

Abstract
It has been long recognized that NOD1 and NOD2 are critical players in the host immune response, primarily by their sensing bacterial peptidoglycan-conserved motifs. Significant advances have been made from efforts that characterize their upstream activators, assembly of signaling complexes, and activation of downstream signaling pathways. Disruption in NOD1 and NOD2 signaling has also been associated with impaired host defense and resistance to the development of inflammatory diseases. In this review, we will describe how NOD1 and NOD2 sense microbes and cellular stress to regulate host responses that can affect disease pathogenesis and outcomes.
Keywords: immunity, inflammation, innate, NOD1, NOD2, Nod-like receptor
1 |. INTRODUCTION
Pattern recognition receptors (PRRs) have a central role in regulating immune and inflammatory responses, and the magnitude of their responses can affect the outcome of infections, autoimmune and inflammatory diseases, and cancer risk. Among the most well-characterized PRRs are the nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs). The NLRs are cytosolic receptors which sense microbial motifs, endogenous byproducts of tissue injury, and environmental signals; however, mechanisms by which each NLR member recognizes different signals are still not well-understood. Once activated, they mediate various effector functions via signal transduction pathways that regulate processes such as cellular death and proliferation, autophagy, tissue repair, and inflammation. Consequently, NLRs are important in maintaining tissue and immune homeostasis that when dysregulated can lead to inflammatory diseases. NOD1 and NOD2 are two seminal NLRs that have been shown to sense specific bacterial ligands and have diverse immunomodulatory effects that are important for both host defense and tissue homeostasis. In this review, we will describe host responses regulated by NOD1 and NOD2 and their impact on host-microbial interactions and inflammatory disease pathogenesis.
2 |. STRUCTURE OF NOD1 AND NOD2 RECEPTORS
NLRs are characterized by a tripartite domain structure consisting of a C-terminal leucine-rich repeat (LRR) domain required for ligand sensing, a central long nucleotide-binding NACHT domain (NBD domain) that mediates oligomerization, and a variable N-terminal effector domain important for interactions with downstream effector proteins. NLRs are further divided into four subfamilies based on the nature of their N-terminal domain: (i) NLRP contain a pyrin domain (PYD), (ii) NLRA contain an acidic transcriptional activation (TA) domain, (iii) NLRB have a baculovirus IAP repeat (BIR) domain, and (iv) NLRC have a caspase activation and recruitment domain (CARD), respectively. As founding members of the NLRs, NOD1 and NOD2 have often retained their original nomenclature; however, since they contain an N-terminal CARD domain, they belong to the NLRC sub-family of NLRs and are technically designated as NLRC1 and NLRC2, respectively.1 NOD1 and NOD2 have similar domain architectures, but differ in the number of CARD domains: NOD1 contains one, whereas two tandem CARD domains are found in NOD2 (Figure 1). Systematic mutagenesis experiments have been informative in delineating the functions of the various domains in the activity of NOD1 and NOD2. The CARD domain is critical for interaction with the downstream adapter protein RIP2 with crucial residues in both CARD domains of Nod2 important for this activity.2–5 Within the centrally located NBD are Walker A- and B box motifs that contain residues important for ATP binding and hydrolysis as well as ligand-dependent activation of downstream signaling pathways, notably NF-κB.5,6 ATP binding by NOD2 enhances both ligand binding and oligomerization, and mutations in this region that disrupt this can abrogate downstream signaling.6,7 On the other hand, inhibition of ATP hydrolysis and stabilization of binding results in increased activity. Interestingly, mutations in a highly conserved patch of acidic residues (extended Walker B box) in NOD2 that have been associated with autoinflammatory diseases, such as early-onset sarcoidosis and Blau syndrome, result in constitutive activation of NOD2, but inactivation of NOD1, suggesting that despite strong similarities in domain structure, different mechanisms are involved in their activation.6
FIGURE 1.
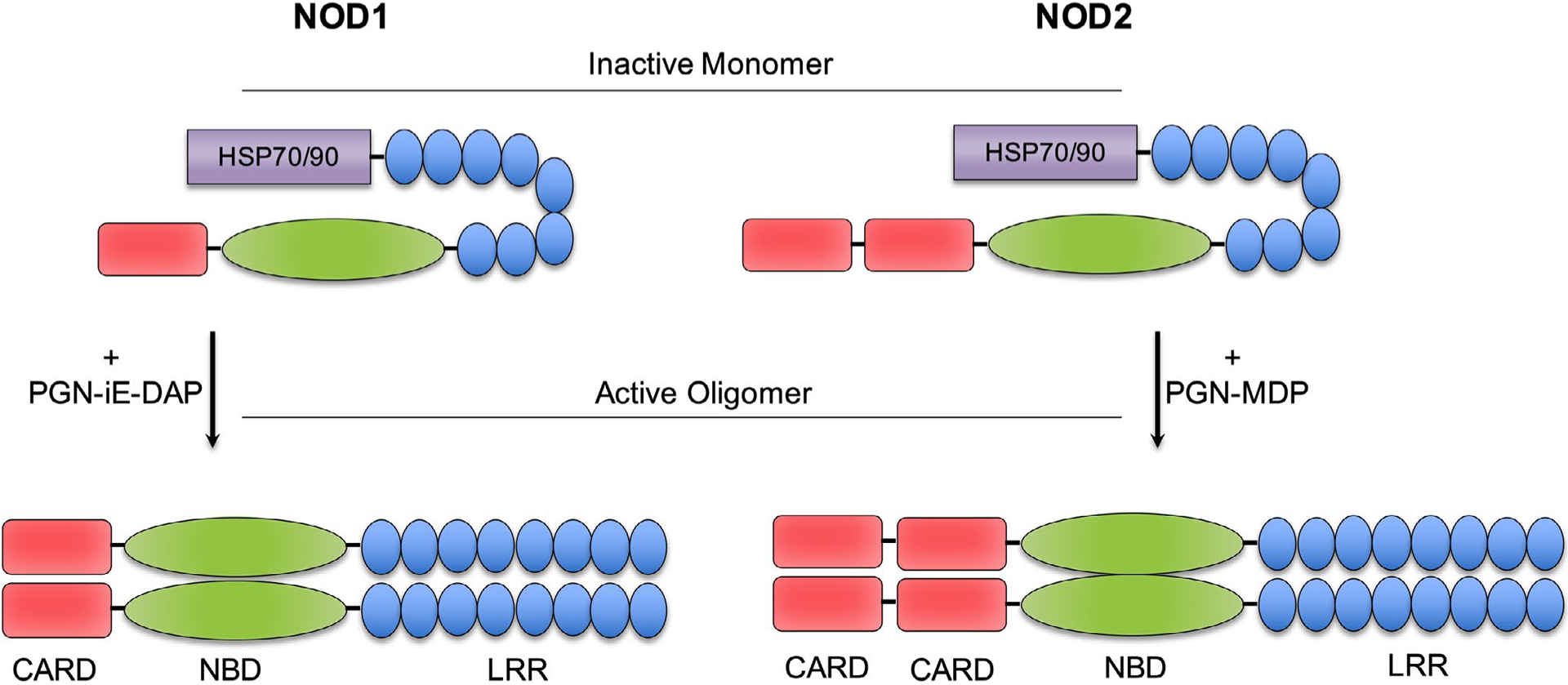
Schematic representation of NOD-like receptors 1 and 2. NOD1 consists of one N-terminal caspase activation and recruitment domain (CARD), and NOD2 has two in tandem. In both receptors, the domain CARD is followed by a core nucleotide-binding domain (NBD) and a C-terminal leucine-rich repeat domain (LRR). In the absence of ligand, NOD1 and NOD2 are in an inactive monomeric form, maintained by the binding of LRR domain into NBD and stabilized by chaperone proteins, such as HSP70 or HSP90. Upon recognition of PGN (peptidoglycan) ligands, a conformational change occurs, resulting in homo-oligomerization of two NOD molecules that once activated trigger inflammatory signaling pathways
The C-terminal LRR domain differs in size between NOD1 and NOD2 and is crucial for ligand recognition and binding.8,9 Comparative genomic studies show high conservation particularly within the LRR among different species in residues especially along a predicted concave surface of the LRRs to form a ligand binding site,10 which is also consistent with the crystal structure of NOD2.11 Furthermore, the LRR is solely responsible for dictating ligand specificity.5,8
3 |. ACTIVATORS OF NOD1 AND NOD2
NOD1 and NOD2 recognize distinct fragments of peptidoglycan (PGN) that is a major component of the bacterial cell wall of Gram-positive bacteria, outside the cytoplasmic membrane, providing them structure, rigidity, and protection.12,13 A thin layer of PGN is also found in Gram-negative bacteria within the periplasmic space. PGN consists of a polymeric chain of alternating sugar residues of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) that form the backbone with short peptides covalently bound to MurNAc to create a muramyl peptide, which can be cross-linked to form the lattice structure of peptidoglycan 14 (Figure 2). The short peptide chains in the PGN contain 3–5 amino acids that are differentially found in Gram-negative and Gram-positive bacteria and are also differentially recognized by NOD1 and NOD2.14 Muropeptides are peptidoglycan fragments that can be generated by degradation of PGN either by host or by bacterial enzymes. NOD1 senses muropeptides containing the minimal γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP) dipeptide core, which is predominantly found in Gram-negative, but also in a few Gram-positive bacteria such as Listeria monocytogenes and Bacillus spp.15,16 Interestingly, screening of bacteria derived from soil revealed that the genus Bacillus bacteria has the strongest NOD1-stimulatory effect associated with culture supernatants rather than bacterial cell extracts and is also highly stable, more so than Toll-like receptor 4 (TLR4) or NOD2 ligands, suggesting the potential for environmental stimuli to contribute to homeostatic regulation of the immune system and that NOD1 agonists can be produced and released by bacteria to activate NOD1.17 Indeed, muramyl peptides spontaneously shed by the Shigella are able to trigger a NOD1-mediated response, and Listeria lacking streptolysin O, which is required to escape from the phagosome into the cytosol, is still able to activate NOD1.17,18 Furthermore, the synthetic addition of lipophilic acyl residues greatly enhanced Nod1 activation and may be related to interaction with the cell membrane to facilitate translocation into the intracellular compartment of the host cell for recognition by NOD1.19 Although a direct interaction between NOD1 and the ligand l-Ala-γ-D-Glu-meso-diaminopimelic acid (TriDAP) that is LRR-dependent has been demonstrated by surface plasmon resonance and atomic microscopy,20 it remains unclear whether this is true for all ligands and whether other co-factors are involved in ligand binding.
FIGURE 2.
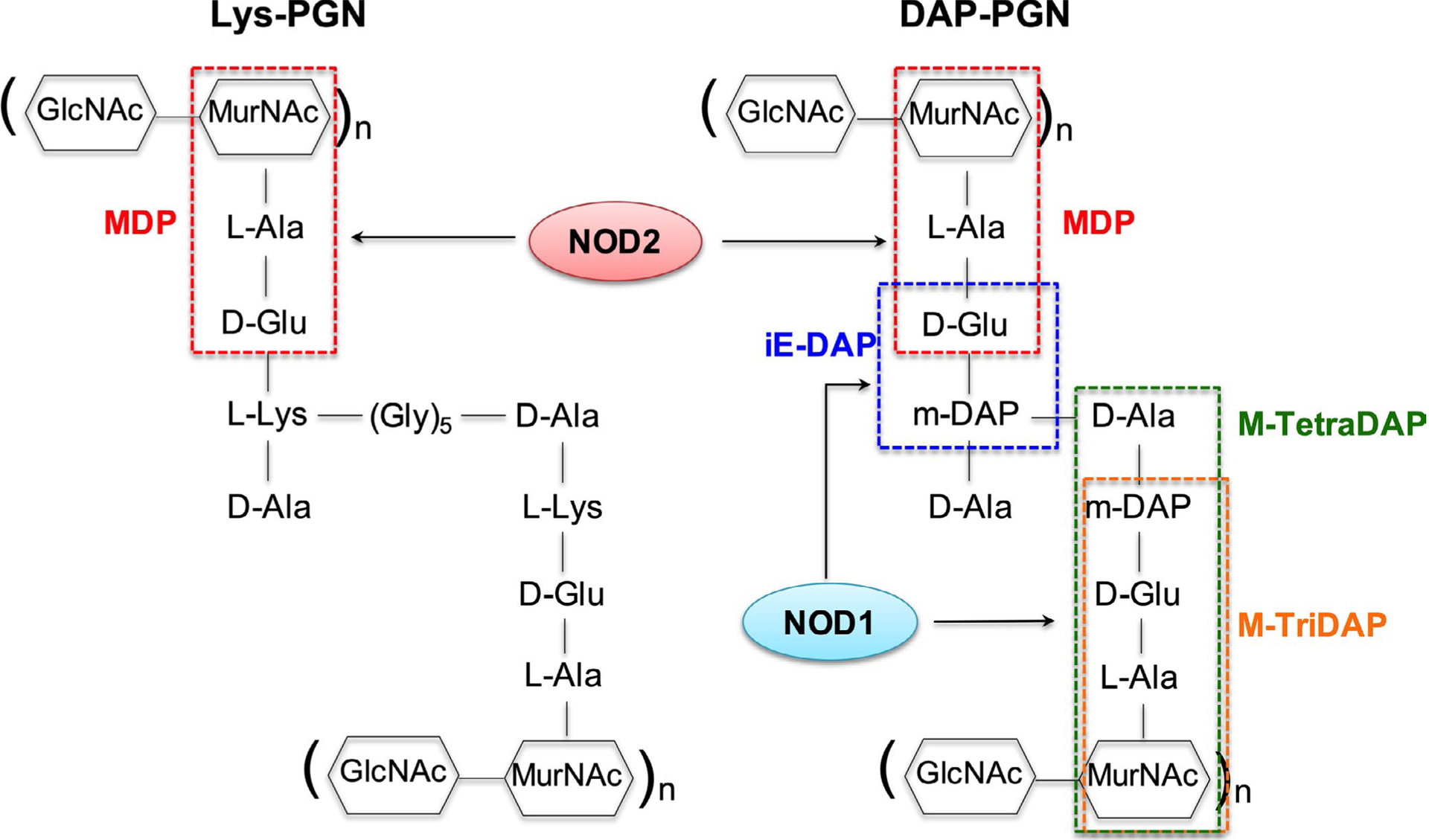
Basic structure of peptidoglycan (PGN) motifs recognized by NOD1 and NOD2. Lys-PGN is present in Gram-positive bacteria, whereas DAP-PGN constitutes the cell wall of Gram-negative bacteria. Abbreviations: D-Ala: D-alanine; D-Glu: D-glutamic acid; GlcNAc: N-acetylglucosamine; L-Ala: L-alanine; L-Lys: L-lysine; mDAP: meso-diaminopimelic acid; MurNAc: N-acetylmuramic acid; iE-DAP: γ-D-glutamyl-meso-diaminopimelic acid; M-Tetra-DAP: MurNAc-L-Ala-D-Glu-mDAP-D-Ala; M-TriDAP: MurNAc-L-Ala-D-Glu-mDAP
In contrast to NOD1, NOD2 requires PGN fragments containing an intact MurNac ring structure and an attached sugar to the dipeptide moiety and has been shown to directly bind muramyl dipeptide (MDP) that is broadly expressed in both Gram-positive and Gram-negative bacteria.21–23 To activate NOD2, MDP may have an intact MurNAc ring structure, and the sugar must be attached to the peptide moiety.23 Unlike NOD1, the length of the peptide moiety is not critical for recognition, and therefore, meso-DAP-muropeptides can be recognized by both NOD1 and NOD2, allowing NOD2 to be activated by a broader range of muropeptides from both Gram-negative and Gram-positive bacteria.14 Although several studies have demonstrated a direct interaction between NOD2 and MDP,7,22,24 it remains to be determined whether direct binding occurs with larger MDP-containing PGN fragments and whether additional accessory proteins are involved. For example, it has been shown that glucosaminyl-MDP is capable of binding to YB1, which can form a complex with NOD2 leading to cooperative activation of downstream signaling pathways.25
There is some evidence to suggest that in addition to bacterial peptidoglycan fragments, NOD1 and NOD2 can recognize other types of ligands such as viral RNA. Both in vitro experiments with overexpressed proteins and in vivo studies using Nod2-deficient mice demonstrated the ability of viral single-stranded RNA (ssRNA) to activate NOD2 that is dependent on the presence of both the NBD and LRR domains. However, ssRNA is unable to activate NOD1.26 Rather, at least in vitro in a hepatocyte cell line, synthetic dsRNA (polyI:C) or dsRNA generated by the RNA polymerase of hepatitis C virus induces NOD1 expression and interacts with NOD1, resulting in activation of downstream signaling pathways.27 Interestingly, this interaction occurred independently of the LRR, but requires an intact NBD for full activity.27 Similarly, zebrafish NOD1 overexpressed in the carp fish cell line epithelioma papulosum cyprinid (EPC) was capable of binding to the dsRNA virus, spring viremia of carp virus (SVCV), as well as polyI:C that was CARD-dependent, suggesting perhaps the involvement of an intermediary protein for this interaction.28
Besides pathogen-associated molecular patterns (PAMPs), there is increasing evidence that NOD1 and NOD2, like other NLRs, can also respond to danger signals or damage-associated molecular patterns (DAMPs). Notably, disturbances in endoplasmic reticulum (ER) function can lead to ER stress with accumulation of misfolded proteins within the ER lumen, which, if left unchecked, can lead to cellular injury and apoptosis. Thus, ER stress, which can be induced by various cellular stressors including microbial infection, can be construed as a danger signal indicative of cellular dysfunction. As a means of restoring ER function and cellular homeostasis, ER stress triggers the activation of various signaling pathways including inflammation that constitute the unfolded protein response (UPR).29 Chemical inducers of ER stress, such as thapsigargin and dithiothreitol, resulted in upregulation of the inflammatory cytokine IL-6, which was in part dependent on the presence of NOD1 and NOD2.30 In addition, infection with B abortus which induces ER stress also promoted an inflammatory response in bone marrow-derived macrophages and in mice, which was reversed by the ER stress inhibitor TUCDA and was impaired in the absence of NOD1 and NOD2. Together, these results strongly suggest a PGN-independent pathway involving ER stress can activate NOD1 and NOD2 although the precise upstream signal sensed by NOD1 and NOD2 remains unclear. However, it has also been posited based on results from another study that ER stress per se does not activate NOD1 and NOD2; rather, cellular perturbations related to Ca2+ flux may be the main activating stimulus as thapsigargin, which induces ER stress by inhibiting ER calcium ATPase and Ca2+ accumulation in the ER lumen, resulted in NOD1/2-dependent cytokine production in HCT116 colon epithelial cells.31 On the other hand, other ER inducers not associated with changes in intracellular Ca2+ levels, such as tunicamycin, did not induce an inflammatory response via NOD1 or NOD2.31 Furthermore, inhibitors of Ca2+ flux partially recapitulated the effects of thapsigargin. Therefore, it was hypothesized that increases in intracellular Ca2+ can lead to endocytosis and internalization of extracellular PGN fragments present in either media or serum to activate NOD1 or NOD2 to explain results from previous studies although this has not been definitely proven 31
PGN fragments from non-invasive bacteria can be transported into the eukaryotic cytosol through bacterial secretion systems where they are sensed by both NOD1 and NOD2 receptors. However, bacterial effector proteins belonging to the type III secretion system can also activate NOD1 and NOD2 independently of PGN stimulation that may be related to perturbations in the cytoskeleton. In particular, NOD1 and NOD2 can sense the activation state of Rho GTPases, which regulate the actin cytoskeleton and various signal transduction pathways.32 For example, bacterial effector proteins such as the Salmonella type III secretion system protein SopE can act as a guanine nucleotide exchange factor and efficiently activate the Rho GTPases CDC42 and Rac1, which then interacts with NOD1 resulting in activation of downstream signaling pathways, specifically NF-κB.33,34 More specifically, the presence of SopE introduces membrane ruffles and recruitment of NOD1 into a multiprotein complex containing SopE, Rac1, CDC42, Hsp90, and NOD1. SopE containing a mutation that is unable to activate Rac1 or CDC42 did not result in membrane ruffling and recruitment of NOD1. Similarly, Salmonella SipA, another type III secretion system effector protein, can colocalize with NOD1 and NOD2 and activate NF-κB in a NOD1- and NOD2-dependent manner, but independently of PGN.35 Shigella flexneri effector proteins, such as OspB and IpgB2, can also induce membrane ruffling resulting in recruitment of the guanidine exchange factor, GEF-H1, which, in turn, activates the rho GTPase, RhoA, leading to NF-κB activation that requires NOD1.36 NOD2 can also interact with Rho GTPases; specifically, it was demonstrated that NOD2 can be co-immunoprecipitated together with Rac1 in HT29 colon epithelial cells with localization of NOD2 to membrane ruffles induced by active Rac1.37 The ESX-1 bacterial secretion system of Mycobacterium tuberculosis can also cause pore formation and cell membrane damage, resulting in activation of NOD2.38,39 It is, therefore, possible that under these conditions, what activates NOD1 and NOD2 are disturbances in the actin cytoskeleton structure. Consistently, treatment of the HT29 colon epithelial cell line with cytochalasin D resulted in NF-κB activation that required NOD2 in the absence of PGN.37
4 |. CELLULAR LOCALIZATION OF NOD1 AND NOD2
Although NOD1 and NOD2 are cytosolic receptors, their activation is associated with localization to the plasma membrane, bacteria-containing phagosomes, and endosomes likely reflecting points of bacterial and ligand entry.40–43 Association with the plasma membrane by NOD1 or NOD2 also typically requires an intact LRR domain, and mutations in the LRR domain resulted in localization largely to the cytosol.42 How recruitment to intracellular membranes occurs remains to be fully elucidated, but may involve interaction with the actin cytoskeleton or interactions with proteins that can associate with the membrane.37,42,43 For example, NOD2 colocalizes with vimentin on the cell membrane44 and is also capable of binding to Erbin and FRMPD2,45,46 a negative and positive regulator of NOD2 signaling, respectively, on the basolateral membrane of intestinal epithelial cells. Rho GTPases, which regulate the actin cytoskeleton, are capable of interacting with NOD1 or NOD2 for their activation.33,44–46 It was also demonstrated that recruitment to the plasma membrane requires lipid modifications for their recruitment to the cell membrane. Specifically, S-palmitoylation mediated by the ZDHHC5 enzyme, which has been associated with the endosomal system and is constitutively localized to phagosomes, was required for membrane recruitment to mount an effective immune response to PGN.47
5 |. MECHANISMS OF LIGAND DELIVERY
Given their intracellular location, efficient NOD1 and NOD2 activation requires ligands to be delivered into the cytosol and entry be achieved by multiple routes (Figure 3, Table 1). Polymeric PGN undergoes phagocytosis and lysosomal digestion48 and may, there-after, be transported to the cytosol. Ligands can be internalized by host cells through phagocytosis of whole bacteria, endocytosis, and uptake of outer membrane vesicle (OMVs).49
FIGURE 3.
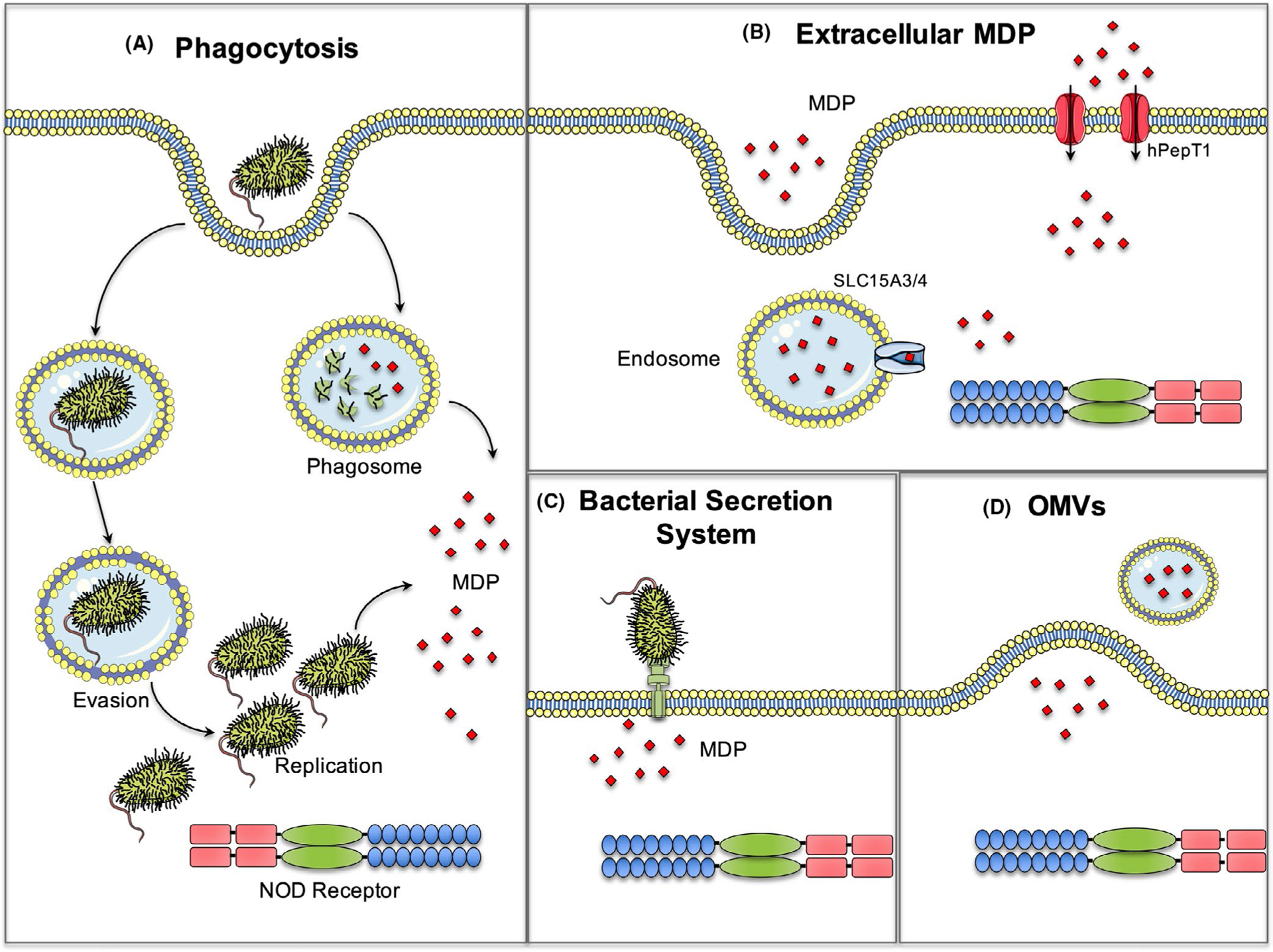
Mechanisms of PGN entrance into host cells to trigger NOD signaling. Host cells can internalize PGN, such as MDP through different mechanisms: (A) Phagocytosis of bacteria can release PGN in the cytoplasm after degradation of bacteria on the phagosome. Some pathogenic bacteria can evade the phagosome and replicate in the host cell, thus releasing PGN in the cytoplasm; (B) extracellular PGN fragments can enter the host cell through endocytosis and transported to the cytosol through SLC15A3/4, lysosomal membrane transporters. Alternatively, the dipeptide transporter hPepT1, expressed in the intestine, can be also carrier-free PGN fragments toward the host cell; (C) some bacteria can deliver PGN into the host cell cytoplasm through its secretion system; (D) uptake of outer membrane vesicles (OMVs) released by Gram-negative bacteria facilitates internalization of PGN
TABLE 1.
Mechanisms of bacterial entry into cells and recognition by NOD1 and NOD2
| Transport system | Microbe/ligand | Receptor | Outcome(s) | Reference(s) |
|---|---|---|---|---|
| Uptake of PGN-containing OMVs | Gram-negative bacteria including H pylori, P aeruginosa, V cholerae, A actinomycetemcomitans, B fragilis, and Neisseria gonorrhea | NOD1/2 | Autophagy; migration of NOD1/PGN-OMVs/RIPK2 to early endosome; NF-κB and MAPK; IL-8 upregulation; adaptive and immune responses | 40,61–63,66,181 |
| SLC15A3/4 peptide transporters | MDP and Salmonella | NOD2 | MDP transport across endosomal membranes; NF-κB/MAPK activation | 41 |
| Peptide transporter hPepT1 | MDP | NOD2 | NF-κB activation in epithelial cell and IL-8 release | 56 |
| Peptide transporter PepT2 | iE-DAP | NOD1 | N F-κB/MAPK activation | 57,58 |
| Type III secretion system-dependent invasive bacteria | S flexneri; S Typhimurium | NOD1 | NF-κB signaling; RhoA activation; SGT1-dependent I L-8 secretion | 36,113,122 |
| Type IV bacterial secretion system | MDP/Brucella abortus | NOD1/2 | ER stress-induced inflammation | 30 |
| Type VII ESX-1 secretion system | MDP/M tuberculosis | NOD2 | Type I IFN expression in macrophages | 39 |
There are multiple potential mechanisms by which PGN fragments can enter the cell to activate NOD1 and NOD2, such as bacterial phagocytosis as well as endocytosis dependent on clathrin and dynamin.50–52 Although certain bacterial secretion systems can activate NOD1 and NOD2 independent of PGN, delivery of PGN can also occur via bacterial secretion systems as is the case with Helicobacter pylori, whose detection by NOD1 is dependent on an intact type IV secretion system and delivery of PGN.53 Also, invasive bacteria, such as Shigella flexneri, shed ligands into the cytosol.18
Membrane transport systems can also transport PGN fragments across the cellular surface into the cytosol. The peptide transporters of the SLC15 family were shown to mediate ligand delivery.51,52 The delivery process occurs through either micropinocytosis in a G protein-coupled calcium-sensing receptor (CaSR)-dependent manner or macropinocytosis in phagocytes.54 PepT1 (SLC15A1), which is highly expressed in the small intestinal epithelium as well as in the colon during inflammation, transports specifically MDP, but not NOD1-activating molecules.55,56 On the other hand, PepT2 (SLC15A2), which is also expressed in epithelial cells, such as in the lung, was capable of actively transporting the NOD1 ligand iE-DAP, but not MDP, to activate inflammatory responses in a NOD1-dependent manner.57,58 The relative importance in the utilization of these transporters may also be dependent on cell type as PepT1, although important for transport of MDP into intestinal epithelial cells, was not required for MDP entry into macrophages, and similarly, transport of NOD1 ligands did not involve PepT2 in HEK293T cells.51,52 Although PepT2 was not important for MDP entry into lung epithelial cells, there was reduced activation of NOD2 in response to NOD2 agonists, such as MDP in Pept2-deficient bone marrow macrophages.57,59 Transport of NOD1 and NOD2 ligands out of endosomes is mediated by additional transporters, including SLC15A3 and SLC15A4 (Pht1) that are specifically expressed in macrophages and dendritic cells.41,52,59,60 These studies demonstrate the importance of transporters for intracellular peptidoglycan delivery and activation of NOD1 and NOD2, but the events occurring downstream of peptide transporters remain to be fully elucidated.
Gram-negative bacteria can release outer membrane vesicles (OMVs) that are 10–300 nm in diameter and can contain PGN fragments, which can be internalized to activate NOD1 and NOD2. Both pathogens and commensal bacteria can produce OMVs and may be a mechanism by which non-phagocytic cells, such as epithelial cells, internalize PGN to enable NOD1 or NOD2 signaling. Pathogens, such as Vibrio cholerae, Helicobacter pylori, Pseudomonas aeruginosa, Neisseria gonorrhea, and Aggregatibacter actinomycetemcomitans, can produce PGN-containing OMVs that activate either NOD1 or NOD2.61–63 Besides pathogenic bacteria, OMVs from commensal bacteria such as the gut microbiota are also capable of activating NOD1 and NOD2 in intestinal epithelial cells.64 Although it was shown that OMVs can enter the host cytosol via lipid rafts located on the cell surface62 and that NOD1 can colocalize with OMVs at early endosomes,40,64 the precise mechanism of OMV entry into the cytosol and their localization within the cell can differ depending on the bacterial strain and host cell type.65,66
6 |. SIGNALING PATHWAYS DOWNSTREAM OF NOD1 AND NOD2
NOD1 and NOD2 regulate multiple pathways involved in a variety of cellular responses, including inflammatory responses via activation of NF-κB, MAPK, and type I IFNs, and autophagy (see Figure 4). In the absence of any stimulation, both NOD1 and NOD2 exist in an inactive “autoinhibited” state in which the LRR domain folds onto the NBD and CARD domain preventing oligomerization and engagement of proteins involved in downstream signaling pathways (see Figure 4).11 Consistently deletion of the LRR domain or mutations that enforce oligomerization in NOD1 is sufficient to induce NF-κB activation.4,67 Upon sensing their cognate ligands, NOD1 and NOD2 self-oligomerize via their NACHT domain to recruit and likewise bring into close proximity molecules of the adapter protein receptor-interacting serine-threonine kinase 2 (RIPK2), which contains an N- and C-terminal CARD domain, via homotypic CARD-CARD interactions.3,4,67–70 This subsequently leads to K63-linked polyubiquitination of RIP2 within its kinase domain, which is required for activation of NF-κB.71 Multiple E3 ligases are involved in the polyubiquitination of RIPK2 and include cIAP-TRAF complexes and Pellino3.71–73 Polyubiquitination of RIPK2 is necessary for recruitment of transforming growth factor B-activated kinase 1 (TAK1) and TAK1-binding proteins (TAB), which subsequently lead to MAPK and NF-κB activation. Non-K63 ubiquitination of NOD2 can also occur via XIAP and leads to the recruitment of the linear ubiquitin chain assembly complex (LUBAC), which produces M1-linked ubiquitin chains that, together with K63-linked chains, facilitate the activation of NF-κB.74 On the other hand, deubiquination of RIPK2 by the deubiquitinases A20, OTULIN, or cylindromatosis protein (CYLD) results in downregulation of NOD1 and/or NOD2 signaling.71,75–79
FIGURE 4.
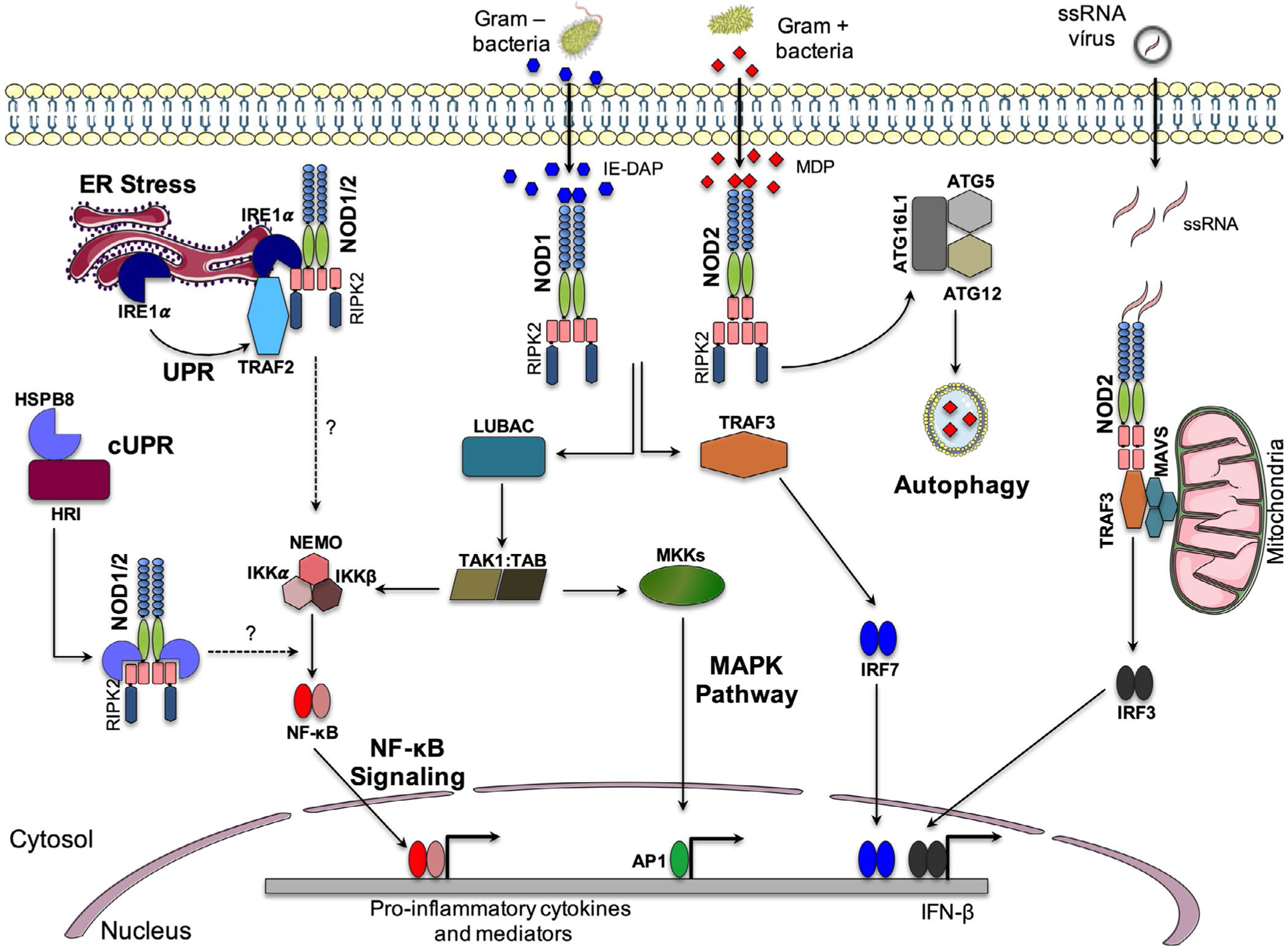
NOD1 and NOD2 signaling pathways. Sensing of iE-DAP and/or MDP occurs via the LRR domains by NOD1 and NOD2, respectively. Once activated, NOD receptors recruit the kinase RIP2 which can interact with LUBAC and the kinase complex TAK1:TAB. This complex of proteins can activate two different pathways: (1) activation of the IKK complex (NEMO: IKKα: IKKβ) which in turn activates NF-κB nuclear translocation; (2) activation of MAP kinases (eg, p38, ERK, and JNK) will activate the transcription factor AP1. Both NF-κB and MAPK pathways will induce gene expression of pro-inflammatory cytokines and mediators, including anti-microbial peptides (AMP). Alternatively, ER stress triggers the unfolded protein response (UPR) caused by accumulation of unfolded or misfolded proteins and bacterial infection. ER stress activates IRE1α (inositol-requiring enzyme 1α) which recruits TRAF2, NOD receptors, and RIPK2 to the ER membrane and initiates inflammatory response through NF-κB signaling; however, the exact mechanism of NF-κB signaling activation by ER stress is still unclear. A cytosolic UPR (cUPR) has been described that is required for NOD1 and NOD2 complex formation and activation of NF-κB. Upon PGN recognition, the heat-shock protein HSPBB8 is released from the complex with HRI and binds NOD1 or NOD2 allowing the folding and release of these receptors from endomembranes. This pathway is associated with NF-κB activation, although the mechanism is not totally known. At the bacterial entry site on the plasma membrane, NOD2 can recruit the autophagy protein ATG16L1, leading to elimination of intracellular pathogens. NOD1 and NOD2 signaling can also induce type I interferon expression. NOD2-RIPK2 activates TRAF3 which recruits TBK1 and induces the IFN-β transcription factor IRF7. In addition, NOD2 is also activated by sensing of virus-derived single-strand RNA (ssRNA). Binding of NOD2/TRAF3 to mitochondrial antiviral signaling (MAVS) induces activation of IRF3 which induces IFN-β gene expression
RIPK2 is critical for activation of both MAPK and NF-κb pathways. RIPK2 binds to both TAK1 and the NF-κB essential modulator kinase (NEMO) also known as IKKγ.71 The colocalization of NEMO and TAK1 promotes the subsequent phosphorylation of the IκKβ subunit of the inhibitor of kappa B kinase (IKK) complex by TAK1, which results in the phosphorylation and degradation of IκBα from the IKK complex. The IKK complex then drives phosphorylation of signal-responsive serine residues of inhibitors of kappa B (IκBs), which are bound to NF-κB dimers in the cytosol. IκBs undergo pro-teasomal degradation to allow the cytoplasmic release and nuclear translocation of NF-κB followed by transcription upregulation of pro-inflammatory mediators.80 NF-κB subsequently binds to kappa B (κB) elements, activating transcription of inflammatory and immune response components. Initiation of RIPK signaling and formation of the TAK1 kinase complex also result in phosphorylation of MKK6 and subsequent activation of the MAPKs, including the p38, extracellular signal–regulated protein kinase (ERK), and c-Jun N-terminal kinase (JNK) pathways.69,81–84 Phosphorylated MAPKs then translocate into the nucleus and phosphorylate AP-1 transcription factors, such as ATF, c-fos, c-Jun, and JDP family members, and mediate cytokines, chemokines, and anti-microbial peptide expression. Thus, the engagement of RIPK2 and TAK1 allows NOD1 and NOD2 to upregulate inflammatory responses via both NF-κB and MAPKs.
Formation of the NOD1/2-RIPK2 multiprotein complex after ligand recognition is a critical event required for the activation NF-κB/MAPK. Upon ligand-induced oligomerization of NOD1/2, RIPK2 is recruited via CARD-CARD interactions. Structural and mutagenesis studies of RIPK2 demonstrated that it forms long filamentous structures that are bound by NOD2 and that the polymerization of RIPK2 is important for NOD2 activation.68 The assembly and stability of NOD1/2-RIPK2 complexes require the heme-regulated inhibitor (HIR), an eIF2a kinase, and the associated heat-shock protein, HSPB8A, and the absence of these components results in impaired NF-κB-mediated production of inflammatory cytokines.85 As HIR promotes the solubility of NOD1 oligomers after activation, it has been proposed that the HIR/HSPB8 axis is necessary to promote the stability of large complexes including the NOD1 or NOD2 signalosome complex formation.85
NOD1 and NOD2 have also been shown to induce type I interferon (IFN) signaling, which plays an important role in antiviral immunity.86 For example, upon exposure to viral ssRNA, NOD2, but not NOD1, interacts with MAVS (mitochondrial antiviral signaling) via its CARD and NBD domains, resulting in activation of interferon regulatory factor-3 (IRF3) in a TRAF3-dependent manner and induction of IFNβ production.26,87 Overexpression and knockdown of NOD2 also affected type I IFN production in response to CMV, a dsRNA virus, which was also mediated, in part, by IRF3.88 The addition of MDP can potentiate type I IFN responses to CMV infection likely via induction of TBK1/IRF3/7 pathway.89 MDP alone has also been shown to induce type I IFN responses,90 and this occurs via RIPK2-mediated activation of TBK1 and IRF5 in the context of Mycobacterium tuberculosis infection of macrophages.39
In the case of NOD1, although not responsive to ssRNA like NOD2, NOD1 has been shown to interact with dsRNA and can augment IFN-β gene expression in certain cell types mediated by RIG-I or MDA5/MAVS.27,28 NOD1 can also participate in type I IFN signaling independently of MAVS. Stimulation of intestinal epithelial cells or hepatocytes with NOD1 ligand (ie, iE-DAP or TriDAP) induces IFN-β production that is dependent on RIPK2.91,92 In particular, stimulation of cells with NOD1 ligand results in RIP2 binding to TRAF3, which then leads to recruitment and activation of TANK-binding kinase 1 (TBK1), IκB kinase e (IKKε), and IRF7 to induce IFN-β production and signaling via the ISGF3 pathway.92
Activation of NOD1 and NOD2 can also lead to initiation of autophagy pathways. Specifically, stimulation of various cell types with NOD1 or NOD2 agonists induced autophagy.93–95 This phenomenon also occurred in vivo in peritoneal macrophages when mice were injected intraperitoneally with either NOD1 or NOD2 agonists. Induction of autophagy occurred independently of RIPK2 and NEMO, a component of NF-κB signaling; rather, NOD1 and NOD2 interacted with the autophagy regulator ATG16L1 at the plasma membrane, triggering autophagy, which can promote the sequestration of intracellular bacteria into autophagosomes to promote their subsequent clearance.93
7 |. NOD1 AND NOD2 IN ADAPTIVE IMMUNITY
Both NOD1 and NOD2 can participate in adaptive immunity. NOD1 and NOD2 agonists can act as adjuvants to enhance antibody production96 and T cell responses. Immunization of NOD1-deficient mice with complete Freud’s adjuvant, which contain both TLR4 and NOD1 agonists, resulted in impaired antigen-specific antibody production and reduced numbers of IL-17, IL-4, and IFN-γ-producing T cells.97 This may, in part, be due to the ability of NOD1 ligand to induce/stimulate DC responses alone and cooperatively with TLR4 agonists.97,98 In addition, immunization of mice with NOD1 ligand and ovalbumin (OVA) results in an enhanced Th2-polarized antigen-specific response that required NOD1 function in non-hematopoietic cells, possibly as a result of the NOD1-mediated induction of chemokines, including the Th2-promoting factor, MCP.97 Similar to what occurs with NOD1 ligand, systemic release of MCP occurs after administration of MDP to mice, and immunization of mice with MDP and OVA results in a Th2-polarized antigen-specific T and B cell response that requires NOD2 function in non-hematopoietic cells and in CD11c+ DCs.99,100 In particular, both NOD1 and NOD2 promote the production of thymic stromal lymphopoietin protein (TSLP) production by non-hematopoietic cells and upregulate OX40 ligand (OX40L) surface expression on DCs that are important for Th2 immunity.100,101 In addition, NOD2 is also capable of synergizing with TLR4 agonists to enhance the production of Th1-polarizing cytokines by DCs and prime Th1 cells as occurs with CFA immunization.99 Thus, while co-stimulation with NOD1 and/or NOD2 collaborates to a Th2 response, co-stimulation with TLR agonists can synergize to induce Th1, Th2, or Th17 immune responses.97–99 NOD1 and NOD2 may also promote specific T cell responses via other mechanisms that affect the priming function of DCs. For example, NOD2 stimulation induces autophagy in DCs, which is required from MHC class II (MHC-II) antigen presentation and antigen-specific CD4+ T cell responses.102 NOD2 can also affect miRNA expression in DCs that can affect the production of specific Th17 cell–polarizing cytokines.103 Injection of NOD1 or NOD2 ligands into mice also increases DC-mediated cross-presentation by enhancing antigen presentation and co-stimulatory molecule expression on DCs.104 Altogether, these studies provide strong evidence for a role of NOD1 and NOD2 signaling in regulating adaptive immune responses.
NOD1 and NOD2 are also expressed in B and T cells, although level of expression is dependent on cellular location. There are some data to suggest that NOD1 and NOD2 ligands can enhance B and T cells after B-cell receptor (BCR) and T-cell receptor (TCR) engagement.105–107 Specifically, in human tonsillar B cells, NOD1 or NOD2 activation alone by treatment with their respective ligands was insufficient to trigger B cell activation or proliferation whereas the combination of NOD1 and NOD2 ligands and BCR activation via IgM or IgD stimulation resulted in enhanced cellular proliferation in vitro, which is further augmented by concomitant TLR stimulation.105 Stimulation of purified CD3+ human tonsillar T cells with NOD1 or NOD2 ligands alone or after TCR activation with anti-CD3/CD28 did not induce cellular proliferation or T-cell cytokine production. Nonetheless, in mixed cultures of tonsillar mononuclear cells, enhanced cellular proliferation of T cells was observed after anti-CD3/CD28 stimulation together with NOD1/NOD2 agonists although the effect was relatively weak.106 With purified murine splenic CD3+ T cells, it was also demonstrated that NOD1 ligand can act as a co-stimulatory molecule to enhance IFN-γ production after stimulation with anti-CD3 alone; however, the possibility of a contributing factor by potential contaminating antigen-presenting cells was not ruled out.107 Regardless, as these studies were all performed in vitro, the physiologic significance of these findings remains to be determined in vivo.
8 |. ROLE OF NOD1 AND NOD2 IN HOST DEFENSE
As sensors of peptidoglycan and regulators of key inflammatory pathways, such as NF-κB and MAPK, NOD1 and NOD2 play important roles in bacterial recognition and the activation of immune responses important for bacterial killing and clearance. Specifically, NOD1 and NOD2 promote host defense by inducing: (i) inflammatory cytokines that have bacterial killing activity, (ii) chemokines that lead to the recruitment of neutrophils and macrophages to the sites of infection, (iii) anti-microbial peptide production by epithelial cells, (iv) type I IFNs, (v) reactive oxygen species, (vi) adaptive immune responses,53,108 and (vii) autophagy.
Largely through recognition of their respective peptidoglycan ligands, but also via peptidoglycan-independent mechanisms, NOD1 and/or NOD2 are capable of sensing L monocytogenes,50,96,109 Streptococcus pneumoniae,110,111 Salmonella,35,47,85,108,112,113 Mycobacterium tuberculosis,94,114 Staphylococcus aureus,115,116 E coli,117–119 C pneumoniae,120 Shigella flexeri, and Borrelia burgdorferi.18,36,121–123
Unlike NOD2 which is expressed primarily in myeloid cells, monocytes, macrophages, and dendritic cells,3 NOD1 is ubiquitously expressed such as in epithelial cells.4 Thus, a common mechanism of NOD1-mediated host defense against bacterial pathogens is the induction of chemokines and anti-microbial peptides by epithelial cells that promote the recruitment of immune cells such as neutrophils and bacterial killing. In vitro stimulation of intestinal epithelial cells with NOD1 synthetic ligands results in the production of CXCL1, CCL2, and IL-8 that are important for recruitment of neutrophils.124 Moreover, in vivo NOD1 stimulation can induce neutrophil recruitment into the peritoneal cavity after i.p. administration of NOD1 ligand; this response was abolished in NOD1-deficient mice.124 The ability of epithelial cells to respond to bacterial stimulation via NOD1 can be particularly important in the intestine where intestinal epithelial cells have low TLR expression and are less responsive to TLR stimulation with lipopolysaccharide.117,125 In the case of Clostridium difficile which contains high NOD1- but low NOD2-stimulatory activity, particularly in mesothelial cells, intraperitoneal injection resulted in significant neutrophil recruitment in the peritoneum that was NOD1-dependent, and NOD1-deficient mice had increased mortality to C difficile colitis associated with impaired production of CXCL1, decreased neutrophil infiltration in the colon lamina propria, and reduced bacterial clearance after oral gavage.126 Besides the intestine, NOD1 signaling in the gastric epithelium is important in host defense against Helicobacter pylori infection, which, despite being a Gram-negative bacteria, does not activate TLR4 signaling in the gastric mucosa.53,127 Peptidoglycan-containing H pylori can activate both NF-κb and MAPK pathways that are NOD1-dependent.53,128 NOD1-deficient mice had increased susceptibility to H pylori infection resulting in reduced production of the chemokine MIP-2 and increased bacterial loads.53 Other mechanisms besides chemokine production likely contribute to clearance of H pylori including production of anti-microbial peptides such as β-defensins,129,130 induction of autophagy-induced inflammatory responses,40,131 and synthesis of type I IFNs by epithelial cells.92 NOD1 also promotes adaptive immune responses to H pylori as NOD1-deficient mice exhibit reduced IgG levels in response to H pylori OMVs,62 which may be related to a role for NOD1 in regulating release of processed IL-33,132 a key driver of Th2 immune responses, and NOD1-dependent type I IFN production can lead to enhanced Th1 chemokine secretion.92 Thus, NOD1 engages multiple pathways to mediate host resistance to pathogens.
NOD2 has a more restrictive expression pattern than NOD1 with expression primarily in monocytes and macrophages. Besides the production of inflammatory cytokines, like NOD1, NOD2 can also upregulate chemokine production via NF-κB and MAPK pathways to recruit immune cells important for eradication of bacterial pathogens.69,133 For example, in vivo, NOD2 promotes the CC-chemokine ligand 2 (CCL2)-CCR2-dependent recruitment of Ly6Chigh monocytes in the intestine and clearance of Citrobacter rodentium-infected mice.134 S pneumoniae also induces CCL2 via NOD2, leading to the recruitment of inflammatory macrophages that play a role in bacterial clearance in the lung.111
An important function of NOD1 and NOD2 is to act cooperatively with Toll-like receptors (TLRs) to enhance immune responses against pathogenic infection as well as promote immune responses after tolerization by TLR ligands. For example, bone marrow-derived macrophages (BMDMs) or mice stimulated with the TLR4 ligand, lipopolysaccharide (LPS), in combination with MDP, resulted in enhanced activation of NF-κB and increased production of IL-6 and TNF-α. In addition, BMDMs made insensitive or tolerant to TLRs by previous exposure to TLR ligands, such as LPS, exhibited enhanced response to NOD1 and NOD2 stimulation.135 The significance of this was demonstrated in vivo in which Nod1−/−Nod2−/− mice previously infected with E coli had decreased bacterial clearance and mouse survival after subsequent infection with Listeria monocytogenes, which contains both NOD1 and NOD2 ligands. Similarly, although activation of NF-κB and MAPK and production of inflammatory cytokines in BMDMs to M tuberculosis were not dependent on NOD1, immune responses to M tuberculosis were partly mediated by NOD1 signaling after tolerization of TLRs by LPS pretreatment.136 These results demonstrate the importance of NOD1 and NOD2 signaling in host defense especially in the setting of secondary bacterial infections when TLR responses are impaired as a result of tolerization.135
NOD1 and NOD2 signaling are also important for antiviral immunity via different mechanisms. Nod2−/− mice have been known to have increased susceptibility against a variety of viral pathogens.26,87,137 NOD2 is capable of interacting with ssRNA, and infection of NOD2-expressing cells in vitro or mice in vivo with VSV, RSV, and influenza leads to upregulation of type I IFNs that are NOD2-dependent.26 Infection of Nod2−/− mice with RSV develops worse lung pathology compared to that of wildtype mice.26 Nod2−/− mice also exhibit reduced survival and clearance of influenza virus after infection, which is associated with impaired DC responses and reduced priming of CD8+ T cells to influenza.87 NOD2 is also capable of limiting influenza-induced lung pathology, by the induction of autophagy, and more specifically mitophagy, resulting in decreased mitochondrial damage and suppression of inflammasome activation that can contribute to immunopathology and morbidity.138 Although NOD1 mice can similarly respond to specific viruses in vitro, whether direct recognition of viruses of NOD1 affects disease outcomes remains to be determined.
Host antiviral responses can also be potentiated by stimulation of NOD1 and NOD2 by their respective PGN ligand. For example, MDP treatment of mice infected with influenza resulted in the induction of type I IFNs as well as CCL2 resulting in increased recruitment of inflammatory monocytes, reduced viral loads, improved lung pathology, and decreased mortality that was NOD2-dependent.139 Stimulation of NOD2 by MDP also inhibited cytomegalovirus (CMV) replication in vitro and was associated with the synergistic induction of type I IFNs by CMV and MDP.89 Liver sinusoidal endothelial cells stimulated with the NOD1 ligand diaminopimelic acid (DAP) resulted in maturation of liver sinusoidal endothelial cells and increased T cell responses in vitro.140 Consequently, administration of DAP in vivo resulted in increased hepatitis B virus (HBV)-specific T cell responses in vivo and replication of HBV.140 Mice pretreated with the NOD1 ligand iE-DAP also exhibited reduced CMV load after infection,91 and the combination of iE-DAP and MDP enhanced inhibition of CMV replicative activity which required type I IFNs.91 These results suggest that exposure to bacteria and stimulation of NOD1 and NOD2 can augment antiviral immunity. Conversely, it has also been demonstrated that type I IFNs can induce expression of NOD1 and NOD2 in BMDMs resulting in enhanced NF-κB/MAPK activation and inflammatory cytokine production. Consistently, infection of bone marrow macrophages by murine norovirus (MNV) resulted in increased NOD1- and NOD2-dependent activation of NF-κB/MAPK activation and production of TNF-α and IL-6. Although this phenomenon may have been designed to enhance immune responses against a secondary bacterial infection, in vivo infection by bacteria such as E coli after MNV infection resulted in increased inflammation and mortality in mice, which is reminiscent of the negative consequences of secondary bacterial infections in humans due to an overexuberant inflammatory response.137
A role for NOD1 and NOD2 in fungal and parasitic infections has been less well-studied. Addition of Aspergillus fumigatus to immortalized human corneal epithelial cells results in increased expression of NOD1 and RIPK2.141 However, although knockdown of Nod1 results in reduced production of inflammatory cytokines in response to Aspergillus,141 in vivo infection in NOD1-deficient mice was associated with increased survival, reduced inflammation, and improved clearance, suggesting that NOD1 signaling, in fact, suppresses immune responses against Aspergillus, possibly due to NOD1-mediated reduction in Dectin-1, a C-type lectin pattern recognition receptor, which is important for Aspergillus killing.142 Similarly, NOD2-deficient mice were also protected against invasive Aspergillus and negatively regulated Dectin-1 expression 143. NOD2 also has negative regulatory role in the immune response against chitin, a polysaccharide of the fungal cell wall.144 Fungal chitin, which forms a component of PGN, induces the release of the anti-inflammatory cytokine IL-10 by BMDMs via NOD2 and TLR9 whereas the production of TNF-α was dependent on TLR2 and Dectin-1.144 Intracellular delivery of chitin was required and mediated by the mannose receptor, a C-type lectin PRR, which is capable of actin remodeling and mediating both phagocytosis and endocytosis.145 Importantly, the chitin-induced anti-inflammatory response may play a critical role during the late phase of infection to allow resolution of inflammation.144
Both NOD1 and NOD2 have also been shown to sense certain parasitic infections. In particular, NOD1-deficient mice were more susceptible to Trypanosoma cruzi infection compared to that of wildtype (WT) mice. NOD2 was also shown to be important for resistance against Toxoplasma gondii infection by inducing Th1 responses.146 NOD2 deficiency also resulted in increased susceptibility to T gondii-induced ileitis and cerebral inflammation after oral infection.147 The impairment in mounting Th1 responses in NOD2-deficient mice against T gondii was not due to a defect in DC priming, but T cell–intrinsic role for NOD2 in regulating IL-2-deficient mice via non-canonical NF-κB signaling.146 However, a separate study was unable to demonstrate a protective role for NOD2 against T gondii infection or a T-cell–intrinsic function,148 which was posited to be due to either insufficient backcrossing of NOD2-deficient mice used in the previous study or differences in the gut microbiota in mice in different mouse facilities. NOD2 was also suggested to recognize the malarial pigment hemozoin,149 and Nod1−/−Nod2−/− mice exhibited reduced inflammatory cytokine production in response to Plasmodium berghei; however, there were no differences in survival or susceptibility to cerebral malaria.150 Thus, how NOD1 and NOD2 sense parasites and whether they play a role in disease susceptibility remain to be fully elucidated.
9 |. NOD1 AND NOD2 IN INTESTINAL HOMEOSTASIS
Through their regulation of pro-inflammatory cytokines/chemokines, anti-microbial peptides, adaptive immunity, and autophagy, NOD1 and NOD2 can promote intestinal homeostasis by enhancing epithelial barrier function 151,152 and resistance to pathogen invasion that can lead to infectious colitis. Activation of NOD1 and NOD2 by Salmonella infection in the intestine increases clearance and limits the severity of Salmonella colitis in mice.108 NOD2-mediated regulation of CCL2 secretion and Th17 responses protects mice against Citrobacter rodentium-induced colitis.108,134 NOD1 signaling promotes neutrophil recruitment via the induction of chemokines which is important for reducing the severity of Clostridium difficile colitis.126 Intestinal bacterial infections by L monocytogenes, S flexeri, and Helicobacter hepaticus are also ameliorated as a result of NOD1 and NOD2 activation via the production of pro-inflammatory mediators, anti-microbial peptides, and/or bacterial autophagy.93,96,153
NOD1 and NOD2 also play other important functions to help maintain intestinal homeostasis. NOD2 is constitutively expressed in LGR5+ intestinal stem cells (ISCs) for example. Activation of NOD2 is associated with increased intestinal organoid-forming efficiency, and in vivo treatment of mice with MDP improved intestinal epithelial regeneration and healing after doxorubicin-induced injury associated with greater stem cell activity 154 although the precise mechanism for this protection remains to be determined. NOD1 signaling in non-hematopoietic cells regulates the development of isolated lymphoid follicles (ILFs) in the intestine, and treatment of germfree mice with a NOD1, but not NOD2 agonist, resulted in increased ILF formation, particularly in the ileum via NOD1-dependent induction of CCL20 and beta-defensin 3, both of which promote the formation of ILFs.155 How NOD1-mediated induction of ILFs affects susceptibility to intestinal disease is not clear; however, the ileal bio-film of NOD1-deficient mice contained a greater bacterial load than that of WT mice as well as an expansion of Gram-negative bacteria, including Bacteroides and Enterobacteriaceae. As ILFs are important sites of IgA induction,156 it is possible that an effect on IgA production may have contributed to the altered microbiome composition as IgA-deficient mice exhibit similar changes.155,157 Whether these effects on the gut microbiota lead to disease susceptibility remains to be determined.
Significant evidence pointing to the importance of NOD2 in maintaining intestinal homeostasis comes from studies pointing to a link between NOD2 dysfunction and inflammatory bowel disease (IBD). IBD is a chronic, relapsing-remitting inflammatory disorder involving the gastrointestinal tract. There are two forms of IBD, ulcerative colitis and Crohn’s disease, and they are distinguished from each other based on clinical characteristics and histopathologic characteristics. Ulcerative colitis affects just the colon, particularly the rectum, and only the surface mucosal layer. In contrast, Crohn’s disease can affect any part of the GI tract, although it occurs commonly in the ileum, and inflammation is transmural and not necessarily contiguous (“skip” lesions). Although the pathogenesis of IBD is still not fully understood, the prevailing model is that combined genetic and environment factors lead to disruption of the integrity of the epithelial barrier, pathologic changes in the composition of the gut microbiome, referred to as dysbiosis, and aberrant immune responses to the gut microbiota.158 NOD2 has been identified as a major susceptibility gene for Crohn’s disease, and multiple polymorphisms, including loss-of-function mutations, have been associated with increased risk of developing Crohn’s disease159,160 (Figure 5A, Table 2). Individuals harboring NOD2 homozygous or compound heterozygous for NOD2 variants exhibit 20- to 40-fold increased risk of developing CD.161 Although conferring CD risk, the majority of individuals carrying NOD2-associated polymorphisms do not develop disease.
FIGURE 5.
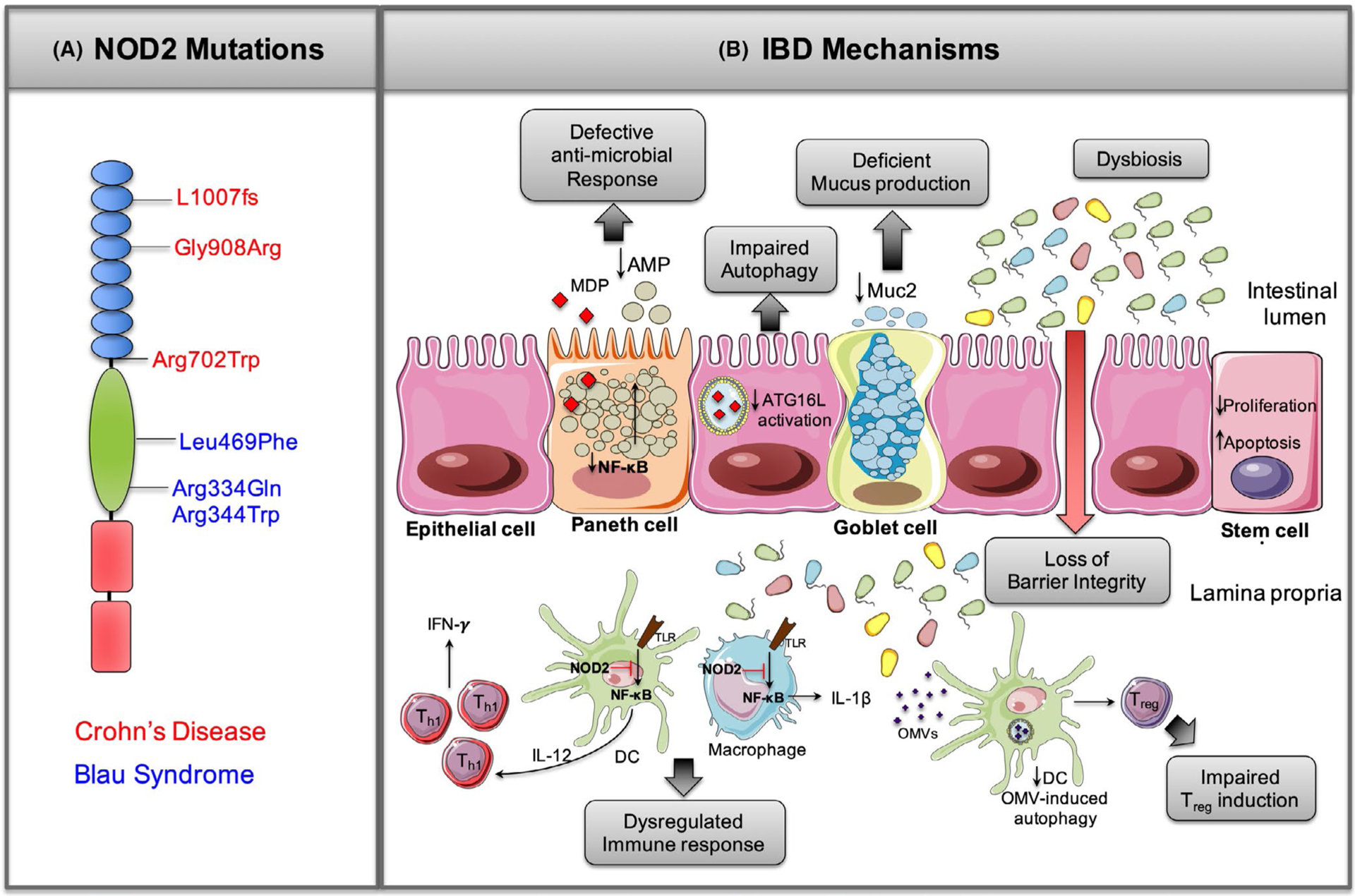
NOD2 Polymorphisms in IBD. (A) Mutations in the NBD are associated with Blau syndrome, and single nucleotide polymorphisms in the LRR domain are associated with Crohn’s disease. (B) NOD2 mutations result in poor sensing of MDP fragments impairing NF-κB activation and decrease production of anti-microbial peptides (AMPs) by Paneth cells. NOD2 variants also fail to recruit ATG16L1 resulting in impaired autophagy by epithelial cells. Dysregulation of these mechanisms leads to reduced bacterial clearance and loss of mucosal barrier function. NOD2 is also important for maintenance of the goblet cell number and mucus secretion. In addition, loss of commensal bacteria homeostasis possibly related to defects in NOD2 surveillance can lead to dysbiosis, which is associated with increased mucosal adherence and consequent bacterial translocation. NOD2 stimulation by MDP also maintains stem cell survival through protection against oxidative stress-mediated cell death, and NOD2 depletion results in reduced stem cell survival and proliferation. Finally, NOD2 variants can cause dysregulation of immune responses in the lamina propria. Defective NOD2 expressed in macrophages and DC is not able to suppress TLR2 signaling leading to overactivation of NF-κB and increased expression of IL-12, IL-1β, and IFN-γ, which in turn can lead to damage of the epithelial layer. In addition, DC autophagy-induced Treg cells are reduced favoring a dysregulated inflammation. Abbreviations: ATG16L1: autophagy-related protein 16-like 1; DC: dendritic cell; MDP: muramyl dipeptide; NBD: Nucleotide-binding domain; NOD2: nucleotide-binding oligomerization domain 2; NF-κB: nuclear factor kappa B; Th1: T-helper 1; TLR: Toll-like receptor; Treg: T regulatory cell
TABLE 2.
Causal genetic variants of NOD1 and NOD2 that are associated with granulomatous autoinflammatory diseases, Crohn’s disease, asthma, and infectious diseases
| Gene | Blau syndrome/early-onset sarcoidosis | Crohn’s disease | Asthma | Infectious diseases |
|---|---|---|---|---|
| NOD1 | G796A255 | ND1+32656247 | Peptic ulceration in H pylori | |
| Rs2975632 | ||||
| Rs2075822 | E266K 256 | |||
| Rs2907749 | Cytomegalovirus | |||
| Rs2907748 244 | rs2284358 | |||
| rs2970500 | ||||
| rs10267377 91 | ||||
| NOD2 | R334W/Q223,227 | R702W159,272 | Leprosy | |
| E383K226,257 | G908R159,272 | rs8057431273 | ||
| R587C227,228 | Fs1007insC159,162,272 | rs9302752274,275 | ||
| H480R258 | R311W, | rs7194886 | ||
| L469F223 | S431L, | rs8057341 | ||
| Y563H259 | R703C, | rs3135499275 | ||
| E667K260 | N852S, | Tuberculosis | ||
| T605N261 | M863V272 | Ala725Gly276 | ||
| G464W262 | R138Q, | Arg587Arg277 | ||
| E600A263 | W157R, | H pylori-associated lymphoma | ||
| D512H259 | N289S, | |||
| E383D264 | D291N, | |||
| C495Y227 | L348V, | R702W278 | ||
| G481D265 | 558delLG, | |||
| R426H221,225 | A612T, | |||
| T605P225,266 | A612V, | |||
| E498G267 | R713C, | |||
| H496L225,266 | E843K231 | |||
| W490L227,228 | ||||
| E338D268 | ||||
| E499_L500 delinsV269 | ||||
| H520Y268,270 | ||||
| N670K225 | ||||
| D390V268 | ||||
| M513R/T225,271 | ||||
| Q809K268 |
How NOD2 regulates susceptibility to developing IBD has been the focus of significant investigation, but several mechanisms have been proposed (Figure 5B). The most common CD-associated Nod2 variant is an L1007fs frameshift insertion at nucleotide 3020 (3020insC) in the LRR domain that generates a truncated protein.3,159,162 Monocytes from homozygous individuals showed impaired cytokine response after MDP stimulation,163 which was similarly observed in mice BMDMs carrying the Nod2 mutation homologous to the human L1007fsinsC (Nod22939iCstop)164 or in cells transfected with mutant NOD2 in response to MDP.121,162 This mutant version of NOD2 does not localize to the plasma membrane43,165 and consequently does not recruit ATG16L, thereby preventing the formation of autophagosomes and impairing bacterial clearance.93 The important of the NOD2-ATG16L pathway is further supported by the observation that individuals homozygous for the single nucleotide polymorphism rs2241880 in the ATG16L gene are associated with increased risk for Crohn’s disease,166,167 and cells from these individuals have defective autophagy after MDP stimulation.93 Crohn’s disease variants in NOD2 have also been shown to have defective S-palmitoylation, which is required for plasma membrane association and optimal bacterial sensing.47 DCs isolated from individual harboring either the 1007fsinsC Nod2 or the Crohn’s variant ATG16L T300A exhibited defective autophagy, MHC-II processing, and antigen-specific CD4+ T cell responses against bacteria, which can reduce mucosal immunity.102 In addition, DCs harboring Crohn’s disease Nod2 variants have impaired levels of mir-29 as a result of impaired NOD2 signaling, resulting in enhanced release of IL-23 and IL12p40 which can contribute to disease.103 It has also been shown that BMDMs from a different mouse strain that harbors the same homologous 1007fsinsC (Nod22939ic) allele exhibited increased NF-κB activation and heightened cytokine responses consistent with the observation that Crohn’s disease patients have increased production of inflammatory cytokines.168 Nod22939ic mice also exhibited increased susceptibility to chemically induced colitis with dextran sulfate sodium (DSS),168 a commonly used mouse model that has features that recapitulate that of human IBD. The difference in BMDM responses to MDP between the two different mouse strains is not clear, but it was postulated that this discrepancy may be due to the addition of amino acids after the frameshift mutation in Nod22939ic mice that are not present in the wildtype NOD2 protein or in the truncated Crohn’s disease-related mutant protein that is present in Nod22939ic mice.164
Mutations in NOD2 have been linked to disease localization to the ileum.169,170 Notably, Paneth cells are located in the crypts of the small intestine, and abnormalities in Paneth cells have been noted in Crohn’s disease patients.171 Paneth cells are important for the production of anti-microbial peptides (AMPs). Decreased alpha-defensin levels have been observed in Crohn’s disease patients with Nod2 mutations.172 In mice, NOD2 deficiency is associated with decrease in AMP production, specifically cryptdins, resulting in impaired gut barrier function and increased susceptibility to bacterial infection.96 Under steady-state conditions, AMPs, such as lysozyme, are synthesized in the ER and transported to the Golgi, where they are packed into immature secretory dense core vesicles (DCV) in Paneth cells. NOD2 stimulation allows the proper lysozyme sorting during DCV maturation, and in the absence of NOD2, lysozyme degradation and depletion are observed, which can contribute to decrease bacterial clearance.173,174 Other abnormalities in the small intestinal epithelium have been observed with NOD2 deficiency, including decreased MUC2 expression and reduced goblet cell numbers, which may lead to reduced mucus production and barrier function.175
Mice that lack NOD2 or harbor the 1007fsinsC mutation do not develop spontaneous colitis,96,168 consistent with the fact that most individuals who have this mutation do not develop disease, suggesting that other factors, including environmental, likely contribute to disease pathogenesis. There is growing evidence that pathologic changes in the gut microbiome, or dysbiosis, can directly contribute to IBD. In particular, the microbiome of IBD patients typically exhibits a loss of microbial diversity, expansion of Enterobacteriaceae, and loss of Faecalibacterium prausnitzii.176 NOD2 deficiency in mice has been associated with changes in microbiome composition, which may be related to defects in mucosal barrier function and microbial sensing. NOD2 knockout mice exhibited increased bacterial loads in the ileum as well as higher abundance of Bacteroidetes and decreased levels of Firmicutes in the intestine, similar to what has been observed in Crohn’s disease patients harboring SNP13 variants, suggesting a role for NOD2 in regulating gut microbiome composition, which in turn can affect susceptibility to colitis.177–179 Consistently, fecal transplantation of stool from NOD2-deficient mice into germfree mice resulted in increased susceptibility to DSS-induced colitis compared to that of germfree mice harboring microbiota from WT mice.180 In a separate study, NOD2-deficient mice were found to exhibit an expansion of Bacteroides vulgatus, which can promote piroxicam-induced small intestinal inflammation, resulting in further changes in the microbiota, including increases in Proteobacteria and decreases in Bacteroidetes and Clostridiales.175 NOD2 polymorphisms may also promote disease through defects in sensing protective signals from the microbiome that are important for gut homeostasis. For example, it has been shown that OMVs from the commensal Bacteroides fragilis OMVs can trigger the autophagy pathway in DCs, which, in turn, induces regulatory T cells in the gut associated with protection against chemically induced colitis that is NOD2-dependent.181 Thus, the loss of NOD2 function may lead to impairment of bacterial recognition and clearance resulting in aberrant inflammation through other inflammatory pathways, including TLRs. As NOD2 has also been shown to directly negatively regulate TLR signaling, defective NOD2 signaling may further enhance immune responses to dysbiosis and increase colitis susceptibility.182–184
As mentioned above, multiple mouse studies have shown a protective effect of NOD2 against multiple models of intestinal inflammation,175,180,182 and treatment of mice with MDP resulted in protection from both DSS- and trinitrobenzene sulfonic acid (TNBS)-induced colitis.185 However, there have also been studies demonstrating no effect of NOD2 in ameliorating DSS-induced colitis.186 In addition, in the SAMP1Yit/Fc (SAMP) mouse model of spontaneous ileitis, NOD2 deletion improved intestinal inflammation in the presence and absence of DSS and was associated with reduced Th2 responses.187 No significant differences in overall microbiome composition were observed at least on a community level between SAMP1Yit/Fc and wildtype mice.187 As the gut microbiome can drive host immune responses in the gut, the discrepancy in findings from in vivo mouse studies is likely dependent not only on the strain of mice, but also on microbiome differences between mouse facilities. It is also important to note that it is unclear whether previous studies used littermate WT and Nod2−/− mice, and therefore, it is also possible that any observed effects in colitis susceptibility were related to differences in the gut microbiota from vertical transmission not related to NOD2 deficiency. Future studies using littermate mice or conventionalized GF WT and NOD2-deficient mice will be necessary to further delineate the effects of NOD2 on microbiome and colitis susceptibility.
NOD1 has also been associated with IBD although to a much lesser extent with gene polymorphisms associated with IBD risk identified in only specific populations.188–191 Although there is strong evidence that NOD1 can limit infectious colitis in mice by specific pathogens, such as C difficile, there are limited studies evaluating the role of NOD1 in colitis. In a mouse model of colitis-associated tumorigenesis which combines the injection of the experimental carcinogen, azoxymethane (AOM), with multiple rounds of DSS to induce chronic, relapsing colitis, NOD1-deficient mice were found to have increased severity off AOM/DSS-induced colonic inflammation compared to WT mice, which was associated with reduced epithelial barrier integrity and increased intestinal epithelial permeability.192 Other studies evaluating Nod1−/−Nod2−/− mice also demonstrated increased chemically induced colitis severity in these mice compared to that of wildtype mice.193 Additional studies using littermate or germfree Nod1−/− mice to evaluate NOD1-specific effects on colitis susceptibility and any microbiome-dependent contributions will need to be performed to further understand NOD1 function in intestinal homeostasis.
10 |. NOD1 AND NOD2 IN SYSTEMIC INFLAMMATORY DISEASE
10.1 |. Type 1 diabetes
Predisposing genetic background and environmental insults can give rise to autoimmune disorders, such as type 1 diabetes (T1D).194–196 T1D is characterized by chronic inflammation involving the pancreatic islets of Langerhans and irreversible elimination of pancreatic beta-cells mediated by autoreactive T cells leading to insulin deficiency and hyperglycemia. Besides the well-known involvement of adaptive immunity in the pathogenesis T1D, there are studies to suggest a potential role for innate immunity, including NOD1 and NOD2, although this has been less well-studied. Environmental factors that affect gut permeability and alterations in the composition of the intestinal microbiota all can affect NOD1 and NOD2 signaling to trigger inflammation and promote T1D.196–198 For example, in a mouse model of streptozocin-induced T1D mice exhibited changes in the gut microbiota and translocation of bacteria into pancreatic lymph nodes, which preceded the development of T1D199 and led to upregulation of NOD2-dependent inflammatory responses in macrophages and DCs. Treatment of mice with MDP in this model promoted the development of hyperglycemia.200 On the other hand, NOD2-deficient non-obese diabetic (NOD) mice had increased susceptibility to developing T1D although this was microbiota-dependent. Clearly, additional studies are necessary to clarify the importance of NOD1 and NOD2 in T1D.
10.2 |. Type 2 diabetes
There is a compelling evidence that dysregulated NOD1 and NOD2 signaling can also affect risk for developing type 2 diabetes (T2D), a chronic inflammatory condition that is typically associated with insulin resistance and obesity, although studies are conflicting in terms of their protective versus detrimental roles.201–204 Endotoxemia due to changes in intestinal permeability and translocation of PAMPs to the circulation is observed in mouse models of T2D, which can give rise to inflammation and insulin resistance.205 Individuals with metabolic syndrome, who are at high risk for developing insulin resistance and T2D, exhibit high expression and activity of NOD1.206 Increased intestinal permeability and translocation of bacteria systemically in mice on a high-fat diet (HFD) led to increased circulating levels of NOD1 agonists, and deficiency in NOD1 resulted in a protective effect with reduced insulin resistance, glucose intolerance, and HFD-induced inflammation.204,207 Administration of NOD1-activating muropeptides also worsened glucose tolerance.207 ER stress is also highly associated with insulin resistance since mice deficient for XBP-1, a regulator of the unfolded protein response, develop diet-induced insulin resistance.208 Saturated fatty acids, which can be sensed by NOD1, also can activate the unfolded protein response and induce ER stress, via ER calcium depletion, to induce inflammation and insulin resistance.209 Therefore, it is possible NOD1 is linked to ER stress that occurs during the development of HFD-induced obesity that potentiates systemic inflammation and predisposes to T2D.210
On the other hand, NOD2-deficient mice developed increased insulin resistance and adipose tissue compared to WT mice on HFD.203 In a separate study, Nod2−/− mice on HFD exhibited increased intestinal bacterial translocation into metabolic tissues (eg, hepatic and adipose), which was associated with inflammation and insulin resistance due to the microbiota changes in NOD2-deficient mice.211 Accordingly, mice injected with the NOD2 ligand MDP showed decreased insulin resistance in obese mice.202 Altogether, these results suggest divergent roles of NOD1 and NOD2 in T2D development; however, the mechanisms involved still remain unclear.
10.3 |. Atherosclerosis
Atherosclerosis is a vascular disease characterized by atheromatous plaques in the intima as well as systemic inflammation. The cell death of vascular smooth muscle cells (VSMCs) in the necrotic core of advanced plaques may imperil the atherosclerotic lesion and thin the protective fibrous cap due to decreased collagen production, leading to cellular debris accumulation and intimal inflammation, causing plaque vulnerability which can lead to myocardial infarction.212,213 ER stress has been observed in VSMCs in the atherosclerotic plaques after perturbation.214,215 NOD2 is expressed in VSMCs216 and plays protective effects in a vascular injury model of neointima hyperplasia by promoting VSMC proliferation, migration, and neointimal formation after vascular injury.217 NOD2-deficient VSMCs also enhance ER stress-induced cell death.218 Finally, NOD2 ablation promoted disruption of atherosclerotic lesions in ApoE−/− mice under an atherogenic diet.218 These data demonstrated a possible protective role of NOD2 during ER stress-induced VSMC death in processes related to plaque necrosis and progression of atherosclerotic lesions.
11 |. NOD1 AND NOD2 IN AUTOINFLAMMATORY DISEASES
11.1 |. Blau syndrome/early-onset sarcoidosis
The autoinflammatory granulomatous diseases Blau syndrome (BS) and early-onset sarcoidosis (EOS) are associated with excessive inflammation and granuloma formation. Both diseases are phenotypically similar, characterized by arthritis, skin rash, and uveitis, and histologically by the occurrence of non-caseating epithelioid granulomas that typically present between 3 and 4 years of age.219,220 BS and EOS share a common genetic etiology, namely gain-of-function single nucleotide polymorphisms (SNPs) in the NOD2 gene leading to an autoactivation of NOD2-mediated NF-κB signaling.221–224 NOD2-related BS/EOS polymorphisms were shown to cluster into the ATP/Mg2+-binding site and helical domain 1 (HD1) in the NOD2 NOD domain, which may dysregulate ATP hydrolysis and NOD2 autoinhibition, respectively225 (Table 2). Complementary mutations in NOD1, however, do not mirror the NOD2 phenotype, which suggest that NOD1 and NOD2 are activated and regulated by distinct methods.222 The mechanism by which NOD2 mutations participate in granulomatous inflammation still remains to be clarified.
While most of NOD2 mutations associated with CD susceptibility are located between the NBD and LRR domains (Figure 5), BS mutations primarily affect the NBD domain with the missense mutation at position 334 (either p.R334Q or p.R334W) found to be the most common and severe.223,226–230 These mutations enhance the self-oligomerization of NOD2 increasing its activity, even in the absence of MDP. Consistently, it was reported that IL-1β, IL-6, and TNF-α levels in the plasma of BS patients were significantly higher than those of healthy controls.227 Overexpression of disease-causing mutations in cell lines was also associated with enhanced NF-κB activation.225,231
SNPs in the NOD2 associated with loss of function have also been described222,232 (Table 1). However, there is only weak evidence pointing to NOD2 loss of function in BS development. Notably, in vitro NF-κB activity related to the overexpression of BS NOD2 mutations was not considered a good predictor of BS/EOS severity although the number of patients evaluated was small.233 Also, mice carrying the BS/EOS p.Arg314Gln mutation do not develop spontaneous disease, but instead show reduced circulating inflammatory cytokines after MDP stimulation, accompanied by decreased macrophage activity and RIPK2, MAPK, and NF-κB activation in these cells.234 Thus, BS/EOS results primarily from the overactivation of NOD2, rather than a loss of function of NOD2 as observed in Crohn’s disease.
11.2 |. Sarcoidosis
In contrast to BS/EOS, the primary site of sarcoidosis granuloma formation is in the lung. Although BS and EOS were reported to share identical NOD2 mutations, no association has been reported between NOD2 and sarcoidosis.235 Instead, NOD1 polymorphisms were found in patients with sarcoidosis236 (Table 2). The Nod1 796-allele was shown to diminish NF-κB activation in response to intracellular Cutibacterium acnes, a bacterium that has been isolated from sarcoid lesions and has a slightly higher odds of being detected in sarcoidosis patients than in healthy individuals.236–238 Regardless, sarcoidosis is considered a non-infectious granulomatous disease, and its etiology remains unknown. NOD1 activation can also contribute to heightened inflammation in sarcoidosis. Sustained activation of p38 MAPK in response to NOD1 agonist and a lack of negative feedback loop via mitogen-activated protein kinase (MKP1) have also been shown to lead to inflammation in chronic sarcoidosis. Consistently, overexpression of MKP1 or chemical inhibition of p38 activation abrogated NOD1-mediated Th1 cytokine production.239 Additional studies evaluating bacterial contributions and the function of NOD1 in the pathogenesis of sarcoid are still needed.
11.3 |. Asthma
NOD1/NOD2/RIPK2 signaling has also been implicated in the development of Th2 immune responses and might play a critical role in the pathology of Th2-mediated conditions such as asthma and atopy. NOD2 is significantly upregulated in asthma patient tissues, and its overexpression in human airway smooth muscle cells (HASMC) promoted proliferation and pro-inflammatory cytokine release in a manner dependent of TSLP.240 Mice immunized with OVA and NOD1 or NOD2 agonist exhibit Th2-skewed T cell responses.99,100 In a mouse model of house dust mite (HDM)-induced asthma, ablation of RIPK2 was sufficient to diminish Th2 inflammatory responses and pathology in the lung, indicating that NOD1 and NOD2 signaling can contribute to allergic airway inflammation.241 It was also demonstrated that NOD1-mediated exacerbation of allergic asthma requires the DC-derived pro-Th2 chemokine CCL17.242 These data suggest that NOD1 and NOD2 activation may influence the development of asthma. Importantly, polymorphisms in Nod1, Nod2, and Ripk2 genes were associated with allergy and asthma,243–246 and a genome-wide association study points to a link between NOD1 polymorphisms, asthma, and high levels of serum IgE247 (Table 2). How these polymorphisms affect NOD1 function and disease pathogenesis remains to be elucidated.
12 |. CNS INFLAMMATORY DISEASE
Both NOD1 and NOD2 have been shown to play a role in the development of autoinflammatory disease involving the CNS, specifically multiple sclerosis, an inflammatory demyelinating disease, via recognition of PGN.248 PGN is present within antigen-presenting cells (APCs) in the CNS of multiple sclerosis (MS) patients249 as well as in CNS lesions in animal models of MS,250,251 suggesting that the presence of PGN, possibly as a result of prior bacterial infection, may contribute to the pathogenesis of multiple sclerosis.252 In experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, PGN can induce disease by stimulating dendritic cells to promote autoimmunity250 and also able to induce EAE in mice.250,253 Importantly, recognition of PGN via NOD1 and NOD2 promoted EAE progression, and NOD1−/−, NOD2−/−, and RIPK2−/− mice were resistant to EAE progression reduced numbers of T cells and activated DCs in the CNS of knockout mice.248 APCs isolated from RIPK2−/− mice with EAE were defective in their ability to drive Th17 effector cell differentiation, whereas RIPK2 function in T cells was not required for disease progression. Altogether, these findings suggest that activation of NOD1 and NOD2 signaling by PGN is required for the activation of CNS-infiltrating DCs, resulting in reactivation of myelin-specific T cells, which, in turn, mediate pathogenesis of EAE. The clinical implication of these findings is significant as they point to a potential therapeutic target in the treatment of multiple sclerosis; however, it remains to determine the significance of NOD1 and NOD2 signaling in the pathogenesis of multiple sclerosis as there have been no polymorphisms in NOD1 or NOD2 identified that are associated with human disease.254
13 |. CONCLUSION
It has become clear that NOD1 and NOD2, as sensors of peptidoglycan and cellular stress and activators of multiple signaling pathways involved in immunity and tissue repair, are key players in the maintenance of health and resistance to infectious and inflammatory diseases. Despite significant advances that have been made using mice models and in vitro systems, questions still remain regarding the precise mechanism by which NOD1 and NOD2 signaling contribute to disease pathogenesis. In particular, carefully controlled studies that dissect and separate potential interactions between NOD1/NOD2 and the gut microbiota and how this crosstalk affects the development, maintenance, regulation, and activation of the innate and adaptive immune system in health and in disease are still needed. Epigenetic mechanisms of NOD1 and NOD2 molecular signaling and the identity of NOD1 and NOD2 interacting partners that are involved in recognition of upstream activators also remain to be fully elucidated. Such studies could lead to preventive and/or therapeutic options for health maintenance or diseases related to dysfunction of NOD1/NOD2 signaling pathways.
ACKNOWLEDGMENTS
We would like to thank Dr Patricia A. Assis for assisting in the conceptualization and preparation of Figures. Work in the laboratory of GYC is supported by National Institutes of Health grant R01DK122812, National Institute of Food and Agriculture grant NIFA 2018-67017-27520, and U.S. Department of Defense grant CA170922 to GYC.
Funding information
National Institutes of Health, Grant/Award Number: R01DK122812; U.S. Department of Defense, Grant/Award Number: CA170922; National Institute of Food and Agriculture, Grant/Award Number: 2018-67017-27520
Footnotes
CONFLICTS OF INTEREST
None.
REFERENCES
- 1.Chen G, Shaw MH, Kim YG, Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. 2009;4:365–398. [DOI] [PubMed] [Google Scholar]
- 2.Bertin J, Nir WJ, Fischer CM, et al. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-kappaB. J Biol Chem. 1999;274(19):12955–12958. [DOI] [PubMed] [Google Scholar]
- 3.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276(7):4812–4818. [DOI] [PubMed] [Google Scholar]
- 4.Inohara N, Koseki T, del Peso L, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem. 1999;274(21):14560–14567. [DOI] [PubMed] [Google Scholar]
- 5.Tanabe T, Chamaillard M, Ogura Y, et al. Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J. 2004;23(7):1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zurek B, Proell M, Wagner RN, Schwarzenbacher R, Kufer TA. Mutational analysis of human NOD1 and NOD2 NACHT domains reveals different modes of activation. Innate Immun. 2012;18(1):100–111. [DOI] [PubMed] [Google Scholar]
- 7.Mo J, Boyle JP, Howard CB, Monie TP, Davis BK, Duncan JA. Pathogen sensing by nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J Biol Chem. 2012;287(27):23057–23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Girardin SE, Jehanno M, Mengin-Lecreulx D, Sansonetti PJ, Alzari PM, Philpott DJ. Identification of the critical residues involved in peptidoglycan detection by Nod1. J Biol Chem. 2005;280(46):38648–38656. [DOI] [PubMed] [Google Scholar]
- 9.Werts C, Girardin SE, Philpott DJ. TIR, CARD and PYRIN: three domains for an antimicrobial triad. Cell Death Differ. 2006;13(5):798–815. [DOI] [PubMed] [Google Scholar]
- 10.Boyle JP, Mayle S, Parkhouse R, Monie TP. Comparative Genomic and Sequence Analysis Provides Insight into the Molecular Functionality of NOD1 and NOD2. Front Immunol. 2013;4:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maekawa S, Ohto U, Shibata T, Miyake K, Shimizu T. Crystal structure of NOD2 and its implications in human disease. Nat Commun. 2016;7:11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inohara N, Ogura Y, Chen FF, Muto A, Nunez G. Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J Biol Chem. 2001;276(4):2551–2554. [DOI] [PubMed] [Google Scholar]
- 13.Bertsche U, Mayer C, Gotz F, Gust AA. Peptidoglycan perception–sensing bacteria by their common envelope structure. Int J Med Microbiol. 2015;305(2):217–223. [DOI] [PubMed] [Google Scholar]
- 14.Pashenkov MV, Dagil YA, Pinegin BV. NOD1 and NOD2: Molecular targets in prevention and treatment of infectious diseases. Int Immunopharmacol. 2018;54:385–400. [DOI] [PubMed] [Google Scholar]
- 15.Girardin SE, Boneca IG, Carneiro LA, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300(5625):1584–1587. [DOI] [PubMed] [Google Scholar]
- 16.Chamaillard M, Hashimoto M, Horie Y, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4(7):702–707. [DOI] [PubMed] [Google Scholar]
- 17.Hasegawa M, Yang K, Hashimoto M, et al. Differential release and distribution of Nod1 and Nod2 immunostimulatory molecules among bacterial species and environments. J Biol Chem. 2006;281(39):29054–29063. [DOI] [PubMed] [Google Scholar]
- 18.Nigro G, Fazio LL, Martino MC, et al. Muramyl peptide shedding modulates cell sensing of Shigella flexneri. Cell Microbiol. 2008;10(3):682–695. [DOI] [PubMed] [Google Scholar]
- 19.Hasegawa M, Kawasaki A, Yang K, et al. A role of lipophilic peptidoglycan-related molecules in induction of Nod1-mediated immune responses. J Biol Chem. 2007;282(16):11757–11764. [DOI] [PubMed] [Google Scholar]
- 20.Laroui H, Yan Y, Narui Y, et al. L-Ala-gamma-D-Glu-meso-diaminopimelic acid (DAP) interacts directly with leucine-rich region domain of nucleotide-binding oligomerization domain 1, increasing phosphorylation activity of receptor-interacting serine/threonine-protein kinase 2 and its interaction with nucleotide-binding oligomerization domain 1. J Biol Chem. 2011;286(35):31003–31013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278(8):5509–5512. [DOI] [PubMed] [Google Scholar]
- 22.Grimes CL, Ariyananda Lde Z, Melnyk JE, O’Shea EK. The innate immune protein Nod2 binds directly to MDP, a bacterial cell wall fragment. J Am Chem Soc. 2012;134(33):13535–13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girardin SE, Travassos LH, Hervé M, et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem. 2003;278(43):41702–41708. [DOI] [PubMed] [Google Scholar]
- 24.Lauro ML, D’Ambrosio EA, Bahnson BJ, Grimes CL. Molecular recognition of muramyl dipeptide occurs in the leucine-rich repeat domain of Nod2. ACS Infect Dis. 2017;3(4):264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laman AG, Lathe R, Shepelyakovskaya AO, et al. Muramyl peptides activate innate immunity conjointly via YB1 and NOD2. Innate Immun. 2016;22(8):666–673. [DOI] [PubMed] [Google Scholar]
- 26.Sabbah A, Chang TH, Harnack R, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10(10):1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vegna S, Gregoire D, Moreau M, et al. NOD1 participates in the innate immune response triggered by hepatitis C virus polymerase. J Virol. 2016;90(13):6022–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu XM, Zhang J, Li PW, et al. NOD1 promotes antiviral signaling by binding viral RNA and regulating the interaction of MDA5 and MAVS. J Immunol. 2020;204(8):2216–2231. [DOI] [PubMed] [Google Scholar]
- 29.Hetz C The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. [DOI] [PubMed] [Google Scholar]
- 30.Keestra-Gounder AM, Byndloss MX, Seyffert N, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature. 2016;532(7599):394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molinaro R, Mukherjee T, Flick R, Philpott DJ, Girardin SE. Trace levels of peptidoglycan in serum underlie the NOD-dependent cytokine response to endoplasmic reticulum stress. J Biol Chem. 2019;294(22):9007–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol. 2016;17(8):496–510. [DOI] [PubMed] [Google Scholar]
- 33.Keestra AM, Winter MG, Auburger JJ, et al. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature. 2013;496(7444):233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friebel A, Ilchmann H, Aepfelbacher M, Ehrbar K, Machleidt W, Hardt WD. SopE and SopE2 from Salmonella typhimurium activate different sets of RhoGTPases of the host cell. J Biol Chem. 2001;276(36):34035–34040. [DOI] [PubMed] [Google Scholar]
- 35.Keestra AM, Winter MG, Klein-Douwel D, et al. A salmonella virulence factor activates the NOD1/NOD2 signaling pathway. MBio. 2011;2(6), e00266–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukazawa A, Alonso C, Kurachi K, et al. GEF-H1 mediated control of NOD1 dependent NF-kappaB activation by Shigella effectors. PLoS Pathog. 2008;4(11):e1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Legrand-Poels S, Kustermans G, Bex F, Kremmer E, Kufer TA, Piette J. Modulation of Nod2-dependent NF-kappaB signaling by the actin cytoskeleton. J Cell Sci. 2007;120(Pt 7):1299–1310. [DOI] [PubMed] [Google Scholar]
- 38.Conrad WH, Osman MM, Shanahan JK, et al. Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc Natl Acad Sci U S A. 2017;114(6):1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pandey AK, Yang Y, Jiang Z, et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog. 2009;5(7):e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irving A, Mimuro H, Kufer T, et al. The immune receptor NOD1 and kinase RIP2 interact with bacterial peptidoglycan on early endosomes to promote autophagy and inflammatory signaling. Cell Host Microbe. 2014;15(5):623–635. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura N, Lill JR, Phung Q, et al. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature. 2014;509(7499):240–244. [DOI] [PubMed] [Google Scholar]
- 42.Kufer TA, Kremmer E, Adam AC, Philpott DJ, Sansonetti PJ. The pattern-recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell Microbiol. 2008;10(2):477–486. [DOI] [PubMed] [Google Scholar]
- 43.Barnich N, Aguirre JE, Reinecker HC, Xavier R, Podolsky DK. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-{kappa}B activation in muramyl dipeptide recognition. J Cell Biol. 2005;170(1):21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stevens C, Henderson P, Nimmo ER, et al. The intermediate filament protein, vimentin, is a regulator of NOD2 activity. Gut. 2013;62(5):695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lipinski S, Grabe N, Jacobs G, et al. RNAi screening identifies mediators of NOD2 signaling: implications for spatial specificity of MDP recognition. Proc Natl Acad Sci U S A. 2012;109(52): 21426–21431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McDonald C, Chen FF, Ollendorff V, et al. A role for Erbin in the regulation of Nod2-dependent NF-kappaB signaling. J Biol Chem. 2005;280(48):40301–40309. [DOI] [PubMed] [Google Scholar]
- 47.Lu Y, Zheng Y, Coyaud É, et al. Palmitoylation of NOD1 and NOD2 is required for bacterial sensing. Science. 2019;366(6464): 460–467. [DOI] [PubMed] [Google Scholar]
- 48.Shimada T, Park BG, Wolf AJ, et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe. 2010;7(1):38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al Nabhani Z, Dietrich G, Hugot JP, Barreau F. Nod2: The intestinal gate keeper. PLoS Pathog. 2017;13(3):e1006177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herskovits AA, Auerbuch V, Portnoy DA. Bacterial ligands generated in a phagosome are targets of the cytosolic innate immune system. PLoS Pathog. 2007;3(3):e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marina-García N, Franchi L, Kim Y-G, et al. Clathrin- and dynamin-dependent endocytic pathway regulates muramyl dipeptide internalization and NOD2 activation. J Immunol. 2009;182(7):4321–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee J, Tattoli I, Wojtal KA, Vavricka SR, Philpott DJ, Girardin SE. pH-dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J Biol Chem. 2009;284(35):23818–23829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Viala J, Chaput C, Boneca IG, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5(11):1166–1174. [DOI] [PubMed] [Google Scholar]
- 54.Canton J, Schlam D, Breuer C, Gutschow M, Glogauer M, Grinstein S. Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat Commun. 2016;7:11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ismair MG, Vavricka SR, Kullak-Ublick GA, Fried M, Mengin-Lecreulx D, Girardin SE. hPepT1 selectively transports muramyl dipeptide but not Nod1-activating muramyl peptides. Can J Physiol Pharmacol. 2006;84(12):1313–1319. [DOI] [PubMed] [Google Scholar]
- 56.Vavricka SR, Musch MW, Chang JE, et al. hPepT1 transports muramyl dipeptide, activating NF-kappaB and stimulating IL-8 secretion in human colonic Caco2/bbe cells. Gastroenterology. 2004;127(5):1401–1409. [DOI] [PubMed] [Google Scholar]
- 57.Swaan PW, Bensman T, Bahadduri PM, et al. Bacterial peptide recognition and immune activation facilitated by human peptide transporter PEPT2. Am J Respir Cell Mol Biol. 2008;39(5):536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takano M, Takeuchi T, Kuriyama S, Yumoto R. Role of peptide transporter 2 and MAPK signaling pathways in the innate immune response induced by bacterial peptides in alveolar epithelial cells. Life Sci. 2019;229:173–179. [DOI] [PubMed] [Google Scholar]
- 59.Hu Y, Song F, Jiang H, Nunez G, Smith DE. SLC15A2 and SLC15A4 mediate the transport of bacterially derived Di/tripeptides to enhance the nucleotide-binding oligomerization domain-dependent immune response in mouse bone marrow-derived macrophages. J Immunol. 2018;201(2):652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sasawatari S, Okamura T, Kasumi E, et al. The solute carrier family 15A4 regulates TLR9 and NOD1 functions in the innate immune system and promotes colitis in mice. Gastroenterology. 2011;140(5):1513–1525. [DOI] [PubMed] [Google Scholar]
- 61.Thay B, Damm A, Kufer TA, Wai SN, Oscarsson J Aggregatibacter actinomycetemcomitans outer membrane vesicles are internalized in human host cells and trigger NOD1- and NOD2-dependent NF-kappaB activation. Infect Immun. 2014;82(10): 4034–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaparakis M, Turnbull L, Carneiro L, et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol. 2010;12(3):372–385. [DOI] [PubMed] [Google Scholar]
- 63.Chatterjee D, Chaudhuri K. Vibrio cholerae O395 outer membrane vesicles modulate intestinal epithelial cells in a NOD1 protein-dependent manner and induce dendritic cell-mediated Th2/Th17 cell responses. J Biol Chem. 2013;288(6):4299–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Canas MA, Fabrega MJ, Gimenez R, Badia J, Baldoma L. Outer membrane vesicles from probiotic and commensal escherichia coli activate NOD1-mediated immune responses in intestinal epithelial cells. Front Microbiol. 2018;9:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bielig H, Dongre M, Zurek B, Wai SN, Kufer TA. A role for quorum sensing in regulating innate immune responses mediated by Vibrio cholerae outer membrane vesicles (OMVs). Gut Microbes. 2011;2(5):274–279. [DOI] [PubMed] [Google Scholar]
- 66.Bielig H, Rompikuntal PK, Dongre M, et al. NOD-like receptor activation by outer membrane vesicles from Vibrio cholerae non-O1 non-O139 strains is modulated by the quorum-sensing regulator HapR. Infect Immun. 2011;79(4):1418–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Inohara N, Koseki T, Lin J, et al. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275(36):27823–27831. [DOI] [PubMed] [Google Scholar]
- 68.Pellegrini E, Desfosses A, Wallmann A, et al. RIP2 filament formation is required for NOD2 dependent NF-kappaB signalling. Nat Commun. 2018;9(1):4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park J-H, Kim Y-G, McDonald C, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178(4):2380–2386. [DOI] [PubMed] [Google Scholar]
- 70.Kobayashi K, Inohara N, Hernandez LD, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416(6877):194–199. [DOI] [PubMed] [Google Scholar]
- 71.Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. 2008;27(2):373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009;30(6):789–801. [DOI] [PubMed] [Google Scholar]
- 73.Yang S, Wang B, Humphries F, et al. Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and protective effects in colitis. Nat Immunol. 2013;14(9):927–936. [DOI] [PubMed] [Google Scholar]
- 74.Damgaard R, Nachbur U, Yabal M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. 2012;46(6):746–758. [DOI] [PubMed] [Google Scholar]
- 75.Hitotsumatsu O, Ahmad R-C, Tavares R, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28(3):381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fiil B, Damgaard R, Wagner S, et al. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol Cell. 2013;50(6):818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Draber P, Kupka S, Reichert M, et al. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. 2015;13(10):2258–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hrdinka M, Fiil B, Zucca M, et al. CYLD limits Lys63- and Met1-linked ubiquitin at receptor complexes to regulate innate immune signaling. Cell Rep. 2016;14(12):2846–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wex K, Schmid U, Just S, et al. Receptor-interacting protein kinase-2 inhibition by CYLD Impairs Antibacterial Immune Responses In Macrophages. Front Immunol. 2015;6:650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–362. [DOI] [PubMed] [Google Scholar]
- 81.Windheim M, Lang C, Peggie M, Plater LA, Cohen P. Molecular mechanisms involved in the regulation of cytokine production by muramyl dipeptide. Biochem J. 2007;404(2):179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412(6844):346–351. [DOI] [PubMed] [Google Scholar]
- 83.da Silva CJ, Miranda Y, Leonard N, Hsu J, Ulevitch RJ. Regulation of Nod1-mediated signaling pathways. Cell Death Differ. 2007;14(4):830–839. [DOI] [PubMed] [Google Scholar]
- 84.Hsu Y-M, Zhang Y, You Y, et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol. 2007;8(2):198–205. [DOI] [PubMed] [Google Scholar]
- 85.Abdel-Nour M, Carneiro LAM, Downey J, et al. The heme-regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science. 2019;365(6448):eaaw4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seth RB, Sun L, Chen ZJ. Antiviral innate immunity pathways. Cell Res. 2006;16(2):141–147. [DOI] [PubMed] [Google Scholar]
- 87.Lupfer C, Thomas PG, Kanneganti TD. Nucleotide oligomerization and binding domain 2-dependent dendritic cell activation is necessary for innate immunity and optimal CD8+ T Cell responses to influenza A virus infection. J Virol. 2014;88(16):8946–8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kapoor A, Forman M, Arav-Boger R. Activation of nucleotide oligomerization domain 2 (NOD2) by human cytomegalovirus initiates innate immune responses and restricts virus replication. PLoS One. 2014;9(3):e92704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kapoor A, Fan YH, Arav-Boger R. Bacterial muramyl dipeptide (MDP) restricts human cytomegalovirus replication via an IFN-beta-dependent pathway. Sci Rep. 2016;6:20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Leber JH, Crimmins GT, Raghavan S, Meyer-Morse NP, Cox JS, Portnoy DA. Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog. 2008;4(1):e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fan Y-H, Roy S, Mukhopadhyay R, et al. Role of nucleotide-binding oligomerization domain 1 (NOD1) and its variants in human cytomegalovirus control in vitro and in vivo. Proc Natl Acad Sci U S A. 2016;113(48):E7818–E7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Watanabe T, Asano N, Fichtner-Feigl S, et al. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest. 2010;120(5):1645–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Travassos LH, Carneiro LAM, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11(1):55–62. [DOI] [PubMed] [Google Scholar]
- 94.Juárez E, Carranza C, Hernández-Sánchez F, et al. Nucleotide-oligomerizing domain-1 (NOD1) receptor activation induces pro-inflammatory responses and autophagy in human alveolar macrophages. BMC Pulm Med. 2014;14:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology. 2010;139(5):1630–1641, 41 e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307(5710):731–734. [DOI] [PubMed] [Google Scholar]
- 97.Fritz JH, Le Bourhis L, Sellge G, et al. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity. 2007;26(4):445–459. [DOI] [PubMed] [Google Scholar]
- 98.Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect Immun. 2005;73(12):7967–7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Magalhaes JG, Fritz JH, Le Bourhis L, et al. Nod2-dependent Th2 polarization of antigen-specific immunity. J Immunol. 2008;181(11):7925–7935. [DOI] [PubMed] [Google Scholar]
- 100.Magalhaes JG, Rubino SJ, Travassos LH, et al. Nucleotide oligomerization domain-containing proteins instruct T cell helper type 2 immunity through stromal activation. Proc Natl Acad Sci U S A. 2011;108(36):14896–14901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ito T, Wang Y-H, Duramad O, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202(9):1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16(1):90–97. [DOI] [PubMed] [Google Scholar]
- 103.Brain O, Owens B, Pichulik T, et al. The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity. 2013;39(3):521–536. [DOI] [PubMed] [Google Scholar]
- 104.Asano J, Tada H, Onai N, et al. Nucleotide oligomerization binding domain-like receptor signaling enhances dendritic cell-mediated cross-priming in vivo. J Immunol. 2010;184(2):736–745. [DOI] [PubMed] [Google Scholar]
- 105.Petterson T, Jendholm J, Mansson A, Bjartell A, Riesbeck K, Cardell LO. Effects of NOD-like receptors in human B lymphocytes and crosstalk between NOD1/NOD2 and Toll-like receptors. J Leukoc Biol. 2011;89(2):177–187. [DOI] [PubMed] [Google Scholar]
- 106.Petterson T, Mansson A, Riesbeck K, Cardell LO. Nucleotide-binding and oligomerization domain-like receptors and retinoic acid inducible gene-like receptors in human tonsillar T lymphocytes. Immunology. 2011;133(1):84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhan Y, Seregin SS, Chen J, Chen GY. Nod1 limits colitis-associated tumorigenesis by regulating IFN-gamma production. J Immunol. 2016;196(12):5121–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Geddes K, Rubino S, Streutker C, et al. Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect Immun. 2010;78(12):5107–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mosa A, Trumstedt C, Eriksson E, et al. Nonhematopoietic cells control the outcome of infection with Listeria monocytogenes in a nucleotide oligomerization domain 1-dependent manner. Infect Immun. 2009;77(7):2908–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Opitz B, Puschel A, Schmeck B, et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem. 2004;279(35):36426–36432. [DOI] [PubMed] [Google Scholar]
- 111.Davis KM, Nakamura S, Weiser JN. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J Clin Invest. 2011;121(9):3666–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124(4):993–1000. [DOI] [PubMed] [Google Scholar]
- 113.Bhavsar AP, Brown NF, Stoepel J, et al. The Salmonella type III effector SspH2 specifically exploits the NLR co-chaperone activity of SGT1 to subvert immunity. PLoS Pathog. 2013;9(7):e1003518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ferwerda G, Girardin SE, Kullberg B-J, et al. NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog. 2005;1(3):279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Deshmukh HS, Hamburger JB, Ahn SH, McCafferty DG, Yang SR, Fowler VG Jr. Critical role of NOD2 in regulating the immune response to Staphylococcus aureus. Infect Immun. 2009;77(4):1376–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kapetanovic R, Nahori M-A, Balloy V, et al. Contribution of phagocytosis and intracellular sensing for cytokine production by Staphylococcus aureus-activated macrophages. Infect Immun. 2007;75(2):830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim JG, Lee SJ, Kagnoff MF. Nod1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by toll-like receptors. Infect Immun. 2004;72(3):1487–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Theivanthiran B, Batra S, Balamayooran G, et al. NOD2 signaling contributes to host defense in the lungs against Escherichia coli infection. Infect Immun. 2012;80(7):2558–2569. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 119.Hruz P, Zinkernagel AS, Jenikova G, et al. NOD2 contributes to cutaneous defense against Staphylococcus aureus through alpha-toxin-dependent innate immune activation. Proc Natl Acad Sci U S A. 2009;106(31):12873–12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shimada K, Chen S, Dempsey PW, et al. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog. 2009;5(4):e1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278(11):8869–8872. [DOI] [PubMed] [Google Scholar]
- 122.Girardin SE, Tournebize R, Mavris M, et al. CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2001;2(8):736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Oosting M, Berende A, Sturm P, et al. Recognition of Borrelia burgdorferi by NOD2 is central for the induction of an inflammatory reaction. J Infect Dis. 2010;201(12):1849–1858. [DOI] [PubMed] [Google Scholar]
- 124.Masumoto J, Yang K, Varambally S, et al. Nod1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J Exp Med. 2006;203(1):203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174(8):4453–4460. [DOI] [PubMed] [Google Scholar]
- 126.Hasegawa M, Yamazaki T, Kamada N, et al. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol. 2011;186(8):4872–4880. [DOI] [PubMed] [Google Scholar]
- 127.Backhed F, Rokbi B, Torstensson E, et al. Gastric mucosal recognition of Helicobacter pylori is independent of Toll-like receptor 4. J Infect Dis. 2003;187(5):829–836. [DOI] [PubMed] [Google Scholar]
- 128.Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol. 2009;183(12):8099–8109. [DOI] [PubMed] [Google Scholar]
- 129.Grubman A, Kaparakis M, Viala J, et al. The innate immune molecule, NOD1, regulates direct killing of Helicobacter pylori by antimicrobial peptides. Cell Microbiol. 2010;12(5):626–639. [DOI] [PubMed] [Google Scholar]
- 130.Boughan PK, Argent RH, Body-Malapel M, et al. Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection. J Biol Chem. 2006;281(17):11637–11648. [DOI] [PubMed] [Google Scholar]
- 131.Tang B, Li N, Gu J, et al. Compromised autophagy by MIR30B benefits the intracellular survival of Helicobacter pylori. Autophagy. 2012;8(7):1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tran LS, Tran D, De Paoli A, et al. NOD1 is required for Helicobacter pylori induction of IL-33 responses in gastric epithelial cells. Cell Microbiol. 2018;20(5):e12826. [DOI] [PubMed] [Google Scholar]
- 133.Jeong Y-J, Kang M-J, Lee S-J, et al. Nod2 and Rip2 contribute to innate immune responses in mouse neutrophils. Immunology. 2014;143(2):269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kim Y-G, Kamada N, Shaw M, et al. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity. 2011;34(5):769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim YG, Park JH, Shaw MH, Franchi L, Inohara N, Nunez G. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity. 2008;28(2):246–257. [DOI] [PubMed] [Google Scholar]
- 136.Lee J-Y, Hwang E-H, Kim D-J, et al. The role of nucleotide-binding oligomerization domain 1 during cytokine production by macrophages in response to Mycobacterium tuberculosis infection. Immunobiology. 2016;221(1):70–75. [DOI] [PubMed] [Google Scholar]
- 137.Kim Y-G, Park J-H, Reimer T, et al. Viral infection augments Nod1/2 signaling to potentiate lethality associated with secondary bacterial infections. Cell Host Microbe. 2011;9(6):496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lupfer C, Thomas PG, Anand PK, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14(5):480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Coulombe F, Fiola S, Akira S, Cormier Y, Gosselin J. Muramyl dipeptide induces NOD2-dependent Ly6C(high) monocyte recruitment to the lungs and protects against influenza virus infection. PLoS One. 2012;7(5):e36734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang S, Zou S, Chen M, et al. Local stimulation of liver sinusoidal endothelial cells with a NOD1 agonist activates T cells and suppresses hepatitis B virus replication in mice. J Immunol. 2018;200(9):3170–3179. [DOI] [PubMed] [Google Scholar]
- 141.Zhang Y, Wu J, Xin Z, Wu X. Aspergillus fumigatus triggers innate immune response via NOD1 signaling in human corneal epithelial cells. Exp Eye Res. 2014;127:170–178. [DOI] [PubMed] [Google Scholar]
- 142.Gresnigt MS, Jaeger M, Subbarao Malireddi RK, et al. The absence of NOD1 enhances killing of aspergillus fumigatus through modulation of dectin-1 expression. Front Immunol. 2017;8:1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gresnigt MS, Cunha C, Jaeger M, et al. Genetic deficiency of NOD2 confers resistance to invasive aspergillosis. Nat Commun. 2018;9(1):2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wagener J, Malireddi RKS, Lenardon MD, et al. Fungal chitin dampens inflammation through IL-10 induction mediated by NOD2 and TLR9 activation. PLoS Pathog. 2014;10(4):e1004050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rajaram MVS, Arnett E, Azad AK, et al. Tuberculosis-initiated human mannose receptor signaling regulates macrophage recognition and vesicle trafficking by FcRgamma-chain, Grb2, and SHP-1. Cell Rep. 2017;21(1):126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shaw MH, Reimer T, Sánchez-Valdepeñas C, et al. T cell-intrinsic role of Nod2 in promoting type 1 immunity to Toxoplasma gondii. Nat Immunol. 2009;10(12):1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Heimesaat MM, Dunay IR, Alutis M, et al. Nucleotide-oligomerization-domain-2 affects commensal gut microbiota composition and intracerebral immunopathology in acute Toxoplasma gondii induced murine ileitis. PLoS One. 2014;9(8):e105120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Caetano BC, Biswas A, Lima-Junior DS, et al. Intrinsic expression of Nod2 in CD4+ T lymphocytes is not necessary for the development of cell-mediated immunity and host resistance to Toxoplasma gondii. Eur J Immunol. 2011;41(12):3627–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Corbett Y, Parapini S, D’Alessandro S, et al. Involvement of Nod2 in the innate immune response elicited by malarial pigment hemozoin. Microbes Infect. 2015;17(3):184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Finney CA, Lu Z, LeBourhis L, Philpott DJ, Kain KC. Disruption of Nod-like receptors alters inflammatory response to infection but does not confer protection in experimental cerebral malaria. Am J Trop Med Hyg. 2009;80(5):718–722. [PubMed] [Google Scholar]
- 151.Nighot PK, Hu CA, Ma TY. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J Biol Chem. 2015;290(11):7234–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wu Y, Tang L, Wang B, Sun Q, Zhao P, Li W. The role of autophagy in maintaining intestinal mucosal barrier. J Cell Physiol. 2019;234(11):19406–19419. [DOI] [PubMed] [Google Scholar]
- 153.Biswas A, Liu Y-J, Hao L, et al. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci U S A. 2010;107(33):14739–14744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nigro G, Rossi R, Commere PH, Jay P, Sansonetti PJ. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe. 2014;15(6):792–798. [DOI] [PubMed] [Google Scholar]
- 155.Bouskra D, Brézillon C, Bérard M, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456(7221):507–510. [DOI] [PubMed] [Google Scholar]
- 156.Knoop KA, Newberry RD. Isolated lymphoid follicles are dynamic reservoirs for the induction of intestinal IgA. Front Immunol. 2012;3:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. 2002;298(5597):1424–1427. [DOI] [PubMed] [Google Scholar]
- 158.Ramos GP, Papadakis KA. Mechanisms of disease: inflammatory bowel diseases. Mayo Clin Proc. 2019;94(1):155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hugot J-P, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603. [DOI] [PubMed] [Google Scholar]
- 160.Hampe J, Cuthbert A, Croucher PJP, et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet. 2001;357(9272):1925–1928. [DOI] [PubMed] [Google Scholar]
- 161.Hugot J-P, Zaccaria I, Cavanaugh J, et al. Prevalence of CARD15/NOD2 mutations in Caucasian healthy people. Am J Gastroenterol. 2007;102(6):1259–1267. [DOI] [PubMed] [Google Scholar]
- 162.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411(6837):603–606. [DOI] [PubMed] [Google Scholar]
- 163.van Heel DA, Ghosh S, Butler M, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet. 2005;365(9473):1794–1796. [DOI] [PubMed] [Google Scholar]
- 164.Kim YG, Shaw MH, Warner N, et al. Cutting edge: Crohn’s disease-associated Nod2 mutation limits production of proinflammatory cytokines to protect the host from Enterococcus faecalis-induced lethality. J Immunol. 2011;187(6):2849–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lécine P, Esmiol S, Métais J-Y, et al. The NOD2-RICK complex signals from the plasma membrane. J Biol Chem. 2007;282(20):15197–15207. [DOI] [PubMed] [Google Scholar]
- 166.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39(5):596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–211. [DOI] [PubMed] [Google Scholar]
- 168.Maeda S, Hsu LC, Liu H, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307(5710):734–738. [DOI] [PubMed] [Google Scholar]
- 169.Cuthbert AP, Fisher SA, Mirza MM, et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology. 2002;122(4):867–874. [DOI] [PubMed] [Google Scholar]
- 170.Buning C, Genschel J, Buhner S, et al. Mutations in the NOD2/CARD15 gene in Crohn’s disease are associated with ileocecal resection and are a risk factor for reoperation. Aliment Pharmacol Ther. 2004;19(10):1073–1078. [DOI] [PubMed] [Google Scholar]
- 171.VanDussen KL, Liu TC, Li D, et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology. 2014;146(1):200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wehkamp J, Harder J, Weichenthal M, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53(11):1658–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wang H, Zhang X, Zuo Z, et al. Rip2 is required for Nod2-mediatedlysozyme sorting in paneth cells. J Immunol. 2017;198(9):3729–3736. [DOI] [PubMed] [Google Scholar]
- 174.Zhang Q, Pan Y, Yan R, et al. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol. 2015;16(9):918–926. [DOI] [PubMed] [Google Scholar]
- 175.Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity. 2014;41(2):311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14(10):573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Petnicki-Ocwieja T, Hrncir T, Liu Y-J, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A. 2009;106(37):15813–15818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rehman A, Sina C, Gavrilova O, et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut. 2011;60(10):1354–1362. [DOI] [PubMed] [Google Scholar]
- 179.Frank DN, Robertson CE, Hamm CM, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17(1):179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Couturier-Maillard A, Secher T, Rehman A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123(2):700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chu H, Khosravi A, Kusumawardhani IP, et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science. 2016;352(6289):1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Watanabe T, Kitani A, Murray PJ, Wakatsuki Y, Fuss IJ, Strober W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 2006;25(3):473–485. [DOI] [PubMed] [Google Scholar]
- 183.Udden SMN, Peng L, Gan J-L, et al. NOD2 suppresses colorectal tumorigenesis via downregulation of the TLR pathways. Cell Rep. 2017;19(13):2756–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Watanabe T, Kitani A, Strober W. NOD2 regulation of Toll-like receptor responses and the pathogenesis of Crohn’s disease. Gut. 2005;54(11):1515–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Watanabe T, Asano N, Murray PJ, et al. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118(2):545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jing X, Zulfiqar F, Park SY, Nunez G, Dziarski R, Gupta D. Peptidoglycan recognition protein 3 and Nod2 synergistically protect mice from dextran sodium sulfate-induced colitis. J Immunol. 2014;193(6):3055–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Corridoni D, Rodriguez-Palacios A, Di Stefano G, et al. Genetic deletion of the bacterial sensor NOD2 improves murine Crohn’s disease-like ileitis independent of functional dysbiosis. Mucosal Immunol. 2017;10(4):971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lu WG, Zou YF, Feng XL, et al. Association of NOD1 (CARD4) insertion/deletion polymorphism with susceptibility to IBD: a meta-analysis. World J Gastroenterol. 2010;16(34):4348–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Verma R, Ahuja V, Paul J. Frequency of single nucleotide polymorphisms in NOD1 gene of ulcerative colitis patients: a case-control study in the Indian population. BMC Med Genet. 2009;10:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.McGovern DPB, Hysi P, Ahmad T, et al. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum Mol Genet. 2005;14(10):1245–1250. [DOI] [PubMed] [Google Scholar]
- 191.Huebner C, Ferguson LR, Han DY, et al. Nucleotide-binding oligomerization domain containing 1 (NOD1) haplotypes and single nucleotide polymorphisms modify susceptibility to inflammatory bowel diseases in a New Zealand caucasian population: a case-control study. BMC Res Notes. 2009;2:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chen GY, Shaw MH, Redondo G, Nunez G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 2008;68(24):10060–10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Natividad JMM, Petit V, Huang X, et al. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1−/−; Nod2−/− mice. Inflamm Bowel Dis. 2012;18(8):1434–1446. [DOI] [PubMed] [Google Scholar]
- 194.Bach J-F, Bendelac A, Brenner MB, et al. The role of innate immunity in autoimmunity. J Exp Med. 2004;200(12):1527–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Beyan H, Buckley LR, Yousaf N, Londei M, Leslie RD. A role for innate immunity in type 1 diabetes? Diabetes Metab Res Rev. 2003;19(2):89–100. [DOI] [PubMed] [Google Scholar]
- 196.Antonela B, Ivana G, Ivan K, et al. Environmental risk factors for type 1 diabetes mellitus development. Exp Clin Endocrinol Diabetes. 2017;125(8):563–570. [DOI] [PubMed] [Google Scholar]
- 197.Stene LC, Magnus P, Lie RT, Sovik O, Joner G; Norwegian Childhood Diabetes Study Group. Birth weight and childhood onset type 1 diabetes: population based cohort study. BMJ. 2001;322(7291):889–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rewers M, Ludvigsson J. Environmental risk factors for type 1 diabetes. Lancet. 2016;387(10035):2340–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Miranda MCG, Oliveira RP, Torres L, et al. Frontline science: abnormalities in the gut mucosa of non-obese diabetic mice precede the onset of type 1 diabetes. J Leukoc Biol. 2019;106(3):513–529. [DOI] [PubMed] [Google Scholar]
- 200.Costa FRC, Françozo MCS, de Oliveira GG, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med. 2016;213(7):1223–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chen L, Chen R, Wang H, Liang F. Mechanisms linking inflammation to insulin resistance. Int J Endocrinol. 2015;2015:508409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Cavallari JF, Fullerton MD, Duggan BM, et al. Muramyl dipeptide-based postbiotics mitigate obesity-induced insulin resistance via IRF4. Cell Metab. 2017;25(5):1063–1074.e3. [DOI] [PubMed] [Google Scholar]
- 203.Denou E, Lolmede K, Garidou L, et al. Defective NOD2 peptidoglycan sensing promotes diet-induced inflammation, dysbiosis, and insulin resistance. EMBO Mol Med. 2015;7(3):259–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chan KL, Tam TH, Boroumand P, et al. Circulating NOD1 activators and hematopoietic NOD1 contribute to metabolic inflammation and insulin resistance. Cell Rep. 2017;18(10):2415–2426. [DOI] [PubMed] [Google Scholar]
- 205.Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. [DOI] [PubMed] [Google Scholar]
- 206.Zhou Y-J, Liu C, Li C-L, et al. Increased NOD1, but not NOD2, activity in subcutaneous adipose tissue from patients with metabolic syndrome. Obesity (Silver Spring). 2015;23(7):1394–1400. [DOI] [PubMed] [Google Scholar]
- 207.Schertzer JD, Tamrakar AK, Magalhaes JG, et al. NOD1 activators link innate immunity to insulin resistance. Diabetes. 2011;60(9):2206–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. [DOI] [PubMed] [Google Scholar]
- 209.Zhou YJ, Tang YS, Song YL, Li A, Zhou H, Li Y. Saturated fatty acid induces insulin resistance partially through nucleotide-binding oligomerization domain 1 signaling pathway in adipocytes. Chin Med Sci J. 2013;28(4):211–217. [DOI] [PubMed] [Google Scholar]
- 210.Keestra-Gounder AM, Tsolis RM. NOD1 and NOD2: beyond peptidoglycan sensing. Trends Immunol. 2017;38(10):758–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Rodriguez-Nunez I, Caluag T, Kirby K, Rudick CN, Dziarski R, Gupta D. Nod2 and Nod2-regulated microbiota protect BALB/c mice from diet-induced obesity and metabolic dysfunction. Sci Rep. 2017;7(1):548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Clarke MCH, Figg N, Maguire JJ, et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12(9):1075–1080. [DOI] [PubMed] [Google Scholar]
- 213.Clarke M, Bennett M. The emerging role of vascular smooth muscle cell apoptosis in atherosclerosis and plaque stability. Am J Nephrol. 2006;26(6):531–535. [DOI] [PubMed] [Google Scholar]
- 214.Myoishi M, Hao H, Minamino T, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116(11):1226–1233. [DOI] [PubMed] [Google Scholar]
- 215.Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV. Role of endoplasmic reticulum stress in atherosclerosis and diabetic mac-rovascular complications. Biomed Res Int. 2014;2014:610140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Oh H-M, Lee H-J, Seo G-S, et al. Induction and localization of NOD2 protein in human endothelial cells. Cell Immunol. 2005;237(1):37–44. [DOI] [PubMed] [Google Scholar]
- 217.Kwon M-Y, Liu X, Lee S-J, et al. Nucleotide-binding oligomerization domain protein 2 deficiency enhances neointimal formation in response to vascular injury. Arterioscler Thromb Vasc Biol. 2011;31(11):2441–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kwon MY, Hwang N, Back SH, Lee SJ, Perrella MA, Chung SW. Nucleotide-binding oligomerization domain protein 2 deficiency enhances CHOP expression and plaque necrosis in advanced atherosclerotic lesions. FEBS J. 2020;287(10):2055–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Janssen CE, Rose CD, De Hertogh G, et al. Morphologic and immunohistochemical characterization of granulomas in the nucleotide oligomerization domain 2-related disorders Blau syndrome and Crohn disease. J Allergy Clin Immunol. 2012;129(4):1076–1084. [DOI] [PubMed] [Google Scholar]
- 220.James G Blau’s syndrome and sarcoidosis. Lancet. 1999;354(9183):1035. [DOI] [PubMed] [Google Scholar]
- 221.Girardelli M, Loganes C, Pin A, et al. Novel NOD2 mutation in early-onset inflammatory bowel phenotype. Inflamm Bowel Dis. 2018;24(6):1204–1212. [DOI] [PubMed] [Google Scholar]
- 222.Parkhouse R, Boyle JP, Monie TP. Blau syndrome polymorphisms in NOD2 identify nucleotide hydrolysis and helical domain 1 as signalling regulators. FEBS Lett. 2014;588(18):3382–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29(1):19–20. [DOI] [PubMed] [Google Scholar]
- 224.Tromp G, Kuivaniemi H, Raphael S, et al. Genetic linkage of familial granulomatous inflammatory arthritis, skin rash, and uveitis to chromosome 16. Am J Hum Genet. 1996;59(5):1097–1107. [PMC free article] [PubMed] [Google Scholar]
- 225.Kanazawa N, Okafuji I, Kambe N, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105(3):1195–1197. [DOI] [PubMed] [Google Scholar]
- 226.Saulsbury FT, Wouters CH, Martin TM, et al. Incomplete penetrance of the NOD2 E383K substitution among members of a pediatric granulomatous arthritis pedigree. Arthritis Rheum. 2009;60(6):1804–1806. [DOI] [PubMed] [Google Scholar]
- 227.Aróstegui JI, Arnal C, Merino R, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56(11):3805–3813. [DOI] [PubMed] [Google Scholar]
- 228.Rosé CD, Aróstegui JI, Martin TM, et al. NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: study of an international registry and a national cohort in Spain. Arthritis Rheum. 2009;60(6):1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Snyers B, Dahan K. Blau syndrome associated with a CARD15/NOD2 mutation. Am J Ophthalmol. 2006;142(6):1089–1092. [DOI] [PubMed] [Google Scholar]
- 230.Pillai P, Sobrin L. Blau syndrome-associated uveitis and the NOD2 gene. Semin Ophthalmol. 2013;28(5–6):327–332. [DOI] [PubMed] [Google Scholar]
- 231.Chamaillard M, Philpott D, Girardin SE, et al. Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc Natl Acad Sci U S A. 2003;100(6):3455–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Szymanski AM, Ombrello MJ. Using genes to triangulate the pathophysiology of granulomatous autoinflammatory disease: NOD2, PLCG2 and LACC1. Int Immunol. 2018;30(5):205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Okafuji I, Nishikomori R, Kanazawa N, et al. Role of the NOD2 genotype in the clinical phenotype of Blau syndrome and early-onset sarcoidosis. Arthritis Rheum. 2009;60(1):242–250. [DOI] [PubMed] [Google Scholar]
- 234.Dugan J, Griffiths E, Snow P, et al. Blau syndrome-associated Nod2 mutation alters expression of full-length NOD2 and limits responses to muramyl dipeptide in knock-in mice. J Immunol. 2015;194(1):349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Schurmann M, Valentonyte R, Hampe J, Muller-Quernheim J, Schwinger E, Schreiber S. CARD15 gene mutations in sarcoidosis. Eur Respir J. 2003;22(5):748–754. [DOI] [PubMed] [Google Scholar]
- 236.Tanabe T, Ishige I, Suzuki Y, et al. Sarcoidosis and NOD1 variation with impaired recognition of intracellular Propionibacterium acnes. Biochim Biophys Acta. 2006;1762(9):794–801. [DOI] [PubMed] [Google Scholar]
- 237.Yamada T, Eishi Y, Ikeda S, et al. In situ localization of Propionibacterium acnes DNA in lymph nodes from sarcoidosis patients by signal amplification with catalysed reporter deposition. J Pathol. 2002;198(4):541–547. [DOI] [PubMed] [Google Scholar]
- 238.Esteves T, Aparicio G, Garcia-Patos V. Is there any association between Sarcoidosis and infectious agents?: a systematic review and meta-analysis. BMC Pulm Med. 2016;16(1):165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Rastogi R, Du W, Ju D, et al. Dysregulation of p38 and MKP-1 in response to NOD1/TLR4 stimulation in sarcoid bronchoalveolar cells. Am J Respir Crit Care Med. 2011;183(4):500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ni G, Chen Y, Wu F, Zhu P, Song L. NOD2 promotes cell proliferation and inflammatory response by mediating expression of TSLP in human airway smooth muscle cells. Cell Immunol. 2017;312:35–41. [DOI] [PubMed] [Google Scholar]
- 241.Miller MH, Shehat MG, Alcedo KP, Spinel LP, Soulakova J, Tigno-Aranjuez JT. Frontline Science: RIP2 promotes house dust mite-induced allergic airway inflammation. J Leukoc Biol. 2018;104(3):447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Yahia SA, Azzaoui I, Everaere L, et al. CCL17 production by dendritic cells is required for NOD1-mediated exacerbation of allergic asthma. Am J Respir Crit Care Med. 2014;189(8):899–908. [DOI] [PubMed] [Google Scholar]
- 243.Weidinger S, Klopp N, Rummler L, et al. Association of NOD1 polymorphisms with atopic eczema and related phenotypes. J Allergy Clin Immunol. 2005;116(1):177–184. [DOI] [PubMed] [Google Scholar]
- 244.Weidinger S, Klopp N, Rummler L, et al. Association of CARD15 polymorphisms with atopy-related traits in a population-based cohort of Caucasian adults. Clin Exp Allergy. 2005;35(7):866–872. [DOI] [PubMed] [Google Scholar]
- 245.Nakashima K, Hirota T, Suzuki Y, et al. Association of the RIP2 gene with childhood atopic asthma. Allergol Int. 2006;55(1):77–83. [DOI] [PubMed] [Google Scholar]
- 246.Cai X, Xu Q, Zhou C, Zhou L, Dai W, Ji G. The association of nucleotide-binding oligomerization domain 2 gene polymorphisms with the risk of asthma in the Chinese Han population. Mol Genet Genomic Med. 2019;7(6):e00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hysi P, Kabesch M, Moffatt MF, et al. NOD1 variation, immunoglobulin E and asthma. Hum Mol Genet. 2005;14(7):935–941. [DOI] [PubMed] [Google Scholar]
- 248.Shaw PJ, Barr MJ, Lukens JR, et al. Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity. 2011;34(1):75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Schrijver IA, van Meurs M, Melief MJ, et al. Bacterial peptidoglycan and immune reactivity in the central nervous system in multiple sclerosis. Brain. 2001;124(Pt 8):1544–1554. [DOI] [PubMed] [Google Scholar]
- 250.Visser L, Jan de Heer H, Boven LA, et al. Proinflammatory bacterial peptidoglycan as a cofactor for the development of central nervous system autoimmune disease. J Immunol. 2005;174(2):808–816. [DOI] [PubMed] [Google Scholar]
- 251.Visser L, Melief M-J, van Riel D, et al. Phagocytes containing a disease-promoting Toll-like receptor/Nod ligand are present in the brain during demyelinating disease in primates. Am J Pathol. 2006;169(5):1671–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Buljevac D, Flach HZ, Hop WCJ, et al. Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain. 2002;125(Pt 5):952–960. [DOI] [PubMed] [Google Scholar]
- 253.Dube JY, McIntosh F, Zarruk JG, David S, Nigou J, Behr MA. Synthetic mycobacterial molecular patterns partially complete Freund’s adjuvant. Sci Rep. 2020;10(1):5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Enevold C, Oturai AB, Sorensen PS, Ryder LP, Koch-Henriksen N, Bendtzen K. Multiple sclerosis and polymorphisms of innate pattern recognition receptors TLR1–10, NOD1–2, DDX58, and IFIH1. J Neuroimmunol. 2009;212(1–2):125–131. [DOI] [PubMed] [Google Scholar]
- 255.Molnar T, Hofner P, Nagy F, et al. NOD1 gene E266K polymorphism is associated with disease susceptibility but not with disease phenotype or NOD2/CARD15 in Hungarian patients with Crohn’s disease. Dig Liver Dis. 2007;39(12):1064–1070. [DOI] [PubMed] [Google Scholar]
- 256.Hofner P, Gyulai Z, Kiss ZF, et al. Genetic polymorphisms of NOD1 and IL-8, but not polymorphisms of TLR4 genes, are associated with Helicobacter pylori-induced duodenal ulcer and gastritis. Helicobacter. 2007;12(2):124–131. [DOI] [PubMed] [Google Scholar]
- 257.van Duist MM, Albrecht M, Podswiadek M, et al. A new CARD15 mutation in Blau syndrome. Eur J Hum Genet. 2005;13(6):742–747. [DOI] [PubMed] [Google Scholar]
- 258.Kim W, Park E, Ahn YH, et al. A familial case of Blau syndrome caused by a novel NOD2 genetic mutation. Korean J Pediatr. 2016;59(Suppl 1):S5–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Jesus AA, Fujihira E, Watase M, et al. Hereditary autoinflammatory syndromes: a Brazilian multicenter study. J Clin Immunol. 2012;32(5):922–932. [DOI] [PubMed] [Google Scholar]
- 260.Jain L, Gupta N, Reddy MM, et al. A novel mutation in helical domain 2 of NOD2 in sporadic blau syndrome. Ocul Immunol Inflamm. 2018;26(2):292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Milman N, Ursin K, Rodevand E, Nielsen FC, Hansen TV. A novel mutation in the NOD2 gene associated with Blau syndrome: a Norwegian family with four affected members. Scand J Rheumatol. 2009;38(3):190–197. [DOI] [PubMed] [Google Scholar]
- 262.Khubchandani RP, Hasija R, Touitou I, Khemani C, Wouters CH, Rose CD. Blau arteritis resembling Takayasu disease with a novel NOD2 mutation. J Rheumatol. 2012;39(9):1888–1892. [DOI] [PubMed] [Google Scholar]
- 263.Ebrahimiadib N, Samra KA, Domina AM, et al. A novel NOD2-associated mutation and variant blau syndrome: phenotype and molecular analysis. Ocul Immunol Inflamm. 2018;26(1):57–64. [DOI] [PubMed] [Google Scholar]
- 264.Ong LT, Nachbur U, Rowczenio D, Ziegler JB, Fischer E, Lin MW. A novel nucleotide oligomerisation domain 2 mutation in a family with Blau syndrome: Phenotype and function. Innate Immun. 2017;23(7):578–583. [DOI] [PubMed] [Google Scholar]
- 265.Okada S, Konishi N, Tsumura M, et al. Cardiac infiltration in early-onset sarcoidosis associated with a novel heterozygous mutation, G481D, in CARD15. Rheumatology (Oxford). 2009;48(6):706–707. [DOI] [PubMed] [Google Scholar]
- 266.Kanazawa N, Matsushima S, Kambe N, Tachibana T, Nagai S, Miyachi Y. Presence of a sporadic case of systemic granulomatosis syndrome with a CARD15 mutation. J Invest Dermatol. 2004;122(3):851–852. [DOI] [PubMed] [Google Scholar]
- 267.Chauhan K, Michet C. A case of Blau syndrome. Case Rep Rheumatol. 2014;2014:216056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Carreno E, Guly CM, Chilov M, et al. Optic nerve and retinal features in uveitis associated with juvenile systemic granulomatous disease (Blau syndrome). Acta Ophthalmol. 2015;93(3):253–257. [DOI] [PubMed] [Google Scholar]
- 269.Sakai H, Ito S, Nishikomori R, et al. A case of early-onset sarcoidosis with a six-base deletion in the NOD2 gene. Rheumatology (Oxford). 2010;49(1):194–196. [DOI] [PubMed] [Google Scholar]
- 270.Sharma SM, Martin TM, Rose CD, Dick AD, Ramanan AV. Distinguishing between the innate immune response due to ocular inflammation and infection in a child with juvenile systemic granulomatous disease treated with anti-TNFalpha monoclonal antibodies. Rheumatology (Oxford). 2011;50(5):990–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Jimenez-Martinez MC, Cruz F, Groman-Lupa S, Zenteno JC. Immunophenotyping in peripheral blood mononuclear cells, aqueous humour and vitreous in a Blau syndrome patient caused by a novel NOD2 mutation. Int J Immunogenet. 2011;38(3):233–242. [DOI] [PubMed] [Google Scholar]
- 272.Rivas MA, Beaudoin M, Gardet A, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43(11):1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Sales-Marques C, Salomão H, Fava VM, et al. NOD2 and CCDC122-LACC1 genes are associated with leprosy susceptibility in Brazilians. Hum Genet. 2014;133(12):1525–1532. [DOI] [PubMed] [Google Scholar]
- 274.Grant AV, Alter A, Huong NT, et al. Crohn’s disease susceptibility genes are associated with leprosy in the Vietnamese population. J Infect Dis. 2012;206(11):1763–1767. [DOI] [PubMed] [Google Scholar]
- 275.Zhang F-R, Huang W, Chen S-M, et al. Genomewide association study of leprosy. N Engl J Med. 2009;361(27):2609–2618. [DOI] [PubMed] [Google Scholar]
- 276.Austin CM, Ma X, Graviss EA. Common nonsynonymous polymorphisms in the NOD2 gene are associated with resistance or susceptibility to tuberculosis disease in African Americans. J Infect Dis. 2008;197(12):1713–1716. [DOI] [PubMed] [Google Scholar]
- 277.Zhao M, Jiang F, Zhang W, et al. A novel single nucleotide polymorphism within the NOD2 gene is associated with pulmonary tuberculosis in the Chinese Han, Uygur and Kazak populations. BMC Infect Dis. 2012;12:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Rosenstiel P, Hellmig S, Hampe J, et al. Influence of polymorphisms in the NOD1/CARD4 and NOD2/CARD15 genes on the clinical outcome of Helicobacter pylori infection. Cell Microbiol. 2006;8(7):1188–1198. [DOI] [PubMed] [Google Scholar]