

Abstract
We report a new reversible lysine conjugation that features a novel diazaborine product and much slowed dissociation kinetics in comparison to the previously known iminoboronate chemistry. Incorporating the diazaborine-forming warhead RMR1 to a peptide ligand gives potent and long-acting reversible covalent inhibitors of the staphylococcal sortase. The efficacy of sortase inhibition is demonstrated biochemical and cell-based assays. A comparative study of RMR1 and an iminoboronate-forming warhead highlights the significance and potential of modulating bond dissociation kinetics in achieving long-acting reversible covalent inhibitors.
Graphical Abstract
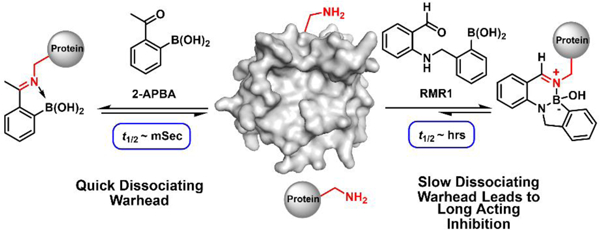
There has been an increasing interest in developing reversible covalent inhibitors1–5 as the reversibility can minimize off-target reactions and also avoid permanent modification of the target proteins.1 Conceptually, a reversible covalent inhibitor first binds its target through noncovalent interactions, and then the warhead forges a covalent bond to enhance binding and inhibition (Figure 1a).To minimize off-target reactions, it is desirable for the warhead to have a modest on-rate and fast off-rate (high koff) so that inadvertent off-target conjugations can be quickly reversed. This combination, however, would lead to reduced potency (higher Kd (=koff/kon)). In other words, we wish to have low off-rate to maximize the thermodynamic benefit of the warhead. Furthermore, a low off-rate is also desirable to achieve long-lasting inhibition. These paradoxical considerations highlight the necessity of optimizing the kinetics of reversible covalent warheads. An elegant example of such endeavors comes from the work of Taunton and coworkers, which demonstrates that a slow dissociating warhead can give highly potent and long-acting inhibitors of important kinases.5–7
Figure 1.
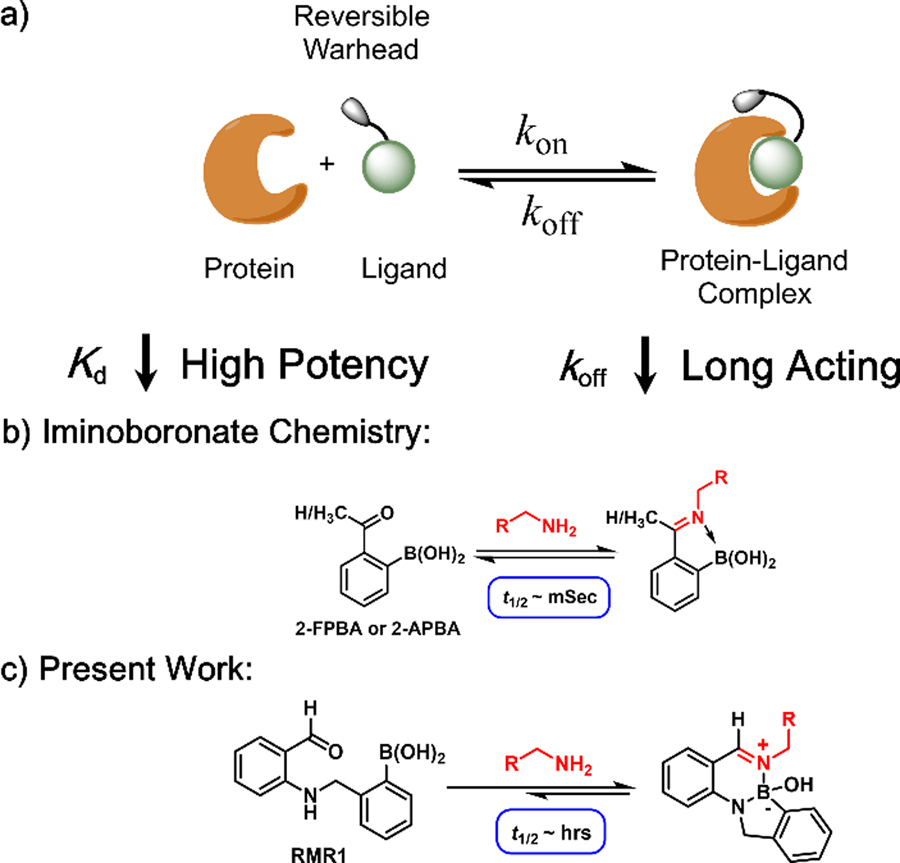
Reversible covalent drugs: design considerations (a) and suitable chemistries for targeting lysines (b, c)
Only a handful of papers describe lysine-reactive reversible covalent inhibitors.8–10 Essentially all reported work is based on some variant of the imine chemistry as exemplified by the anti-sickling drug voxelotor.8 A recent development ushers in the iminoboronate chemistry,11 which gives imines of improved stability due to N-B coordination (Figure 1b). The iminoboronate formation elicited by 2-FPBA/APBA has shown promise in lysine-targeted molecular probes12–13 and inhibitors.9, 14 In our continued search for novel reversible lysine-conjugation chemistries (Figure 1c), we were inspired by the diazaborine formation of hydrazides, which were recently developed as ultrafast bioorthogonal conjugations.15–17 Diazaborines and related B-N heterocycles have also been explored as enzyme inhibitors18–19 and as reversible linkers for drug delivery to cancer cells.20
While some diazaborines exhibit a flat aromatic structure, others display a hydrated boron with a nonplanar tetrahedral geometry (Figure 2a). These hydrated diazaborines are known to undergo quick hydrolysis.16, 21 We postulated that a lysine side chain amine, analogous to the hydrazides, could be induced to undergo reversible conjugation to give hydrolysis-prone diazaborines. Towards this end, we designed a new warhead RMR1 (Figure 2b), a benzaldehyde derivative that installs an ortho-aminomethyl phenyl boronic acid (AMPB) moiety. As a result of N-B coordination, AMPB adopts a closed conformation in neutral aqueous media,22 in which the boron center is brought close to the aldehyde to facilitate RMR1 conjugation with a lysine. This proposed reaction cascade (supported by DFT calculations, Figure S1) yields a hydrated diazaborine, although a different regioisomer from those shown in Figure 2a.
Figure 2.
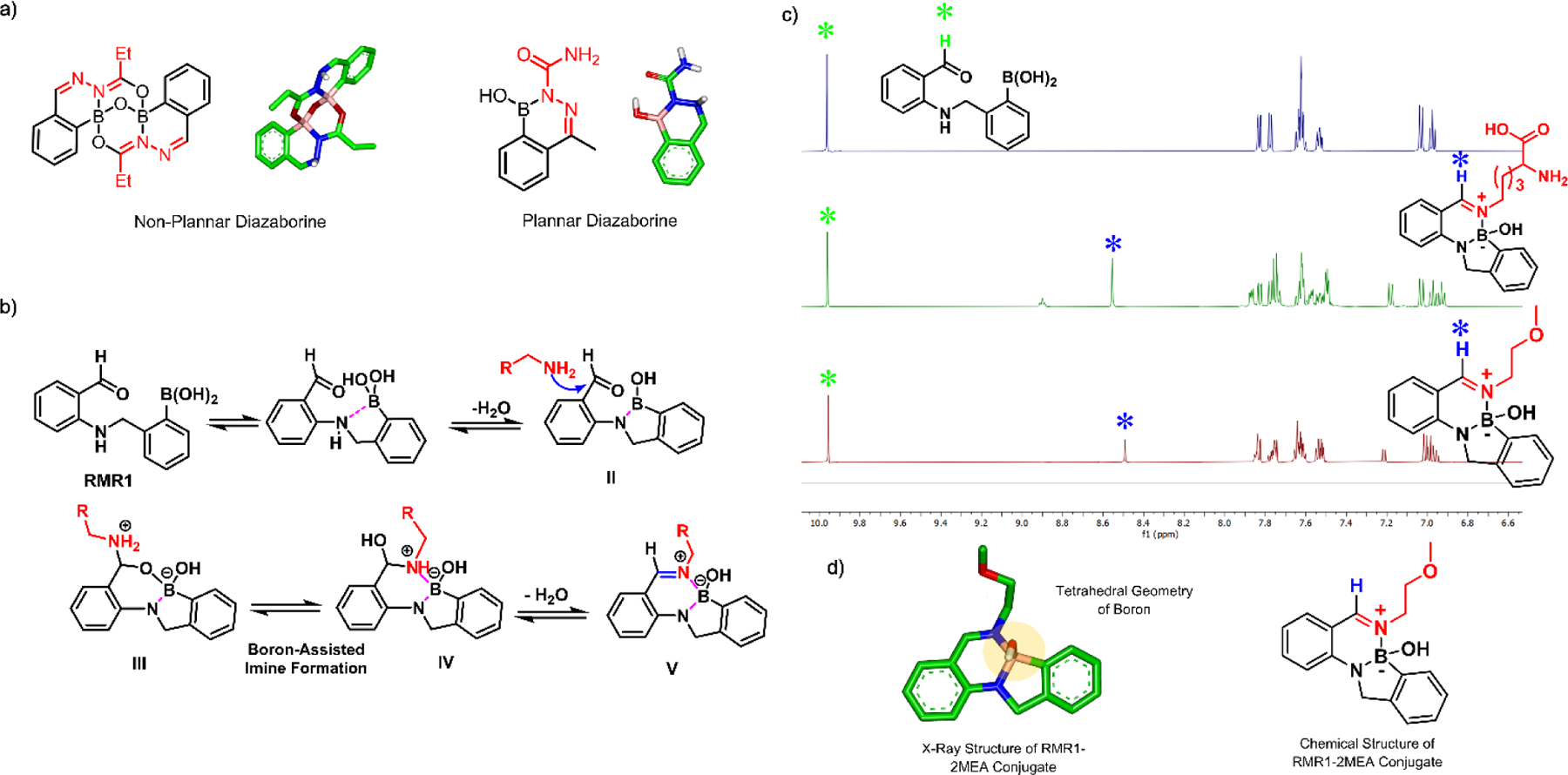
Reversible formation of diazaborines. (a) Previously reported diazaborine structures with (left) and without (right) reversibility. (b) A postulated reaction cascade for RMR1-amine conjugation to give a hydrated diazaborine.(c) 1H-NMR spectra showcasing the conjugation of RMR1 to lysine and 2-methoxyethylamine. Samples were prepared in a PBS buffer containing 40% DMSO for solubility. (d) Crystal structure of the RMR1-MEA conjugate.
The details of RMR1 synthesis are given in the SI (Scheme S1-S2). Excitingly, when mixed with equimolar concentrations of 2-methoxyethylamine (MEA) or lysine (10 mM reactant, pH 7.4), the aldehyde peak (~9.9 ppm) of RMR1 dropped in intensity and a new singlet characteristic of aldimine was observed at 8.5 ppm (Figure 2c). The diazaborine formation was confirmed by X-ray crystallography. While the lysine conjugate failed to crystalize, we were able to crystalize the RMR1-MEA conjugate from a water-methanol mixture. The crystal structure (Figure 2d) revealed a B-N heterocyclic core with a tetrahedral boron, which was consistently supported by 11B-NMR and mass spectrometry analyses (Figure S4, S11). The RMR1 induced diazaborine formation was further investigated via comparison to two control molecules, RMR2 and RMR3 (Figure S5-S6). Neither control molecule afforded any conjugation with lysine. Interestingly, the RMR1 induced diazaborine formation appears to be sensitive to steric hindrance as minimal (<2%) conjugation was observed with the α-amine of lysine (Figure S7-S8).
The reversibility of the diazaborine conjugation was confirmed via a dilution experiment. A 10 mM mixture of RMR1 and lysine was incubated overnight, and then diluted 20 times (Figure 3a). The 1H-NMR data show that the RMR1-lysine conjugate dropped from 50% to 13% over time, confirming the autonomous reversibility of the conjugation. We then determined the thermodynamic and kinetic parameters of RMR1-amine conjugation via a set of UV-vis experiments. First, we measured the apparent Kd values for RMR1 binding lysine, MEA and a glycine amide respectively (Figure 3b, c). All three amines gave a comparable Kd around 1 mM (Figure 3e, S13). The dissociation rate of the diazaborines was determined by diluting a 10 mM solution to 50 µM and monitoring the relaxation kinetics using UV-vis spectroscopy (Figure 3d, S14). As the diluted concentration is much below the Kd values, the relaxation kinetics should be dictated by the dissociation rate. Exponential curve fitting gives comparable dissociation rate constants (k-1) for the three amines with a value of 2.6×10−5 s−1 for lysine (Figure 3e). The slow dissociation of the diazaborines was further corroborated by 1H-NMR analysis of diluted samples (Figure S15-S16). With the Kd and k-1 values, we estimated the forward reaction rate of the RMR1-lysine conjugation to be 2.1×10−2 M−1s-1. In comparison to the iminoboronate formation, which shows instantaneous equilibrium (Figure S14, S17),21, 23 the RMR1-lysine conjugation exhibits slower kinetics but more favorable thermodynamic profile. This is confirmed by a competition experiment (Figure S9-S10), in which the iminoboronate conjugates of lysine slowly exchanged with RMR1 to give the diazaborine as the predominant end product.
Figure 3.
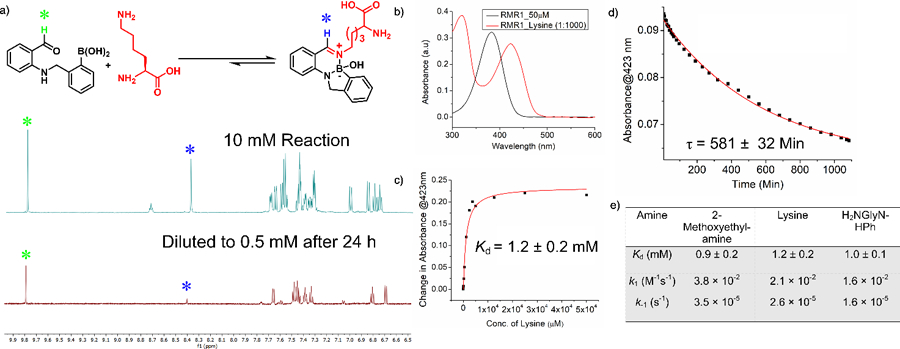
Thermodynamic and kinetic characterizations of the RMR1-mediated diazaborine formation. (a) A dilution experiment demonstrating the reversibility of RMR1-lysine conjugation. (b) UV-vis absorption changes of RMR1 upon lysine conjugation. (c) A titration experiment revealing the Kd value of RMR1-lysine conjugation. (d) Dissociation kinetics of the diazaborine conjugate of RMR1 and lysine. (e) Tabulated thermodynamic and kinetic parameters of RMR1 conjugation with small molecule amines.
The distinct kinetic profiles of the two reversible lysine conjugations present an opportunity to compare the fast and slow dissociating warheads towards developing lysine-targeted reversible covalent inhibitors. Towards this end, we resorted to an important enzyme of Staphylococcus aureus, sortase A (SrtA) as a model system. SrtA is an appealing anti-Staph target as SrtA inhibition is not bactericidal and hence does not force the acquisition of resistance mechanisms.24–25 Through phage display, we identified a cyclic peptide W7 that binds SrtA with low micromolar potency (Figure 4a, S21). Computational docking indicates that W7 binds to the SrtA active site with the N-terminus and C-terminus projected away from the protein and potentially suitable for warhead incorporation (Figure S22-S23).
Figure 4.
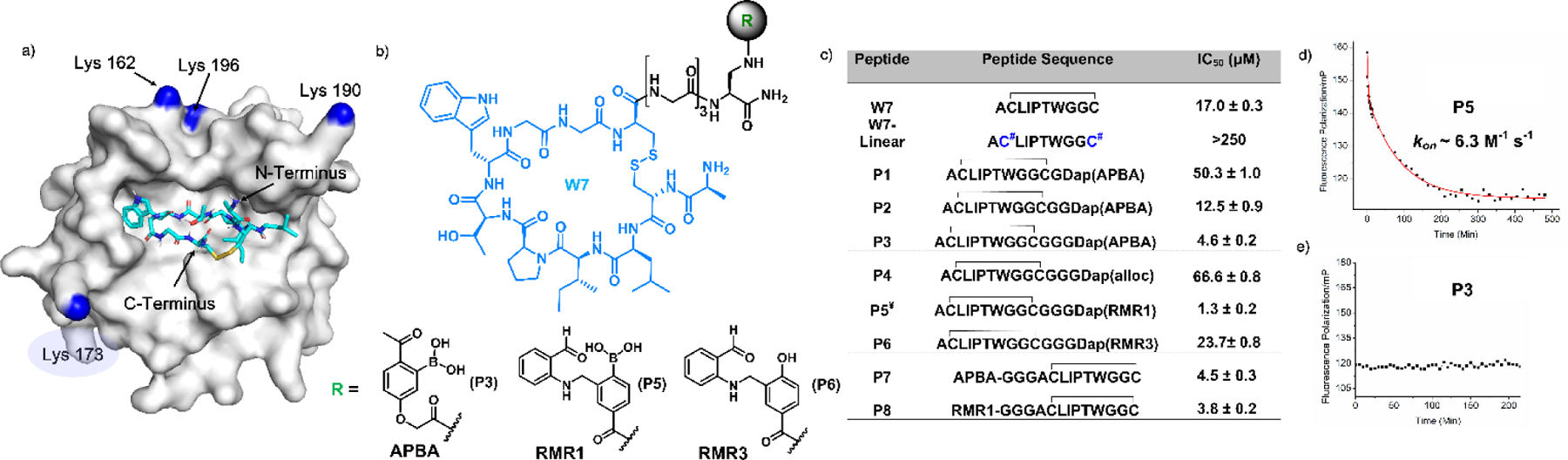
Reversible covalent inhibition of SrtA. (a) SrtA structure with docked W7 (PDB: 1t2w). The peptide’s termini and the proximal lysine residues are labeled with the lysine ε-amines colored blue. K173 (in light blue shade) was found to be the conjugation site for P5. (b) Chemical structure of W7 and the warheads for its modification. (c) IC50 values of various peptides measured through competition against W7-F. C# represent iodoacetamide-alkylated cysteines. ¥P5 was N-acetylated to prevent RMR1 conjugation with the N-terminus. (d, e) SrtA binding kinetics of P3 and P5 recorded via a competition assay using W7-F as a fluorescence reporter.
For peptide conjugation, we synthesized a -COOH derivative of RMR1 and 2-APBA respectively (Scheme S5-S10). We did not explicitly study 2-FPBA due to its similarity to 2-APBA in iminoboronate formation (Figure S9-S10). These -COOH derivatives were either conjugated directly to the W7 N-terminus or to the C-terminus via an orthogonally protected diaminopropionic acid (Dap) residue (Figure 4b). The resulting peptides were assessed for SrtA binding through a fluorescence polarization-based competition assay, in which a fluorophore labeled W7 (W7-F) was used as a reporter. Fitting the titration curves yielded the IC50 values (Figure 4c, S24), which can be approximated as Kd’s as the reporter peptide was used at much lower concentrations (0.2 µM). Interestingly, RMR1 installed on the C-terminus was found to undergo slow intramolecular conjugation with the N-terminal amine (Figure S25), and hence, the peptide P5 was N-acetylated to avoid cyclization (Figure S26). Comparative studies of W7 and P3 shows that N-acetylation has little effect on their SrtA binding potencies (Figure S27).
The unmodified W7 (no warhead) gave an IC50 of 17 µM. Installing Dap(APBA) onto the C-terminus with a single glycine spacer (P1) diminished the SrtA-binding potency to 50 µM. Interestingly, extending the spacer length regained the potency, yielding an IC50 of 12.5 and 4.6 µM for P2 and P3 respectively. The control peptide P4 afforded a 15-fold higher IC50 than P3, highlighting the importance of the APBA warhead. Replacing the APBA warhead with RMR1 (P5) gave even greater potency with an IC50 of 1.3 µM. Remarkably, P5 is 18 times more potent than the control peptide P6, which incorporates RMR3 as a non-reactive RMR1 analogue. Conjugating APBA or RMR1 onto the N-terminus of W7 significantly improves the peptide’s binding to SrtA as well, yielding an IC50 of 4.5 and 3.8 µM for P7 and P8 respectively. Finally, the postulated covalent binding of P5 to SrtA was confirmed by LC-MS (Figure S28). To pinpoint the site of conjugation, we performed peptide mapping experiments, in which the SrtA-P5 conjugate was subjected to trypsin digestion and then LC-MS analysis (Figure S29). The results allowed identification of K173 as the conjugation site for P5, consistent with the fact that K173 exhibits the shortest distance to the peptide’s C-terminus as seen in the results of the docking studies (Figure S23).
Kinetic characterization of the P5-SrtA binding (Figure 4d) yielded a kon of 6.3 M−1 s−1, which is ~300 times faster than the RMR1-lysine conjugation. The koff was determined to be 8.2×10−6 s−1, 4 times slower than the RMR1-lysine conjugation. The faster on-rate and slower off-rate are expected for the cooperative action between the covalent warhead and the W7 peptide that binds SrtA noncovalently. In contrast to P5, the P3-SrtA binding reached equilibrium instantaneously (Figure 4e), consistent with the rapid kinetics of iminoboronate formation.11,21
We further tested the peptides for SrtA inhibition on live S. aureus cells. A fluorescently labeled SrtA substrate (FAM-GSLPETGGS) was mixed with S. aureus in LB media with or without a peptide inhibitor (Figure 5a). The SrtA-mediated bacterial labeling was assessed using fluorescence microscopy and flow cytometry. Without an inhibitor, the bacterial cells exhibited strong fluorescence staining after incubation with the SrtA substrate (Figure 5b) The peptide inhibitors inhibited SrtA-mediated fluorescence labeling of the cells in a concentration-dependent manner (Figure 5c). Curve fitting yielded an IC50 of 10.7 µM for W7, which is on par with the IC50 determined from enzyme binding studies. Also consistent with the earlier results, both P3 and P5 exhibited higher potency of inhibition, yielding IC50 of 3.7 and 2.9 µM respectively.
Figure 5.
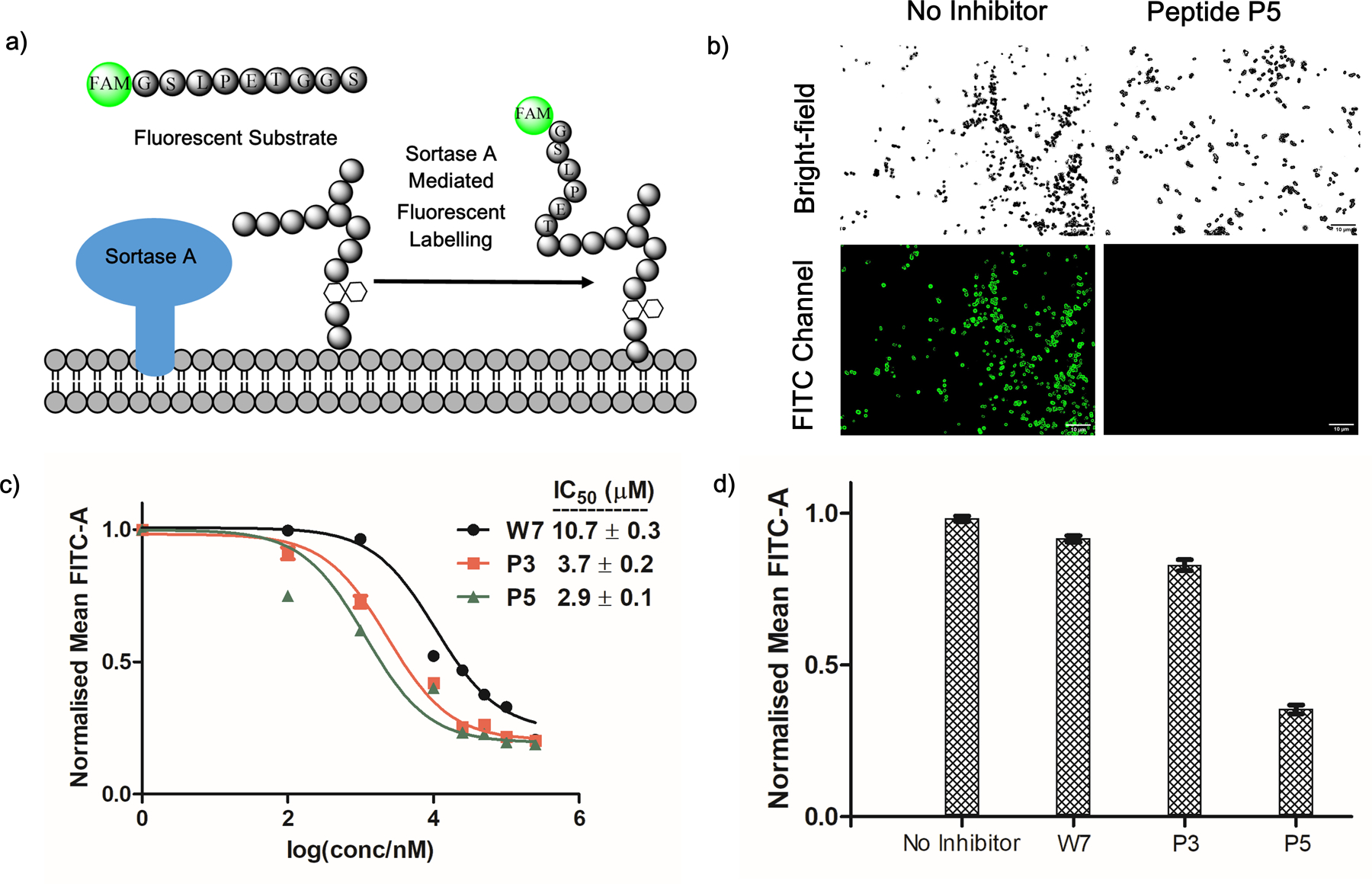
SrtA inhibition on live S. aureus cells. (a) Schematic representation of SrtA-mediated fluorescence labeling of S. aureus cells. (b) Fluorescence microscopy images showing SrtA inhibition by peptide P5 on live cells, leading to diminished cell staining. P5 concentration, 250 µM; Scale bar, 10 µm. (c) Concentration profiles of W7, P3 and P5 for SrtA inhibition showing the enhanced potency resulting from the reversible covalent warheads. (d) Comparative studies showing that, after washing out the unbound inhibitors, the covalently bound P5 sustained SrtA inhibition over the time course of 6 hours. In contrast, marginal inhibition was observed for W7 and P3 under the same experimental conditions.
Although a similar IC50 was observed for P3 and P5 in our SrtA inhibition assay, we reasoned that the slow dissociation of P5 could afford kinetic benefit, which manifests as longer residence time in the enzyme leading to long-lasting inhibition. To test this hypothesis, we treated the S. aureus cells with P5 and then the unbound inhibitor was washed away before the fluorescent SrtA substrate was added. The S. aureus cells were subjected to flow cytometry analysis after 6 hours of incubation. The results (Figure 5d) show that, even with washing, P5 efficiently inhibited the SrtA-mediated fluorescence labeling of S. aureus. In sharp contrast, W7 and P3 only elicited marginal reduction of the fluorescence of the cells. These results nicely demonstrate the long-lasting SrtA inhibition by P5, which presumably results from its slow dissociation kinetics.
In summary, this contribution reports a novel lysine conjugation chemistry, in which a warhead RMR1 reversibly conjugates with lysines to give a hydrated diazaborine. In contrast to the iminoboronate chemistry, a known lysine conjugation with fast dissociation kinetics, the RMR1-induced lysine conjugation shows a much-slowed reverse reaction, with dissociation happening on the time scale of hours. Using the Staphylococcal sortase as a model system, we show that RMR1 can be grafted onto a peptide scaffold to create potent reversible covalent inhibitors, the efficacy of which is demonstrated against the recombinant protein as well as on live bacterial cells. Importantly, a RMR1-bearing inhibitor affords long-lasting SrtA inhibition hours after the inhibitor clearance, which presents the first demonstration of the kinetic benefit of lysine-targeted reversible covalent inhibitors. We envision the RMR1 warhead can be tuned to give a wide range of kinetic profiles for reversible lysine conjugation, which will greatly expand the warhead repertoire for the creation of lysine-targeted reversible covalent inhibitors. Work towards this end is currently underway.
Supplementary Material
ACKNOWLEDGMENT
We thank Professor Jia Niu and Ms. Qiwen Su for help with flow cytometry analysis, and Dr. Bo Li for solving the crystal structure of the diazaborine conjugate.
Funding Sources
National Institutes of Health (GM102735), National Science Foundation (CHE-1904874) and the Ono Pharma Foundation.
Footnotes
ASSOCIATED CONTENT
Details of synthesis and characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Bandyopadhyay A; Gao J “Targeting biomolecules with reversible covalent chemistry” Curr. Opin. Chem. Biol 2016, 34, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Guo W-H; Qi X; Yu X; Liu Y; Chung C-I; Bai F; Lin X; Lu D; Wang L; Chen J; Su LH; Nomie KJ; Li F; Wang MC; Shu X; Onuchic JN; Woyach JA; Wang ML; Wang J “Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry” Nat. Commun 2020, 11, 4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fairhurst RA; Knoepfel T; Buschmann N; Leblanc C; Mah R; Todorov M; Nimsgern P; Ripoche S; Niklaus M; Warin N; Luu VH; Madoerin M; Wirth J; Graus-Porta D; Weiss A; Kiffe M; Wartmann M; Kinyamu-Akunda J; Sterker D; Stamm C; Adler F; Buhles A; Schadt H; Couttet P; Blank J; Galuba I; Trappe J; Voshol J; Ostermann N; Zou C; Berghausen J; Del Rio Espinola A; Jahnke W; Furet P “Discovery of Roblitinib (FGF401) as a Reversible-Covalent Inhibitor of the Kinase Activity of Fibroblast Growth Factor Receptor 4” J. Med. Chem 2020, 63, 12542–12573. [DOI] [PubMed] [Google Scholar]
- (4).Gabizon R; Shraga A; Gehrtz P; Livnah E; Shorer Y; Gurwicz N; Avram L; Unger T; Aharoni H; Albeck S; Brandis A; Shulman Z; Katz B-Z; Herishanu Y; London N “Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs” J. Am. Chem. Soc 2020, 142, 11734–11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Serafimova IM; Pufall MA; Krishnan S; Duda K; Cohen MS; Maglathlin RL; McFarland JM; Miller RM; Frodin M; Taunton J “Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles” Nat. Chem. Biol 2012, 8, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Krishnan S; Miller RM; Tian B; Mullins RD; Jacobson MP; Taunton J “Design of reversible, cysteine-targeted Michael acceptors guided by kinetic and computational analysis” J. Am. Chem. Soc 2014, 136, 12624–12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bradshaw JM; McFarland JM; Paavilainen VO; Bisconte A; Tam D; Phan VT; Romanov S; Finkle D; Shu J; Patel V; Ton T; Li X; Loughhead DG; Nunn PA; Karr DE; Gerritsen ME; Funk JO; Owens TD; Verner E; Brameld KA; Hill RJ; Goldstein DM; Taunton J “Prolonged and tunable residence time using reversible covalent kinase inhibitors” Nat. Chem. Biol 2015, 11, 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Oksenberg D; Dufu K; Patel MP; Chuang C; Li Z; Xu Q; Silva-Garcia A; Zhou C; Hutchaleelaha A; Patskovska L; Patskovsky Y; Almo SC; Sinha U; Metcalf BW; Archer DR “GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease” Br. J. Haematol 2016, 175, 141–153. [DOI] [PubMed] [Google Scholar]
- (9).Akcay G; Belmonte MA; Aquila B; Chuaqui C; Hird AW; Lamb ML; Rawlins PB; Su N; Tentarelli S; Grimster NP; Su Q “Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain” Nat. Chem. Biol 2016, 12, 931–936. [DOI] [PubMed] [Google Scholar]
- (10).Dal Corso A; Catalano M; Schmid A; Scheuermann J; Neri D “Affinity Enhancement of Protein Ligands by Reversible Covalent Modification of Neighboring Lysine Residues” Angew. Chem. Int. Ed. Engl 2018, 57, 17178–17182. [DOI] [PubMed] [Google Scholar]
- (11).Cal PM; Vicente JB; Pires E; Coelho AV; Veiros LF; Cordeiro C; Gois PM “Iminoboronates: A New Strategy for Reversible Protein Modification” J. Am. Chem. Soc 2012, 134, 10299–10305. [DOI] [PubMed] [Google Scholar]
- (12).Bandyopadhyay A; McCarthy KA; Kelly MA; Gao J “Targeting bacteria via iminoboronate chemistry of amine-presenting lipids” Nat. Commun 2015, 6, 6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).McCarthy KA; Kelly MA; Li K; Cambray S; Hosseini AS; van Opijnen T; Gao J “Phage Display of Dynamic Covalent Binding Motifs Enables Facile Development of Targeted Antibiotics” J. Am. Chem. Soc 2018, 140, 6137–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Quach D; Tang G; Anantharajan J; Baburajendran N; Poulsen A; Wee J; Retna P; Li R; Liu B; Tee D; Kwek P; Joy J; Yang W-Q; Zhang C-J; Foo K; Keller T; Yao SQ “Strategic Design of Catalytic Lysine-Targeting Reversible Covalent BCR-ABL Inhibitors” Angew. Chem. Int. Ed. Engl 2021, 60, 17131–17137. [DOI] [PubMed] [Google Scholar]
- (15).Cambray S; Bandyopadhyay A; Gao J “Fluorogenic diazaborine formation of semicarbazide with designed coumarin derivatives” Chem. Commun 2017, 53, 12532–12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gu H; Chio TI; Lei Z; Staples RJ; Hirschi JS; Bane S “Formation of hydrazones and stabilized boron-nitrogen heterocycles in aqueous solution from carbohydrazides and ortho-formylphenylboronic acids” Org. Biomol. Chem 2017, 15, 7543–7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Stress CJ; Schmidt PJ; Gillingham DG “Comparison of boron-assisted oxime and hydrazone formations leads to the discovery of a fluorogenic variant” Org. Biomol. Chem 2016, 14, 5529–5533. [DOI] [PubMed] [Google Scholar]
- (18).António JPM; Goncalves LM; Guedes RC; Moreira R; Gois PMP “ Diazaborines as New Inhibitors of Human Neutrophil Elastase “ ACS Omega 2018, 3, 7418–7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Montalbano F; Cal PMSD; Carvalho MABR; Gonçalves LM; Lucas SD; Guedes RC; Veiros LF; Moreira R; Gois PMP “Discovery of new heterocycles with activity against human neutrophile elastase based on a boron promoted one-pot assembly reaction’’ Org. Biomol. Chem 2013, 11, 4465–4472. [DOI] [PubMed] [Google Scholar]
- (20).Lopes RMRM; Ventura AE; Silva LC; Faustino H; Gois PMP “ N, O-Iminoboronates: Reversible Iminoboronates with Improved Stability for Cancer Cells Targeted Delivery” Chem. Eur. J 2018, 24, 12495–12499 [DOI] [PubMed] [Google Scholar]
- (21).Bandyopadhyay A; Gao J “Iminoboronate Formation Leads to Fast and Reversible Conjugation Chemistry of alpha-Nucleophiles at Neutral pH” Chem. Eur. J 2015, 21, 14748–14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Collins BE; Sorey S; Hargrove AE; Shabbir SH; Lynch VM; Anslyn EV “Probing Intramolecular B−N Interactions in Ortho-Aminomethyl Arylboronic Acids” J. Org. Chem 2009, 74, 4055–4060. [DOI] [PubMed] [Google Scholar]
- (23).Bandyopadhyay A; Gao J “Iminoboronate-Based Peptide Cyclization That Responds to pH, Oxidation, and Small Molecule Modulators” J. Am. Chem. Soc 2016, 138, 2098–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Cascioferro S; Raffa D; Maggio B; Raimondi MV; Schillaci D; Daidone G “Sortase A Inhibitors: Recent Advances and Future Perspectives” J. Med. Chem 2015, 58, 9108–9123. [DOI] [PubMed] [Google Scholar]
- (25).Zhang J; Liu H; Zhu K; Gong S; Dramsi S; Wang Y-T; Li J; Chen F; Zhang R; Zhou L; Lan L; Jiang H; Schneewind O; Luo C; Yang C-G “Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase” Proc. Natl. Acad. Sci. U.S.A 2014, 111, 13517–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.