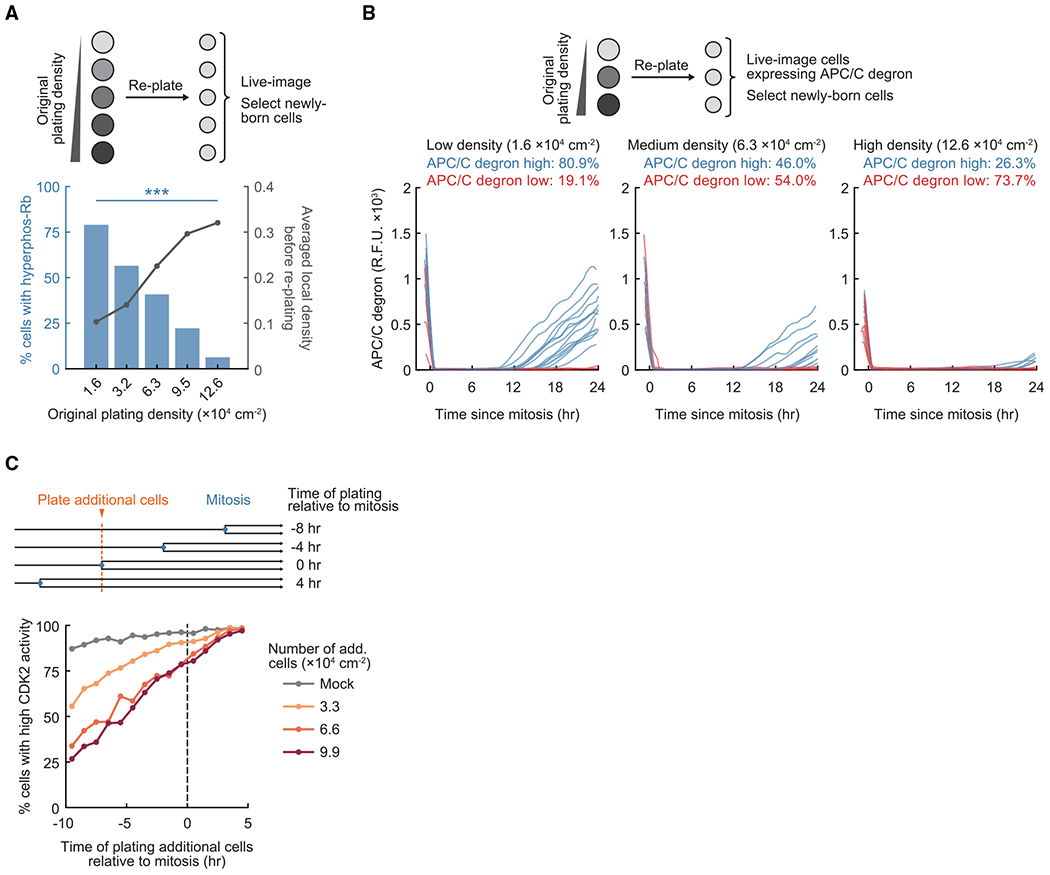
(A) Top: schematic of the experimental setup. Cells plated at different densities in 12-well plates were re-plated onto 96-well plates at the lowest density and imaged. Newborn cells were gated in silico for analysis. Bottom: average local density measured before re-plating (n ≥ 1,100 cells per condition) and percentage of newborn cells with hyperphosphorylated Rb (n ≥ 120 cells per condition). Data are representative of 3 independent experiments. One-way ANOVA of 3 replicates, p = 2.2 × 10−5.
(B) Top: schematic of the experimental setup. Cells born within 2 h after re-plating were gated in silico for analysis. Bottom: n = 20 random sample traces of APC/C degron intensity in cells that were originally plated at different densities (total n ≥ 95 cells per condition). Data are representative of 3 independent experiments.
(C) Top: schematic of the experimental setup. Asynchronously cycling cells expressing H2B-mTurquoise and the CDK2 activity reporter were imaged; additional non-fluorescent cells were plated to acutely increase cell density. Bottom: percentage of newborn cells with high CDK2 activity(n ≥ 20 cells per data point). Data are representative of 4 independent experiments.
See also Figure S3.
In an additional approach to test for the fate of daughter cells, we repeated the re-plating assay and used a live-cell marker that measures whether cells have passed the G1/S checkpoint and entered S phase (Figure 2B, top schematic). APC/CCdh1 inactivation is part of the irreversible commitment at the G1/S transition and can be measured using the APC/C degron reporter (Cappell et al., 2016). We found that daughter cells born from mothers that experienced higher cell density were less likely to inactivate APC/CCdh1 (Figure 2B, bottom panels; Figure S3B) and, thus, fail to enter the next cell cycle.
We further validated that a memory of local cell density is passed on from mothers to daughters by testing how an acute increase in cell density in mother cells changes the proliferation state of daughters. In this live-cell analysis, non-fluorescent cells (which adhered and spread out within 20–30 min; Figure S3C) were added to asynchronously cycling cells expressing the CDK2 reporter, and cell cycle entry or exit in daughters was tracked by measuring the CDK2 reporter signal. We selected cells for analysis where the increase in cell density through addition of unlabeled cells occurred at different time points before and after mitosis (Figure 2C, top panel). This analysis showed that an increase in cell density during the preceding cell cycle reduced the percentage of newborn cells that activated CDK2 after mitosis in a dose-dependent manner (Figure 2C, bottom panel). In contrast, the effect of increased cell density on CDK2 activity was much smaller when cell density increased after mitosis. As an additional control, we found that increasing cell density in mother cells by plating additional cells also suppressed Rb hyperphosphorylation in newborn cells (Figure S3D). Our results argue that newborn cells retain a memory of high cell density experienced by mother cells, which suppresses Rb hyperphosphorylation, CDK2 activation, and APC/CCdh1 inactivation after mitosis and directs cells to quiescence.
High cell density in mother cells promotes exit to quiescence by increasing p27 and reducing cyclin D1 expression in newborn cells
Newborn cells dephosphorylate Rb to a hypophosphorylated state and enter a prolonged G1 phase or exit to quiescence when they fail to activate CDK4/6 in G1 phase (Chung et al., 2019; Moser et al., 2018; Yang et al., 2017). To determine how mother cells instruct newborn cells to dephosphorylate Rb, we combined live-cell microscopy and immunofluorescence to measure the nuclear protein levels of the CDK4/6 activator cyclin D1 and the CDK inhibitors p21 and p27. This analysis allows us to match the cell density experienced by mother cells to the expression of cyclin D1, p21, and p27 in newborn daughters. We focused on these three regulators of CDK4/6 and CDK2 activity because they are known to be direct regulators of CDK activities and cell cycle entry in G1 phase (Morgan, 1997; Satyanarayana and Kaldis, 2009; Sherr and Roberts, 1999). Although p27 has been implicated in regulating contact inhibition (Polyak et al., 1994; St Croix et al., 1998), it was not known whether and how p27, p21, and cyclin D1 contribute to density-dependent exit to quiescence in newborn cells. As a complication for understanding their potential contribution, p21 and p27 can function as inhibitors that form inactive trimers with cyclin D-CDK4/6 and cyclin E/A-CDK2, but p21 and especially p27 may also function as assembly factors and activators of cyclin D-CDK4/6 when phosphorylated on particular sites (Guiley et al., 2019; James et al., 2008; Sherr and Roberts, 1999). These open regulatory questions motivated us to determine whether and how the local cell density experienced by mother cells regulates the expression levels of cyclin D1, p21, and p27 in newborn cells and how these levels control Rb hyperphosphorylation.
We first plotted the nuclear protein levels of cyclin D1, p21, and p27 in each newborn cell as a function of the local density experienced by its mother. Analysis of thousands of individual mother and newborn cells showed that the level of the CDK4/6 activator cyclin D1 in newborn cells was proportionally lower when the local cell density experienced by its mother was higher, whereas the level of the CDK inhibitor p27 increased as the local cell density experienced by its mother increased (Figure 3A). We also found similar correlations (1) when we normalized cyclin D1 and p27 levels to a nuclear DNA stain (Hoechst) to account for potential changes in nuclear geometry associated with changes in local density (Figure S4A) and (2) when we repeated the same experiments using MCF10A cells (Figure S4C). In contrast, the level of the CDK inhibitor p21 was not correlated with local density in mother cells (Figure S4B). Knockdown of cyclin D1 and overexpression of p27 showed that cyclin D1 and p27 function primarily as activators and inhibitors of Rb hyperphosphorylation, respectively, during density-dependent cell cycle exit (Figure S4D, orange bars). We also confirmed that CDK4/6 activity is rate limiting for Rb hyperphosphorylation during G1 phase in newborn RPE-1 cells (Figures S4G and S4G’), consistent with previous results in MCF10A cells and mouse embryonic fibroblasts (Chung et al., 2019).
Figure 3. An activator-inhibitor balance between cyclin D1 and p27 directs newborn cells to quiescence.
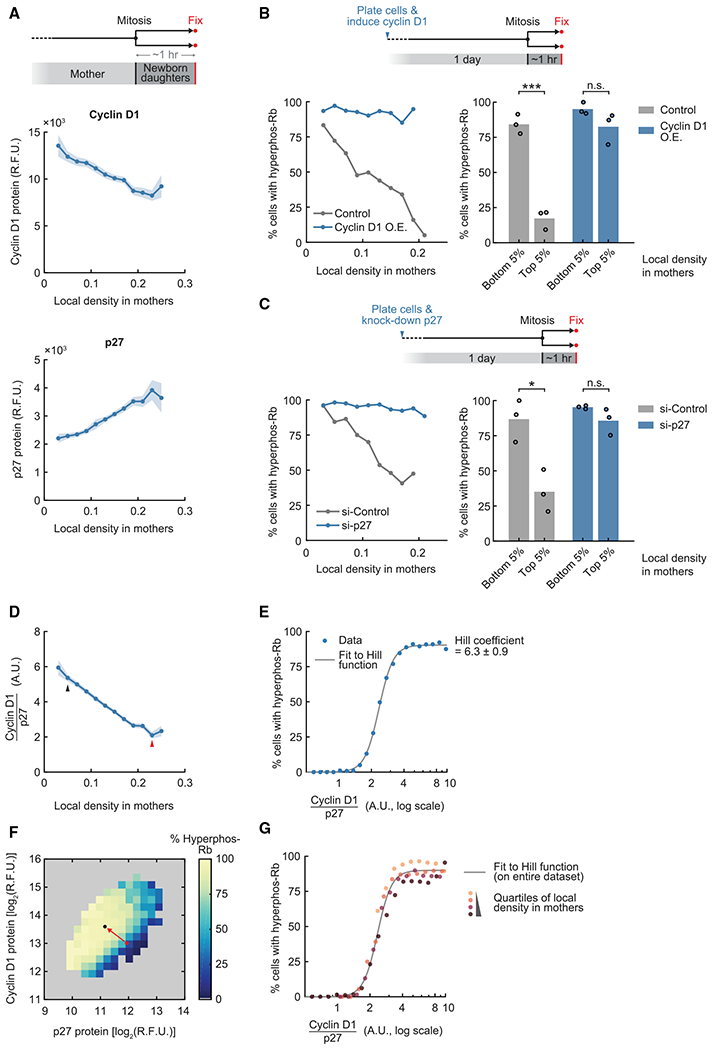
(A) Top: schematic of the experimental setup. Center: correlation between nuclear cyclin D1 level in newborn cells and local density in mothers. Pearson correlation coefficient, −0.193, p = 2.6 × 10−73. Bottom: correlation between nuclear p27 level in newborn cells and local density in mothers. Pearson correlation coefficient, 0.257, p = 6.2 × 10−130. Line plots are population medians in each bin; shaded error bars are 95% confidence intervals (n ≥ 50 cells per bin, n = 8,574 cells total). Data are representative of 4 independent experiments.
(B) Top: schematic of the experimental setup. Cyclin D1 overexpression (O.E.) was driven by a doxycycline-inducible promoter. Bottom left: correlation of Rb hyperphosphorylation in newborn cells and local density in mothers (n ≥ 20 cells per bin; control, n = 694 cells total; cyclin D1 O.E., n = 973 cells total). Data are representative of 3 independent experiments. Bottom right: percentage of newborn cells with hyperphosphorylated Rb when gating for cells whose mothers experienced different local densities (n ≥ 25 cells per data point). The same gating was used for control and cyclin D1 O.E. Student’s t test; control conditions, p = 2.9 × 10−4; cyclin D1 O.E. conditions, p = 0.14 (n = 3 independent experiments).
(C) Top: schematic of the experimental setup. Bottom left: correlation of Rb hyperphosphorylation in newborn cells and local density in mothers (n ≥ 25 cells per bin; si-Control, n = 1,063 cells total; si-p27, n = 1,622 cells total). Data are representative of 3 independent experiments. Bottom right: percentage of newborn cells with hyperphosphorylated Rb when gating for cells whose mothers experienced different local densities (n ≥ 28 cells per data point). The same gating was used for si-Control and si-p27. Student’s t test; si-Control conditions, p = 0.014; si-p27 conditions, p = 0.15 (n = 3 independent experiments).
(D) Cyclin D1/p27 ratio in newborn cells as a function of local cell density in mothers (the ratio is expressed in arbitrary units and not indicative of actual stoichiometry; data from A). Pearson correlation coefficient, −0.423, p < 1 × 10−130. Line plots are population medians in each bin; shaded error bars are 95% confidence intervals (n ≥ 50 cells per bin, n = 8,574 cells total). Arrows indicate data points represented by circles in (F). Data are representative of 4 independent experiments. a.u., arbitrary unit.
(E) Percentage of newborn cells with Rb hyperphosphorylation as a function of cyclin D1/p27 ratio (n ≥ 20 cells per bin, n = 8,669 total, data from A). The Hill function was fitted onto the linear scale data. Hill coefficient, 6.3 ± 0.9 (mean ± s.t.d. of 4 independent experiments).
(F) Percentage of newborn cells with Rb hyperphosphorylation as a function of cyclin D1 and p27 protein level (n ≥ 10 cells per bin, n = 8,255 total, data from A). Data are representative of 4 independent experiments.
(G) Percentage of newborn cells with Rb hyperphosphorylation as a function of cyclin D1/p27 ratio after binning cells into quartiles by local density in mothers (n ≥ 20 cells per bin, n ≥ 2,000 total for each quartile, data from A). The Hill curve was fitted to the entire dataset (the same curve as in E).
See also Figure S4.
We next tested whether regulation of cyclin D1 and p27 in newborn cells by the history of local cell density in mother cells is rate limiting for regulating Rb hyperphosphorylation. We increased cyclin D1 protein levels in newborn cells to approximately 2.5-fold through overexpression (Figure S4E) or reduced p27 protein levels approximately 3-fold using small interfering RNA (siRNA) knockdown (Figure S4F). Importantly, increased expression of cyclin D1 or reduced expression of p27 abolished the inhibitory effect of high local density in mothers on Rb hyperphosphorylation in newborn cells (Figures 3B and 3C). These results argue that cyclin D1 and p27 have opposing rate-limiting roles in regulating Rb hyperphosphorylation in newborn cells.
We conclude that density-dependent exit of newborn cells to quiescence is largely controlled by changes in nuclear cyclin D1 and p27 levels, which, in turn, controls CDK4/6 activation and Rb hyperphosphorylation. This raises the question of how cyclin D1 and p27 work together to control Rb hyperphosphorylation in newborn cells.
High cell density triggers a shift in the cyclin D1/p27 ratio that forces Rb dephosphorylation in an ultrasensitive manner
We hypothesized that density-dependent cell cycle exit is the result of a competition where opposing changes in the concentration of the activator cyclin D1 and inhibitor p27 jointly dictate whether CDK4/6 is activated and Rb is hyperphosphorylated in newborn cells. When we measured the ratio between nuclear cyclin D1 and p27 levels in the same newborn cell, we found that the cyclin D1/p27 ratio was negatively correlated with local cell density in mothers (Figure 3D; Figure S4C, right panel). The dynamic range of the ratio was approximately 3-fold when compared between cells that experienced highest versus lowest local densities. This limited window of regulation raises the question whether and how a relatively small change in a single parameter can be robustly converted into a binary decision to enter the next cell cycle or exit to quiescence.
To determine the relationship between the cyclin D1/p27 ratio and Rb hyperphosphorylation, we performed multi-color immunofluorescence and measured these three parameters in the same newborn cell. Strikingly, this analysis revealed an ultrasensitive response of Rb hyperphosphorylation to the cyclin D1/p27 ratio (Figure 3E; Figure S4H), which can be quantified by a Hill coefficient of ~6. A small increase in the level of cyclin D1 relative to p27 can shift the cell from hypophosphorylated to hyperphosphorylated Rb (Figure 3F, red arrow). We further binned cells into quartiles, with each quartile representing a different range of local cell density in mothers. This analysis showed that the relationship between Rb hyperphosphorylation and cyclin D1/p27 ratio largely collapsed onto the same ultrasensitive curve (Figure 3G), suggesting that the balance between cyclin D1 and p27 is the primary mechanism that regulates exit to quiescence over different ranges of cell density.
We conclude that the multiple cell density-sensing pathways that originate in mother cells are integrated into a single activator-inhibitor balance between cyclin D1 and p27 in newborn cells, which controls CDK4/6 activation and Rb hyperphosphorylation in an ultrasensitive manner. Because of the highly ultrasensitive shape of the response curve in newborn cells, a small shift in the cyclin D1/p27 ratio can be amplified into a large change in the percentage of newborn cells that are directed to quiescence.
High cell density suppresses the MEK-ERK pathway to force newborn cells to quiescence
The MEK-ERK signaling pathway is required for proliferation of epithelial cells and many other cell types (Meloche and Pouysségur, 2007; Figure 4A) and can be regulated by cell density and cell compaction (Aoki et al., 2013, 2017; Hino et al., 2020; Moreno et al., 2019; Viñals and Pouysségur, 1999). We first determined whether ERK activity is regulated in mother cells by local cell density. To track ERK activity and cell density changes, we used a nuclear-localized fluorescence resonance energy transfer (FRET)-based sensor, EKAREV-NLS (Komatsu et al., 2011). We observed cell-to-cell variability in ERK activity within a cell population and indeed found that time-averaged ERK activity was negatively correlated with local cell density (Figure 4B). Control experiments using a specific MEK inhibitor (Figure 4B), RAF inhibitor (Figure S5A), or ERK inhibitor (Figure S5B) showed suppression of ERK activity across the entire range of local cell density and a loss of correlation with cell density. Additional control experiments showed a similar correlation between ERK activity and local cell density when we used a kinase translocation reporter (KTR) specific to ERK activity (Regot et al., 2014; Figure S5C) or when we repeated the same experiments using MCF10A cells (Figure S5D). When we measured the frequency, amplitude, and absolute height of ERK pulses (Albeck et al., 2013; Aoki et al., 2013; Toettcher et al., 2013), we observed a similar negative correlation with local cell density as we did for time-averaged ERK activity (Figures S5E and S5F).
Figure 4. Changes in ERK activity in mother cells control exit to quiescence in newborn cells.
(A) Schematic of the ERK signaling pathway in mammalian cells.
(B) Top: schematic of the experimental setup. ERK activity was measured by the EKAEV-NLS FRET sensor, which was used throughout unless stated otherwise. Bottom left: sample traces of ERK activity at different local density and with DMSO or MEK inhibitor (PD0325901, 100 nM) treatment. Shaded regions indicate ERK activity above baseline (average activity when treated with the MEK inhibitor). Bottom right: correlation between time-averaged ERK activity and local density. Linear regression slope, −1.09 versus −0.41. Arrows indicate data points represented by sample traces. Line plots are population medians in each bin; shaded error bars are 95% confidence intervals (n ≥ 39 cells per bin; DMSO, n = 11,693 cells total; MEK inhibitor, n = 12,447 cells total). Data are representative of 3 independent experiments.
(C) Cell density was acutely increased by plating additional non-fluorescent cells (1.32 × 105 cells/cm2). Line plots are population medians of 140 or more cells; shaded error bars are 95% confidence intervals. Student’s t test comparing ERK activity in mock versus treatment: 15 min before treatment, p = 0.21; 30 min after treatment, p = 1.5 × 10−26. Data are representative of 3 independent experiments.
(D) Left: schematic of the experimental setup. Cell traces are shown on the bottom. Right: ERK activity after releasing cells from confinement, measured by the ERK KTR. Line plots are population medians of 92 or more cells; shaded error bars are SEM. Student’s t test comparing ERK activity in released (periphery) versus unreleased cells: 15 min before release, p = 0.29; 3 h after release, p = 8.0 × 10−8. Data are representative of 2 independent experiments.
(E) Top: schematic of the experimental setup. Bottom: percentage of newborn cells with Rb hyperphosphorylation after inhibition of the MEK-ERK pathway (MEK inhibitor: PD0325901, 100nM; ERK inhibitor: SCH772984, 1 μM) (n ≥ 785 cells per condition). Student’s t test; MEK inhibitor versus DMSO, p = 6.5 × 10−5 (n = 3 independent experiments); ERK inhibitor versus DMSO, p = 4.4 × 10−4 (n = 2 independent experiments).
(F) Top: schematic of the experimental setup. Constitutively active MEK1 (MEK1-CA: S218D/S222D) O.E. was driven by a doxycycline-inducible promoter. Center: correlation of Rb hyperphosphorylation in newborn cells and local density in mothers (n ≥ 16 cells per bin; control, n = 2,068 cells total; MEK1-CA O.E., n = 1,984 cells total). Data are representative of 4 independent experiments. Bottom: percentage of newborn cells with hyperphosphorylated Rb when gating for cells whose mothers experienced different local densities(n ≥ 10 cells per data point). The same gating was used for control and MEK1-CA O.E. Student’s t test; control conditions, p = 2.4 × 10−5; MEK1-CA O.E. conditions, p = 0.020; control versus MEK1-CA O.E., high density, p = 0.010 (n = 4 independent experiments).
(G) Top: schematic of the experimental setup. Bottom: mRNA and protein levels after drug treatment, measured by RNA FISH and immunofluorescence, respectively (MEK inhibitor: PD0325901, 100 nM). Error bars are 95% confidence intervals. Kolmogorov-Smirnov test; cyclin D1 mRNA, p = 7.9 × 10−99; p27 mRNA, p = 1.6 × 10−17; cyclin D1 protein, p = 1.5 × 10−59; p27 protein, p = 4.6 × 10−67 (n ≥ 320 cells per condition). Data are representative of 3 independent experiments.
See also Figure S5.
To determine whether a fast change in local cell density causes a change in ERK activity, we acutely increased cell density by adding non-fluorescent cells (1.32 × 105 cells/cm2) and found that ERK activity was rapidly downregulated within an hour (Figure 4C). We also found a time delay of less than an hour between changes in local cell density and ERK activity in a temporal cross-correlation analysis of unperturbed cells that experienced changing local cell density by naturally occurring cell movement (Figure S5G). Control experiments showed that this is a one-way regulation because inhibition of MEK-ERK activity using a MEK inhibitor did not affect local cell density (Figure S5H).
Conversely, we determined whether an acute reduction in local cell density upregulates ERK activity. We cultured an island of contact-inhibited cells within a polydimethylsiloxane (PDMS) barrier (Figure 4D). Upon removal of the barrier, cells on the periphery migrated into the open space and upregulated ERK activity within 3 h (Figure 4D), arguing that lowering cell density can also rapidly activate ERK. We note that there was an initial spike of ERK activity upon removal of the barrier. This increase was likely induced by shear stress associated with physically removing the barrier because it was short-lived, and cells in the middle of the island, which did not migrate outward, also showed a similar ERK activity spike.
Having confirmed a causal link between local cell density and ERK activity, we next investigated whether regulation of ERK activity is a major mechanism through which cell density in mother cells regulates Rb hyperphosphorylation in newborn cells. We first showed that ERK activity in mother cells was a strong predictor of Rb hyperphosphorylation in newborn cells (Figure S5I). We then tested whether this correlation reflects a causal relationship. Indeed, addition of selective MEK, ERK, or RAF inhibitors in mother cells completely suppressed Rb hyperphosphorylation in newborn cells (Figure 4E; Figure S5J). To determine whether ERK activity is rate limiting for density-dependent suppression of Rb hyperphosphorylation in newborn cells, we overexpressed a constitutively active form of MEK1 (MEK1-CA; S218D/S222D) (Mansour et al., 1994). Persistent MEK-ERK activation largely but not completely abolished regulation of Rb hyperphosphorylation in newborn cells by local cell density in mother cells (Figure 4F; Figure S5K). We conclude that rapid regulation of ERK activity by local cell density changes in mother cells is a major signaling mechanism regulating Rb hyperphosphorylation in newborn cells.
ERK activity has been shown in other systems to increase expression of cyclin D1 (Lavoie et al., 1996; Robinson and Cobb, 1997) and reduce expression of p27 (Delmas et al., 2001; Sakakibara et al., 2005). We treated mother cells with a MEK inhibitor and measured mRNA and protein-level changes in single newborn cells using fluorescence in situ hybridization (FISH) and immunofluorescence, respectively. Inhibition of the MEK-ERK pathway downregulated cyclin D1 mRNA and upregulated p27 mRNA in newborn cells (Figure 4G, top panel) but did not substantially affect an unrelated mRNA RPL10 used as a negative control (Figure S5L). Using quantitative reverse-transcriptase PCR (qRT-PCR) and intron-specific primers that detect nascently transcribed pre-mRNA as a readout for instantaneous transcriptional activity, we further showed that MEK-ERK activity rapidly regulates the rate of nascent transcription of cyclin D1 and p27 within an hour (Figure S5M). This result argues that changes in the two mRNA levels in newborn cells are mediated at least partially by a change in mRNA synthesis rates. Consistent with the direction of mRNA-level changes, we also found that MEK inhibition in mother cells resulted in lower cyclin D1 and higher p27 protein levels in newborn cells (Figure 4G, bottom panel).
We conclude that regulation of ERK activity is a primary and fast-acting mechanism by which the memory of local cell density in mother cells controls Rb hyperphosphorylation in newborn cells. Increasing local cell density rapidly suppresses ERK activity in mother cells to downregulate and upregulate expression of cyclin D1 and p27 in newborn cells, respectively, which, in turn, controls whether newborn cells dephosphorylate Rb and exit to quiescence.
Competition between cell density and mitogen signals in mother cells controls exit to quiescence in newborn cells
Previous studies showed that mitogen and cell contact signals compete to regulate proliferation (Kim et al., 2009), raising the question of how newborn cells integrate previous cell density and mitogen signals to control Rb hyperphosphorylation and cell cycle exit. We tested the hypothesis that competing cell density and mitogen signals are integrated via ERK signaling into opposing shifts in the cyclin D1/p27 ratio in newborn cells to control Rb hyperphosphorylation and cell proliferation.
We first measured the correlation between ERK activity and local cell density while lowering the concentration of mitogen-containing serum. We found that cells maintained cell density-regulated ERK activity at lower mitogen levels, but the curve was shifted toward lower average levels of ERK activity (Figure 5A, top panel). Heatmap analysis also shows competition between the effects of cell density and mitogen stimuli on time-averaged ERK activity (Figure 5A, bottom panel). Similar results were obtained when we measured the frequency, amplitude, or absolute height of ERK pulses (Figures S6A–S6C).
Figure 5. Competing cell density and mitogen signals shift the same activator-inhibitor balance to decide whether newborn cells exit to quiescence.
(A) ERK activity was time-averaged over 4 h. Top: line plots are population medians of 25 or more cells per bin; shaded error bars are 95% confidence intervals. Bottom: population medians of ERK activity (n ≥ 200 cells per condition). Data are representative of 3 independent experiments.
(B) Top: line plots are population medians of 30 or more cells per bin; shaded error bars are 95% confidence intervals. Bottom: population medians of cyclin D1/p27 ratio and percentage of cells with Rb hyperphosphorylation (n ≥ 250 cells per condition). Data are representative of 3 independent experiments.
(C) Percentage of newborn cells with Rb hyperphosphorylation is plotted as a function of cyclin D1/p27 ratio (n ≥ 20 cells per bin, n ≥ 320 total for each condition). The Hill curve was fitted to the entire dataset. The color-coded schematic in the legend corresponds to the heatmaps in (B).
See also Figure S6.
We further found that the competition between cell density and mitogen signaling controls cyclin D1 and p27 expression (Figure S6D). Importantly, when we compared the cyclin D1/p27 ratio versus local cell density in mothers as we reduced the strength of mitogen stimuli, we found a parallel reduction of the cyclin D1/p27 ratio and the percentage of newborn cells with hyperphosphorylated Rb (Figure 5B). This striking result argues that a single parameter, the cyclin D1/p27 ratio, controls Rb hyperphosphorylation independent of whether the same cyclin D1/p27 ratio is generated by high cell density and high mitogen stimuli or low cell density and low mitogen stimuli. Indeed, when we analyzed three different conditions with similar cyclin D1/p27 ratios, we found that the relationship between Rb hyperphosphorylation and the cyclin D1/p27 ratio fell onto the same ultrasensitive curve (Figure 5C). We observed similar results when plotting all density and serum conditions (Figure S6E). To test whether the competition of cell density and mitogen signals also applies to other epithelial cells, we titrated epidermal growth factor (EGF) to stimulate MCF10A cells and observed similar opposing effects of cell density and EGF stimulation on regulation of ERK activity (Figure S6F) and Rb hyperphosphorylation (Figure S6G).
These different lines of evidence argue that cells use a simple code to integrate local cell density and mitogen signals in individual mother cells, which is based on ongoing competition that ultimately shifts a single parameter in newborn cells, the cyclin D1/p27 ratio, above or below a sharp threshold to control the binary cell fate of each newborn cell.
DISCUSSION
An ultrasensitive activator-inhibitor balance controls exit to quiescence in response to memory of local cell density inherited from mother cells
Our study shows that newborn cells integrate the history of local cell density experienced by mother cells to decide whether to exit to quiescence or keep proliferating (Figure 6A). We combined live-cell microscopy and fixed-cell immunofluorescence to link changes in local cell density to three molecular events that have been shown to be tightly coupled to cell cycle entry or exit at the single-cell level in daughter cells: Rb hyperphosphorylation (Chung et al., 2019; Moser et al., 2018), CDK2 activity bifurcation (Schwarz et al., 2018; Spencer et al., 2013), and APC/CCdh1 inactivation (Cappell et al., 2016).
Figure 6. Regulation of the proliferation-quiescence decision by cell density.
(A) Model.
(B) Schematic of the ultra sensitive response curve of Rb hyperphosphorylation as a function of the cyclin D1/p27 ratio. In this example, the Hill function has EC50 = 1 (the ratio at which 50% of cells have hyperphosphorylated Rb) and Hill coefficient nH = 6.3 (measured in Figure 3E). EC90/EC10 = 2.0, EC95/EC5 = 2.5.
(C) Cell-to-cell variability in the cyclin D1/p27 ratio in cells that experienced similar local density and mitogen conditions. A histogram of the cyclin D1/p27 ratio in newborn cells when local cell density experienced by mothers is between 0.18 and 0.20 (n = 767 cells, data from Figure 3D) is overlaid with the Hill function fitted to the entire dataset (data from Figure 3E, nH = 6.3).
A striking finding of our study was that the levels of cyclin D1 and p27 control Rb hyperphosphorylation in newborn cells in an ultrasensitive manner. The steep shape of the response curve dictates a tight control of Rb hyperphosphorylation based on the ratio of nuclear cyclin D1/p27 levels (Figure 3E; Hill coefficient, ~6). This argues that the complex integration of cell density and mitogen signals can be reduced to a single ratio parameter of two CDK regulators that decides the cell fate of newborn cells. Only a 2-fold difference in the cyclin D1/p27 ratio is needed to switch the percentage of cells with hyperphosphorylated Rb from 10% to 90% (EC10 and EC90) (Figure 6B). The cyclin D1/p27 ratio changed around 3-fold between cells that experienced highest versus lowest local densities (Figure 3D). Because the observed distributions of the cyclin D1/p27 ratio were near the critical threshold, even a small downward shift in the ratio can force cells to dephosphorylate Rb and exit to quiescence.
The finding of an ultrasensitive control of Rb hyperphosphorylation by cyclin D1 and p27 restricts plausible models of how p27 regulates CDK4/6 activity. Some previous studies showed that p27 functions as an assembly factor and activator of cyclin D-CDK4/6 when phosphorylated on certain sites (Blain et al., 1997; Guiley et al., 2019; James et al., 2008; Sherr and Roberts, 1999). Nevertheless, our study shows that cells with relatively higher nuclear levels of p27 over cyclin D1 consistently lost Rb hyperphosphorylation. Furthermore, we found that p27 overexpression suppressed Rb hyperphosphorylation, whereas p27 knockdown enhanced Rb hyperphosphorylation (Figure S4D, right panel). These results support a model where p27 primarily functions as an inhibitor of cyclin D-CDK4/6 activity and Rb hyperphosphorylation during cell density-mediated cell cycle exit.
Knockout mouse models also provide support for the hypothesis that p27, cyclin D1, and CDK4/6 kinases are rate limiting for control of tissue cell number and tissue size. p27 knockout mice have a larger body size and enlarged tissues with increased numbers of cells (Fero et al., 1996; Kiyokawa et al., 1996; Nakayama et al., 1996). Cyclin D1 knockout mice (Fantl et al., 1995; Sicinski et al., 1995), CDK4 knockout mice (Martín et al., 2003; Rane et al., 1999; Tsutsui et al., 1999), and CDK6 knockout mice (Malumbres et al., 2004) show reduced body sizes. These results are consistent with rate-limiting competition between p27 and cyclin D-CDK4/6 in controlling the number of cell divisions and tissue size.
Additional signaling pathways that sense cell density and control proliferation
In addition to ERK signaling (Aoki et al., 2013; Viñals and Pouysségur, 1999), known mediators of cell density-dependent regulation of proliferation include receptor tyrosine kinase signaling and cell-cell adhesion molecules (Curto et al., 2007; Perrais et al., 2007; Qian et al., 2004), the Hippo-YAP pathway (Aragona et al., 2013; Koo et al., 2020; Zhao et al., 2007), and other signaling pathways. Our study shows that ERK signaling in mother cells is a rate-limiting step controlling density-dependent proliferation in newborn cells. In addition, regulation of ERK activity by local density and regulation of cyclin D1 and p27 transcription by ERK activity are rapid (within 1 h), consistent with the timescale of the maternal memory of local density.
We note that hyperactivating MEK-ERK largely but incompletely abolished regulation of newborn cell proliferation by the memory of cell density in mother cells (Figure 4F). This suggests that, although ERK signaling is a primary and fast-acting mediator of this regulation, additional mechanisms are also involved. Such additional signaling mechanisms may also be important to robustly maintain the quiescent state over long time periods, in parallel to the rapid response of the ERK pathway to faster changes in cell density.
Model for cell density control through probabilistic regulation of proliferation by the memory of local cell density from mother cells
Maintaining proper cell density is critical for epithelial homeostasis and function and is achieved through density-regulated proliferation as well as cell death and extrusion (Eisenhoffer and Rosenblatt, 2013; Macara et al., 2014). Our study provides two conceptual insights into how cell density may be robustly controlled in a two-dimensional cell sheet. First we show that there is remaining cell-to-cell variability in the cyclin D1/p27 ratio even when cells experienced the same local cell density and mitogen signal (Figure 6C). As a result, instead of an all-or-none response where all cells in a population stop proliferating at a specific density, the activator-inhibitor balance controls a probabilistic outcome. The percentage of cells that proliferate in a population in turn controls cell number and density.
Similar evidence of a stochastic mechanism controlling cell density has been reported in vivo. Intravital imaging of mouse epidermal stem cells has shown that cell proliferation is triggered by a decrease in local cell density (Mesa et al., 2018). Consistent with a probabilistic regulation model, neighboring cells that experienced a similar decrease in local cell density could adopt different fates (Mesa et al., 2018). A potential advantage of probabilistic regulation of proliferation is to prevent cell density from overshooting the optimal range, which is more likely if all cells in a sheet were to exit or enter the cell cycle in response to a sharply defined cell density.
The second conceptual insight is that cell density regulation of proliferation occurs primarily in the mother rather than the daughter. Consistent with such a memory mechanism, sister cells in the basal layer of adult mouse epidermis are more likely to share the same fate (proliferation versus differentiation) than expected by chance (Mesa et al., 2018; Rompolas et al., 2016). Extending the time period when cells can integrate cell density inputs may provide more accurate measurements of density in a noisy environment and more robustly regulate proliferation and cell density in an in vivo cell population.
Conclusion
Our study shows that newborn daughter cells inherit the memory of local cell density from their mothers. We show that high cell density rapidly suppresses ERK activity, which, in turn, controls the ratio between cyclin D1 and p27 levels in newborn cells. Small changes in the cyclin D1/p27 ratio are then translated by an ultrasensitive response that controls CDK4/6 activity and Rb hyperphosphorylation, which dictate cell fate by directing cells to proliferate or exit to quiescence. Finally, our findings argue for a regulatory mechanism where competing cell density and mitogen signals are integrated in mother cells to shift the cyclin D1/p27 ratio in newborn cells in opposite directions across a sharp threshold in the activator-inhibitor balance that decides cell fate.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tobias Meyer (tobias1@stanford.edu).
Materials availability
Newly generated materials in this study are available from the Lead Contact upon request.
Data and code availability
Microscopy and all other data reported in this paper will be shared by the Lead Contact upon request.
The code for the image analysis pipeline is publicly available at https://github.com/scappell/Cell_tracking. Additional original code has been deposited at Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-cyclin D1 mAb [SP4] | Abcam | Cat#ab16663; RRID: AB_443423 |
| Mouse anti-p27 mAb [SX53G8.5] | Cell Signaling Technology | Cat#3698; RRID: AB_2077832 |
| Rabbit anti-phospho-Rb (Ser807/811) mAb [D20B12] | Cell Signaling Technology | Cat#8516; RRID: AB_11178658 |
| Rabbit anti-phospho-Rb (Ser807/811) mAb (Alexa Fluor 488 conjugate) [D20B12] | Cell Signaling Technology | Cat#4277; RRID: AB_2797605 |
| Mouse anti-N-cadherin mAb [H-2] | Santa Cruz Biotechnology | Cat#sc-393933; RRID: AB_2832921 |
| Mouse anti-HA tag mAb [16B12] | Abcam | Cat#ab130275; RRID: AB_11156884 |
| Normal Rabbit IgG antibody | Cell Signaling Technology | Cat#2729; RRID: AB_1031062 |
| Chemicals, peptides, and recombinant proteins | ||
| PD0325901 | Selleck Chemicals | Cat#S1036; CAS#391210-10-9 |
| SCH772984 | Selleck Chemicals | Cat#S7101; CAS#942183-80-4 |
| LY3009120 | Selleck Chemicals | Cat#S7842; CAS#1454682-72-4 |
| PD-0332991 (palbociclib) | Selleck Chemicals | Cat#S1116; CAS#827022-32-2 |
| Cdk1/2 Inhibitor III | Millipore Sigma | Cat#217714; CAS#443798-55-8 |
| Doxycycline hyclate | Sigma-Aldrich | Cat#D9891; CAS#24390-14-5 |
| Bovine plasma fibronectin | Sigma-Aldrich | Cat#F1141 |
| Bovine collagen (type I) | Advanced BioMatrix | Cat#5005 |
| Critical commercial assays | ||
| ViewRNA ISH Cell Assay Kit | Thermo Fisher | Cat#QVC0001 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat#1725122 |
| Experimental models: Cell lines | ||
| Human: RPE-1 hTERT | ATCC | Cat#CRL-4000; RRID: CVCL_4388 |
| Human: MCF10A | ATCC | Cat#CRL-10317; RRID: CVCL_0598 |
| Oligonucleotides | ||
| Non-targeting siRNA | Dharmacon | Cat#D-001810-10-05 |
| CCND1 siRNA: 5′-GCAUGUAGUCACUUUAUAA-3′ | Dharmacon | Cat#J-003210-17 |
| CCND1 siRNA: 5′-GCGUGUAGCUAUGGAAGUU-3′ | Dharmacon | Cat#J-003210-18 |
| CDKN1B siRNA: 5′-ACGUAAACAGCUCGAAUUA-3′ | Dharmacon | Cat#J-003472-07 |
| CCND1 RNA FISH probe | Thermo Fisher | Cat#VA1-11978 |
| CDKN1B RNA FISH probe | Thermo Fisher | Cat#VA1-12174 |
| RPL10RNA FISH probe | Thermo Fisher | Cat#VA1-12078 |
| Pre-CCND1 RT-qPCR forward primer: 5′-TTTGTCATCGGCCAGAAATA-3′ | This manuscript | N/A |
| Pre-CCND1 RT-qPCR reverse primer: 5′-GACCTTCAGAGCACAGACCA-3′ | This manuscript | N/A |
| Pre-CDKN1B RT-qPCR forward primer: 5′-CGCAGGAATAAGGAAGCGAC-3′ | This manuscript | N/A |
| Pre-CDKN1B RT-qPCR reverse primer: 5′-GAATACGCCGAAAAGCAAGC-3′ | This manuscript | N/A |
| EEF1A1 RT-qPCR forward primer: 5′-GATGGCCAGTAGTGGTGGAC-3′ | This manuscript | N/A |
| EEF1A1 RT-qPCR reverse primer: 5′-TTTTTCGCAACGGGTTTG-3′ | This manuscript | N/A |
| Recombinant DNA | ||
| pLV-EF1a-H2B-mTurquoise-IRES-PuroR | Spencer et al., 2013 | N/A |
| CSII-hDHB (amino acids 994-1087)-mVenus | Spencer et al., 2013 | N/A |
| CSII-pEF1a-mCherry-Geminin (amino acids 1-110) | Cappell et al., 2016 | N/A |
| pLV-EF1a-CaaX-iRFP-IRES-Puro | Bisaria et al., 2020 | N/A |
| pPBbsr2-EKAR-NLS | Yang et al., 2017 | N/A |
| ERK KTR | Regot et al., 2014 | N/A |
| pCW-TRE-PGK promoter-cyclin D1-PuroR-T2A-rtTA | This manuscript | N/A |
| pCW-TRE-PGK promoter-p27-BlastR-T2A-rtTA | This manuscript | N/A |
| pCW-TRE-PGK promoter-HA-MEK1-S218D/S222D-PuroR-T2A-rtTA | This manuscript | N/A |
| Software and algorithms | ||
| Microscopy image analysis: cell tracking | Cappell et al., 2016 | https://github.com/scappell/Cell_tracking |
| Microscopy image analysis: custom code for measuring local cell density | This manuscript | Zenodo: https://doi.org/10.5281/zenodo.4960626 |
| Other | ||
| Sylgard Silicone Elastomer Kit 184 | Dow | SKU#DC4019862 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
RPE-1 hTERT cells (obtained from ATCC, CRL-4000, RRID: CVCL_4388, female) were cultured in phenol red-free DMEM/F12 (Invitrogen) supplemented with 10% fetal bovine serum (Atlanta Biologicals), 10ug/mL hygromycin B (InvivoGen), 50 U/mL penicillin, and 50 μg/mL streptomycin (Thermo Fisher). MCF10A cells (obtained from ATCC, CRL-10317, RRID: CVCL_0598, female) were cultured in phenol red-free DMEM/F12 (Invitrogen) supplemented with 5% horse serum (ATCC), 20 ng/mL EGF (PeproTech), 10 μg/mL insulin (Sigma-Aldrich), 0.5 μg/mL hydrocortisone (Sigma-Aldrich), 100 ng/mL cholera toxin (Sigma-Aldrich), 50 U/mL penicillin and 50 μg/mL streptomycin (Thermo Fisher); for imaging experiments, MCF10A cells were cultured in phenol red-free DMEM/F12 supplemented with 0.3% bovine serum albumin (Sigma-Aldrich), 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin, and the indicated concentration of EGF. All cells were cultured at 37°C with 5% CO2.
METHOD DETAILS
Cell culture
All experiments were performed using RPE-1 hTERT cells unless noted otherwise. RPE-1 hTERT cells (obtained from ATCC, CRL-4000) were cultured in phenol red-free DMEM/F12 (Invitrogen) supplemented with 10% fetal bovine serum (Atlanta Biologicals), 10ug/mL hygromycin B (InvivoGen), 50 U/mL penicillin, and 50 μg/mL streptomycin (Thermo Fisher). MCF10A cells (obtained from ATCC, CRL-10317) were cultured in phenol red-free DMEM/F12 (Invitrogen) supplemented with 5% horse serum (ATCC), 20 ng/mL EGF (PeproTech), 10 μg/mL insulin (Sigma-Aldrich), 0.5 μg/mL hydrocortisone (Sigma-Aldrich), 100 ng/mL cholera toxin (Sigma-Aldrich), 50 U/mL penicillin and 50 μg/mL streptomycin (Thermo Fisher); for imaging experiments, MCF10A cells were cultured in phenol red-free DMEM/F12 supplemented with 0.3% bovine serum albumin (Sigma-Aldrich), 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin, and the indicated concentration of EGF. All cells were cultured at 37°C with 5% CO2.
Constructs and stable cell lines
Cells were transduced with lentiviral vectors pLV-EF1a-histone 2B (H2B)-mTurquoise-IRES-PuroR and CSII-hDHB (amino acids 994-1087)-mVenus (Spencer et al., 2013), CSII-pEF1a-mCherry-Geminin (amino acids 1-110) (Cappell et al., 2016; Sakaue-Sawano et al., 2008), and pLV-EF1a-CaaX-iRFP-IRES-Puro (Bisaria et al., 2020) as previously described. Cells were co-transfected with pPBbsr2-EKAR-NLS and a piggyBac transposase as previously described (Yang et al., 2017). ERK-KTR cDNA (Regot et al., 2014) tagged with mVenus was cloned into a pLV-EF1a backbone. To construct doxycycline-inducible expression constructs, human cyclin D1 cDNA and human MEK1-S218D/S222D (a gift from David Sabatini & Kris Wood, Addgene plasmid #64604) were cloned into the lentiviral backbone pCW-TRE-PGK promoter-PuroR-T2A-rtTA, derived from pCW-Cas9-Puro (a gift from Eric Lander & David Sabatini, Addgene plasmid #50661); human p27 cDNA was cloned into the lentiviral backbone pCW-TRE-PGK promoter-BlastR-T2A-rtTA, derived from pCW-Cas9-Blast (a gift from Mohan Babu, Addgene plasmid #83481).
Chemicals
PD0325901 (Selleck Chemicals S1036, used at 100 nM), SCH772984 (Selleck Chemicals S7101, used at 1 μM), LY3009120 (Selleck Chemicals S7842, used at 4.096 μM), PD-0332991/palbociclib (Selleck Chemicals S1116, used at 1 μM), Cdk1/2 Inhibitor III (Millipore Sigma 217714, used at 1 μM), doxycycline hyclate (Sigma-Aldrich D9891, used at 1 μg/mL).
siRNA transfection
siRNAs were reverse transfected into cells using DharmaFect 1 (Dharmacon) according to manufacturer instructions, at a final concentration of 40 nM. Cells were changed into growth media approximately 6 hours after transfection. siRNAs were obtained from Dharmacon: non-targeting control (D-001810-10-05), cyclin D1 (J-003210-17: GCAUGUAGUCACUUUAUAA; J-003210-18: GCGUGUAGCUAUGGAAGUU), and p27 (J-003472-07: ACGUAAACAGCUCGAAUUA).
Immunofluorescence
Cells were fixed in 4% paraformaldehyde (PBS solution) for 15 minutes at room temperature, and washed three times in PBS. Cells were permeabilized in 0.2% Triton X-100 (PBS solution) for 15 minutes and blocked with blocking buffer (10% fetal bovine serum, 1% bovine serum albumin, 0.1% Triton X-100, and 0.01% NaN3 in PBS) for 1 hour. Cells were stained overnight at 4°C with primary antibodies (diluted in blocking buffer), washed three times in PBS, and stained with fluorophore-conjugated secondary antibodies (Thermo Scientific, 1:1000) for 1 hour. Cells were subsequently stained with Hoechst 33342 (Thermo Fisher Scientific, 1:10,000 in PBS) for 10 minutes and washed three times in PBS. To stain with a fluorophore-conjugated primary antibody, cells were treated with rabbit IgG (Cell Signaling Technology 2729, 1:100 in PBS) for 1 hour to block binding to secondary antibodies, stained with the primary antibody for 3 hours at 4°C, and washed three times in PBS. Antibodies used in the study: rabbit anti-cyclin D1 (Abcam ab16663, 1:500, validated with knock-down and overexpression), mouse anti-p27 (Cell Signaling Technology 3698, 1:500, validated with knock-down and overexpression), rabbit anti-phospho-Rb (Ser807/811) (Cell Signaling Technology 8516, 1:2500, validated with knock-down and CDK4/6 inhibitor palbociclib treatment), rabbit anti-phospho-Rb (Ser807/811) (Alexa Fluor 488 conjugate) (Cell Signaling Technology 4277, 1:2500, validated with CDK4/6 inhibitor palbociclib treatment), mouse anti-N-cadherin (Santa Cruz Biotechnology sc-393933, 1:250, validated with knock-down), and mouse anti-HA tag (Abcam ab130275, 1:1000, validated with overexpression).
RNA FISH
Cells were fixed in 4% paraformaldehyde (PBS solution) for 15 minutes at room temperature, and washed three times in PBS. Cells were permeabilized in 0.2% Triton X-100 (PBS solution) for 15 minutes and washed three times in PBS. RNA in situ hybridization was carried out using the ViewRNA ISH cell assay (Thermo Fisher) according to manufacturer instructions. Cells were subsequently stained with Hoechst 33342 (1:10,000 in PBS) for 10 minutes and washed three times in PBS. Hybridization probes used in this study: CCND1 (VA1-11978), CDKN1B (VA1-12174), RPL10 (VA1-12078). Probe specificity for CCND1 and CDKN1B was confirmed through siRNA knock-downs.
PDMS barrier assay
Polydimethylsiloxane (Sylgard Silicone Elastomer Kit 184) was prepared as previously described (Thery and Piel, 2009). Briefly, the elastomer base and curing agent were mixed at a 10:1 ratio, degassed, and cured at 65°C for 3 hours. PDMS was cut into rings with an outer diameter ~4 mm, inner diameter ~2 mm, and height ~3mm using a commercial leather belt hole punch. Glass-bottom 96-well plates (Cellvis P96-1.5H-N) were coated with 100 μg/mL bovine plasma fibronectin (Sigma-Aldrich F1141), washed with sterile water, and dried. PDMS rings were sterilized with UV irradiation and placed inside wells of 96-well plates, firmly adhering to the bottom. Cells were plated inside the PDMS ring at 6,000 cells per well (~1.9 × 105 cm−2), in a mixture of non-fluorescent and fluorescent cells (5:1 ratio) to facilitate tracking at high density. Cells were maintained for two days in growth media, and PDMS barriers were removed with forceps on the day of the experiment.
Microscopy
Cells were plated onto glass-bottom 96-well plates (Cellvis P96-1.5H-N), which were coated overnight with 20 μg/mL bovine plasma fibronectin (Sigma-Aldrich F1141) for RPE-1 hTERT cells or with 60 μg/mL bovine collagen I (Advanced BioMatrix #5005-B) for MCF10A cells. To probe a wide range of cell density, cells were plated at multiple densities for each experiment. Plating density ranged between 5,000 cells to 16,000 cells per well (1.7 × 104cm−2 ~5.3 × 104cm−2). Cells were imaged on an ImageXpress Micro microscope (Molecular Devices) or Eclipse Ti2 microscope (Nikon) in humidified 37°C chambers with 5% CO2 and with the appropriate filter sets. 10x objective (0.3 N.A.) with no binning was used for live-cell imaging of H2B, CDK2 activity reporter, APC/C degron reporter, and ERK kinase translocation reporter; 20x objective (0.75 N.A.) with 2-by-2 pixel binning was used for live-cell FRET imaging and fixed-cell immunofluorescence and RNA FISH imaging. During live-cell imaging, images were taken every 12 min or 15 min, with the total light exposure under 300 ms for each multi-color image.
RT-qPCR
RNA was extracted from cells using RNeasy Mini Kit and QIAshredder (QIAGEN) and depleted of genomic DNA using RNase-free DNase (QIAGEN). Reverse transcription was performed using RevertAid reverse transcriptase (Thermo Scientific) and random hexamer primers (Thermo Scientific) according to manufacturer instructions. Quantitative PCR (qPCR) was performed using iTaq Universal SYBR Green Supermix (Bio-Rad) on a LightCycler 480 II (Roche) according to manufacturer instructions. Measurements were normalized to the housekeeping control (EEF1A1). Primers used in this study: pre-CCND1-F: 5′- TTTGTCATCGGCCAGAAATA-3′, pre-CCND1-R: 5′- GACCTTCAGAGCACAGACCA-3′, pre-CDKN1B-F: 5′-CGCAGGAATAAGGAAGCGAC-3′, pre-CDKN1B-R: 5′- GAATACGCCGAAAAGCAAGC-3′, EEF1A1-F: 5′- GATGGCCAGTAGTGGTGGAC-3′, EEF1A1-R: 5′- TTTTTCGCAACGGGTTTG-3′. PCR program: 95°C 5 min, 45 cycles of 95°C 15 s, 60°C 15 s, 72°C 15 s.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image analysis
Image analysis was performed with a custom MATLAB pipeline as previously described (Cappell et al., 2016; Chung et al., 2019). Briefly, optical illumination bias was corrected by measuring background autofluorescence signal for each imaging session, which was used to flatten the raw images. Global background subtraction was then performed on all images. Cell nuclei were segmented based on H2B-mTurquoise (live-cell imaging), EKAREV-NLS (live-cell imaging of EKAREV FRET sensor), or Hoechst (fixed-cell imaging, immunofluorescence and RNA FISH).
Local cell density was measured by examining a 50-μm circle around the nuclear centroid (results robust to exact circle size): the fraction of pixels covered by neighboring nuclei was used as the metric “local cell density.” In experiments combining live-cell tracking and fixed-cell immunofluorescence, newborn cells that underwent a round of mitosis (anaphase occurred 1~1.5 hr before fixation) were selected in silico for analysis. Local density in mothers was averaged during −3 hr to 0 hr relative to mitosis, which is used throughout the paper unless stated otherwise.
FRET signals were quantified by taking the ratio between the median nuclear intensity of the FRET channel (CFP excitation, YFP emission) and the median nuclear intensity of the CFP channel (CFP excitation, CFP emission). The EKAREV-NLS FRET signal may vary between experiments, thus ERK activity is only compared between different conditions of the same experiment.
To measure the CDK2 activity reporter and ERK kinase translocation reporter, the cytoplasm was sampled by expanding a ring outside the nucleus (with inner radius of 0.65 μm and outer radius of 3.25 μm) without overlapping with cytoplasm from a neighboring cell. The activity was calculated by taking the ratio between the median cytoplasmic intensity and the median nuclear intensity.
For immunofluorescence quantification, DNA content was calculated as the total nuclear Hoechst intensity; nuclear fluorescence signals (cyclin D1, p27, p21, phospho-Rb-Ser807/811) were calculated as the median nuclear intensity; cytoplasmic signal (HA-MEK1-CA) was calculated as the median cytoplasmic intensity (cytoplasm was sampled as described above). The measured cyclin D1/p27 ratio may vary between experiments due to variability in immunofluorescence staining, thus the ratio is only compared between different conditions of the same experiment. The ratio is not indicative of actual stoichiometry between the two proteins.
RNA FISH quantification was adapted from Chung et al. (2019): the cytoplasm was sampled by expanding a ring outside the nucleus (with inner radius of 1.3 μm and outer radius of 7.15 μm, results robust to exact ring size) without overlapping with cytoplasm from a neighboring cell; FISH puncta were identified as the foreground by thresholding on background-subtracted images; RNA puncta count was calculated as the number of foreground pixels within the sampled cytoplasm.
Statistical analysis
Statistical analyses were performed using Fisher’s exact test (MATLAB fishertest), one-way ANOVA (MATLAB anova1), Student’s two-sample t test (MATLAB ttest2) or paired/one-sample t test (MATLAB ttest), and two-sample Kolmogorov-Smirnov test (MATLAB kstest2). Logistic regression was performed using MATLAB’s fitglm function. The likelihood ratio test for regression slopes is based on the chi-square statistic of the full model and the constant model (where the slope is zero). Cross-validation was performed by randomly partitioning the data into k equally sized subsamples (k-fold cross-validation), training the model on k-1 subsamples and testing the model on the remaining subsample, and repeating the process over different testing subsamples. Further details, including test used, number of n, and measure of center and dispersion, could be found in the figure legends. The following p value convention is used throughout the paper: n.s. p > 0.05, * p < 0.05, ** p < 0.01, *** p < 0.001
Supplementary Material
Highlights.
Memory of local cell density from mother cells regulates proliferation in daughters
Memory of local cell density shifts the expression ratio of cyclin D1 over p27
The cyclin D1-p27 balance controls proliferation in an ultrasensitive manner
Competing cell density and mitogen signals converge on the cyclin D1-p27 balance
ACKNOWLEDGMENTS
We thank Damien Garbett, Hee Won Yang, Nalin Ratnayeke, Leighton Daigh, Lindsey Pack, Mingyu Chung, and Anjali Bisaria for help with reagents and cell lines; Mingyu Chung, Lindsey Pack, Damien Garbett, Katie Ferrick, and Leighton Daigh for critical reading of the manuscript; members of the Meyer lab for feedback and technical support; and the Stanford Shared FACS Facility for cell sorting. This work was funded by a National Institute of General Medical Sciences (NIGMS) R35 grant (GM127026). Y.F. was supported by the Stanford Graduate Fellowship and the Stanford Center for Systems Biology.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109436.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Albeck JG, Mills GB, and Brugge JS (2013). Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell 49, 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K, Kumagai Y, Sakurai A, Komatsu N, Fujita Y, Shionyu C, and Matsuda M (2013). Stochastic ERK activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol. Cell 52, 529–540. [DOI] [PubMed] [Google Scholar]
- Aoki K, Kondo Y, Naoki H, Hiratsuka T, Itoh RE, and Matsuda M (2017). Propagating Wave of ERK Activation Orients Collective Cell Migration. Dev. Cell 43, 305–317.e5. [DOI] [PubMed] [Google Scholar]
- Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, and Piccolo S (2013). A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154, 1047–1059. [DOI] [PubMed] [Google Scholar]
- Arora M, Moser J, Phadke H, Basha AA, and Spencer SL (2017). Endogenous Replication Stress in Mother Cells Leads to Quiescence of Daughter Cells. Cell Rep. 19, 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novák B, and Bakal C (2017). DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun 8, 14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Skotheim JM, and de Bruin RA (2013). Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol 14, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaria A, Hayer A, Garbett D, Cohen D, and Meyer T (2020). Membrane-proximal F-actin restricts local membrane protrusions and directs cell migration. Science 368, 1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blain SW, Montalvo E, and Massagué J (1997). Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J. Biol. Chem 272, 25863–25872. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, and Hannon GJ (1995). Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377, 552–557. [DOI] [PubMed] [Google Scholar]
- Cappell SD, Chung M, Jaimovich A, Spencer SL, and Meyer T (2016). Irreversible APC(Cdh1) Inactivation Underlies the Point of No Return for Cell-Cycle Entry. Cell 166, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung M, Liu C, Yang HW, Köberlin MS, Cappell SD, and Meyer T (2019). Transient Hysteresis in CDK4/6 Activity Underlies Passage of the Restriction Point in G1. Mol. Cell 76, 562–573.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curto M, Cole BK, Lallemand D, Liu CH, and McClatchey AI (2007). Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J. Cell Biol 177, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas C, Manenti S, Boudjelal A, Peyssonnaux C, Eychène A, and Darbon JM (2001). The p42/p44 mitogen-activated protein kinase activation triggers p27Kip1 degradation independently of CDK2/cyclin E in NIH 3T3 cells. J. Biol. Chem 276, 34958–34965. [DOI] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, and Leder P (1995). Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82, 675–684. [DOI] [PubMed] [Google Scholar]
- Eisenhoffer GT, and Rosenblatt J (2013). Bringing balance by force: live cell extrusion controls epithelial cell numbers. Trends Cell Biol. 23, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantl V, Stamp G, Andrews A, Rosewell I, and Dickson C (1995). Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 9, 2364–2372. [DOI] [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, et al. (1996). A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 85, 733–744. [DOI] [PubMed] [Google Scholar]
- Guiley KZ, Stevenson JW, Lou K, Barkovich KJ, Kumarasamy V, Wijeratne TU, Bunch KL, Tripathi S, Knudsen ES, Witkiewicz AK, et al. (2019). p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 366, eaaw2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn AT, Jones JT, and Meyer T (2009). Quantitative analysis of cell cycle phase durations and PC12 differentiation using fluorescent biosensors. Cell Cycle 8, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hino N, Rossetti L, Marín-Llauradó A, Aoki K, Trepat X, Matsuda M, and Hirashima T (2020). ERK-Mediated Mechanochemical Waves Direct Collective Cell Polarization. Dev. Cell 53, 646–660.e8. [DOI] [PubMed] [Google Scholar]
- Hitomi M, and Stacey DW (1999). Cellular ras and cyclin D1 are required during different cell cycle periods in cycling NIH 3T3 cells. Mol. Cell. Biol 19, 4623–4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James MK, Ray A, Leznova D, and Blain SW (2008). Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol. Cell. Biol 28, 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kushiro K, Graham NA, and Asthagiri AR (2009). Tunable interplay between epidermal growth factor and cell-cell contact governs the spatial dynamics of epithelial growth. Proc. Natl. Acad. Sci. USA 106, 11149–11153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NG, Koh E, Chen X, and Gumbiner BM (2011). E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 108, 11930–11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, and Koff A (1996). Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 85, 721–732. [DOI] [PubMed] [Google Scholar]
- Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, and Matsuda M (2011). Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 22, 4647–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JH, Plouffe SW, Meng Z, Lee DH, Yang D, Lim DS, Wang CY, and Guan KL (2020). Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev. 34, 72–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie JN, L’Allemain G, Brunet A, Müller R, and Pouysségur J (1996). Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem 271, 20608–20616. [DOI] [PubMed] [Google Scholar]
- Macara IG, Guyer R, Richardson G, Huo Y, and Ahmed SM (2014). Epithelial homeostasis. Curr. Biol 24, R815–R825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, and Barbacid M (2001). To cycle or not to cycle: a critical decision in cancer. Nat. Rev. Cancer 1, 222–231. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Sotillo R, Santamaría D, Galán J, Cerezo A, Ortega S, Dubus P, and Barbacid M (2004). Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118, 493–504. [DOI] [PubMed] [Google Scholar]
- Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, and Ahn NG (1994). Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265, 966–970. [DOI] [PubMed] [Google Scholar]
- Martín J, Hunt SL, Dubus P, Sotillo R, Néhmé-Pélluard F, Magnuson MA, Parlow AF, Malumbres M, Ortega S, and Barbacid M (2003). Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene 22, 5261–5269. [DOI] [PubMed] [Google Scholar]
- McClatchey AI, and Yap AS (2012). Contact inhibition (of proliferation) redux. Curr. Opin. Cell Biol 24, 685–694. [DOI] [PubMed] [Google Scholar]
- Meloche S, and Pouysségur J (2007). The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 26, 3227–3239. [DOI] [PubMed] [Google Scholar]
- Mesa KR, Kawaguchi K, Cockburn K, Gonzalez D, Boucher J, Xin T, Klein AM, and Greco V (2018). Homeostatic Epidermal Stem Cell Self-Renewal Is Driven by Local Differentiation. Cell Stem Cell 23, 677–686.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min M, Rong Y, Tian C, and Spencer SL (2020). Temporal integration of mitogen history in mother cells controls proliferation of daughter cells. Science 368, 1261–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E, Valon L, Levillayer F, and Levayer R (2019). Competition for Space Induces Cell Elimination through Compaction-Driven ERK Downregulation. Curr. Biol 29, 23–34.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO (1997). Cyclin-dependent kinases: engines, clocks, and micro-processors. Annu. Rev. Cell Dev. Biol 13, 261–291. [DOI] [PubMed] [Google Scholar]
- Moser J, Miller I, Carter D, and Spencer SL (2018). Control of the Restriction Point by Rb and p21. Proc. Natl. Acad. Sci. USA 115, E8219–E8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, and Nakayama K (1996). Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85, 707–720. [DOI] [PubMed] [Google Scholar]
- Nevins JR (2001). The Rb/E2F pathway and cancer. Hum. Mol. Genet 10, 699–703. [DOI] [PubMed] [Google Scholar]
- Perrais M, Chen X, Perez-Moreno M, and Gumbiner BM (2007). E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol. Biol. Cell 18, 2013–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, and Koff A (1994). p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 8, 9–22. [DOI] [PubMed] [Google Scholar]
- Puliafito A, Hufnagel L, Neveu P, Streichan S, Sigal A, Fygenson DK, and Shraiman BI (2012). Collective and single cell behavior in epithelial contact inhibition. Proc. Natl. Acad. Sci. USA 109, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Karpova T, Sheppard AM, McNally J, and Lowy DR (2004). E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 23, 1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, and Barbacid M (1999). Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat. Genet 22, 44–52. [DOI] [PubMed] [Google Scholar]
- Regot S, Hughey JJ, Bajar BT, Carrasco S, and Covert MW (2014). High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MJ, and Cobb MH (1997). Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol 9, 180–186. [DOI] [PubMed] [Google Scholar]
- Rompolas P, Mesa KR, Kawaguchi K, Park S, Gonzalez D, Brown S, Boucher J, Klein AM, and Greco V (2016). Spatiotemporal coordination of stem cell commitment during epidermal homeostasis. Science 352, 1471–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin SM (2013). Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci 38, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara K, Kubota K, Worku B, Ryer EJ, Miller JP, Koff A, Kent KC, and Liu B (2005). PDGF-BB regulates p27 expression through ERK-dependent RNA turn-over in vascular smooth muscle cells. J. Biol. Chem 280, 25470–25477. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Satyanarayana A, and Kaldis P (2009). Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 28, 2925–2939. [DOI] [PubMed] [Google Scholar]
- Schwarz C, Johnson A, Kõivomägi M, Zatulovskiy E, Kravitz CJ, Doncic A, and Skotheim JM (2018). A Precise Cdk Activity Threshold Determines Passage through the Restriction Point. Mol. Cell 69, 253–264.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ (1993). Mammalian G1 cyclins. Cell 73, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, and Roberts JM (1999). CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, and Weinberg RA (1995). Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82, 621–630. [DOI] [PubMed] [Google Scholar]
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, and Meyer T (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Croix B, Sheehan C, Rak JW, Flørenes VA, Slingerland JM, and Kerbel RS (1998). E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1). J. Cell Biol 142, 557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streichan SJ, Hoerner CR, Schneidt T, Holzer D, and Hufnagel L (2014). Spatial constraints control cell proliferation in tissues. Proc. Natl. Acad. Sci. USA 111, 5586–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery M, and Piel M (2009). Adhesive micropatterns for cells: a microcontact printing protocol. Cold Spring Harb. Protoc 2009, pdb.prot5255. [DOI] [PubMed] [Google Scholar]
- Toettcher JE, Weiner OD, and Lim WA (2013). Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell 155, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, and Kiyokawa H (1999). Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol. Cell. Biol 19, 7011–7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viñals F, and Pouysségur J (1999). Confluence of vascular endothelial cells induces cell cycle exit by inhibiting p42/p44 mitogen-activated protein kinase activity. Mol. Cell. Biol 19, 2763–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Chung M, Kudo T, and Meyer T (2017). Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature 549, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Cappell SD, Jaimovich A, Liu C, Chung M, Daigh LH, Pack LR, Fan Y, Regot S, Covert M, and Meyer T (2020). Stress-mediated exit to quiescence restricted by increasing persistence in CDK4/6 activation. eLife 9, e44571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, and Guan KL (2015). Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 163, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. (2007). Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microscopy and all other data reported in this paper will be shared by the Lead Contact upon request.
The code for the image analysis pipeline is publicly available at https://github.com/scappell/Cell_tracking. Additional original code has been deposited at Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-cyclin D1 mAb [SP4] | Abcam | Cat#ab16663; RRID: AB_443423 |
| Mouse anti-p27 mAb [SX53G8.5] | Cell Signaling Technology | Cat#3698; RRID: AB_2077832 |
| Rabbit anti-phospho-Rb (Ser807/811) mAb [D20B12] | Cell Signaling Technology | Cat#8516; RRID: AB_11178658 |
| Rabbit anti-phospho-Rb (Ser807/811) mAb (Alexa Fluor 488 conjugate) [D20B12] | Cell Signaling Technology | Cat#4277; RRID: AB_2797605 |
| Mouse anti-N-cadherin mAb [H-2] | Santa Cruz Biotechnology | Cat#sc-393933; RRID: AB_2832921 |
| Mouse anti-HA tag mAb [16B12] | Abcam | Cat#ab130275; RRID: AB_11156884 |
| Normal Rabbit IgG antibody | Cell Signaling Technology | Cat#2729; RRID: AB_1031062 |
| Chemicals, peptides, and recombinant proteins | ||
| PD0325901 | Selleck Chemicals | Cat#S1036; CAS#391210-10-9 |
| SCH772984 | Selleck Chemicals | Cat#S7101; CAS#942183-80-4 |
| LY3009120 | Selleck Chemicals | Cat#S7842; CAS#1454682-72-4 |
| PD-0332991 (palbociclib) | Selleck Chemicals | Cat#S1116; CAS#827022-32-2 |
| Cdk1/2 Inhibitor III | Millipore Sigma | Cat#217714; CAS#443798-55-8 |
| Doxycycline hyclate | Sigma-Aldrich | Cat#D9891; CAS#24390-14-5 |
| Bovine plasma fibronectin | Sigma-Aldrich | Cat#F1141 |
| Bovine collagen (type I) | Advanced BioMatrix | Cat#5005 |
| Critical commercial assays | ||
| ViewRNA ISH Cell Assay Kit | Thermo Fisher | Cat#QVC0001 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat#1725122 |
| Experimental models: Cell lines | ||
| Human: RPE-1 hTERT | ATCC | Cat#CRL-4000; RRID: CVCL_4388 |
| Human: MCF10A | ATCC | Cat#CRL-10317; RRID: CVCL_0598 |
| Oligonucleotides | ||
| Non-targeting siRNA | Dharmacon | Cat#D-001810-10-05 |
| CCND1 siRNA: 5′-GCAUGUAGUCACUUUAUAA-3′ | Dharmacon | Cat#J-003210-17 |
| CCND1 siRNA: 5′-GCGUGUAGCUAUGGAAGUU-3′ | Dharmacon | Cat#J-003210-18 |
| CDKN1B siRNA: 5′-ACGUAAACAGCUCGAAUUA-3′ | Dharmacon | Cat#J-003472-07 |
| CCND1 RNA FISH probe | Thermo Fisher | Cat#VA1-11978 |
| CDKN1B RNA FISH probe | Thermo Fisher | Cat#VA1-12174 |
| RPL10RNA FISH probe | Thermo Fisher | Cat#VA1-12078 |
| Pre-CCND1 RT-qPCR forward primer: 5′-TTTGTCATCGGCCAGAAATA-3′ | This manuscript | N/A |
| Pre-CCND1 RT-qPCR reverse primer: 5′-GACCTTCAGAGCACAGACCA-3′ | This manuscript | N/A |
| Pre-CDKN1B RT-qPCR forward primer: 5′-CGCAGGAATAAGGAAGCGAC-3′ | This manuscript | N/A |
| Pre-CDKN1B RT-qPCR reverse primer: 5′-GAATACGCCGAAAAGCAAGC-3′ | This manuscript | N/A |
| EEF1A1 RT-qPCR forward primer: 5′-GATGGCCAGTAGTGGTGGAC-3′ | This manuscript | N/A |
| EEF1A1 RT-qPCR reverse primer: 5′-TTTTTCGCAACGGGTTTG-3′ | This manuscript | N/A |
| Recombinant DNA | ||
| pLV-EF1a-H2B-mTurquoise-IRES-PuroR | Spencer et al., 2013 | N/A |
| CSII-hDHB (amino acids 994-1087)-mVenus | Spencer et al., 2013 | N/A |
| CSII-pEF1a-mCherry-Geminin (amino acids 1-110) | Cappell et al., 2016 | N/A |
| pLV-EF1a-CaaX-iRFP-IRES-Puro | Bisaria et al., 2020 | N/A |
| pPBbsr2-EKAR-NLS | Yang et al., 2017 | N/A |
| ERK KTR | Regot et al., 2014 | N/A |
| pCW-TRE-PGK promoter-cyclin D1-PuroR-T2A-rtTA | This manuscript | N/A |
| pCW-TRE-PGK promoter-p27-BlastR-T2A-rtTA | This manuscript | N/A |
| pCW-TRE-PGK promoter-HA-MEK1-S218D/S222D-PuroR-T2A-rtTA | This manuscript | N/A |
| Software and algorithms | ||
| Microscopy image analysis: cell tracking | Cappell et al., 2016 | https://github.com/scappell/Cell_tracking |
| Microscopy image analysis: custom code for measuring local cell density | This manuscript | Zenodo: https://doi.org/10.5281/zenodo.4960626 |
| Other | ||
| Sylgard Silicone Elastomer Kit 184 | Dow | SKU#DC4019862 |