

Accurate experimentally determined structure models of biological macromolecules are used by a large and diverse community of researchers. It is an established practice to base the assessment of the structure model quality on both, expectations of correct stereochemistry and, most importantly, on examination of the model's fit to the primary experimental evidence. In the case of X‐ray crystallography, the primary evidence is provided by the electron density map. The worldwide Protein Data Bank (wwPDB 1 ) is a global repository of macromolecular models and the accompanying experimental data that allow to examine agreement between the electron density and structural model using programs such as Coot, 2 Chimera, 3 Pymol, 4 or Molstack. 5 Throughout its 50‐year history, the PDB has accumulated over 180,000 macromolecular structures, and gained the reputation of the gold standard in structural biology and of the most reliable data resource in biomedical research in general. 6
Recently, the PDB has seen an influx of many depositions from large‐scale crystallographic fragment screening projects using a complex computational procedure called Pan‐Dataset Density Analysis (PanDDA 7 ). Based on a sophisticated multi‐data‐set analysis of reference models and potential ligand‐complex crystals, 7 such group depositions serve the purpose of identifying very low‐occupancy small molecule ligands in macromolecular complexes. In brief, the idea of PanDDA consists of partial background solvent inclusion and subtraction of a virtual “multi‐crystal ground state,” to produce so‐called “event map,” revealing the supposed ligands in each of a multitude of different ligand data sets. As a result, the PDB accumulates large numbers of “group depositions” of many putative ligand complexes of the same protein target that presently do not conform to the primary objective of the PDB as a repository of high‐quality structure models and data. In particular, the maps that can be retrieved for such deposits have dubious agreement with the models (Figure 1).
FIGURE 1.
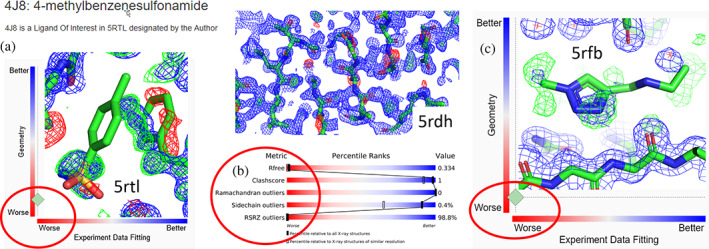
Discrepancies between the atomic model coordinates and electron density maps of representative PanDDA depositions in the PDB. (a) Model of 4‐methylbenzenesulfonamide in SARS‐CoV‐2 NSP3 macrodomain (PDB ID 5rtl) does not fit the electron density, accordant with the poor real space PDB metrics shown in the panel frame as provided by the RCSB PDB site. Map coefficients were calculated from downloaded “Data from final refinement with ligand” and the deposited model. The misplaced phenylalanine residue (to the right of the ligand) indicates that no refinement of the binding site or ligand occurred. (b) PDBe‐downloaded electron density map and model of the nominal highest‐resolution (0.85 Å) structure of endothiapepsin, PDB ID 5rdh. R‐free and % of real space fit (RSRZ) outliers in the PDB slider panel are extremely high and indicate poor agreement with the electron density, including unmodeled solvent, which is indicative of incomplete refinement. Despite the ultrahigh resolution, the model is not useful as a quality reference for pepsin‐like enzymes. (c) Electron density generated from RCSB PDB site map coefficients, and model of SARS‐CoV‐2 main protease, 3CLpro (PDB ID 5rfb). The map provides scant evidence, with real space correlation coefficient of the ligand (0.65) at the noise level. No useful conclusions can be derived by PDB users from this ligand modeled at 0.4 occupancy. 2mFo‐DFc maps (blue) are contoured at 1σ, mFo‐DFc difference maps are contoured at +3σ (green) and −3σ (red)
While suited for fragment screening and lead discovery, 8 the group deposition models dumped en masse into the PDB do not conform to the quality standards 9 , 10 expected of PDB entries. In particular, PanDDA deposits confuse most biomedical researchers as their data structure is different than that of other PDB deposits and the quality of the structure models, despite occasional high nominal resolution, is often questionable.
The presence of group deposits that do not conform to PDB standards of data retrieval and model quality, but nevertheless are presented on a par with conventional entries, degrades the PDB integrity. For standard entries, the PDB‐provided map coefficients or density maps are calculated from the supplied experimental and model data, allowing validation of the model against experiment. This procedure is not possible for PanDDA entries, as the “event maps” 7 have a completely different nature and purpose. Consequently, group deposition entries are difficult or even impossible to individually validate and assess. They can mislead PDB users when selected as models to underpin further studies and may also mislead systems like AlphaFold2 11 during selection of optimal templates for structure prediction. Presence of nonconforming entries is particularly problematic for automated data‐mining projects, including applications of Artificial Intelligence, as the presence of such data adds unexpected levels of noise during the learning and testing stages.
The gold standard of structural biology, that is, the agreement of a structure model with underlying electron density, fails for group deposition models that severely disagree with the user‐accessible electron density maps (Figure 1).
If group deposition entries violating accepted quality metrics 9 , 10 become primary references for their protein families due to their reported very high resolution, the effect could be disastrous (Figure 1b). The presence of such deposits will affect the reliability of the PDB and its reputation as the most reliable repository in life sciences. While even the community of structural biologists may already have problems with understanding the limitations of group deposits, biomedical researchers, and bioinformaticians generally assume that the same quality standards and data structure are consistently applied to all models in the PDB. Nonstructural research communities rarely verify the model by comparison with the electron density, and thus rely entirely on structural biology standard metrics, such as reported resolution, and expect that PDB structure deposits represent uniformly valid and verified models.
We postulate that PDB group depositions from large‐scale ligand screening projects that do not present fully refined macromolecular models should be, as a minimum, very clearly marked as members of this special category. Ideally, however, such models should be relocated from the PDB into a separate database dedicated to group depositions. The inventors of the PanDDA 7 ligand‐screening methodology have already developed database capabilities ideally suited to storing and handling fragment screening data. 12 They could, therefore, collaborate with the PDB to establish such a database, equipped with tools needed for proper examination and evaluation of fragment‐screening entries. Clear annotation and separation of nonstandard entries will minimize contamination of the PDB by suboptimal structures; and importantly, structure‐informed biomedical research will remain based on validated and verified experimental evidence.
AUTHOR CONTRIBUTIONS
Mariusz Jaskolski, Alexander Wlodawer, Zbigniew Dauter, Wladek Minor, Bernhard Rupp: Conceptualization (equal); formal analysis (equal); writing – original draft (equal); writing – review and editing (equal).
Jaskolski M, Wlodawer A, Dauter Z, Minor W, Rupp B. Group depositions to the Protein Data Bank need adequate presentation and different archiving protocol. Protein Science. 2022;31:784–786. 10.1002/pro.4271
Funding information Narodowe Centrum Nauki, Grant/Award Number: 2020/01/0/NZ1/00134; National Cancer Institute; National Institutes of Health (USA), Grant/Award Number: GM132595; Austrian Science Fund (FWF), Grant/Award Number: I‐5152
Contributor Information
Mariusz Jaskolski, Email: mariuszj@amu.edu.pl.
Bernhard Rupp, Email: br@hofkristallamt.org.
REFERENCES
- 1. Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10:980. 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 2. Casañal A, Lohkamp B, Emsley P. Current developments in coot for macromolecular model building of electron cryo‐microscopy and crystallographic data. Protein Sci. 2020;29:1069–1078. 10.1002/pro.3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goddard TD, Huang CC, Meng EC, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeLano WL. The PyMOL molecular graphics system. New York, NY: Schrödinger, LLC, 2015. [Google Scholar]
- 5. Porebski PJ, Sroka P, Zheng H, Cooper DR, Minor W. Molstack‐interactive visualization tool for presentation, interpretation, and validation of macromolecules and electron density maps. Protein Sci. 2018;27:86–94. 10.1002/pro.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonvin AMJJ. 50 years of PDB: A catalyst in structural biology. Nat Methods. 2021;18:448–449. 10.1038/s41592-021-01138-y. [DOI] [PubMed] [Google Scholar]
- 7. Pearce NM, Krojer T, Bradley AR, et al. A multi‐crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat Commun. 2017;8:15123. 10.1038/ncomms15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomas SE, Collins P, James RH, et al. Structure‐guided fragment‐based drug discovery at the synchrotron: Screening binding sites and correlations with hotspot mapping. Philos Trans A Math Phys Eng Sci. 2019;377:20180422. 10.1098/rsta.2018.0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Laskowski RA, Jabłońska J, Pravda L, Vařeková RS, Thornton JM. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018;27:129–134. 10.1002/pro.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adams PD, Aertgeerts K, Bauer C, et al. Outcome of the first wwPDB/CCDC/D3R ligand validation workshop. Structure. 2016;24:502–508. 10.1016/j.str.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krojer T, Talon R, Pearce N, et al. The XChemExplorer graphical workflow tool for routine or large‐scale protein‐ligand structure determination. Acta Crystallogr Sec D. 2017;73:267–278. 10.1107/S2059798316020234. [DOI] [PMC free article] [PubMed] [Google Scholar]