

Abstract
The analysis of (trace) contaminants in environmental samples represents an important tool for exposure assessment and for the evaluation of potential risks to human health. Currently, mass spectrometric detection using triple quadrupole (TQMS) systems is the established method of choice. However, screening methods using high resolution mass spectrometry (HRMS) find increasing application as they provide advantages such as enhanced selectivity. A complex composition of environmental samples is known to have enormous effects on mass analyzers. The present work therefore compares the impact of a highly matrix-loaded sample material like house-dust on the performance of mass spectrometric detection of the emerging indoor contaminant group of mycotoxins by quadrupole time-of-flight (QTOF) and TQMS after ultrahigh-performance liquid chromatographic separation. Furthermore, the role of ionization efficiencies of different ion sources in instrument sensitivity was compared using an electrospray ionization source and a newly developed heated electrospray ion source (Bruker VIP-HESI) during QTOF experiments. Finally, it was evaluated whether an additional dimension of separation enables increased sensitivity in QTOF-HRMS detection by applying mycotoxins in house-dust to an (trapped) ion mobility spectrometry instrument. The sensitivity of the QTOF detection was positively influenced by the application of the VIP-HESI ion source, and overall HRMS instruments provided enhanced selectivity resulting in simplified data evaluation compared to the TQMS. However, all performed experiments revealed strong signal suppression due to matrix components. QTOF results showed more severe effects, enabling a more sensitive detection of mycotoxins in house-dust by applying TQMS detection.
The analysis of contaminants of emerging concern (CECs) in environmental samples can be a valuable biomonitoring tool for epidemiologists.1 Semi- and non-volatile compounds are deposited in settled dust, which therefore is a particularly suitable medium for the estimation of human exposure to indoor contaminants.2 Routine screenings for known indoor pollutants in house-dust include contaminants like polychlorinated biphenyls, phthalates, nicotine, pesticides, and others.2−4
Mycotoxins have also been frequently detected in house-dust over the past decades and represent an emerging group of environmental contaminants.5,6 They are secondary metabolites produced by various molds and are commonly known for their occurrence in food and feed.7 An indoor mycotoxin contamination of building material, air, and dust may be associated with mold infestation by Aspergillus, Penicillium, and Stachybotrys species.6,8−10 Detectable mycotoxins included highly toxic compounds such as macrocyclic trichothecenes (strong inhibitors of protein biosynthesis),11 sterigmatocystin (a potential precursor of carcinogenic aflatoxin B1),10−12 aflatoxins,13 and others. Available data on human indoor exposure to mycotoxins are still limited due to analytical and sampling issues. However, published studies imply that mycotoxins potentially evoke health problems of inhabitants of mold-infested housing.14
Approaches for the analysis of CECs in environmental matrices consisting of chromatographic separation in combination with different mass spectrometric detection techniques are widespread. Triple quadrupole mass spectrometric (TQMS) methods are applied for targeted and quantitative measurements as they are affordable and provide fast and sensitive detection of the analytes of interest.15,16 Limitations in TQMS analysis can exist concerning the number of simultaneously detectable targets. Furthermore, selectivity is restricted due to unit resolution of most instruments.17,18
Some of the limitations of TQMS detection can be compensated by the application of high resolution mass spectrometry (HRMS) instruments.19 These instrument types are especially suitable for untargeted, qualitative screenings17 and therefore applicable in the analysis of CECs as this analyte group covers a large variety of compounds. Furthermore, the investigation of newly discovered CECs is simplified, as retrospective data analysis is enabled in HRMS detection. Modern quadrupole time-of-flight (QTOF) instruments provide additional (fragmentation) experiments for enhanced selectivity in comparison to TQMS instruments resulting in a more secure identification of target analytes in complex samples.20 An even further improved selectivity can be achieved by introducing an additional dimension of separation prior to mass spectrometric detection. Ion mobility spectrometry (IMS) for instance enables the separation of ions according to their gas-phase motion.21 Ion mobility (IM) in combination with HRMS is predominantly used for proteomics applications for an improved identification of proteins in biological materials22 but has also been utilized for the analysis of small molecules.23,24
The complex composition of (environmental) samples is known to have a substantial influence on mass spectrometric detection.25,26 House-dust is an especially inhomogeneous and variable mixture of both organic particles and inorganic shares.27 Its abundant matrix is reported to interfere with TQMS detection of contaminants like mycotoxins. These interferences manifest themselves in severe signal suppression, partially to less than 10% of the expected intensity, resulting in high limits of detection (LODs) and a potential underestimation of the indoor exposure to mycotoxins.28,29
The presented study describes the development of TQMS and QTOF-HRMS approaches for the detection of mycotoxins in residential dust after ultra-high performance liquid chromatographic (UHPLC) separation. All methods cover ≥34 secondary metabolites from a variety of molds such as Aspergillus, Alternaria, Fusarium, and Penicillium species. Furthermore, a special focus is set on mycotoxins derived from the particularly toxic indoor mold Stachyborys spp. as almost all available data on indoor exposure to mycotoxins are restricted to the compound class of macrocyclic trichothecenes.11 As a supplement to this, phenylspirodrimanes, which are a group of secondary metabolites produced by all Stachybotrys species in high concentrations,30,31 are included in the analyte spectrum of the presented study. The effects of the highly matrix-contaminated sample material on both QTOF-HRMS and TQMS analysis are directly compared, and the suitability of the techniques for house-dust analysis is evaluated. This is carried out on the basis of the results of method validation experiments. Additionally, an electrospray ionization (ESI, Apollo II) source and a newly developed high-performance heated electrospray ion source (HESI, Bruker VIP-HESI) were applied during QTOF-HRMS experiments in order to investigate influences of ionization efficiencies on the sensitivity. Finally, it was evaluated whether an additional dimension of separation enables increased sensitivity in QTOF-HRMS detection by applying mycotoxins in house dust to an IMS instrument. The analysis of a small set of residential dust samples, partially derived from naturally mold-infested housing, was carried out using the detection technique showing the highest sensitivity.
Experimental Section
Chemicals, Materials, and Mycotoxin Standards
Acetonitrile (MeCN) in LC–MS-grade purity and formic acid (FA) were obtained from Fisher Scientific (Schwerte, Germany) and Merck (Darmstadt, Germany). Ultrapure water (ASTM type 1 grade) was produced in-house by a Purelab Flex 2 system (Veolia Water Technologies, Celle, Germany). In the study, included mycotoxins were either purchased commercially or obtained in the course of previous research projects. Citrin (CIT), aflatoxins B1, B2, G1, and G2 (AFB1/2, AFG1/2), enniatins A, A1, B, and B1 (ENA, ENA1, ENB, ENB1), and beauvericin (BEA) were acquired from Sigma-Aldrich (Taufkirchen, Germany). Gliotoxin (GTX) and sterigmatocystin (STG) were purchased from Cayman Chemicals (Ann Arbor, Michigan, USA). Following mycotoxins derived from various genera of fungi were isolated from respective fungal culture: altenuene (ALT), alternariol monomethyl ether (AME), alternariol (AOH), deoxynivalenol (DON), fumonisin B1 (FB1), ochratoxin A (OTA), penitrems A and E (PEN A/E), T-2 toxin (T-2), HT-2 toxin (HT-2), and zearalenone (ZEN).32−39 Secondary metabolites derived from the toxic indoor fugus Stachybotrys spp. were also isolated from fungal culture by Jagels et al. and comprised stachybotrychromenes A and B (STCHR A/B),40 satratoxins G and H (SAT G/H) and the phenylspirodrimanes stachybotrydial (STDIAL), stachybotrydial acetate (STDIAL AC), 2α-acetoxystachybotrydial acetate (ACDIAL AC), L-671,667 (L-671), stachybotrysin B (ST B), stachybotrysin C (ST C), stachybonoid D (STBON D), stachybotrylactam (STLAC), stachybotrylactam acetate (STLAC AC), and stachybotryamid (STAM).31,40 2′R-OTA was produced by Cramer et al. by thermal degradation of OTA.41 The chemical structures of all compounds under study can be found in Table S1 (Supporting Information).
A separate working solution containing all analytes was prepared in MeCN for (IM-)QTOF-HRMS and TQMS detection at a 100-fold concentration of the highest calibration point. All working solutions were stored at −18 °C.
Sample Collection and Preparation
All samples (n = 21) were residential dust samples from vacuum cleaner bags from various households (flats, houses, student dormitories, etc.) in Germany, which were analyzed in duplicate. Visible mold infestation occurred in 6 of the respective households. The samples were stored at −18 °C until further preparation. In the first step, the dust samples were homogenized in a two-stage sieving process with a mesh size of 2 mm and 63 μm. From each sample, 50 mg of the fine dust fraction (Ø < 63 μm) was weighed in a 4 mL glass vial, and 500 μL of acetonitrile/water (MeCN/H2O, 85/15 v/v) was added. This step was followed by homogenization on a vortex mixer and extraction in an ultrasonic bath for 30 min at room temperature. Finally, samples were centrifugated at 13,000g at RT for 5 min and diluted 1:10 with UHPLC solvent (initial conditions). An equal mixture of 5 different blank dusts was prepared in the previously described manners. The obtained extracts were applied in matrix-matched calibration solutions. Recovery experiments were carried out using the <63 μm fraction of the dust mixture. Attempts to reduce matrix components in the injection solutions by application of an elaborated sample preparation were not successful. Several approaches based on different liquid–liquid and solid phase extraction techniques were tested, but none of the methods was able to ensure a simultaneous recovery of all mycotoxins of the complex analyte spectrum. Detailed information on tested sample preparation methods is given in Table S2 (Supporting Information).
UHPLC Apparatus and Conditions
Prior to mass spectrometric sample analysis, chromatographic separation was realized using Elute HPG 1300 UHPLC-systems (Bruker Daltonics, Bremen, Germany). In QTOF-HRMS and TQMS analysis, 30 μL of the diluted dust extract was injected using PAL HTC-xt and RSI autosamplers (CTC Analytics AG, Zwingen, Switzerland). In IM-QTOF-HRMS analysis, the maximum injection volume was limited to 27 μL due to technical limitations of the applied Elute autosampler (Bruker Daltonics, Bremen, Germany). Chromatographic separation was performed on Nucleodur C18 Gravity-SB columns (75 × 2 mm, 1.8 μm, Macherey-Nagel GmbH & Co. KG, Düren, Germany) equipped with a KrudKatcher Ultra (Phenomenex, Aschaffenburg, Germany) or a Nucleodur C18 Gravity-SB pre-column (1.8 μm, Macherey-Nagel GmbH & Co. KG, Düren, Germany) at 40 °C in Elute column oven systems (Bruker Daltonics, Bremen, Germany). Gradient elution conditions using MeCN + 0.1% FA (A) and H2O + 0.1% FA (B) were applied as follows: 0.0 min 5% A, 2.0 min 5% A, 3.9 min 25% A, 6.5 min 70% A, 7.5 min 70% A, 9.0 min 95% A, 12.0 min 95% A, 12.2 min 5% A, and 15.0 min 5% A. Additionally, a flow gradient of 350 μL/min respective 450 μL/min (3.9–12.2 min) was included. The first 2 min of each run were directed into waste. Software Compass HyStar (versions 4.1, 5.1, and 6.0) (Bruker Daltonics, Bremen, Germany) was used for the operation of the UHPLC systems.
QTOF-HRMS Apparatus and Conditions
Impact II QTOF mass spectrometers (Bruker Daltonics, Bremen, Germany) were utilized for QTOF-HRMS experiments. Ionization in the mass spectrometers was performed using an Apollo II ESI source and a vacuum-insulated probe HESI (VIP-HESI) source (Bruker Daltonics, Bremen, Germany), which were each operated under optimized conditions. The parameters for the Apollo II source were set as follows: ionization was performed in the positive and negative mode at 4.5 and −3.0 kV, respectively. The optimum ionization mode for each mycotoxin in the given sample material was determined individually. Dry gas temperature was set to 220 °C at a flow rate of 10.0 L/min. Nebulizer gas pressure was 2 bar. The VIP-HESI ion source was operated at ±4 kV with a probe gas temperature of 450 °C at a flow rate of 4 L/min. The dry gas temperature was set to 300 °C at a flow rate of 9.5 L/min. The nebulizer gas pressure was 3 bar, and the active exhaust was turned on during measurements. A mass range of 50–1000 m/z was covered, and the full scan and MS2 data were recorded at a spectra rate of 4 Hz. Data-independent acquisition in the broadband collision-induced dissociation (bbCID) mode was chosen for MS/MS experiments. Fragmentation took place in a collision-induced dissociation cell using nitrogen. Spectral acquisition was performed at alternating collision energies (CEs) of 24 and 36 eV. Sodium formate cluster ions were applied for instrument mass calibration and for re-calibration of individual raw data files. Software Compass otofControl (software versions 4.1 and 6.3, Bruker Daltonics, Bremen, Germany) was used for the operation of the mass spectrometer and for data acquisition. Data processing was executed using software TASQ (software versions 2.1 and 2.2, Bruker Daltonics, Bremen, Germany). Evaluation criteria included retention time and the detection of the principal ion and at least one confirmatory fragment ion with a maximum mass deviation of 5 ppm. Suitable mycotoxin fragments in the matrix were determined experimentally and are presented alongside further MS parameters in Table S3 (Supporting Information).
IM-QTOF-HRMS Apparatus and Conditions
Experiments on whether an additional IM separation step increases not only the selectivity but also the sensitivity of the HRMS detection of mycotoxins in house-dust were performed on a timsTOF Pro 2 equipped with a VIP-HESI ion source (Bruker Daltonics, Bremen, Germany). As the construction of the mass spectrometer shows similarities to impact II, many acquisition parameters were transferable. Ion-transfer parameters were partially adapted, and a fixed collision energy of 30 eV was set for bbCID acquisition. TIMS accumulation time was automatically regulated by activating ion charge control in order to prevent an overload of the TIMS cartridge due to a high matrix load. A value of 7.5 mio counts was set. Effective in-run accumulation times varied between 1 and 10 ms. Instrument mass and mobility calibration was performed using a mixture of sodium formate and the ESI tune mix (positive mode: G2431A, negative mode: G1969-85000, Agilent Technologies, Waldbronn, Germany). Compass timsControl (software version 3.0, Bruker Daltonics, Bremen, Germany) was used for the operation of the mass spectrometer and for data acquisition. Processing and evaluation of data were performed in TASQ 2021 b applying the criteria described above and also taking IM into consideration. Individual collisional cross section (CCS) values of analyzed mycotoxins were determined according to the guidelines (single field) of the Unified CCS Compendium proposed by Picache et al.(42) The obtained list is presented in Table S3 (Supporting Information). The values were obtained from the recalibrated data files of a high calibration level in a solvent using the previously described UHPLC setup and are in good accordance with previously published data.43 The substances angiotensin I, cortisol, d-glucose, levomefolic acid, and uric acid (LGC Standards GmbH, Wesel, Germany) and different amino acids (AAS18, Sigma-Aldrich, Taufkirchen, Germany) served as quality assessment compounds during measurements.
TQMS Apparatus and Conditions
TQMS analysis was performed on an EVOQ Elite triple quadrupole mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with the standard HESI source. Source parameters were optimized resulting in an ion spray voltage of 5.5 kV in the positive and −4.5 kV in the negative ionization mode. The cone gas was heated to 250 °C, and the temperature of the heated probe was set to 500 °C. Gas flows of 20, 50, and 60 psi were set for the cone gas, heated probe gas, and nebulizer gas, respectively. Active exhaust was turned on during measurements. Argon was used as collision gas, and CEs and analyte-dependent multiple reaction monitoring transitions (MRMs) were optimized to enable in-run detection in the scheduled multiple reaction monitoring (sMRM) mode. MRM optimization was performed both in the solvent and in the matrix in order to select 2 respectively 4 suitable transitions for each compound. Additionally, the resolutions of quadrupoles 1 and 3 (Q1 and Q3) were adapted individually for each mycotoxin. A heightened resolution of Q1 was set to remove matrix components, and the resolution of Q3 was lowered in order to achieve a higher transmission of analyte fragments. MS Workstation version 8.2.1 (Bruker Daltonics, Bremen, Germany) was used for system control of the mass spectrometer and for data acquisition. Data processing was carried out in TASQ 2.1 (Bruker Daltonics, Bremen, Germany) taking the presence and the ratios of the MRMs into consideration as well as the retention time. Detailed MS parameters for all analytes are listed in Table S4 (Supporting Information).
Method Validation
In-house validation experiments concerning LODs, limits of quantitation (LOQs), and linearity were performed for all MS systems for performance comparison. For TQMS analysis, validation was extended by determination of extraction efficiencies (EEs), apparent recoveries (RAs), and intra- and interday repeatability. The experiments were designed for the detection of mycotoxins at trace amounts as the performance of HRMS and TQMS instruments in a realistic concentration range of naturally contaminated samples is a main concern. During validation experiments, the previously described blank matrix consisting of 5 different house-dusts was deployed for determination of LODs, LOQs, for matrix-matched calibrations, and for determination of recoveries by adding mycotoxin standard solution to either the blank matrix extract or the dry blank matrix. LODs and LOQs were determined based on signal to noise (S/N) ratios in the matrix extract applying a value of 3 for LODs and 10 for LOQs. Calibration curves for mycotoxins were generated of a maximum of 7 and a minimum of 4 points for QTOF-HRMS analysis (Table S5, Supporting Information). TQMS calibration curves consisted of 5–8 calibration levels within the working range depending on the mycotoxin (Table S6, Supporting Information).
Influences of the matrix house-dust on the different instruments and detection techniques were assessed by calculating the matrix-induced signal suppression and enhancement [SSE (%)] by dividing the slope of the matrix-matched calibration by the slope of the solvent calibration and multiplying by factor 100 for each mycotoxin. Values below 100% imply a negative effect of coeluting matrix components and therefore a reduction of analyte signals, whereas increased SSE values of >100% show a positive matrix influence and signal enhancement. A direct comparison of determined LOD and SSE values of all investigated mycotoxins is presented in Table S7 of the Supporting Information.
EE and RA experiments were performed at a medium concentration level. Three independent replicates were prepared, analyzed, and quantitated. Calculation of the EEs was achieved by quantitation via respective matrix-matched calibrations. Quantitation using solvent calibrations enabled the calculation of RAs. In order to evaluate the precision of the developed TQMS detection method, intra- and interday repeatability (calculated as relative standard deviation) were investigated applying the previously described recovery samples. Intraday repeatability was determined by analyzing 12 independent samples on 1 day. Twelve additional recovery samples were used for the assessment of interday repeatability: in the course of 2 weeks, the samples were analyzed in quadruplicate on 3 different days. Both intra- and interday repeatability were calculated by quantitation via matrix-matched calibration. Detailed results of the TQMS validation experiments are presented in Table S9 of the Supporting Information. In order to evaluate the suitability of the developed method for the quantitation of mycotoxins in the heterogenous matrix house-dust, EE and RA experiments were additionally carried out applying different dusts. For this purpose, three dusts from the dust mixture mentioned above, three blank dust samples, and one commercially available standard reference dust (NIST SRM 2583, Sigma-Aldrich, Taufkirchen, Germany) were utilized in the previously described manner. The results are presented in the Supporting Information in Table S10 (results of individual samples) and Table S11 (ratio between results of the spiked individual samples and spiked dust mixture).
Application
The developed and validated UHPLC-TQMS method was successfully utilized for the analysis of the previously described 21 house-dust samples from various German households. The six samples originating from buildings with visible mold infestation were especially suited to enable a realistic evaluation of the applied system for the detection of mycotoxins in dust. The samples were analyzed in duplicate determination, and the results are shown in Tables S12 and S13 of the Supporting Information.
Results and Discussion
HRMS Analysis
The determination of LODs was successful for all analyzed mycotoxins (see Table S1 for chemical structures) and revealed valuable information for QTOF-HRMS detection of mycotoxins in the complex sample material house-dust. Lowest LODs were observed for certain aflatoxins and for the investigated enniatins with values between 40.9 μg/kg (ENB1) and 89.8 μg/kg dust (AFB1). For the majority of the other analytes, LODs were in the three-digit μg/kg range, whereas LODs of GTX, HT-2, SAT G, and the stachybotrychromenes reached values of more than 1 mg/kg (see the Table S7 column “Apollo II-QTOF-HRMS”, Supporting Information). While comparing the determined LODs using an HRMS detection technique with available data in the literature, it became apparent that as expected, the sensitivity of previous studies, which analyzed mycotoxins in settled dust by HPLC-TQMS, was not reached for many compounds.11,28,44
The heightened LODs for the detection of mycotoxins in residential dust by QTOF-HRMS in the presented study can additionally be caused by severe matrix effects. Calculation of SSE values was performed for 34 analytes. The results are presented in Figure 1 and in the Supporting Information (Table S7). In total, all mycotoxins underwent strong signal suppression by the matrix. For 32 analytes, the signals were suppressed by more than factor 2, including a number of mycotoxins for which only 20% of the expected peak areas were observable. As mentioned before, strong interferences of house-dust in TQMS detection are known.28,29 However, the extent of signal suppression that was determined in QTOF-HRMS analysis exceeds the one described in the literature by far.
Figure 1.

SSE [%] observed during UHPLC-QTOF-HRMS analysis (Apollo II) of mycotoxins in residential dust. The detailed data are presented in Table S7 of the Supporting Information.
A reduction of sensitivity in LC-MS can occur due to an impairment of ionization efficiency by coeluting matrix components in the ion source.25 In sophisticated ionization sources, thermal energy in the form of heated gas is applied during the vaporization process in order to improve desolvation of the LC eluent and therefore improve the ionization efficiency.45 To investigate the impact of the ionization process on the sensitivity of the QTOF-HRMS analysis of mycotoxins in residential dust, the standard Apollo II ESI source was substituted by a newly developed HESI source, the Bruker Vacuum-Insulated Probe-HESI (VIP-HESI). The source has a different, optimized geometry including a new, active exhaust system, which is intended to reduce chemical background during measurements of samples with a high matrix load. The most substantial difference in comparison to the Apollo II source, however, is that thermal energy is not only provided from the mass spectrometer inlet side (dry gas) but the probe gas passing around the capillary is heated as well. Due to the vacuum insulation of the capillary, additional heating of the non-vaporized solvent is omitted. According to the manufacturer, this design should prevent the decomposition of analytes in the flow medium in the spray capillary. Differences in sensitivity caused by the two ion sources were evaluated by comparing the LODs of mycotoxins in house-dust. In Figure 2, the effects of the Apollo II and VIP-HESI ionization on the sensitivity of QTOF-HRMS detection are presented by dividing the LODs after Apollo II ionization by LODs after VIP-HESI ionization.
Figure 2.

Effects of Apollo II and VIP-HESI ionization on the sensitivity of QTOF-HRMS detection of mycotoxins in residential dust. Shown is the ratio between the LODApollo II/LODVIP-HESI. A ratio of <0.8 was classified as a reduction in sensitivity by application of the VIP-HESI source (red). A ratio between 0.8 and 1.2 was accounted as equal sensitivities (gray), and values >1.2 were considered as enhanced sensitivity using VIP-HESI (green). The detailed data are presented in Table S7 of the Supporting Information.
An increase in sensitivity was observable for 26 out of 36 mycotoxins after applying the VIP-HESI ion source. Comparable LODs were determined for five analytes and a decreased sensitivity for five compounds as well. Reasons for elevated LODs in ionization under the application of heated gas by VIP-HESI are a thermolability of analytes and/or increased matrix interferences due to a likewise increased ionization efficiency of coeluting matrix components. The fact that matrix components can also undergo an enhanced ionization process is reflected in the determined SSE values of the UHPLC-VIP-HESI-QTOF-HRMS experiments (see Table S7 of the Supporting Information), which show an even stronger effect on mycotoxin signals in the matrix in comparison to ionization with the Apollo II source for the majority of compounds. Nevertheless, the application of the VIP-HESI source in this complex matrix resulted in a significant increase in sensitivity for the majority of the investigated mycotoxins as the LODs are reduced by factor 2.5 on average. In case of the highly indoor-relevant mycotoxin STG, the tremendous increase in signal intensity and the strong improvement in sensitivity after VIP-HESI ionization are demonstrated in Figure 3 on the basis of the steepness of the matrix-matched calibration curves of both ion sources. The identification of STG at lower calibration levels using VIP-HESI in comparison to Apollo II ionization (Figure 4) additionally underlines the applicability of the VIP-HESI ion source in complex matrices.
Figure 3.
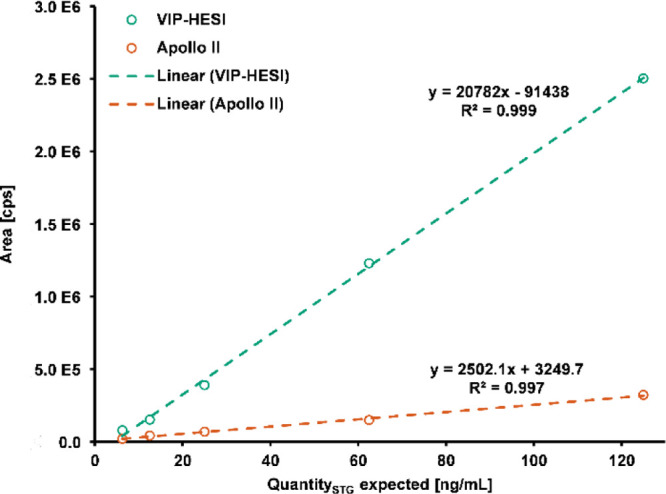
Matrix-matched calibration curves of STG in UHPLC-QTOF-HRMS analysis after ionization by Apollo II and VIP-HESI sources.
Figure 4.

Extracted ion chromatograms of STG ([M + H]+m/z 325.0707) and its qualifier bbCID fragment ion (m/z 310.0473) in QTOF-HRMS analysis at a low-level (0.625 ng/mL) matrix-matched solution after applying VIP-HESI (left) and Apollo II (right) sources for ionization.
IM-HRMS Analysis
For many mycotoxins in dust samples, interferences in the HRMS extracted ion chromatograms hampered quantitation in the low concentration range. An additional IM separation prior to mass spectrometric detection could enable the reduction of background noise as analyte signals can be distinguished from isomeric and isobaric matrix compounds. Therefore, investigations on whether an improved selectivity leads to an increased sensitivity were carried out using a VIP-HESI-IM-QTOF-HRMS instrument. The determination of LODs was performed as previously described (see Table S7 in the Supporting Information for detailed results). An enormous reduction of noise by applying the obtained individual analyte mobilities as an additional criterion was observable in both bbCID spectra and in chromatograms as exemplarily shown in Figure 5. For almost all analytes, the improved selectivity resulted in a facilitated identification process. However, the strong matrix load introduced into the TIMS cartridge forced the system to lower the injection time into the trapping unit of the IM device. Consequently, a low accumulation time was accomplished, resulting in an increased sensitivity for only 9 of 34 mycotoxins. Comparable LODs were observed for 4 analytes, and detection was less sensitive for 21 of the considered mycotoxins.
Figure 5.

Mobility filtered extracted ion chromatograms of STG ([M + H]+m/z 325.0707) and its qualifier bbCID fragment ion (m/z 310.0473) in VIP-HESI-IM-QTOF-HRMS (left) compared to VIP-HESI-QTOF-HRMS (right) analysis at a low-level (0.625 ng/mL) matrix-matched solution.
In addition to the limited accumulation times, the recording mode used for VIP-HESI-IM-QTOF-HRMS did not allow individual optimization of ion-transfer parameters and CEs for mycotoxins of varying m/z (stepping mode). Thus, compromises had to be found, resulting in minor but perceptible decreases in sensitivity and collision efficiency for certain mycotoxins. In summary, VIP-HESI-IM-QTOF-HRMS can therefore be described as a valuable tool for the interference-free analysis of mycotoxins in complex sample matrices, but its power is currently limited by the amount of the sample matrix introduced into the system, which can most probably only be overcome by future technical developments.
TQMS
The previously described HRMS approaches demonstrate the advantages and disadvantages of different QTOF instruments for analyzing complex non-purified samples such as household-dust extracts. To complete method comparison, an analytical method based on modern TQMS detection was established as well. The TQMS method used polarity switching for optimized ionization and is based on multiple reaction monitoring (two transitions) for quantitation. On applying a TQ system with these parameters in combination with the identical chromatographic separation as described before, TQMS analysis resulted in an increased sensitivity for 32 of 36 matching mycotoxins compared to VIP-HESI-QTOF-HRMS detection. A tabular listing of all LODs in the matrix is presented in the Supporting Information in Table S7 along with the determined SSE values. An enhanced sensitivity of TQMS compared to HRMS detection has also been previously reported for mycotoxins in other matrices.17 Interestingly, in our studies, the difference in sensitivity was significantly reduced, when mycotoxins in neat solvents were analyzed using the two mentioned approaches. In this case, LODs of TQMS detection were only lowered for 19 out of 36 mycotoxins (Table S8, Supporting Information). Thus, the impact of coeluting matrix compounds appears to have a higher impairment on HRMS instruments compared to TQMS. This trend is supported when regarding the ratios between the LODs of both instruments in the solvent and in house-dust (Figure 6), where a clear shift to heightened sensitivities of the TQMS instrument (green) in the more complex sample solution is observable. The gap in sensitivity is therefore clearly amplified in the matrix.
Figure 6.

Comparison of LODs of investigated mycotoxins in residential dust and neat solvent solution determined using VIP-HESI-QTOF-HRMS and TQMS detection after UHPLC separation. Shown is the ratio between the LODVIP-HESI-QTOF-HRMS/LODTQMS in the solvent (fully colored columns) and in the dust matrix (striped columns) independently. Ratios >1.2 represent an increased sensitivity of the TQMS instrument compared to the QTOF-HRMS (green). A ratio between 0.8 and 1.2 was accounted as equal sensitivities (gray), and values of <0.8 were classified as a reduction in sensitivity by detection by TQMS (red).
A closer examination of the calculated matrix effects (Table S7, Supporting Information) also confirmed this observation: for 10 mycotoxins, minimal effects of coeluting matrix components (SSE 100 ± 30%) were identifiable. For additional 12 mycotoxins, the SSE value ranged between 50 and 70%, corresponding to only moderate matrix effects. Only for eight analytes, the signal intensity dropped by >50%. Signal enhancement occurred for the group of enniatins, FB1, Pen A, ST B, and STCHR A. In summary, the signal intensity of specific sMRMs is therefore less influenced by the matrix concerning signal suppression compared to the detection of all ions in a broad m/z range using the above-mentioned QTOF-HRMS systems.
Besides the determination of LODs and SSE values, an elaborated method validation was performed for TQMS analysis. The determined EE lay between 70 and 130% for 19 mycotoxins. Lowest values were calculated for FB1 and STDIAL AC. An EE of >100% was also observed. Due to its calculation, the apparent recovery (RA) can be affected by matrix effects. RAs of 100 ± 30% were achieved for 8 analytes. For other mycotoxins, more severe losses or increased RA values were observed. Intraday repeatability was (significantly) below <20% for 32 of the 38 mycotoxins, but repeatability was also lowered by matrix interferences for certain analytes. Interday repeatability was on average somewhat higher in comparison to intraday repeatability. Complete results of the experiments are shown in Table S9 of the Supporting Information. Spiking experiments applying different (house) dusts were performed to determine the extent, to which the used dust mixture is a suitable model for the matrix-matched quantitation of mycotoxins in this heterogenous matrix. As the EE and RA values in Table S10 (results of individual samples) and especially in Table S11 (ratio between results of the spiked individual samples and spiked dust mixture) in the Supporting Information indicate, the applied workflow enables an adequate quantitation of the majority of mycotoxins in the investigated individual dust samples. Concerning certain mycotoxins like ACDIAL AC, T-2, and the group of enniatins, analyte-specific higher deviations were recognizable. Furthermore, the values obtained for measurements of the spiked dusts, which were also a part of the dust mixture applied in method validation experiments (matrix dust 1–3), showed overall higher compliance with the results presented in Table S9 (Supporting Information). The matrix of the spiked standard reference material and samples was not represented equally well by the applied house-dust mixture. However, the deviations were still classified as acceptable regarding the extremely heterogenous composition of house-dust and the lack of alternative calibration approaches such as internal standards.
Application
Performed method validation experiments revealed the UHPLC-TQMS analysis of mycotoxins in residential dust to provide the most elaborated sensitivity compared to measurements on QTOF-HRMS instruments. Therefore, this detection approach was applied for the analysis of a small set (n = 21) of house-dust samples. Six samples were derived from households with a present mold infestation, but positive samples were not exclusively derived from said households as an entry of mycotoxins can occur through various other sources like air or indoor plants.
About 80% of the analyzed samples were positive for the cyclic Fusarium depsipeptides BEA and enniatins ENA, ENB, and ENB1 as they are detectable even in low quantities due to signal enhancement. The compound group shows a number of biological effects and has cytotoxic activity against different human tumor cell lines.46 STG, which is a secondary metabolite of Aspergillus versicolor and classified as possibly carcinogenic to humans by the International Agency for Research on Cancer (IARC),47,48 was detectable in 24% of the samples. One sample showed a high contamination of more than 3 mg/kg settled dust. Two dusts from independent households were positive for the phenylspirodrimanes L-671 and STBON D, which are derived from the indoor fungus Stachybotrys. Phenylspirodrimanes show cytotoxic effects in in vitro studies using human tumor cells.49 Finally, PEN A, a neurotoxin produced by Penicillium species, which causes tremors in humans,50 was detected in one of the analyzed samples. Detailed results are presented in Tables S12 and S13 of the Supporting Information.
Conclusions
Sensitive detection of mycotoxins can be achieved using different mass spectrometers with specific advantages and disadvantages. For a long time, a TQMS instrument was regarded as the only suitable instrument for sensitive mycotoxin trace analysis. Our data demonstrate that improvements of ionization sources such as the Bruker VIP-HESI source push QTOF instruments further into the field of trace analysis. However, when extremely challenging matrices like residential house-dust extracts without purification are analyzed, sensitivity of QTOF instruments is stronger affected compared to TQMS.
Besides high S/N ratios and the chance of low detection limits, the selectivity of the mass spectrometric detection is a crucial factor in every analytical laboratory. For the complex matrix house-dust, the application of TIMS drastically removed noise and interferences, but also, the application of HRMS alone compared to TQMS gave more selectivity. Consequently, peak integration and detection were easier, faster, and better applicable for automated algorithms. Based on these data, it is likely that in the future, the use of HRMS instruments will be more widespread in many areas of (quantitative) application as QTOF-HRMS and especially IM-QTOF-HRMS instruments bring additional advantages in speed, nontarget screening capabilities, retrospective analysis, and multi-analyte detection.
Acknowledgments
The authors thank Bruker Daltonics GmbH (Bremen, Germany) for providing the EVOQ Elite TQMS instrument and the associated setup and for enabling and supporting the measurements using the VIP-HESI ion source and the timsTOF Pro 2. The authors are especially thankful for the technical support of Karin Wendt, Birgit Schneider, and Dr. Carsten Baessmann. Additionally, the authors thank Joana Katharina Hillen for her support of the project.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.1c04254.
Information on investigated mycotoxins; sample preparation methods; applied HRMS settings; applied TQMS settings; calibration levels of HRMS analysis; calibration levels of TQMS analysis; LODs and SSEs in house-dust; LODs in neat solvent solution; validation data of TQMS detection; EE and RA determined in spiked individual dust samples; EE and RA for spiked individual dust samples compared to the spiked dust mixture; overview of mycotoxins in the investigated sample set; and determined mycotoxin concentration in house-dust samples (PDF)
Author Contributions
V.L. performed the experiments. V.L. and B.C. wrote the article. B.C. and H.-U.H supervised the project. All authors have given approval to the final version of the article.
The authors declare no competing financial interest.
Supplementary Material
References
- Whitehead T.; Metayer C.; Buffler P.; Rappaport S. M. Estimating exposures to indoor contaminants using residential dust. J. Expo. Sci. Environ. Epidemiol. 2011, 21, 549–564. 10.1038/jes.2011.11. [DOI] [PubMed] [Google Scholar]
- Butte W.; Heinzow B. Pollutants in house dust as indicators of indoor contamination. Rev. Environ. Contam. Toxicol. 2002, 175, 1–46. [PubMed] [Google Scholar]
- Hilton D. C.; Jones R. S.; Sjödin A. A method for rapid, non-targeted screening for environmental contaminants in household dust. J. Chromatogr. A 2010, 1217, 6851–6856. 10.1016/j.chroma.2010.08.039. [DOI] [PubMed] [Google Scholar]
- Mercier F.; Gilles E.; Saramito G.; Glorennec P.; Le Bot B. A multi-residue method for the simultaneous analysis in indoor dust of several classes of semi-volatile organic compounds by pressurized liquid extraction and gas chromatography/tandem mass spectrometry. J. Chromatogr. A 2014, 1336, 101–111. 10.1016/j.chroma.2014.02.004. [DOI] [PubMed] [Google Scholar]
- Székács A. Mycotoxins as emerging contaminants. Introduction to the special issue “Rapid Detection of Mycotoxin Contamination”. Toxins 2021, 13, 475. 10.3390/toxins13070475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevalainen A.; Täubel M.; Hyvärinen A. Indoor fungi. Companions and contaminants. Indoor Air 2015, 25, 125–156. 10.1111/ina.12182. [DOI] [PubMed] [Google Scholar]
- Bennett J. W.; Klich M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. 10.1128/cmr.16.3.497-516.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagels A.; Stephan F.; Ernst S.; Lindemann V.; Cramer B.; Hübner F.; Humpf H. U. Artificial vs. natural stachybotrys infestation – Comparison of mycotoxin production on various building materials. Indoor Air 2020, 30, 1268–1282. 10.1111/ina.12705. [DOI] [PubMed] [Google Scholar]
- Gottschalk C.; Bauer J.; Meyer K. Detection of satratoxin G and H in indoor air from a water-damaged building. Mycopathologia 2008, 166, 103–107. 10.1007/s11046-008-9126-z. [DOI] [PubMed] [Google Scholar]
- Vishwanath V.; Sulyok M.; Weingart G.; Kluger B.; Täubel M.; Mayer S.; Schuhmacher R.; Krska R. Evaluation of settled floor dust for the presence of microbial metabolites and volatile anthropogenic chemicals in indoor environments by LC-MS/MS and GC-MS methods. Talanta 2011, 85, 2027–2038. 10.1016/j.talanta.2011.07.043. [DOI] [PubMed] [Google Scholar]
- Bloom E.; Bal K.; Nyman E.; Must A.; Larsson L. Mass spectrometry-based strategy for direct detection and quantification of some mycotoxins produced by Stachybotrys and Aspergillus spp. in indoor environments. Appl. Environ. Microbiol. 2007, 73, 4211–4217. 10.1128/aem.00343-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhart S.; Loock A.; Skutlarek D.; Sagunski H.; Lommel A.; Färber H.; Exner M. Occurrence of toxigenic Aspergillus versicolor isolates and sterigmatocystin in carpet dust from damp indoor environments. Appl. Environ. Microbiol. 2002, 68, 3886–3890. 10.1128/aem.68.8.3886-3890.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polizzi V.; Delmulle B.; Adams A.; Moretti A.; Susca A.; Picco A. M.; Rosseel Y.; Kindt R. t.; van Bocxlaer J.; De Kimpe N.; van Peteghem C.; De Saeger S. JEM Spotlight: Fungi, mycotoxins and microbial volatile organic compounds in mouldy interiors from water-damaged buildings. J Environ Monit 2009, 11, 1849–1858. 10.1039/b906856b. [DOI] [PubMed] [Google Scholar]
- Robbins C. A.; Swenson L. J.; Nealley M. L.; Kelman B. J.; Gots R. E. Health effects of mycotoxins in indoor air. A critical review. Appl. Occup. Environ. Hyg. 2000, 15, 773–784. 10.1080/10473220050129419. [DOI] [PubMed] [Google Scholar]
- Herebian D.; Zühlke S.; Lamshöft M.; Spiteller M. Multi-mycotoxin analysis in complex biological matrices using LC-ESI/MS: Experimental study using triple stage quadrupole and LTQ-Orbitrap. J. Sep. Sci. 2009, 32, 939–948. 10.1002/jssc.200800589. [DOI] [PubMed] [Google Scholar]
- Jensen T.; de Boevre M.; Preußke N.; de Saeger S.; Birr T.; Verreet J. A.; Sönnichsen F. D. Evaluation of high-resolution mass spectrometry for the quantitative analysis of mycotoxins in complex feed matrices. Toxins 2019, 11, 531. 10.3390/toxins11090531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tittlemier S. A.; Cramer B.; Dall’Asta C.; Iha M. H.; Lattanzio V. M. T.; Maragos C.; Solfrizzo M.; Stranska M.; Stroka J.; Sumarah M. Developments in mycotoxin analysis. An update for 2018-19. World Mycotoxin J. 2020, 13, 3. 10.3920/wmj2019.2535. [DOI] [Google Scholar]
- Tittlemier S. A.; Brunkhorst J.; Cramer B.; DeRosa M. C.; Lattanzio V. M. T.; Malone R.; Maragos C.; Stranska M.; Sumarah M. W. Developments in mycotoxin analysis. An update for 2019-2020. World Mycotoxin J. 2021, 14, 3–26. 10.3920/wmj2020.2664. [DOI] [Google Scholar]
- Kaufmann A. The current role of high-resolution mass spectrometry in food analysis. Anal. Bioanal. Chem. 2012, 403, 1233–1249. 10.1007/s00216-011-5629-4. [DOI] [PubMed] [Google Scholar]
- Gómez-Ramos M. M.; Ferrer C.; Malato O.; Agüera A.; Fernández-Alba A. R. Liquid chromatography-high-resolution mass spectrometry for pesticide residue analysis in fruit and vegetables. Screening and quantitative studies. J. Chromatogr. A 2013, 1287, 24–37. 10.1016/j.chroma.2013.02.065. [DOI] [PubMed] [Google Scholar]
- Michelmann K.; Silveira J. A.; Ridgeway M. E.; Park M. A. Fundamentals of trapped ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 14–24. 10.1007/s13361-014-0999-4. [DOI] [PubMed] [Google Scholar]
- Yu F.; Haynes S. E.; Teo G. C.; Avtonomov D. M.; Polasky D. A.; Nesvizhskii A. I. Fast quantitative analysis of timsTOF PASEF data with MSFragger and IonQuant. Mol. Cell. Proteomics 2020, 19, 1575–1585. 10.1074/mcp.tir120.002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapthorn C.; Pullen F.; Chowdhry B. Z. Ion mobility spectrometry-mass spectrometry (IMS-MS) of small molecules. Separating and assigning structures to ions. Mass Spectrom. Rev. 2013, 32, 43–71. 10.1002/mas.21349. [DOI] [PubMed] [Google Scholar]
- Righetti L.; Bergmann A.; Galaverna G.; Rolfsson O.; Paglia G.; Dall’Asta C. Ion mobility-derived collision cross section database. Application to mycotoxin analysis. Anal. Chim. Acta 2018, 1014, 50–57. 10.1016/j.aca.2018.01.047. [DOI] [PubMed] [Google Scholar]
- Taylor P. J. Matrix effects. The Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin. Biochem. 2005, 38, 328–334. 10.1016/j.clinbiochem.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Steiner D.; Krska R.; Malachová A.; Taschl I.; Sulyok M. Evaluation of matrix effects and extraction efficiencies of LC–MS/MS methods as the essential part for proper validation of multiclass contaminants in complex feed. J. Agric. Food Chem. 2020, 68, 3868–3880. 10.1021/acs.jafc.9b07706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyntelberg F.; Suadicani P.; Nielsen J. W.; Skov P.; Valbjorn O.; Nielsen P. A.; Schneider T.; Jorgensen O.; Wolkoff P.; Wilkins C. K.; Gravesen S.; Norn S. Dust and the sick building syndrome. Indoor Air 1994, 4, 223–238. 10.1111/j.1600-0668.1994.00003.x. [DOI] [Google Scholar]
- Vishwanath V.; Sulyok M.; Labuda R.; Bicker W.; Krska R. Simultaneous determination of 186 fungal and bacterial metabolites in indoor matrices by liquid chromatography/tandem mass spectrometry. Anal. Bioanal. Chem. 2009, 395, 1355–1372. 10.1007/s00216-009-2995-2. [DOI] [PubMed] [Google Scholar]
- Jaderson M.; Park J.-H. Evaluation of matrix effects in quantifying microbial secondary metabolites in indoor dust using ultraperformance liquid chromatograph–tandem mass spectrometer. Saf. Health Work 2019, 10, 196–204. 10.1016/j.shaw.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis B. B.; Salemme J.; Morals A. Stachybotrys toxins. 1. Nat. Toxins 1995, 3, 10–16. 10.1002/nt.2620030104. [DOI] [PubMed] [Google Scholar]
- Jagels A.; Lindemann V.; Ulrich S.; Gottschalk C.; Cramer B.; Hübner F.; Gareis M.; Humpf H. U. Exploring secondary metabolite profiles of Stachybotrys spp. by LC-MS/MS. Toxins 2019, 11, 11. 10.3390/toxins11030133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretz M.; Beyer M.; Cramer B.; Humpf H.-U. Stable isotope dilution analysis of the Fusarium mycotoxins deoxynivalenol and 3-acetyldeoxynivalenol. Mol. Nutr. Food Res. 2006, 50, 251–260. 10.1002/mnfr.200500230. [DOI] [PubMed] [Google Scholar]
- Hübner F.; Harrer H.; Fraske A.; Kneifel S.; Humpf H.-U. Large scale purification of B-type fumonisins using centrifugal partition chromatography (CPC). Mycotoxin Res. 2012, 28, 37–43. 10.1007/s12550-011-0114-7. [DOI] [PubMed] [Google Scholar]
- Cramer B.; Harrer H.; Nakamura K.; Uemura D.; Humpf H.-U. Total synthesis and cytotoxicity evaluation of all ochratoxin A stereoisomers. Bioorg. Med. Chem. 2010, 18, 343–347. 10.1016/j.bmc.2009.10.050. [DOI] [PubMed] [Google Scholar]
- Beyer M.; Ferse I.; Humpf H.-U. Large-scale production of selected type A trichothecenes. The use of HT-2 toxin and T-2 triol as precursors for the synthesis of d3-T-2 and d3-HT-2 toxin. Mycotoxin Res. 2009, 25, 41–52. 10.1007/s12550-009-0006-2. [DOI] [PubMed] [Google Scholar]
- Cramer B.; Bretz M.; Humpf H.-U. Stable isotope dilution analysis of the Fusarium mycotoxin zearalenone. J. Agric. Food Chem. 2007, 55, 8353–8358. 10.1021/jf0717283. [DOI] [PubMed] [Google Scholar]
- Bittner A.; Cramer B.; Humpf H.-U. Matrix binding of ochratoxin A during roasting. J. Agric. Food Chem. 2013, 61, 12737–12743. 10.1021/jf403984x. [DOI] [PubMed] [Google Scholar]
- Hickert S.; Bergmann M.; Ersen S.; Cramer B.; Humpf H.-U. Survey of Alternaria toxin contamination in food from the German market, using a rapid HPLC-MS/MS approach. Mycotoxin Res. 2016, 32, 7–18. 10.1007/s12550-015-0233-7. [DOI] [PubMed] [Google Scholar]
- Kalinina S. A.; Jagels A.; Cramer B.; Geisen R.; Humpf H. U. Influence of environmental factors on the production of penitrems A–F by Penicillium crustosum. Toxins 2017, 9, 210. 10.3390/toxins9070210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagels A.; Hövelmann Y.; Zielinski A.; Esselen M.; Köhler J.; Hübner F.; Humpf H.-U. Stachybotrychromenes A-C: novel cytotoxic meroterpenoids from Stachybotrys sp. Mycotoxin Res. 2018, 34, 179–185. 10.1007/s12550-018-0312-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer B.; Königs M.; Humpf H.-U. Identification and in vitro cytotoxicity of ochratoxin A degradation products formed during coffee roasting. J. Agric. Food Chem. 2008, 56, 5673–5681. 10.1021/jf801296z. [DOI] [PubMed] [Google Scholar]
- Picache J. A.; Rose B. S.; Balinski A.; Leaptrot K. L.; Sherrod S. D.; May J. C.; McLean J. A. Collision cross section compendium to annotate and predict multi-omic compound identities. Chem. Sci. 2019, 10, 983–993. 10.1039/c8sc04396e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Righetti L.; Dreolin N.; Celma A.; McCullagh M.; Barknowitz G.; Sancho J. V.; Dall’Asta C. Travelling wave ion mobility-derived collision cross section for mycotoxins: Investigating interlaboratory and interplatform reproducibility. J. Agric. Food Chem. 2020, 68, 10937–10943. 10.1021/acs.jafc.0c04498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom E.; Nyman E.; Must A.; Pehrson C.; Larsson L. Molds and mycotoxins in indoor environments--a survey in water-damaged buildings. J. Occup. Environ. Hyg. 2009, 6, 671–678. 10.1080/15459620903252053. [DOI] [PubMed] [Google Scholar]
- Manisali I.; Chen D. D. Y.; Schneider B. B. Electrospray ionization source geometry for mass spectrometry. Past, present, and future. Trends Anal. Chem. 2006, 25, 243–256. 10.1016/j.trac.2005.07.007. [DOI] [Google Scholar]
- Firáková S.; Proksa B.; Sturdíková M. Biosynthesis and biological activity of enniatins. Pharmazie 2007, 62, 563–568. [PubMed] [Google Scholar]
- IARC IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Some Naturally Occurring Substances; WHO: Lyon, 1976. [Google Scholar]
- IARC Overall Evaluations of Carcinogenicity: An Updating of IARC Monographs Volumes 1 to 42; WHO: Lyon, 1987. [PubMed] [Google Scholar]
- Ma X.-H.; Zheng W.-M.; Sun K.-H.; Gu X.-F.; Zeng X.-M.; Zhang H.-T.; Zhong T.-H.; Shao Z.-Z.; Zhang Y.-H. Two new phenylspirodrimanes from the deep-sea derived fungus Stachybotrys sp. MCCC 3A00409. Nat. Prod. Res. 2018, 33, 386. 10.1080/14786419.2018.1455041. [DOI] [PubMed] [Google Scholar]
- Lewis P. R.; Donoghue M. B.; Cook L.; Granger L. V.; Hocking A. D. Tremor syndrome associated with a fungal toxin: Sequelae of food contamination. Med. J. Aust. 2005, 182, 582–584. 10.5694/j.1326-5377.2005.tb06819.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.