

Abstract
Proteolysis Targeting Chimeras (PROTACs), heterobifunctional compounds that consist of protein-targeting ligands linked to an E3 ligase recruiter, have arisen as a powerful therapeutic modality for targeted protein degradation (TPD). Despite the popularity of TPD approaches in drug discovery, only a small number of E3 ligase recruiters are available for the >600 E3 ligases that exist in human cells. Here, we have discovered a cysteine-reactive covalent ligand, EN106, that targets FEM1B, an E3 ligase recently discovered as the critical component of the cellular response to reductive stress. By targeting C186 in FEM1B, EN106 disrupts recognition of the key reductive stress substrate of FEM1B, FNIP1. We further establish that EN106 can be used as a covalent recruiter for FEM1B in TPD applications by demonstrating that a PROTAC linking EN106 to the BET Bromodomain inhibitor JQ1 or the kinase inhibitor dasatinib leads to the degradation of BRD4 and BCR-ABL, respectively. Our study showcases a covalent ligand that targets a natural E3 ligase-substrate binding site and highlights the utility of covalent ligand screening in expanding the arsenal of E3 ligase recruiters suitable for TPD applications.
Keywords: activity-based protein profiling, targeted protein degradation, Proteolysis Targeting Chimeras, PROTAC, cysteine, covalent ligand, chemoproteomics, induced proximity, FEM1B, FNIP1, E3 ligase
Graphical Abstract
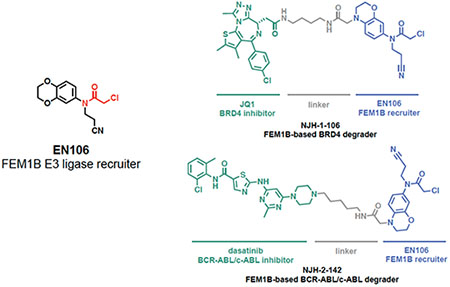
INTRODUCTION
Targeted protein degradation (TPD) is based on molecular glues and heterobifunctional compounds, which induce the proximity of E3 ubiquitin ligases to target proteins of interest and thereby elicit their ubiquitylation and proteasome-mediated degradation. TPD has become a sought-after therapeutic modality in drug discovery because of its potential ability to specifically eliminate any disease-causing protein in the cell, including classically undruggable targets 1. However, a major bottleneck with TPD platforms is the relatively small number of E3 ligase recruiters that are available, despite the >600 E3 ligases that exist in human cells. In fact, most efficient E3 ligase recruiters are based on thalidomide-type immunomodulatory drugs (IMiD) that attract the CUL4 adaptor Cereblon or ligands that recruit the CUL2 adaptor VHL 2–5.
Indications that chemoproteomics-enabled covalent ligand discovery platforms could be used to discover novel covalent E3 ligase recruiters for PROTAC applications were initially revealed by Backus et al. demonstrating the ligandability of E3 ligases with cysteine-reactive covalent ligands 6. This discovery enabled the development of bifunctional molecules containing electrophiles that react with the E3 ligases DCAF16, RNF4, and RNF114, allowing them to be recruited to model proteins and trigger their degradation 7–9. Since these initial proof-of-concept studies, similar strategies have been to discover and apply additional covalent ligands that recruit RNF114 and DCAF11 as protein degradation triggers 10,11. Overall, this previous work has highlighted the utility of covalent PROTACs that irreversibly bind the E3 ligase 7–10 and has showcased how chemoproteomic approaches and cysteine-targeting ligands can be used to expand the arsenal of E3 ligase recruiters for TPD applications.
The reactivity of cysteine residues in cells is maintained by specific signaling pathways that are often centered on E3 ligases and maintain the cellular redox state. Under optimal redox balance, the CUL3 E3 ligase Kelch-like ECH associated protein (KEAP1) sequesters the nuclear transcription factor NRF2 in the cytosol to induce its ubiquitylation and proteasome-mediated degradation. Under conditions of oxidative stress, redox-sensing cysteines on KEAP1 become oxidized to prevent KEAP1-mediated ubiquitylation and degradation of NRF2, subsequently allowing NRF2 accumulation and antioxidant gene expression.
Recently, the CUL2 E3 ligase FEM1B was discovered as a critical regulator of the cellular response to persistent depletion of reactive oxygen species (ROS), a condition referred to as reductive stress 12. Reductive stress arises from prolonged antioxidant signaling or mitochondrial inactivity and can block stem cell differentiation or lead to diseases, such as cardiomyopathy, diabetes, or cancer 13. Manford et al. discovered that FEM1B recognizes reduced cysteines on its substrate FNIP1 under reductive stress conditions, leading to FEM1B-dependent FNIP1 ubiquitylation and degradation to restore mitochondrial activity, redox homeostasis and stem cell integrity 12. The authors also demonstrated through structural biology studies that a key cysteine residue C186 was critical for FEM1B substrate recognition through a zinc-mediated molecular glue interaction between FNIP1 and the FEM1B C186 12,14. This suggests that one could target C186 with a cysteine-reactive covalent ligand to develop a FEM1B recruiter for TPD applications. In this study, we screened a library of cysteine-reactive covalent ligands to discover a covalent FEM1B recruiter and demonstrated that this recruiter can be incorporated into PROTACs to degrade specific protein targets in cells.
RESULTS AND DISCUSSION
To identify a covalent FEM1B recruiter, we screened a library of 566 cysteine-reactive covalent ligands in a competitive fluorescence polarization assay using a TAMRA-conjugated FNIP1562-591 degron and recombinant mouse FEM1B (Figure 1a–1b, Table S1). Through this screen, we identified the chloroacetamide EN106 that inhibited FEM1B-FNIP1 degron fluorescence polarization with a 50 % inhibitory concentration (IC50) of 2.2 μM (Figure 1c–1d). From the initial screen, EN106 showed the most significant inhibition of FEM1B interactions with the FNIP1 degron (Figure 1b). EN106 showed competition against labeling of FEM1B with a cysteine-reactive rhodamine-conjugated iodoacetamide (IA-rhodamine) probe by gel-based ABPP, confirming a direct interaction of EN106 with a cysteine on FEM1B (Figure 1e). Analysis of EN106 reactivity with recombinant FEM1B by liquid-chromatography-tandem mass spectrometry (LC-MS/MS) analysis of FEM1B tryptic digests revealed an EN106 adduct only on C186—the site that was previously shown to be critical for FEM1B substrate recognition (Figure 1f) 12. We also demonstrated that a non-reactive version of EN106, NJH-2-082, does not inhibit FEM1B interactions with the FNIP1 degron, confirming the importance of the covalent interactions of the cysteine-reactive warhead with C186 (Figure S1).
Figure 1. Discovering a FEM1B recruiter.
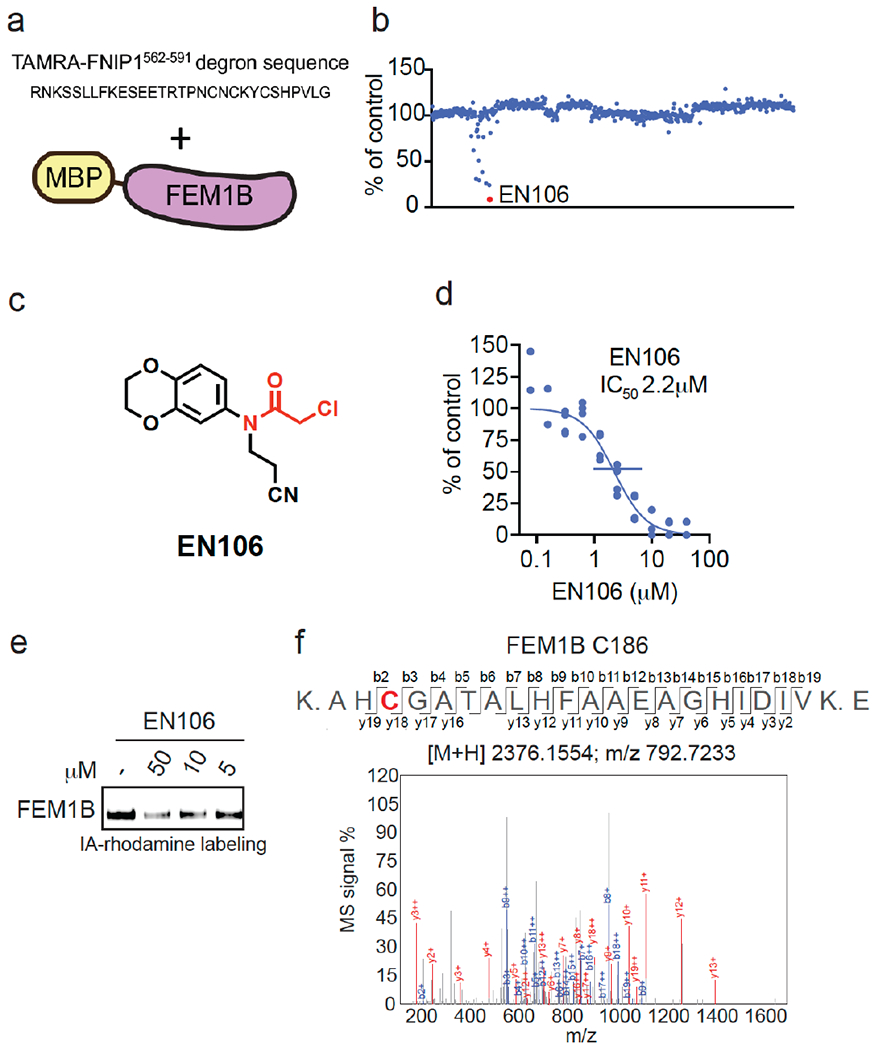
(a, b) Screening a cysteine-reactive covalent ligand library in a fluorescence polarization assay with TAMRA-conjugated FNIP1562-591 degron with recombinant MBP-tagged FEM1B (a). FEM1B was pre-incubated with DMSO vehicle or covalent ligand (50 μM) for 1 h prior to addition of the TAMRA-conjugated degron (b). Data is in Table S1. (c) Structure of EN106 with covalent chloroacetamide handle in red. (d) Dose-response of EN106 inhibition of FEM1B and TAMRA-conjugated FNIP1 interaction assessed by fluorescence polarization expressed as percent fluorescence polarization compared to DMSO control. (e) Gel-based ABPP of EN106. 50 nM pure FEM1B protein was pre-treated with DMSO or EN106 for 30 min at room temperature prior to addition of IA-rhodamine (500 nM, 30 min) at room temperature, after which protein was resolved on SDS/PAGE and visualized by in-gel fluorescence. (f) Site of modification of EN106 on FEM1B. FEM1B was labeled with EN106 (50 μM) for 30 min, and FEM1B tryptic digests were analyzed by LC-MS/MS for the EN106 adduct. Data in (b) show average from n=2/group. Data in (d) shows individual data replicates from n=2-4/group. Data in (e) shows representative gel from n=3/group.
To confirm that EN106 engaged FEM1B in cells, we synthesized NJH-2-030, an alkyne-functionalized derivative of EN106 (Figure 2a). To maintain engagement of C186, the alkyne was positioned distal to the chloroacetamide by exchanging the benzodioxan for a dihydro[1,4]benzoxazine scaffold. The starting benzoxazine was Boc-protected to give 1 before reduction of the nitro group to provide aniline 2. Alkylation of 2 with acrylonitrile provided the propionitrile-substituted compound, which was acylated to obtain the chloroacetamide 3. Boc deprotection and acylation with hex-5-ynoyl chloride provided alkyne probe NJH-2-030 (Figure 2a).
Figure 2. Characterization of EN106 binding with FEM1B.
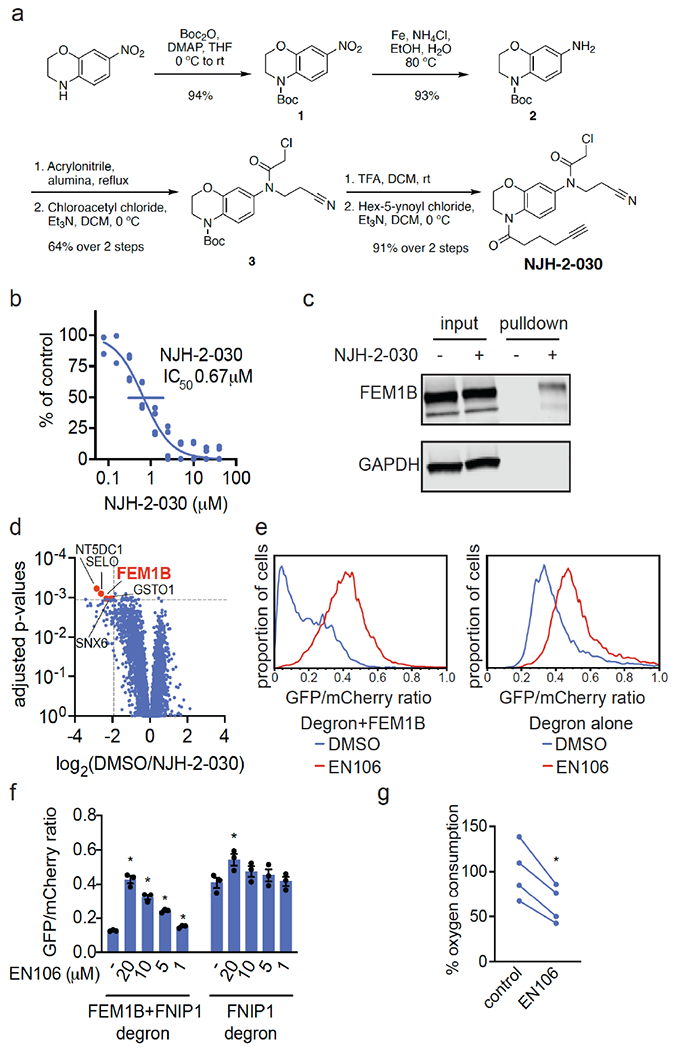
(a) Synthesis of NJH-2-030. (b) Dose-response of NJH-2-030 inhibition of FEM1B and TAMRA-conjugated FNIP1 interaction assessed by fluorescence polarization. (c, d) NJH-2-030 engagement of FEM1B in cells. HEK293T cells were treated with DMSO or NJH-2-030 (10 μM) for 4 h. Cell lysates were subjected to CuAAC with biotin picolyl azide, probe-modified proteins were avidin-enriched, eluted, and analyzed by SDS/PAGE and Western blotting for FEM1B or loading control GAPDH (c) or analyzed by TMT-based quantitative proteomic profiling (d). Data are from n=3/group. Proteins annotated and highlighted in red showed >2-fold enrichment with probe over DMSO with adjusted p-values=0.001 or p-values<0.001. (e, f) Flow cytometry analysis of GFP-FNIP1 degron levels compared to mCherry levels with EN106 treatment in HEK293T cells for 12 h with either basal levels of FEM1B or with transient FEM1B overexpression. Representative flow cytometry traces of DMSO and EN106 (20 μM) treatment groups shown in (e) and quantified data shown in (f). (g) EN106 inhibits oxygen consumption in HEK293T cells. HEK293T cells were treated with DMSO vehicle or EN106 (10 μM) for 16 h after which mitochondrial oxygen consumption was read out with MitoXpress Xtra reagent. Shown in (b, f, g) are individual biological replicate values and/or average ± sem for n=2-4/group. Significance in (f, g) is expressed as *p<0.05 compared to vehicle-treated controls in each group in (f) or compared to each paired control in (g).
NJH-2-030 maintained inhibitory activity against FEM1B recognition of the FNIP1 degron with an IC50 of 0.67 μM (Figure 2b). The improved potency with the amide substituent may indicate additional favorable contacts within the FEM1B substrate recognition domain. The NJH-2-030 probe showed FEM1B engagement in HEK293T cells, as demonstrated by FEM1B enrichment from NJH-2-030 treatment in cells by subsequent appendage of biotin-azide by copper-catalyzed azide-alkyne cycloaddition (CuAAC) in cell lysates, avidin-pulldown, and blotting for FEM1B in HEK293T cells compared to vehicle-treated controls (Figure 2c). An unrelated target GAPDH was not enriched by NJH-2-030 treatment and pulldown (Figure 2c).
To assess the proteome-wide cysteine-reactivity of EN106, we also performed a competitive isotopic tandem orthogonal proteolysis-ABPP (isoTOP-ABPP) study to quantitatively assess proteome-wide cysteine reactivity of EN106 in cells (Figure S2; Table S2). While we did not capture FEM1B in our chemoproteomics experiment, we only observed two targets of EN106 in cells—C63 of HNRNPA3 and C127 of PRDX3—across 1465 quantified cysteines (Figure S2). Neither of these off-targets of EN106 were E3 ligases. Given that we did not detect C186 of FEM1B in isoTOP-ABPP experiments, we also used our NJH-2-030 probe in situ in HEK293T cells to perform pulldown quantitative proteomic experiments to further confirm target engagement and proteome-wide selectivity of this close EN106 derivative (Figure 2d; Table S3). FEM1B was one of the most significantly enriched targets by the NJH-2-030 probe compared to DMSO controls with four additional off-targets detected—GSTO1, SNX6, SELO, and NT5DC1—of which none of these proteins were E3 ligases (Figure 2d; Table S3). These data collectively showed that EN106 or its derivatives functionally engaged FEM1B in cells without detectable off-target effects on other endogenous degradation pathway components.
To further demonstrate that EN106 disrupted substrate recognition by FEM1B in cells, we monitored the degradation of GFP linked to a FNIP1 degron compared to IRES driven expression of mCherry from the same plasmid in HEK293T cells by flow cytometry. EN106 treatment significantly stabilized FNIP1 degron-GFP levels, compared to vehicle-treated controls in a dose-responsive manner in FEM1B overexpressing cells (Figure 2e, f). EN106 increased FNIP1 reporter levels in cells lacking exogenously expressed FEM1B to a similar extent as previously observed upon deletion of FEM1B 12, indicating that this compound can target the endogenous E3 ligase (Figure 2e–2f). EN106 did not affect the pomalidomide-induced degradation of an unrelated E4F1 degron by the E3 ligase cereblon (Figure S3). These findings thus indicate that EN106 not only engages, but also inhibits CUL2FEM1B dependent ubiquitylation. Stabilization of the mitochondrial pool of FNIP1 impairs mitochondrial activity, as being read out by the oxygen consumption rate 12,14. In line with engaging endogenous FEM1B and stabilizing FNIP1, we showed that EN106 significantly reduced cellular mitochondrial oxygen consumption in HEK293T cells (Figure 2g).
To demonstrate that EN106 could be used as a covalent FEM1B recruiter in TPD applications, we next synthesized a series of FEM1B-based BET bromodomain degraders by linking EN106 to the BET bromodomain inhibitor JQ1 that targets BRD4 as well as other BET family proteins via 6 different linkers (Figure 3a, Figure S5). Maintaining the core benzoxazine of the alkyne probe NJH-2-030, we first attached an acetate spacer to provide methyl ester 4. The nitro group was reduced, the resulting aniline 5 mono-alkylated with acrylonitrile, and acylated to provide the chloroacetamide intermediate 6. The methyl ester was hydrolyzed under mild basic conditions and coupled to amines 7a-7f, JQ1 derivatives with different linker attachments, to provide the bifunctional degraders NJH-2-088, NJH-1-106, NJH-2-090, NJH-2-091, NJH-2-092, and NJH-2-093 (scheme for NJH-1-106 shown in Figure 3a; Figure S4). These compounds all degraded BRD4 in HEK293T cells to varying extent with NJH-1-106 showing the best degradation potency with a DC50 of 250 nM and 94 % maximal degradation of BRD4 (Figure 3b–3c, Figure S4). NJH-1-106 maintained inhibitory activity against FEM1B recognition of the FNIP1 degron with an IC50 of 1.5 μM (Figure 3d). This BRD4 degradation was time-dependent with significant degradation observed by 4 h of treatment with NJH-1-106 (Figure 3e–3f). NJH-1-106 also degraded BRD4 in cancer cell lines including the 231MFP breast cancer and HAP1 leukemia cancer cell lines (Figure S5a–S5b).
Figure 3. FEM1B-based BRD4 degrader.
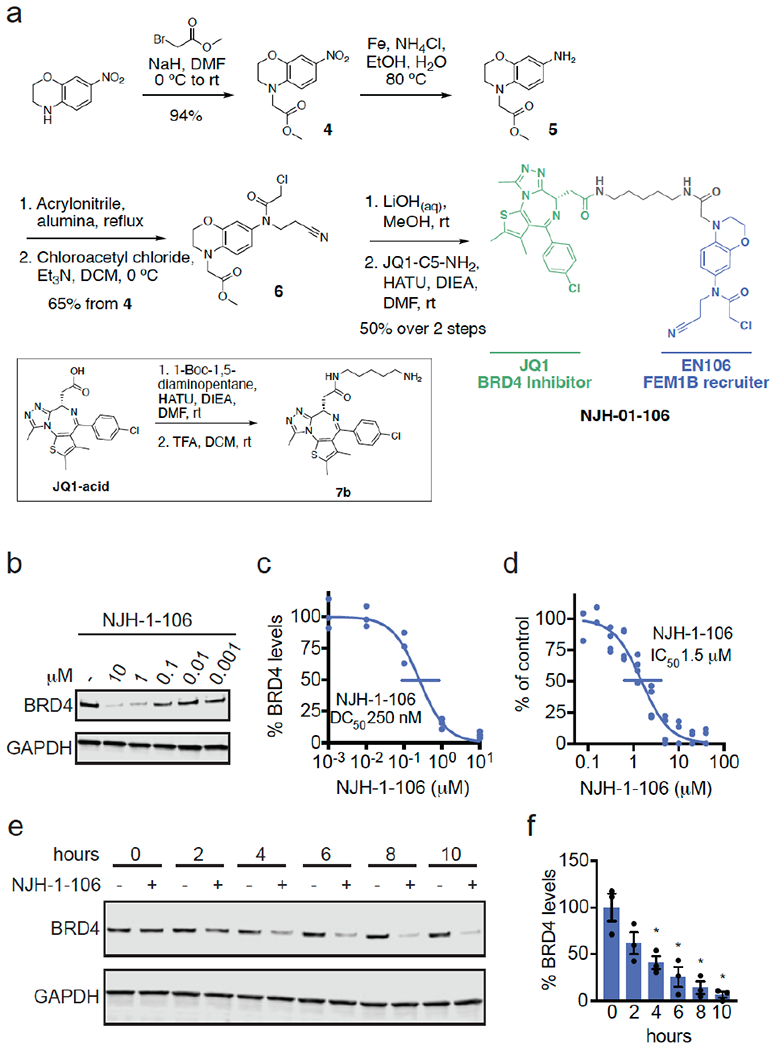
(a) Synthesis of FEM1B-based BRD4 degrader NJH-1-106 linking EN106 to JQ1. (b) Degradation of BRD4 by NJH-1-106. NJH-1-106 was treated in HEK293T cells for 8 h and BRD4 and loading control GAPDH levels were detected by Western blotting. (c) Quantification of BRD4 degradation from experiment in (b) and 50 % degradation concentration value (DC50). Individual biological replicate values shown in (b) from n=2-4/group. Gel shown in (b) is representative of n=3 / group which are shown in (c). (d) Dose-response of NJH-1-106 inhibition of FEM1B and TAMRA-conjugated FNIP1 interaction assessed by fluorescence polarization expressed as percent fluorescence polarization compared to DMSO vehicle-treated control. (e) Time course of BRD4 degradation with NJH-1-106 treatment. HEK293T cells were treated with DMSO vehicle or NJH-1-106 (10 μM) for 0, 2, 4, 6, 8, and 10 h and BRD4 and loading control GAPDH levels were detected by Western blotting. Gel shown is representative of n=3 / group. (f) Quantification of BRD4 degradation shown in bar graph as average ± sem with individual biological replicate points shown from experiment designed in (e). Data shown in (c, d, f) are averages with sem in (f) and individual biological replicate values from n=2-4/group
Consistent with the necessity of the covalent warhead, the non-reactive version of NJH-1-106, NJH-2-105, did not inhibit FEM1B interactions with the FNIP1 degron and did not degrade BRD4 (Figure S6a–S6d). Loss of BRD4 was attenuated by proteasome and NEDDylation inhibitors, consistent with a proteasome- and Cullin E3 ligase-dependent mechanism of BRD4 degradation (Figure 4a–4b). FEM1B levels remained unaltered by NJH-1-106 treatment in HEK293T cells (Figure 4a–4b). BRD4 degradation was also attenuated by pre-treatment of cells with EN106 or JQ1, showing the necessity of the ternary complex and both ends of the molecule to degrade BRD4 (Fig. 4c). In addition, BRD4 degradation was attenuated in FEM1B knockout (KO) cells compared to wild-type (WT) cells, further demonstrating FEM1B-dependent degradation of BRD4 (Fig. 4d). The incomplete rescue we observe in FEM1B KO cells could be due to residual wild-type cells in the FEM1B KO population or due to other potential off-target E3 ligases. Global proteomic profiling in HEK293T cells treated with NJH-1-106 also showed selective degradation of BRD4 amongst 4446 quantified proteins with only the largely uncharacterized PNMAL1 as an apparent off-target (Fig. 4e; Table S4). PNMAL1, however, was a likely false-positive of our proteomic analysis (Figure S7). While JQ1 is a pan BET bromodomain inhibitor, we only observed degradation of BRD4, but not BRD2 or BRD3 (Table S4). However, we cannot rule out that these other bromodomains could be degraded under longer treatments. The observed smaller fold change for BRD4 compared to the Western blot data likely reflect the well-known fold change suppression in TMT-based quantitative proteomics 15.
Figure 4. Characterization of FEM1B-based BRD4 degrader.
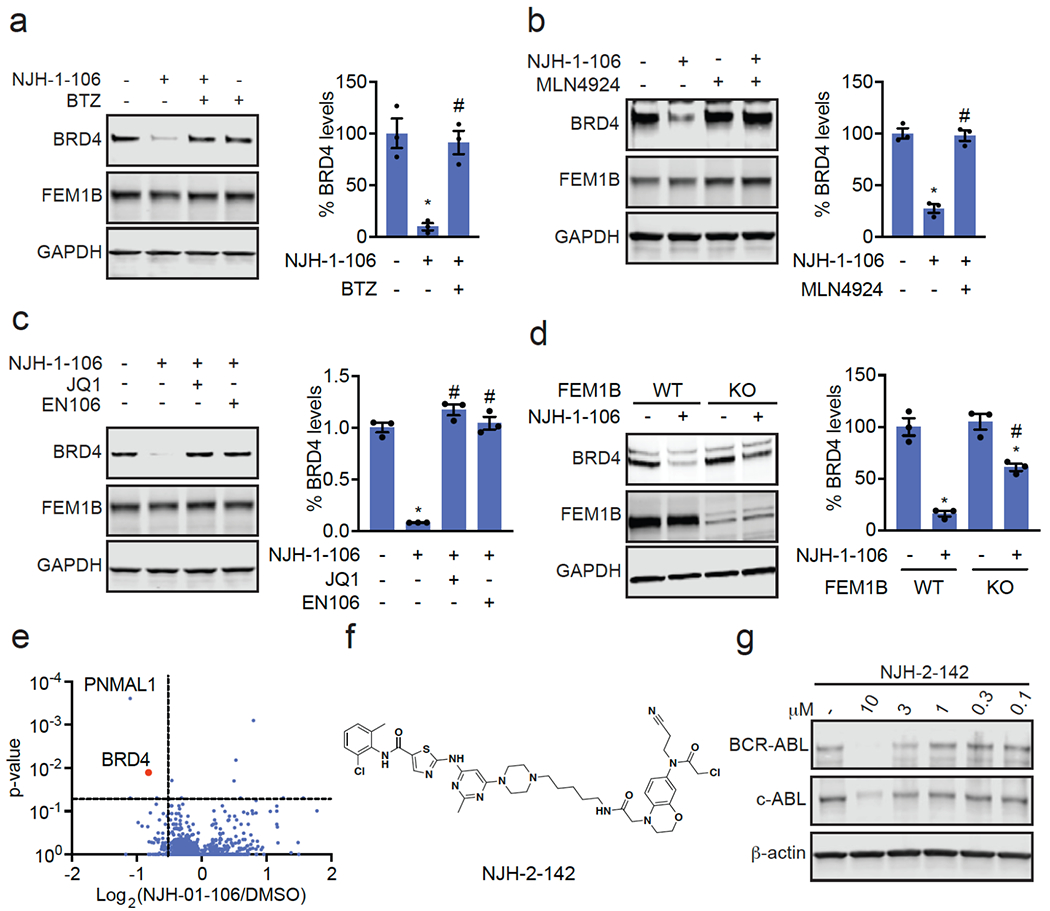
(a, b) Proteasome and NEDDylation-dependence of NJH-1-106-mediated degradation of BRD4. HEK293T cells were pre-treated with DMSO vehicle, proteasome inhibitor bortezomib (BTZ) (1 μM), or NEDDylation inhibitor MLN4924 (0.2 μM) for 2 h prior to treatment with DMSO vehicle, NJH-1-106 (10 μM), or MZ1 (1 μM) for 8 h. (c) Attenuation of BRD4 degradation by EN106 and JQ1. HEK293T cells were pre-treated with DMSO vehicle, EN106 (50 μM), or JQ1 (50 μM) for 2 h prior to treatment with DMSO vehicle or NJH-1-106 (10 μM) for 8 h. (d) BRD4 degradation by NJH-1-106 in FEM1B wild-type (WT) and knockout (KO) HEK293T cells. Cells were treated with DMSO vehicle or NJH-1-106 (1 μM) for 8 h. (e) Proteomic profiling of NJH-1-106 treatment in HEK293T cells. HEK293T cells were treated with DMSO vehicle or NJH-1-106 (1 μM) for 12 h. Protein level changes in cell lysate were quantitatively assessed by TMT-based proteomic profiling. (f) Structure of FEM1B-based BCR-ABL/c-ABL degraders linking EN106 to dasatinib. (g) K562 cells were treated with DMSO vehicle or NJH-2-142 at the designated concentrations for 24 h. shown in (a-e, g) are from n=3 /group. Gels shown in (a-d, g) are representative gels from n=3 /group. Individual replicate and average ± sem values for (a-d) are shown in bar graphs. Significance is shown as *p<0.05 compared to vehicle-treated groups, and #p<0.05 compared to NJH-1-106-treated groups in (a-c) and NJH-1-106-treated WT group in (d).
We further demonstrated that this FEM1B recruiter can be used to degrade additional targets beyond BRD4. A FEM1B-based kinase degrader linking EN106 to dasatinib, an inhibitor of BCR-ABL and c-ABL among other kinases, exhibited degradation of BCR-ABL in K562 leukemia cancer cells (Figure 4f–4g, Figure S8a–S8b). Together, these results document that covalent modification of a cysteine residue in the CUL2 adaptor FEM1B can lead to the development of specific E3 ligase recruiters for targeted protein degradation.
CONCLUSIONS
In this study, we discovered the covalent recruiter EN106 against FEM1B, the E3 ligase controlling the reductive stress response, that can be used for TPD applications. While zinc has been discovered to be a molecular glue that brings together FEM1B with its endogenous substrate FNIP1 14, EN106 represents the first synthetic small-molecule ligand against FEM1B. While EN106 is an early hit tool compound with low micromolar potency against FEM1B with a metabolically unstable chloroacetamide warhead and requires further medicinal chemistry efforts to improve potency, selectivity, and drug-like properties, we demonstrate that EN106 targets a cysteine residue in FEM1B that is essential for substrate recognition. We show that EN106 can be used as a FEM1B recruiter in bifunctional degraders to recruit potential neo-substrates to the physiological target recognition site of FEM1B, likely placing them in an optimal manner for ubiquitylation through CUL2-RBX1. Surprisingly, we observed BRD4 degradation with all of the FEM1B-based BRD4 PROTACs regardless of linker length, although there were preferences for optimal Dmax and DC50s. These data could potentially suggest that positive cooperativity is less crucial with a covalent FEM1B degrader as long as protein clashes are avoided. Future studies will be needed to determine whether EN106 or its derivatives can act as molecular glue degraders to recruit potential neo-substrates for FEM1B-dependent ubiquitylation and degradation. More broadly, it will also be of future interest to determine whether cell-state specific degraders can be developed. Additionally, determining whether EN106 and more potent derivatives can be used therapeutically to inhibit CUL2FEM1B and disrupt reductive stress signaling through stabilization on FNIP1 in certain cancer settings would be of future interest. Overall, our study underscores the utility of covalent ligand screening in expanding the scope of E3 ligase recruiters for TPD applications.
EXPERIMENTAL SECTION
Materials
Cysteine-reactive covalent ligand libraries were purchased from Enamine. Cysteine-reactive covalent ligand libraries are stored as 50 mM DMSO stock solutions and arrayed into single-use plates for screening. Compound integrity of our stock solutions have been confirmed through spot-checking, and degraded compounds have been replaced with repurchased material, Primary antibodies used were: BRD4 (Cell Signaling Technologies (CST) #13440), GAPDH (ProteinTech 60004-1-Ig), FEM1B (ProteinTech 19544-1-AP), b-Actin (CST 8H10D10 #3700), c-Abl (CST #2862).
Fluorescence polarization assay
Fluorescence polarization assays were performed with purified mouse MBP-FEM1B 12, TAMRA-labeled FNIP1 peptide (5,6-TAMRA-RNKSSLLFKESEETRTPNCNCKYCSHPVLG, Koch Institute/MIT Biopolymers lab). For the screen, 0.5 μl of 2.5 mM compounds were spotted into 384 well non-binding plates (Greiner, 781900). 12.5 μl of 250 nM MBP-FEM1B in binding buffer (40 mM HEPES 7.5, 150 mM NaCl, 0.2 % NP40 substitute, and 100μM TCEP (Tris(2-carboxyethyl)phosphine hydrochloride)) was added to each well and incubated for 1 hour at room temperature. After the incubation, 12.5 μl of 100 nM FNIP1 peptide diluted in binding buffer was added bringing the final concentration to 50 nM for the peptide and 125 nM for MBP-FEM1B. After 1 hour of incubation plates were measured on a Perkin Elmer 2104 Envision plate reader. Data was calculated from mP values (1000*(S-G*P)/(S+G*P), S = 595s channel 2 and P = 595p channel 1, G=1.1) subtracted from peptide only plate. Dose response assays were performed as above, but with 250 nM MBP-FEM1B treated with indicated concentrations of compound (relative to the final reaction volume) or DMSO in separate tubes for 1 hour at room temperature. 12.5 μl of treated MBP-FEM1B (125 nM final) was then added to 12.5 μl of peptide (10 nM final) and incubated with gentle rocking for 30 minutes before measuring fluorescence polarization.
Gel-Based ABPP
Recombinant MBP-FEM1B1-377 (0.1μg/sample) was pre-treated with either DMSO vehicle or EN106 or at 37°C for 30 min in 25 μL of PBS, and subsequently treated with of IA-Rhodamine (concentrations designated in figure legends) (Setareh Biotech) at room temperature for 1 h. The reaction was stopped by addition of 4×reducing Laemmli SDS sample loading buffer (Alfa Aesar). After boiling at 95°C for 5 min, the samples were separated on precast 4–20% Criterion TGX gels (Bio-Rad). Probe-labeled proteins were analyzed by in-gel fluorescence using a ChemiDoc MP (Bio-Rad).
Cell Culture
HEK293T, K562, and HAP1 cells were obtained from the UC Berkeley Cell Culture Facility and cultured in DMEM (Gibco) containing 10% (v/v) fetal bovine serum (FBS) and maintained at 37 °C with 5% CO2. 231MFP cells were obtained from Benjamin Cravatt and were generated from explanted tumor xenografts of MDA-MB-231 cells as previously described 16. The FEM1B knockout HEK293T cell line was generated as described by Manford et al 12.
Oxygen consumption measurements
HEK293T cells were plated into two black 96 well clear bottom plates at 100,000 cells per well in 200 μl of DMEM 10% Fetal Bovine Serum. 8 hours after plating cells were treated as indicated for 16 hours. After the 16 hours, the media was changed 3 x with fresh media leaving a final volume 90 μl. The cells were equilibrated back to 37°C for 15min, after which 10 μl of prewarmed MitoXpress Xtra reagent (MX-200-4, Agilent) was added to each well. Prewarmed mineral oil was quickly layered on top of all analysis wells and the plate was measured over time using Perkin Elmer 2104 Envision plate reader at 37°C using time-resolved fluorescence measurement. 5-6 wells for each condition were analyzed (occasional wells with negative slopes were omitted) and the average rate (RFU/hour) was normalized to the cell count of three wells from the second 96 well plate.
NJH-2-030 Pulldown and Blotting for FEM1B or for TMT-based Quantitative Proteomics
HEK293T cells were treated with DMSO or 10 μM NJH-2-030 in situ 8h. Cells were harvested and lysed by sonication. Pulldown experiments for blotting and TMT-based quantitative proteomic experiments were performed as described previously17.
Flow Cytometry Analysis of GFP-FNIP1 degron/mCherry
HEK293T cells/well were seeded into 6 well plates. The next day the cells were transfected with 0.1 μg of pCS2-GFP-FNIP1562-591-IRES-mCherry or 0.1 μg of pCS2-E4F123432-GFP-IRES-mCherry, with 0.075 μg pCS2-3xFLAG-FEM1B as indicated. Empty pCS2 was added to 2 μg total DNA for each transfection in 300 μl Opti-MEM (Thermo Fisher, 31985-070) with 12 μg polyethyleneimine (PEI, Polysciences 23966-1). Each well was transfected with 65 μl of the transfection mix. 12 hours post-transfection, indicated concentrations of EN106 or DMSO was added. After 12 hours of EN106 treatment, cells were trypsinized, spun down, resuspended in DMEM + 10% FBS and analyzed on Fortessa X20. Data was processed using FlowJo and all quantifications are the median GFP/mCherry ratios. For the pomalidomide treated cells, 10 μM pomalidomide (Med-ChemExpress, HY-10984) was added for 4 hours before analyzing.
Cell Lysis Protocol
Pelleted cells were lysed with RIPA lysis buffer (50mM Tris-HCl, 165mM NaCl, 12mM sodium deoxycholate, 1% Triton X-100, 0.01% SDS), protein concentration normalized using a BCA assay (Thermo).
Western Blot Protocol
Proteins were resolved by SDS–PAGE (4–20% TGX gels, Bio-Rad Laboratories, Inc.) and transferred to nitrocellulose membranes using the Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked with 5% BSA in Tris-buffered saline containing Tween 20 (TBST) solution for 1 h at room temperature, washed in TBST and probed with primary antibody diluted in diluent, as recommended by the various manufacturers, overnight at 4 °C. Primary antibodies used were: BRD4 (Cell Signaling Technologies #13440), GAPDH (ProteinTech 60004-1-Ig), FEM1B (ProteinTech 19544-1-AP). Following washes with TBST, the blots were incubated in the dark with secondary antibodies purchased from Li-Cor Biosciences and used at 1:10,000 dilution in 5% BSA in TBST at room temperature for 1 h. Blots were visualized using an Odyssey Li-Cor scanner after additional washes. Protein intensity was quantified using ImageJ software.
IsoTOP-ABPP Chemoproteomic Experiments
Quantitative TMT Proteomics Analysis
Quantitative TMT-based proteomic analysis of protein level changes were 19.
Supplementary Material
Table S1. Structures of screened covalent ligands
Table S2. isoTOP-ABPP data
Table S3. NJH-2-030 pulldown quantitative proteomics data
Table S4. Quantitative proteomic profiling of NJH-01-106
ACKNOWLEDGMENT
We thank the members of the Nomura and Rape labs for critical reading of the manuscript. We want to thank Durga Kolla for the E4F1 degron reporter construct. We also want to thank Eddie Wehri and the Henry Wheeler Center for Emerging and Neglected Diseases (CEND) UC Berkeley Drug Discovery Center for providing assistance and equipment for fluorescence polarization assay and screen. We also want to extend our gratitude to the UC Berkeley Cancer Research Laboratory Flow Cytometry Facility. We thank Drs. Hasan Celik, Alicia Lund, and UC Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance.
FUNDING SOURCES
This work was supported by Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies (NB-CPACT for NJH, SNB, JNS, JMK, JT, MS, EK, DKN), the Mark Foundation for Cancer Research (ASPIRE Award for DKN, NJH, JNS, EZ), the National Institutes of Health (R01CA240981 for DKN), and Howard Hughes Medical Institute (for AGM, MR). MR is an investigator of the Howard Hughes Medical Institute. Instruments in the College of Chemistry-NMR Facility were supported in part by NIH S10OD024998.
COMPETING FINANCIAL INTEREST STATEMENT
JAT, JMK, MS, SMB are employees of Novartis Institutes for BioMedical Research. This study was funded by the Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies. MR is a co-founder, shareholder, and adviser for Nurix Therapeutics, a member of the scientific advisory board of Monte Rosa Therapeutics, and iPartner for The Column Group. DKN is a co-founder, shareholder, and adviser for Frontier Medicines and Vicinitas Therapeutics. DKN is also on the scientific advisory board of the Mark Foundation for Cancer Research and Photys Therapeutics.
Footnotes
Supporting Information
The Supporting Information is available free of charge at:
Supporting Table Legends; Supporting Figures; Synthetic Characterization and Methods;
Supporting Figures
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- (1).Burslem GM; Crews CM Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181 (1), 102–114. 10.1016/jxell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Burslem GM; Crews CM Small-Molecule Modulation of Protein Homeostasis. Chem. Rev 2017, 117 (17), 11269–11301. 10.1021/acs.chemrev.7b00077. [DOI] [PubMed] [Google Scholar]
- (3).Lai AC; Crews CM Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discov 2017, 16 (2), 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bondeson DP; Mares A; Smith IED; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol 2015, 11 (8), 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE DRUG DEVELOPMENT. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348 (6241), 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Backus KM; Correia BE; Lum KM; Forli S; Horning BD; González-Páez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF Proteome-Wide Covalent Ligand Discovery in Native Biological Systems. Nature 2016, 534 (7608), 570–574. 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang X; Crowley VM; Wucherpfennig TG; Dix MM; Cravatt BF Electrophilic PROTACs That Degrade Nuclear Proteins by Engaging DCAF16. Nat. Chem. Biol 2019, 15 (7), 737–746. 10.1038/s41589-019-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Spradlin JN; Hu X; Ward CC; Brittain SM; Jones MD; Ou L; To M; Proudfoot A; Ornelas E; Woldegiorgis M; Olzmann JA; Bussiere DE; Thomas JR; Tallarico JA; McKenna JM; Schirle M; Maimone TJ; Nomura DK Harnessing the Anti-Cancer Natural Product Nimbolide for Targeted Protein Degradation. Nat. Chem. Biol 2019, 15 (7), 747–755. 10.1038/s41589-019-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ward CC; Kleinman JI; Brittain SM; Lee PS; Chung CYS; Kim K; Petri Y; Thomas JR; Tallarico JA; McKenna JM; Schirle M; Nomura DK Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol 2019, 14 (11), 2430–2440. 10.1021/acschembio.8b01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang X; Luukkonen LM; Eissler CL; Crowley VM; Yamashita Y; Schafroth MA; Kikuchi S; Weinstein DS; Symons KT; Nordin BE; Rodriguez JL; Wucherpfennig TG; Bauer LG; Dix MM; Stamos D; Kinsella TM; Simon GM; Baltgalvis KA; Cravatt BF DCAF11 Supports Targeted Protein Degradation by Electrophilic Proteolysis-Targeting Chimeras. J. Am. Chem. Soc 2021, 143 (13), 5141–5149. 10.1021/jacs.1c00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Luo M; Spradlin JN; Boike L; Tong B; Brittain SM; McKenna JM; Tallarico JA; Schirle M; Maimone TJ; Nomura DK Chemoproteomics-Enabled Discovery of Covalent RNF114-Based Degraders That Mimic Natural Product Function. Cell Chem. Biol 2021, 28 (4), 559–566.e15. 10.1016/j.chembiol.2021.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Manford AG; Rodríguez-Pérez F; Shih KY; Shi Z; Berdan CA; Choe M; Titov DV; Nomura DK; Rape M A Cellular Mechanism to Detect and Alleviate Reductive Stress. Cell 2020, 183 (1), 46–61.e21. 10.1016/j.cell.2020.08.034. [DOI] [PubMed] [Google Scholar]
- (13).Bellezza I; Giambanco I; Minelli A; Donato R Nrf2-Keap1 Signaling in Oxidative and Reductive Stress. Biochim. Biophys. Acta Mol. Cell Res 2018, 1865 (5), 721–733. 10.1016/j.bbamcr.2018.02.010. [DOI] [PubMed] [Google Scholar]
- (14).Manford AG; Mena EL; Shih KY; Gee CL; McMinimy R; Martínez-González B; Sherriff R; Lew B; Zoltek M; Rodríguez-Párez F; Woldesenbet M; Kuriyan J; Rape M Structural Basis and Regulation of the Reductive Stress Response. Cell 2021, 184 (21), 5375–5390.e16. 10.1016/j.cell.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Savitski MM; Mathieson T; Zinn N; Sweetman G; Doce C; Becher I; Pachl F; Kuster B; Bantscheff M Measuring and Managing Ratio Compression for Accurate ITRAQ/TMT Quantification. J. Proteome Res 2013, 12 (8), 3586–3598. 10.1021/pr400098r. [DOI] [PubMed] [Google Scholar]
- (16).Jessani N; Humphrey M; McDonald WH; Niessen S; Masuda K; Gangadharan B; Yates JR; Mueller BM; Cravatt BF Carcinoma and Stromal Enzyme Activity Profiles Associated with Breast Tumor Growth in Vivo. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (38), 13756–13761. 10.1073/pnas.0404727101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Boike L; Cioffi AG; Majewski FC; Co J; Henning NJ; Jones MD; Liu G; McKenna JM; Tallarico JA; Schirle M; Nomura DK Discovery of a Functional Covalent Ligand Targeting an Intrinsically Disordered Cysteine within MYC. Cell Chem. Biol 2021, 28 (1), 4–13.e17. 10.1016/j.chembiol.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Weerapana E; Wang C; Simon GM; Richter F; Khare S; Dillon MBD; Bachovchin DA; Mowen K; Baker D; Cravatt BF Quantitative Reactivity Profiling Predicts Functional Cysteines in Proteomes. Nature 2010, 468 (7325), 790–795. 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Spradlin JN; Hu X; Ward CC; Brittain SM; Jones MD; Ou L; To M; Proudfoot A; Ornelas E; Woldegiorgis M; Olzmann JA; Bussiere DE; Thomas JR; Tallarico JA; McKenna JM; Schirle M; Maimone TJ; Nomura DK Harnessing the Anti-Cancer Natural Product Nimbolide for Targeted Protein Degradation. Nat. Chem. Biol 2019, 15 (7), 747–755. 10.1038/s41589-019-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Grossman EA; Ward CC; Spradlin JN; Bateman LA; Huffman TR; Miyamoto DK; Kleinman JI; Nomura DK Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-Cancer Natural Products. Cell Chem. Biol 2017, 24 (11), 1368–1376.e4. 10.1016/j.chembiol.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xu T; Park SK; Venable JD; Wohlschlegel JA; Diedrich JK; Cociorva D; Lu B; Liao L; Hewel J; Han X; Wong CCL; Fonslow B; Delahunty C; Gao Y; Shah H; Yates JR Pro-LuCID: An Improved SEQUEST-like Algorithm with Enhanced Sensitivity and Specificity. J. Proteomics 2015, 129, 16–24. 10.1016/j.jprot.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Käll L; Canterbury JD; Weston J; Noble WS; MacCoss MJ Semi-Supervised Learning for Peptide Identification from Shotgun Proteomics Datasets. Nat. Methods 2007, 4 (11), 923–925. 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Structures of screened covalent ligands
Table S2. isoTOP-ABPP data
Table S3. NJH-2-030 pulldown quantitative proteomics data
Table S4. Quantitative proteomic profiling of NJH-01-106
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.