

Abstract
Tyrosine phosphorylation by protein tyrosine kinases (PTKs) is a type of post-translational modification. Tec kinases, which are a subfamily of non-receptor PTKs, were originally discovered in the hematopoietic system and include five members: Tec, Btk, Itk/Emt/Tsk, Etk/Bmx, and Txk/Rlk. With the progression of modern research, certain members of the Tec family of kinases have been found to be expressed outside the hematopoietic system and are involved in the development and progression of a variety of diseases. The role of Tec family kinases in cardiovascular disease is receiving increasing attention. Tec kinases are involved in the occurrence and progression of ischemic heart disease, atherosclerosis, cardiac dysfunction associated with sepsis, atrial fibrillation, myocardial hypertrophy, coronary atherosclerotic heart disease, and myocardial infarction and post-myocardial. However, no reviews have comprehensively clarified the role of Tec kinases in the cardiovascular system. Therefore, this review summarizes research on the role of Tec kinases in cardiovascular disease, providing new insights into the prevention and treatment of cardiovascular disease.
Subject terms: Medical research, Diseases, Biochemistry
Introduction
Protein phosphorylation: refers to the process catalyzed by protein kinase to transfer the phosphate group of ATP to the substrate protein amino acid residues (serine, threonine, tyrosine), or bind GTP under the action of signal. It is a common regulation mode in organisms and plays an important role in the process of cellular signal transduction. Protein phosphorylation is the most basic, universal, and important mechanism for regulating and controlling protein viability and function. The main role of serine phosphorylation is to allosteric proteins to activate the activity of proteins, mainly referring to enzyme activity. In addition to allosteric and activating the activity of the protein, tyrosine phosphorylation can bind proteins to provide a structural gene to promote their interaction with other proteins to form multiprotein complexes. The formation of protein complexes further promotes the phosphorylation of proteins. Cycling, the signal generated by the initial protein phosphorylation turns on step by step. If a signal is initially generated that stimulates cell growth, this signal is eventually transferred to the nucleus, resulting in DNA replication and cell division. The enzymes that perform this modification are protein tyrosine kinases (PTKs), which catalyze the transfer of adenosine triphosphate to tyrosine residues on protein substrates [1]. Two classes of PTK exist in cells: transmembrane receptor PTKs and non-receptor PTKs [2].
Ninety unique tyrosine kinase genes have been identified in the human genome, of which 58 receptor PTKs are distributed in 20 subfamilies, and 32 non-receptor PTKs are distributed in 10 subfamilies [3, 4]. Their functions include cell growth, cell differentiation, and signal transmission. And it plays an important role in a variety of human diseases.
Tec kinases, which are a subfamily of non-receptor PTKs represented by the first member, Tec, are key players in intracellular signaling in lymphocytes [5]. This family consists of five members: Tec, Btk, Itk/Emt/Tsk, Etk/Bmx, and Txk/Rlk. While Btk, Itk, and Txk are selectively expressed in hematopoietic cells, the expression patterns of Bmx and Tec are more widespread (Table 1). Many cells may express several Tec kinases, and it has been reported that they may functionally complement each other [6, 7]. PTK signaling plays an important role in the development of many diseases, including cancer, Alzheimer’s disease and diabetes mellitus, and the use of PTK inhibitors to treat disease is receiving increasing attention [8–10]. Cardiovascular system disease pathogenesis is complex, and multiple factors play a role in its development [11–13]. In recent years, much attention has been paid to cardiovascular disease and the role of PTKs in cardiovascular disease. At the same time Tec family kinases have also received more attention. Tec kinases are not only associated with immune system diseases and hematological diseases, but they are also involved in inflammatory diseases, infectious diseases, and cardiovascular diseases (Table 2) (Fig. 1). Some Tec kinase inhibitors have shown good application prospects in the treatment of cardiovascular disease (Table 3).
Table 1.
Chromosome assignment and expression of Tec family kinases.
| Kinase | Chromosome assignment | Expression |
|---|---|---|
| TEC | 4p12 | T cells, B cells |
| Liver, heart, kidney and ovary | ||
| BTK | Xq22 | T cells, B cells, innate immune cells, megakaryocytes and platelets |
| BMX | Xp22.2 | Granulocytic monocyte lineage, endothelium of the endocardium and arteries |
| ITK | 5q31–32 | T cells, mast cells, natural killer cells |
| TXK | 4p12 | T cells |
Table 2.
The Tec family kinases functions and regulation.
| Kinase | Mechanisms | Biological response | References |
|---|---|---|---|
| TEC | Ischemic injury as well as purines can stimulate Tec kinase translocation in cardiomyocytes. Tec regulates proteins associated with lipid membranes, and is involved in the temporal response of the heart to ischemia-reperfusion. | Ischaemic heart disease | [33, 34] |
| Tec may play a cardioprotective role by binding to ROCK1. | Cardiac pressure overload hypertrophy | [33, 35] | |
| BTK | BTK promotes ox-LDL-induced ER stress, oxidative stress, and inflammatory responses in macrophages, and BTK knockdown can inhibit ox-LDL-induced B signaling activation and inhibit M1 polarization in NK-κ macrophages.BTK regulates the NLRP3 inflammasome.Btk is involved in bt/VWF (Botrocetin/von Willebrand factor) -mediated lectin-induced TxA2 production and GPIb-dependent arterial thrombosis. | Atherosclerosis | [41–56] |
| Btk plays a role in Toll-like receptor signaling and NLRP3 inflammasome activation. | Sepsis-related cardiac dysfunction | [46, 47, 57, 68] | |
| BTK promotes NLRP3 inflammasome activation. | Atrial fibrillation | [46, 47, 69] | |
| BMX | The effect of Bmx on cardiac hypertrophy is mediated through the endothelial cells of the coronary vessels. | Cardiac hypertrophy | [73, 77] |
| Bmx regulates VEGFR2 expression and VEGF-induced angiogenesis, and Bmx is also a downstream effector of VEGFR2. It is involved in ischemia-induced arteriogenesis and angiogenesis. Bmx is also involved in cell adhesion, migration and survival in TNF-induced angiogenesis. | Coronary atherosclerotic heart disease | [78–83] | |
| Bmx is involved in the mechanism of NO-induced PKC-ε signaling and VEGF-dependent lymphangiogenesis signaling. | Myocardial Infarction and Post-Myocardial Infarction | [84–87] | |
| ITK | None | None | None |
| TXK | None | None | None |
Fig. 1. The regulation and functions of Tec family kinases in cardiac and vascular disease.

Ox-LDL oxidized low-density lipoprotein, ROCK1 Rho kinase 1, ER endoplasmic reticulum, bt/VWF botrocetin/von Willebrand factor-mediated, TXA2 thromboxane A2, GPIb glycoprotein Ib, VEGF Vascular endothelial growth factor, VEGFR2 VEGF receptor 2, PKC-ε protein kinase C -ε.
Table 3.
So far, studies of Btk inhibitors related to cardiovascular disease.
Structural features of Tec family kinases
Tec kinases have a highly conserved carboxy-terminal kinase domain and a relatively long and unique amino-terminal domain (Fig. 2). The amino-terminal domain can be further divided into a pleckstrin homology (PH) domain [14] and a Tec homology (TH) domain [15], which are characteristic of Tec kinases. The PH domain binds to phospholipids to mediate membrane association (the PH domain recruits Tec kinases to the cell membrane by binding certain phospholipids) [16], but the PH domain of Txk is replaced by a cysteine motif, which mediates membrane association by palmitoylation [17]. The TH domain includes an array of zinc-binding Btk homology (BH) motifs and one or two proline-rich (PR) motifs. The BH motif is involved in the binding site that constitutes the guanosine triphosphate-binding “active” Gα subunit of the heterotrimeric G protein. The PR motif can bind to the Src homology 3 (SH3) domain in an intramolecular manner, and this action may be important for protein targeting and enzyme activation [3]. Structurally, the TH domain of Txk lacks the BH sequence, the TH domain of Bmx lacks the PR sequence, and the SH3 domain shows a slightly different sequence. In all five kinases, the TH domain is followed by an SH3 domain and an SH2 domain [18]. The SH3 domain is primarily responsible for mediating protein–protein interactions [19, 20]. The SH3 domain of non-receptor PTKs appears to be implicated in the negative regulation of PTK activity [21]. SH2 can induce hyperphosphorylation of docking proteins [22], and it also stabilizes the intramolecular interactions between the SH3 domain and the PR motif [3]. SH2 is followed by a catalytic kinase domain, also known as the SH1 domain [23]. Tec family non-receptor PTKs are also unique in that they possess autophosphorylation sites inside their molecules. In studies on Btk, Itk, and Txk, all autophosphorylation sites were identified in the SH3 domain [24–27].
Fig. 2. Schematic representation of Tec family kinases and their domains.
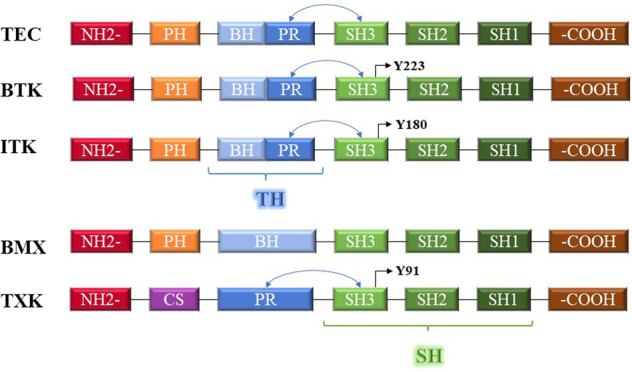
PH pleckstrin homology, CS cysteine, BH Btk homology, PR proline-rich, TH Tec homology, SH Src homology, Y223, Y180, Y91 autophosphorylation sites; The PR motif can bind to the SH3 domain in an intramolecular manner.
Tec
Tec function
Tec is very widely expressed in the human body and serves multiple functions. Tec tyrosine kinase plays an important role in the immune system which is involved in T cell development, cytokine production, T helper cell differentiation and B-cell development and activation [28, 29]. Tec, along with Btk, plays an important role in bone immunity and bone differentiation [30, 31]. Tec is also expressed in the liver, heart, kidney, and ovary [32], and is involved in the progression of a variety of diseases. Even in hepatoma cells, the expression of Tec has been found [32]. In recent years, the link between Tec and cardiovascular disease has attracted much attention, especially in ischemic heart disease and cardiac pressure overload hypertrophy.
The role of Tec in ischemic heart disease
According to the World Health Organization, ischemic heart disease is caused by myocardial damage due to an imbalance between coronary blood flow and myocardial demand consequent of changes in the coronary circulation. The most common cause of ischemic heart disease is coronary artery stenosis and occlusion caused by coronary atherosclerosis. Insufficient blood supply and oxygen supply cause myocardial injury, which in turn develops into coronary heart disease or myocardial infarction. Tec translocates to Triton X-100 (TX-100)-insoluble fractions after ischemia and reperfusion. Adenosine and other purine nucleotides are released during ischemia-reperfusion. It has been proved that purinergic stimulation, but not hypoxia, induces translocation of Tec in cardiac cells [33]. Tec regulates proteins associated with lipid membranes. Tec is clearly involved in the temporal response of the heart to ischemia–reperfusion, and plays a protective role in myocardial ischemia [33]. HIF-1α, identified as a Tec interactor [33], is an important mediator of adenosine nucleoside-induced neuroprotection in hypoxia-stimulated PC12 cells [34]. Since Tec is activated by adenosine nucleotide stimulation in cardiomyocytes, it may participate in HIF-1α signaling during cardiac protection.
The role of Tec in cardiac pressure overload hypertrophy
Rho kinase 1 (ROCK1), an immediate downstream kinase effector of RhoA GTPase, was be found as a Tec interactor [33]. A study [35] has indicated a critical role for ROCK1 in reactive fibrosis during pressure-overload hypertrophy. Tec may play a cardioprotective role by binding to ROCK1. However, the specifics of how Tec may contribute to this process are unclear.
Btk
Btk function
In 1993, Btk began to receive attention after it was reported that mutations in its gene result in X-linked agammaglobulinemia (XLA) in humans. Btk is the most extensively studied kinase among the Tec family of kinases. Btk plays an important role in immune function; is involved in many autoimmune diseases [36]. Btk primarily mediates B cell receptor signaling [37, 38]. Btk is also found in innate immune cells [38] and T cells [39] and is expressed in megakaryocytes and platelets [40]. Btk is involved in the inflammatory response in vivo and may play an important role at multiple points during the development and function of the bone marrow lineage. There is currently no definitive evidence that Btk is expressed in cardiomyocytes and vascular endothelial cells. What’s interesting is that Btk is closely related to a variety of cardiovascular diseases, such as atherosclerosis, cardiac dysfunction associated with sepsis and atrial fibrillation. And the mechanism of Btk in cardiovascular disease is complex.
The role of Btk in atherosclerosis
Atherosclerosis is a chronic metabolic disease of the arterial wall characterized by lipid deposition and persistent aseptic inflammation. Atherosclerosis causes arterial stenosis, resulting in tissue and organ ischemia and hypoxia. Atherosclerosis is considered to underlie a variety of cardiovascular and cerebrovascular diseases. The effect of Btk on atherosclerosis manifests in the following three ways.
In the initial stages of atherosclerosis, reactive oxygen species signaling through Btk-p300-STAT1-PPARγ activates cholesterol crystal-induced CD36 expression and foam cell formation [41]. Btk promotes oxidized low-density lipoprotein (ox-LDL)-induced endoplasmic reticulum stress, oxidative stress, and the inflammatory response in macrophages, and Btk knockdown can inhibit ox-LDL-induced signaling activation and M1 polarization in NK-κ macrophages [42]. Btk can also promote atherosclerosis progression.
The NLRP3 inflammasome is an innate immune signaling complex. NLRP3 inflammasome activation helps drive atherosclerosis development and progression of vascular inflammatory responses and is also a major producer of cleaved interleukin (IL)-1 cytokines [43]. The NLRP3 inflammasome is activated by cholesterol crystals and is required for atherogenesis [44]. IL-1 cytokines are closely related to various cardiovascular diseases, such as atherosclerosis and myocardial infarction [45]. Btk is important for NLRP3 inflammasome activation. Btk regulates the assembly and, hence, activation of the NLRP3 inflammasome by binding to the ASC (apoptosis-associated speck-like protein containing a CARD) component [46, 47]. Inhibition of BTK by pharmacological or genetic means severely impairs activation of the NLRP3 inflammasome [47]. Targeting the NLRP3 inflammasome using Btk has received attention as another feasible approach for the treatment of cardiovascular disease [48].
Platelet activation and aggregation are key events in acute arterial thrombosis during plaque formation, which is one of the key factors affecting prognosis. Btk is involved in botrocetin/von Willebrand factor-mediated and lectin-induced TXA2 production and also in platelet membrane glycoprotein Ib-IX-V complex-dependent arterial thrombosis [49, 50]. Although Tec has been found to compensate for lack of Btk in patients with XLA leaving patients without a bleeding phenotype [51], Tec cannot compensate for the missing function of Btk during platelet activation and aggregation caused by atherosclerotic plaques [49]. Atherosclerotic plaques stimulate static platelet aggregation and platelet thrombosis through collagen I and III of the collagen receptor glycoprotein VI (GPVI). It has long been shown that Btk is important for collagen signaling through GPVI. GPVI is coupled to the Fc receptor gamma chain (FcRgamma). The FcRgamma-chain contains a consensus sequence known as the immune-receptor tyrosine-based activation motif (ITAM). Tyrosine phosphorylation of the ITAM upon GPVI stimulation is the initial step in the regulation of phospholipase C gamma2 (PLCγ2) isoforms via the tyrosine kinase p72 in platelets. Collagen and a collagen-related peptide (CRP) binds to GPVI and induced Btk tyrosine phosphorylation in platelets. Btk plays a crucial role during activation of platelets by CRP and collagen by regulating tyrosine phosphorylation and activation of PLCγ2 [52]. But Btk is not essential for thrombin-mediated platelet activation [52]; that is, Btk does not affect the physiological hemostatic process. Studies with multiple Btk inhibitors, such as acalabrutinib and ONO/GS-4059, have shown that Btk inhibitors specifically block the formation of platelet thrombi in atherosclerotic plaques, but do not affect physiological hemostasis [53–55]. Moreover, low-dose irreversible Btk inhibitors have unique potential as antiplatelet agents in atherothrombosis [56].
The role of Btk in cardiac dysfunction associated with sepsis
Sepsis leads to a complex intramyocardial inflammatory response and sepsis-induced myocardial dysfunction [57, 58]. Toll-like receptor 3 (TLR3) mediates antiviral response by recognizing double-stranded RNA. Its cytoplasmic domain is tyrosine phosphorylated upon ligand binding and initiates downstream signaling via the adapter TIR-containing adapter inducing interferon-β (TRIF). Bruton’s tyrosine kinase phosphorylates Toll-like receptor 3 to initiate antiviral response [59], which facilitates recovery of cardiac dysfunction associated with sepsis.
The activation of NF-κB plays an important role in the cardiac dysfunction in sepsis [60, 61]. Inhibition of the activity of NF-κB attenuates the cardiac dysfunction in sepsis [62, 63]. Inhibition of BTK activity with ibrutinib or acalabrutinib reduces both the activation of NF-κB in septic hearts and the cardiac dysfunction caused by cecal ligation and puncture (CLP)-sepsis [64].
The activation of the NLRP3 inflammasome is a key component in the cardiac dysfunction [65] and the pathophysiology of sepsis [66]. This effect may result from activation of IL-1β and IL-18 via NLRP3 inflammasome [67, 68]. As previously stated BTK regulates the assembly and, hence, activation of the NLRP3 inflammasome by binding to the ASC component [46, 47]. Inhibition of BTK activity reduces both the assembly and subsequent activation of the NLRP3 inflammasome in septic hearts, resulting in reduced serum levels of IL-1β, and this contributes to the improvement of cardiac function [64].
The role of Btk in atrial fibrillation
Atrial fibrillation (AF) is a type of arrhythmia caused by abnormal cardiac electrical conduction. However, an enhanced inflammatory response is frequently observed in AF patients. Btk promotes activation of the NLRP3 inflammasome as described above [46, 47]. Studies have shown that increases in NLRP3-inflammasome in cardiomyocytes might contribute to the evolution of atrial remodeling that promotes AF induction and maintenance. CM-specific activation of NLRP3 promotes abnormal sarcoplasmic reticulum (SR) Ca2+ release and electrical remodeling. Genetic inhibition of NLRP3 prevents spontaneous AF in CREM-transgenic mice (a well-characterized mouse model of spontaneous AF), which is sufficient to illustrate the importance of NLRP3 in atrial fibrillation [69]. But the role and mechanism of Btk in atrial fibrillation needs to be further studied.
Thus, through NLRP3 inflammasome, Btk is closely linked to atherosclerosis, cardiac dysfunction associated with sepsis, and atrial fibrillation. NLRP3 inflammasome is a hub where Btk is linked to cardiovascular disease (Fig. 3). Some Btk kinase inhibitors have shown good application prospects in the treatment of cardiovascular disease (Table 3).
Fig. 3. NLRP3-centered, cardiovascular disease network diagram related to Btk.

CC cholesterol crystals, ASC apoptosis-associated speck-like protein containing a CARD, NLRP3 Human NACHT, LRR, and PYD domain-containing protein 3, IL interleukin.
Bmx
Bmx function
Bmx was initially found to be expressed in the granulocytic monocyte lineage within the hematopoietic system and is involved in the immune response. Bmx is also involved in certain inflammatory diseases, enhances IL-8 secretion, and is an important inflammatory mediator in vivo [70]. Bmx expression has also been demonstrated in a variety of cancers, such as prostate and breast cancer [71]. Bmx is also specifically expressed in the endothelium of the endocardium and arteries [72]; is involved in the growth, differentiation, apoptosis, and proliferation of epithelial cells; and plays an important role in cardiovascular disease.
The role of Bmx in cardiac hypertrophy
The heart undergoes hypertrophy in response to hemodynamic overload to increase contractility and reduce ventricular wall stress. This adaptive hypertrophy will eventually transform into heart failure through pathological remodeling. Bmx knockout mice showed reduced cardiac hypertrophy in a transverse aortic constriction model [73] and reduced vascular endothelial growth factor-beta (VEGF-β) transgene-induced cardiac hypertrophy [74]. In an angiotensin II (Ang II)-induced cardiac hypertrophy model, the occurrence of cardiac hypertrophy was significantly reduced in Bmx-deficient or Bmx-inactivated mice. Bmx inactivation inhibits myocardial expression of genes associated with Ang II-induced inflammation and the extracellular matrix response, while in Bmx-inactivated hearts, Ang II administration maintains the expression of RNA encoding mitochondrial proteins [75]. Moreover, Bmx expression is high in arterial endothelial cells and minimal in cardiomyocytes, and Bmx RNA is detected in cardiac fibroblasts. These results strongly suggest that the effects of Bmx on cardiac hypertrophy are mediated through endothelial cells of the coronary vessels [75]. Mitochondria are particularly important for maintaining a healthy myocardium. STAT3 activation is an important cardioprotective factor associated with cardiac hypertrophy [76, 77], and the role of BMX gene silencing is to inhibit downstream STAT3 signaling [75].
The role of Bmx in coronary atherosclerotic heart disease
Coronary atherosclerosis causes narrowing or obstruction of the vascular lumen, resulting in myocardial ischemia, hypoxia, or necrosis, causing coronary atherosclerotic heart disease. Long-term narrowing of the coronary arteries causes vascular remodeling to compensate for an insufficient blood supply to the heart. Vascular endothelial growth factor (VEGF) is an important proangiogenic factor [78]. Nuclear localization of Bmx mediates VEGF receptor 2 (VEGFR2) expression and regulates VEGF-induced angiogenesis [79]. Bmx is also a downstream effector of VEGFR2 [80] and plays an important role in ischemia-induced arteriogenesis and angiogenesis. Bmx is also involved in cell adhesion, migration, and survival in tumor necrosis factor-induced angiogenesis [81]. In a study using VEGF-2 gene therapy for coronary heart disease, patients had reduced angina pectoris and significantly improved perfusion and function [82]. It has also been shown that deletion of endothelial Bmx tyrosine kinase reduces tumor angiogenesis and growth [83]. Thus, Bmx may be a new target for the treatment of vascular diseases, such as coronary artery disease.
The role of Bmx in myocardial infarction and post-myocardial infarction
Myocardial infarction is myocardial necrosis caused by acute or chronic, persistent ischemia and hypoxia of the coronary arteries. It can be complicated by ventricular aneurysm, cardiac rupture, mural thrombosis, arrhythmia, heart failure, cardiogenic shock, and post-myocardial infarction syndrome, which often endanger the patient’s life. Treatment and prognosis of myocardial infarction are equally important. Protein kinase C (PKC)-ε is a central modulator of cardioprotective signal transduction and can be used to prevent and treat myocardial infarction [84]. Moreover, Bmx plays a cardioprotective role by participating in nitric oxide-induced PKC-ε signaling [85]. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis and improves cardiac function after myocardial infarction [86]. Moreover, Bmx is involved in mediating the mechanism of VEGF-dependent lymphangiogenesis signaling [87]. Thus, the application of Bmx in the prevention, treatment, and prognosis of myocardial infarction demonstrates promise.
The role of Bmx in cardiac hypertrophy, atherosclerotic heart disease, and myocardiun infarction are all associated with VEGF signaling. Here we summarize the link of Bmx to cardiovascular disease mediated through VEGF signaling (Fig. 4).
Fig. 4. The link of Bmx to cardiovascular disease mediated through VEGF signaling.

VEGF vascular endothelial growth factor, VEGF-β vascular endothelial growth factor-beta, VEGFR2 vascular endothelial growth factor receptor 2.
Itk and Txk
Itk and Txk function
In T cells, four Tec kinases are expressed: Itk, Rlk, Tec, and Btk. These Tec kinases are important in T cell development and mature T cell differentiation, especially in TCR signaling [88]. Among them, Itk has the highest level of expression and is also known as Emt. It is expressed in mast cells [89] and natural killer cells [90] and is involved in certain autoimmune diseases, such as asthma [91] and atopic dermatitis [92]. Itk is also associated with inflammatory reactions [93], certain cancers [94] and viral infection [95]. Itk inhibitors have achieved beneficial results in many studies on disease treatment [96, 97]. Txk, also known as Rlk, is mainly detected in T cells and some myeloid cell lineages [98]. Txk has been implicated in immune-inflammatory diseases, such as rheumatoid arthritis, bronchial asthma, and atopic dermatitis [99, 100]. However, no association between Itk and Txk and cardiovascular disease has been found thus far.
Conclusions and prospects
Tec is a stress-activated kinase. Upon cardiac ischemic injury, Tec kinase is translocated and exerts cardioprotective effects by eliciting downstream signaling through binding to H1F-1α and ROCK1. Tec kinases are functionally diverse and expressed in a variety of organs, with clear evidence of expression in the heart. Tec kinase plays an important role in the growth and development of immune cells. If the expression of Tec gene is inhibited in the heart, will it have an effect on the function of the heart? Currently, only a clear association between Tec and Ischemic heart disease was found in the current study. So it is unknown whether Tec has a corresponding relationship with other cardiovascular diseases and whether it also plays a protective role in it.
Btk promotes the progression of atherosclerosis by participating in the Btk–p300–STAT1–PPARγ pathway and promoting a series of responses induced by ox-LDL and NLRP3. Btk promotes platelet activation mediated by atherosclerotic plaques. Although Btk initiates an antiviral response by activating TLR3, Btk aggravates the cardiac dysfunction associated with sepsis by promoting the conduction of NF-κB and NLRP3 and their downstream signals. Btk contributes to the development of atrial fibrillation by affecting NLRP3. Many studies have demonstrated that Bmx can reduce the occurrence of cardiac hypertrophy, acting through the coronary endothelium and STAT3. Overall, Btk inhibitors have a good application prospect in the treatment of Atherosclerosis and cardiac dysfunction associated with sepsis, providing us with new ideas for cardiovascular disease treatment. However, it is unknown whether Btk inhibitors can be used in the clinical treatment of cardiovascular disease and whether they can have the expectant effect of treatment or prevention. A large number of studies are needed to explore the clinical significance of Btk inhibitors. Interestingly, while there is no evidence for Btk expression in the heart and blood vessels, we can see that through NLRP3, Btk is closely linked to three diseases: Atherosclerosis, Cardiac dysfunction associated with sepsis, and Atrial fibrillation. Are NLRP3 involved in other cardiovascular diseases? Are Btk involved in other cardiovascular diseases via NLRP3? We also need to further explore and clarify the interaction mechanism of Btk with NLRP3.
Bmx knockout can reduced VEGF-β transgene-induced cardiac hypertrophy. Mitochondria are particularly important for maintaining a healthy myocardium. The role of BMX gene silencing is to inhibit downstream STAT3 signaling which is an important cardioprotective factor associated with cardiac hypertrophy. Bmx exhibits positive and negative effects in cardiac hypertrophy. If the expression of Bmx is inhibited, it still needs to be clarified how it will eventually have an effect on cardiac hypertrophy. Bmx is involved in VEGF-induced angiogenesis and plays a beneficial role in coronary atherosclerotic heart disease. Bmx protects the heart in myocardial infarction by participating in nitric oxide-evoked PKC-ε signaling, which is involved in mediating VEGF-dependent lymphopoiesis signaling. We note that the effect of Bmx on cardiovascular disease, is mediated through VEGF signaling, which is very similar to the link between Btk and cardiovascular disease. VEGF signaling is associated with a variety of cardiovascular diseases, then it is unknown whether Bmx is involved in other cardiovascular diseases through VEGF signaling.
The expression of Tec kinases is widespread. In recent years, the association of Tec kinases, especially Btk and Bmx, with cardiovascular diseases has received extensive attention. To explore the relationship between Tec family tyrosine kinases and cardiovascular diseases, would provide us with new ideas for cardiovascular disease treatment.
Supplementary information
Author contributions
NZ performed the research, YS and YZ designed the research study, ZY, YMZ, DW, XYH and SJX wrote paper and LC revised it. ZY drew graphs and tables.
Funding
This study was supported by National Natural Science Foundation of China (82171572, 82171571, 81970211, 81900355 and 81900372) and National Postdoctoral Innovative Talents Support Program (BX2021376).
Data availability
The [DATA TYPE] data used to support the findings of this study are available from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ying Zhang, Email: yzhang02@cmu.edu.cn.
Yingxian Sun, Email: yxsun@cmu.edu.cn.
Naijin Zhang, Email: njzhang@cmu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41420-022-00927-4.
References
- 1.Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–98. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 2.Neet K, Hunter T. Vertebrate non-receptor protein-tyrosine kinase families. Genes Cells. 1996;1:147–69. doi: 10.1046/j.1365-2443.1996.d01-234.x. [DOI] [PubMed] [Google Scholar]
- 3.Andreotti AH, Bunnell SC, Feng S, Berg LJ, Schreiber SL. Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature. 1997;385:93–7. doi: 10.1038/385093a0. [DOI] [PubMed] [Google Scholar]
- 4.Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–57. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 5.Mano H. The Tec family protein-tyrosine kinases: a subset of kinases for a subset of signalings. Int J Hematol. 1999;69:6–12. [PubMed] [Google Scholar]
- 6.Mao J, Xie W, Yuan H, Simon MI, Mano H, Wu D. Tec/Bmx non-receptor tyrosine kinases are involved in regulation of Rho and serum response factor by Galpha12/13. EMBO J. 1998;17:5638–46.. doi: 10.1093/emboj/17.19.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomlinson MG, Kurosaki T, Berson AE, Fujii GH, Johnston JA, Bolen JB. Reconstitution of Btk signaling by the atypical tec family tyrosine kinases Bmx and Txk. J Biol Chem. 1999;274:13577–85. doi: 10.1074/jbc.274.19.13577. [DOI] [PubMed] [Google Scholar]
- 8.Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang YS. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 2018;17:36. doi: 10.1186/s12943-018-0801-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Norman P. Investigational Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Expert Opin Investig Drugs. 2016;25:891–9. doi: 10.1080/13543784.2016.1182499. [DOI] [PubMed] [Google Scholar]
- 10.Gotlib J. Tyrosine kinase inhibitors in the treatment of eosinophilic neoplasms and systemic mastocytosis. Hematol Oncol Clin North Am. 2017;31:643–61. doi: 10.1016/j.hoc.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 11.Wu B, You S, Qian H, Wu S, Lu S, Zhang Y, et al. The role of SIRT2 in vascular-related and heart-related diseases: a review. J Cell Mol Med. 2021;25:6470–8. doi: 10.1111/jcmm.16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang P, Zhang N, Wu B, Wu S, Zhang Y, Sun Y. The role of mitochondria in vascular calcification. J Transl Int Med. 2020;8:80–90. doi: 10.2478/jtim-2020-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Qian H, Wu B, You S, Wu S, Lu S, et al. E3 Ubiquitin ligase NEDD4 family‑regulatory network in cardiovascular disease. Int J Biol Sci. 2020;16:2727–40.. doi: 10.7150/ijbs.48437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemmon MA, Ferguson KM, Schlessinger J. PH domains: diverse sequences with a common fold recruit signaling molecules to the cell surface. Cell. 1996;85:621–4. doi: 10.1016/s0092-8674(00)81022-3. [DOI] [PubMed] [Google Scholar]
- 15.Vihinen M, Nilsson L, Smith CI. Tec homology (TH) adjacent to the PH domain. FEBS Lett. 1994;350:263–5. doi: 10.1016/0014-5793(94)00783-7. [DOI] [PubMed] [Google Scholar]
- 16.Harlan JE, Hajduk PJ, Yoon HS, Fesik SW. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature. 1994;371:168–70. doi: 10.1038/371168a0. [DOI] [PubMed] [Google Scholar]
- 17.Debnath J, Chamorro M, Czar MJ, Schaeffer EM, Lenardo MJ, Varmus HE, et al. rlk/TXK encodes two forms of a novel cysteine string tyrosine kinase activated by Src family kinases. Mol Cell Biol. 1999;19:1498–507. doi: 10.1128/mcb.19.2.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mano H. Tec family of protein-tyrosine kinases: an overview of their structure and function. Cytokine Growth Factor Rev. 1999;10:267–80. doi: 10.1016/s1359-6101(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 19.Andersen TCB, Kristiansen PE, Huszenicza Z, Johansson MU, Gopalakrishnan RP, Kjelstrup H, et al. The SH3 domains of the protein kinases ITK and LCK compete for adjacent sites on T cell-specific adapter protein. J Biol Chem. 2019;294:15480–94. doi: 10.1074/jbc.RA119.008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guinamard R, Fougereau M, Seckinger P. The SH3 domain of Bruton’s tyrosine kinase interacts with Vav, Sam68 and EWS. Scand J Immunol. 1997;45:587–95. doi: 10.1046/j.1365-3083.1997.d01-447.x. [DOI] [PubMed] [Google Scholar]
- 21.Yamashita Y, Miyazato A, Ohya K, Ikeda U, Shimada K, Miura Y, et al. Deletion of Src homology 3 domain results in constitutive activation of Tec protein-tyrosine kinase. Jpn J Cancer Res. 1996;87:1106–10. doi: 10.1111/j.1349-7006.1996.tb03118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayer BJ, Hirai H, Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr Biol. 1995;5:296–305.. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- 23.Yang WC, Collette Y, Nunès JA, Olive D. Tec kinases: a family with multiple roles in immunity. Immunity. 2000;12:373–82. doi: 10.1016/s1074-7613(00)80189-2. [DOI] [PubMed] [Google Scholar]
- 24.Park H, Wahl MI, Afar DE, Turck CW, Rawlings DJ, Tam C, et al. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity. 1996;4:515–25. doi: 10.1016/s1074-7613(00)80417-3. [DOI] [PubMed] [Google Scholar]
- 25.Joseph RE, Fulton DB, Andreotti AH. Mechanism and functional significance of Itk autophosphorylation. J Mol Biol. 2007;373:1281–92. doi: 10.1016/j.jmb.2007.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joseph RE, Severin A, Min L, Fulton DB, Andreotti AH. SH2-dependent autophosphorylation within the Tec family kinase Itk. J Mol Biol. 2009;391:164–77. doi: 10.1016/j.jmb.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kashiwakura J, Suzuki N, Takeno M, Itoh S, Oku T, Sakane T, et al. Evidence of autophosphorylation in Txk: Y91 is an autophosphorylationsite. Biol Pharm Bull. 2002;25:718–21. doi: 10.1248/bpb.25.718. [DOI] [PubMed] [Google Scholar]
- 28.Lucas JA, Miller AT, Atherly LO, Berg LJ. The role of Tec family kinases in T cell development and function. Immunol Rev. 2003;191:119–38. doi: 10.1034/j.1600-065x.2003.00029.x. [DOI] [PubMed] [Google Scholar]
- 29.Kitanaka A, Mano H, Conley ME, Campana D. Expression and activation of the nonreceptor tyrosine kinase Tec in human B cells. Blood. 1998;91:940–8. [PubMed] [Google Scholar]
- 30.Boyce BF, Xing L. Bruton and Tec: new links in osteoimmunology. Cell Metab. 2008;7:283–5. doi: 10.1016/j.cmet.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 31.Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132:794–806. doi: 10.1016/j.cell.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 32.Mano H, Ishikawa F, Nishida J, Hirai H, Takaku F. A novel protein-tyrosine kinase, tec, is preferentially expressed in liver. Oncogene. 1990;5:1781–6. [PubMed] [Google Scholar]
- 33.Zhang MJ, Franklin S, Li Y, Wang S, Ru X, Mitchell-Jordan SA, et al. Stress signaling by Tec tyrosine kinase in the ischemic myocardium. Am J Physiol Heart Circ Physiol. 2010;299:H713–22. doi: 10.1152/ajpheart.00273.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.zur Nedden S, Tomaselli B, Baier-Bitterlich G. HIF-1 alpha is an essential effector for purine nucleoside-mediated neuroprotection against hypoxia in PC12 cells and primary cerebellar granule neurons. J Neurochem. 2008;105:1901–14. doi: 10.1111/j.1471-4159.2008.05275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang YM, Bo J, Taffet GE, Chang J, Shi J, Reddy AK, et al. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. FASEB J. 2006;20:916–25. doi: 10.1096/fj.05-5129com. [DOI] [PubMed] [Google Scholar]
- 36.Liang C, Tian D, Ren X, Ding S, Jia M, Xin M, et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini-review. Eur J Med Chem. 2018;151:315–26. doi: 10.1016/j.ejmech.2018.03.062. [DOI] [PubMed] [Google Scholar]
- 37.Corneth OBJ, Klein Wolterink RGJ, Hendriks RW. BTK signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol. 2016;393:67–105. doi: 10.1007/82_2015_478. [DOI] [PubMed] [Google Scholar]
- 38.Crofford LJ, Nyhoff LE, Sheehan JH, Kendall PL. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev Clin Immunol. 2016;12:763–73. doi: 10.1586/1744666X.2016.1152888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia S, Liu X, Cao X, Xu S. T-cell expression of Bruton’s tyrosine kinase promotes autoreactive T-cell activation and exacerbates aplastic anemia. Cell Mol Immunol. 2020;17:1042–52. doi: 10.1038/s41423-019-0270-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Futatani T, Watanabe C, Baba Y, Tsukada S, Ochs HD. Bruton’s tyrosine kinase is present in normal platelets and its absence identifies patients with X-linked agammaglobulinaemia and carrier females. Br J Haematol. 2001;114:141–9.. doi: 10.1046/j.1365-2141.2001.02905.x. [DOI] [PubMed] [Google Scholar]
- 41.Kotla S, Singh NK, Rao GN. ROS via BTK-p300-STAT1-PPARγ signaling activation mediates cholesterol crystals-induced CD36 expression and foam cell formation. Redox Biol. 2017;11:350–64. doi: 10.1016/j.redox.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiu J, Fu Y, Chen Z, Zhang L, Li L, Liang D, et al. BTK promotes atherosclerosis by regulating oxidative stress, mitochondrial injury, and ER stress of macrophages. Oxid Med Cell Longev. 2021;2021:9972413. doi: 10.1155/2021/9972413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ Res. 2018;122:1722–40. doi: 10.1161/CIRCRESAHA.118.311362. [DOI] [PubMed] [Google Scholar]
- 44.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfeiler S, Winkels H, Kelm M, Gerdes N. IL-1 family cytokines in cardiovascular disease. Cytokine. 2019;122:154215. doi: 10.1016/j.cyto.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 46.Liu X, Pichulik T, Wolz OO, Dang TM, Stutz A, Dillen C, et al. Human NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome activity is regulated by and potentially targetable through Bruton tyrosine kinase. J. Allergy Clin Immunol. 2017;140:1054–1067.e10. doi: 10.1016/j.jaci.2017.01.017. [DOI] [PubMed] [Google Scholar]
- 47.Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun. 2015;6:7360. doi: 10.1038/ncomms8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber ANR. Targeting the NLRP3 Inflammasome via BTK. Front Cell Dev Biol. 2021;9:630479. doi: 10.3389/fcell.2021.630479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu J, Fitzgerald ME, Berndt MC, Jackson CW, Gartner TK. Bruton tyrosine kinase is essential for botrocetin/VWF-induced signaling and GPIb-dependent thrombus formation in vivo. Blood. 2006;108:2596–603. doi: 10.1182/blood-2006-01-011817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jamasbi J, Megens RT, Bianchini M, Münch G, Ungerer M, Faussner A, et al. Differential inhibition of human atherosclerotic plaque-induced platelet activation by dimeric GPVI-Fc and anti-GPVI antibodies: functional and imaging studies. J Am Coll Cardiol. 2015;65:2404–15. doi: 10.1016/j.jacc.2015.03.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oda A, Ikeda Y, Ochs HD, Druker BJ, Ozaki K, Handa M, et al. Rapid tyrosine phosphorylation and activation of Bruton’s tyrosine/Tec kinases in platelets induced by collagen binding or CD32 cross-linking. Blood. 2000;95:1663–70. [PubMed] [Google Scholar]
- 52.Quek LS, Bolen J, Watson SP. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8:1137–40. doi: 10.1016/s0960-9822(98)70471-3. [DOI] [PubMed] [Google Scholar]
- 53.Busygina K, Jamasbi J, Seiler T, Deckmyn H, Weber C, Brandl R, et al. Oral Bruton tyrosine kinase inhibitors selectively block atherosclerotic plaque-triggered thrombus formation in humans. Blood. 2018;131:2605–16. doi: 10.1182/blood-2017-09-808808. [DOI] [PubMed] [Google Scholar]
- 54.Lindner JR. Btk inhibitors in atherosclerosis. Blood. 2018;131:2601–2. doi: 10.1182/blood-2018-04-841916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Denzinger V, Busygina K, Jamasbi J, Pekrul I, Spannagl M, Weber C, et al. Optimizing platelet GPVI inhibition versus haemostatic impairment by the Btk inhibitors ibrutinib, acalabrutinib, ONO/GS-4059, BGB-3111 and evobrutinib. Thromb Haemost. 2019;119:397–406. doi: 10.1055/s-0039-1677744. [DOI] [PubMed] [Google Scholar]
- 56.Busygina K, Denzinger V, Bernlochner I, Weber C, Lorenz R, Siess W. Btk Inhibitors as first oral atherothrombosis-selective antiplatelet drugs? Thromb Haemost. 2019;119:1212–21. doi: 10.1055/s-0039-1687877. [DOI] [PubMed] [Google Scholar]
- 57.Walley KR. Sepsis-induced myocardial dysfunction. Curr Opin Crit Care. 2018;24:292–9. doi: 10.1097/MCC.0000000000000507. [DOI] [PubMed] [Google Scholar]
- 58.Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med. 2007;35:1599–608. doi: 10.1097/01.CCM.0000266683.64081.02. [DOI] [PubMed] [Google Scholar]
- 59.Lee KG, et al. Bruton’s tyrosine kinase phosphorylates Toll-like receptor 3 to initiate antiviral response. Proc Natl Acad Sci USA. 2012;109:5791–6. doi: 10.1073/pnas.1119238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee KG, Xu S, Kang ZH, Huo J, Huang M, Liu D, et al. Activation of NF-kappaB varies in different regions of the gastrointestinal tract during endotoxemia. Shock. 2000;14:118–22. doi: 10.1097/00024382-200014020-00007. [DOI] [PubMed] [Google Scholar]
- 61.Liu SF, Ye X, Malik AB. Pyrrolidine dithiocarbamate prevents I-kappaB degradation and reduces microvascular injury induced by lipopolysaccharide in multiple organs. Mol Pharm. 1999;55:658–67. [PubMed] [Google Scholar]
- 62.Al Zoubi S, Chen J, Murphy C, Martin L, Chiazza F, Collotta D, et al. Linagliptin attenuates the cardiac dysfunction associated with experimental sepsis in mice with pre-existing type 2 diabetes by inhibiting NF-κB. Front Immunol. 2018;9:2996. doi: 10.3389/fimmu.2018.02996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen J, Kieswich JE, Chiazza F, Moyes AJ, Gobbetti T, Purvis GS, et al. IκB kinase inhibitor attenuates sepsis-induced cardiac dysfunction in CKD. J Am Soc Nephrol. 2017;28:94–105. doi: 10.1681/ASN.2015060670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Riordan CE, Purvis GSD, Collotta D, Chiazza F, Wissuwa B, Al Zoubi S, et al. Bruton’s tyrosine kinase inhibition attenuates the cardiac dysfunction caused by cecal ligation and puncture in mice. Front Immunol. 2019;10:2129. doi: 10.3389/fimmu.2019.02129. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Zhang W, Xu X, Kao R, Mele T, Kvietys P, Martin CM, et al. Cardiac fibroblasts contribute to myocardial dysfunction in mice with sepsis: the role of NLRP3 inflammasome activation. PLoS ONE. 2014;9:e107639. doi: 10.1371/journal.pone.0107639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee S, Nakahira K, Dalli J, Siempos II, Norris PC, Colas RA, et al. NLRP3 inflammasome deficiency protects against microbial sepsis via increased lipoxin B(4) synthesis. Am J Respir Crit Care Med. 2017;196:713–26. doi: 10.1164/rccm.201604-0892OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vanden Berghe T, Demon D, Bogaert P, Vandendriessche B, Goethals A, Depuydt B, et al. Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am J Respir Crit Care Med. 2014;189:282–91. doi: 10.1164/rccm.201308-1535OC. [DOI] [PubMed] [Google Scholar]
- 68.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci USA. 2001;98:2871–6. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yao C, Veleva T, Scott L, Jr, Cao S, Li L, Chen G, et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation. 2018;138:2227–42. doi: 10.1161/CIRCULATIONAHA.118.035202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gottar-Guillier M, Dodeller F, Huesken D, Iourgenko V, Mickanin C, Labow M, et al. The tyrosine kinase BMX is an essential mediator of inflammatory arthritis in a kinase-independent manner. J Immunol. 2011;186:6014–23. doi: 10.4049/jimmunol.1002813. [DOI] [PubMed] [Google Scholar]
- 71.Guo L, Guo Y, Xiao S. Expression of Etk/Bmx tyrosine kinase in intrahepatic cholangiocarcinoma. J Surg Oncol. 2008;97:428–32. doi: 10.1002/jso.20983. [DOI] [PubMed] [Google Scholar]
- 72.Ekman N, Lymboussaki A, Västrik I, Sarvas K, Kaipainen A, Alitalo K. Bmx tyrosine kinase is specifically expressed in the endocardium and the endothelium of large arteries. Circulation. 1997;96:1729–32. doi: 10.1161/01.cir.96.6.1729. [DOI] [PubMed] [Google Scholar]
- 73.Mitchell-Jordan SA, Holopainen T, Ren S, Wang S, Warburton S, Zhang MJ, et al. Loss of Bmx nonreceptor tyrosine kinase prevents pressure overload-induced cardiac hypertrophy. Circ Res. 2008;103:1359–62. doi: 10.1161/CIRCRESAHA.108.186577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bry M, Kivelä R, Holopainen T, Anisimov A, Tammela T, Soronen J, et al. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122:1725–33. doi: 10.1161/CIRCULATIONAHA.110.957332. [DOI] [PubMed] [Google Scholar]
- 75.Holopainen T, Räsänen M, Anisimov A, Tuomainen T, Zheng W, Tvorogov D, et al. Endothelial Bmx tyrosine kinase activity is essential for myocardial hypertrophy and remodeling. Proc Natl Acad Sci USA. 2015;112:13063–8. doi: 10.1073/pnas.1517810112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ye S, Luo W, Khan ZA, Wu G, Xuan L, Shan P, et al. Celastrol Attenuates angiotensin II-induced cardiac remodeling by targeting STAT3. Circ Res. 2020;126:1007–23. doi: 10.1161/CIRCRESAHA.119.315861. [DOI] [PubMed] [Google Scholar]
- 77.Zouein FA, Booz GW, Altara R. STAT3 and endothelial cell-cardiomyocyte dialog in cardiac remodeling. Front Cardiovasc Med. 2019;6:50. doi: 10.3389/fcvm.2019.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 79.Liu T, Li Y, Su H, Zhang H, Jones D, Zhou HJ, et al. Nuclear localization of the tyrosine kinase BMX mediates VEGFR2 expression. J Cell Mol Med. 2020;24:126–38. doi: 10.1111/jcmm.14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.He Y, Luo Y, Tang S, Rajantie I, Salven P, Heil M, et al. Critical function of Bmx/Etk in ischemia-mediated arteriogenesis and angiogenesis. J Clin Investig. 2006;116:2344–55. doi: 10.1172/JCI28123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pan S, An P, Zhang R, He X, Yin G, Min W. Etk/Bmx as a tumor necrosis factor receptor type 2-specific kinase: role in endothelial cell migration and angiogenesis. Mol Cell Biol. 2002;22:7512–23. doi: 10.1128/MCB.22.21.7512-7523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Freedman SB. Clinical trials of gene therapy for atherosclerotic cardiovascular disease. Curr Opin Lipido. 2002;13:653–61. doi: 10.1097/00041433-200212000-00009. [DOI] [PubMed] [Google Scholar]
- 83.Holopainen T, López-Alpuche V, Zheng W, Heljasvaara R, Jones D, He Y, et al. Deletion of the endothelial Bmx tyrosine kinase decreases tumor angiogenesis and growth. Cancer Res. 2012;72:3512–21. doi: 10.1158/0008-5472.CAN-11-1070. [DOI] [PubMed] [Google Scholar]
- 84.Ping P, Zhang J, Pierce WM, Jr, Bolli R. Functional proteomic analysis of protein kinase C epsilon signaling complexes in the normal heart and during cardioprotection. Circ Res. 2001;88:59–62. doi: 10.1161/01.res.88.1.59. [DOI] [PubMed] [Google Scholar]
- 85.Zhang J, Ping P, Wang GW, Lu M, Pantaleon D, Tang XL, et al. Bmx, a member of the Tec family of nonreceptor tyrosine kinases, is a novel participant in pharmacological cardioprotection. Am J Physiol Heart Circ Physiol. 2004;287:H2364–6. doi: 10.1152/ajpheart.00416.2004. [DOI] [PubMed] [Google Scholar]
- 86.Henri O, Pouehe C, Houssari M, Galas L, Nicol L, Edwards-Lévy F, et al. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation. 2016;133:1484–97. doi: 10.1161/CIRCULATIONAHA.115.020143. [DOI] [PubMed] [Google Scholar]
- 87.Jones D, Xu Z, Zhang H, He Y, Kluger MS, Chen H, et al. Functional analyses of the bone marrow kinase in the X chromosome in vascular endothelial growth factor-induced lymphangiogenesis. Arterioscler Thromb Vasc Biol. 2010;30:2553–61. doi: 10.1161/ATVBAHA.110.214999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Readinger JA, Mueller KL, Venegas AM, Horai R, Schwartzberg PL. Tec kinases regulate T-lymphocyte development and function: new insights into the roles of Itk and Rlk/Txk. Immunol Rev. 2009;228:93–114. doi: 10.1111/j.1600-065X.2008.00757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol. 2010;2:a002287. doi: 10.1101/cshperspect.a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Khurana D, Arneson LN, Schoon RA, Dick CJ, Leibson PJ. Differential regulation of human NK cell-mediated cytotoxicity by the tyrosine kinase Itk. J Immunol. 2007;178:3575–82. doi: 10.4049/jimmunol.178.6.3575. [DOI] [PubMed] [Google Scholar]
- 91.Ferrara TJ, Mueller C, Sahu N, Ben-Jebria A, August A. Reduced airway hyperresponsiveness and tracheal responses during allergic asthma in mice lacking tyrosine kinase inducible T-cell kinase. J Allergy Clin Immunol. 2006;117:780–6. doi: 10.1016/j.jaci.2005.12.1330. [DOI] [PubMed] [Google Scholar]
- 92.Matsumoto Y, Oshida T, Obayashi I, Imai Y, Matsui K, Yoshida NL, et al. Identification of highly expressed genes in peripheral blood T cells from patients with atopic dermatitis. Int Arch Allergy Immunol. 2002;129:327–40. doi: 10.1159/000067589. [DOI] [PubMed] [Google Scholar]
- 93.Huang W, Morales JL, Gazivoda VP, August A. Nonreceptor tyrosine kinases ITK and BTK negatively regulate mast cell proinflammatory responses to lipopolysaccharide. J Allergy Clin Immunol. 2016;137:1197–205. doi: 10.1016/j.jaci.2015.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu Y, Wang X, Deng L, Ping L, Shi Y, Zheng W, et al. ITK inhibition induced in vitro and in vivo anti-tumor activity through downregulating TCR signaling pathway in malignant T cell lymphoma. Cancer Cell Int. 2019;19:32. doi: 10.1186/s12935-019-0754-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Youssefian L, Vahidnezhad H, Yousefi M, Saeidian AH, Azizpour A, Touati A, et al. Inherited interleukin 2-inducible T-cell (ITK) kinase deficiency in siblings with epidermodysplasia verruciformis and Hodgkin lymphoma. Clin Infect Dis. 2019;68:1938–41. doi: 10.1093/cid/ciy942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kaur M, Bahia MS, Silakari O. Inhibitors of interleukin-2 inducible T-cell kinase as potential therapeutic candidates for the treatment of various inflammatory disease conditions. Eur J Pharm Sci. 2012;47:574–88. doi: 10.1016/j.ejps.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 97.Cho HS, Shin HM, Haberstock-Debic H, Xing Y, Owens TD, Funk JO, et al. A small molecule inhibitor of ITK and RLK impairs Th1 differentiation and prevents colitis disease progression. J Immunol. 2015;195:4822–31. doi: 10.4049/jimmunol.1501828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kashiwakura J, Suzuki N, Nagafuchi H, Takeno M, Takeba Y, Shimoyama Y, et al. Txk, a nonreceptor tyrosine kinase of the Tec family, is expressed in T helper type 1 cells and regulates interferon gamma production in human T lymphocytes. J Exp Med. 1999;190:1147–54. doi: 10.1084/jem.190.8.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mihara S, Suzuki N. Role of Txk, a member of the Tec family of tyrosine kinases, in immune-inflammatory diseases. Int Rev Immunol. 2007;26:333–48. doi: 10.1080/08830180701690835. [DOI] [PubMed] [Google Scholar]
- 100.Suzuki N, Nara K, Suzuki T. Skewed Th1 responses caused by excessive expression of Txk, a member of the Tec family of tyrosine kinases, in patients with Behcet’s disease. Clin Med Res. 2006;4:147–51. doi: 10.3121/cmr.4.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The [DATA TYPE] data used to support the findings of this study are available from the corresponding author upon request.