

Abstract
A critical step of pre-mRNA splicing is the recruitment of U2 snRNP to the branch point sequence of an intron. U2 snRNP conformation changes extensively during branch helix formation, and several RNA-dependent ATPases are implicated in the process. However, the molecular mechanisms involved remain to be fully dissected. We took advantage of the differential nucleotide triphosphates requirements for DExD/H-box enzymes to probe their contributions to in vitro spliceosome assembly. Both ATP and GTP hydrolysis support the formation of A-complex, indicating the activity of a DEAH-enzyme because DEAD-enzymes are selective for ATP. We immunodepleted DHX15 to assess its involvement, and although splicing efficiency decreases with reduced DHX15, A-complex accumulation incongruently increases. DHX15 depletion also results in the persistence of the atypical ATP-independent interaction between U2 snRNP and a minimal substrate that is otherwise destabilized in the presence of either ATP or GTP. These results lead us to hypothesize that DHX15 plays a quality control function in U2 snRNP's engagement with an intron. In efforts to identify the RNA target of DHX15, we determined that an extended polypyrimidine tract is not necessary for disruption of the atypical interaction between U2 snRNP and the minimal substrate. We also examined U2 snRNA by RNase H digestion and identified nucleotides in the branch binding region that become accessible with both ATP and GTP hydrolysis, again implicating a DEAH-enzyme. Together, our results demonstrate that multiple ATP-dependent rearrangements are likely involved in U2 snRNP addition to the spliceosome and that DHX15 may have an expanded role in maintaining splicing fidelity.
Keywords: splicing, spliceosome, U2 snRNP, A-complex, DExH NTPase, DHX15, PRP43
INTRODUCTION
Pre-mRNA splicing by the spliceosome is an essential step in eukaryotic gene expression and must be highly accurate to generate functional messenger RNAs. The boundaries of an intron are initially designated by base-pairing interactions between U1 snRNA and the 5′ splice site and U2 snRNA and the branch point sequence. U2 snRNA recognition of the branch point sequence takes place in the context of a small ribonucleoprotein particle (snRNP), which also contains ten core proteins, three SF3A proteins, and seven SF3B proteins. These events signal the rest of the spliceosome to assemble into a catalytic entity.
A collection of RNA-dependent ATPases that drive rearrangements required during spliceosome assembly have also been linked to enforcing splice site fidelity in S. cerevisiae (Villa and Guthrie 2005; Mayas et al. 2006; Xu and Query 2007; Koodathingal et al. 2010; Koodathingal and Staley 2013; Yang et al. 2013; Toroney et al. 2019). Most of these enzymes, classified as either DEAD (DDX) or DEAH (DHX), are considered helicases because they can disrupt RNA base-pairing interactions; however, they exhibit some mechanistic differences (Jankowsky 2011; Gilman et al. 2017). DDX-enzymes have a Q-motif that enforces a selectivity for ATP as the source of energy for RNA unwinding (Tanner et al. 2003). In contrast, DHX-enzymes lack this motif and can utilize other nucleotides, with a preference for ATP and GTP (He et al. 2010; Walbott et al. 2010).
During in vitro spliceosome assembly, recruitment of U2 snRNP to the branch point sequence is the first ATP-dependent step, and results in formation of a stable entity referred to as A-complex. The mechanistic basis of the requirement for ATP is not fully understood, but striking differences between recent cryo-EM structures of U2 snRNP and A-complex imply several large-scale molecular rearrangements of interactions within the snRNP and with the intron. Several RNA-dependent ATPases are associated with U2 snRNP: three DDX-enzymes (DDX46, DDX39B, DDX42) and DHX15. The DDX-enzymes display the expected nucleotide preferences for ATP (O'Day et al. 1996; Uhlmann-Schiffler et al. 2006; Shen et al. 2007) and DHX15 is an NTPase with a preference for ATP and GTP (Robert-Paganin et al. 2017). Both DDX46 (aka Prp5) and DDX39B (aka UAP56 or Sub2) have been linked to A-complex assembly, and likely mediate some of the rearrangements. For example, DDX46 is localized near the branch stem–loop (BSL) in the 17S U2 snRNP structure (Zhang et al. 2020). In yeast, BSL mutations suppress an amino-terminal deletion of the DDX46 ortholog Prp5, which links it to BSL unwinding (Perriman and Ares 2010). DDX46 is also proposed to proofread the branch between U2 snRNA and the branch point sequence of an intron (Xu and Query 2007; Liang and Cheng 2015; Zhang et al. 2021). UAP56 interacts with U2AF, a factor that binds the polypyrimidine tract (PYT) downstream from the branch to help recruit U2 snRNP (Fleckner et al. 1997). DDX42 associates exclusively with a form of U2 snRNP that lacks the SF3A and SF3B complexes and is unlikely to be directly involved in spliceosome assembly (Will et al. 2002). DHX15 (aka Prp43) is best known for its role in disassembly of the intron lariat spliceosome (ILS) at the end of splicing, although studies differ on whether it interacts directly with the intron or U6 snRNA (Fourmann et al. 2016; Toroney et al. 2019). DHX15 also has a role in ribosome biogenesis, and its specificity appears to be regulated by different G-patch cofactors that direct it to the intron lariat (Wen et al. 2008) or pre-rRNA (Memet et al. 2017). Notably, DHX15 is present in pulldowns of U2 snRNP and A-complex spliceosomes, along with G-patch proteins RBM5, RBM10, RBM17, CHERP, and SUGP1 (Agafonov et al. 2011).
For human introns, the sequences that define splice sites are quite variable, especially the branch point sequence. Quality control of branch point selection is likely important to maintain cellular health as alterations in the players involved is evidenced to lead to cancer (Bonnal et al. 2020). Unsurprisingly, these include mutations in several proteins associated with U2 snRNP. For example, specific point mutations in SF3B1, a U2 snRNP protein that directly contacts the branch point sequence in early spliceosome assembly, are enriched in cells from cancers and dysplasia syndromes, particularly in hematological lineages (Dvinge et al. 2016). In addition, SF3B1 mutations result in altered branch point selection for some introns (Darman et al. 2015; DeBoever et al. 2015; Alsafadi et al. 2016; Wang et al. 2016; Kesarwani et al. 2017).
In this study, we use an in vitro splicing system to investigate the possibility of a role for DHX15 in mediating ATP-dependent rearrangements involved in U2 snRNP's engagement with an intron. Depletion of DHX15 results in increased A-complex formation, but lower splicing efficiency. The effect is amplified when we assemble spliceosomes on introns incapable of forming complexes competent to complete splicing, leading us to propose that DHX15 may have an additional splicing related function in quality control of branch sequence recognition. Using a powerful affinity-tagged version of the core component SNRPB2, we also characterize changes in U2 snRNP that show differential selectivity for ATP and GTP, suggesting involvement of different DDX-and DHX-enzymes in a growing constellation of rearrangements that may impact how the spliceosome finds introns with high fidelity.
RESULTS
Both ATP and GTP can support spliceosome assembly and splicing
To test the hypothesis that molecular rearrangements during A-complex assembly are promoted by one or more of the RNA-dependent ATPases associated with U2 snRNP, we used nucleotide preference to differentiate between the activity of DDX- and DHX-enzymes. DDX-enzymes require ATP to disrupt RNA interactions and are often locked onto their target when bound to a nonhydrolyzable ATP analog (Gilman et al. 2017). In contrast, DHX-enzymes are processive RNA helicases that can also utilize GTP (Jankowsky 2011). If a DHX-enzyme is involved in the initial rearrangement, then GTP should support A-complex formation prior to the first rearrangement that requires the activity of a DDX-enzyme. Alternatively, if ATP-binding is sufficient for an initial DDX-mediated rearrangement, then nonhydrolyzable AMP–PNP will support assembly to that point.
We carried out in vitro splicing assays on a radiolabeled pre-mRNA substrate in the presence or absence of ATP, GTP, and AMP–PNP and excluded creatine phosphate to prevent recycling of nucleotides. We used native gels to assess spliceosome assembly on the pre-mRNA and denaturing gels to measure splicing efficiency (Fig. 1A,B). As expected, ATP confers robust spliceosome assembly and splicing chemistry, which are both lost with no added nucleotide. AMP–PNP does not support assembly nor splicing, leading us to conclude that binding of a target RNA by a DDX-enzyme is not sufficient to promote the first rearrangement needed for U2 snRNP to engage an intron, with the caveat that AMP–PNP binding is slower than ATP (Polach and Uhlenbeck 2002). Surprisingly, GTP promotes both a significant amount of spliceosome assembly and splicing, although less efficiently than with ATP. We considered that nucleoside-diphosphate kinase (NME1) could be active in our extracts and transfer a phosphate from GTP to any ADP that remains after extract preparation. To address this possibility, we passed nuclear extract over a G-50 size exclusion column (SEC) to remove endogenous nucleotides (Anderson and Moore 2000). Post SEC, we still observe spliceosome assembly and splicing with the addition of ATP or GTP in the nucleotide-depleted extract (Supplemental Fig. 1). B-complex and splicing products accumulate more slowly with GTP in the nucleotide-depleted extracts relative to those cycled at 30°C. This difference indicates that a small amount of ADP recycling over an extended incubation may contribute to a portion of the B-complex formation and splicing activity. However, GTP is clearly sufficient to support robust A-complex assembly.
FIGURE 1.
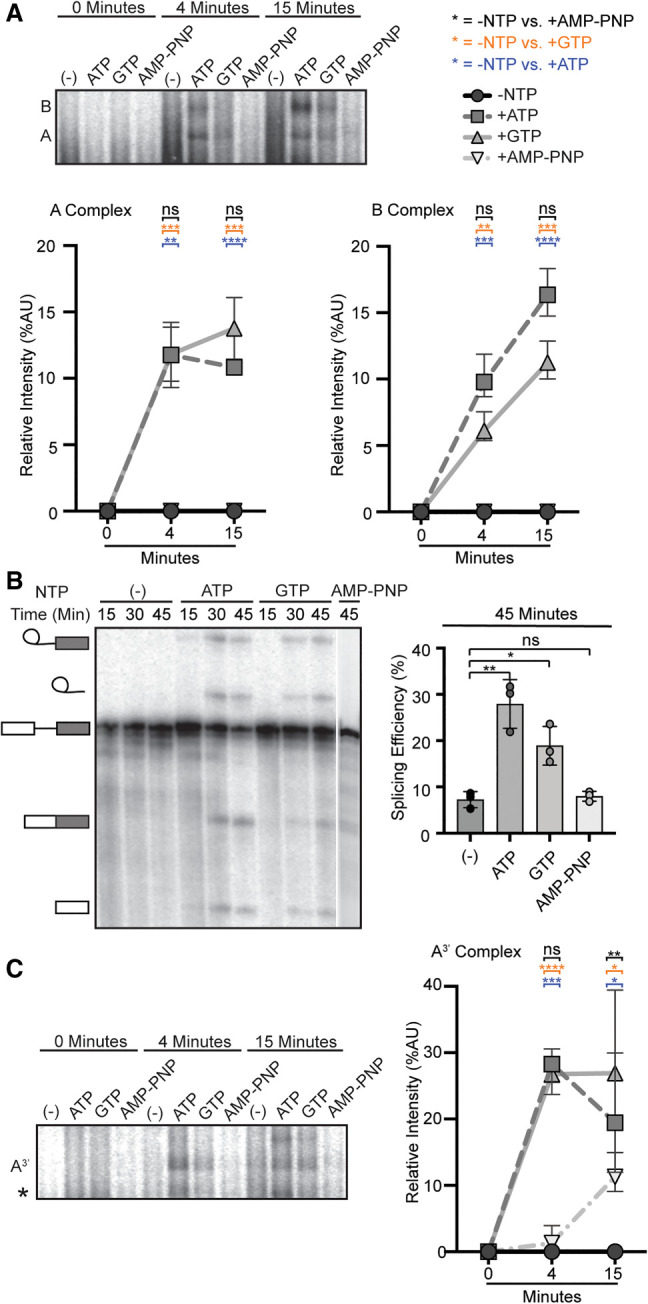
Both ATP and GTP can support spliceosome assembly and splicing. (A, top) Representative native gel analysis of in vitro spliceosome assembly with a radiolabeled full-length substrate with different added NTPs at the indicated timepoints. A- and B-complex band positions are labeled. (Bottom) Normalized band intensity relative to the entire lane for the indicated timepoints of three independent trials. Statistical differences were examined by unpaired Student's t-test with (***) P < 0.001, (**) P < 0.01, (*) P < 0.05. (B, left) Representative denaturing gel analysis of radiolabeled RNA isolated at the indicated timepoints from in vitro splicing reactions using a full-length substrate with different added NTPs. The RNA band identities are illustrated on the left as (top to bottom): lariat intron intermediate, lariat intron, pre-mRNA substrate, mRNA, and 5′ exon intermediate. (Right) Splicing efficiency is measured as intensity of the mRNA band over total RNA bands and shown for the 45-min timepoint for three independent trials. Statistical differences were examined as in A. (C) Same as A, except that an A3′-substrate was used for spliceosome assembly.
We also tested nucleotide requirements of early spliceosome assembly in the context of a truncated substrate (A3′ substrate) that recapitulates U2 snRNP interactions with an intron (Konarska and Sharp 1986). We find that both ATP and GTP addition results in formation of the A3′-complex, but not AMP–PNP (Fig. 1C). Because GTP supports A- and A3′-complex assembly, we conclude that a DHX-enzyme can promote an initial rearrangement that allows U2 snRNP to engage with an intron. We hypothesize that the higher accumulation of B-complex with ATP reflects the contribution of a DDX-enzyme, potentially by capturing a transient rearrangement or conformation that allows further spliceosome assembly. Based on their direct association with U2 snRNP, DHX15 and DDX46 are the most-likely candidates for the two activities.
Depletion of DHX15 reduces splicing efficiency, but not spliceosome assembly
To determine whether DHX15 has a role in U2 snRNP addition to the spliceosome, we immunodepleted the protein from HeLa nuclear extract. Despite the high abundance of DHX15, 55%–72% of the protein was removed in three independent immunodepletions relative to mock depletion as determined by western blot analysis (Fig. 2A; Supplemental Fig. 2). We used DHX15- and mock-depleted extracts for in vitro spliceosome assembly on the A3′ substrate to focus on U2 snRNP incorporation. Contrary to the prediction that loss of DHX15 would inhibit assembly, the relative band intensity of A-complex in DHX15-depeleted extract is increased compared to the mock-depleted extract (Fig. 2B). We repeated the experiment with a full-length substrate to determine if the difference is due to the truncated substrate. A small but consistent increase in the relative band intensities of both A- and B- complex spliceosomes persist with DHX15-depleted extracts (Fig. 2C). Surprisingly, overall splicing efficiency is lower in the DHX15-depleted extracts compared to mock-depleted extract (Fig. 2D).
FIGURE 2.

Depletion of DHX15 reduces splicing efficiency, but not spliceosome assembly. (A) Western blot analysis of three independent immunodepletions of DHX15 from HeLa nuclear extract. Depletions were quantified by comparing the band intensity of DHX15 (α-15) to a nonspecific band (NS), both normalized to the intensity of the entire lane. Samples are shown for untreated nuclear extract (Input), control depletion with 1X PBS or IgG (Mock) and depletion with anti-DHX15 (dDHX15). (B, top) Representative native gel analysis of in vitro spliceosome assembly with a radiolabeled A3′-substrate in Mock and dDHX15 nuclear extracts at the indicated timepoints. (Bottom) Normalized band intensity relative to the entire lane from three independent trials. Statistical differences were examined by paired Student's t-test with P-values as in Figure 1A. (C) Same as B, but with a full-length splicing substrate. (D, left) Representative denaturing gel analysis of radiolabeled RNA isolated at the indicated timepoints from in vitro splicing reactions using Mock or dDHX15 nuclear extracts. The RNA band identities are as described in Figure 1B. (Right) Splicing efficiency determined as in Figure 1B.
In the context of our original hypothesis, these results indicate that DHX15 may not be responsible for the initial rearrangement that allows U2 snRNP to interact with an intron, although we cannot rule out that the remaining DHX15 after immunodepletion is sufficient to carry out that role. However, the opposing increase in spliceosome assembly and decreased splicing efficiency, in tandem with the significant increase of A3′-complexes, which cannot complete assembly, lead us to suspect that DHX15 may have a role in quality control of early spliceosome assembly. With reduced DHX15 activity, unproductive complexes accumulate and potentially sequester spliceosome components, which results in overall less splicing efficiency. Alternatively, the decrease in splicing efficiency may indirectly result from the loss of spliceosome disassembly in the ILS and/or another role for DHX15 promoting splicing chemistry after B-complex formation. As we are currently unable to add back purified DHX15, we also cannot rule out the possibility that a codepleted factor is responsible for the observed effects.
Reduction of DHX15 stabilizes the ATP-independent interaction between U2 snRNP and a minimal intron
Query et al. (1997) showed that a minimal RNA (Amin substrate) containing only a branch point sequence followed by a PYT interacts with U2 snRNP in the absence of ATP to form the Amin-complex. Notably, in the presence of ATP, the Amin-complex is destabilized by an unknown entity (Newnham and Query 2001). To determine whether DHX15 could be responsible, we incubated the Amin substrate in both DHX15-depleted and mock-depleted HeLa nuclear extract with and without ATP. In the absence of ATP, the expected Amin-complex band forms in both extracts (Fig. 3A). Addition of ATP to the mock-depleted extract results in the loss of most Amin-complex and the appearance of a faster migrating complex of unknown composition (*) that decreases over time (Query et al. 1997). In DHX15-depleted extracts with ATP, the Amin-complex persists, indicating that DHX15 and/or a co-depleted factor is responsible for the ATP-dependent loss. The faster migrating complex (*) is largely unaffected.
FIGURE 3.
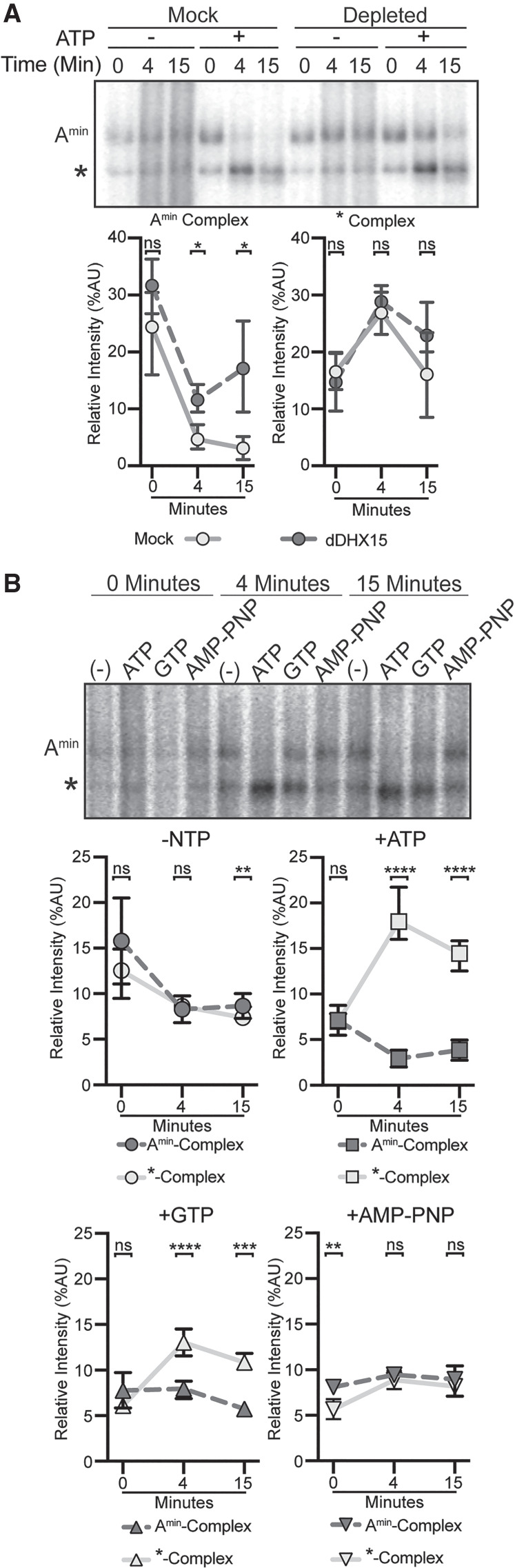
Reduction of DHX15 stabilizes the ATP-independent interaction between U2 snRNP and minimal intron. (A, top) Representative native gel analysis of in vitro spliceosome assembly with a radiolabeled Amin-substrate in control (Mock) and DHX15-immunodepleted (dDHX15) nuclear extracts at the indicated timepoints. (Bottom) Normalized band intensity relative to the entire lane from three independent trials. Statistical differences were examined by paired Student's t-test. (B, top) Representative native gel analysis of in vitro spliceosome assembly with Amin-substrate with different added NTPs at the indicated timepoints. Statistical differences were examined by unpaired Student's t-test.
If DHX15 is responsible for disrupting the unproductive Amin-complex, then GTP should also promote its disassembly in normal nuclear extract. We compared the addition of ATP, GTP, and AMP–PNP with the Amin substrate, and found that both ATP and GTP result in the loss of Amin-complex, while AMP–PNP does not (Fig. 3B). Taken together, these results support a model in which DHX15 disrupts an unproductive interaction between U2 snRNP and the intron in Amin-complex as a quality control mechanism.
Amin destabilization does not occur via an extended PYT
In the Amin-complex, there are only two possible RNAs for DHX15 to target: U2 snRNA and the short Amin substrate. Given that DHX-enzymes act by translocating from 3′ to 5′ on a single RNA strand (Sengoku et al. 2006; Yang and Jankowsky 2006; Yang et al. 2007; Mallam et al. 2012; Tauchert et al. 2017; Hamann et al. 2019), the 3′ end of the Amin substrate is a likely DHX15 target. We tested this possibility by truncating the 3′ end of the intron in 4 nt increments to see if loss of the putative target sequence results in Amin-complex stabilization despite added ATP (Fig. 4A). We stopped at 10 nt downstream from the branch point sequence because too short of a PYT interferes with spliceosome assembly (Bessonov et al. 2010). The truncations did not interfere with Amin assembly in the absence of ATP (Fig. 4B,C), and complexes were all destabilized with added ATP. We conclude an extended PYT is not necessary for Amin destabilization, meaning that DHX15 either interacts with the remaining 10 nt of the PYT or recognizes another RNA feature within the Amin-complex.
FIGURE 4.

Amin destabilization does not occur via an extended PYT. (A) Schematic of the Amin RNA substrate with branch point sequence (BPS) and PYT. The arrows indicate the 3′ end of the PYT in each RNA. (B) Representative native gel analysis of in vitro spliceosome assembly with truncated Amin substrate ±ATP at the indicated timepoints. PYT length is indicated on the right. (C) Background-corrected band intensity of Amin- and *-complexes from two independent trials. Graphs correspond to the adjacent native gel images in B.
U2 snRNA accessibility is regulated by NTP hydrolysis
The other RNA in Amin is U2 snRNA, of which nucleotides 32–46 are of special interest because they both interact with the intron in an extended branch helix and form the mutually exclusive branch stem–loop (BSL) structure (Fig. 5A; Perriman and Ares 2010; Zhang et al. 2020). In the context of the U2 snRNP in nuclear extract, this region was shown to become accessible for base-pairing with a complementary DNA oligonucleotide in an ATP-dependent manner (Black et al. 1985), although the factor responsible for the ATP-dependence was unknown. To determine whether a DHX-enzyme could be involved, we tested whether GTP or AMP–PNP could also enable base-pairing of different regions in the 5′ half of U2 snRNA. We created a series of overlapping DNA oligonucleotides complementary to U2 snRNA (nt 1–15, nt 12–26, nt 24–38, nt 32–46, Fig. 5A), which we added to HeLa nuclear extract treated with various NTP's. If the targeted region is accessible for base-pairing, endogenous RNase H in the extracts will induce cleavage of the RNA/DNA hybrid. We mapped the specific cleavage sites using primer extension (Fig. 5B) and quantified overall digestion of U2 snRNA induced by the oligonucleotides (Fig. 5C).
FIGURE 5.
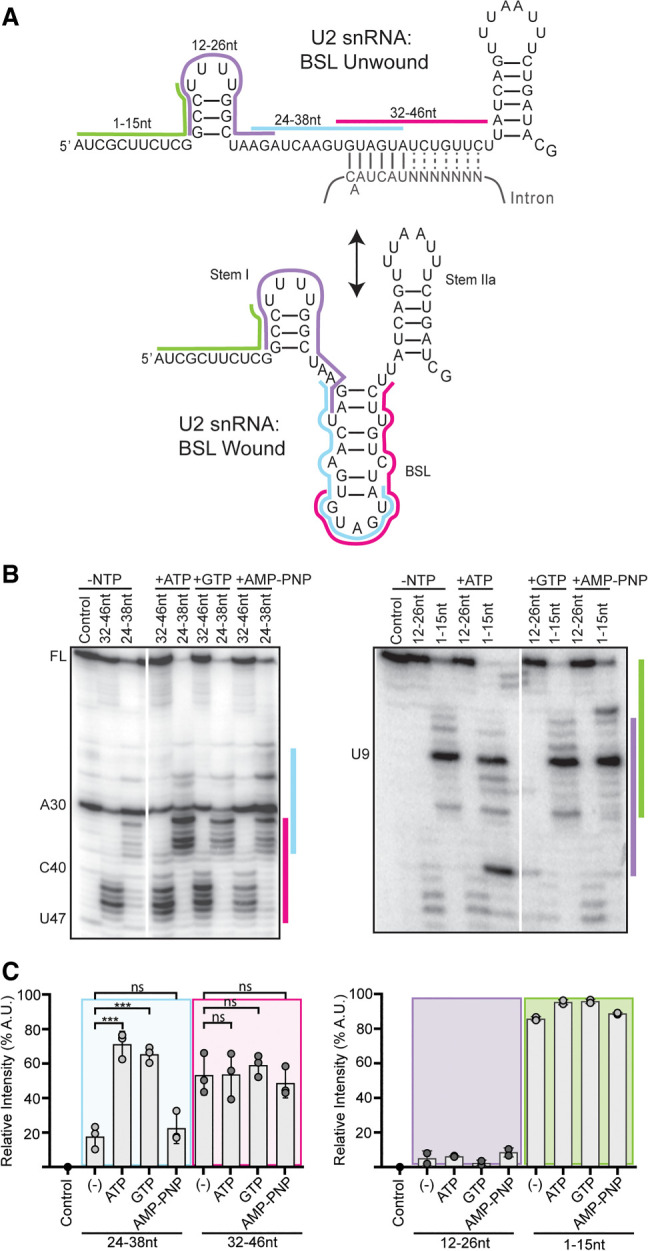
U2 snRNA accessibility is regulated by NTP hydrolysis. (A) Secondary structure of the U2 snRNA with the BSL (bottom) or unwound interaction with the gray intron (top). The position of complementary DNA oligonucleotides used for RNase H digestion are indicated with colored bars. (B) Primer extension analysis of U2 snRNA isolated from nuclear extract after RNase H digestion. Extracts were treated with ATP, GTP, or AMP–PNP, or no NTP. Control digestions were carried out with a noncomplementary DNA oligonucleotide. (C) Cleavage efficiency was determined as band intensity in the region complementary to the oligonucleotide over the intensity of the entire lane of three independent trials. Statistical differences relative to the NTP sample were examined by unpaired Student's t-test.
With an oligonucleotide that targets the 5′ half of the BSL (nt 24–28), over 60% of U2 snRNA molecules in the extract are cleaved after both ATP and GTP treatment, compared with ∼20% cleaved with no added NTP or AMP–PNP. Specifically, most cleavage occurs after nucleotides 32–36 (GUGUA) with some cleavage after nucleotide 37. Using an oligonucleotide targeting the 3′ half of the BSL (nt 32–46), around half of U2 snRNA molecules are cleaved, primarily after nucleotides 42–46 independent of added NTP, although there is a small increase in cleavage after nucleotides 37 and 38 with ATP and GTP. Cleavage in the presence of the other oligonucleotides is not influenced by NTP treatment. With an oligonucleotide targeting the beginning of U2 snRNA (nt 1–15), nearly all the U2 snRNA molecules are cleaved after U9 indicating that while the 5′ end of U2 snRNA is available for base-pairing, nucleotides 10–15 are protected, likely because of their participation in Stem I (Fig. 5A). An oligonucleotide that targets nucleotides 12–26 also does not induce cleavage, further supporting the stability of Stem I.
Because both ATP and GTP hydrolysis results in increased accessibility, we conclude that a DHX-enzyme mediates the rearrangement that makes U2 snRNA nucleotides 24–38 available for base-pairing. The enzyme may unwind an RNA structure, such as the BSL, or dislodge a protein that protects the RNA.
U2 snRNP composition is affected by NTP hydrolysis
To determine if a change in protein interactions is responsible for the NTP-dependent change in U2 snRNA accessibility, we generated a HeLa cell line with a stably integrated transgene encoding a V5-tagged SNRPB2, a core U2 snRNP protein, for pulldown studies (Khandelia et al. 2011; Kim 2019). Western analysis of nuclear extract shows that over half the SNRPB2 expressed in these cells carries the tag (input lane), and that we can specifically immunoprecipitate (IP) the V5-tagged protein (Fig. 6A; Supplemental Fig. 3). We analyzed anti-V5 IP's for RNA and see enrichment of U2 snRNA indicating that the tagged protein incorporates into U2 snRNP (Fig. 6B). IP's of in vitro splicing reactions using a radiolabeled full-length pre-mRNA reveal higher levels of pre-mRNA coeluting with the V5-tagged SNRPB2 relative to IgG control, which is further enhanced by addition of ATP to the splicing reactions (Fig. 6C). We repeated the IP's with the Amin substrate, and in this case more RNA coelutes from splicing reactions without added ATP (Fig. 6D). These results parallel our observations by native gel analysis, indicating that the V5-tagged U2 snRNP incorporates splicing complexes.
FIGURE 6.
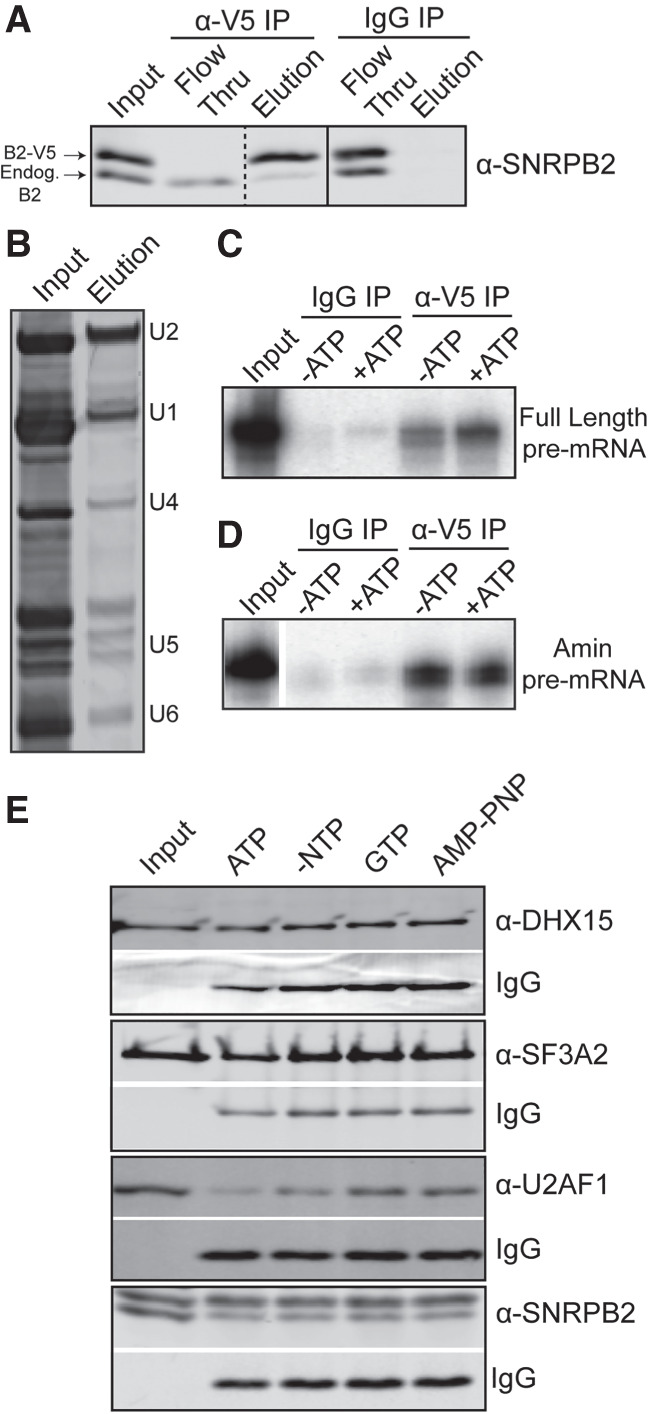
ATP hydrolysis alters U2 snRNP protein composition. (A) Western blot analysis of IP with anti-V5 and mouse IgG from HeLa nuclear extract containing V5-tagged SNRPB2 and probed with anti-SNRPB2, which detects both endogenous (endog) and V5-tagged SNRPB2 (V5-B2). (B) Denaturing gel analysis of RNA isolated from anti-V5 IP eluate stained with SYBR Gold. (C) Denaturing gel analysis of RNA isolated anti-V5 IP of in vitro splicing reactions ±ATP using a radiolabeled full-length RNA substrate in the conditions. (D) Same as C, except with an Amin substrate. (E) Western analysis anti-V5 of IP's from nuclear extract incubated under in vitro splicing conditions in the presence of ATP, GTP, or AMP–PNP. Blots were probed with the indicated antibody. Bands labeled with IgG show the amount of anti-V5 antibody in the elutions.
To test whether protein composition of U2 snRNP is altered by NTP hydrolysis, we IP'd V5-tagged U2 snRNP from nuclear extract incubated with ATP, GTP, or AMP–PNP. Western analysis of eluates showed that the NTP treatments do not alter the immunoprecipitation of V5-tagged SNRPB2, or the association of the 17S U2 snRNP protein SF3A2 (Fig. 6E). We detect DHX15 in the eluates under all conditions. In contrast, U2AF1 is reduced after ATP treatment. This change is more likely due to the activity of a DDX-enzyme because GTP treatment does not result in the same decrease. AMP–PNP also has no effect. Relating these results to the increased U2 snRNA accessibility in the snRNP after both ATP and GTP treatment, the ATP-specificity of U2AF1 loss means it is not responsible for blocking access to the branch binding region. Because DHX15 remains with the snRNP, it could be responsible removing a different protein to make the branch binding region of U2 snRNA available for base-pairing interactions. It is tempting to speculate that DHX15 may be disrupting unproductive spliceosomes in a similar manner.
DISCUSSION
Spliceosome assembly relies on an assortment of DDX- and DHX-enzymes to both drive molecular rearrangements and promote splicing fidelity. Both DDX46 (yeast Prp5) and DDX39B (UAP56, yeast Sub2) play roles in U2 snRNP's addition to the branch point sequence. Recent cryo-EM structures show that DDX46 interacts with U2 snRNP near both the BSL and an unproductive branch-helix, consistent with its proposed roles in BSL rearrangements and fidelity of branch point selection (Perriman and Ares 2010; Liang and Cheng 2015; Zhang et al. 2020, 2021). However, many questions remain open for both enzymes. For example, does DDX46 directly unwind the BSL, or does it dislodge HTATSF1 to destabilize the BSL? For DDX39B as well, what it targets, what it rearranges, and how it is regulated is still not clear. Using the differential nucleotide triphosphate selectivity of DDX- and DHX-enzymes, our study supports added roles for a DHX-enzyme(s) in regulating U2 snRNP structure and early assembly of human spliceosomes. We show that GTP can substitute for ATP in overcoming whatever blocks U2 snRNP from binding an intron with an anchor sequence. GTP can also mediate remodeling of the U2 snRNP to expose the branch binding sequence. Finally, GTP also promotes the destabilization of the unproductive interaction between U2 snRNP and an anchorless minimal intron, which we linked to the presence of DHX15. Additionally, we identified an ATP-specific loss of U2AF1 from U2 snRNP, which suggests the activity of a DDX-enzyme. DDX39B is a strong candidate because it has been shown to directly interact with U2AF1 (Shen et al. 2007).
DHX15 and its yeast ortholog Prp43 are ubiquitous nuclear RNA-dependent NTPases with disparate cellular functions and are best characterized as promoting ribosomal RNA biogenesis and spliceosome disassembly (Wen et al. 2008; Wild et al. 2010). DHX15's specificity is regulated by G-patch cofactors (Heininger et al. 2016), with NKRF controlling rRNA biogenesis (Memet et al. 2017) and TFIP11 mediating spliceosome disassembly (Studer et al. 2020). In this manuscript, we define a new role for DHX15 in early spliceosome assembly that may provide higher eukaryotes an additional quality control mechanism to ensure splicing fidelity in the face of divergent branch sequences. Our rationale for quality control stems from the increased accumulation of unproductive A-like complexes on three different substrates, in combination with decreased splicing efficiency for a full-length pre-mRNA (Fig. 2). In recent years, links between recognition of the branch point region and cancer have dramatically increased (Agrawal et al. 2018). For example, hematological malignancies frequently select for specific point mutations in the U2 snRNP-associated proteins SF3B1 and U2AF1 (Hautin et al. 2020). Notably, DHX15 is also often misregulated in blood cancers (Pan et al. 2017; Jing et al. 2018).
Our working model is that DHX15 performs quality control of the initial interaction between U2 snRNP and an intron by disassembling complexes that are incompetent to form a productive branch helix (Fig. 7). It could work in concert with DDX46, which is proposed to directly proofread the branch helix. The yeast ortholog Prp5 releases from A-complex less readily with a mutant versus consensus branch helix, with its release being necessary for continued spliceosome assembly (Liang and Cheng 2015). If we extend this model to mammalian introns, which have more variable branch point sequences, DDX46 is more likely to stall on nonproductive branch helices. As a result, the more complex intronic landscape necessitates a mechanism for disassembly. We suspect that DHX15 fulfills this role through a disassembly mechanism that parallels disruption of U2 snRNA interactions with the intron in the ILS.
FIGURE 7.

Model of DDX- and DHX-enzyme contributions to early spliceosome assembly. Structural models are adapted from PDB entries: 6Y5Q, 7OQE, and 6G90.
If DHX15 destabilizes stalled A-complex, how it is recognized comes into question. So far, DHX15's RNA targets are imposed by the identity of the complex being remodeled. For example, the enzyme promotes ILS disassembly by binding the 3′ end of U6 snRNA (Toroney et al. 2019), although how the branch helix between U2 snRNA and the intron is disrupted remains unclear. A constitutively active DHX15 can also interact with the intron to promote disassembly of Bact spliceosomes (Fourmann et al. 2016). In the context the early spliceosome, DHX15's target is not yet defined. One option is the intron. With the limited real estate of the minimal intron substrate, we were able to narrow down the potential RNA handle to 10 nt downstream from the branch point sequence. U2 snRNA is the other possibility, and we find that the branch binding region of U2 snRNA becomes accessible upon addition of both ATP and GTP. Because DHX15 associates with U2 snRNP, it is a good candidate for mediating the change, which could mimic removal of an intron in either the context of the ILS or A-complex.
Our model also requires a G-patch partner to direct DHX15 to stalled spliceosomes and may further dictate its RNA target. Notably, five different G-patch proteins associate with U2 snRNP and/or A-complex: RBM5, RBM10, RBM17, CHERP, and SUGP1. Of these, SUGP1 binds to SF3B1 to prevent use of cryptic branch points, although its DHX-enzyme partner was not identified (Zhang et al. 2019). Mutations in SUGP1 are also associated with aberrant splicing related to branch point selection in cancer cells that also correlate with SF3B1 mutations (Liu et al. 2020).
MATERIALS AND METHODS
HeLa nuclear extract
Nuclear extract was generated as previously described (Dignam et al. 1983) from HeLa S3 cells grown in DMEM/F-12 1:1 and 5% (v/v) newborn calf serum. HeLa cells containing an integrated transgene for V5-tagged SNRPB2 were generated using the HILO-RMCE cassette and RMCE acceptor cells (generously provided by E. Makeyev) as described in Khandelia et al. (2011). Expression of V5-tagged SNRPB2 was induced with doxycycline 48 h prior to harvest (Kim 2019). Prior to use for the NTP studies, HeLa nuclear extract was either incubated at 30°C for 15 min to cycle residual ATP or passed through a G-50 size exclusion column to deplete residual nucleotides.
T7 run-off RNA transcription
Full-length pre-mRNA sequence is derived from the adenovirus major late (AdML) transcript with uracil substituted at the last four bases of the 5′ exon, and a UACUAAC branch point sequence. Sequences for the A3′ and Amin RNA substrates are provided in Supplemental Figure 4. Double stranded DNA oligos (Eurofins) were generated as templates for PYT truncations. All RNA substrates were generated by T7 run-off transcription reactions supplemented with 32P-UTP. Full-length substrate was capped by G(5′)ppp(5′)G and gel purified. A3′, Amin, and PYT truncation substrates were GMP-capped and purified by a size-exclusion spin column.
Native gel analysis of in vitro splicing reactions
Splicing reactions containing 20 nM RNA substrate, 40 mM potassium glutamate, 2 mM magnesium acetate, 0.05 mg/mL tRNA, 40% (v/v) HeLa nuclear extract, and 2 mM ATP, GTP, AMP–PNP, or no added NTP were incubated at 30°C for 0–15 min. 2× native loading dye (20 mM Tris, 20 mM glycine, 25% glycerol, 0.1% bromophenol blue, 0.1% cyan blue, 1 mg/mL heparin) was added directly to each reaction, which were separated on native gels with 1.9% low melting temp agarose (Invitrogen, 15517-014) in 20 mM Tris, 20 mM glycine for 3 h at 72 volts. Gels were dried on Whatman paper and visualized by phosphorimaging (Amershan Typhoon).
Denaturing gel analysis of in vitro splicing reactions
Splicing reactions were generated as described above, except that pre-mRNA substrate was at 6 nM and incubation time was extended to 45 min. Reactions were stopped with addition of splicing dilution buffer (100 mM Tris pH 7.5, 10 mM EDTA, 1% SDS, 150 mM sodium chloride, and 0.3 M sodium acetate pH 5.2) followed by phenol:chloroform:isoamyl alcohol (25:24:1) extraction and ethanol precipitation. RNA was resuspended in FEB (95% formamide, 20 mM EDTA, 0.01% bromophenol blue and 0.01% cyan blue), separated by 15% urea-PAGE, and visualized by phosphorimaging.
DHX15 depletion
A total of 10 µg DHX15 antibody (Abcam, ab70454), IgG (Epredia, NC748P) or 1× PBS was incubated with protein A beads (NEB, S1425S) with agitation for 1 h at 4°C. HeLa nuclear extract with 500 mM potassium chloride was added to the antibody bound beads and rotated end-over-end for 2 h at 4°C. The depleted nuclear extract was then dialyzed into Buffer E for 5 h (100 mM potassium chloride, 20% glycerol, 20 mM Tris, pH 7.9, 1.5 mM magnesium chloride, 0.5 mM DTT) in small dialysis cups (Thermo Scientific, 69570).
RNase H digestion and primer extension
HeLa nuclear extract was supplemented with 2 mM magnesium acetate and 2 mM ATP, GTP, AMP–PNP, or no NTP. DNA oligos (Eurofins) complementary to the U2 snRNA nt 1–15, nt 12–26, nt 24–38, and nt 32–46 or a control oligo (Supplemental Fig. 5) was added at 5 µM and incubated for 15 min at 30°C to allow for cleavage by endogenous RNase H. The oligonucleotides were degraded by addition of 1 µL RQ1 DNase (Promega, M6101) for 10 min at 30°C. RNA was isolated by phenol:chloroform:isoamyl alcohol (25:24:1) and chloroform:isoamyl alcohol (24:1) extraction, followed by ethanol precipitation and resuspended in water. For primer extension, 10 picomoles of DNA oligonucleotides complementary to either U2 snRNA nt 97–117 or nt 28–42 (U2L15 [Black et al. 1985]) were labeled with γ-32P ATP and purified via Sephadex G-25 (MilliporeSigma Supelco G258010G) column. The isolated RNA and radiolabeled oligonucleotide were annealed by incubation at 95°C for 2 min, 53°C for 5 min, and on ice for 5 min and then added to reverse transcription reactions containing 50 mM Tris pH 7.9, 75 mM potassium chloride, 7 mM DTT, 3 mM magnesium chloride, 1 mM dNTPs, and 0.5 µg reverse transcriptase (MMLV variant). Reactions were incubated for 30 min at 53°C, and the DNA was isolated by the addition of 0.3 M sodium acetate pH 5.2, 0.5 mM EDTA, and 0.05% SDS followed by ethanol precipitation. The labeled DNA was resuspended in FEB and separated on a 9.6% urea:PAGE. The gel was dried on Whatman paper and visualized by phosphorimaging.
Immunoprecipitations
For IP's in Figure 6A,C and D, 1.5 µg of anti-V5 antibody (Invitrogen, R960-25) or IgG (Epredia, NC748P) was added directly to splicing reactions containing 6 nM full-length RNA substrate, 2 mM magnesium acetate, 40 mM potassium glutamate, 0.1 mg/mL tRNA, and 40% V5-SNRPB2 HeLa ±2 mM ATP incubated at 30°C for 10 min. For other co-IP's, 3 µg of anti-V5 antibody (GenScript, A01724) was added to 2 mM magnesium acetate, 20 mM potassium glutamate, 0.1 mg/mL tRNA, and 60% V5-SNRPB2 HeLa nuclear extract ±2 mM ATP, GTP, or AMP–PNP and incubated at 30°C for 10 min. All samples were incubated with antibody for 13.5 h at 4°C with rocking. Samples were then added to protein A magnetic beads (NEB, S1425S) and rotated at 4°C for 4 h. The beads were washed three or more times with IP wash buffer (100 mM Tris pH 7.5, 120 mM potassium chloride, 1 mM EGTA, 0.1% NP-40, or IGEPAL). For western blots, samples were eluted with 0.1 M glycine, pH 2.5 and quenched with equal volume 1 M Tris, pH 7.9. For RNA analysis, samples were eluted with splicing dilution buffer followed by precipitation with phenol:chloroform:isoamyl alcohol (25:24:1) and ethanol precipitation. RNA samples were analyzed by 15% urea-PAGE as described for in vitro splicing reactions.
Western blot analysis
Samples were prepared in 5× Laemmli buffer (62.5 mM Tris, 25% glycerol, 6.25% SDS, 0.1% bromophenol blue, 5% beta-mercaptoethanol) and heated at 95°C for 2 min prior to separation by 10% SDS-PAGE. Gels were transferred to PVDF membrane (Bio-Rad Mini Trans-Blot Cell) and blocked in 1% nonfat milk in 1× Tris-buffered saline with Tween 20 (TBST) for 1 h at room temperature while rocking. The following antibodies were added directly to the blocking buffer at the indicated concentrations and incubated at 4°C overnight while rocking. From Proteintech: DHX15 (12265-1-AP, 1:1000), SNRPB2 (13512-1-AP, 1:2500), and U2AF1 (60289-1-Ig, 1:1000). From Santa Cruz Biotech: SF3A2 (sc-390444, 1:1000) and HDAC1 (sc-7872, 1:5000). Blots were washed three times in 1× TBST, and corresponding LICOR secondaries (1:15000) were added in blocking buffer and rocked at room temperature for 1 h. The blots were again washed three times and then imaged on a LICOR Imaging System. Images were quantified and processed utilizing LICOR Image Studio Lite.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01GM72649 and University of California Cancer Research Coordinating Committee, grant ID CRR-17-421968 to M.S.J.
Author contributions: H.M.N., A.A., J.K., M.M., and M.J. conceived and designed the experiments. H.M.N., A.A., T.S., J.K., M.M., and B.P. performed the experiments. H.M.N., A.A., J.K., and M.J. analyzed the data. H.M.N., A.A., and M.J. wrote the paper.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.078977.121.
MEET THE FIRST AUTHOR
Hannah M. Maul-Newby.

Meet the First Author(s) is a new editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Hannah Maul-Newby is the first author of this paper, “A model for DHX15 mediated disassembly of A-complex spliceosomes.” Hannah is a graduate student in Dr. Melissa Jurica's laboratory in the Center for Molecular Biology of RNA and the Department of Molecular, Cell and Developmental Biology at the University of California, Santa Cruz. Her research focuses on uncovering the role of DHX15 in early spliceosome assembly.
What are the major results described in your paper and how do they impact this branch of the field?
This paper uncovers a new role for the RNA helicase, DHX15, in early spliceosome assembly. DHX15 is traditionally associated with the disassembly of the intron lariat spliceosome at the end of each splicing cycle and ribosome biogenesis. It is also associated with early spliceosome assembly, but does not have a defined function. To understand which helicases may be playing specific roles in early spliceosome assembly, I focused on which steps require ATP or other NTPs. Since there are both DDX and DHX helicases associated with these steps, I focused on ATP and GTP and found that usage of both resulted in robust A-complex assembly. Therefore, I hypothesized that DHX15 must be playing a role in this process as it can utilize any NTP. I depleted the protein and found that complexes accumulate in native assembly assays; however, splicing efficiency decreases. These results led me to further hypothesize that the accumulated complexes may be unproductive and DHX15 may be disassembling these complexes. Through further experimentation, I propose a new model for early A-complex assembly in higher eukaryotes (which have divergent branch sequences) in which DHX15 participates in a quality control step and mediates disassembly of unproductive complexes. This pathway may be a mechanism for the cell to remove deleterious transcripts from being translated and may provide insight into why DHX15 is regularly associated with cancer.
What led you to study RNA or this aspect of RNA science?
Prior to coming to graduate school, I worked in a crystallography laboratory as an undergraduate at Gonzaga University. After I received my bachelor's degree, I worked as a staff research associate at UCLA in Dr. Alison Frand's laboratory, where we studied the molting cycle in C. elegans. At UCLA, the majority of my work centered around FBN-1, a protein that has many isoforms generated through different splicing events between exons 14 and 19. One specific deletion in exon 8 that I worked on results in attenuation over time through what is hypothesized to be alternative splicing. One generation would be near death and the next completely healthy, yet the worms still carried the mutation. I thought that this was so cool! Therefore, I wanted to pursue my PhD in a laboratory that mixed the biochemical aspects that I enjoyed as an undergraduate with the biological aspects I thrived on at UCLA. To that end, I chose to pursue my PhD in a laboratory that studies splicing, in order to learn more about this process at a mechanistic level.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
One of the first experiments I performed was to deplete DHX15 from HeLa nuclear extract and ask what happens. This process proved very difficult because DHX15 is an essential protein that is highly abundant and involved in many different cellular processes. As a result, I was only able to obtain a partial depletion which made interpretation of my data more difficult in comparison to full depletion. Therefore, when I first observed accumulation of assembled complexes and decreased splicing efficiency in partially depleted extracts, I was very surprised as these results contradicted my initial hypothesis that more complexes would yield more splicing. As such, this result made me start to think about the current models of A-complex assembly and how they did not fit my data. After many trials and different experiments, I realized that it was not my data that were inconsistent, but that the field's understanding of complex assembly and disassembly needed to be expanded, which was a really exciting realization and led to the proposed model in the paper.
If you were able to give one piece of advice to your younger self, what would that be?
If I could give my younger self one piece of advice, it would be to unapologetically pursue your passions without fear.
What are your subsequent near- or long-term career plans?
Currently, I am starting the search for a post-doctoral position with a start date sometime in late summer/early fall 2022. My long-term goal is to become a professor in an academic setting whose laboratory focuses on enzymes that disrupt protein/RNA interactions and specifically to decipher what roles these relationships play in human disease.
REFERENCES
- Agafonov DE, Deckert J, Wolf E, Odenwalder P, Bessonov S, Will CL, Urlaub H, Luhrmann R. 2011. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol 31: 2667–2682. 10.1128/MCB.05266-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal AA, Yu L, Smith PG, Buonamici S. 2018. Targeting splicing abnormalities in cancer. Curr Opin Genet Dev 48: 67–74. 10.1016/j.gde.2017.10.010 [DOI] [PubMed] [Google Scholar]
- Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. 2016. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 7: 10615. 10.1038/ncomms10615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K, Moore MJ. 2000. Bimolecular exon ligation by the human spliceosome bypasses early 3′ splice site AG recognition and requires NTP hydrolysis. RNA 6: 16–25. 10.1017/s1355838200001862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessonov S, Anokhina M, Krasauskas A, Golas MM, Sander B, Will CL, Urlaub H, Stark H, Luhrmann R. 2010. Characterization of purified human Bact spliceosomal complexes reveals compositional and morphological changes during spliceosome activation and first step catalysis. RNA 16: 2384–2403. 10.1261/rna.2456210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL, Chabot B, Steitz JA. 1985. U2 as well as U1 small nuclear ribonucleoproteins are involved in premessenger RNA splicing. Cell 42: 737–750. 10.1016/0092-8674(85)90270-3 [DOI] [PubMed] [Google Scholar]
- Bonnal SC, López-Oreja I, Valcárcel J. 2020. Roles and mechanisms of alternative splicing in cancer—implications for care. Nat Rev Clin Oncol 17: 457–474. 10.1038/s41571-020-0350-x10.1101/542506 [DOI] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. 2015. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep 13: 1033–1045. 10.1016/j.celrep.2015.09.053 [DOI] [PubMed] [Google Scholar]
- DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. 2015. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol 11: e1004105. 10.1371/journal.pcbi.1004105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RD. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11: 1475–1489. 10.1093/nar/11.5.1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. 2016. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 16: 413–430. 10.1038/nrc.2016.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckner J, Zhang M, Valcarcel J, Green MR. 1997. U2AF65 recruits a novel human DEAD box protein required for the U2 snRNP-branchpoint interaction. Genes Dev 11: 1864–1872. 10.1101/gad.11.14.1864 [DOI] [PubMed] [Google Scholar]
- Fourmann JB, Dybkov O, Agafonov DE, Tauchert MJ, Urlaub H, Ficner R, Fabrizio P, Lührmann R. 2016. The target of the DEAH-box NTP triphosphatase Prp43 in Saccharomyces cerevisiae spliceosomes is the U2 snRNP-intron interaction. Elife 5: e15564. 10.7554/eLife.15564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman B, Tijerina P, Russell R. 2017. Distinct RNA-unwinding mechanisms of DEAD-box and DEAH-box RNA helicase proteins in remodeling structured RNAs and RNPs. Biochem Soc Trans 45: 1313–1321. 10.1042/BST20170095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann F, Enders M, Ficner R. 2019. Structural basis for RNA translocation by DEAH-box ATPases. Nucleic Acids Res 47: 4349–4362. 10.1093/nar/gkz150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautin M, Mornet C, Chauveau A, Bernard D, Corcos L, Lippert E. 2020. Splicing anomalies in myeloproliferative neoplasms: paving the way for new therapeutic venues. Cancers (Basel) 12: E2216. 10.3390/cancers12082216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Andersen GR, Nielsen KH. 2010. Structural basis for the function of DEAH helicases. EMBO Rep 11: 180–186. 10.1038/embor.2010.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heininger AU, Hackert P, Andreou AZ, Boon KL, Memet I, Prior M, Clancy A, Schmidt B, Urlaub H, Schleiff E, et al. 2016. Protein cofactor competition regulates the action of a multifunctional RNA helicase in different pathways. RNA Biol 13: 320–330. 10.1080/15476286.2016.1142038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky E. 2011. RNA helicases at work: binding and rearranging. Trends Biochem Sci 36: 19–29. 10.1016/j.tibs.2010.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Y, Nguyen MM, Wang D, Pascal LE, Guo W, Xu Y, Ai J, Deng FM, Masoodi KZ, Yu X, et al. 2018. DHX15 promotes prostate cancer progression by stimulating Siah2-mediated ubiquitination of androgen receptor. Oncogene 37: 638–650. 10.1038/onc.2017.371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesarwani AK, Ramirez O, Gupta AK, Yang X, Murthy T, Minella AC, Pillai MM. 2017. Cancer-associated SF3B1 mutants recognize otherwise inaccessible cryptic 3′ splice sites within RNA secondary structures. Oncogene 36: 1123–1133. 10.1038/onc.2016.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelia P, Yap K, Makeyev EV. 2011. Streamlined platform for short hairpin RNA interference and transgenesis in cultured mammalian cells. Proc Natl Acad Sci 108: 12799–12804. 10.1073/pnas.1103532108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. 2019. A pipeline for tagging snRNP associated proteins in HeLa. University of California, Santa Cruz, CA. [Google Scholar]
- Konarska MM, Sharp PA. 1986. Electrophoretic separation of complexes involved in the splicing of precursors to mRNAs. Cell 46: 845–855. 10.1016/0092-8674(86)90066-8 [DOI] [PubMed] [Google Scholar]
- Koodathingal P, Staley JP. 2013. Splicing fidelity: DEAD/H-box ATPases as molecular clocks. RNA Biol 10: 1073–1079. 10.4161/rna.25245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koodathingal P, Novak T, Piccirilli JA, Staley JP. 2010. The DEAH box ATPases Prp16 and Prp43 cooperate to proofread 5' splice site cleavage during pre-mRNA splicing. Mol Cell 39: 385–395. 10.1016/j.molcel.2010.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WW, Cheng SC. 2015. A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev 29: 81–93. 10.1101/gad.253708.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhang J, Sun Y, Perea-Chamblee TE, Manley JL, Rabadan R. 2020. Pan-cancer analysis identifies mutations in SUGP1 that recapitulate mutant SF3B1 splicing dysregulation. Proc Natl Acad Sci 117: 10305–10312. 10.1073/pnas.1922622117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallam AL, Del Campo M, Gilman B, Sidote DJ, Lambowitz AM. 2012. Structural basis for RNA-duplex recognition and unwinding by the DEAD-box helicase Mss116p. Nature 490: 121–125. 10.1038/nature11402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayas RM, Maita H, Staley JP. 2006. Exon ligation is proofread by the DExD/H-box ATPase Prp22p. Nat Struct Mol Biol 13: 482–490. 10.1038/nsmb1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memet I, Doebele C, Sloan KE, Bohnsack MT. 2017. The G-patch protein NF-κB-repressing factor mediates the recruitment of the exonuclease XRN2 and activation of the RNA helicase DHX15 in human ribosome biogenesis. Nucleic Acids Res 45: 5359–5374. 10.1093/nar/gkx013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newnham CM, Query CC. 2001. The ATP requirement for U2 snRNP addition is linked to the pre-mRNA region 5′ to the branch site. RNA 7: 1298–1309. 10.1017/S1355838201010561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Day CL, Dalbadie-McFarland G, Abelson J. 1996. The Saccharomyces cerevisiae Prp5 protein has RNA-dependent ATPase activity with specificity for U2 small nuclear RNA. J Biol Chem 271: 33261–33267. 10.1074/jbc.271.52.33261 [DOI] [PubMed] [Google Scholar]
- Pan L, Li Y, Zhang HY, Zheng Y, Liu XL, Hu Z, Wang Y, Wang J, Cai YH, Liu Q, et al. 2017. DHX15 is associated with poor prognosis in acute myeloid leukemia (AML) and regulates cell apoptosis via the NF-kB signaling pathway. Oncotarget 8: 89643–89654. 10.18632/oncotarget.20288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perriman R, Ares M Jr. 2010. Invariant U2 snRNA nucleotides form a stem loop to recognize the intron early in splicing. Mol Cell 38: 416–427. 10.1016/j.molcel.2010.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach KJ, Uhlenbeck OC. 2002. Cooperative binding of ATP and RNA substrates to the DEAD/H protein DbpA. Biochemistry 41: 3693–3702. 10.1021/bi012062n [DOI] [PubMed] [Google Scholar]
- Query CC, McCaw PS, Sharp PA. 1997. A minimal spliceosomal complex A recognizes the branch site and polypyrimidine tract. Mol Cell Biol 17: 2944–2953. 10.1128/MCB.17.5.2944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert-Paganin J, Halladjian M, Blaud M, Lebaron S, Delbos L, Chardon F, Capeyrou R, Humbert O, Henry Y, Henras AK, et al. 2017. Functional link between DEAH/RHA helicase Prp43 activation and ATP base binding. Nucleic Acids Res 45: 1539–1552. 10.1093/nar/gkw1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. 2006. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell 125: 287–300. 10.1016/j.cell.2006.01.054 [DOI] [PubMed] [Google Scholar]
- Shen J, Zhang L, Zhao R. 2007. Biochemical characterization of the ATPase and helicase activity of UAP56, an essential pre-mRNA splicing and mRNA export factor. J Biol Chem 282: 22544–22550. 10.1074/jbc.M702304200 [DOI] [PubMed] [Google Scholar]
- Studer MK, Ivanović L, Weber ME, Marti S, Jonas S. 2020. Structural basis for DEAH-helicase activation by G-patch proteins. Proc Natl Acad Sci 117: 7159–7170. 10.1073/pnas.1913880117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner NK, Cordin O, Banroques J, Doère M, Linder P. 2003. The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol Cell 11: 127–138. 10.1016/s1097-2765(03)00006-6 [DOI] [PubMed] [Google Scholar]
- Tauchert MJ, Fourmann JB, Lührmann R, Ficner R. 2017. Structural insights into the mechanism of the DEAH-box RNA helicase Prp43. Elife 6: e21510. 10.7554/eLife.21510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toroney R, Nielsen KH, Staley JP. 2019. Termination of pre-mRNA splicing requires that the ATPase and RNA unwindase Prp43p acts on the catalytic snRNA U6. Genes Dev 33: 1555–1574. 10.1101/gad.328294.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann-Schiffler H, Jalal C, Stahl H. 2006. Ddx42p—a human DEAD box protein with RNA chaperone activities. Nucleic Acids Res 34: 10–22. 10.1093/nar/gkj403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa T, Guthrie C. 2005. The Isy1p component of the NineTeen Complex interacts with the ATPase Prp16p to regulate the fidelity of pre-mRNA splicing. Genes Dev 19: 1894–1904. 10.1101/gad.1336305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walbott H, Mouffok S, Capeyrou R, Lebaron S, Humbert O, van Tilbeurgh H, Henry Y, Leulliot N. 2010. Prp43p contains a processive helicase structural architecture with a specific regulatory domain. EMBO J 29: 2194–2204. 10.1038/emboj.2010.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S, Hergert S, Yin S, Freeman SS, Levin JZ, et al. 2016. Transcriptomic characterization of SF3B1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Cancer Cell 30: 750–763. 10.1016/j.ccell.2016.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tannukit S, Paine ML. 2008. TFIP11 interacts with mDEAH9, an RNA helicase involved in spliceosome disassembly. Int J Mol Sci 9: 2105–2113. 10.3390/ijms9112105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild T, Horvath P, Wyler E, Widmann B, Badertscher L, Zemp I, Kozak K, Csucs G, Lund E, Kutay U. 2010. A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol 8: e1000522. 10.1371/journal.pbio.1000522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Urlaub H, Achsel T, Gentzel M, Wilm M, Luhrmann R. 2002. Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J 21: 4978–4988. 10.1093/emboj/cdf480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YZ, Query CC. 2007. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol Cell 28: 838–849. 10.1016/j.molcel.2007.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Jankowsky E. 2006. The DEAD-box protein Ded1 unwinds RNA duplexes by a mode distinct from translocating helicases. Nat Struct Mol Biol 13: 981–986. 10.1038/nsmb1165 [DOI] [PubMed] [Google Scholar]
- Yang Q, Del Campo M, Lambowitz AM, Jankowsky E. 2007. DEAD-box proteins unwind duplexes by local strand separation. Mol Cell 28: 253–263. 10.1016/j.molcel.2007.08.016 [DOI] [PubMed] [Google Scholar]
- Yang F, Wang XY, Zhang ZM, Pu J, Fan YJ, Zhou J, Query CC, Xu YZ. 2013. Splicing proofreading at 5′ splice sites by ATPase Prp28p. Nucleic Acids Res 41: 4660–4670. 10.1093/nar/gkt149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ali AM, Lieu YK, Liu Z, Gao J, Rabadan R, Raza A, Mukherjee S, Manley JL. 2019. Disease-causing mutations in SF3B1 alter splicing by disrupting interaction with SUGP1. Mol Cell 76: 82–95.e7. 10.1016/j.molcel.2019.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Will CL, Bertram K, Dybkov O, Hartmuth K, Agafonov DE, Hofele R, Urlaub H, Kastner B, Lührmann R, et al. 2020. Molecular architecture of the human 17S U2 snRNP. Nature 583: 310–313. 10.1038/s41586-020-2344-3 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Rigo N, Dybkov O, Fourmann JB, Will CL, Kumar V, Urlaub H, Stark H, Lührmann R. 2021. Structural insights into how Prp5 proofreads the pre-mRNA branch site. Nature 596: 296–300. 10.1038/s41586-021-03789-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.