

Abstract
Von Willebrand A Domain-Containing Protein 8 (VWA8) is a poorly-characterized, mitochondrial matrix-targeted protein with a AAA ATPase domain and ATPase activity that rises in abundance in livers of high fat fed mice. This study was undertaken to use CRISPR/Cas9 to delete VWA8 in cultured mouse hepatocytes and gain insight into its function. Unbiased omics techniques and bioinformatics were used to guide subsequent assays, including assessment of oxidative stress and determination of bioenergetic capacity. Metabolomics analysis showed VWA8 null cells had higher oxidative stress and protein degradation; assays of hydrogen peroxide production revealed higher production of reactive oxygen species (ROS). Proteomics and transcriptomics analyses showed VWA8 null cells had higher expression of mitochondrial proteins (electron transport chain Complex I, ATP synthase), peroxisomal proteins, and lipid transport proteins. The pattern of higher protein abundance in the VWA8 null cells could be explained by higher hepatocyte nuclear factor 4 alpha (HNF4a) expression. Bioenergetic assays showed higher rates of carbohydrate oxidation and mitochondrial and non-mitochondrial lipid oxidation in intact and permeabilized cells. Inhibitor assays localized sites of ROS production to peroxisomes and NOX1/4. Rescue of VWA8 protein restored the wildtype phenotype and treatment with antioxidants lowered HNF4a expression. Thus, loss of VWA8 produces a mitochondrial defect that may be sensed by NOX4, leading to a rise in ROS that results in higher HNF4a. The compensatory HNF4a response results in higher oxidative capacity and even higher ROS production. We hypothesize that VWA8 is a AAA ATPase protein that plays a role in mitochondrial protein quality.
Keywords: VWA8, hepatocytes, bioenergetics, Hnf4a
Graphical Abstract
Introduction
High fat feeding in the mouse produces obesity, systemic and hepatic insulin resistance, fatty liver, and inflammatory responses 2–5; 6–13. Using mass spectrometry-based global proteomics methods, we found that KIAA0564, the protein product of the KIAA0564 gene, was more abundant in livers from wildtype C57Bl6/J mice fed a 60 percent fat diet than in lean mice fed standard chow 14. KIAA0564 is poorly characterized but is widely expressed in highly energetic tissues such as liver, kidney, heart, pancreas, and skeletal muscle, and is targeted exclusively to mitochondria15. These data suggested that KIAA0564 may play a heretofore unknown role in metabolism or bioenergetics. The recommended name for KIAA0564 is VWA8 (Von Willebrand A Domain-Containing Protein 8, Uniprot Accession ID Q8CC88), based on homology of a region of the protein to VWA domains. However, VWA8 also contains a AAA ATPase domain, Walker A and B motifs, ATPase activity and an N-terminal mitochondrial matrix targeting sequence 15. Because VWA8 expression rises in liver with overfeeding, it is important to define the role of VWA8 in the mitochondrion of the hepatocyte.
This study was undertaken to further define the cellular function of VWA8 by using CRISPR/Cas9 to delete the VWA8 gene in differentiated AML12 mouse liver cells. An unbiased approach using metabolomics, proteomics, and transcriptomics techniques was used to define the phenotype of the VWA8 null cells and gain insight into the function of VWA8. This phenotype was further refined using bioinformatics approaches and bioenergetic assays. VWA8 null cells were found to have higher oxidative stress, higher production of reactive oxygen species, and higher expression of electron transport chain Complex I and ATP synthase proteins, accompanied and likely explained by higher hepatocyte nuclear factor 4 alpha (HNF4a) expression. As a result, the VWA8 null cells had higher rates of carbohydrate oxidation and mitochondrial and non-mitochondrial lipid oxidation. Sites of ROS production were localized to peroxisomes and NOX1/4. We conclude that loss of VWA8 produces a mitochondrial defect that may be sensed by NOX4, leading to a rise in ROS that results in higher HNF4a. The compensatory HNF4a response results in higher oxidative capacity and even higher ROS production.
Materials and Methods
AML12 Hepatocyte culture and cloning.
AML12 mouse hepatocytes purchased from ATCC (ATCC CRL-2254) were cultured in DMEM/F12 with 10% FBS, 1% Penicillin/Streptomycin, and 0.005 mg/ml insulin, 0.005 mg/ml transferrin, 5 ng/ml selenium, and 40 ng/ml dexamethasone. To establish a monoclonal parent AML12 cell line, polyclonal AML12 cells were cloned by limiting dilution in 96-well plates. The monoclonal cell lines derived by limiting dilution were differentiated in this manner, and one (monoclonal parent cell line 6) expanded and differentiated well. All cells were differentiated by lowering the serum concentration to 0.2% for 3–7 days. Differentiation was assessed by expression of hepatocyte markers. Monoclonal AML12 cell line 6 was selected as the parent cell line for deleting the VWA8 gene.
CRISPR knockout of VWA8.
The VWA8 gene was targeted and deleted using CRISPR/Cas9 as described 16. Two target sites within Exon One of the mouse VWA8 gene were selected (http://crispr.mit.edu/). The reverse-strand guide sequence of first target site was AGGCGATCGGCGAGCGGGACAGG (Score 92%) and the forward-strand guide sequence of second site was CACTCTGGGGCGACCGACTCAGG, putatively deleting a 237 base pair region. Guide sequences were inserted into pSpCas9(BB)-2A-GFP (Addgene plasmid ID: 48138) and the plasmids with guide sequence were confirmed by sequencing. This scheme is shown in Figure S1. Parent #6 AML12 cells were transfected with the plasmids with guide sequence and transfected cells with GFP were sorted by fluorescence activated cell sorting (FACS). The sorted cells were further cloned by limiting dilution in 96-well plates at a density of one cell in 100 μl medium per well. Colonies originating from single cells were transferred to 24 well plates and then 60 mm dishes for expansion. The monoclonal cell lines were tested by Surveyor PCR for deletion of the selected region on VWA8, using primers that yielded a 664 bp fragment in the wild type gene and a 427 bp fragment in a successful deletion (664 – 237 base pairs); the heterozygous knockout has both bands (664 base pairs and 427 base pairs). For control, wildtype cells, we chose to use a cell line that had undergone all procedures but did not yield a deletion of VWA8, as confirmed by the Surveyor assay, PCR, and immunoblotting. In subsequent assays, we found that this “CRISPR wild type” cell line was indistinguishable from the parent cell line 6. For further experiments, we chose monoclonal cell line 6–8 as the CRISPR wild type (C-WT#6–8) and line 6–10 as the homozygous CRISPR knockout (C-KO 6–10) for subsequent experiments.
Metabolomics Analysis.
C-WT 6–8 and C-KO 6–10 cells were seeded in 150 mm plates. Cells at 75–80% confluence were differentiated for 5 days using reduced FBS. For metabolomics analysis with exogenous lipid fuel, cells were treated with palmitate-BSA (175 μM palmitate conjugated to 29.75 μM bovine serum albumin), carnitine (0.5 mM) and glucose (2.5 mM) for 30 min (C-WT 6–8, n = 6; C-KO 6–10, n = 6 each). Metabolomics analysis of cells exposed to carbohydrate as exogenous fuel used glucose (10 mM), glutamine (2 mM), and pyruvate (1 mM) for 30 min (C-WT 6–8, n = 6; C-KO 6–10, n = 6). After treatment, cells were gently scraped, pelleted by centrifugation, flash-frozen and stored at −80°C. Metabolomics analysis was performed by Metabolon (Research Triangle Park, North Carolina), where samples were inventoried and immediately stored at −80°C until they were prepared using the automated MicroLab STAR® system from Hamilton Company.
Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectroscopy (UPLC-MS/MS).
All Metabolomics methods utilized a Waters ACQUITY ultra-performance liquid chromatography (UPLC) and a Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer interfaced with a heated electrospray ionization (HESI-II) source and Orbitrap mass analyzer operated at 35,000 mass resolution. The sample extract was dried then reconstituted in appropriate solvents. Each reconstitution solvent contained a series of standards at fixed concentrations to ensure injection and chromatographic consistency. One aliquot was analyzed using acidic positive ion conditions, chromatographically optimized for more hydrophilic compounds. In this method, the extract was gradient eluted from a C18 column (Waters UPLC BEH C18–2.1×100 mm, 1.7 μm) using water and methanol, containing 0.05% perfluoropentanoic acid (PFPA) and 0.1% formic acid (FA). A second aliquot was also analyzed using acidic positive ion conditions and was chromatographically optimized for more hydrophobic compounds. In this method, the extract was gradient eluted from the same afore mentioned C18 column using methanol, acetonitrile, water, 0.05% PFPA and 0.01% FA and was operated at an overall higher organic content. A third aliquot was analyzed using basic negative ion optimized conditions using a separate dedicated C18 column. The basic extracts were gradient eluted from the column using methanol and water, however with 6.5mM Ammonium Bicarbonate at pH 8. The fourth aliquot was analyzed via negative ionization following elution from a HILIC column (Waters UPLC BEH Amide 2.1×150 mm, 1.7 μm) using a gradient consisting of water and acetonitrile with 10mM Ammonium Formate, pH 10.8. The MS analysis alternated between MS and data-dependent MSn scans using dynamic exclusion. The scan range varied slightly between methods but covered 70–1000 m/z. Raw data files are archived and extracted as described below.
Metabolomic Bioinformatics.
The informatics system consisted of four major components, the Laboratory Information Management System (LIMS), the data extraction and peak-identification software, data processing tools for QC and compound identification, and a collection of information interpretation and visualization tools for use by data analysts. The hardware and software foundations for these informatics components were the LAN backbone, and a database server running Oracle 10.2.0.1 Enterprise Edition. Raw data was extracted, peak-identified and QC processed using Metabolon’s hardware and software. Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities. Peaks were quantified using area-under-the-curve. For studies spanning multiple days, a data normalization step was performed to correct variation resulting from instrument inter-day tuning differences.
Metabolomics Statistical Methods and Terminology.
Standard statistical analyses were performed in ArrayStudio on log transformed data. For those analyses not standard in ArrayStudio, the programs R (http://cran.r-project.org/) or JMP were used. Statistical comparison of selected metabolite concentrations was performed using a two-way analysis of variance.
Proteomics Analysis.
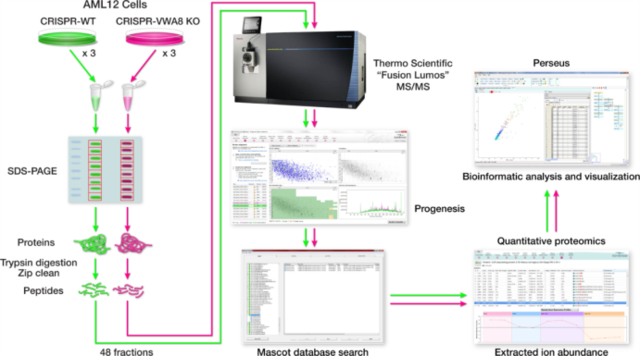
The proteomics workflow is shown in Figure S2. One 100 mm plate for each biological replicate was lysed in 1.0 ml of lysis buffer 17. AML12 cell lysates were then incubated on ice for 30 minutes followed by centrifugation at 4° for 30 minutes at 14,000 rpm. 20 μL of the AML12 cell lysate supernatant was separated on a 10% SDS-PAGE gel and stained with Bio-Safe Coomassie G-250 Stain. Each lane of the SDS-PAGE gel was cut into eight slices, placed in a 0.6 mL LoBind polypropylene tube (Eppendorf, Hauppauge, NY), destained twice with 375 μl of 50% acetonitrile (CAN) in 40 mm NH4HCO3 and dehydrated with 100% CAN for 15 min. After removal of the CAN by aspiration, the gel pieces were dried in a vacuum centrifuge at 60 °C for 30 min. Trypsin (500 ng; Thermo Scientific) in 50 μl of 100 mm NH4HCO3 was added, and the samples were maintained at 4 °C for 15 min prior to the addition of 50–150 μL 100 mm NH4HCO3. The digestion was allowed to proceed at 37 °C overnight and was terminated by addition of 10 μl of 5% formic acid (FA). After further incubation at 37 °C for 30 min and centrifugation for 1 min, each supernatant was transferred to a clean LoBind polypropylene tube. The extraction procedure was repeated using 40 μl of 0.5% FA, and the two extracts were combined and dried down to approximately 5–10 μL followed by the addition of 10 μL 0.05% heptafluorobutyric acid:5% FA (v/v) and incubation at room temperature for 15min. The resulting peptide mixtures were loaded on a solid phase C18 ZipTip (Millipore, Billerica, MA) and washed with 35 μL 0.005% heptafluorobutyric acid:5%FA (v/v) followed by elution first with 4 μl of 50% CAN:1% FA (v/v) and then a more stringent elution with 4 μl of 80% CAN:1% FA (v/v). The eluates were combined and dried completely by vacuum centrifugation and 6 μl of 0.1% FA (v/v) was added followed by sonication for 2 min. 2.5 μl of the final sample was then analyzed by mass spectrometry.
Mass spectrometry and proteomics database search.
HPLC-ESI-MS/MS was performed in positive ion mode on a Thermo Scientific Orbitrap Fusion Lumos tribrid mass spectrometer fitted with an EASY-Spray Source (Thermo Scientific, San Jose, CA). NanoLC was performed using a Thermo Scientific UltiMate 3000 RSLCnano System with an EASY Spray C18 LC column (Thermo Scientific, 75cm x 75 μm inner diameter, packed with PepMap RSLC C18 material, 2 μm, cat. # ES805); loading phase for 15 min at 0.300 μL/min; mobile phase, linear gradient of 1–25% Buffer B in 89 min at 0.220 μL/min followed by another linear gradient of 25–40% Buffer B in 30 min at 0.220 μL/min followed by a step to 95% Buffer B over 4 min at 0.220 μL/min, hold 5 min at 0.300 μL/min, and then a step to 1% Buffer B over 5 min at 0.300 μL/min and a final hold for 9 min at 0.200 μL/min (total run 159 min); Buffer A = 0.1% FA/H2O; Buffer B = 0.1% FA in 80% ACN. All solvents were liquid chromatography mass spectrometry grade. Spectra were acquired using XCalibur, version 2.3 (Thermo Scientific). A “top speed” data-dependent MS/MS analysis was performed. Dynamic exclusion was enabled with a repeat count of 1, a repeat duration of 30 sec, and an exclusion duration of 60 sec. Tandem mass spectra were extracted from Xcalibur ‘RAW’ files and charge states were assigned using the ProteoWizard 3.0 msConvert script using the default parameters. The fragment mass spectra were then searched against the mouse SwissProt_2016_10 database (16838 entries) using Mascot (Matrix Science, London, UK; version 2.6.0) using the default probability cut-off score. The search variables that were used were: 10 ppm mass tolerance for precursor ion masses and 0.5 Da for product ion masses; digestion with trypsin; a maximum of two missed tryptic cleavages; variable modifications of oxidation of methionine and phosphorylation of serine, threonine, and tyrosine. Cross-correlation of Mascot search results with X! Tandem was accomplished with Scaffold (version Scaffold_4.8.2; Proteome Software, Portland, OR, USA). Probability assessment of peptide assignments and protein identifications were made through the use of Scaffold. Only peptides with ≥ 95% probability were considered.
Label-free peptide/protein quantification and identification.
Progenesis QI for proteomics software (version 2.4, Nonlinear Dynamics Ltd., Newcastle upon Tyne, UK) was used to perform ion-intensity based label-free quantification. In brief, in an automated format, .raw files were imported and converted into two-dimensional maps (y-axis = time, x-axis =m/z) followed by selection of a reference run for alignment purposes. An aggregate data set containing all peak information from all samples was created from the aligned runs, which was then further narrowed down by selecting only +2, +3, and +4 charged ions for further analysis. A peak list of fragment ion spectra from only the top eight most intense precursors of a feature was exported in Mascot generic file (.mgf) format and searched against the mouse SwissProt_2016_10 database using Mascot (Matrix Science, London, UK; version 2.4). The search variables that were used were: 10 ppm mass tolerance for precursor ion masses and 0.5 Da for product ion masses; digestion with trypsin; a maximum of two missed tryptic cleavages; variable modifications of oxidation of methionine and phosphorylation of serine, threonine, and tyrosine; 13C=1. The resulting Mascot .xml file was then imported into Progenesis, allowing for peptide/protein assignment, while peptides with a Mascot Ion Score of <25 were not considered for further analysis. Protein quantification and logarithmic transformation was performed using only non-conflicting peptides and precursor ion-abundance values were normalized in a run to those in a reference run (not necessarily the same as the alignment reference run). Statistical analysis of protein abundance was performed in transformed peak area data [asinh(peak area)] using analysis of variance in Progenesis QI.
Transcriptomics Analysis.
To determine changes in gene expression in AML12 liver cells associated with deletion of VWA8 (n=4 each). Total RNA was isolated from cells using RNAStat solution (Tel-Test Inc., Friendswood, TX) and the total RNA was purified with RNeasy and DNase I treatment (Qiagen, Valencia, CA). RNA was prepared for hybridization to Affymetrix Mouse 2.0 arrays according to manufacturer’s instructions at the University of Arizona Genomics Core Facility (National Institutes of Health Cancer Center Support Grant P30CA023074). CEL files were imported into the open source R package. The Affy package and Linear Models for Microarray Data version 3.22.0 were used for analysis. The data were background corrected using normal exponent, quantile normalized, and an unweighted linear model was performed to generate fold changes between groups. The fold changes were log transformed. Expression values obtained were evaluated by a moderated t-statistic (nominal P value) and adjusted using the Benjamini-Hochberg multiple testing correction (adjusted P value). Microarray data have been deposited in the Gene Expression Omnibus (GSE109610).
Assessment of oxidative stress.
For measurement of net H2O2 production the method of Wong et al. was used18. CRISPR VWA8 null (C-KO 6–10) and CRISPR wild type (C-WT 6–8) AML12 cells were differentiated for 5 days in the presence of 0.2% serum in a 96-well plate. Cells were washed with Krebs Ringer Bicarbonate (KRB) medium (MgCl2,0.49 mM; KCl, 4.56 mM; NaCl, 120 mM; Na2HPO4, 0.7 mM; NaH2PO4, 1.3 mM; NaHCO3, 15.0 mM, with fresh added 0.1% BSA plus 10 mM glucose or 0.1% BSA plus 175 μM palmitate and 2.5 mM glucose) once and then incubated in KRB at 37°C for 30 min. Cells again were washed with KRB and 75 μl of Reaction Buffer (25 μM Amplex UltraRed, 5 U/mL HRP and 25 U/mL SOD1 in KRB) was added to each well. Carbohydrate or lipid fuels were included in KRB. Changes in fluorescence (Ex 540/Em 590) were recorded for 31 cycles at 37 °C using a BioTek Synergy H1 plate reader. Data were normalized to cell protein. All statistical analyses were performed using non-paired t-tests.
Sites of superoxide production were probed using the method of Quinlan and colleagues19. For this purpose, experiments included inhibitors of electron transport chain Complex I (S1, 50 μM), Complex III (S3, 50 μM), NOX1/4 (GKT137831, 10 μM), NOX1 (ML171, 10 μM), and peroxisomal beta oxidation (thioridazine, used at concentrations indicated).
Bioenergetic assays.
Monoclonal cell line Parent 6, C-WT 6–8, or C-KO 6–10 were seeded in Seahorse cell culture microplates (Seahorse Bioscience #100777–004, 24-well plates), at 20,000 cells/well. Cells at 75 % confluency were differentiated by lowering FBS to 0.2% for 5 days. All bioenergetic assays (oxygen consumption rates, or OCR) followed Seahorse Bioscience protocols using a machine XFe24 Extracellular Flux Analyzer (Seahorse Bioscience). Intact and permeabilized (perfringolysin) cells were studied.
To assay palmitate oxidation in intact cells, fatty acid oxidation assay medium (FAO, as recommended by Seahorse protocols) supplemented with 2.4 mM glucose and 0.5 mM carnitine; with or without Etomoxir (40 μM) to inhibit carnitine palmitoyl transferase; palmitate conjugated to bovine serum albumin (BSA; 175 μM palmitate/29.75 μM BSA) or BSA alone as control (29.75 μM) were used. After basal respiration was determined, the following were added sequentially: 2.0 μM oligomycin to inhibit Complex V and estimate phosphorylative OCR, 2.5 μM FCCP to uncouple mitochondria and estimate maximal respiratory capacity, and 2.0/4.0 μM rotenone/antimycin A to inhibit Complex I and Complex III respectively, thus allowing an estimate of non-mitochondrial respiration. Respiration due to proton leak can be estimated as the difference between the OCR after oligomycin addition minus the OCR after addition of rotenone/antimycin A.
To assay oxidation of carbohydrate (glucose) in intact cells, XF Base Medium (Seahorse Bioscience #102353–100) with 10 mM Glucose, 2.0 mM Glutamine and 1.0 mM Pyruvate was used. Other test components included 2.0 μM oligomycin, 2.5 μM FCCP, and 2.0/4.0 μM rotenone/antimycin A as above. In intact cell experiments (palmitate oxidation and glucose oxidation), respiration (OCR) in each well was normalized to cell number, measured with CyQUANT kit (Invitrogen #C7026) following the manufacturer’s protocol. Normalization using cell protein resulted in similar results.
For assays using cells with permeabilized membranes (essentially in situ mitochondria) the Agilent Seahorse XF Plasma Membrane Permeabilizer (PMP) Quickstar procedure was followed. Oxidation of palmitoyl-carnitine was used to monitor lipid oxidation independent of the CPT1 step and was performed using MAS Medium with 40 μM palmitoyl-carnitine and 1.0 mM malate. Oxidation of palmitoyl-CoA was used to monitor lipid oxidation that at least in part depended on CPT-1. This was performed using MAS Medium with 40 μM palmitoyl–CoA, 0.5 mM carnitine and 1.0 mM malate. To monitor oxidation of carbohydrate in permeabilized cells a combination of 5 mM pyruvate, 1.0 mM malate, and 2 mM glutamine. All of above three assays used test components 2 mM ADP, 2.0 μM oligomycin, 2.0 μM FCCP, 2.0/2.0 μM rotenone/antimycin A. In experiments using permeabilized cells, the OCR of each well was expressed as pmol oxygen consumed per minute, as estimation of cell number was not feasible.
To test the specificity of the effects of VWA8 deletion, VWA8a (long isoform) or VWA8b (short isoform lacking the VWA domain) were restored to wildtype levels using adenoviral expression. The empty adenoviral vector was used as a control for these experiments. Statistical comparison of wildtype and VWA8 null cells were performed using non-paired t-tests.
Transcription Factor Enrichment Analysis.
To assess potential transcription factor binding site motif enrichment in promoter regions, Refseq nucleotide sequences for proteins and genes of interest were submitted to PScan Software 20.
Immunoblot analysis.
Immunoblot analysis was performed using commercial antibodies, as described 15.
Results
CRISPR/Cas9 effectively deletes VWA8.
A monoclonal parent AML12 cell line was produced from AML12 mouse hepatocytes obtained from commercial sources (American Type Culture Collection, ATCC). This allowed for deletion of VWA8 in cell lines that would be otherwise as genetically identical as possible to control, wild type AML12 cells (see Methods). Figure 1A shows the strategy used, with most of Exon 1 being excised. DNA analysis showed deletion of the desired portion of Exon 1 (Figure 1B) and immunoblot and mass spectrometry analyses confirmed absence of the protein (Figure 1C). Of the monoclonal cell lines that were developed during this process, line 6–10 had the clearest genomic deletion (Figure 1B), so we used that cell line in all subsequent experiments. AML12 parent and CRISPR cell lines had relatively low mitochondrial content and markers of hepatocyte differentiation, so in order to study these cells in a more differentiated, hepatocyte-like state, the serum concentration in the medium was lowered to 0.2% as recommended by ATCC. Under these conditions, immunoblot analysis showed that hepatocyte and mitochondrial marker content rose in wild type and VWA8 null cells over 3–7 days (Figure 1D), consistent with differentiation into a more hepatocyte-like cell. All subsequent experiments with differentiated cells were performed after 5 days of differentiation, since by day 7 cells showed reduced adherence. For a wild type cell line, preliminary experiments found no difference between the parent monoclonal cell line and “CRISPR wildtype” cell line 6–8, which we then used as a control. We define CRISPR wildtype as cell lines that were derived from parent cells treated in the same way as the knockout cells, but where deletion of the gene was unsuccessful.
Figure 1.

CRISPR/Cas9 deletion of VWA8. A. 237 nucleotides of exon 1 were deleted, which deletes VWA8a and VWA8b. B. Surveyor DNA assay showing deletion of 237 nucleotides. #6, Parent AML12 cells; #6–8, “CRISPR wildtype”; #6–10, knockout cell line; #6–13, knockout cell line. C. Immunoblots and number of mass spectra observed for VWA8a, showing deletion of all VWA8a protein. No antibody exists for VWA8b, but spectra for this protein were absent. D. AML12 cells differentiated to a more hepatocyte-like phenotype, with higher mitochondrial content, when cell culture serum is reduced to 0.2% (see text). CPS-1, carbamoyl phosphate synthase-1; MTCO2, cytochrome c oxidase subunit 2; SOD2, superoxide dismutase 2.
Metabolomic analysis reveals deletion of VWA8 raises oxidative stress and protein degradation.
To determine whether changes in metabolic intermediates could reveal a phenotype produced by deletion of VWA8, we exposed differentiated wildtype and VWA8 null AML12 hepatocytes to palmitate or glucose and subjected cell lysates to metabolomic analysis. Data for all metabolites detected and quantified (657 in total) are given in Table S1. A summary of statistical comparisons is given in Table S2.
To confirm validity of the glucose vs. palmitate treatments, metabolic intermediates in carbohydrate and lipid metabolism were measured. When treated with glucose, both wildtype and VWA8 null cells had higher levels of glycolytic intermediates relative to palmitate treated cells (Table 1). In contrast palmitate-treated cells of both genotypes had higher levels of palmitate and palmitate derivatives such as palmitoyl carnitine relative to the glucose-treated cells (Table 1).
Table 1.
Effect of deletion of VWA8 on selected fuel metabolites.
| Glucose | Palmitate | |||
|---|---|---|---|---|
|
| ||||
| WT | KO | WT | KO | |
|
|
||||
| Glucose | 1.88±0.35** | 1.99±0.21** | 0.86±0.02 | 1.02±0.04 |
| Glucose 6-phosphate | 6.03±0.28** | 2.95±0.52**§ | 2.12±0.47 | 0.25±0.06§ |
| 2-Phospho-glycerate | 2.02±0.59** | 1.43±0.66** | 0.10±0.02 | 0.08±0.0 |
| Pyruvate | 1.75±0.23** | 1.37±0.19** | 0.97±0.13 | 0.88±0.15 |
| Palmitate | 0.09±0.29** | 0.17±0.11** | 1.31±0.15 | 1.20±0.35 |
| Palmitoyl-carnitine | 0.003±0.001** | 0.002±0.0001** | 1.52±0.11 | 1.99±0.30 |
Data are raw area counts normalized to protein concentration. WT, CRISPR wildtype AML12 cells; KO, VWA8 null AML12 cells.
P<0.01 Glucose vs. Palmitate
P<0.01 KO vs. WT; WT, wildtype; KO, VWA8 null cells. Statistical comparison of wildtype and VWA8 null cells was performed using non-paired t-tests.
With regard to other significantly different metabolites, it was notable that gamma-glutamyl amino acids were significantly lower in VWA8 null cells in the presence of palmitate or glucose, indicating that higher oxidative stress was present in the KO cells (Figure 2A). Moreover, deletion of VWA8 raised other indicators of oxidative stress such as allantoin and methionine sulfoxide, especially in the presence of palmitate as a fuel (Figure 2B,C, respectively). In the presence of palmitate, dipeptides also were higher as a group, suggesting higher protein breakdown in the VWA8 null cells (Figure 2D).
Figure 2.

Deletion of VWA8 decreases concentrations of gamma-glutamyl amino acids (A) and raises levels of allantoin (B), methionine sulfoxide (C), and dipeptides (D). Data are expressed relative to the average value of the wildtype cells (shown in blue). Values for VWA8 null cells are shown in orange. *P<0.05, **P<0.01 vs. wildtype by non-paired t-test.
To confirm the finding of higher oxidative stress in the VWA8 null cells, we incubated cells with glucose or palmitate, as described for the metabolomics experiments, and monitored the rise in fluorescence of Amplex UltraRed as an indicator of cellular stress (net H2O2 production).
When data were expressed relative to the average value of H2O2 production in wildtype cells exposed to glucose, VWA8 null cells had significantly higher rates of net H2O2 production in the presence of either glucose or palmitate (Figure 3A). H2O2 production was higher in VWA8 null cells in the presence of palmitate even before differentiation and rose throughout 7 days after the cell culture serum concentration was dropped to 0.2% to induce differentiation (Figure 3B).
Figure 3.

Net rates of hydrogen peroxide production by wildtype and VWA8 null AML12 cells, measured using Amplex Red fluorescence. A. H2O2 net production in wildtype (closed bars) and VWA8 null cells (open bars) in response to glucose or palmitate as fuel (n=12, **P<0.01 vs. wildtype). B. Time course (days) of the rise in H2O2 production during differentiation of wildtype (closed bars) and VWA8 null cells (open bars) AML12 cells in the presence of palmitate. Data are given as Mean ± SD (n=12, **P<0.01 vs wildtype). Statistical analysis was performed using non-paired t-tests.
Deletion of VWA8 alters the hepatocyte transcriptome and proteome.
To obtain a global picture of gene expression changes induced by deletion of VWA8 null cells, Affymetrix microarray analysis of gene expression was carried out using RNA isolated from wildtype and VWA8-null AML12 cells. This analysis found 4585 probes to be different at a nominally significant level between the genotypes (Table S3). Of the 4585 significant probes, 2318 were higher in VWA8 null cells, and 2267 were higher in wildtype cells. Probes that mapped to a gene were analyzed using Panther to determine over-represented GO classifications (Table S4). For genes that increased in the VWA8 null cells, significant GO classifications included fatty acid beta-oxidation, amino acid catabolic process, sulfur compound metabolic process, peroxisome, mitochondrion, mitochondrial inner membrane, hydro-lyase activity and oxidoreductase activity.
In order to determine whether mRNA changes produced corresponding changes in protein abundance that can explain the dramatically altered bioenergetic phenotype, VWA8 null and wildtype AML12 cells (n = 3 each) were subjected to label-free quantitative proteomics analysis. A total of 6544 proteins were quantifiable (Table S5). Of these, 2577 were significantly different between the two groups. Figure 4A shows a dendrogram and heat map of all 2577 significantly different proteins, which divide into two main branches representing VWA8 null and wildtype AML12 cells. The patterns of directional differences were highly consistent among the three replicate experiments using each condition. Immunoblots showing examples of alterations in protein abundance revealed by proteomics analysis are illustrated in Figure 4B, with quantification shown in Figure 4C. For illustration, we chose IRS-1 and Acadm, which were approximately 5- and 2-fold higher by immunoblots and 3.7- and 2.1-fold higher by proteomics analysis. To determine concordance between changes in the transcriptome and changes in the proteome, fold-changes in mRNA expression and protein abundance induced by deletion of VWA8 were plotted against each other for the approximately 1000 genes and proteins that were higher or lower in the VWA8 null cells. Changes in gene expression and protein abundance produced by deletion of VWA8 were highly correlated (Figure 4D; r = 0.87, P < 0.00001).
Figure 4.

Proteomics and transcriptomics analyses. A. Three biological replicates were analyzed for protein abundance (see Methods) and highly replicable changes in protein abundance were found. B. Immunoblots illustrating protein abundance differences in wildtype (WT) and VWA8 null (KO) cells. C. Quantification of immunoblots. D. Correlation between fold changes in mRNA expression (X-axis) and protein abundance (Y-axis) between KO and WT cells.
Gene Ontology (GO) over-representation analysis for all significantly different proteins was conducted separately for proteins that were higher or lower in abundance in VWA8 null cells. Complete data for all significant GO classifications including cellular component, biological process, and molecular function are given in Table S6. Significantly over-represented pathways of note among proteins that were higher after VWA8 deletion included peroxisome, ATP synthase complex, mitochondrial inner membrane, fatty acid beta oxidation, and amino acid catabolic process. Among proteins that were higher in abundance in wildtype cells, GO categories that were significantly over-represented included nuclear transport, mRNA processing, mRNA splicing, and a number of other GO categories related to DNA and RNA processes. These results were consistent with the transcriptomics results.
Of individual proteins that were significantly higher in the VWA8 null cells, 248 also were 2-fold greater than wildtype values. There also were 376 proteins that were statistically significant and 2-fold lower than wildtype in VWA8 null cells. These robustly and significantly altered proteins are given in Tables S7 and S8, respectively. Notable among those proteins that were higher in the VWA8 null cells, in light of the phenotype of increased basal, non-mitochondrial, and maximal respiration, were components of omega oxidation of fatty acids (Cyp4a12a, Adh1), peroxisomal proteins (Pipox, Pxmp4, Phyh, Pex11a, Decr2, Ehhadh, and Pxmp2), proteins involved in fatty acid oxidation (Acot4, Acaa2, Hadh, Acot1, Acadm), and proteins involved in intra- and extracellular lipid transport, such as microsomal triglyceride transport protein (Mtp), liver-specific fatty acid binding protein (Fabpl), and apolipoproteins (Apob, Apoa1, Apoe). In addition, there were higher levels of carbamoyl phosphate synthase (Cps1) and ornithine carbamoyltransferase (Otc). Proteins that were 2-fold higher in wildtype cells primarily were represented by pathways related to DNA and RNA processing and replication and cell division, along with several transcription factors. Changes in the proteome mirrored those observed with transcriptomic analysis.
Deletion of VWA8 raises oxidative capacity during hepatocyte differentiation.
In light of the observations that deletion of VWA8 raises oxidative stress and mRNA and protein expression in pathways related to mitochondrial and non-mitochondrial oxidative metabolism, fuel supply and oxidation, experiments were conducted to determine whether deletion of VWA8 produces a bioenergetic or mitochondrial phenotype in intact AML12 cells. Seahorse XF24e metabolic analyzer assays of respiration (oxygen consumption rate, OCR) in response to added lipid fuel (palmitate conjugated to BSA) over the time course of cell differentiation showed that the wildtype and VWA8 null cells behaved similarly in the undifferentiated state. However, the genotypes diverged during differentiation, with the most striking phenotypic differences seen by day 5 (Figure 5), where the VWA8 null cells had higher maximum respiration stimulated by the protonophore FCCP (carbonylcyanide p-trifluoromethoxyphenylhydrazine) in response to lipid fuel. Responses of maximal respiration to carbohydrate fuel (glucose) were increased similarly.
Figure 5.

Time course of changes in cellular respiration in wildtype (parent #6, C-WT #6–8) and VWA8 null cells (KO #6–10, KO #6–13) in response to palmitate fuel. After Day 0 (undifferentiated cells), the cell culture serum concentration was reduced to 0.2% and additional measurements were made at 3, 5, and 7 days of differentiation. Oligo (oligomycin); FCCP (carbonylcyanide p-trifluoromethoxyphenylhydrazine); Rot (Rotenone); AA, Antimycin A.
In intact cells, the sequential additions of oligomycin, FCCP, and the electron transport chain (ETC) inhibitors rotenone plus antimycin A (as shown in Figure 5) enable the estimation of basal cellular respiration, the fractional contributions of proton leak and ATP-linked respiration to basal respiration, peak (FCCP-induced) intact cell OCR, and non-mitochondrial respiration 21–23. The basal ATP turnover linked to oxidative phosphorylation of intact cells is estimated by the net fall in OCR after addition of oligomycin (“basal phosphorylative respiration”). Proton leak is estimated as mitochondrial OCR remaining after addition of oligomycin to inhibit ATP synthase. These data are given in Table 2 for both glucose and palmitate added as fuels.
Table 2.
Comparison of Seahorse respiration results in intact, differentiated wildtype and VWA8-null AML12 hepatocytes.
| Glucose | Palmitate | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| WT | KO | KO/WT (fold) | WT | KO | KO/WT (fold) | |
| Basal Phosphorylative respiration | 94 ± 4 | 286 ± 9*** | 3.04 | 89 ± 3 | 218 ± 10*** | 2.42 |
| Proton leak | 66 ± 4 | 122 ± 7** | 1.89 | 70 ± 6 | 160 ± 20* | 2.30 |
| Maximum (FCCP) | 801 ± 24 | 1157 ± 60** | 1.45 | 562 ± 116 | 1047 ± 71* | 1.86 |
| Non-mitochondrial | 69 ± 6 | 119 ± 7** | 1.72 | 64 ± 12 | 162 ± 6** | 2.52 |
Rates of respiration in wildtype (WT) and VWA8 null (KO) differentiated AML12 cells Data are given as Means ± SEM in units of pmol O2 consumption per minute. N = 4 experiments
P<0.01
P<0.001
P<0.0001 vs. wildtype. Statistical analysis was performed using non-paired t-tests. Glucose (10 mM), glutamine (2 mM), and pyruvate (1 mM) were used to monitor carbohydrate induced respiration; palmitate conjugated to BSA (175 μM), carnitine (0.5 mM), and glucose (2.4 mM) were used to monitor fat oxidation.
All data were corrected for non-mitochondrial respiration estimated by inhibition of electron transport with antimycin A plus rotenone. Non-mitochondrial respiration also is given in Table 2. Respiration was higher in permeabilized cells than in intact cells under all conditions. The rise in respiration in intact cells was generally greater when palmitate was provided as a fuel, compared to addition of glucose. However, Seahorse experiments conducted in cells that had permeabilized plasma membranes (removing fuel delivery as a factor), generally had greater increases in respiration when carbohydrate (pyruvate) was supplied, compared to lipid (palmitoyl carnitine) (Table S9). Taken together, data from these experiments suggest deletion of VWA8, in addition to enhancing respiration, also raises lipid delivery to mitochondria in intact cells. These findings were consistent with changes observed in the proteome of VWA8 null cells.
Because of the consistent and apparently coordinated pattern of gene expression, protein abundance, and metabolic changes in VWA8 null cells, we sought to determine whether the promoter regions of genes whose proteins were robustly and significantly affected in the VWA8 null cells have common transcription factor binding motifs. The promoter regions of genes coding for proteins that were significantly altered in VWA8 null cells were analyzed for enrichment in putative transcription factor binding motifs. The transcription factors that were most significantly enriched in these promoters are given in Table 3.
Table 3.
Transcription factor binding motif enrichment in genes for proteins with significantly higher abundance in VWA8 null cells.
| Transcription factor | Bonferroni P | Protein fold difference | Significance of Protein Difference |
|---|---|---|---|
|
| |||
| HnfG | 3.52 E-11 | ND | |
| HnfA | 6.90 E-11 | 3.8 | 0.01 |
| NR2F1 | 3.14 E-07 | 1.19 | NS |
| RXRB | 1.15 E-05 | 1.07 | NS |
| RXRA | 1.32 E-05 | 2.67 | 0.05 |
| CEBPA | 2.28 E-05 | ND | |
| NR2F6 | 2.46 E-05 | 1.3 | NS |
| SP1 | 9.66 E-05 | 0.67 | 0.05 |
| SP2 | 1.06 E-04 | 0.67 | 0.05 |
Transcription factor binding motif analysis was performed using the −950 to +50 base 5’ untranslated regions of the genes coding for proteins that were significantly higher in VWA8-null cells compared to wildtype. PScan software, with the JASPAR 2016 database was used for analysis and statistical comparisons. Protein fold difference for VWA8 null compared to wild type cells are from proteomic results. ND = not detected in proteomic analysis.
Also shown in Table 3 are data indicating whether protein abundance levels for these transcription factors were altered in the VWA8 null cells. For proteins that were higher in abundance in VWA8 null cells, hepatocyte nuclear factor 4 alpha (Hnf4a, 3.8-fold higher in VWA8 null cells by proteomic analysis) binding motifs also were highly significantly enriched in the promoters of genes for increased proteins (P = 6.9 X 10−11). A number of known Hnf4a target genes 24 also had higher protein abundance in VWA8 null cells (Table 4).
Table 4.
Known Hnf4a target genes with increased protein abundance in VWA8 null cells.
| Protein | Gene | Fold Increase | P |
|---|---|---|---|
|
| |||
| Apolipoprotein A1 | ApoA1 | 6.64 | 0.00059 |
| Apolipoprotein B | ApoB | 3.06 | 0.0000070 |
| Apolipoprotein E | ApoE | 3.96 | 0.00099 |
| Serotransferrin | Tf | 3.10 | 0.00587 |
| Transthyretin | Ttr | 2.70 | 0.036 |
| Alpha-1-antitrypsin | Serpin1c | 2.58 | 0.00024 |
| Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | Acadm | 2.07 | 0.0017 |
| Hydroxymethylglutaryl-CoA synthase, mitochondrial | Hmgcs2 | 2.51 | 0.00045 |
| 3-ketoacyl-CoA thiolase, mitochondrial | Acaa2 | 2.26 | 0.00067 |
| Aldehyde dehydrogenase, mitochondrial | Aldh2 | 1.64 | 0.00072 |
| Aldehyde dehydrogenase X, mitochondrial | Aldh1b1 | 32.6 | 0.0029 |
| Cytochrome P450 2D6 | Cyp2d6 | 5.87 | 0.00003 |
| Aldolase B | Aldob | 4.65 | 0.00028 |
| Ornithine carbamoyltransferase, mitochondrial | Otc | 2.81 | 0.0027 |
| Antithrombin-III | Ant3 | 1.86 | 0.047 |
| Angiotensinogen | Angt | 5.6 | 0.0012 |
| Solute carrier family 2, facilitated glucose transporter member 2 | Slc2a2 | 4.12 | 0.000030 |
Protein abundance changes in VWA8 null cells in known Hnf4 targets. Proteomic results were used to obtain the fold-increase and statistical significance.
In light of the potential central role of Hnf4a in development of the phenotype seen in VWA8 null cells, the higher expression of HNF4a, and to more closely define the differences in Hnf4a abundance in the null knockout, immunoblot analysis of Hnf4a was performed in wildtype and VWA8 null AML12 cells during the time course of differentiation following serum reduction. Neither wildtype nor VWA8 null cells had significant Hnf4a protein levels in the undifferentiated state, but differentiation of AML12 cells rapidly and dramatically raised Hnf4a protein to a much greater degree in the VWA8 null cells (Figure 6). The rise in HNF4a in both genotypes during differentiations is consistent with the role of HNF4a as a hepatocyte differentiation factor, but the rise in AML12 cells null for VWA8 was much greater.
Figure 6.

A. Immunoblot showing the rise in HNF4a protein during differentiation of AML12 CRISPR wildtype (C-WT #6–8) and CRISPR knockout (C-KO #6–10) AML12 cells. B. Quantification of immunoblot.
Rescue of VWA8 protein in VWA8 null cells restores the wildtype bioenergetic phenotype and lowers oxidative stress, Hnf4a abundance, and expression of mitochondrial proteins.
To determine whether the phenotype associated with deletion of VWA8 was specific to the loss of that protein, we used an adenoviral vector to express the protein in VWA8 null cells. Rescue of VWA8 protein in VWA8 null cells restored the wildtype bioenergetic phenotype at the same time Hnf4a expression was reduced to wildtype levels, suggesting the effects of VWA8 deletion on the phenotype observed in differentiated cells of overall higher oxidative capacity are mediated via Hnf4a (Figure 7A). Expression of the shorter isoform, VWA8b, which does not contain the VWA domain, also restored the bioenergetic phenotype to the wildtype state (not shown). Restoration of VWA8 protein (VWA8a or VWA8b) reduced net superoxide production (Figure 7B) and restored abundance of a number of key proteins, including Hnf4a and mitochondrial superoxide dismutase 2 (SOD2) to the levels observed in wildtype cells (Figure 7C, D).
Figure 7.

A. Expression of VWA8 in knockout cells lowers respiration to wildtype levels. Average data (± SD) are shown for wildtype (closed circles), knockout (open circles), knockout with VWA8 rescue (closed diamonds), and knockout with empty adenoviral vector control (open diamonds). Palmitate was used as fuel. B. Quantification of superoxide production (n=12, **p<0.01 vs. Ad-Vec). C. Immunoblots showing rescue of VWA8a (Ad-VWA8a) or VWA8b (Ad-VWA8b) restores protein abundance of marker proteins to wildtype levels. D. Quantification of immunoblots (n=3). No antibody is available for VWA8b, so an HA tag was used CPS-1, carbamoyl phosphate synthase-1; MTCO2, mitochondrial cytochrome c oxidase subunit 2; SOD2, superoxide dismutase 2; Cyto-C, cytochrome C reductase. Statistical comparisons of hydrogen peroxide production were made using one-way analysis of variance.
Addition of an antioxidant mixture (AOH, L-ascorbate, L-glutathione, and alpha tocopherol acetate) lowered H2O2 production, as expected (Figure 8A). To test whether rescue of VWA8 protein also lowered ROS production, VWA8 null AML12 cells were transfected with adenoviral vector for VWA8a, VWA8b, or empty vector as a control. The sites of H2O2 production were probed using inhibitors, as described by Quinlan et al.19 (Figure 8B, C). Inhibitors of superoxide production from Complex I (S1, Iq inhibitor19) and Complex III (S3, IIIQ inhibitor19) had no effect on H2O2 production, but the NOX inhibitors GKT136901 (NOX1/4) and ML171 (mainly NOX1) lowered H2O2 production by more than 50% (Figure 8B). Thioridazine, an inhibitor of peroxisomal beta oxidation of very long chain fatty acids25, however, also lowered H2O2 production significantly (Figure 8C).
Figure 8.

A. Antioxidant mixture (AOH, L-ascorbate, L-glutathione, and alpha tocopherol acetate) lowered net H2O2 production in differentiated VWA8 null AML12 cells using palmitate as fuel. B. Effect of inhibitors of Complex I (S1), Complex III (S3), and NOX isoforms (GKT13783, NOX1/4 inhibitor and ML171, NOX1 inhibitor1) were used in differentiated VWA8 null AML12 cells using palmitate as fuel. C. Effect of thioridazine, an inhibitor of peroxisomal beta oxidation, on H2O2 production in VWA8 null AML12 cells in response to palmitate fuel. Statistical comparisons were made using analysis of variance.
Discussion
VWA8 is a previously uncharacterized protein that is higher in abundance in livers from high fat-fed obese, insulin resistant mice, is targeted to mitochondria and possesses ATPase activity, presumably due to its AAA ATPase domain 14, 15. In the present experiments, unbiased metabolomics, transcriptomics, and proteomics approaches in differentiated AML12 hepatocytes in which VWA8 had been deleted using CRISPR/Cas9 to gain insight into the function of VWA8. Results from the metabolomics analysis revealed robust changes in markers of oxidative stress, especially in cells offered lipid fuel. For example, gamma-glutamyl amino acids, as a class, were lower in the VWA8 null cells regardless of the fuel used. Lower levels of gamma-glutamyl amino acids are markers of oxidative stress, and gamma-glutamyl cysteine, which was lower in VWA8 cells, is directly involved in detoxification of hydrogen peroxide26. Allantoin, which is the product of the reaction of uric acid with reactive oxygen species, and methionine sulfoxide, which is the product of the interaction of methionine with reactive oxygen species, both were significantly higher in the VWA8 null cells when palmitate was used as fuel, but not glucose. It is not clear why the metabolomics data suggest the rise in reactive oxygen species was higher in the presence of lipid fuel. However, as measured more directly by net H2O2 production, the rise in oxidative stress in the VWA8 null cells appeared to be similar whether carbohydrate or lipid fuels are provided. Taken together, the data indicate that reactive oxygen species are higher in the VWA8 null cells before differentiation and that this becomes more exaggerated as the cells differentiate and other phenotypes develop. The rise in reactive oxygen species thus appears to be the earliest detectable change in the VWA8 null cells.
Although the methods used here measured net H2O2 production, some insight as to whether the higher levels were due to production or breakdown can be gained from other data. Data using inhibitors of various oxidative processes known to produce ROS19 suggest that one of the sites of higher ROS production is likely to be NADPH oxidase (NOX) and possibly NOX4, which is a mitochondrial protein and energy sensor27. Consistent with this, transcriptomics results showed that NOX4 mRNA was approximately 60% higher in VWA8 null cells. Proteomics results showed that levels of abundance of protein subunits of the electron transport chain Complex I, also an important site of ROS production19, also were higher. However, inhibition of Complex I at a site known to be a major contributor to mitochondrial superoxide production19 did not inhibit the higher ROS production in the VWA8 null cells. Inhibition of superoxide production from Complex III also had no effect on ROS production, and Complex III proteins were not higher in abundance in the knockout, as assessed by the proteomics analysis. On the other hand, thioridazine, an inhibitor of peroxisomal beta oxidation of very long chain fatty acids, also inhibited ROS production. Higher ROS production from peroxisomes would be predicted by the higher abundance of peroxisomal proteins assessed by proteomics analysis. Therefore, up to half of the rise in net ROS production could be accounted for by NOX isoforms, likely predominantly NOX1 and NOX4, and half from peroxisomes, although higher concentrations of thioridazine potentially inhibited more than half of H2O2 putatively produced by peroxisomal beta oxidation of fatty acids. Because measurement of H2O2 in cells with Amplex Red fluorescence reflects both ROS production and detoxification, it also is relevant to discuss the latter aspect of net ROS release. Regarding detoxification of ROS, glutathione peroxidase was somewhat higher in wildtype cells, but the much more abundant enzyme catalase was higher in VWA8 null cells (Table S4). Higher catalase in the knockout cells would raise H2O2 detoxification and implies that ROS production in the VWA8 null cells may have been even higher than what was indicated by higher net H2O2. Therefore, we conclude that the earliest detectable effect of deletion of VWA8 is higher ROS production, likely from mitochondrial NOX4, NOX1, and peroxisomes, although specificity of these inhibitors is imperfect1. The rise in peroxisomal ROS production may arise during differentiation and may not reflect the initial defect, but rather by a byproduct of a compensatory response. Higher ROS production from NOX4 can arise from lower ATP binding to NOX4, so NOX 4 can serve as a sensor of a mitochondrial defect27. Since the first detectable effect of deletion of VWA8 is the rise in ROS levels and this probably occurs before a rise in HNF4a leads to higher peroxisomal abundance (and higher ROS from peroxisomes), it can be speculated that deletion of VWA8 induces a defect in mitochondrial quality that is sensed by NOX4.
Analysis of protein and mRNA expression changes produced by deletion of VWA8 showed that as the cells differentiated, the expression of proteins involved in oxidative metabolism rose. This was widespread, including mitochondrial, peroxisomal, and other non-mitochondrial sources of oxidative metabolism as well as fuel supply. To determine whether there was an underlying transcription factor or factors that might be altered in the VWA8 null cells and producing this pattern of changes, the promoter regions of genes coding for proteins that were higher in the null cells were examined for transcription factor binding site enrichment. There was clear and significant evidence that HNF4a binding sites were enriched in these promoters, and HNF4a abundance was higher by almost 4-fold in the differentiated VWA8 null cells. Therefore, a rise in HNF4a appears to be responsible for the overall higher level of mitochondrial, peroxisomal, and non-mitochondrial oxidative metabolism, as measured in the bioenergetic experiments, in the differentiated VWA8 null cells. This may be a compensatory effort to correct a mitochondrial defect. With regard to the cause of the greater rise in HNF4a VWA8 null cells, this may be linked to higher levels of reactive oxygen species. In C. elegans, for example, the HNF4 counterpart NHR-49 is a transcriptional regulator of lipid metabolism that is required for the compensatory transcriptional response to oxidative stress28, 29. In mammalian cells, HNF4a also is involved in the response to reactive oxygen species30. Therefore, we hypothesize that deletion of VWA8 produces a mitochondrial quality defect that induces higher ROS production which in turn raises HNF4a activity, producing a compensatory rise in mitochondrial and other proteins involved in energy metabolism. In support of this hypothesis, rescue of VWA8 protein or treatment of VWA8 null cells with antioxidants restored cells to a more wildtype condition, with lower HNF4a expression and a more wildtype bioenergetic profile. As the VWA8 null cells differentiate, ROS production rises even more relative to the wildtype cells. This is likely a by-product of the compensatory HNF4a response. Greater mitochondrial capacity (“more mitochondria”) is created by the compensatory response, but these mitochondria still lack VWA8. Therefore, there may be more mitochondria, but these new mitochondria still have a quality defect, so ROS are produced at ever-increasing rates as the HNF4a compensatory response proceeds.
These data allow some speculation regarding the function of VWA8. VWA8 contains a putative AAA ATPase domain and ATPase activity and has an N-terminal mitochondrial matrix targeting sequence15. AAA ATPase proteins have diverse functions but often are motor proteins, chaperones involved in protein folding, or complex disassembly31. These proteins also typically form hexamers, similar to the mitochondrial chaperone ClpX32. The rise in levels of dipeptides in the VWA8 null cells, indicative of higher protein breakdown, suggest that VWA8 may be involved in regulating protein quality in mitochondria. Thus, the loss of VWA8 would produce a mitochondrial defect resulting in bioenergetic changes that are sensed by NOX4, resulting in higher ROS, higher HNF4a expression, and an attempt to compensate for lower mitochondrial quality through generation of mitochondrial and peroxisomal proteins. Since the compensatory effort results in greater oxidative capacity at the expense of the continued lower mitochondrial quality (the lack of VWA8 remains), a vicious cycle ensues where oxidative stress from defective mitochondria continues to rise and is exacerbated by the compensatory rise in peroxisome activity. Taken together, the data from these experiments indicates that VWA8 is a AAA ATPase protein that plays an important role in maintenance of mitochondrial protein quality.
Protein Accession IDs
The Uniprot IDs for proteins cited in the manuscript are given here. Uniprot accessions for all 6545 proteins assigned in the proteomics experiments are given in Table S5.
| Protein | Uniprot ID |
|---|---|
|
| |
| VWA8 | Q8CC88 |
| HNF4a | P49698 |
| Cyp4a12a | Q91WL5 |
| Adh1 | P00329 |
| Pipox | Q9D826 |
| Pxmp4 | Q9JJW0 |
| Phyh | Q35386 |
| Pex11a | Q9Z211 |
| Decr2 | Q9W768 |
| Ehhadh | Q9BBM2 |
| Pxmp2 | P42925 |
| Acot4 | Q8BWN8 |
| Acaa2 | Q8BWT1 |
| Hadh | Q61425 |
| Acot1 | O55137 |
| Acadm | P45952 |
| Mtp | O08601 |
| Fabpl | P12710 |
| Apob | E9Q414 |
| Apoa1 | Q00623 |
| Apoe | P08226 |
| Cps1 | Q8C196 |
| Otc | P11725 |
| Nox1 | Q8CIZ9 |
| Nox4 | Q9JHI8 |
Supplementary Material
Figure S1. Cell cloning scheme for CRISPR/Cas9 deletion of VWA8.
Figure S2. Proteomics workflow for unbiased, label-free comparison of protein abundance in wildtype and VWA8 null AML12 differentiated hepatocytes.
Table S9. Comparison of Seahorse respiration results in differentiated, permeabilized wildtype and VWA8-null AML12 hepatocytes.
Table S2. Metabolomics data. Shown are mean values and statistical significance for all metabolites, all contrasts.
Table S1. Raw and normalized data for all metabolites detected in VWA8-null (KO) and wildtype AML12 differentiated hepatocytes. Intact cells were exposed to palmitate/BSA, palmitate/BSA/etomoxir, or glucose/BSA. N=6 each condition.
Table S4. Gene Ontology (GO) categories for which probes that were either higher or lower in VWA8-null (KO) cells were significantly over-represented. Panther Pathway Analysis software was used in this analysis.
Table S6. Gene Ontology (GO) categories for which proteins that were either higher or lower in VWA8-null (KO) cells were significantly over-represented. Panther Pathway Analysis software was used in this analysis.
Table S5. All quantifiable proteins. VWA8-null and wildtype AML12 differentiated hepatocytes (n=3 each) were subjected to mass spectrometry analysis and quantification using Progenesis software. Proteins are ranked by most significant (lowest P value). Normalized abundance values are given as asinh(ion intensity).
Table S7. Proteins that were 2-fold higher and significantly different by Progenesis analysis in VWA8-null (KO) than wildtype (WT AML12 cells).
Table S8. Proteins that were 2-fold lower and significantly different by Progenesis analysis in VWA8-null (KO) than wildtype (WT AML12 cells).
Table S3. Transcriptome results. VWA8-null (KO) and wildtype (WT) AML12 differentiated hepatocytes were lysed for isolation of RNA. CEL files were imported into the free open source R package. Affy package and Linear Models for Microarray Data (Limma) version 3.22.0 was used for the analysis. The data were background corrected using normal exponent, quantile normalized, and an unweighted linear model was performed to generate fold changes between groups. The fold changes were log transformed. Expression values obtained were evaluated by a moderated t-statistic (nominal P value), and adjusted using the Benjamini-Hochberg multiple testing correction (adj.P.Val).
Acknowledgements
This study was supported in part by NIH R01DK047936 (LJM).
References
- [1].Altenhofer S, Radermacher KA, Kleikers PW, Wingler K, and Schmidt HH (2015) Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement, Antioxid Redox Signal 23, 406–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Li Y, Luo Z, Wu X, Zhu J, Yu K, Jin Y, Zhang Z, Zhao S, and Zhou L (2018) Proteomic Analyses of Cysteine Redox in High-Fat-Fed and Fasted Mouse Livers: Implications for Liver Metabolic Homeostasis, J Proteome Res 17, 129–140. [DOI] [PubMed] [Google Scholar]
- [3].Qiao JT, Cui C, Qing L, Wang LS, He TY, Yan F, Liu FQ, Shen YH, Hou XG, and Chen L (2018) Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease, Metabolism 81, 13–24. [DOI] [PubMed] [Google Scholar]
- [4].Sabrautzki S, Kaiser G, Przemeck GKH, Gerst F, Lorza-Gil E, Panse M, Sartorius T, Hoene M, Marschall S, Haring HU, Hrabe de Angelis M, and Ullrich S (2017) Point mutation of Ffar1 abrogates fatty acid-dependent insulin secretion, but protects against HFD-induced glucose intolerance, Mol Metab 6, 1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xu JL, Li LY, Wang YQ, Li YQ, Shan M, Sun SZ, Yu Y, and Wang B (2018) Hepatocyte-specific deletion of BAP31 promotes SREBP1C activation, promotes hepatic lipid accumulation, and worsens IR in mice, J Lipid Res 59, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chung B, Stadion M, Schulz N, Jain D, Scherneck S, Joost HG, and Schurmann A (2015) The diabetes gene Zfp69 modulates hepatic insulin sensitivity in mice, Diabetologia 58, 2403–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hasebe T, Tanaka H, Sawada K, Nakajima S, Ohtake T, Fujiya M, and Kohgo Y (2017) Bone morphogenetic protein-binding endothelial regulator of liver sinusoidal endothelial cells induces iron overload in a fatty liver mouse model, Journal of Gastroenterology 52, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim K, Boo K, Yu YS, Oh SK, Kim H, Jeon Y, Bhin J, Hwang D, Kim KI, Lee JS, Im SS, Yoon SG, Kim IY, Seong JK, Lee H, Fang S, and Baek SH (2017) RORalpha controls hepatic lipid homeostasis via negative regulation of PPARgamma transcriptional network, Nat Commun 8, 162–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Willebrords J, Maes M, Pereira IVA, da Silva TC, Govoni VM, Lopes VV, Crespo Yanguas S, Shestopalov VI, Nogueira MS, de Castro IA, Farhood A, Mannaerts I, van Grunsven L, Akakpo J, Lebofsky M, Jaeschke H, Cogliati B, and Vinken M (2017) Protective effect of genetic deletion of pannexin1 in experimental mouse models of acute and chronic liver disease, Biochim Biophys Acta 1864, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Benard O, Lim J, Apontes P, Jing X, Angeletti RH, and Chi Y (2016) Impact of high-fat diet on the proteome of mouse liver, Journal of Nutritional Biochemistry 31, 10–19. [DOI] [PubMed] [Google Scholar]
- [11].Liao BM, Raddatz K, Zhong L, Parker BL, Raftery MJ, and Schmitz-Peiffer C (2014) Proteomic analysis of livers from fat-fed mice deficient in either PKCdelta or PKCepsilon identifies Htatip2 as a regulator of lipid metabolism, Proteomics 14, 2578–2587. [DOI] [PubMed] [Google Scholar]
- [12].Sabido E, Wu Y, Bautista L, Porstmann T, Chang CY, Vitek O, Stoffel M, and Aebersold R (2013) Targeted proteomics reveals strain-specific changes in the mouse insulin and central metabolic pathways after a sustained high-fat diet, Mol Syst Biol 9, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schafer A, Neschen S, Kahle M, Sarioglu H, Gaisbauer T, Imhof A, Adamski J, Hauck SM, and Ueffing M (2015) The Epoxyeicosatrienoic Acid Pathway Enhances Hepatic Insulin Signaling and is Repressed in Insulin-Resistant Mouse Liver, Mol Cell Proteomics 14, 2764–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Luo M, Mengos AE, Stubblefield TM, and Mandarino LJ (2012) High Fat Diet-Induced Changes in Hepatic Protein Abundance in Mice, Journal of Proteomics and Bioinformatics 5, 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Luo M, Mengos AE, Ma W, Finlayson J, Bustos RZ, Xiao Zhu Y, Shi CX, Stubblefield TM, Willis WT, and Mandarino LJ (2017) Characterization of the novel protein KIAA0564 (Von Willebrand Domain-containing Protein 8), Biochem Biophys Res Commun 487, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013) Genome engineering using the CRISPR-Cas9 system, Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hwang H, Bowen BP, Lefort N, Flynn CR, De Filippis EA, Roberts C, Smoke CC, Meyer C, Hojlund K, Yi Z, and Mandarino LJ (2010) Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes, Diabetes 59, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wong HS, Benoit B, and Brand MD (2019) Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts, Free Radic Biol Med 130, 140–150. [DOI] [PubMed] [Google Scholar]
- [19].Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, and Brand MD (2013) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates, Redox Biol 1, 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zambelli F, Pesole G, and Pavesi G (2009) Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes, Nucleic Acids Res 37, W247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gerencser AA, Neilson A, Choi SW, Edman U, Yadava N, Oh RJ, Ferrick DA, Nicholls DG, and Brand MD (2009) Quantitative microplate-based respirometry with correction for oxygen diffusion, Anal Chem 81, 6868–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mookerjee SA, Gerencser AA, Nicholls DG, and Brand MD (2017) Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements, J Biol Chem 292, 7189–7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, and Murphy AN (2011) High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria, PLoS One 6, e21746–e21757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bolotin E, Liao H, Ta TC, Yang C, Hwang-Verslues W, Evans JR, Jiang T, and Sladek FM (2010) Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays, Hepatology 51, 642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shi R, Zhang Y, Shi Y, Shi S, and Jiang L (2012) Inhibition of peroxisomal beta-oxidation by thioridazine increases the amount of VLCFAs and Abeta generation in the rat brain, Neurosci Lett 528, 6–10. [DOI] [PubMed] [Google Scholar]
- [26].Quintana-Cabrera R, and Bolanos JP (2013) Glutathione and gamma-glutamylcysteine in hydrogen peroxide detoxification, Methods Enzymol 527, 129–144. [DOI] [PubMed] [Google Scholar]
- [27].Shanmugasundaram K, Nayak BK, Friedrichs WE, Kaushik D, Rodriguez R, and Block K (2017) NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance, Nat Commun 8, 997–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hu Q, D’Amora DR, MacNeil LT, Walhout AJM, and Kubiseski TJ (2018) The Caenorhabditis elegans Oxidative Stress Response Requires the NHR-49 Transcription Factor, G3 (Bethesda) 8, 3857–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Goh GYS, Winter JJ, Bhanshali F, Doering KRS, Lai R, Lee K, Veal EA, and Taubert S (2018) NHR-49/HNF4 integrates regulation of fatty acid metabolism with a protective transcriptional response to oxidative stress and fasting, Aging Cell 17, e12743–e12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Darsigny M, Babeu JP, Seidman EG, Gendron FP, Levy E, Carrier J, Perreault N, and Boudreau F (2010) Hepatocyte nuclear factor-4alpha promotes gut neoplasia in mice and protects against the production of reactive oxygen species, Cancer Res 70, 9423–9433. [DOI] [PubMed] [Google Scholar]
- [31].Ogura T, and Wilkinson AJ (2001) AAA+ superfamily ATPases: common structure--diverse function, Genes Cells 6, 575–597. [DOI] [PubMed] [Google Scholar]
- [32].Baker TA, and Sauer RT (2012) ClpXP, an ATP-powered unfolding and protein-degradation machine, Biochim Biophys Acta 1823, 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cell cloning scheme for CRISPR/Cas9 deletion of VWA8.
Figure S2. Proteomics workflow for unbiased, label-free comparison of protein abundance in wildtype and VWA8 null AML12 differentiated hepatocytes.
Table S9. Comparison of Seahorse respiration results in differentiated, permeabilized wildtype and VWA8-null AML12 hepatocytes.
Table S2. Metabolomics data. Shown are mean values and statistical significance for all metabolites, all contrasts.
Table S1. Raw and normalized data for all metabolites detected in VWA8-null (KO) and wildtype AML12 differentiated hepatocytes. Intact cells were exposed to palmitate/BSA, palmitate/BSA/etomoxir, or glucose/BSA. N=6 each condition.
Table S4. Gene Ontology (GO) categories for which probes that were either higher or lower in VWA8-null (KO) cells were significantly over-represented. Panther Pathway Analysis software was used in this analysis.
Table S6. Gene Ontology (GO) categories for which proteins that were either higher or lower in VWA8-null (KO) cells were significantly over-represented. Panther Pathway Analysis software was used in this analysis.
Table S5. All quantifiable proteins. VWA8-null and wildtype AML12 differentiated hepatocytes (n=3 each) were subjected to mass spectrometry analysis and quantification using Progenesis software. Proteins are ranked by most significant (lowest P value). Normalized abundance values are given as asinh(ion intensity).
Table S7. Proteins that were 2-fold higher and significantly different by Progenesis analysis in VWA8-null (KO) than wildtype (WT AML12 cells).
Table S8. Proteins that were 2-fold lower and significantly different by Progenesis analysis in VWA8-null (KO) than wildtype (WT AML12 cells).
Table S3. Transcriptome results. VWA8-null (KO) and wildtype (WT) AML12 differentiated hepatocytes were lysed for isolation of RNA. CEL files were imported into the free open source R package. Affy package and Linear Models for Microarray Data (Limma) version 3.22.0 was used for the analysis. The data were background corrected using normal exponent, quantile normalized, and an unweighted linear model was performed to generate fold changes between groups. The fold changes were log transformed. Expression values obtained were evaluated by a moderated t-statistic (nominal P value), and adjusted using the Benjamini-Hochberg multiple testing correction (adj.P.Val).