

Abstract
Bimolecular homolytic substitution (SH2) is an open-shell mechanism that is implicated across a host of biochemical alkylation pathways. Surprisingly, however, this radical substitution manifold has not been generally deployed as a design element in synthetic C–C bond formation. Here, we demonstrate that the SH2 mechanism can be leveraged to enable a biomimetic sp3-sp3 cross-coupling platform that furnishes quaternary sp3-carbon centers, a longstanding challenge in organic molecule construction. This heteroselective radical-radical coupling combines the capacity of iron porphyrin to readily distinguish between the SH2 bond-forming roles of open-shell primary and tertiary carbons, and photocatalysis to generate both radical classes simultaneously from widely abundant functional groups. Mechanistic studies confirm the intermediacy of a primary alkyl–Fe(III) species prior to coupling and provide evidence for the SH2 displacement pathway in the critical quaternary sp3-carbon bond formation step.
Over the past five decades, transition metal-catalyzed cross-coupling has comprehensively transformed the landscape of molecule construction in the applied sciences, especially with respect to pharmaceuticals, agrochemicals and functional materials (1, 2). In particular, the combination of three mechanistic steps—oxidative addition, transmetalation, and reductive elimination—has served as a robust catalytic paradigm for C–C bond formation, enabling a highly modular, yet general approach to fragment coupling (Fig. 1A). While this paradigm has proven to be exceptionally successful for forging C(sp2)–C(sp2) bonds, it is important to recognize that each of these three elementary steps is less efficient when transition metals engage with secondary or tertiary alkyl fragments, limiting the development of a C(sp3)–C(sp3) cross-coupling platform of broad utility (3–6).
Fig. 1. Biomimetic C(sp3)–C(sp3) cross-coupling via dual iron/photoredox catalysis.
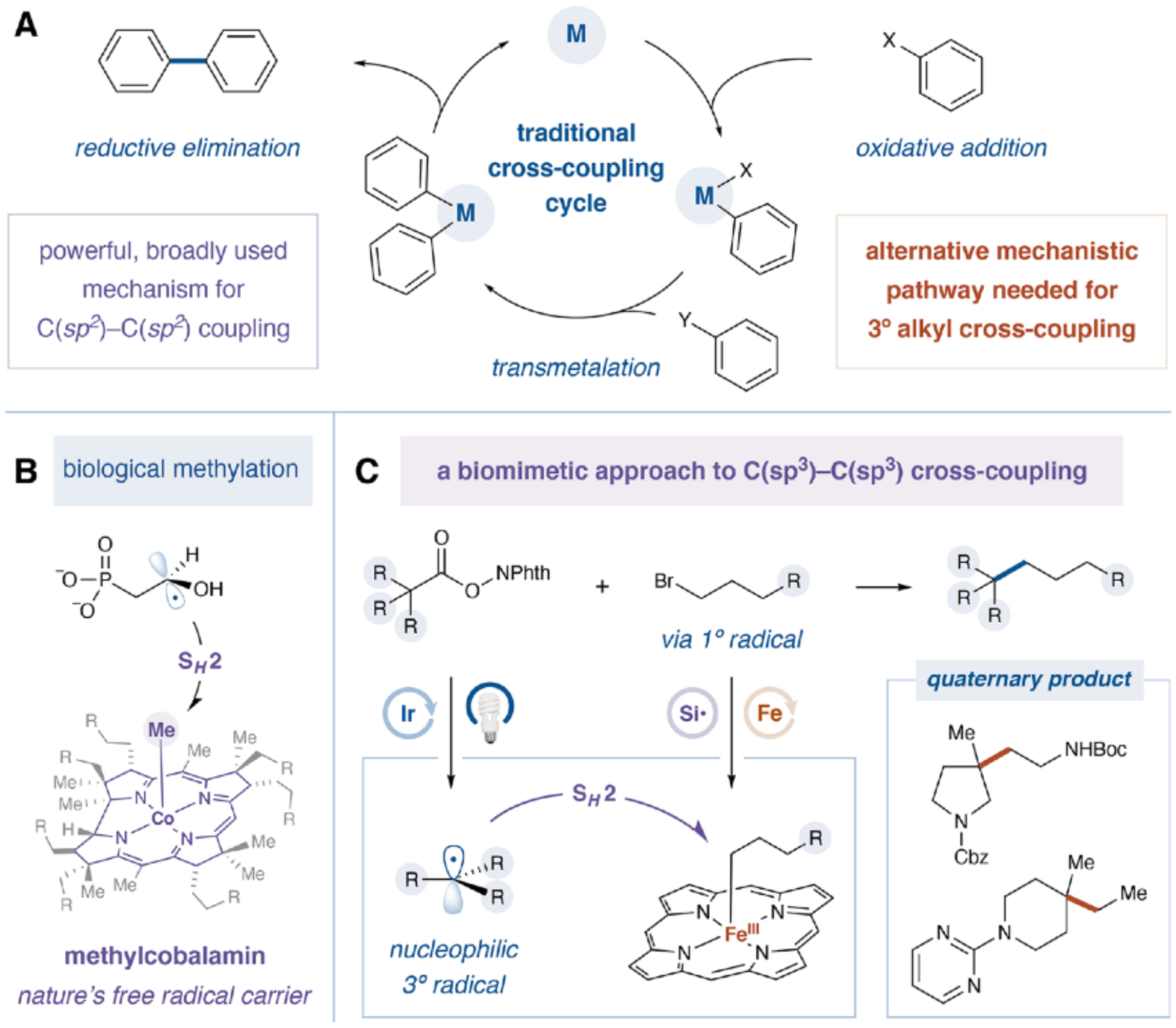
R, alkyl group; Phth, phthalimide; Boc, tert-butoxycarbonyl group; Cbz, benzyloxycarbonyl group.
It is intriguing to consider that enzymatic formation of C(sp3)–C(sp3) bonds proceeds by fundamentally different open-shell pathways to achieve pivotal alkylation reactions (7, 8). As one canonical example, methylcobalamin systems serve as nature’s “free radical carrier” by stabilizing otherwise highly reactive methyl radicals (9, 10). As such, in cobalamin-dependent radical SAM methyltransferases, transiently-generated carbon-centered radicals can react with these alkyl–cobalt complexes via bimolecular homolytic substitution (SH2) (Fig. 1B) (11). Although cobalamin provides critical stabilization to the reactive methyl radical, the methyl-cobalt bond remains notably weak (bond dissociation energy (BDE) = ~37 kcal/mol), which underpins the kinetic preference for the SH2 mechanism and heteroselective carbon-carbon bond formation (12). Elegant biosynthetic studies have shown that the rates of such enzymatic SH2 reactions are extremely fast (~ 108 s−1) and enable the formation of sterically congested quaternary C(sp3) centers (11). However, despite broad biochemical relevance, SH2-based cross-coupling paradigms remain effectively unknown within the laboratory setting outside of stoichiometric organonickel methylation or intramolecular SH-cyclizations from seminal contributions of Sanford, Zhang and others (13–18). Indeed, as Johnson stated in 1983 with respect to C–C bond formation, the SH2 mechanism is “seldom postulated, rarely discussed, and frequently discarded as improbable” (19).
We recently questioned if a homolytic SH2 pathway in combination with photoredox catalysis might be exploited to render an alternative catalysis paradigm for C(sp3)–C(sp3) bond formation (Fig. 1C). Previous bioinorganic studies have demonstrated that both cobalt and iron porphyrins can serve as model systems of cobalamin, given that their respective alkyl-metal complexes possess weak metal-carbon bonds (20, 21). These metalloporphyrins capture and release alkyl radicals reversibly and the equilibrium is governed by the well-established BDE of the metal-carbon bond (22). With this in mind, we recognized that such metalloporphyrin catalysts might effectively partition the roles of primary and tertiary radicals in a cross-coupling SH2 reaction (Fig. 2A). More specifically, electron-rich tertiary radical 3 should be favored to induce SH2 displacement of the primary alkyl fragment from 1° alkyl–Fe porphyrin 5 to generate hetero-coupled C(sp3)–C(sp3) tertiary-primary linkages. However, the same 1° alkyl–Fe porphyrin 5 would be less susceptible to displacement by primary alkyl radical 2 given the reduced SOMO-nucleophilicity of primary radicals (23), a feature that should suppress the formation of 1°-1° homocoupled dimers. At the same time, 3° alkyl-metal porphyrin complex 7 is not formed in measurable equilibrium concentrations at room temperature (24), and its SH2 displacement with other radicals (1°, 2°, or 3°) is kinetically slow toward due to induced non-bonding interactions (i.e., the pyramidalization of the 3° alkyl–Fe(III) intermediate) (25). As such, we postulated that the simultaneous generation of both primary and tertiary alkyl radicals in the presence of Fe-porphyrin complexes should lead to heteroselective C(sp3)–C(sp3) bond formation in lieu of a statistical combination of open-shell processes.
Fig. 2. Reaction design and development.

(A) Proposed mechanism for the metallaphotoredox sp3–sp3 cross-coupling using iron porphyrin. (B) Representative reaction scheme. t-Bu, tert-butyl group; Me, methyl group; Et, ethyl group.
Traditionally, alkyl-Fe or -Co systems are generated using Grignard reagents for alkyl transfer or via SN2 pathways between low-valent metal porphyrins and alkyl halides, a viable yet relatively slow substitution step (k = ~102 s−1 for iron) (26). Furthermore, these alkyl-metal complexes are often heat- and oxygen-sensitive, restricting the options for open-shell alkyl nucleophile generation. As part of our design strategy, we recognized that photoredox catalysis should allow simultaneous generation of both primary and tertiary open-shell intermediates from widely abundant functional groups under mild conditions. For this first study, we selected a silyl radical-mediated halogen abstraction-radical capture (HARC) strategy (27–29) for the facile oxidative generation of alkyl radicals from primary alkyl bromides, while access to electron-rich tertiary radicals from redox-active esters (readily derived from carboxylic acids) via reduction would ensure a net redox-neutral pathway (30).
It has long been recognized within medicinal chemistry that cyclic, quaternary centers are conformationally restricted, a structural feature that is often linked to superior potency and metabolic stability in drug candidates (31, 32). However, only a limited number of sp3–sp3 cross-coupling reports to date involve the formation of all aliphatic quaternary carbons, and these methods typically rely on highly reactive tertiary Grignard reagents or alkyl iodide electrophiles (33–36). We felt that the use of readily available redox-active esters and alkyl bromides as modular coupling fragments, in conjunction with the capacity of SH2 for mechanistic partitioning, should lead to a generically useful C(sp3)–C(sp3) cross-coupling method, thereby expanding the chemical space of sp3-rich scaffolds that can be readily explored by medicinal chemists (37).
A description of our proposed mechanism for cross-coupling is outlined in Fig. 2A (see fig. S1 for a detailed proposal). Upon visible light excitation, the photocatalyst [Ir(FMeppy)2(dtbbpy)][PF6] [FMeppy = 2-(4-fluorophenyl)-5-(methyl)pyridine; dtbbpy = 4,4’-di-tert-butyl-2,2’-bipyridine)] (11) would access a long-lived triplet excited state species (lifetime τ = 1.1 μs) (38). This oxidizing Ir complex [E1/2red (*IrIII/IrII) = +0.77 V versus saturated calomel electrode (SCE) in CH3CN] can undergo single electron transfer (SET) with the aminosilane reagent (Epox = +0.86 V versus SCE in DMA/tert-amyl alcohol) to generate a reduced Ir(II) complex (39). The oxidized silane reagent would generate a reactive silyl radical, which readily abstracts a bromine atom from alkyl bromide 1 (39). The resulting primary alkyl radical 2 is expected to be captured by the Fe(II) porphyrin catalyst 6 at near diffusion-controlled rates to furnish 1° alkyl–Fe(III) intermediate 5 (40). Concurrently, the reduced Ir(II) complex [E1/2red(IrIII/IrII) = −0.94 V versus SCE in CH3CN) can reduce redox-active ester 4 via SET to furnish tertiary radical 3 upon extrusion of carbon dioxide and phthalimide (30). This matched combination of tertiary radical 3 with 1° alkyl–Fe(III) radicalphile 5 would lead to a successful SH2 reaction, affording cross-coupled product and regenerating the Fe(II) catalyst.
With this mechanistic proposal in mind, we examined the cross-coupling between tertiary redox-active ester 8 and primary alkyl bromide 9, both of which were selected on the basis of medicinal chemistry relevance (Fig. 2B). To our delight, we identified that the commercial complex Fe(OEP)Cl [OEP = 2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphine] as an effective SH2 catalyst, in tandem with photocatalyst 11 and the aminosilane reagent (TMS)3SiNHAdm to deliver the quaternary carbon-bearing alkylation adduct 10 in 70% yield upon blue light irradiation. Control experiments revealed that all of the employed components were necessary for optimal reaction performance and, importantly, without Fe(OEP)Cl only 13% yield of the desired product was observed, a result of free-radical background coupling (fig. S2). Initial kinetic studies revealed that the reaction is 0th order in both of the fragment coupling substrates and 1st order in photocatalyst and light intensity (see supplementary materials). However, an intriguing inverse order in the SH2 catalyst, Fe(OEP)Cl, was observed. We subsequently determined that the iron porphyrin catalyst acts as an optical filter due to strong absorbance at 450 nm, thereby decreasing the photonic power available for the photoredox cycle in a reciprocal relationship to the concentration of the SH2 catalyst. Indeed, with this information in hand, we recognized that similar levels of reaction efficiency should be achieved when the Fe(OEP)Cl loading is decreased (2 mol%) in proportion to light intensity, a hypothesis that was readily substantiated (fig. S4). Importantly, the use of lower Fe porphyrin loadings allows for this coupling protocol to be scaled without loss in efficiency, a useful insight especially when a lower light intensity apparatus is employed.
With optimal conditions in hand, we next examined the generality of our cross-coupling protocol with respect to the carboxylic acid component (Fig. 3). Bulky α-substitutions on pyrrolidine such as isopropyl and benzyl groups were well-tolerated to furnish tertiary amine-bearing cross-coupled adducts in excellent yield (15 and 16, 71% and 80% yield), underscoring the capacity of the SH2 mechanism to generally construct sterically-hindered centers. Redox-active esters containing electron-deficient backbones (i.e., azetidine and difluoropyrrolidine) were found to be viable coupling partners and underwent alkylation in good yields (17 to 19, 46% to 69% yield). In addition, bulky α-functionalized exocyclic or acyclic amines could be accessed via cross-coupling in good efficiencies (20 and 21, 64% and 51% yield). Furthermore, the use of tertiary redox-active esters enabled the formation of quaternary carbons, as represented in the acyclic tert-butyl moiety and a cyclic β-substituted pyrrolidine, as well as a medicinally important cyclic sulfone (22 to 24, 47% to 63% yield) (41). For α-oxy esters, radicals generated adjacent to both phenoxy and methoxy substituents can participate in cross-coupling with respectable efficiencies, providing a new entry to the synthesis of hindered ethers (25 and 26, 50% and 68% yield). Finally, secondary redox-active esters can also be used in our metallaphotoredox protocol to couple with primary bromides in good yields (27 to 29, 50% to 65% yield). On the other hand, tertiary benzylic radical was found to be a challenging SH2 reaction partner, presumably due to the diminished radical nucleophilicity (for additional examples and limitations of substrate scope, see fig. S12).
Fig. 3. Photoredox and iron-catalyzed C(sp3)–C(sp3) cross-coupling: Redox-active ester and alkyl bromide scope.
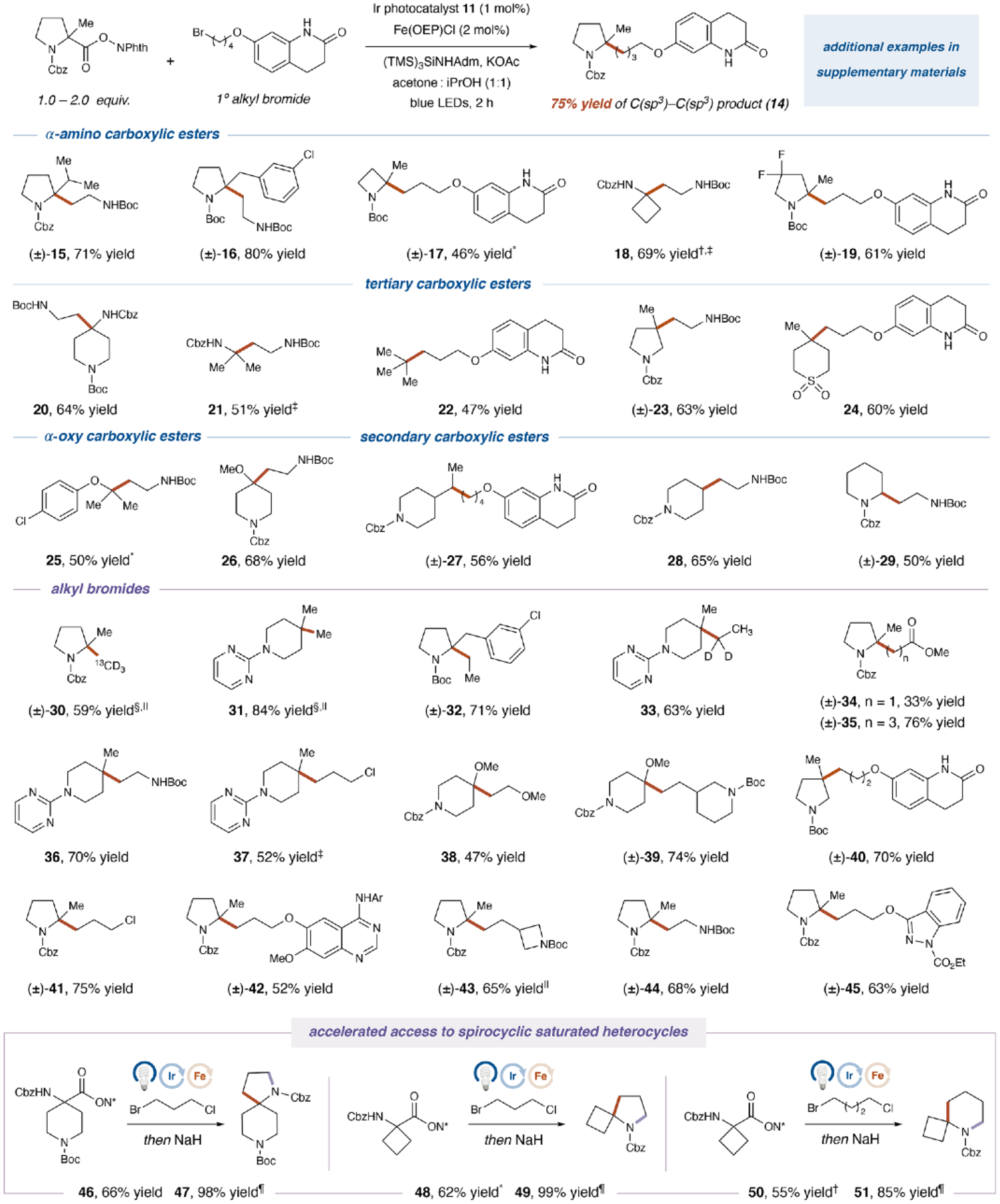
All yields are isolated. See supplementary materials for detailed reaction conditions. TMS, trimethylsilyl group; Adm, 1-adamantyl; KOAc, potassium acetate; iPrOH, isopropanol; Ar, 3-chloro-4-fluorophenyl; N*, phthalimide. *With KOAc and Zn(OAc)2 as bases. †With Zn(OAc)2 as the base. ‡With Ir(ppy)2(dtbbpy)PF6 as the photocatalyst. §With 2 equivalents of methyl p-toluenesulfonate and tetrabutylammonium bromide. ‖With Ir[dF(CF3)ppy]2(dtbbpy)PF6 as the photocatalyst. ¶Sodium hydride, 60°C.
Next, we examined the scope of alkyl bromides using both α-heteroatom and tertiary redox-active esters as the representative coupling partners. Small alkyl fragments such as methyl and ethyl can be introduced into sp3-scaffolds in high efficiencies (30 to 33, 59% to 84% yield). Methyl bromide was generated in-situ via the combination of methyl tosylate and tetrabutylammonium bromide, whereas the use of methyl iodide led to diminished reactivity. Importantly, no isomerization was observed in the alkylated product 33 when 1-bromoethane-1,1-d2 was subjected to our reaction, demonstrating the orthogonality of the SH2 mechanism to reductive elimination for C–C bond formation. Furthermore, 2-methylproline-derived redox-active esters could be alkylated with α- and γ-haloesters, providing straightforward access to homologated amino acids inaccessible via conjugate addition (34 and 35, 33% and 76% yield). A wide variety of functionalized alkyl bromides were successful coupling partners and furnished value-added products in good to high yields (36 to 45, 47% to 75% yield). The core heterocyclic fragments in aripiprazole (40), gefitinib (42) and benzydamine (45) were well-tolerated in the cross-coupling, demonstrating the applicability of our method to medicinal chemistry campaigns. Finally, we employed 1,3- and 1,4-bromo, chloro-alkyls as bifunctional linkers which, after decarboxylative coupling, readily underwent intramolecular cyclization to directly construct medicinally-relevant, spirocyclic structures. This formal decarboxylative cycloaddition strategy was successfully applied to the synthesis of [5.6], [4.5], and [4.6] ring systems (47, 49 and 51), providing a new and general approach to these synthetically challenging heterocycles from simple starting materials (42).
Finally, we performed detailed mechanistic experiments to support the proposed catalytic cycle and the intermediacy of 1° alkyl–Fe(III) species (Fig. 4 and fig. S9). Fluorescence quenching experiments confirmed the reductive quenching of the excited iridium photocatalyst 12 by aminosilane reagent 52 at a near diffusion-controlled rate (k = 6.7 × 108 M−1 s−1), whereas the α-amino redox-active ester 53 was not found to be an effective quencher (fig. S9). In order to probe the intermediacy of the proposed alkyl–Fe(III) species, we employed photoNMR techniques to monitor the cross-coupling between 1-bromobutane and α-amino redox-active ester 53 under our standard reaction conditions (Fig. 4A). By comparing these in situ spectra with an independently prepared n-Bu–Fe(OEP) complex (43), we directly observed the formation of the alkyl–iron porphyrin intermediate, and the concentration of this species was observed to slowly increase upon light exposure and persist throughout the reaction. Furthermore, to demonstrate the catalytic relevance of the observed alkyl–Fe(III) species, we investigated the use of 10 mol% of the previously isolated n-Bu–Fe(OEP) adduct as both a catalytic intermediate and precatalyst in the cross-coupling of redox-active ester 53 and primary bromide 55 (Fig. 4B). Gratifyingly, the desired product 14 was observed in 64% yield, similar to the efficiencies observed when Fe(OEP)Cl was used as the SH2 precatalyst, and notably, the n-butyl group was also incorporated into the alkylated product (54), providing direct evidence for the participation of the Fe(III)–alkyl species in the cross-coupling reaction. Finally, given that the alkyl–Fe bonds of porphyrin complexes are known to homolyze under light irradiation to release alkyl radicals, it was unclear if the carbon–carbon formation proceeds through free radical-radical coupling or the proposed SH2 pathway. We sought to determine if light is required in the C–C bond formation (Fig. 4C). When the independently generated n-Bu–Fe(OEP) complex was subjected to an α-amino radical arising from redox-active ester 53 under non-photonic conditions (i.e., using zinc as the single-electron reductant) (44), the corresponding alkylation product was observed in good yield, indicating that the C–C bond formation is not dependent upon photoexcitation of the SH2 catalyst. Additionally, performing the same experiment under blue light irradiation led to the identical level of product formation, a result which aligns with the mechanistic interpretation that blue light is only required for the photoredox cycle, yet is not involved in the C–C bond formation step. Furthermore, the iron porphyrin catalyst was able to achieve a degree of diastereocontrol during the cross-coupling of a β-chiral alkyl bromide 56 with redox-active ester 53 (Fig. 4D). While free radical coupling without iron led to unselective diastereomer formation (1 to 1.1 d.r.), the addition of iron porphyrin catalyst favored one major diastereomer in 3.2 to 1 diastereocontrol, providing further evidence for a concerted SH2 mechanism and the participation of the iron-bound alkyl complex in the critical C–C bond forming event.
Fig. 4. Mechanistic studies for the proposed catalytic cycle and evidence of the intermediacy of alkyl–Fe(III) species.

n-Bu, n-butyl group; ZnCl2, zinc chloride; d.r., diastereomeric ratio.
Supplementary Material
ACKNOWLEDGMENTS
Funding:
Research reported in this publication was supported by the NIH National Institute of General Medical Sciences (R35 GM134897-02) and gifts from Janssen, Merck, Bristol Myers Squibb, GenMab, and Pfizer. C.A.G. thanks the Beckman Foundation for a postdoctoral fellowship.
Footnotes
Competing interests: D.W.C.M. declares a financial interest with respect to the Integrated Photoreactor.
Data and materials availability:
Data are available in the supplementary materials.
REFERENCES AND NOTES
- 1.De Meijere A, Diederich F, Metal-Catalyzed Cross-Coupling Reactions, Vol. 1 (Wiley-VCH, 2004). [Google Scholar]
- 2.Uehling MR, King RP, Krska SW, Cernak T, Buchwald SL, Pharmaceutical diversification via palladium oxidative addition complexes. Science 363, 405–408 (2019). doi: 10.1126/science.aac6153 [DOI] [PubMed] [Google Scholar]
- 3.Choi J, Fu GC, Transition metal-catalyzed alkyl-alkyl bond formation: Another dimension in cross-coupling chemistry. Science 356, eaaf7230 (2017). doi: 10.1126/science.aaf7230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tasker SZ, Standley EA, Jamison TF, Recent advances in homogeneous nickel catalysis. Nature 509, 299–309 (2014). doi: 10.1038/nature13274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS, A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science 352, 801–805 (2016). doi: 10.1126/science.aaf6123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnston CP, Smith RT, Allmendinger S, MacMillan DWC, Metallaphotoredox-catalysed sp3–sp3 cross-coupling of carboxylic acids with alkyl halides. Nature 536, 322–325 (2016). doi: 10.1038/nature19056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Q, van der Donk WA, Liu W, Radical-mediated enzymatic methylation: A tale of two SAMS. Acc. Chem. Res 45, 555–564 (2012). doi: 10.1021/ar200202c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauerle MR, Schwalm EL, Booker SJ, Mechanistic diversity of radical Sadenosylmethionine (SAM)-dependent methylation. J. Biol. Chem 290, 3995–4002 (2015). doi: 10.1074/jbc.R114.607044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halpern J, Mechanisms of coenzyme B12-dependent rearrangements. Science 227, 869–875 (1985). doi: 10.1126/science.2857503 [DOI] [PubMed] [Google Scholar]
- 10.Daikh BE, Finke RG, The persistent radical effect: A prototype example of extreme, 105 to 1, product selectivity in a free-radical reaction involving persistent. cntdot.Coll[macrocycle] and alkyl free radicals. J. Am. Chem. Soc 114, 2938–2943 (1992). doi: 10.1021/ja00034a028 [DOI] [Google Scholar]
- 11.Wang Y, Begley TP, Mechanistic Studies on CysS - A Vitamin B12-Dependent Radical SAM Methyltransferase Involved in the Biosynthesis of the tert-Butyl Group of Cystobactamid. J. Am. Chem. Soc 142, 9944–9954 (2020). doi: 10.1021/jacs.9b06454 [DOI] [PubMed] [Google Scholar]
- 12.Martin BD, Finke RG, Methylcobalamin’s full- vs. “half”-strength cobalt-carbon sigma bonds and bond dissociation enthalpies: A >10(15) Co-CH3 homolysis rate enhancement following one-antibonding-electron reduction of methlycobalamin. J. Am. Chem. Soc 114, 585–592 (1992). doi: 10.1021/ja00028a027 [DOI] [PubMed] [Google Scholar]
- 13.Bour JR, Ferguson DM, McClain EJ, Kampf JW, Sanford MS, Connecting Organometallic Ni(III) and Ni(IV): Reactions of Carbon-Centered Radicals with High-Valent Organonickel Complexes. J. Am. Chem. Soc 141, 8914–8920 (2019). doi: 10.1021/jacs.9b02411 [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Wen X, Cui X, Zhang XP, Enantioselective Radical Cyclization for Construction of 5-Membered Ring Structures by Metalloradical C-H Alkylation. J. Am. Chem. Soc 140, 4792–4796 (2018). doi: 10.1021/jacs.8b01662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Ke J, Zhu Y, Deb A, Xu Y, Zhang XP, Asymmetric Radical Process for General Synthesis of Chiral Heteroaryl Cyclopropanes. J. Am. Chem. Soc 143, 11121–11129 (2021). doi: 10.1021/jacs.1c04655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie J, Xu P, Zhu Y, Wang J, Lee WC, Zhang XP, New Catalytic Radical Process Involving 1,4-Hydrogen Atom Abstraction: Asymmetric Construction of Cyclobutanones. J. Am. Chem. Soc 143, 11670–11678 (2021). doi: 10.1021/jacs.1c04968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou M, Lankelma M, van der Vlugt JI, de Bruin B, Catalytic Synthesis of 8-Membered Ring Compounds via Cobalt(III)-Carbene Radicals. Angew. Chem. Int. Ed 59, 11073–11079 (2020). doi: 10.1002/anie.202002674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walton JC, Homolytic Substitution: A Molecular Ménage à Trois. Acc. Chem. Res 31, 99–107 (1988). doi: 10.1021/ar970259v [DOI] [Google Scholar]
- 19.Johnson MD, Bimolecular homolytic displacement of transition-metal complexes from carbon. Acc. Chem. Res 16, 343–349 (1983). doi: 10.1021/ar00093a005 [DOI] [Google Scholar]
- 20.Collman JP, Boulatov R, Sunderland CJ, Fu L, Functional analogues of cytochrome c oxidase, myoglobin, and hemoglobin. Chem. Rev 104, 561–588 (2004). doi: 10.1021/cr0206059 [DOI] [PubMed] [Google Scholar]
- 21.Riordan CG, Halpern J, Kinetics, mechanism and thermodynamics of iron carbon bond dissociation in organoiron porphyrin complexes. Inorg. Chim. Acta 243, 19–24 (1996). doi: 10.1016/0020-1693(95)04887-1 [DOI] [Google Scholar]
- 22.Halpern J, Determination of transition metal-alkyl bond dissociation energies from kinetic measurements. Polyhedron 7, 1483–1490 (1988). doi: 10.1016/S0277-5387(00)81776-3 [DOI] [Google Scholar]
- 23.Parsaee F, Senarathna MC, Kannangara PB, Alexander SN, Arche PDE, Welin ER, Radical philicity and its role in selective organic transformations. Nat. Rev. Chem 5, 486–499 (2021). doi: 10.1038/s41570-021-00284-3 [DOI] [PubMed] [Google Scholar]
- 24.Balch AL, Hart RL, Latos-Grazynski L, Traylor TG, Nuclear magnetic resonance studies of the formation of tertiary alkyl complexes of iron(III) porphyrins and their reactions with dioxygen. J. Am. Chem. Soc 112, 7382–7388 (1990). doi: 10.1021/ja00176a044 [DOI] [Google Scholar]
- 25.Dodd D, Johnson MD, Lockman BL, Kinetics and mechanism of apparent alkyl transfer from alkylcobaloximes to cobaloxime(I), cobaloxime(II), and cobaloxime(III) reagents. J. Am. Chem. Soc 99, 3664–3673 (1977). doi: 10.1021/ja00453a026 [DOI] [Google Scholar]
- 26.Lexa D, Mispelter J, Savéant JM, Electroreductive alkylation of iron in porphyrin complexes. Electrochemical and spectral characteristics of. sigma.- alkylironporphyrins. J. Am. Chem. Soc 103, 6806–6812 (1981). doi: 10.1021/ja00413a004 [DOI] [Google Scholar]
- 27.Zhang P, Le CC, MacMillan DWC, Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc 138, 8084–8087 (2016). doi: 10.1021/jacs.6b04818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le C, Chen TQ, Liang T, Zhang P, MacMillan DWC, A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 360, 1010–1014 (2018). doi: 10.1126/science.aat4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavagnino MN, Liang T, MacMillan DWC, HARC as an open-shell strategy to bypass oxidative addition in Ullmann-Goldberg couplings. Proc. Natl. Acad. Sci. U.S.A 117, 21058–21064 (2020). doi: 10.1073/pnas.2011831117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okada K, Okamoto K, Morita N, Okubo K, Oda M, Photosensitized decarboxylative Michael addition through N-(acyloxy)phthalimides via an electron-transfer mechanism. J. Am. Chem. Soc 113, 9401–9402 (1991). doi: 10.1021/ja00024a074 [DOI] [Google Scholar]
- 31.Talele TT, Opportunities for Tapping into Three-Dimensional Chemical Space through a Quaternary Carbon. J. Med. Chem 63, 13291–13315 (2020). doi: 10.1021/acs.jmedchem.0c00829 [DOI] [PubMed] [Google Scholar]
- 32.Lovering F, Bikker J, Humblet C, Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem 52, 6752–6756 (2009). doi: 10.1021/jm901241e [DOI] [PubMed] [Google Scholar]
- 33.Tsuji T, Yorimitsu H, Oshima K, Cobalt-catalyzed coupling reaction of alkyl halides with allylic grignard reagents. Angew. Chem. Int. Ed 41, 4137–4139 (2002). doi: [DOI] [PubMed] [Google Scholar]
- 34.Yang CT, Zhang ZQ, Liang J, Liu JH, Lu XY, Chen HH, Liu L, Copper-catalyzed cross-coupling of nonactivated secondary alkyl halides and tosylates with secondary alkyl Grignard reagents. J. Am. Chem. Soc 134, 11124–11127 (2012). doi: 10.1021/ja304848n [DOI] [PubMed] [Google Scholar]
- 35.Iwasaki T, Takagawa H, Singh SP, Kuniyasu H, Kambe N, Co-catalyzed cross-coupling of alkyl halides with tertiary alkyl Grignard reagents using a 1,3-butadiene additive. J. Am. Chem. Soc 135, 9604–9607 (2013). doi: 10.1021/ja404285b [DOI] [PubMed] [Google Scholar]
- 36.Green SA, Huffman TR, McCourt RO, van der Puyl V, Shenvi RA, Hydroalkylation of Olefins To Form Quaternary Carbons. J. Am. Chem. Soc 141, 7709–7714 (2019). doi: 10.1021/jacs.9b02844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dombrowski AW, Gesmundo NJ, Aguirre AL, Sarris KA, Young JM, Bogdan AR, Martin MC, Gedeon S, Wang Y, Expanding the Medicinal Chemist Toolbox: Comparing Seven C(sp2)-C(sp3) Cross-Coupling Methods by Library Synthesis. ACS Med. Chem. Lett 11, 597–604 (2020). doi: 10.1021/acsmedchemlett.0c00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Malliaras GG, Bernhard S, Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex. Chem. Mater 17, 5712–5719 (2005). doi: 10.1021/cm051312+ [DOI] [Google Scholar]
- 39.Sakai HA, Liu W, Le CC, MacMillan DWC, Cross-Electrophile Coupling of Unactivated Alkyl Chlorides. J. Am. Chem. Soc 142, 11691–11697 (2020). doi: 10.1021/jacs.0c04812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brault D, Bizet C, Morliere P, Rougee M, Land EJ, Santus R, Swallow AJ, One-electron reduction of ferrideuterioporphyrin IX and reaction of the oxidized and reduced forms with chlorinated methyl radicals. J. Am. Chem. Soc 102, 1015–1020 (1980). doi: 10.1021/ja00523a018 [DOI] [Google Scholar]
- 41.Hugelshofer CL, Bao J, Du J, Ashley E, Yu W, Ji T, Hu B, Liu D, Rondla R, Karampuri S, Sharma V, Ethiraj K, Lim YH, Scalable Preparation of 4,4-Disubstituted Six-Membered Cyclic Sulfones. Org. Lett 23, 943–947 (2021). doi: 10.1021/acs.orglett.0c04141 [DOI] [PubMed] [Google Scholar]
- 42.Carreira EM, Fessard TC, Four-membered ring-containing spirocycles: Synthetic strategies and opportunities. Chem. Rev 114, 8257–8322 (2014). doi: 10.1021/cr500127b [DOI] [PubMed] [Google Scholar]
- 43.Cocolios P, Lagrange G, Guilard R, Synthese et caracterisation par RMN 1H d’alkyl(aryl)ferriporphyrines a liaison σ metal carbone. J. Organomet. Chem 253, 65–79 (1983). doi: 10.1016/0022-328X(83)80109-0 [DOI] [Google Scholar]
- 44.Wang J, Cary BP, Beyer PD, Gellman SH, Weix DJ, Ketones from Nickel-Catalyzed Decarboxylative, Non-Symmetric Cross-Electrophile Coupling of Carboxylic Acid Esters. Angew. Chem. Int. Ed 58, 12081–12085 (2019). doi: 10.1002/anie.201906000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perrin DD, Armarego WLF, Purification of Laboratory Chemicals (Pergamon, ed. 3, 1988). [Google Scholar]
- 46.Liu W, Groves JT, Manganese porphyrins catalyze selective C-H bond halogenations. J. Am. Chem. Soc 132, 12847–12849 (2010). doi: 10.1021/ja105548x [DOI] [PubMed] [Google Scholar]
- 47.Burés J, Variable Time Normalization Analysis: General Graphical Elucidation of Reaction Orders from Concentration Profiles. Angew. Chem. Int. Ed 55, 16084–16087 (2016). doi: 10.1002/anie.201609757 [DOI] [PubMed] [Google Scholar]
- 48.Lan Y, Chen Y, Xu X, Qiu Y, Liu S, Liu X, Liu BF, Zhang G, Synthesis and biological evaluation of a novel sigma-1 receptor antagonist based on 3,4-dihydro-2(1H)-quinolinone scaffold as a potential analgesic. Eur. J. Med. Chem 79, 216–230 (2014). doi: 10.1016/j.ejmech.2014.04.019 [DOI] [PubMed] [Google Scholar]
- 49.Du J, Kang Y, Zhao Y, Zheng W, Zhang Y, Lin Y, Wang Z, Wang Y, Luo Q, Wu K, Wang F, Synthesis, Characterization, and in Vitro Antitumor Activity of Ruthenium(II) Polypyridyl Complexes Tethering EGFR-Inhibiting 4-Anilinoquinazolines. Inorg. Chem 55, 4595–4605 (2016). doi: 10.1021/acs.inorgchem.6b00309 [DOI] [PubMed] [Google Scholar]
- 50.Feenstra RW, Stoit A, Terpstra J, Pras-Raves ML, McCreary AC, Van Vliet BJ, Hesselink MB, Kruse CG, Van Scharrenburg GJM, U.S. Pat. Appl. Publ., 20060122189 (2006).
- 51.Melnykov KP, Artemenko AN, Ivanenko BO, Sokolenko YM, Nosik PS, Ostapchuk EN, Grygorenko OO, Volochnyuk DM, Ryabukhin SV, Scalable Synthesis of Biologically Relevant Spirocyclic Pyrrolidines. ACS Omega 4, 7498–7515 (2019). doi: 10.1021/acsomega.9b00896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Webb EW, Park JB, Cole EL, Donnelly DJ, Bonacorsi SJ, Ewing WR, Doyle AG, Nucleophilic (Radio)Fluorination of Redox-Active Esters via Radical-Polar Crossover Enabled by Photoredox Catalysis. J. Am. Chem. Soc 142, 9493–9500 (2020). doi: 10.1021/jacs.0c03125 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available in the supplementary materials.