

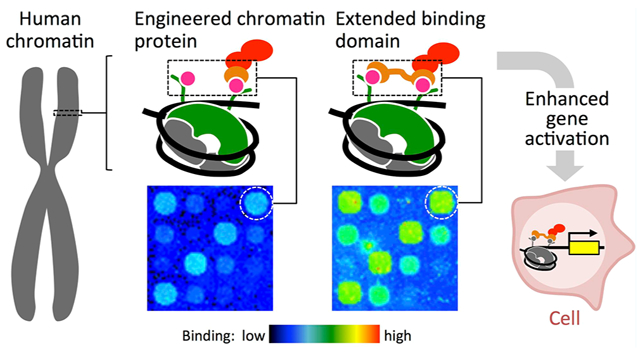
Abstract
Fusion proteins that specifically interact with biochemical marks on chromosomes represent a new class of synthetic transcriptional regulators that decode cell state information rather than DNA sequences. In multicellular organisms, information relevant to cell state, tissue identity, and oncogenesis is often encoded as biochemical modifications of histones, which are bound to DNA in eukaryotic nuclei and regulate gene expression states. We have previously reported the development and validation of the “polycomb-based transcription factor” (PcTF), a fusion protein that recognizes histone modifications through a protein-protein interaction between its polycomb chromodomain (PCD) motif and trimethylated lysine 27 of histone H3 (H3K27me3) at genomic sites. We demonstrated that PcTF activates genes at methyl-histone-enriched loci in cancer-derived cell lines. However, PcTF induces modest activation of a methyl-histone associated reporter compared to a DNA-binding activator. Therefore, we modified PcTF to enhance its binding avidity. Here, we demonstrate the activity of a modified regulator called Pc2TF, which has two tandem copies of the H3K27me3-binding PCD at the N-terminus. Pc2TF has a smaller apparent dissociation constant value in vitro and shows enhanced gene activation in HEK293 cells compared to PcTF. These results provide compelling evidence that the intrinsic histone-binding activity of the PCD motif can be used to tune the activity of synthetic histone-binding transcriptional regulators.
Keywords: chromatin, histone, transcription factor, protein engineering, epigenetics, synthetic biology
Graphical Abstract
INTRODUCTION
The discovery of histone post-translational modifications (PTMs) and the peptides that specifically interact with these marks has enabled scientists and cell engineers to manipulate chromatin, the DNA-protein structure that regulates gene expression states in eukaryotic cells. Structure-based models have informed targeted knock-down of chromatin subunits and the rational design of low molecular weight inhibitor compounds (reviewed in ref 1). DNA-binding domains fused with structural chromatin proteins and histone-modifying enzymes have been used to generate ectopic chromatin conformations at specific loci.2,3 Until recently, scientists had not yet leveraged PTM-binding peptides from natural effector proteins to “read” the rich biological information encoded in histone marks in living cells. Peptides that recognize specific histone PTM signals are essential for synthetic systems that integrate epigenetic regulatory signals. In order to use PTM-binding peptides in synthetic fusion proteins, the peptides must be portable, that is, maintain their intrinsic function within a new protein sequence. Early studies established important foundational knowledge by demonstrating that the interaction of the chromodomain motif (CD) with trimethylated histone H3 lysine 27 (H3K27me3) is an intrinsic activity that is maintained by the CD in the context of recombinant, fusion proteins.4,5 Other protein folds including the bromodomain (BRD) and plant homeodomain finger (PHD) function as isolated peptides6–8 and within fusion proteins7,9 to specifically interact with acetylated histone lysines (BRD) and H3K4me3 (PHD).
We constructed the polycomb-based transcription factor (PcTF) using a histone PTM-binding motif from the natural protein CBX8.10 The CBX8 effector protein binds to histone H3 trimethylated at lysine 27 (H3K27me3) through its N-terminal polycomb chromodomain (PCD) and establishes a silenced transcriptional state. Expression of PcTF, an artificial transcriptional activator with an N-terminal PCD, mCherry tag, and C-terminal VP64 activation domain, led to increased expression of H3K27me3-enriched genes in three different cancer-derived cell lines.11 These results show promise for designing transcription factors that can read chromatin marks to rewire aberrant epigenetic programming. However, binding affinities observed in vitro for isolated PCDs is poor, reported as 5 to >500 μM,12,13 compared to DNA-binding domains with target affinities in the pico- to nanomolar range such as transcription activator-like effectors (TALEs, ~3–220 nM),14 zinc fingers (~0.01–16 nM),15,16 and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated domains (Cas) (~0.5 nM).17 In other work, we observed stronger gene upregulation when mCherry-VP64 was targeted to a promoter via a Gal4 DNA binding domain compared to the PCD histone-binding domain.10 PcTF-mediated gene activation is dose dependent,11 and high PcTF expression levels are required for optimal activity. This limits the usefulness of PcTF for therapeutic applications where barriers to delivery severely limit the number of proteins that ultimately reach the nuclei of target cells. Although pharmacokinetic barriers to DNA and protein delivery in vivo are not trivial, increasing the effective dose of PcTF could significantly advance this technology toward clinical use.
The appearance of tandem histone binding domains within natural proteins suggests that the performance of histone-binding regulators can be customized and tuned through multivalency, defined as contact with more than one histone PTM via multiple domains (reviewed in refs 3, 18, and 19). Multivalent chromatin proteins can engage adjacent PTMs within a single histone tail, such as K4me3 and R8me2 on histone H3 bound by Spindlin120 or K5ac and K12ac on histone H4 bound by TAFII250.21 PTM targets can also reside on two distinct histone tails, such as H4K16ac and H3K4me3 bound by BPTF.7 Dual recognition of histone PTMs is accomplished by tandem protein motifs within the histone-binding protein. Comparisons of natural mono- and divalent proteins,22 as well as histone peptide on- and off-target binding studies,20,23 have produced compelling evidence that tandem motifs contribute to avidity and specificity. The idea that combinatorial avidity allows proteins to read a “histone code” has been the topic of some controversy. Until recently, multivalency had not been demonstrated using a rationally designed composition of binding domains. Tandem histone binding domains have been used to design protein probes to fluorescently label regions that are enriched for specific histone modifications.24,25 To date, multivalency has not been used to design a transcriptional regulator and tandem PCDs have not been reported. In order to compensate for the modest affinity of the CBX8 PCD12 for its target, we added a second copy of H3K27me3-binding PCD to the N-terminus of PcTF to build Pc2TF. Here, we demonstrate that Pc2TF shows stronger avidity for H3K27me3 in vitro. This activity corresponds with enhanced activation of a H3K27me3-repressed gene in cultured cells. Our results have important implications for building and tuning fusion proteins that target sites of polycomb-mediated silencing, which plays a central role in cancer and stem cell plasticity.
RESULTS AND DISCUSSION
Design of a Bivalent Synthetic Chromatin-Based Transcriptional Regulator.
We designed the Pc2TF protein to simultaneously recognize two copies of the histone post-translational modification H3K27me3. The polycomb chromodomain motif (PCD) consists of three β strands packed against a C-terminal α helix and a hydrophobic pocket formed by three aromatic residues that interact with a methyl-lysine side chain13,26 (Figure 1A). The arrangement of histones within the nucleosome octamer suggests that Pc2TF might bind adjacent trimethylated H3K27 residues. A single nucleosome includes eight individual histone proteins. The central tetramer contains two copies of histone H3 and H4. The H3 proteins are oriented in cis so that the unfolded N-terminal tails protrude away from the nucleosome in the same direction27 (Figure 1B). One or both H3 tails28 can become trimethylated at lysine 27 by the enzyme enhancer of zeste (EZH).29 Therefore, tandem PCDs in the multivalent protein Pc2TF might interact with two histone post translational modifications (PTMs) in a single nucleosome (Figure 1B) or single PTMs on adjacent nucleosomes.
Figure 1.
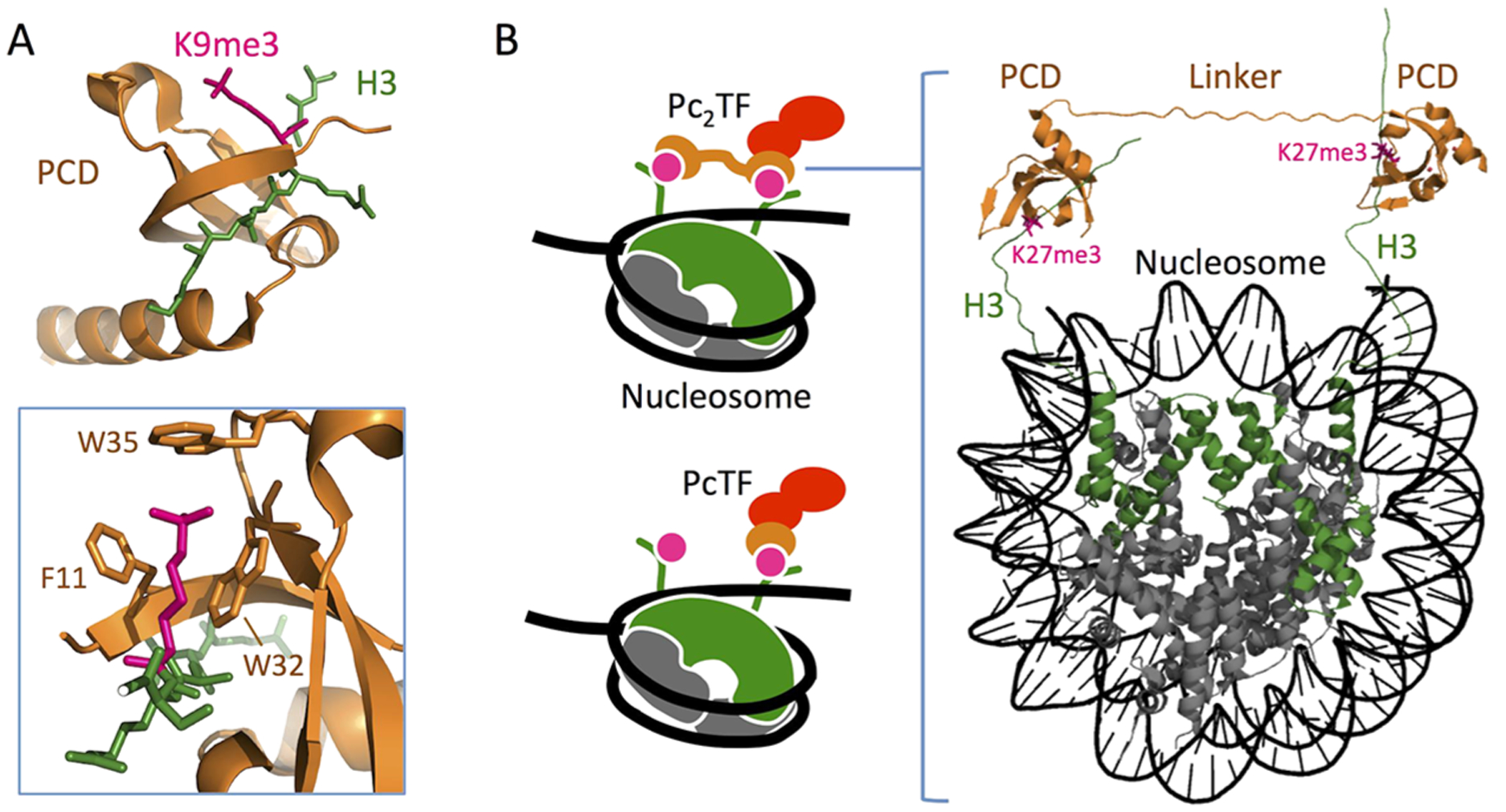
Three-dimensional model layout to show the plausibility of Pc2TF binding to adjacent H3K27me3 marks. (A) PCD (CBX8) in complex with trimethyl lysine (PDB 3I91).37 Three residues form a hydrophobic cage and surround the Kme3 moiety (inset). (B) H3K27me3 recognition by synthetic fusion proteins that carry a single or tandem PCD domains (PcTF and Pc2TF, respectively). The 3D rendering was composed in the PyMOL Molecular Graphics System, version 1.3, Schrödinger, LLC (https://www.pymol.org), using data for CBX8/H3K9me3 (PDB 4X3K)51 and a whole nucleosome assembly (PDB 5AV8)51,52 from the Protein Data Bank.
To quickly and efficiently identify a linker that would allow contact of each PCD with a H3K27me3 ligand, we used an in vitro expression and ELISA procedure to test four Pc2TF variants. Different lengths and physical characteristics were explored by using flexible glycine-serine linkers30 and rigid α-helical31,32 linkers. Glycine and serine, amino acids with small side chains, have been used in a wide range protein engineering applications to build linker peptides that have minimal interference with the function of tethered proteins. However, as was demonstrated by mutagenesis of a rigid linker in the bivalent protein BPTF, added flexibility can destabilize protein-histone interactions.7 Rigid linkers might perform better by stabilizing the distance between PCDs to support interactions with neighboring K27me3 moieties.33,34 The Pc2TF constructs included two tandem copies of the PCD separated by one of four linkers: flexible (GGGGS)4, long flexible (GGGGS)16, rigid (EAAAR)4, and long rigid (EAAAR)16. Based on a simplified layout of the interacting components (PCDs and a nucleosome carrying two H3K27me3 modifications; Figure 1B), we predicted that 20 amino acids would provide sufficient length for adjacent PCDs to bind simultaneously. The 80-amino-acid linkers were used to determine the impact of increased spacing between PCDs.
To expedite the prototyping stage, we used a cell-free expression system.35 Pc2TF variants and a control protein with no binding domain (PcΔTF) were expressed from a pET28 vector (Figure 2A) in TXTL solution. Real-time detection of mCherry fluorescence in a Roche thermal cycler confirmed expression of recombinant proteins. For ELISAs, biotinylated histone peptides were immobilized on a neutravidin-coated 96 well plate. HRP-conjugated anti-mCherry was used for immunodetection of bound fusion proteins. Significantly higher HRP signal was detected compared to background (unmodified K27 and K27ac) for variants that contained the flexible (GGGGS)4, long flexible (GGGGS)16, and long rigid (EAAAR)16 linkers (Figure 2C). The implications of these results are discussed in depth in the Conclusions. Assuming that HRP signal is proportional to Pc-fusion molecules bound, the flexible (GGGGS)4 linker conferred the strongest avidity in this assay. Therefore, we used this variant in subsequent experiments to determine the impact of bivalency on the activity of synthetic, histone-binding effectors.
Figure 2.
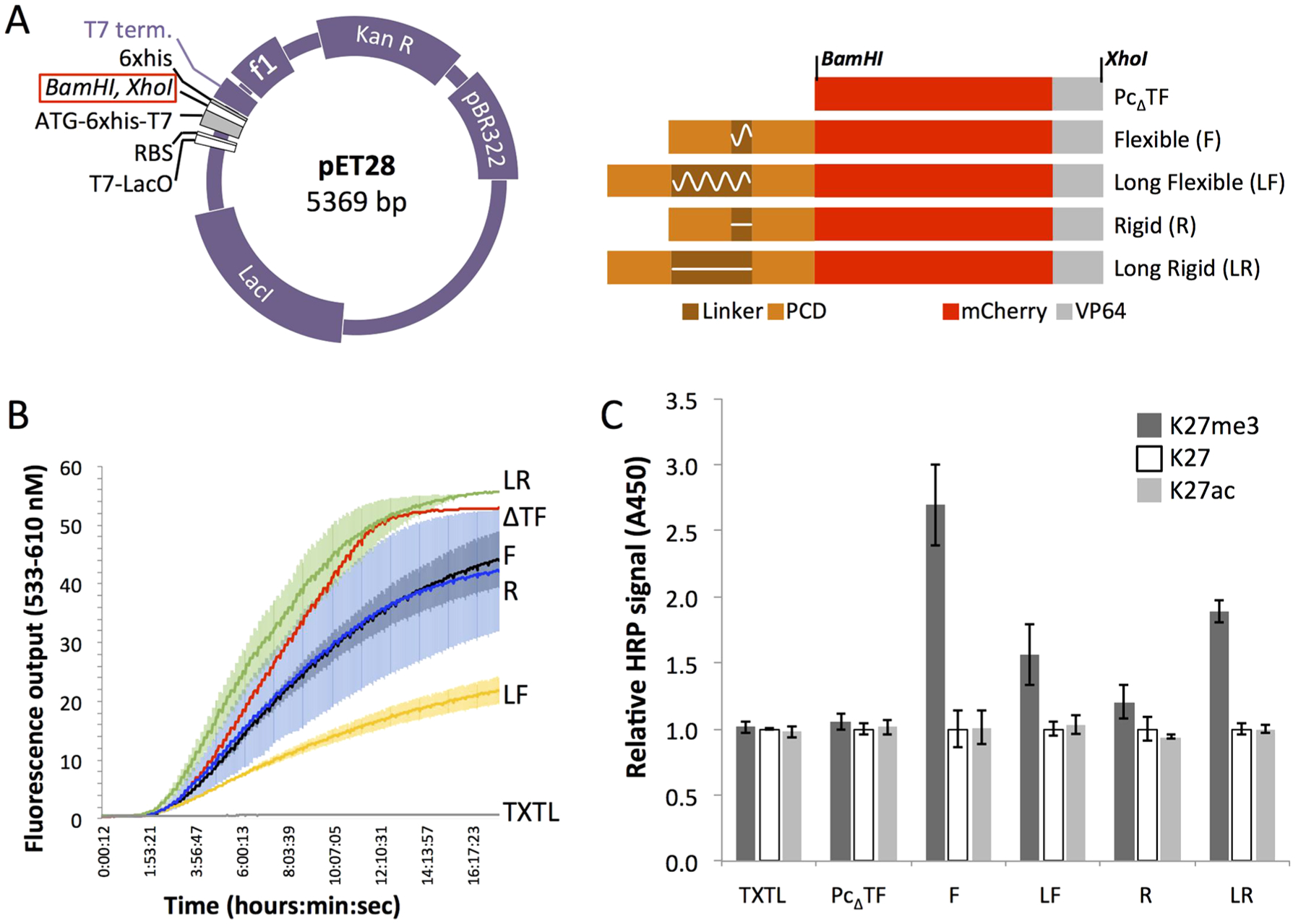
Comparison of Pc2TF variants that were expressed in a bacterial cell-free expression system. (A) Map of the expression vector and open reading frames (ORFs). Fusion-encoding ORFs were cloned in the pET28 vector at BamHI and XhoI. (B) Real-time detection of mCherry fluorescence was used to determine expression of recombinant protein in TXTL in a 96-well PCR plate in a Roche thermal cycler. Each replicate is an independent TXTL reaction in a single well (1 replicate for PcΔTF and TXTL without DNA, 3 replicates for others). Replicates were pooled for ELISAs in panel C. Solid line = mean, shaded regions = SDM. (C) The bar chart shows mean signal from anti-mCherry-HRP signal at an absorbance of 450 nm (3 ELISA wells) from TXTL-expressed fusion proteins or plasmid-free “blank” TXTL captured by tethered trimethyl-K27 (K27me3), unmodified (K27), or modified nontarget (K27ac) histone H3 peptides. For each TXTL product, individual values are normalized to the unmodified H3 mean value within the set (error bars = SDM).
Bivalency Strengthens the Avidity of the Pc-Fusion for H3K27me3.
To compare known concentrations of Pc-fusion proteins in subsequent experiments, we overexpressed and purified recombinant proteins from E. coli. Denaturing polyacrylamide electrophoresis (PAGE) of lysates from isopropyl β-d-1-thiogalactopyranoside (IPTG)-treated and untreated E. coli confirmed inducible production of the proteins at roughly the expected sizes: 37, 44, and 52 kDa for PcΔTF, PcTF, and Pc2TF, respectively (Figure 3A). Nickel-NTA column-purified proteins were soluble in 1× phosphate buffered saline (PBS). The visible red hue under white light, which is typical of the mCherry protein,36 indicated proper protein folding (Figure 3A).
Figure 3.
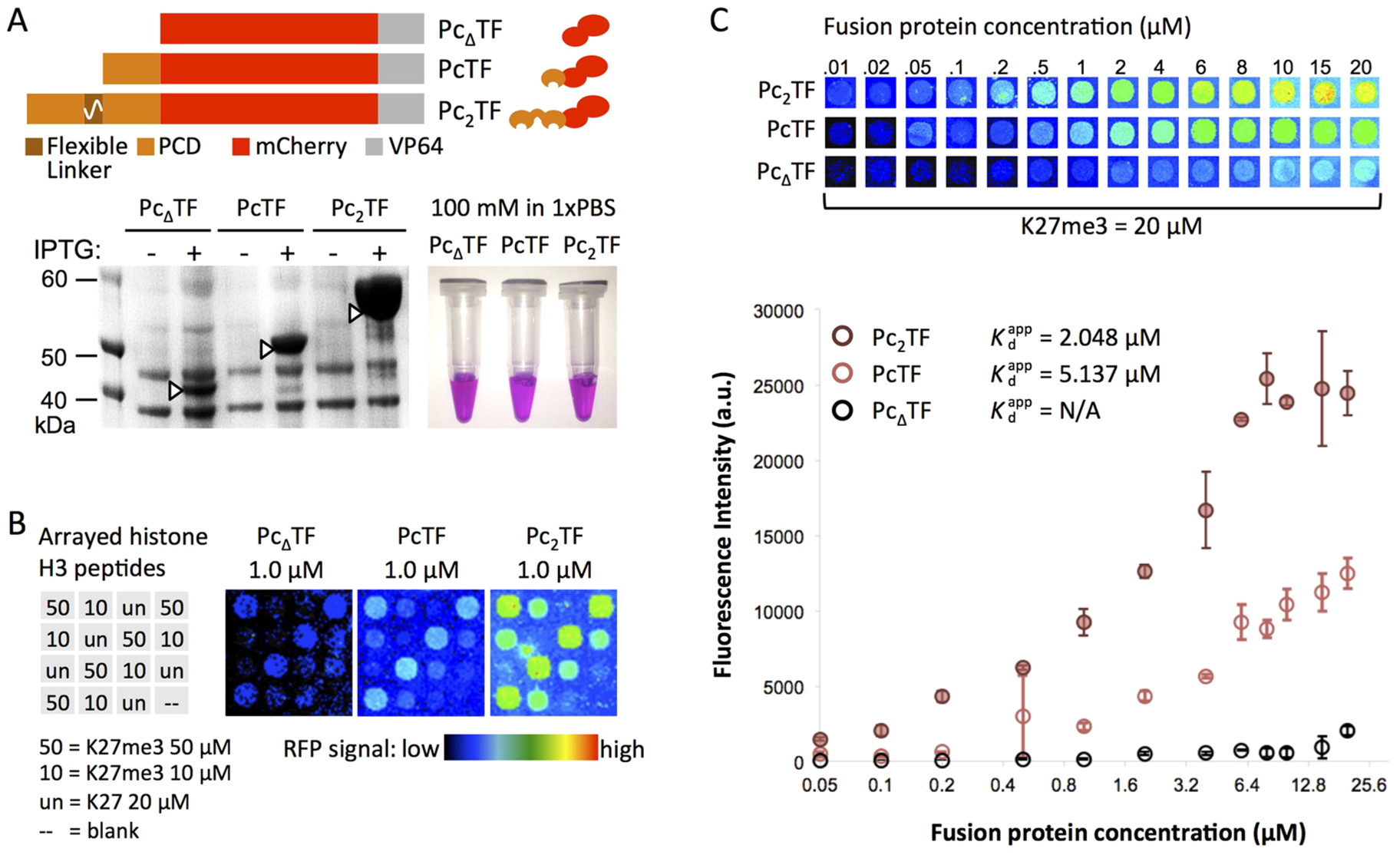
A bivalent PCD fusion peptide shows enhanced avidity for H3K27me3 in microspot array experiments. (A) For high-yield expression, E. coli was transformed with pET28 plasmids encoding PcΔTF (negative control), PcTF (single PCD), and the Pc2TF containing the flexible linker (GGGGS)4. Native polyacrylamide gel electrophoresis (PAGE) of overexpressed proteins purified from E. coli. (B) Test slides were spotted with histone H3 peptides (K27me3 or unmodified K27) as indicated in the grid for qualitative analysis. Pseudocolored images show mCherry signal after an application of 1.0 μM fusion protein to individual arrays. (C) New arrays were spotted with 10, 20, and 50 μM H3K27me3 for quantitative analysis. Fluorescence signal versus the concentration of fusion protein applied to the array was used to calculate the apparent dissociation constant (, not applicable for PcΔTF). Each point in the graph is the mean signal from four spots in one application (error bars = SDM). The data displayed in the graph are from representative applications (out of four total) for 20 μM immobilized H3K27me3.
To determine impact of the additional PCD domain on PcTF avidity, we exposed tethered histone peptides to varying concentrations of soluble PcTF and Pc2TF. Liquid phase ligand (H3K27me3 peptide) binding assays (fluorescence polarization, FP) reported by other groups have determined affinities of N-terminal PCD motifs from the Drosophila Pc protein (residues 1–90, Kd = 5.0 ± 1 μM13) and mammalian CBX8 protein (mouse residues 1–62, Kd = 165 ± 20 μM;12 human residues 8–61, Kd > 500 μM37). The amino acid sequence of the PCD in our fusion proteins (human CBX8 residues 1–62) is identical to the mouse ortholog. To acquire data that is relevant to the full-length fusion proteins (295 to 445 residues) that we had previously tested as gene regulators in cancer cell lines,11 we used a histone peptide microspot array. We used a mathematical model to predict relative RFP signal levels after Pc-fusion binding and subsequent washing of the microarray. PcTF has a higher mCherry/PCD ratio than Pc2TF (1:1 versus 1:2). Therefore, PcTF should show higher relative RFP signal when all targets (H3K27me3 peptides) within a microspot are saturated by PCD binding (Figure S1). Assuming that bivalency supports an additive increase in avidity, a higher fraction of Pc2TF molecules should remain bound at the microspot during washing, resulting in higher total RFP signal. Results from a test array were consistent with this prediction (Figure 3B). We tested concentrations of the recombinant proteins over 2 orders of magnitude (0.2–20 μM) to determine the apparent dissociation constant of each protein for 10, 20, and 50 μM tethered H3K27me3 ligand (Figure S2). We detected no interaction with unmodified histone H3 peptides and very little signal above background for the PcΔTF negative control. The of monovalent PcTF was 5.14–8.95 μM for four independent trials (Figure 3C and S2). The micromolar values are comparable to Kd values from the aforementioned FP experiments, although the wash steps in microspot assay may bias toward the off kinetics of the binding process. We conclude that PCD retains its intrinsic affinity for H3K27me3 as an N-terminal motif within a fusion protein.
Overall, analysis of the microspot array data suggest that at 10 and 20 μM of H3K27me3 the of Pc2TF is roughly 2-fold smaller than PcTF (Figure S2). Assuming that the second PCD fold (PCD2) maintains its intrinsic affinity, PCD2 should approximately double the overall association rate for Pc2TF since there is twice the chance of a PCD-H3K27me3 collision. Avidity is related to the inverse of the equilibrium constant, and the equilibrium constant is proportional to the ratio of association rate over dissociation rate. Thus, the effect we observed is most likely due to increasing the association rate or decreasing the dissociation rate, which would decrease the value (compared to PcTF) roughly 2-fold.
Bivalent Pc2TF Shows Cooperative, On-Target Binding with Solid Phase Target Ligands.
Next, we investigated the binding properties of the mono- and bivalent PCD proteins over a range of target ligand densities to approximate dynamic distributions of H3K27me3 that may occur in chromatin. We assumed that random distribution within each mixture would decrease the spacing between H3K27me3 targets as their concentration was increased. We applied dilutions of the target ligand (0–100% H3K27me3 mixed with unmodified H3K27, 1000 nM final concentration) to ELISA wells and exposed the immobilized ligands to the highest concentration of fusion proteins that produced minimal background signal in preliminary ELISA trials (0.1 μM). At 0–15% H3K27me3, HRP signal for PcTF or Pc2TF was not significantly greater than the negative control fusion protein (PcΔTF). In this range, the number of fixed H3K27me3 ligands may not have captured enough fusion proteins to yield detectable signal after washing. At 20–30% and higher, the HRP signal from the PcTF and Pc2TF wells increased with H3K27me3 concentration.
A Hill slope of 2.75 from the nonlinear regression (R2 = 0.90) for Pc2TF (Figure 4B) indicates that binding scales nonlinearly with the concentration of its ligand. It is difficult to fit a Hill curve to the data for PcTF (R2 = 0.64) because the increase in HRP signal is interrupted by a plateau at 30–80% H3K27me3. The cause of the plateau is unclear; however it is possible that the increase observed above 80% is due to the binding avidity between PcTF and H3K27me3 being exceeded at these concentrations. Overall, we can conclude from the ELISA data that Pc2TF binding is cooperative.
Figure 4.
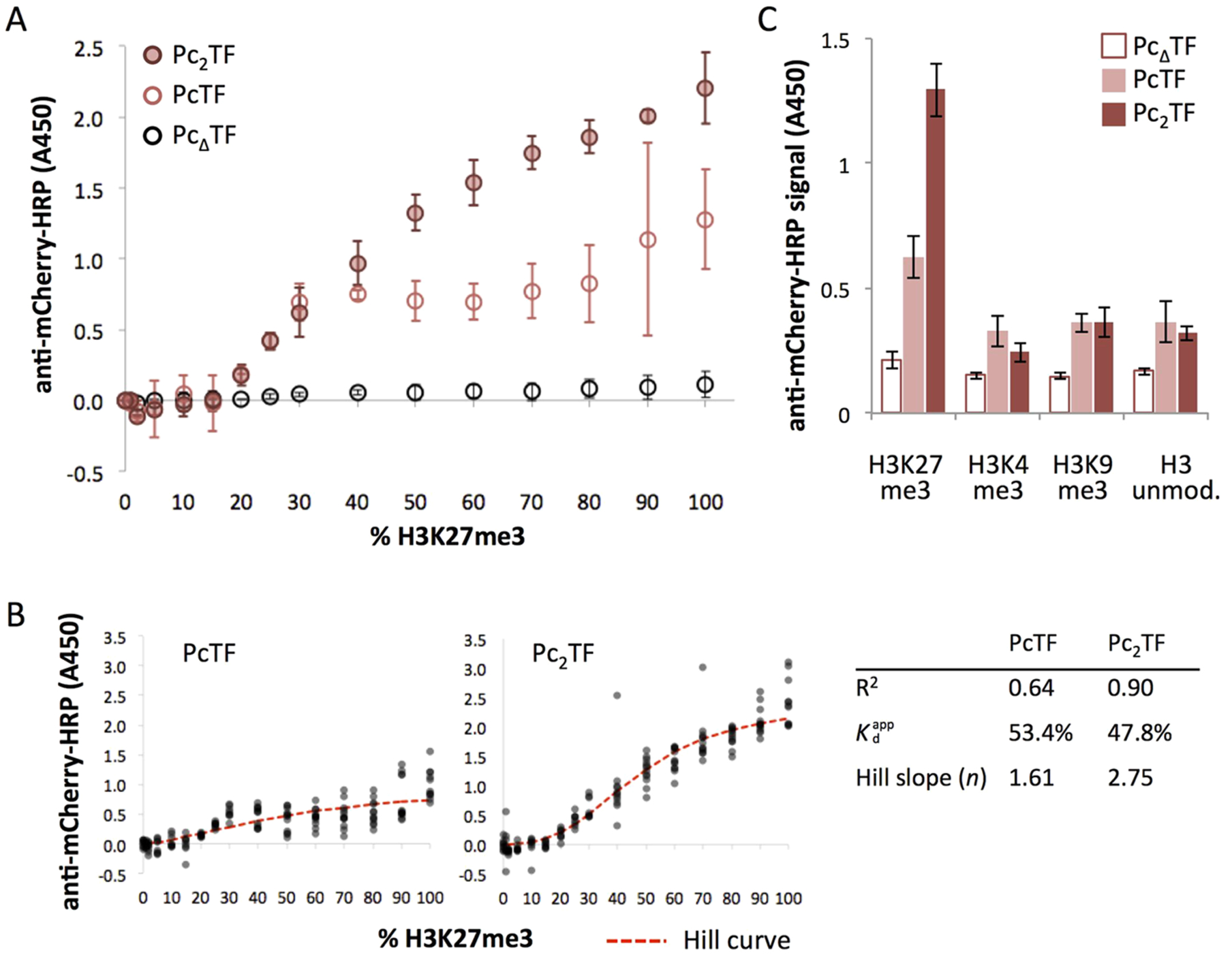
Bivalent Pc2TF shows cooperative and on-target binding with H3K27me3 ligands. (A) The scatter plot shows mean HRP signal at an absorbance of 450 nm (one ELISA trial, means of four technical replicate wells, error bars = SDM) from wells in which 0.1 μM purified protein (Pc2TF, PcTF, or PcΔTF) was allowed to bind with different proportions of H3K27me3 biotinylated peptides (0–100%) mixed with unmodified H3 and tethered to neutravidin-coated surfaces. (B) Hill curves were fit to data for three ELISA trials (dots = technical replicate wells from all trials). (C) ELISA was used to detect interaction of 0.05 μM purified proteins with immobilized histone H3 peptides that were trimethylated at lysine 27, 4, or 9 or unmodified. The bar chart shows mean signal values from anti-mCherry-HRP at an absorbance of 450 nm (4 technical replicates, bars = SDM).
To investigate ligand selectivity, 50 nM purified PcTF, Pc2TF, or PcΔTF was tested for interaction with histone peptides that were trimethylated at different lysine residues. PcTF and Pc2TF showed significant binding with H3K27me3 peptides compared to the control protein PcΔTF (Figure 4C). HRP signal from the H3K27me3 wells was significantly higher than what appears to be nonspecific binding with unmodified H3. This was not the case for off-target ligands H3K4me3 and H3K9me3, suggesting that the Pc-fusions can discriminate between the different methyl marks. No significant increase in HRP signal in the off-target wells was observed for Pc2TF, suggesting that target preference was not lost as avidity was enhanced. One might expect cross-reactivity with H3K9me3 since this PTM appears within a similar motif (ARKS) as H3K27me3.13,37 Others have reported that in vitro, chromodomain peptides from different orthologues (CBX1–8) have varying preferences for the two histone modifications.12,37,38 CBX8, the PCD used for PcTF in our work, has shown weak affinity for H3K27me3 and none for H3K9me3,12,37 which is consistent with our results.
The results from the assays with purified proteins led us to ask what is the biological consequence of increased binding in living cells where the physical distribution of H3K27me3 is much different? In the cellular chromatin environment, H3K27me3 can occur in cis on the radial surface of a single nucleosome (Figure 1B), in trans where DNA bending brings the H3 tails of neighboring nucleosomes close together, or sparsely distributed across many nucleosomes. Furthermore, H3K27me3 marks in living cells are dynamic. The enzyme EZH1/2 adds methyl groups to H3K27, and the enzymes KDM6A (UTX) and KDM6B (JMJD3) remove these marks.39 We set out to compare Pc2TF to PcTF in a cellular milieu.
Bivalent Pc2TF Activates a Target Gene in a Partially Silenced State.
Previously, we demonstrated that PcTF activated a reporter gene near ectopic H3K27me3 in HEK293 cells.10 Here, we determined the biological significance of PCD bivalency by comparing the gene-regulation activities of Pc2TF and PcTF at the same reporter. Doxycycline (dox)-mediated induction of Gal4-EED in HEK293 Gal4-EED/luc cells leads to accumulation of H3K27me3 at and silencing of a chromosomally integrated Tk-luciferase transgene (Figure 5A).40,41 Tk-luciferase repression reaches steady state at 96 h,40 and repression is maintained by epigenetic inheritance after loss of Gal4-EED.41
Figure 5.
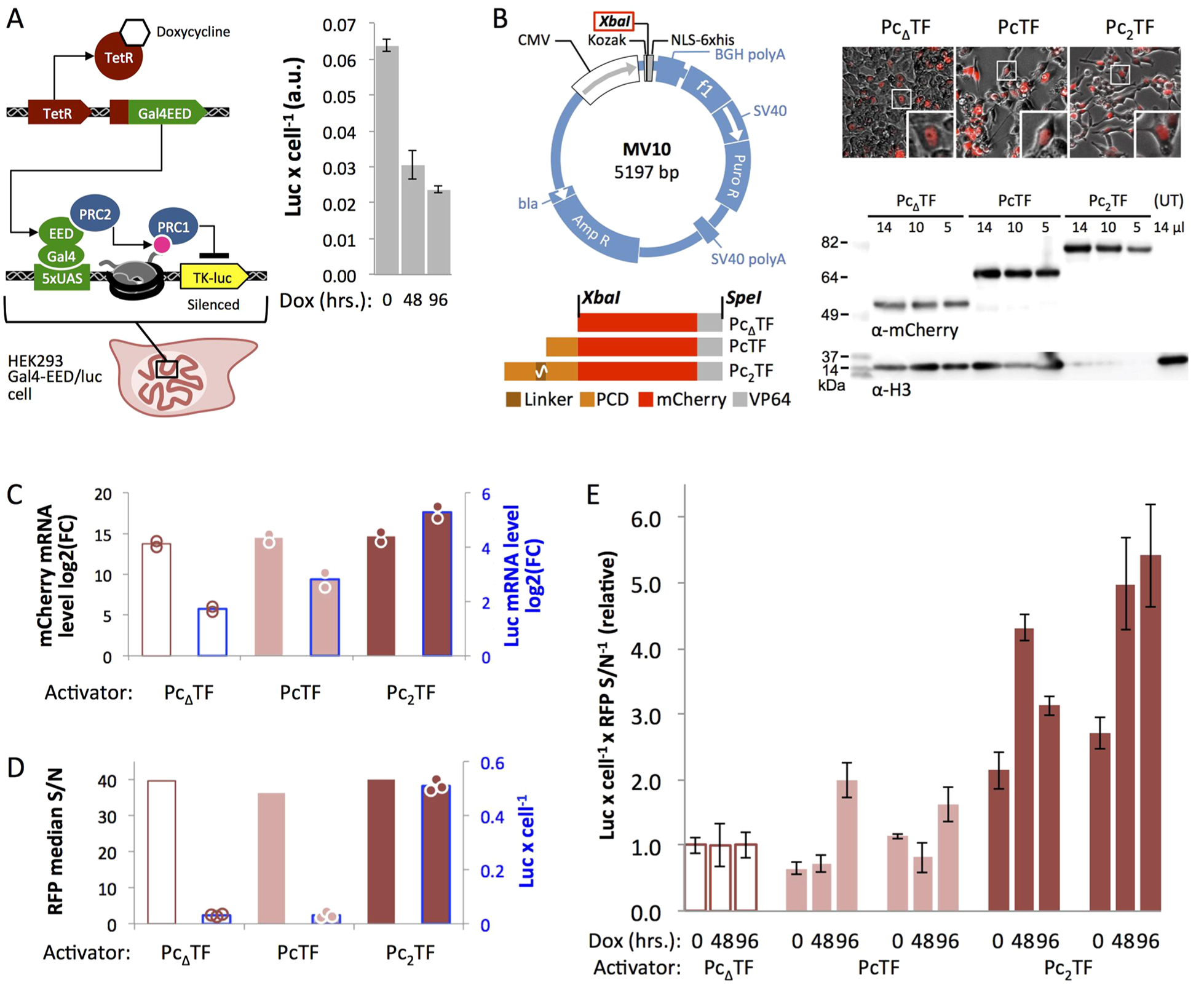
Pc2TF stimulates expression at a polycomb-silenced reporter gene. (A) An engineered HEK293 cell line, Gal4-EED/luc, was used for doxycycline-mediated control of H3K27me3 and PRC-mediated silencing at a Tk-luciferase reporter. Expression is partially silenced prior to dox treatment, as demonstrated previously40 and becomes fully repressed at 96 h. (B) Fusion constructs were cloned into the MV10 vector at XbaI. Fluorescence microscopy confirmed nuclear localization of the fusion proteins in transfected cells. The same samples were used for Western blots to confirm cellular expression of full length proteins, RT-qPCR to measure mRNA levels (C), and flow cytometry to measure RFP signal (D). Changes in the expression states of Tk-luciferase in 96-h dox-treated cells were determined by RT-qPCR (C) and luc activity assays (D) (circles = replicates, described in Methods). (E) Fusion proteins were expressed in cells treated with dox for 0, 48, or 96 h to determine the activity of the fusion activators at intermediate repressed states (bars = mean values for 3 luciferase assays from one transfected sample, each scaled to mean PcΔTF luc/cell; error bars = SDM).
We transfected dox-treated cells (96 h) with the PcΔTF negative control, PcTF, or Pc2TF cloned into a mammalian expression vector (Figure 5B). Fluorescence microscopy and Western blots confirmed nuclear localization and expression of full length proteins. Using reverse transcription followed by quantitative PCR (RT-qPCR), we detected higher luciferase transcript levels in Pc2TF and PcTF-expressing cells compared to PcΔTF (Figure 5C). These results indicate that bivalent Pc2TF is a stronger activator than PcTF. Luciferase (luc) activity levels detected by an enzymatic assay (Figure 5D) and normalized to RFP signal-to-noise ratios from flow cytometry corroborated the RT-qPCR results; Pc2TF stimulated greater luc expression than PcTF. We did not detect significantly higher luc activity for PcTF-expressing cells versus the negative control in this trial (Figure 5D), but did so in additional experiments (Figure 5E).
Expression of fusion regulators in cells that were treated with dox for 0, 48, and 96 h showed that Pc2TF had roughly twice the activity as PcTF (96 h) and that Pc2TF activated Tk-luciferase without prior dox-induced silencing (Figure 5E). The latter result can be explained by an intermediate, partially silenced level of Tk-luciferase expression40 compared to fully active Tk-luciferase in a “Luc14” parental cell line that lacks the Gal4-EED gene, as previously observed.40 H3K27me3 was detected via chromatin immunoprecipitation (ChIP) coupled with qPCR (ChIP-qPCR) near the luciferase promoter (Tk) in uninduced Gal4-EED/luc cells at significantly higher levels than in Luc14 cells. Dox treatment resulted in a further decrease in Tk-luciferase expression and a significant increase in H3K27me3 accumulation. In the experiments reported here, basal Tk-luciferase expression (Figure 5A) agrees with independent experiments from our prior study (0.02–0.07 luciferase activity per cell, au).40 The uninduced state may have low levels of H3K27me3 at nucleosomes near the reporter gene in all cells or high levels of H3K27me3 at the reporter gene in a small proportion of cells in the population. In contrast to Pc2TF, monovalent PcTF only activated Tk-luciferase after silenced chromatin had been induced for 96 h. These results suggest that Pc2TF is more tolerant of low levels of H3K27me3 in cellular chromatin.
CONCLUSIONS
This report presents the first demonstration of modular, synthetic multivalency of a chromatin-derived histone-binding protein with gene-regulating activity. In our previous work, we have demonstrated the use of a monovalent synthetic effector to activate chromatin-silenced genes in live cells. Natural bivalent chromatin proteins that recognize two histone post-translational modifications at once suggest a broader design space for synthetic chromatin effectors. Our application of bivalency to design a synthetic fusion protein produced two important advances for engineering synthetic chromatin effectors. First, we determined that synthetic linkers allow tethered histone PTM-binding peptides to function within the context of a fusion protein in vitro and in live cells. Second, we have established that doubling the valency with tandem PCDs strengthens avidity and increases gene regulation activity by at least 2-fold.
Here, we demonstrated that different synthetic linkers allow tethered histone PTM-binding peptides to bind in vitro to varying degrees. We observed weaker binding for the longer flexible linker (80 amino acids) compared to the shorter linker in our ELISA experiment. This result is likely due to lower production of the long flexible linker variant in TXTL. Given that both variants showed binding above background, GGGGS-repeat number may not significantly affect bivalent PCD engagement with H3K27me3 in vitro. For the rigid linker-tethered PCDs, only the longer length (80 amino acids) appeared to support binding. Assuming that this variant protein was properly folded, lack of binding over background for the shorter EAAAR-repeat variant could be caused by suboptimal rotation, that is, in trans instead of in cis, of the second PCD away from the 2-D binding surface in the ELISA well. This mechanism was demonstrated with mutated α-helical linkers in bivalent BPTF7 and with tandem zinc finger DNA binding domains.32 In the context of cellular chromatin where looping and folding occurs, H3K27me3 would not necessarily be constrained to one face of the Pc2TF protein. Valuable insights and perhaps greater Pc2TF performance might be acquired by exploring additional linker variants in cells as well as in vitro. Such work is beyond the scope of the studies reported here, which accomplished a major step by identifying a functional bivalent synthetic effector protein that specifically interacts with its target H3K27me3.
We have established that tandem PCDs strengthen avidity for H3K27me3 in a cooperative manner in vitro and increase gene regulation activity in live cells by at least 2-fold compared to a monovalent PCD. The wide distribution of multivalency within bromodomain family6 and other effector proteins7 suggests that multivalent engagement has an important, evolutionarily conserved biological role. Multivalency appears to largely be represented by cell-cycle and gene-activating effectors. Relatively few multivalent proteins that recognize silencing marks have been studied in biophysical detail. Examples include the chromodomains of the Arabidopsis protein CMT342 and the mammalian protein HP1β;43 as bivalent homodimers, these proteins show enhanced interaction with their respective ligands H3K9meK27me and H3K9me3. Pc2TF is novel in its composition of histone-binding motifs: adjacent, identical polycomb chromodomains within a single peptide. Therefore, its activity in vitro and in cells provides new insights into the recognition of histone marks by effector proteins.
In the context of cellular chromatin, Pc2TF appears to be active at the target gene prior to full repression (Figure 5E, 0 h dox), whereas detectable activity of monovalent PcTF required a prolonged period of induced repression at the target (Figure 5E, 96 h dox). Our previous ChIP mapping data40 confirm that compared to the fully active and fully silenced states, intermediate levels of H3K27me3 appear at Tk-luciferase (on average) without the addition of doxycycline. It is likely that in the pretreated state, leaky Gal4-EED expression causes a few cells in the population have one or two H3K27me3 marks at a nucleosome near the Tk promoter. Stronger avidity, supported by the additional PCD module, may increase the likelihood of an activation event at the target in this small population of cells. This idea is consistent with the behavior of synthetic zinc finger-based DNA-binding regulators, where stronger affinity of the regulator for its DNA target is associated with stronger gene activation.44 Similar behavior can also be observed for multivalent receptor-binding peptides, which bind with high avidity and specificity to a small number of receptor-positive cells.45
Further engineering efforts to achieve greater nonlinear enhancement of PcTF/Pc2TF may require changes within the PCD binding motif. The hydrophobic interaction between the methylammonium cage and the methyl-lysine moiety (Figure 1A) depends upon proper positioning of PCD residues that appear discontinuously in the primary sequence; this positioning requires specific intramolecular contacts of peptide residues within the PCD fold. Reverse engineering and de novo design of a new binding pocket through randomization of sequences would likely yield many nonfunctional proteins. K27-adjacent interactions that contribute to interactions with the histone tail13 could be leveraged to enhance affinity. However, increasing the stability by introducing additional hydrogen bonding could overwhelm the hydrophobic, K27me3-specific interaction and allow PCD to recognize unmodified tails or off-target modifications. Trade-offs between affinity and specificity pose formidable challenges to enhancing PCD affinity. Therefore, the most practical strategy for identifying alternative PCDs is to leverage H3K27me3-specific orthologues and paralogues from various species.46 It will be important to determine cross-reactivity with different histone modifications since certain CBX PCD peptides have been shown to bind H3K9me3.12
Multivalent engagement of combinatorial histone marks has recently become a key line of evidence to support the controversial histone code hypothesis. Rationally designed synthetic multivalency will advance this important area of research by exploring functions beyond the limits of pre-existing natural multivalent proteins. Furthermore, engineered chromatin effectors provide a practical tool to support artificial regulation of gene expression states through direct engagement with highly conserved components of chromatin, that is, histone tails and their modifications. Therapeutic synthetic gene regulators that leverage this mechanism could help circumvent the shortcomings of epigenetic inhibitors, which target chromatin enzymes that can gain drug-resistant mutations.47,48 In conclusion, our findings demonstrate that synthetic biology is a powerful tool for fundamental investigations of chromatin biology and epigenetic engineering.
METHODS
Plasmid Constructs for TXTL and Bacterial Expression.
Constructs (Figure 2A, Figure 3A) were assembled as BioBrick compatible fragments in vector V0120.49 Fragments were PCR-amplified with Phusion polymerase using primers 1–6 (Table S1) and a protocol adapted from New England Biolabs Phusion High Fidelity DNA Polymerase (98 °C 0:45, [98 °C 0:10, 67 °C 0:20, 72 °C 0:45] × 25, final extension of 5:00), column purified (Qiagen PCR Cleanup Kit), and double-digested with BamHI and XhoI (New England BioLabs). BamHI/XhoI-digested inserts and 50–75 ng of BamHI/XhoI linearized pET28(+) vector were ligated at a 3:1 molar ratio in a 20 μL reaction as described in the New England BioLabs (NEB) protocol for T4 ligase (M0202). Five microliters of each ligation was incubated with 50 μL of Turbo competent DH5-alpha E. coli (NEB) on ice for 5 min, transferred to 45 °C for 45 s, then to ice for 5 min, and allowed to recover in 350 μL of SOC medium at 37 °C with shaking for 30 min. Pelleted cells were resuspended in 50 μL of SOC, plated on LB agar (50 ug/mL kanamycin), and grown at 37 °C overnight. Colony PCR was performed to identify positive ligation results using primers 6 and 7 (Table S1) and the GoTaq Promega protocol. Plasmids were cloned, extracted (Sigma GeneElute Plasmid Miniprep Kit), and Sanger sequenced for verification prior to protein expression in cell-free TXTL or in E. coli. Annotated sequences for all pET28 constructs are available online at Benchling-Hayneslab: Synthetic Chromatin Actuators 2.0 (https://benchling.com/hayneslab/f_/rmSYkAAU-synthetic-chromatin-actuators-2-0).
TXTL: Cell-free Expression.
TXTL reactions were set up with the following conditions as previously described:35 9 μL of lysate, 10 nM final template vector, 0.5 nM σ70-T7 RNA pol vector to a total of 12 μL. A Roche Lightcycler 480 was used to detect mCherry fluorescence with the following protocol: 29 °C for 10 min, bring to 30 °C for 1 s, scan 533–610 nm, repeat 96 times (total 16 h).
E. coli Expression and Purification of Proteins..
All selection media contained 50 μg/mL kanamycin. PcΔTF, PcTF, and Pc2TF in pET28 were transformed into Rosetta 2pLys DE3 cells and plated on LB agar and grown at 37 °C overnight. The next day, a single colony from each was used to inoculate 50 mL of LB and grown overnight at 37 °C at 300 rpm. The next day, 1 L of LB in a baffled Erlenmeyer flask was inoculated to an OD600 of 0.1. The cultures were grown to an OD600 = 0.6, induced with IPTG (1 mM final concentration), and allowed to express PcΔTF and PcTF at 37 °C for 5 h with shaking (220 rpm). Pc2TF expression was carried out overnight at room temperature with shaking (220 rpm) to aid solubility of the protein. Cell disruption and protein purification are described in detail in Supporting Information and Methods. Purification of recombinant protein from E. coli is described in Supporting Information (Methods).
Enzyme-Linked Immunosorbent Assays (ELISAs).
All steps were carried out at room temperature except specifically noted, and all incubations and washes were agitated at 800 rpm on an Eppendorf Thermomixer R. Clear bottom plates (Greiner bio-one #655101) were coated in 50 μL of 20 ng/μL neutravidin in PBS pH 8.0 overnight at 4 °C. The plates were washed the next day 3× with 200 μL of 0.2% PBS-Tween (PBST) with 5 min of shaking at 800 rpm between washes. The plate was blocked for 30 min at 800 rpm at room temperature with 200 μL of 5% BSA in 0.2% PBST followed by 3× washes of 200 μL of 0.2% PBST for 5 min each at 800 rpm. Fifty microliters of 1 μM biotinylated peptides (Anaspec, H3 (21–44), H3K4me3 (1–21), H3K9me3 (1–21), H3K27me3 (21–44), or H3K27Ac (21–43)) in 0.2% PBST) were incubated at room temperature for 1 h at 800 rpm, followed by 3× washes of 200 μL of 0.2% PBST for 5 min at 800 rpm. The plate was blocked with 200 μL of 5% skim milk in 0.2% PBST (room temperature, 800 rpm, 30 min). TxTL, 1.5 μL (Figure 2), or 50 μL of 0.1 μM (Figure 3A) or 0.05 nM (Figure 3C) purified proteins in 50 μL of 5% skim milk in PBST were incubated in each well for 1 h (room temperature, 800 rpm). The wells were washed 3× with 200 μL of 5% skim milk in PBST with 5 min of 800 rpm shaking. After adding 100 μL of 1:3000 chicken polyclonal anti-mCherry (Novus Biologicals #NBP2–25158) in 5% nonfat milk in 0.2% PBST, wells were incubated for 1 h, followed by 3× of 200 μL of 5% skim milk in 0.2% PBST for 5 min each (room temperature, 800 rpm). After addition of 100 μL of 1:3000 rabbit anti-chicken-HRP (RCYHRP Genetel 0.5 mg/mL) in 5% skim milk in 0.2% PBST wells were incubated for 30 min (room temperature, 800 rpm). The plate was washed 5× with 200 μL of 0.2% PBST for 3 min each at 800 rpm. The plate was incubated with 100 μL of 1-step Ultra TMB-ELISA (Thermo-Fisher #34029) for 15 min while protected from light. Reactions were stopped with 100 μL of 2.0 M sulfuric acid, incubated for 2 min, and read at 450 nm. Each plate contained four technical replicates per H3K27me3 concentration per fusion protein. Two ELISA plates (trials) were run for each of two purified protein samples per construct. One trial failed to show significant signal over background (for all recombinant proteins) and was omitted from the final analysis. In Figure 4 “anti-mCherry-HRP (A450)” = HRP signal - mean HRP signal for 0% H3K27me3. The Microsoft Excel Solver tool was used to fit the Hill equation, , to the data by minimizing the sum of the squared errors between the equation and data (varying and n). R2 was calculated as 1 − (SSreg/SStot), where total sum of squares and regression sum of squares .
Peptide Microspot Arrays.
APTES functionalized glass slides were coated with 200 μL of 1:1 (v/v) 40 mg/mL BS3 cross-linking solution and 1 mg/mL neutravidin with a cover slide (Thermo Scientific, #651-2-5251) and incubated overnight at 4 °C. The next day, the cover slide was removed, and the slide was rinsed 3× with 0.2% PBST for 5 min each. Slides were deactivated by incubation with Na2CO3/NaHCO3 buffer, pH 9.4, for 30 min. The slides were quickly rinsed with ddH2O and centrifuged to dry at 1200 rpm for 2 min. Slides were printed with biotinylated peptides (Anaspec) at concentrations of 10, 20, or 50 μM in 20% glycerol and PBS with a pin-printer (spot to spot distance = 600 μm) and incubated at room temperature for 1 h. The slide was rinsed with ddH2O as described above and blocked with superblock for 1 h at room temperature. Proteins were diluted in superblock and incubated on the slide for 1 h at room temperature. The slides were rinsed with 0.2% PBST for 3 min each followed by quick rinsing with ddH2O 3× and centrifuged dry (as described). Red fluorescent protein (mCherry) signal was detected at 50% gain and 50% intensity on a PowerScanner at 635 and 535 nm, 10 μm resolution. Slides were also scanned at 75%–75% and 100%–100% to obtain a suitable signal-to-noise ratio. Arraypro software was used to quantify the median intensity values for each spot and background levels. Graphpad Prism software was used to fit the binding saturation nonlinear regression equation, , to the data, where Bmax is the highest binding value and X is the concentration of protein.
Plasmid Constructs for Mammalian Expression.
MV10 was constructed from pcDNA3.1(+) (Invitrogen) with the following modifications. The CMV promoter was removed via SpeI digestion and T4 ligase recircularization. A dsDNA fragment that encodes Kozak (ribosome binding site), XbaI, a nuclear localization sequence, 6× histidine, and a stop codon (5′-cccgccgccaccatggagtctagacccaagaaaaagcgcaaggtacaccatcaccaccatcacgcgtaaagctgag) with SpeI overhangs at both ends (ctag/t) was inserted at XbaI. CMV (SpeI/XbaI fragment) was reintroduced upstream of Kozak at SpeI. Proper orientation of inserts was confirmed by Sanger sequencing. Constructs PcTF and Pc2TF (Figure 5B) were PCR-amplified (Phusion) with primers 9 and 10 (Table S1), double-digested with XbaI and SpeI, and column-purified (Qiagen PCR Purification, 28104). Construct PcΔTF (Figure 5B) was double-digested with XbaI and SpeI (Thermo Fisher FastDigest) and isolated by electrophoresis and gel purification. XbaI/SpeI fragments and 25 ng of XbaI-linearized, dephosphorylated MV10 vector were ligated at a 2:1 molar ratio in a 10 μL reaction as described in the Roche protocol for Rapid DNA Ligation (11635379001 Roche), using 1.0 μL of NEB T4 ligase instead of the supplied enzyme. All 10 μL of each ligation was incubated with 50 μL of Turbo competent DH5-alpha E. coli (New England Biolabs) on ice for 5 min, transferred to 45 °C for 45 s, then to ice for 5 min. Cells were plated directly on prewarmed LB agar (100 μg/mL ampicillin) without recovery and grown at 37 °C overnight. Plasmid DNA was prepared (Sigma GeneElute Plasmid Miniprep Kit) from 5 mL cultures inoculated with single colonies. Forward orientation of each insert was determined by XbaI and PstI double-digestion of prepped plasmids and Sanger sequencing. Annotated sequences for all MV10 constructs are available online at Benchling-Hayneslab: Synthetic Chromatin Actuators 2.0 (https://benchling.com/hayneslab/f_/rmSYkAAU-synthetic-chromatin-actuators-2-0).
Cell Culture and Transfection.
HEK293 Gal4-EED/luc cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% tetracycline-free fetal bovine serum and 1% penicillin and streptomycin at 37 °C in a humidified CO2 incubator. Silencing of the reporter gene (Tk-luciferase) was induced by supplementing the media with 1 μg/mL of dox for 48 or 96 h. For wash-out of doxycycline (to allow depletion of Gal4-EED), growth medium was removed and replaced with dox-minus medium supplemented with 0.5 μg/mL puromycin to select for the transgenic anti-Gal4-EED shRNA,41 and grown for 5 days. Prior to transfection, dox treated or untreated cells were plated in 12-well culture dishes at 40% confluency (~1.0 × 105 cells per well) in 2 mL of pen/ strep-free growth medium. Transient transfections were carried out by adding 100 μL of DNA/Lipofectamine complexes to each well: 1 μg pDNA or ddH2O for mock transfections (10 μL), 3 μL of Lipofectamine LTX (Invitrogen), 87 μL of Opti-MEM. Forty-eight hours after transfection, cellular mCherry (580/610 excitation/emission) was imaged in culture dishes on a Nikon Eclipse Ti wide field inverted fluorescent microscope (MEA53100) at 200× magnification (eyepiece = 10×; objective = CFI S Plan Fluor ELWD 20×, numerical aperture = 0.45), 25 °C, without oil immersion, and with either phase contrast or an mCherry filter set (TE2000 cube, excitation FF01-562/40-25, emission FF01-641/75-25). Images from each channel were acquired with a digital monochrome camera (Coolsnap ES2 12 bit, 20 MHz) and overlaid using NIS-Elements software. For downstream assays (RT-qPCR, Western blots, and flow cytometry), the growth medium was removed, semiadherent cells were gently collected with 1× PBS washes, pelleted (200 g, room temperature, 5 min), and resuspended in 1× PBS. Six replicate samples (wells) were pooled for assays in Figure 5B,C,D.
RT-qPCR.
Preparation of total RNA, cDNA synthesis, and qPCR were performed as previously described11 using ~1.0 × 106 HEK293 Gal4-EED/luc cells that were pelleted (500 g, room temperature, 5 min) and lysed with 500 μL of TRIzol (Thermo Fisher #15596026). DNA/LNA oligos for qPCR were as follows: mCherry, forward 5′-cctgaagggcgagatcaag, reverse 5′-ttgacctcagcgtcgtagtg, LNA probe #41 (Millipore Sigma #04688007001); luciferase, forward 5′-caggtcttcccgacgatg, reverse 5′-gtctttccgtgctccaaaac, LNA probe #70 (Millipore Sigma #04688937001); GAPDH (reference), Roche human G6PD assay (Millipore Sigma #5046246001). Mean Crossing point (Cp), the first peak of d2y/dx2 (fluorescence over cycle number), was calculated by the Roche LightCycler 480 software for three replicate wells per unique reaction. For each biological replicate (one transfection per fusion protein) two replicate cDNA synthesis reactions (from one RNA prep) were completed. Expression level was calculated as . “mRNA level log2(FC)” = log2(ΔCp-(transfected cells)/ΔCp(mock)).
Western Blots.
Total protein was prepared from roughly 250 000 cells. Sample preparation, polyacrylamide gel electrophoresis (PAGE), and membrane blotting are described in detail in Supporting Information. Immunostaining was carried out with the following: blocking buffer, 5% nonfat dry milk in 1× PBST (1× PBS, 0.1% Tween-20); primary 1, chicken polyclonal anti-mCherry, 1:2000 (Novus Biologicals #NBP2–25158); secondary 1, HRP-conjugated rabbit anti-chicken, 1:2000 (Millipore Sigma #AP162P); primary 2, anti-histone H3, 1:1000 (Abcam #ab1791); secondary 2, HRP-conjugated goat anti-rabbit, 1:2000 (Cell Signalling Technology #7074). Immunostaining was performed at 4 °C overnight (primary) or at room temperature for 1 h (secondary) with nutation in a Parafilm pouch.50 Immunostained blots were washed 4 × 10 min in 1× PBST, with orbital shaking at room temperature. HRP signal was detected using the SuperSignal West Femto substrate kit (Thermo Fisher #34095) and a PXi4 imager (Syngene) with GeneSys software.
Imaging and Flow Cytometry.
Cells were passed through a 35 μm nylon strainer (EMS #64750-25). Red fluorescent signal from mCherry was detected on a BD Accuri C6 flow cytometer (675 nm LP filter) using CFlow Plus software. Data were further analyzed using FlowJo 10.0. One run (~10 000 live cells, gated by forward and side scatter) was completed per sample. “RFP median signal/noise (S/N)” = {median RFP signal from live RFP-positive cells}/{median RFP noise from live untransfected cells}.
Luciferase Assays.
Cell counts (per 100 μL) were determined by flow cytometry (BD Accuri C6). Cells (100 μL) or 1× PBS (blank) were incubated with 100 μL of complete luciferase assay reagent as described in the protocol for the Biotium Firefly Luciferase Assay Kit (89138-960) and in previous work40 in Corning and Costar 96-well Cell Culture Plates, opaque, white (Corning 3789A). Chemiluminescence was detected using a Synergy H1Multi-Mode Reader (Biotek). Replicates included three samples (100 μL each) taken from a single population of transfected cells. “Luc × cell−1 (au)” = [Sample Luciferase signal] − 1× PBS blank signal/[cell count × (100 μL/20 μL)]. For fusion protein-expressing cells, normalization was performed by dividing Luc × cell−1 by the RFP median signal/noise value (from flow cytometry).
Supplementary Material
ACKNOWLEDGMENTS
We thank undergraduate researchers B. Laughlin, C. Gardner, D. Tze, and J. Xu for early contributions to plasmid construction. We also thank V. Noireaux for his generous gift of TXTL reagents. The work was supported by the NIH NCI (Grant K01 CA188164 to K.A.H.). J.L. was supported by the Kleberg Foundation (Grant FP00001994). S.T. and D.V. were supported by the Biological Design PhD Program at ASU. D.V. was also supported by the Western Alliance to Expand Student Opportunities (Grant NSF HRD 1401190). L.S. was supported by the Flinn Foundation (Grant ASUF 30005389).
ABBREVIATIONS
- BPTF
bromodomain PHD finger transcription factor
- ELISA
enzyme-linked immunosorbent assay
- H3K27me3
histone H3 trimethylated at lysine 27
- PCD
polycomb chromodomain motif
- PCR
polymerase chain reaction
- PcTF
monovalent polycomb-based transcription factor
- Pc2TF
bivalent polycomb-based transcription factor
- PRC
polycomb repressive complex
- PTM
post-translational modification
- RFP
red fluorescent protein
- RT-qPCR
reverse transcription followed by quantitative polymerase chain reaction
- TXTL
E. coli-based cell-free transcription-translation system
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.7b00281.
DNA templates and primers used in experiments, predicted Pc-fusion fluorescence after binding and subsequent washing, additional peptide array binding data, methods for purification of recombinant protein from E. coli, and Western blot detailed protocol (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Tekel S, and Haynes KA (2017) Molecular Structures Guide the Engineering of Chromatin. Nucleic Acids Res. 45, 7555–7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cano-Rodriguez D, and Rots MG (2016) Epigenetic Editing: On the Verge of Reprogramming Gene Expression at Will. Curr. Genet. Med. Rep 4, 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Park M, Keung AJ, and Khalil AS (2016) The epigenome: the next substrate for engineering. Genome Biol. 17, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Platero JS, Hartnett T, and Eissenberg JC (1995) Functional analysis of the chromo domain of HP1. EMBO J. 14, 3977–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Messmer S, Franke A, and Paro R (1992) Analysis of the functional role of the Polycomb chromo domain in Drosophila melanogaster. Genes Dev. 6, 1241–1254. [DOI] [PubMed] [Google Scholar]
- (6).Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, Gingras A-C, Arrowsmith CH, and Knapp S (2012) Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou Y, Muir TW, Patel DJ, and Allis CD (2011) Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell 145, 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gatchalian J, Fütterer A, Rothbart SB, Tong Q, Rincon-Arano H, Sánchez de Diego A, Groudine M, Strahl BD, Martínez-A C, van Wely KHM, and Kutateladze TG (2013) Dido3 PHD modulates cell differentiation and division. Cell Rep. 4, 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang R, and You J (2015) Mechanistic analysis of the role of bromodomain-containing protein 4 (BRD4) in BRD4-NUT oncoprotein-induced transcriptional activation. J. Biol. Chem 290, 2744–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Haynes KA, and Silver PA (2011) Synthetic reversal of epigenetic silencing. J. Biol. Chem 286, 27176–27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nyer DB, Daer RM, Vargas D, Hom C, and Haynes KA (2017) Regulation of cancer epigenomes with a histone-binding synthetic transcription factor. npj Genomic Med. 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bernstein E, Duncan EM, Masui O, Gil J, Heard E, and Allis CD (2006) Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol. Cell. Biol 26, 2560–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, and Khorasanizadeh S (2003) Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 17, 1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Meckler JF, Bhakta MS, Kim M-S, Ovadia R, Habrian CH, Zykovich A, Yu A, Lockwood SH, Morbitzer R, Elsäesser J, Lahaye T, Segal DJ, and Baldwin EP (2013) Quantitative analysis of TALE–DNA interactions suggests polarity effects. Nucleic Acids Res. 41, 4118–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jantz D, and Berg JM (2010) Probing the DNA-binding affinity and specificity of designed zinc finger proteins. Biophys. J 98, 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Greisman HA, and Pabo CO (1997) A general strategy for selecting high-affinity zinc finger proteins for diverse DNA target sites. Science 275, 657–661. [DOI] [PubMed] [Google Scholar]
- (17).Sternberg SH, Redding S, Jinek M, Greene EC, and Doudna JA (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ruthenburg AJ, Li H, Patel DJ, and Allis CD (2007) Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol 8, 983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Su Z, and Denu JM (2016) Reading the Combinatorial Histone Language. ACS Chem. Biol 11, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Su X, Zhu G, Ding X, Lee SY, Dou Y, Zhu B, Wu W, and Li H (2014) Molecular basis underlying histone H3 lysine-arginine methylation pattern readout by Spin/Ssty repeats of Spindlin1. Genes Dev. 28, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jacobson RH (2000) Structure and Function of a Human TAFII250 Double Bromodomain Module. Science 288, 1422–1425. [DOI] [PubMed] [Google Scholar]
- (22).Voigt P, and Reinberg D (2011) Histone tails: ideal motifs for probing epigenetics through chemical biology approaches. Chem-BioChem 12, 236–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Oliver SS, Musselman CA, Srinivasan R, Svaren JP, Kutateladze TG, and Denu JM (2012) Multivalent recognition of histone tails by the PHD fingers of CHD5. Biochemistry 51, 6534–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sanchez OF, Mendonca A, Carneiro AD, and Yuan C (2017) Engineering Recombinant Protein Sensors for Quantifying Histone Acetylation. ACS Sens 2, 426–435. [DOI] [PubMed] [Google Scholar]
- (25).Delachat AM-F, Guidotti N, Bachmann AL, Meireles-Filho ACA, Pick H, Lechner CC, Deluz C, Deplancke B, Suter DM, and Fierz B (2018) Engineered Multivalent Sensors to Detect Coexisting Histone Modifications in Living Stem Cells. Cell Chem. Biol 25, 51. [DOI] [PubMed] [Google Scholar]
- (26).Min JR, Zhang Y, and Xu R-M (2003) Structural Basis for specific binding of polycomb chromodomain to histone H3 methylated at K27. Genes Dev. 17, 1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Luger K, Mäder AW, Richmond RK, Sargent DF, and Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- (28).Voigt P, LeRoy G, Drury WJ 3rd, Zee BM, Son J, Beck DB, Young NL, Garcia BA, and Reinberg D (2012) Asymmetrically modified nucleosomes. Cell 151, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, and Reinberg D (2002) Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 16, 2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wriggers W, Chakravarty S, and Jennings PA (2005) Control of protein functional dynamics by peptide linkers. Biopolymers 80, 736–746. [DOI] [PubMed] [Google Scholar]
- (31).Merutka G, Shalongo W, and Stellwagen E (1991) A model peptide with enhanced helicity. Biochemistry 30, 4245–4248. [DOI] [PubMed] [Google Scholar]
- (32).Yan W, Imanishi M, Futaki S, and Sugiura Y (2007) α-Helical Linker of an Artificial 6-Zinc Finger Peptide Contributes to Selective DNA Binding to a Discontinuous Recognition Sequence†. Biochemistry 46, 8517–8524. [DOI] [PubMed] [Google Scholar]
- (33).Shewmake TA, Solis FJ, Gillies RJ, and Caplan MR (2008) Effects of Linker Length and Flexibility on Multivalent Targeting. Biomacromolecules 9, 3057–3064. [DOI] [PubMed] [Google Scholar]
- (34).Chen X, Zaro JL, and Shen W-C (2013) Fusion protein linkers: property, design and functionality. Adv. Drug Delivery Rev 65, 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Caschera F, and Noireaux V (2014) Synthesis of 2.3 mg/mL of protein with an all Escherichia coli cell-free transcription– translation system. Biochimie 99, 162–168. [DOI] [PubMed] [Google Scholar]
- (36).Shen Y, Chen Y, Wu J, Shaner NC, and Campbell RE (2017) Engineering of mCherry variants with long Stokes shift, red-shifted fluorescence, and low cytotoxicity. PLoS One 12, e0171257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kaustov L, Ouyang H, Amaya M, Lemak A, Nady N, Duan S, Wasney GA, Li Z, Vedadi M, Schapira M, Min J, and Arrowsmith CH (2011) Recognition and specificity determinants of the human cbx chromodomains. J. Biol. Chem 286, 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, and Mann M (2010) Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142, 967–980. [DOI] [PubMed] [Google Scholar]
- (39).Swigut T, and Wysocka J (2007) H3K27 demethylases, at long last. Cell 131, 29–32. [DOI] [PubMed] [Google Scholar]
- (40).Daer RM, Cutts JP, Brafman DA, and Haynes KA (2017) The Impact of Chromatin Dynamics on Cas9-Mediated Genome Editing in Human Cells. ACS Synth. Biol 6, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, and Helin K (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol 10, 1291–1300. [DOI] [PubMed] [Google Scholar]
- (42).Lindroth AM, Shultis D, Jasencakova Z, Fuchs J, Johnson L, Schubert D, Patnaik D, Pradhan S, Goodrich J, Schubert I, Jenuwein T, Khorasanizadeh S, and Jacobsen SE (2004) Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. EMBO J. 23, 4146–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hiragami-Hamada K, Soeroes S, Nikolov M, Wilkins B, Kreuz S, Chen C, De La Rosa-Velázquez IA, Zenn HM, Kost N, Pohl W, Chernev A, Schwarzer D, Jenuwein T, Lorincz M, Zimmermann B, Walla PJ, Neumann H, Baubec T, Urlaub H, and Fischle W (2016) Dynamic and flexible H3K9me3 bridging via HP1β dimerization establishes a plastic state of condensed chromatin. Nat. Commun 7, 11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Khalil AS, Lu TK, Bashor CJ, Ramirez CL, Pyenson NC, Joung JK, and Collins JJ (2012) A synthetic biology framework for programming eukaryotic transcription functions. Cell 150, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Rosca EV, Gillies RJ, and Caplan MR (2009) Glioblastoma targeting via integrins is concentration dependent. Biotechnol. Bioeng 104, 408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Senthilkumar R, and Mishra RK (2009) Novel motifs distinguish multiple homologues of Polycomb in vertebrates: expansion and diversification of the epigenetic toolkit. BMC Genomics 10, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Gibaja V, Shen F, Harari J, Korn J, Ruddy D, Saenz-Vash V, Zhai H, Rejtar T, Paris CG, Yu Z, Lira M, King D, Qi W, Keen N, Hassan AQ, and Chan HM (2016) Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene 35, 558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Baker T, Nerle S, Pritchard J, Zhao B, Rivera VM, Garner A, and Gonzalvez F (2015) Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors. Oncotarget 6, 32646–32655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Phillips I, and Silver P (2006) A New Biobrick Assembly Strategy Designed for Facile Protein Engineering. http://dspace.mit.edu/handle/1721.1/32535.
- (50).Quadri SMS (2015) Parafilm-M®, An Available Cost-Effective Alternative for Immuno-blot Pouches. Methods Mol. Biol 1314, 313–323. [DOI] [PubMed] [Google Scholar]
- (51).Ren C, Morohashi K, Plotnikov AN, Jakoncic J, Smith SG, Li J, Zeng L, Rodriguez Y, Stojanoff V, Walsh M, and Zhou M-M (2015) Small-molecule modulators of methyl-lysine binding for the CBX7 chromodomain. Chem. Biol 22, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wakamori M, Fujii Y, Suka N, Shirouzu M, Sakamoto K, Umehara T, and Yokoyama S (2015) Intra- and inter-nucleosomal interactions of the histone H4 tail revealed with a human nucleosome core particle with genetically-incorporated H4 tetra-acetylation. Sci. Rep 5, 17204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.