

Abstract
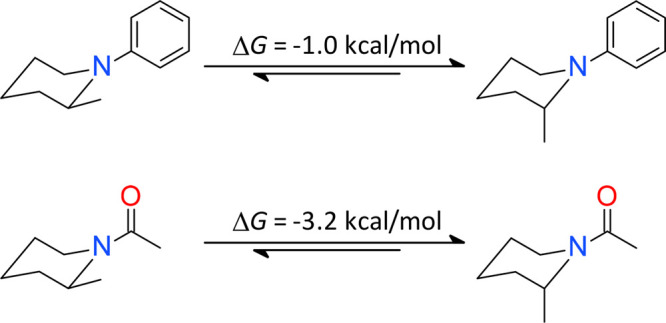
The preference of the axial over the equatorial orientation of 2-substitutent for both phenyl-1-piperidines and N-acylpiperidines is studied at the M06-2X level of theory. For phenyl-1-piperidines, the axial 2-substituent is modestly favored over the equatorial one. In contrast, the pseudoallylic strain in N-acylpiperidines dictates the axial orientation of 2-substituent with a ΔG up to −3.2 kcal/mol. The calculations agree well with the statistics from both the Cambridge Structural Database of small-molecule organic crystal structures and the Protein Data Bank. The equilibrium between the twist-boat and chair conformations for N-acylpiperidines with a 2-substituent was further investigated. The twist-boat conformation is found to be around 1.5 kcal/mol less favorable. Finally, the three-dimensionality in shape resulting from minimization of the pseudoallylic strain is characterized, and its implication in protein–ligand interactions is briefly reviewed.
Introduction
Allylic strain is a type of steric repulsion energy arising from the interaction between a substituent on one end of an alkene and an allylic substituent on the other end.1−4 If the substituents are large enough in size, they can sterically interfere with each other such that one conformer is greatly favored over the other. It has been long recognized in organic chemistry and leveraged as a controlling factor in asymmetric synthesis.5−7
When the piperidine nitrogen is bonded to an aromatic ring8 or gets acylated,9,10 conjugation of its lone pair with the neighboring π-orbital increases its sp2 hybridization and planarity. Consequently, the partial double-bond character of the C–N bond results in a pseudoallylic strain which forces the 2-substituent into an axial orientation. This effect is also applicable to other six-membered rings such as morpholine and piperazine. Given the popularity of those rings in medicinal chemistry, it could be envisioned that the effect of pseudoallylic strain on the conformational preference is frequently encountered. However, the force fields often produce incorrect lowest energy conformers by favoring the equatorial orientation of 2-substituents in the presence of pseudoallylic strain. The transition from the equatorial orientation of 2-substituent to the axial exerts a strong influence on the overall ligand conformation. The high-energy conformers used in molecular docking or the shape-based similarity search would inevitably mislead medicinal chemists. Despite allylic strain being known for decades in organic chemistry1−7 and being partially described in some medicinal chemistry reviews,8,11 there exists a practical need to increase the awareness of its effect on the conformational preference as well as protein–ligand interactions.
Results and Discussion
The free energy of transition from the equatorial to the axial 2-methyl on the piperidine ring in a chair conformation for the six model compounds were compared among different levels of theory in the gas phase and water (Table 1). The error among the different models is within 1 kcal/mol. For the sake of simplicity, we refer to the values calculated at the M06-2X/6-311G(d,p) level for further discussion unless specified otherwise. The optimized geometries are shown in Figure 1.
Table 1. Calculated ΔG (in kcal/mol) at 298 K for the Change from the Equatorial to the Axial 2-Methyl and the Wiberg Bond Order of the C–N Bond.

In the gas phase at the level of B3LYP/6-31G(d).
In water with B3LYP-3D/6-31G(d).
In water with M06-2X/6-311G(d,p).
Wiberg bond order of the C–N bond colored in purple in the axial conformer.
–TΔS.
Figure 1.

Chair conformations of the piperidine ring with the 2-methyl in the equatorial or axial orientation for the six model compounds. Numbers correspond to the dihedral between the four atoms highlighted in the ball representation and the composition of p character in the lone-pair hybrid on the piperidine nitrogen. The black arrow indicates the steric hindrance.
The chair conformer of 1,2-dimethylpiperidine with the equatorial 2-methyl is favored by 1.8 kcal/mol relative to the one in which the methyl is axial. The axial conformer begins being modestly favored for 2-methyl-1-phenylpiperidine with a ΔG of −1.0 kcal/mol. The lone pair of the piperidine nitrogen in the equatorial conformer lies nearly on the phenyl plane showing little overlap with the π-orbital, and as a result, the nitrogen displays the typical pyramidal character. In the axial conformer, the lone pair is largely parallel with the π-orbital of the phenyl and the nitrogen moves toward the sp2 hybridization, which is reflected by the increased composition of p character in its lone pair hybrid from the natural bond orbital (NBO) analysis (Figure 1). The sp2 character of the piperidine nitrogen increases in the axial conformer of N,N,2-trimethylpiperidine-1-carboxamide, which is favored by 1.4 kcal/mol over the equatorial. Interestingly, the axial conformer is favored by 2.1 kcal/mol over the equatorial for N,2-dimethylpiperidine-1-carboxamide which differs in only one carbon atom. This is because the steric hindrance in N,N,2-trimethylpiperidine-1-carboxamide prevents a high level of π-conjugation between the piperidine nitrogen and the carbonyl group. The axial conformer is most favored by 1-(2-methyl-1-piperidyl)ethanone with a ΔG of −3.2 kcal/mol, which has an entropic contribution of −0.5 kcal/mol.
For N-acylpiperidines there exist two conformers in which the 2-methyl and the carbonyl oxygen are in a cis or trans configuration. The trans configuration in the equatorial conformer is destabilized by steric clash with a ΔG of 0.5 kcal/mol for 2-methylpiperidine-1-carboxylate, 1.1 kcal/mol for N,2-dimethylpiperidine-1-carboxamide, and 1.5 kcal/mol for 1-(2-methyl-1-piperidyl)ethanone. In contrast, the free energy difference between the cis and trans configuration in the respective axial conformer is within ±0.1 kcal/mol. Therefore, the switch from the equatorial to the axial 2-methyl for N-acylpiperidines is driven by a conformational entropy too. For the sake of simplicity, the ΔG values in Table 1 were calculated based on the geometries shown in Figure 1 without accounting for the conformational entropy.
The increased preference for the axial orientation of the 2-methyl correlates with the increase in the composition of p character in the lone pair hybrid on the piperidine nitrogen. The dihedral angle between the two exocyclic vectors in the equatorial conformers decreases gradually with an increase in the sp2 character of the piperidine nitrogen, giving rise to a higher steric repulsion. Conversely, the same torsion angle in the axial conformers gradually increases as a way to minimize the steric strain (Figure 1). Note that the transition from the equatorial to the axial orientation places the 2-methyl on the opposite side of the ring so as to keep the same chair conformation.
Next, we looked to the Cambridge Structural Database (CSD) for small-molecule organic crystal structures having either the phenyl-1-piperidine or N-acylpiperidine motif. The two substructures were extended to include both morpholine and piperazine, which are common in medicinal chemistry (Figure 2). To remove ambiguity due to potential steric effects, the neighboring positions are masked by hydrogens. To exclude the anomeric effect12,13 which prefers an axial substituent, the linkage atom of the substituent is further limited to carbon only. For the phenyl-1-piperidine class, there are 12 entries, of which three showed a protonated piperidine nitrogen. These three structures were excluded in the analysis since the phenyl-1-piperidine nitrogen is unlikely to be protonated at the physiological conditions. The remaining nine crystal structures all show a chair conformation of the piperidine ring, and three of them have the 2-substituents in the equatorial orientation (Figure 2A). This observation is in good agreement with the quantum mechanics (QM) estimation that the equatorial orientation is modestly disfavored by 1.0 kcal/mol. The piperidine nitrogen in the three equatorial structures shows rich sp3 character and the N-phenyl plane nearly bisects the piperidine ring, consistent with the QM-optimized geometry in Figure 1.
Figure 2.

Distribution of ring conformations from the nine phenyl-1-piperidine (A) and the 127 N-acylpiperidine entries (B) deposited in the CSD queried by their respective inset structures. The angles α and β are between the symmetry axis of the ring and the C–H or the substituent exit vectors. The chair ring conformations are colored in blue with the twist-boat in orange. Structures showing an equatorial 2-substituent on a chair ring are circled in gray.
For the N-acylpiperidine class, 127 CSD entries totaling to 151 substructures were retrieved, of which 18 (12% which corresponds to roughly a ΔG of 1.4 kcal/mol) showed a twist-boat conformation of the ring (Figure 2B). The QM calculations suggest that the twist-boat conformation is 1.2 kcal/mol more favorable than the chair with the equatorial 2-methyl but 2.0 kcal/mol less favorable than the chair with the 2-methyl axially oriented (Figure 3). This calculation is within about 1 kcal/mol of the experimental statistics from the CSD. It is worth mentioning that the QM calculation of such a simple model compound does not account for intermolecular interactions as a result of the crystal packing. The intramolecular interactions arising in a large molecule, which may stabilize a specific ring conformation, have not been taken into account either. Among the other 133 substructures with a chair ring conformation, only two (1.5%) have an equatorial substituent at the 2-position. The first structure (CSD entry UVIGUC14) features the lipophilic packing of a tert-butyl group against a phenyl permitted by the equatorial orientation of the 2-(4-cyanophenyl). The second one (IROYOF15) corresponds to the drug candidate vestipitant of the N,N-substituted piperazine-1-carboxamide motif. The equatorial orientation is compensated for by the intramolecular π-stacking interaction between the two phenyls (Figure 4). Intriguingly, one structure (PDB entry 4APP(16)) from the Protein Data Bank (PDB) also features an equatorial 2-substituent of the N,N-substituted piperazine-1-carboxamide, where both the salt bridge with Asp458 and the lipophilic burial of the 2-benzyl into the ATP-back pocket stabilize the unfavorable equatorial orientation (Figure 4). Notably, the acylated nitrogen in both cases is rich in sp3 character, and its lone pair is orthogonal to the neighboring carbonyl π-orbital. Combined with what was observed on the phenyl-1-piperidines, the equatorial orientation of the 2-substitutent is concomitant of the rich sp3 character of the piperidine nitrogen. The cis and trans configurations between the 2-substituent and the carbonyl oxygen in the axial conformers have a ratio of 3:2, in a good agreement with the QM estimation.
Figure 3.

Twist-boat and chair ring conformations of 1-[(2S)-2-methyl-1-piperidyl]ethenone.
Figure 4.

Examples of piperazine-1-carboxamides from the CSD (entry IROYOF) and PDB (4APP) showing a chair ring conformation with an equatorial 2-substituent.
Given the strong preference for the axial 2-subsitutent of N-acylpiperidines, the bound ligand conformations in the PDB were queried with the aim to understand to what extent the protein–ligand interactions may overcome the conformational barrier. Among the 294 N-acylpiperidine structures deposited in the PDB, 68 ligand structures (23%) have a twist-boat ring conformations (Figure 5). This nearly doubles that from the CSD. Thus, the thermodynamically less favorable twist-boat ring conformation appears to be stabilized by protein–ligand interactions. However, the potential fitting errors due to poor X-ray resolutions may play a role too.17 For the remaining 226 structures with the piperidine in a chair conformation, only four ligands were found to have an equatorial 2-substituent. Except for the entry 4APP (Figure 4), visual inspection of the other three structures (PDB entry 1K1O,18 5QJ919 chain C, and 7JTW20) reveals incomplete electron density around the ligand, suggesting possible fitting errors. This notion is supported upon comparison of the bound ligand conformations between the structures 5M9621 and 7JTW. Both structures correspond to the retinoic-acid-related orphan receptor γt in complex with similar ligands. The binding affinity of small molecules to a protein target is typically in the range from −7 to −12 kcal/mol, and a conformational penalty of around 3 kcal/mol is unlikely to be rescued by the protein–ligand interactions. The equatorial orientation is thus nearly inaccessible for the 2-substituent of N-acylpiperidines with the few exceptions such as N,N-disubstituted piperidine-1-carboxamides in which the steric hindrance restricts the π-conjugation and hence reduces the double-bond character of the C–N bond.
Figure 5.
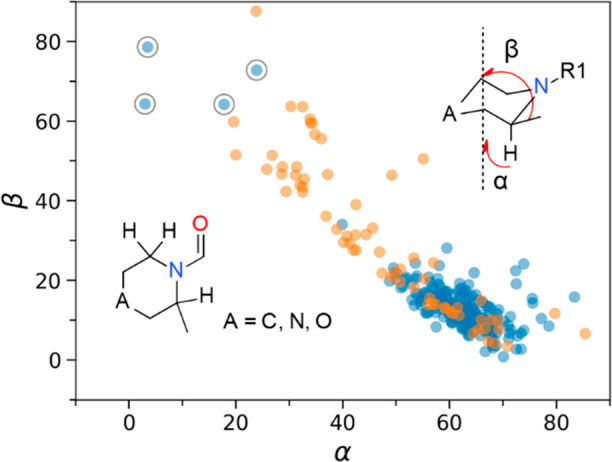
Distribution of ring conformations from the 294 N-acylpiperidine structures deposited in the PDB. The angles α and β are between the ring axis and the C–H or the substituent exit vectors. The chair ring conformations are colored in blue with the twist-boat in orange. Structures showing an equatorial 2-substituent on a chair ring are circled in gray.
Shape complementarity of a ligand with its target is a critical aspect of molecular recognition. It is postulated that the increased three-dimensionality in shape is beneficial for selectivity since many binding sites (e.g., kinases) are cleftlike and have a high tendency to recognize flat structures. The three-dimensionality may also correlate with physical properties such as solubility and meting point.22,23 Increasing saturation is proposed to improve clinical success by escaping from flatland.24 Interestingly, though the fraction of sp3-hybridized carbons decreases from 0.54 for 1-ethyl-2-phenyl-piperidine to 0.46 for 1-(2-phenyl-1-piperidyl)ethanone, the latter molecule is actually more sphere- and disk-shaped arising from the axial orientation of the 2-phenyl (Figure 6). In comparison with the L-shape of the two common motifs 1-benzylpiperidine and 1-(benzenesulfonyl)piperidine, it is characterized by a unique T-shape.
Figure 6.

Shape characterization of the exemplar chemical structures by the principal moments of inertia ternary plot.
The pseudoallylic strain provides a means to modulate conformational preferences by dictating 2-substituents in the axial orientation, and the axial exit vector could allow for exploration of subpockets which otherwise may not be accessible. To illustrate this notion, two examples from the PDB were taken. Filorexant, which was advanced into phase II clinical trials for the treatment of insomnia, represents a special case of directing an axial substituent elsewhere on the piperidine ring by an axial 2-methyl arising from minimization of the pseudoallylic strain. The 5-aryloxymethyl in the axial orientation encourages filorexant to adopt a π-stacked horseshoe conformation which fits well into the binding site of orexin 1 receptor (PDB 6TP6, Figure 7A).25 In contrast, the desmethyl analogues in the series have remarkably lower affinities.26 Avacopan is the first orally administered inhibitor of the complement C5a receptor, approved in 2021 for the treatment of antineutrophil cytoplasmic autoantibody-associated vasculitis. It binds to an allosteric site located on the surface of the transmembrane helical bundle (6C1R, Figure 7B). The 2-phenyl substituent on the piperidine ring is forced in the axial orientation by the pseudoallylic strain and extends into a hotspot which is occupied by chemically diverse allosteric antagonists.27 The overall bound conformation of avacopan displays a characteristic T-shape, allowing it to explore additional subpockets for a higher affinity.
Figure 7.

Examples of modulating conformational preferences by minimization of the pseudoallylic strain toward improved binding interactions with PDB entries 6TP6 (A) and 6C1R (B).
Conclusion
In summary, the preference for the axial 2-substituent of both phenyl-1-piperidines and N-acylpiperidines is studied by the density functional theory. The experimental observations on ligand conformations from both the CSD and PDB agree well with the QM calculations. The pseudoallylic strain arising from the partial double-bond character of the C–N bond dictates the 2-substituent in the axial orientation, which increases the three-dimensionality in shape and affords a unique exit vector to explore the binding site.
Experimental Section
The 3D conformations of piperidines without the 2-methyl were generated by the LigPrep module (Schrödinger release 2021-2), and the 2-methyl was built manually in the equatorial or axial orientation. The QM calculations were carried out with the Gaussian 16 package with methods B3LYP28 and M06-2X.29 The geometry was first optimized in the gas phase at the B3LYP level with a 6-31G(d) basis set. Geometry optimizations were further performed with a PCM implicit water and a D3 empirical dispersion correction,30 followed by a frequency and an NBO analysis. A single point of energy calculation was performed at the M06-2X level with a 6-311G(d,p) basis set and a SMD implicit water.31
The experimentally observed small-molecule structures from both the CSD and PDB were analyzed using the open source cheminformatics software RDKit (www.rdkit.org) with the SMARTS [OR0]=CN1[CH2][CR1X4][N,O,C][CR1X4][CH]1[C,c] for N-acylpiperidines and a[NX3]1[CH2][CR1X4][N,O,C][CR1X4][CH]1[C,c] for phenyl-1-piperidines.
Acknowledgments
The author would like to thank Dr. Per-Ola Norrby for help with QM. The author is grateful to Dr. Jonas Brånalt and Dr. Christian Tyrchan for reading the manuscript.
Glossary
Abbreviations
- CSD
Cambridge Structural Database
- PDB
Protein Data Bank
- QM
quantum mechanics
- NBO
natural bond orbital
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00510.
Optimized geometries of the six model compounds (PDF)
The author declares the following competing financial interest(s): H.Z. is an employee of AstraZeneca, and may own stock or stock options in AstraZeneca.
Supplementary Material
References
- Johnson F.; Malhotra S. K. Steric interference in allylic and pseudo-allylic systems. I. Two stereochemical theorems. J. Am. Chem. Soc. 1965, 87, 5492–5493. 10.1021/ja00951a046. [DOI] [Google Scholar]
- Malhotra S. K.; Johnson F. Steric interference in allylic and pseudo-allylic systems. II. Stereochemistry of exocyclic enolate anion protonation. J. Am. Chem. Soc. 1965, 87, 5493–5495. 10.1021/ja00951a047. [DOI] [Google Scholar]
- Allinger N. L.; Hirsch J. A.; Miller M. A.; Tyminski I. J. Conformational analysis. LXIV. Calculation of the structures and energies of unsaturated hydrocarbons by the Westheimer method. J. Am. Chem. Soc. 1968, 90, 5773–5780. 10.1021/ja01023a021. [DOI] [Google Scholar]
- Broeker J. L.; Hoffmann R. W.; Houk K. N. Conformational Analysis of Chiral Alkenes and Oxonium Ions: Ab Initio Molecular Orbital Calculations and an Improved MM2 Force Field. J. Am. Chem. Soc. 1991, 113, 5006–5017. 10.1021/ja00013a041. [DOI] [Google Scholar]
- Johnson F. Allylic strain in six-membered rings. Chem. Rev. 1968, 68, 375–413. 10.1021/cr60254a001. [DOI] [Google Scholar]
- Hoffmann R. W. Allylic 1,3-strain as a controlling factor in stereoselective transformations. Chem. Rev. 1989, 89, 1841–1860. 10.1021/cr00098a009. [DOI] [Google Scholar]
- Coombs T. C.; Lushington G. H.; Douglas J.; Aube J. 1,3-allylic strain as a strategic diversification element for constructing libraries of substituted 2-arylpiperidines. Angew. Chem., Int. Ed. Engl. 2011, 50, 2734–7. 10.1002/anie.201007133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brameld K. A.; Kuhn B.; Reuter D. C.; Stahl M. Small molecule conformational preferences derived from crystal structure data. A medicinal chemistry focused analysis. J. Chem. Inf. Model. 2008, 48, 1–24. 10.1021/ci7002494. [DOI] [PubMed] [Google Scholar]
- Paulsen H.; Todt K. Magnetic anistropy of the amide group. Angew. Chem., Int. Ed. Engl. 1966, 5, 899–900. 10.1002/anie.196608992. [DOI] [Google Scholar]
- Quick J.; Mondello C.; Humora M.; Brennan T. Synthesis of 2,6-diacetonylpiperidine. X-ray diffraction analysis of its benzoyl derivative. J. Org. Chem. 1978, 43, 2705–2708. 10.1021/jo00407a036. [DOI] [Google Scholar]
- Zheng Y.; Tice C. M.; Singh S. B. Conformational control in structure-based drug design. Bioorg. Med. Chem. Lett. 2017, 27, 2825–2837. 10.1016/j.bmcl.2017.04.079. [DOI] [PubMed] [Google Scholar]
- Juaristi E.; Cuevas G. Recent studies of the anomeric effect. Tetrahedron 1992, 48, 5019–5087. 10.1016/S0040-4020(01)90118-8. [DOI] [Google Scholar]
- Bauerfeldt G. F.; Cardozo T. M.; Pereira M. S.; da Silva C. O. The anomeric effect: the dominance of exchange effects in closed-shell systems. Org. Biomol. Chem. 2013, 11, 299–308. 10.1039/C2OB26818C. [DOI] [PubMed] [Google Scholar]
- Seel S.; Thaler T.; Takatsu K.; Zhang C.; Zipse H.; Straub B. F.; Mayer P.; Knochel P. Highly diastereoselective arylations of substituted piperidines. J. Am. Chem. Soc. 2011, 133, 4774–7. 10.1021/ja201008e. [DOI] [PubMed] [Google Scholar]
- Huang W. X.; Liu L. J.; Wu B.; Feng G. S.; Wang B.; Zhou Y. G. Synthesis of Chiral Piperazines via Hydrogenation of Pyrazines Activated by Alkyl Halides. Org. Lett. 2016, 18, 3082–5. 10.1021/acs.orglett.6b01190. [DOI] [PubMed] [Google Scholar]
- Guo C.; McAlpine I.; Zhang J.; Knighton D. D.; Kephart S.; Johnson M. C.; Li H.; Bouzida D.; Yang A.; Dong L.; Marakovits J.; Tikhe J.; Richardson P.; Guo L. C.; Kania R.; Edwards M. P.; Kraynov E.; Christensen J.; Piraino J.; Lee J.; Dagostino E.; Del-Carmen C.; Deng Y. L.; Smeal T.; Murray B. W. Discovery of pyrroloaminopyrazoles as novel PAK inhibitors. J. Med. Chem. 2012, 55, 4728–39. 10.1021/jm300204j. [DOI] [PubMed] [Google Scholar]
- Wlodawer A.; Dauter Z.; Porebski P. J.; Minor W.; Stanfield R.; Jaskolski M.; Pozharski E.; Weichenberger C. X.; Rupp B. Detect, correct, retract: How to manage incorrect structural models. FEBS J. 2018, 285, 444–466. 10.1111/febs.14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dullweber F.; Stubbs M. T.; Musil D.; Sturzebecher J.; Klebe G. Factorising ligand affinity: a combined thermodynamic and crystallographic study of trypsin and thrombin inhibition. J. Mol. Biol. 2001, 313, 593–614. 10.1006/jmbi.2001.5062. [DOI] [PubMed] [Google Scholar]
- Pearce N. M.; Krojer T.; Bradley A. R.; Collins P.; Nowak R. P.; Talon R.; Marsden B. D.; Kelm S.; Shi J.; Deane C. M.; von Delft F. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 2017, 8, 15123. 10.1038/ncomms15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima R.; Oono H.; Kumazawa K.; Ida T.; Hirata J.; White R. D.; Min X.; Guzman-Perez A.; Wang Z.; Symons A.; Singh S. K.; Mothe S. R.; Belyakov S.; Chakrabarti A.; Shuto S. Discovery of 6-Oxo-4-phenyl-hexanoic acid derivatives as RORgammat inverse agonists showing favorable ADME profile. Bioorg. Med. Chem. Lett. 2021, 36, 127786. 10.1016/j.bmcl.2021.127786. [DOI] [PubMed] [Google Scholar]
- Hintermann S.; Guntermann C.; Mattes H.; Carcache D. A.; Wagner J.; Vulpetti A.; Billich A.; Dawson J.; Kaupmann K.; Kallen J.; Stringer R.; Orain D. Synthesis and Biological Evaluation of New Triazolo- and Imidazolopyridine RORgammat Inverse Agonists. ChemMedChem. 2016, 11, 2640–2648. 10.1002/cmdc.201600500. [DOI] [PubMed] [Google Scholar]
- Ishikawa M.; Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–54. 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Engkvist O.; Llinas A.; Chen H. Beyond size, ionization state, and lipophilicity: influence of molecular topology on absorption, distribution, metabolism, excretion, and toxicity for druglike compounds. J. Med. Chem. 2012, 55, 3667–77. 10.1021/jm201548z. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Rappas M.; Ali A. A. E.; Bennett K. A.; Brown J. D.; Bucknell S. J.; Congreve M.; Cooke R. M.; Cseke G.; de Graaf C.; Dore A. S.; Errey J. C.; Jazayeri A.; Marshall F. H.; Mason J. S.; Mould R.; Patel J. C.; Tehan B. G.; Weir M.; Christopher J. A. Comparison of Orexin 1 and Orexin 2 Ligand Binding Modes Using X-ray Crystallography and Computational Analysis. J. Med. Chem. 2020, 63, 1528–1543. 10.1021/acs.jmedchem.9b01787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman P. J.; Schreier J. D.; Cox C. D.; Breslin M. J.; Whitman D. B.; Bogusky M. J.; McGaughey G. B.; Bednar R. A.; Lemaire W.; Doran S. M.; Fox S. V.; Garson S. L.; Gotter A. L.; Harrell C. M.; Reiss D. R.; Cabalu T. D.; Cui D.; Prueksaritanont T.; Stevens J.; Tannenbaum P. L.; Ball R. G.; Stellabott J.; Young S. D.; Hartman G. D.; Winrow C. J.; Renger J. J. Discovery of [(2R,5R)-5-{[(5-fluoropyridin-2-yl)oxy]methyl}-2-methylpiperidin-1-yl][5-methyl-2 -(pyrimidin-2-yl)phenyl]methanone (MK-6096): a dual orexin receptor antagonist with potent sleep-promoting properties. ChemMedChem. 2012, 7, 415–24. 10.1002/cmdc.201200025. [DOI] [PubMed] [Google Scholar]
- Liu H.; Kim H. R.; Deepak R.; Wang L.; Chung K. Y.; Fan H.; Wei Z.; Zhang C. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 2018, 25, 472–481. 10.1038/s41594-018-0067-z. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5650. 10.1063/1.464913. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–67. 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–96. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.