

Abstract
Background
Bone-marrow-derived haematopoietic stem and progenitor cells (HSPCs) are a prominent part of the highly complex tumour microenvironment (TME) where they localise within tumours and maintain haematopoietic potency. Understanding the role HSPCs play in tumour growth and response to radiation therapy (RT) may lead to improved patient treatments and outcomes.
Methods
We used a mouse model of non-small cell lung carcinoma where tumours were exposed to RT regimens alone or in combination with GW2580, a pharmacological inhibitor of colony stimulating factor (CSF)-1 receptor. RT-PCR, western blotting and immunohistochemistry were used to quantify expression levels of factors that affect HSPC differentiation. DsRed+ HSPC intratumoural activity was tracked using flow cytometry and confocal microscopy.
Results
We demonstrated that CSF-1 is enhanced in the TME following exposure to RT. CSF-1 signaling induced intratumoural HSPC differentiation into M2 polarised tumour-associated macrophages (TAMs), aiding in post-RT tumour survival and regrowth. In contrast, hyperfractionated/pulsed radiation therapy (PRT) and GW2580 ablated this process resulting in improved tumour killing and mouse survival.
Conclusions
Tumours coopt intratumoural HSPC fate determination via CSF-1 signaling to overcome the effects of RT. Thus, limiting intratumoural HSPC activity represents an attractive strategy for improving the clinical treatment of solid tumours.
Subject terms: Translational research, Tumour heterogeneity, Haematopoietic stem cells
Background
The tumour microenvironment (TME) has emerged as one of the predominant defensive weapons with which cancer can resist various forms of therapeutic intervention. The TME is comprised of tumour tissue, stromal cells, immune cells, bone-marrow-derived cells and extra cellular matrix (ECM) proteins. Functional bone-marrow-derived haematopoietic stem and progenitor cells (HSPCs) also take up residence within growing tumours, therefore adding to the growing heterogeneity of the TME [1]. The TME rarely remains static, responding to stimuli from within and outside the tumour to affect tumour growth and survival [2]. Thus, alterations in the TME’s cellular and molecular communication pathways may complicate treatment options and influence patient survival.
Of the various cells comprising the TME, tumour-associated macrophages (TAMs) are particularly abundant and have been shown to have a profound impact on tumour growth [3]. TAMs affect the production of vascular endothelial growth factor (VEGF), tumour necrosis factor alpha, interleukin (IL)-1, IL-6, matrix metalloproteinases and nitric oxide as mechanisms of regulating tumour physiology [4]. Significant correlations between TAM density and poor patient survival have been observed in various types of cancer [5, 6]. In general, TAMs can be subdivided into two phenotypic classes [7]. M1 macrophages exhibit a pro-inflammatory phenotype, promote helper type 1 (Th1) responses, and are known to have anti-tumour activities through the production of reactive oxygen species and various pro-inflammatory cytokines [8]. In contrast, M2 macrophages promote a Th2 response and produce anti-inflammatory factors that promote tumour growth via matrix remodeling, tissue repair, angiogenesis and scavenging [7]. In non-small cell lung carcinoma (NSCLC) patients, tumours expressing high levels of iNOS and possessing a high density of M1 macrophages correlated to more favourable patient outcomes and extended survival in comparison to patients that exhibited lower M1 macrophage content [9]. In contrast, numerous studies have demonstrated that high levels of M2 expressing TAMs were significantly correlated with shorter patient survival, increased disease progression rates and overall poor patient outcomes [10–12].
High energy standard radiation therapy (SRT) is widely used to treat solid tumours. SRT activates several processes and cascades through the oxidation of molecular targets including kinases, phosphatases, cell cycle regulators as well as cell membrane components leading to cell death [13]. However, SRT will exert effects beyond the sole destruction of cancer cells and have a defining influence on the surrounding TME, which can consequently favour tumour survival, radiation resistance and cancer regrowth. For example, SRT induces the migration of various cells from the bone marrow to the site of the tumour which aid in tumour regrowth [14, 15]. SRT also has profound effects on ECM remodeling [16, 17]. In addition, SRT induces the rapid phosphorylation of Akt, ERK and VEGF, which also contribute to tumour survival [18]. Thus, therapies minimising these TME alterations, while promoting cancer cell death could positively influence patient outcomes. Alternate radiation therapy regimens, including pulsed radiation therapy (PRT), in which radiation is fractionated into shorter, separate beams, can enhance tumour ablation by evading DNA damage repair mechanisms [19, 20]. In addition, PRT-treated tumours exhibited differential effects on the TME including a significant maintenance of tumour vasculature, reduced hypoxia, and a decrease in the recruitment of bone-marrow-derived cells [21]. These altered TME effects resulted in significantly decreased tumour volumes compared to tumours receiving SRT. In a recent clinical trial, PRT-treated glioblastoma multiforme tumours demonstrated an improved response, which lead to better overall patient survival compared to the retrospective cohort treated with SRT [22].
An additional mechanism by which the TME resists SRT may involve bone-marrow-derived HSPCs, which play a role in normal tumour biology by directly supporting neovasculogenesis [23]. Bone-marrow-derived HSPCs migrate to growing tumours where they are maintained in a functional state [1]. These intratumoural HSPCs have been shown to increase in number in response to SRT, which directly correlated with significantly enhanced regrowth rates. These findings suggest that the HSPC population is an important TME component that mediates tumour regrowth; however, the molecular mechanisms involved in this response remain undefined.
Given that: (1) TAMs are prominent within the TME and enhance tumour growth, (2) SRT alters TME molecular signaling and (3) HSPCs are present within growing tumours and their numbers are associated with tumour regrowth post-SRT, we hypothesise that HSPCs affect tumour response to therapy through their intratumoural differentiation into TAMs. Overall, our studies indicate a key role of HSPC involvement in tumour response to SRT through TME directed HSPC differentiation into M2 macrophages. Furthermore, the strategy of combining SRT with pharmacologic inhibition of HSPC differentiation into M2 macrophages resulted in significant tumour killing, decreased tumour regrowth post-SRT and longer mouse survival. Together these data identify the activity of HSPCs as a novel therapeutic target in the treatment of solid tumours.
Materials and methods
Lewis lung carcinoma cells
Lewis lung carcinoma (LLC) cells were newly purchased from ATCC (Masassas, VA, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (#11995065, Gibco Life Technologies, Indianapolis, IN, USA), and supplemented with 10% fetal bovine serum (HyClone laboratories, Logan, UT, USA). Cell lines were to be authenticated per the guidelines from the International Cell Line Authentication Committee (iclac.org), and tested for cross-contamination between cell lines every few months of active growth and/or prior to beginning each experimental series using short tandem repeat (STR) profiling.
Mice
C57BL/6 J (stock #000664) and transgenic B6.Cg-Tg(CAG-DsRed*MST)1Nagy/J C57BL/6 J (stock #006051) (DsRed) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All animals were housed at the BRI in an environmentally (temperature, light, and humidity) controlled room and all procedures were approved by the BRI Animal Care and Use Committee.
Tumour inoculation and measurement
Single C57BL/6 J mice were injected with 2 × 106 LLC cells subcutaneously in the right, rear flank. Tumours were measured with digital calipers three times per week starting one week after implant. Tumour volumes were calculated using the equation: [(smaller measurement2) (larger measurement)] (л/6). Treatment groups were randomised based on nominal measurements acquired 1-week after implant. Normalised tumour volumes were calculated using the measurement obtained on the first day of radiation.
Radiation treatment
Irradiation began when tumours were palpable and measurable. Mice that did not grow tumours were excluded from further experimentation. Mice were irradiated at room temperature using a Faxitron X-ray cabinet (160 kVp/4.0 mA at a dose rate of 0.25 Gy/min). Animals were anesthetised with 1–3% isoflurane with oxygen (while on a heating pad) and restrained in a custom-designed irradiation lead-shield, ensuring that only the tumour was in the radiation field. SRT followed a clinically relevant regimen of 5 consecutive days followed by a 2-day break for a daily dose of 2 Gy and weekly dose of 10 Gy. A total of 20 Gy was given over 2 weeks. Treatments were either SRT (continuously delivered dose) or PRT (10 × 0.2 Gy with a 3 min interval between each 0.2 Gy) given at the same dose rate. The numbers of mice in each cohort were based on power calculations. Treatments and measurements were performed on the same days by the lead student (T.M.P.) to minimise confounding issues.
Tumour tissue harvesting
Tumour tissue was collected as indicated or once volumes reached terminal institute standards (tumour volume could not exceed 10% of mouse body weight, affect ambulation or become necrotic). Each tumour was cut into sections. One section was flash frozen in liquid nitrogen and stored in −80 °C for molecular analysis, and another was submerged in zinc buffered formalin before paraffin embedding for immunohistochemical analysis. If required, a third tumour section was taken for fresh tissue analysis via flow cytometry. Mice were anaesthetised with isoflurane followed by euthanasia by cervical dislocation.
Immunohistochemistry (IHC) and confocal microscopy
Paraffin-embedded tumours were cut at 5 µm, placed on microscope slides, and stained with anti-EGR2 (LS-B3577, 1:200; LSBio, Seattle, WA, USA) and anti-CD133 (ab19898, 1:200; Abcam, Cambridge, MA, USA) primary antibodies at 4 °C. Secondary antibody (A32731, 1:1000; ThermoFisher, Waltham, MA, USA) incubation was performed at room temperature shielded from light. Confocal images were obtained using a Nikon C2 + confocal system with NIS-Elements AR 5 imaging software (Melville, NY, USA). Fluorescent IHC images were taken on a Life Technologies Evos Cell Imaging System digital inverted microscope (Austin, TX, USA). ImageJ was used to quantify all images.
Tissue homogenisation and RNA extraction
Frozen tissue sections were homogenised using a gentleMACS™ Dissociator (Miltenyi Biotec, Auburn, CA, USA; #130-093-235) following manufacturer’s protocols. Directly after homogenisation, mRNA was extracted by magnetic labeling of the polyA tails of mRNA using the µMACS mRNA Isolation Kit—Small Scale (Miltenyi Biotec, #130-075-201) following manufacturer’s protocols.
Bone marrow harvest and DsRed CD133+ isolation and implant
Insulin syringes were used to flush bone marrow from the femurs of DsRed transgenic mice using 0.5% BSA in 1× PBS. CD133+ populations were isolated from the bone marrow aspirate via CD133+ Anti-Prominin-1 MicroBeads (Miltenyi Biotec, #130-092-564) following manufacturer’s protocols. 10,000 DsRed CD133+ cells were injected into the tail vein of tumour bearing C57BL/6 J mice. Following the termination of RT, tumour tissues were harvested and processed for confocal microscopy, fluorescent IHC and flow cytometry analysis.
RT-PCR
RNA was quantified and assessed for purity using a Nanodrop. One microgram of RNA was reverse transcribed into cDNA using SuperScript VILO IV Master Mix (Invitrogen, Carlsbad, CA, USA; #11756050) following manufacturer’s protocol. RNA and cDNA quality was confirmed using an absorbance ratio standard of a >1.9 A260/A280 threshold. For each sample, 50 ng cDNA was amplified using Taq MeanGreen Master Mix (Empirical Bioscience, Grand Rapids, MI, USA; #TP-MMWD) and 10µM primers. Primers (IDT) were designed in PrimerBLAST using BioRad validated amplicons, with the following parameters: 40–60% GC content, low self-complementation and Tm close to 60. To verify primer specificity to the gene(s) of interest, all primers were validated using NCBI Primer Blast and were chosen so that the target sequence spanned an intron–exon junction. The efficiency of primers was determined using multiple concentrations of cDNA in various reactions and comparing normalised (to housekeeping genes) relative expression levels across all runs.
TBP-FWD:GACCAGAACAACAGCCTTCCAC
TBP-REV:GCCGTAAGGCATCATTGGACTA
EGR2-FWD:GCACCAGCTGCCTGACA
EGR2-REV:TGCAGAGATGGGAGCGAAG
CSF-1-FWD:AGCGCATGGTCTCATCTATTATGT
CSF-1-REV:GAGACTTCATGCCAGATTGCC
CSF-2-FWD:AGCAGTCTGAGAAGCTGGATT
CSF-2-REV:ATGCGGATTTCATAGACAGCCT
iNOS-FWD:AACTGTGTGCCTGGAGGTTC
iNOS-REV:CCAGGAAGTAGGTGAGGGCT
CD133-FWD:CTCCTTGGAATCAACTGAGATGT
CD133-REV:TGCACATCTTCCTCAACGG
Fpr2-FWD:GCCCATGTCAATTGTTGCTGT
Fpr2-REV:GCCACAACTGCTGTAAGGAC
Reactions were run on a Mastercycler Nexus GSX1 (Eppendorf, San Diego, CA, USA) and amplified using the following conditions: 35 cycles of 95 °C/2 min, 60 °C/30 sec and 72 °C/1 min followed by an elongation step of 72 °C/5 min. Gene expression was normalised to TATA-binding protein (TBP) expression, which maintains stability after ionising radiation [24], and was previously validated in the laboratory. Relative gene expressions were quantified via densitometry analysis using BioRad ImageLab software.
Fluorescent western blotting
Total protein was collected from tumours according to manufacture protocols (Qiagen, #37623), quantified using Bradford standard assays, and assessed for purity using a Nanodrop. Protein was run on 4–20% Mini-PROTEAN TGX Gels (BioRad Laboratories, Hercules, CA, USA; #456-1093) and transferred to PVDF membranes following standard wet transfer conditions. Membrane blotting was performed using the iBind Western Device (ThermoFisher Scientific, #SLF1000) with the iBind Fluorescent Detection Solution Kit (ThermoFisher Scientific, #SLF1019) and primary antibodies: CSF-1 (mouse monoclonal, Santa Cruz), dilution 1:1000; CSF-1R (rat monoclonal, Invitrogen), dilution 1:1000; PhosphoCSF-1R (rabbit monoclonal, Cell Signaling), dilution 1:1000; βactin (chicken monoclonal, ProSci, Poway, CA, USA), dilution 1:1000. Membranes were analysed using the Azure Sapphire Biomolecular Imager (Azure Biosystems, Dublin, CA, USA) laser scanning 4-channel western system using the appropriate fluorescent secondary antibodies (1:2000).
GW2580 treatment
GW2580 (MedKoo, Chapel Hill, NC, USA; #401482) was suspended in 0.5% hydroxypropyl methylcellulose and 0.1% Tween20. Animals received 160 mg/kg of GW2580 daily via oral gavage beginning 2 days prior to RT and ending on the last day of RT.
Flow cytometry analysis
The LLC tumours were dissociated into single-cell suspensions using a Miltenyi GentleMacs Tumor Dissociation Kit, Mouse (Mitenyi Biotec, 130-096-730). Cells were analysed for DsRed HSPCs and FITC-EGR2+ (ThermoFisher Scientific, 12-6691-82) M2 macrophages using a BD FACSCanto II with BDFACSDiva software (BD Biosciences, San Jose, CA, USA).
Statistical analysis
Statistical significance on molecular and tumour growth data was calculated using paired Student’s t-tests and significance is denoted when p < 0.05. Error was calculated via SEM. The Kaplan–Meier Survival curve was generated in GraphPad Prism and a parametric ANOVA statistical analysis was performed in R using the survival package [25]. Survival endpoint was determined when mice reached terminal institute standards, and mice that were euthanised before that point were censored. Significance was determined at p < 0.05.
Results
Effects of radiation therapy on tumour growth and migration of HSPCs to tumours
To understand the kinetic recruitment and involvement of HSPCs in LLC tumour activity upon exposure to radiation therapy, the rear flank of C57BL/6 J mice were inoculated with LLC tumours and exposed to our standard radiation therapy (SRT) protocol for 11 days. Tumours were harvested for analysis 1 day later (day 12) to allow biological effect of the last dose of SRT. During treatment, tumour size and HSPC numbers present in tumours were assessed. SRT resulted in significantly smaller tumours compared to controls that received no radiation therapy (NRT). This indicated that the radiation protocol significantly reduced tumour growth (Fig. 1a).
Fig. 1. SRT modulates tumour growth and levels of intratumoural HSPCs and M2 macrophages.

a Tumour bearing mice treated with SRT produced significantly smaller tumours compared to non-treated controls (n = 15). b, c Analysis of tumours by IHC and image analysis (b) as well as RT-PCR (c) showed an initial increase in HSPC numbers during SRT (n = 9) followed by a decrease at later times (n = 9). A representative image identifies CD133+ cells used for analysis. Scale bar: 15 μm. d Image analysis showed a significant increase in EGR2+ M2 macrophages in tumours during treatment (n = 9). An image is shown to demonstrate EGR2 cell surface staining. Scale bar: 30 μm. e RT-PCR confirmed the increased presence of EGR2 expressing M2 macrophages (n = 9). *p < 0.05 for indicated comparisons, no animals were excluded from analysis, relative gene expression values were normalised to both TBP (housekeeping gene) and NRT day 6 values.
Cell surface CD133 expression from purified murine HSPCs (LSK population) have a distinct propensity to differentiation along the monocyte/macrophage pathway [26]. The presence of CD133+ HSPCs was observed and quantified via both immunohistochemistry (IHC) and RT-PCR during radiation treatment. Tissue staining demonstrated a significant increase in CD133+ HSPC numbers after 1 week of SRT compared to NRT controls (Fig. 1b). Interestingly, in the SRT cohort, these numbers significantly decreased by the conclusion of treatment indicating the loss of intratumoural CD133+ HSPCs after the initial increase in response to SRT. Corroborating results were obtained with tumour CD133 mRNA expression. (Fig. 1c). As expected, nonirradiated tumours (NRT) demonstrated consistent CD133+ HSPC expression throughout the entire radiation procedure. These results support our previous finding using a higher, single dose radiation regimen [1], demonstrate that HSPCs are present in the TME, and that the HSPC population is significantly affected by RT.
Radiation alters the TME to promote the generation of M2 macrophages
Given the significant role that TAMs play in tumour growth, we also assessed how the SRT regimen affected TAM intratumoural density. Early growth response 2 (EGR2) is a potent marker of M2 macrophages [27], so we determined EGR2 expression as a marker of M2 macrophage content in our tumour samples. Both immunostaining and RT-PCR analyses of tumour tissue showed significantly increased levels of cells expressing EGR2 during the course of radiation treatment (Fig. 1d and e). Interestingly, we also observed that CD133+ HSPC numbers concomitantly decreased with increasing M2 macrophage EGR2 expression at day 12 of SRT. This kinetic analysis suggested that as tumours progress through clinically relevant RT, HSPC numbers rise as they are recruited to the TME, then subsequently decrease as they differentiate into M2 macrophages.
The reciprocal changes in HSPC and M2 macrophage content within tumours suggest that these cell populations may be linked and affected by RT. We previously demonstrated that RT can modulate the TME to induce various cellular and molecular alterations that ultimately affect tumour growth and response to therapy [21]. Given that CD133+ HSPCs have the propensity to differentiate into cells of the monocyte/macrophage lineage, we examined whether RT could alter the TME to generate conditions that would favour the production of M2 macrophages from tumour-associated HSPCs. We assessed SRT treated tumours for the expression of factors known to induce the production/polarisation of macrophages and compared to NRT controls. We observed a significant increase in the expression of CSF-1 mRNA and protein. (Figs. 2a and 4b, respectively). Interestingly, we also identified a decrease in CSF-2 and iNOS expression in treated tumours, factors associated with M1 macrophage polarisation (Fig. 2b and c). Analysis of the M1 macrophage marker Fpr2 [27] demonstrated a significant decrease in M1 macrophages following radiation (Fig. 2d). These data indicated that SRT alters the TME to create conditions capable of inducing the directed differentiation of resident HSPCs into tumour supportive M2 macrophages.
Fig. 2. SRT induces TME alterations that favour HSPC differentiation into M2 macrophages.
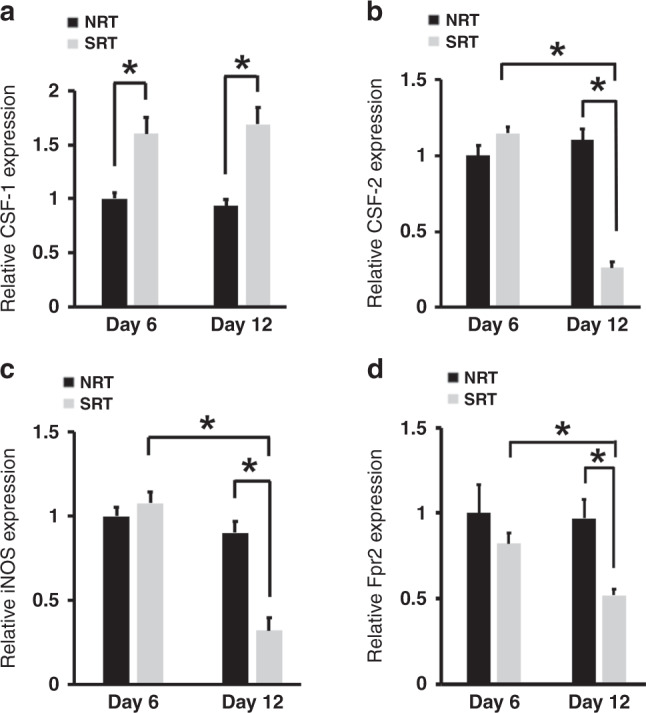
a RT-PCR showed that SRT promoted an increase in the expression of CSF-1 a factor known to promote HSPCs differentiation into M2 polarised macrophages (n = 9). b–d The mRNA levels of CSF-2 (b), iNOS (c) and Fpr2 (d), factors associated with M1 macrophage phenotypes were significantly lower in the TME during treatment (n = 9). *p < 0.05 for indicated comparisons, relative gene expression values were normalised to both TBP (housekeeping gene) and NRT day 6 values.
Fig. 4. Use of the CSF-1R active inhibitor GW2580 prevents differentiation of HSPCs into M2 macrophages after RT reducing tumour regrowth.

a The effects of treating tumour bearing mice with SRT and GW2580 show that each treatment alone or in combination resulted in smaller tumour volumes in comparison to non-treated controls. *p < 0.05 in comparison to NRT-treated tumours; #p < 0.05 in comparison to GW2580 and SRT alone (n = 7). b Western blot analysis demonstrates both the ability of GW2580 to inhibit CSF-1R phosphorylation during dose delivery and the loss of inhibition after dose withdrawal. The fold changes for the densitometry measurements, normalised to β-actin and then compared to NRT controls, are indicated below the corresponding lane. Numbers without brackets compare day 6 changes (n = 3); numbers with brackets compare day 12 changes (n = 6); ND denotes “not detected”. c–e RT-PCR analysis shows that use of GW2580 has no effect on CSF-1 production (c), results in tumour maintenance of CD133+ HSPCs (d), and decreased levels of M2 macrophages (e) (day 6 n = 3, day 12 n = 6). f Tumours significantly regrow after withdrawal of GW2580 from tumour bearing mice (n = 4). *p < 0.05 for indicated comparisons, no animals were excluded from analysis, relative gene expression values were normalised to both TBP (housekeeping gene) and NRT day 6 values.
Stem cell tracking directly demonstrates HSPC differentiation into M2 macrophages following radiation therapy
The significant increase of CSF-1 within tumours during RT indicated the potential for tumour supportive M2 macrophages to be generated from the intratumoural differentiation of HSPCs. To directly demonstrate this activity, we performed stem cell tracking studies wherein CD133+ HSPCs were isolated from the bone marrow of transgenic DsRed C57BL/6 J mice. DsRed CD133+ cell populations were adoptively transplanted into tumour bearing mice and after 24 h tumours were harvested and analysed for the presence of CD133+ HSPCs. Transplanted HSPCs were able to migrate to tumours in as little as 24 h (Fig. 3a). Using this adoptive transplantation strategy, we then tracked the fate of HSPCs in tumours following SRT. Tumours were harvested at day 12 and DsRed cells analysed for the co-expression of EGR2 using flow cytometry. We observed that in the SRT cohort, a significant population of intratumoural DsRed cells co-expressed EGR2 suggesting that the initial transplanted DsRed HSPCs directly differentiated into M2 macrophages within the TME (Fig. 3b and c). Conversely, in NRT controls, the DsRed populations exhibited a significantly lower EGR2 population and a higher maintenance of the CD133+ phenotype, thus suggesting that radiation induces TME conditions that favour the directed differentiation of HSPCs into tumour supportive M2 macrophages. Confocal microscopy of tumour sections was used to visualise the presence of EGR2+ DsRed cells within tumours (representative image; Fig. 3d).
Fig. 3. HSPCs can directly differentiate into M2 macrophages in response to SRT.

a CD133+ HSPCs were isolated from DsRed transgenic mice and transplanted intravenously into tumour bearing mice. Twenty four hours later tumours were harvested and showed that the CD133+DsRed HSPCs had migrated to tumours and taken up residence. Scale bar: 100 μm. b, c Representative flow plots (b) and analysis of dissociated tumours showing a decrease in CD133+ HSPCs and an increase in EGR2+ M2 macrophages with SRT (n = 6) (c). Isotype control plots are shown and depict gating strategy. Gating was established to capture changes in DsRed CD133+ HSPCs and DsRed EGR2+ M2 macrophages. Since tumours were initially transplanted with DsRed cells from transgenic mice, a DsRed population was present in isotype controls. To account for this, horizontal quadrant lines were set to differentiate between DsRed+ and DsRed− cells present in tumours and to minimise false DsRed+ cells from the DsRed− population. Vertical quadrant lines were set to ensure 0.1% or less of events appeared in the upper/lower right quadrants. d An image showing an EGR2+ cell that was derived from the originally transplanted DsRed CD133+ HSPCs. Scale bar: 15 μm. *p < 0.05 for indicated comparison, no animals were excluded from analysis.
Blocking CSF-1/CSF-1R activity with GW2580 reduces tumour growth and prevents M2 macrophage generation
Our studies indicated that HSPC-derived M2 macrophages formed during RT may have a deleterious effect on treatment outcome. To determine if these M2 macrophages were generated in response to increased CSF-1 signaling and affected tumour response to RT, we inhibited the CSF-1/CSF-1R signaling pathway and measured tumour size during radiation therapy. Inhibition was accomplished utilising a pharmacological agent (GW2580), which selectively blocks CSF-1R autophosphorylation and activation [28]. Tumours were grown in mice and exposed to SRT, GW2580 and a combination treatment of SRT + GW2580. We found that all three treatment cohorts resulted in significantly smaller tumours compared to NRT controls beginning at day 9, with the combination of SRT + GW2580 resulting in the smallest tumours at treatment completion (Fig. 4a). Interestingly, comparison between SRT and GW2580-treated tumours indicated that radiation and CSF-1/CSF-1R pathway inhibition had similar deleterious effects on tumour size, signifying HSPC-derived M2 macrophages as potent mediators of tumour growth. To confirm that GW2580 abrogated the CSF-1/CSF-1R cascade, tumour protein expression levels of CSF-1R and phosphorylated CSF-1R were measured. In alignment with our tumour volume data, tumours receiving GW2580 either with or without SRT had significantly lower levels of phosphorylated CSF-1R with no change in CSF-1R expression (Fig. 4b). The decreases in CSF-1R phosphorylation were due to GW2580 treatment because the levels of CSF-1 mRNA and protein present within tumours were similar between SRT and SRT + GW2580 cohorts. (Fig. 4b and c). Importantly, while the tumours in the SRT cohort lost HSPCs along with a significant M2 macrophage (EGR2+) presence increase at day 12, GW2580 treatment alone or in combination with SRT significantly maintained tumour HSPCs (CD133+) with no observable increases of intratumoural M2 macrophages (EGR2) (Fig. 4d and e).
CSF-1R phosphorylation levels were also assessed following the termination of GW2580 delivery in the GW2580 alone treated cohort. GW2580 withdrawal stimulates the restoration of phosphorylated CSF-1R (Fig. 4b). Interestingly, termination of GW2580 treatment also correlated to a significant increase in tumour regrowth within 3 days and enabled tumours to reach the size threshold for euthanasia (Fig. 4f). The dramatic regrowth of these tumours upon GW2580 cessation further demonstrates the significant effect CSF-1/CSF-1R signaling and M2 macrophage generation have in tumour response to treatment and regrowth. Overall, these data confirmed that in vivo treatment with GW2580 decreased tumour volume via inhibition of the CSF-1/CSF-1R signaling pathway.
Hyperfractionated radiation decreases HSPC migration and differentiation into M2 macrophages
In addition to utilising the SRT regimen, we also treated tumours using a dosing schedule of hyperfractionated/pulsed RT (PRT), which we previously showed abrogates cellular repair mechanisms in various cancers [20]. Furthermore, use of PRT in our tumour model altered the TME resulting in deceased migration of bone-marrow-derived cells to tumours and smaller tumours [21]. Based on these findings, we determined if PRT could alter the TME to also prevent M2 macrophage generation from tumour-associated HSPCs. We found that PRT-treated tumours were significantly smaller compared to NRT-treated controls (Fig. 5a). Tumours receiving PRT exhibited significantly lower levels of HSPCs and M2 macrophages following treatment, while exhibiting no significant difference in CSF-1 levels compared to non-treated controls (Fig. 5b–d). These results indicated that the effects of CSF-1 observed following SRT treatment can be attenuated using PRT, thus preventing M2 production from HSPCs. Together with our previous finding that PRT decreases the production of the HSPC chemotactic factor stromal derived factor (SDF)-1 in tumours [21], these results indicate that PRT significantly lowers bone-marrow-derived HSPC migration to tumours. Overall, these results indicate that specialised RT regimens may be used to eliminate HSPC activity resulting in better overall tumour killing.
Fig. 5. Use of fractionated RT decreases HSPC migration and differentiation into M2 macrophages reducing tumour growth.

a The effects of treating tumour bearing mice with combinations of PRT and GW2580 were tested and show that each treatment alone (n = 19) or in combination (n = 10) resulted in smaller tumour volume in comparison to non-treated controls. *p < 0.05 in comparison to NRT-treated tumours (n = 18); δp < 0.05 in comparison to GW2580 alone (n = 13); #p < 0.05 in comparison to GW2580 and PRT alone. b–d Following PRT treatment, lower levels of CD133+ HSPCs (b) and M2 macrophages (c) was observed in tumours in comparison to controls, with no effects on CSF-1 levels (d) (n = 9). e–g RT-PCR analysis shows that use of GW2580 is able to augment PRT effects resulting in lower HSPCs (e), no change in CSF-1 production compared to NRT (f) and decreased levels of M2 macrophages (g) (GW2580: day 6 n = 3, day 12 n = 6; PRT n = 9; PRT + GW2580 n = 3). h Western blot demonstrates the ability of GW2580 to prevent CSF-1R phosphorylation (n = 3). *p < 0.05 for indicated comparisons. i Images demonstrating the drastic effects of treatment on tumour size. No animals were excluded from analysis, relative gene expression values were normalised to both TBP (housekeeping gene) and NRT day 6 values.
Next, we sought to determine if the addition of GW2580 would augment PRT. We exposed tumours to PRT, GW2580, or a combination treatment of PRT + GW2580. The addition of GW2580 resulted in significantly smaller tumours compared to tumours treated with PRT alone, GW2580 alone, or NRT controls (Fig. 5a). Comparison of PRT- and GW2580-treated tumours demonstrated that PRT had a larger suppressive effect on tumour growth. In all instances, PRT-treated tumours showed a significant decrease in HSPCs with or without GW2580 augmentation, while CSF-1 levels were unaffected (Fig. 5e and f). Interestingly, M2 macrophages were observed in tumours treated with PRT alone. However, these values were significantly lower compared to tumours treated with SRT alone (Fig. 5g and Fig. 4e; p < 0.05). This indicated that preventing HSPC migration (due to PRT) produced a profound effect on tumour killing when combined with the GW2580 mediated inhibition of HSPC differentiation into M2 macrophages. Western blot analysis showed that PRT alone treatment did not prevent CSF-1R phosphorylation, thus providing an explanation for why PRT alone treated tumours had significantly more M2 macrophages compared to PRT + GW2580-treated tumours (Fig. 5h). Images of mice with PRT + GW2580-treated tumours visually depict the significant effects of this combination treatment on tumour growth (Fig. 5i). These data suggest that the main effect of PRT is the diminished presence of intratumoural HSPCs leading to fewer supportive M2 macrophages.
Combination treatment enhances mouse survival and diminishes tumour regrowth
Our cumulative results demonstrate that all treatment strategies had a significant effect on tumour growth, albeit to varying degrees. To further assess the various treatment strategies and identify the most efficacious, we terminated all treatments and tracked mouse survival and tumour regrowth. Tumour bearing mice treated with PRT + GW2580 and SRT + GW2580 had the greatest longevity, with survival being observed 17 and 14 days after treatment termination, respectively (Fig. 6a). This aligned with our previous data demonstrating that combination treatments resulted in the smallest tumours following treatment (Figs. 4a and 5a). PRT alone treated mice, the cohort with the next best survival outcomes, survived 12 days after treatment termination, while the SRT alone treated mice survived only an additional 5 days of regrowth. Taken together, these data suggested that PRT was more efficacious than SRT when used either alone or in combination with GW2580, supporting our previous findings [20]. Interestingly, with GW2580 alone cohorts, removal of GW2580 produced similar survival rates compared to non-treated controls (NRT) with mice surviving only 3 days post treatment termination. This suggests that CSF-1R activation can be quickly restored upon elimination of GW2580 resulting in lower survival rates, which may be explained by the observed tumour regrowth (Fig. 4f). This is further supported by the observation that phosphorylated CSF-1R levels were restored to basal levels post GW2580 termination (Fig. 4b). Finally, we analysed the regrowth of tumours for the PRT + GW2580 and SRT + GW2580 cohorts during the survival studies given their robust effects. In agreement with the survival studies, we observed that the PRT + GW2580 group produced the lowest rate of regrowth resulting in significantly smaller tumours in comparison to SRT + GW2580-treated tumours (Fig. 6b).
Fig. 6. Augmentative treatment with GW2580 enhances RT effects and abrogates HSPC activity in the TME.

a The survival of tumour bearing mice treated with SRT, PRT and GW2580 alone or in combination was assessed (n = 4). Kaplan–Meier analysis demonstrated that the combination of PRT + GW2580 provided the best survival (p = 0.0002). b Tumour volumes were tracked upon termination of both the PRT + GW2580 and SRT + GW2580 combination therapies (n = 4). The results show that tumour regrowth is significantly prevented in the PRT + GW2580 cohort. *p < 0.05 for indicated comparisons, no animals were excluded from analysis. c Schematic of the proposed activity of HSPCs in solid tumour growth and regrowth post-RT. HSPCs normally migrate to growing tumours (step 1) where they are maintained in an undifferentiated state (step 2). When tumours are exposed to radiation, there is an increased production of factors such as CSF-1 (step 3), which create a TME that induces HSPC differentiation into M2 macrophages (step 4). These newly formed TAMs aid in tumour regrowth (step 5). Therapies aimed at preventing these processes such as PRT and GW2580 are able to abrogate this supportive mechanism resulting in smaller tumours and enhanced mouse survival.
Discussion
Bone-marrow-derived CD133+ HSPCs migrate to growing tumours where they take up residence within supportive niches. These tumour-associated HSPCs maintain functionality, as they are able to engraft secondary recipient mice and recapitulate haematopoiesis [1]. Interestingly, HSPCs increase their migration to tumours in response to clinically relevant SRT, and their intratumoural numbers correlated directly with tumour regrowth rates. Studies suggested that HSPCs affected tumour response to therapy. However, the cellular and molecular mechanisms modulating this effect remained unknown.
In the present study, we identified a mechanism wherein tumour regrowth after radiation is significantly affected by the differentiation of intratumoural HSPCs into tumour promoting M2 macrophages (Fig. 6c). We found significantly elevated levels of CSF-1 within radiated tumours. CSF-1 is a soluble factor that induces HSPC differentiation into TAMs and is often tied to poor prognosis in cancer patients following radiation [29]. Interestingly, CSF-2, iNOS and Fpr2, factors associated with M1 anti-tumour TAMs [27] and correlated with more positive patient outcomes [30], were significantly decreased within tumours during radiation. It is well known that higher tumour M1/M2 macrophage content ratios can predict better patient overall survival in a variety of cancers [31]. We calculated M1/M2 ratios for our treatment cohorts, taking the average expression levels of the indicated M1- and M2-specific genes. We observed higher M1/M2 ratios in tumours throughout PRT treatment (day 6 = 1.6; day 12 = 1.1) in comparison to SRT treated tumours (day 6 = 0.7; day 12 = 0.2). Concomitantly, a significant decrease of tumour-associated CD133+ HSPCs was observed, suggesting that SRT conditions the TME to favour the differentiation of HSPCs into M2 tumour supportive TAMs, thus decreasing overall M1/M2 ratios.
Cells of the M2 lineage migrate to growing tumours and promote tumour growth and regrowth following radiation [32]. While bone-marrow-derived TAMs have been well studied, our work now identifies an additional source of TAMs, namely tumour-associated HSPCs. Direct stem cell tracking studies revealed that DsRed CD133+ HSPC populations differentiate into M2 macrophages following adoptive transplantation into tumour bearing irradiated mice. This is the first evidence demonstrating that tumours utilise a unique mechanism wherein they recruit normal HSPCs to the TME and exploit their ability to produce M2 macrophages to support their regrowth following therapy. Furthermore, while the role of mutated cancer stem cells in tumour initiation is well documented, this study now identifies a novel role of normal HSPCs in tumour biology. The ability of tumours to hijack the normal process of HSPC migration, homing and differentiation within the TME to promote tumour growth provides an attractive therapeutic target for treatment of solid tumours. Interestingly, HSPCs are known to maintain potency within well-defined bone marrow niches [33]. In conjunction with our previous studies in which tumour-associated HSPCs were isolated and were able to engraft secondary mouse recipients [1], these studies further support the existence of a tumour niche that can maintain HSPC functionality, including the generation of M2 macrophages. Defining this tumour HSPC niche and its ability to maintain HSPC self-renewal and multi-lineage differentiation is the topic of ongoing investigation and will expand our knowledge of cancer and stem cell biology.
CSF-1 expression within tumours has been associated with poor patient prognosis and treatments that target CSF-1 activity has shown promise [34, 35]. In the studies described here, we have identified a novel role of CSF-1 tumour biology. Treatment of tumour bearing mice with the small molecule inhibitor GW2580 inhibited CSF-1R phosphorylation, which enhanced the anti-tumour effect of radiation. GW2580 has shown phospho-inhibition capabilities with no toxicity, making it an ideal agent to target and prevent M2 differentiation from HSPCs [28]. In GW2580-treated tumours, we show a dramatic effect as not only was there a decrease in M2 macrophages within tumours but also a significant decrease in tumour volume, even in the presence of elevated CSF-1 post-SRT. Previous studies have attempted to treat tumours by blocking migration of bone-marrow-derived cells to tumours using the CXCR4 inhibitor AMD3100 [36]. We previously recapitulated these experiments in our model to utilise AMD3100 to block recruitment of CD133+ stem cells to tumours [1], and while these studies demonstrated the efficacy of AMD3100, the present study suggests that solely inhibiting migration is insufficient for eliminating M2 macrophage numbers and their supportive effects within tumours. This is due to M2 macrophage generation from tumour-associated HSPCs in response to increased levels of CSF-1. AMD3100 treatment will not affect M2 generation via this mechanism. Given this understanding, we postulate that augmentation of SRT with GW2580 would provide therapeutic benefit in solid tumour treatment.
GW2580 can prevent TAM migration to various cancers including lung, melanoma and prostate [28]. Herein, we also demonstrate a second action of GW2580, which is the prevention of HSPC differentiation into M2 macrophages. This is directly supported by our adoptive transplantation studies. By transplanting isolated DsRed CD133+ HSPCs into tumour bearing mice, we were able to track HSPC migration to tumours within 24 h. The ability to observe DsRed M2 macrophages (EGR2+) within tumours after 12 days indicated they were derived from the initial transplanted HSPCs. DsRed macrophages present within tumours could not have been bone-marrow-derived as 12 days does not provide enough time for transplanted HSPCs to stably engraft within the bone marrow of recipient mice and begin the production of M2 macrophages [37]. Furthermore, mice did not undergo whole body irradiation, which is necessary for stable HSPC engraftment and subsequent haematopoiesis. Therefore, our data demonstrates that GW2580 effectively inhibits HSPCs within the TME from differentiating into tumour supportive M2 macrophages. This mechanism is distinct from the known role of GW2580 which inhibits recruitment of TAMs to the TME.
We previously found that tumours treated with PRT showed significant tumour size reduction [20]. In addition, we demonstrated compared to SRT, PRT altered the TME at the molecular level. PRT produced TME conditions that exhibited less hypoxia and eliminated tumour supportive mechanisms including angiogenesis and the recruitment of bone-marrow-derived cells. Expanding upon this previous research, we investigated the effect of PRT on the differentiation of HSPCs into M2 macrophages within tumours. We found that PRT treatment resulted in significantly smaller tumours that contained fewer tumour-associated CD133+ HSPCs, similar CSF-1 levels, and fewer M2 macrophages in comparison to NRT controls. The ability to modulate the TME to conditions that decreased M2 macrophages while reducing tumour size represents an additional mechanism by which PRT enhances tumour killing. Administering PRT in conjunction with GW2580 enhanced tumour killing and was the superior strategy used to significantly limit tumour regrowth presumably through the additional inhibition of CSF-1R activation. Mice receiving this combination treatment demonstrated the highest survival rates in comparison to all other treatment strategies. Therefore, the ability to lower HSPC migration (and other bone-marrow-derived cells) to tumours and prevent residual HSPC differentiation into M2 macrophages represents a novel, effective method to treat tumours (Fig. 6c). The data presented here demonstrates that normal HSPCs represent a potent cellular component of the TME that play a significant role in tumour recovery post-RT. Strategies designed to significantly limit HSPC migration to tumours and/or their subsequent differentiation into tumour supportive M2 macrophages post-RT represent viable, attractive clinical strategies for improving cancer treatment.
Supplementary information
Acknowledgements
The BRI animal facility staff and SG assisted with animal husbandry and tumour inoculations.
Author contributions
GJM, GW, LGV-D and TMP conceived the project. TMP, KLB, AH and CP performed the experiments. TMP, KLB, MAB, SEH, RJW, LGV-D, GW and GJM provided resources and analysed data. TMP, GW, LGV-D and GJM were responsible for project management and compliance protocols. GJM, TMP, SEH, RJW and LGV-D wrote the paper. GW and KLB edited the manuscript.
Funding
This work was supported by an OU Center for Biomedical Research grant (GJM) and OU Provost grant (TMP). Flow cytometry was performed in the OU Flow Cytometry Core, supported in part by a NSF-MRI (NSF-1919572) grant (LGV-D and GJM).
Data availability
All data are included in this published article and are available (GJM) upon reasonable request.
Ethics approval and consent to participate
Animal experiments were approved by the BRI IACUC.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-021-01652-y.
References
- 1.Kane J, Krueger SA, Dilworth JT, Torma JT, Wilson GD, Marples B, et al. Hematopoietic stem and progenitor cell migration after hypofractionated radiation therapy in a murine model. Int J Radiat Oncol Biol Phys. 2013;87:1162–70. doi: 10.1016/j.ijrobp.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 2.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320–9. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Cao X. The origin and function of tumor-associated macrophages. Cell Mol Immunol. 2015;12:1–4. doi: 10.1038/cmi.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim J, Bae JS. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm. 2016;2016:6058147. doi: 10.1155/2016/6058147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–65. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 6.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–12. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 7.Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. 2012;4:376–89. [PMC free article] [PubMed] [Google Scholar]
- 8.Jeannin P, Paolini L, Adam C, Delneste Y. The roles of CSFs on the functional polarization of tumor-associated macrophages. FEBS J. 2018;285:680–99. doi: 10.1111/febs.14343. [DOI] [PubMed] [Google Scholar]
- 9.Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur Respir J. 2009;33:118–26. doi: 10.1183/09031936.00065708. [DOI] [PubMed] [Google Scholar]
- 10.Chung FT, Lee KY, Wang CW, Heh CC, Chan YF, Chen HW, et al. Tumor-associated macrophages correlate with response to epidermal growth factor receptor-tyrosine kinase inhibitors in advanced non-small cell lung cancer. Int J Cancer. 2012;131:E227–235. doi: 10.1002/ijc.27403. [DOI] [PubMed] [Google Scholar]
- 11.Ohtaki Y, Ishii G, Nagai K, Ashimine S, Kuwata T, Hishida T, et al. Stromal macrophage expressing CD204 is associated with tumor aggressiveness in lung adenocarcinoma. J Thorac Oncol. 2010;5:1507–15. doi: 10.1097/JTO.0b013e3181eba692. [DOI] [PubMed] [Google Scholar]
- 12.Zhang B, Yao G, Zhang Y, Gao J, Yang B, Rao Z, et al. M2-polarized tumor-associated macrophages are associated with poor prognoses resulting from accelerated lymphangiogenesis in lung adenocarcinoma. Clinics (Sao Paulo) 2011;66:1879–86. doi: 10.1590/S1807-59322011001100006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012;327:48–60. doi: 10.1016/j.canlet.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–20. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klopp AH, Spaeth EL, Dembinski JL, Woodward WA, Munshi A, Meyn RE, et al. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007;67:11687–95. doi: 10.1158/0008-5472.CAN-07-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bigoni-Ordonez GD, Czarnowski D, Parsons T, Madlambayan GJ, Villa-Diaz LG. Integrin alpha6 (CD49f), the microenvironment and cancer stem cells. Curr Stem Cell Res Ther. 2019;14:428–36. doi: 10.2174/1574888X13666181002151330. [DOI] [PubMed] [Google Scholar]
- 17.Jourdan MM, Lopez A, Olasz EB, Duncan NE, Demara M, Kittipongdaja W, et al. Laminin 332 deposition is diminished in irradiated skin in an animal model of combined radiation and wound skin injury. Radiat Res. 2011;176:636–48. doi: 10.1667/RR2422.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cristofaro B, Emanueli C. Possible novel targets for therapeutic angiogenesis. Curr Opin Pharmacol. 2009;9:102–8. doi: 10.1016/j.coph.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 20.Dilworth JT, Krueger SA, Dabjan M, Grills IS, Torma J, Wilson GD, et al. Pulsed low-dose irradiation of orthotopic glioblastoma multiforme (GBM) in a pre-clinical model: effects on vascularization and tumor control. Radiother Oncol. 2013;108:149–54. doi: 10.1016/j.radonc.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 21.Kane JL, Krueger SA, Hanna A, Raffel TR, Wilson GD, Madlambayan GJ, et al. Effect of irradiation on tumor microenvironment and bone marrow cell migration in a preclinical tumor model. Int J Radiat Oncol Biol Phys. 2016;96:170–8. doi: 10.1016/j.ijrobp.2016.04.028. [DOI] [PubMed] [Google Scholar]
- 22.Almahariq MF, Quinn TJ, Arden JD, Roskos PT, Wilson GD, Marples B, et al. Pulsed radiation therapy for the treatment of newly diagnosed glioblastoma. Neuro Oncol. 2020 doi: 10.1093/neuonc/noaa165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madlambayan GJ, Butler JM, Hosaka K, Jorgensen M, Fu D, Guthrie SM, et al. Bone marrow stem and progenitor cell contribution to neovasculogenesis is dependent on model system with SDF-1 as a permissive trigger. Blood. 2009;114:4310–9. doi: 10.1182/blood-2009-03-211342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iyer G, Wang AR, Brennan SR, Bourgeois S, Armstrong E, Shah P, et al. Identification of stable housekeeping genes in response to ionizing radiation in cancer research. Sci Rep. 2017;7:43763. doi: 10.1038/srep43763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.A Package for Survival Analysis in R [computer program]. Version 3.2-7. 2020. https://CRAN.R-project.org/package=survival2020.
- 26.Arndt K, Grinenko T, Mende N, Reichert D, Portz M, Ripich T, et al. CD133 is a modifier of hematopoietic progenitor frequencies but is dispensable for the maintenance of mouse hematopoietic stem cells. Proc Natl Acad Sci USA. 2013;110:5582–7. doi: 10.1073/pnas.1215438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, et al. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE. 2015;10:e0145342. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115:1461–71. doi: 10.1182/blood-2009-08-237412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Y, He MY, Zhu LF, Yang CC, Zhou ML, Wang Q, et al. Tumor-associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J Exp Clin Cancer Res. 2016;35:12. doi: 10.1186/s13046-015-0281-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24:589–602. doi: 10.1016/j.ccr.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 31.Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, Ch’ng ES. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and challenges in routine clinical practice. Front Oncol. 2019;9:1512. doi: 10.3389/fonc.2019.01512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. 2015;36:229–39. doi: 10.1016/j.it.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 33.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–62. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–72. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szilvassy SJ, Meyerrose TE, Ragland PL, Grimes B. Differential homing and engraftment properties of hematopoietic progenitor cells from murine bone marrow, mobilized peripheral blood, and fetal liver. Blood. 2001;98:2108–15. doi: 10.1182/blood.V98.7.2108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are included in this published article and are available (GJM) upon reasonable request.