

Abstract

The synthesis and properties of a series of unsymmetrical thienopentalenes are explored, including both monoareno and diareno derivatives. For the synthesis of monoareno pentalenes, a carbopalladation cascade reaction between alkynes and gem-dibromoolefins was applied. Diareno pentalene derivatives were accessed via gold-catalyzed cyclization of diynes. Thiophene was fused to pentalene in two different geometries via its 2,3 and 3,4 bonds. 2,3-Fusion resulted in increased antiaromaticity of the pentalene unit compared to the 3,4-fusion both in the monoareno and diareno framework. Monothienopentalenes that contained the destabilizing 2,3-fusion could not be isolated. For diareno derivatives, the aromatic character of the different aryl groups fused to the pentalene was not independent. Destabilizing fusion on one side resulted in alleviated aromaticity on the other side and vice versa. The synthesized molecules were characterized experimentally by 1H NMR and UV–vis spectroscopies, cyclic voltammetry, and X-ray crystallography, and their aromatic character was assessed using magnetic (NICS and ACID) and electronic indices (MCI and FLU).
Introduction
Polycyclic conjugated π-electron systems with incorporated antiaromatic [4n]π subunits have been extensively explored recently.1−8 The interest in such materials is twofold. From the perspective of the [4n]π unit,9 these structures provide opportunities to study antiaromatic molecules as π-extension ensures sufficient stabilization of the otherwise unstable [4n]π-electron systems.10−12 From the perspective of the resulting acene-derivatives, [4n]π subunits can improve their stability and processability for organic electronic applications.13−16 Hence, exploring the synthesis and properties of novel π-extended antiaromatics is of considerable interest from both fundamental and applications point of view. Among antiaromatic subunits, pentalene is particularly interesting as this planar 8 π-electron molecule could be extended/stabilized through different fusion and substituent patterns, and the synthesis of its derivatives has advanced a lot recently.2 However, regardless of the available synthetic methodologies for the preparation of derivatives with unsymmetrical conjugated cores,17−26 material properties of linear and symmetric π-extended antiaromatics are explored mostly.27−37 Although the study of symmetric polycyclic conjugated systems is facilitated by their more straightforward syntheses in general, the departure from such structures proved to be fruitful in the case of acenes. Within the class of acenes, the introduction of asymmetry, especially by the unsymmetrical fusion of thiophenes, led to improved stability, good solubility, novel assembly modes, and interesting electronic features such as ambipolar transport properties in several cases.38−42
Unique properties that may emerge from π-extended thieno-antiaromatics having unsymmetrical conjugated cores, along with the potential of asymmetry to tune electronic properties, prompted us to investigate such structures. In this contribution, we explore the synthesis, basic opto-electronic properties, and aromaticity of unsymmetrical thienopentalenes. Special focus is given to derivatives that have a proton attached to the pentalene unit, which could be an important reporter on the extent of antiaromaticity within the molecules.
Results and Discussion
Synthesis
To access unsymmetrical thienopentalenes, we followed two synthetic approaches (Scheme 1), that have been proven to be reliable methods for the synthesis of pentalene derivatives. The methodology developed and optimized by Diederich and co-workers (Scheme 1a)43,44 is based on the cascade carbopalladation between alkynes and gem-dibromoolefines and leads to monoannelated pentalenes. For the synthesis of unsymmetrical diareno[a,e]pentalene-type molecules, we turned to the gold-catalyzed cyclization of diynes developed and optimized by Hashmi and co-workers (Scheme 1b).21 Both methodologies involve the formation of multiple C–C bonds in one-pot, which should be considered in the evaluation of the product yields.
Scheme 1. General Strategies Applied for the Syntheses of Molecules in This Study; (a) Pd-Catalyzed and (b) Au-Catalyzed Cascade Transformation.

We expected that these methodologies could be adapted to synthesize a series of thieno and benzothieno derivatives of pentalene (Figure 1), taken also into account that thiophene can be annelated both through its 2,3(b) and 3,4(c) C–C bonds. It is noted, however, that several reports that deal with synthetic methodology development describe benzo[4,5]pentaleno[2,1-b]thiophene (IV type) derivatives as unstable.17,19−22 Hence, instead of IV, we explored the synthesis of the benzothiophene-fused benzopentalenes (V) only.
Figure 1.

Targeted thienopentalene derivatives. Isolated structures are shown in green (PMP = p-methoxyphenyl).
First, we attempted the synthesis of thienopentalene I (Scheme 2), which was expected to contain the most antiaromatic pentalene unit. The strong antiaromaticity, in this case, comes from the monoannelated structure on one hand and from the strong alkene character of the fused thiophene double bond on the other. The synthesis was started with the bromination of benzo[b]thiophene-2-carboxaldehyde 1 followed by a Sonogashira coupling between the brominated derivative 2 and phenylacetylene. Cross-coupling product 3 was converted to the corresponding gem-dibromoolefin 4 using the procedure reported by Lautens and co-workers.45 The attempted carbopalladation cascade between gem-dibromoolefin 4 and diphenylacetylene did not provide the desired product I. It is noted that the gem-dibromoolefin starting materials were fully consumed during all the cascade carbopalladation reactions in this study, so the lack of isolable products in some cases is most probably not related to the reactivity of these starting materials but rather their non-selective transformations under the reaction conditions.
Scheme 2. Attempted Synthesis of Compound I.

The low stability of I might be the reason of its unsuccessful synthesis. Thus, we instead targeted thienopentalene II (Scheme 3), in which the fused benzothiophene unit was reduced to a single thiophene ring. Although thiophene has a considerable diene character, the bond order of its 2,3(b) double bond is somewhat lower compared to the same bond in benzothiophene. This was expected to contribute to increased stability of II through somewhat decreased antiaromaticity of the pentalene unit compared to that of I.
Scheme 3. Attempted Synthesis of Compound II.

The attempted synthesis of II (Scheme 3) followed a similar route as of I. A Sonogashira coupling on 3-bromothiophene-2-carboxaldehyde 5 is followed by the conversion of the aldehyde in 6 into a gem-dibromoolefin 7, which was the starting material for the pentalene-forming cascade reaction. Unfortunately, as in the case of compound I, no pentalene formation was detected. It has been shown earlier by Kawase and co-workers that thiophene fusion through its 3,4(c) bond strongly reduces the pentalene contribution to the resulting π-system.32 Those molecules (pentaleno[1,2-c:4,5-c′]dithiophene derivatives), in contrast to dibenzo[a,e]pentalene derivatives, were characterized by allowed highest occupied molecular orbital (HOMO)-to-lowest occupied molecular orbital (LUMO) transitions and a level of fluorescence. A reduced bond order of the fused thiophene bond explains such pronounced changes in properties. Based on the work of Kawase and co-workers, we explored the synthesis of III (Scheme 4), where a single thiophene ring is fused to pentalene through its 3,4(c) bond. 3,4-Dibromothiophene 8 was used as a starting material, which was lithiated with nBuLi and quenched with DMF to access bromoaldehyde 9. Subsequently, a Sonogashira coupling with phenylacetylene and gem-dibromoolefination of the resulting aldehyde 10 led to compound 11, which could be converted to III in 11% yield as a bench-stable green material.
Scheme 4. Synthesis of Compound III.

Following the monoannelated thienopentalenes, we turned to the synthesis of thiophene containing diarenopentalene derivatives. Compound 3 from the synthesis of I was chosen as a starting material to access the non-substituted derivative of V [V(H)] (Scheme 5) with two pentalene protons in the molecule, which could be useful to evaluate its antiaromaticity with 1H NMR spectroscopy. The aldehyde group of 3 could be converted to a terminal alkyne via a Seyferth–Gilbert homologation using the Bestmann–Ohira reagent for the transformation. The resulting diacetylene 12 was submitted to gold-catalyzed pentalene-forming conditions. The gold-catalyzed cascade did not provide the expected V(H); instead, a compound was isolated with a structure that was suggested to be 13. Such dimerized side products have been reported under the applied conditions and were associated with the presence of terminal alkynes.46
Scheme 5. Attempted Synthesis of Compound V(H).
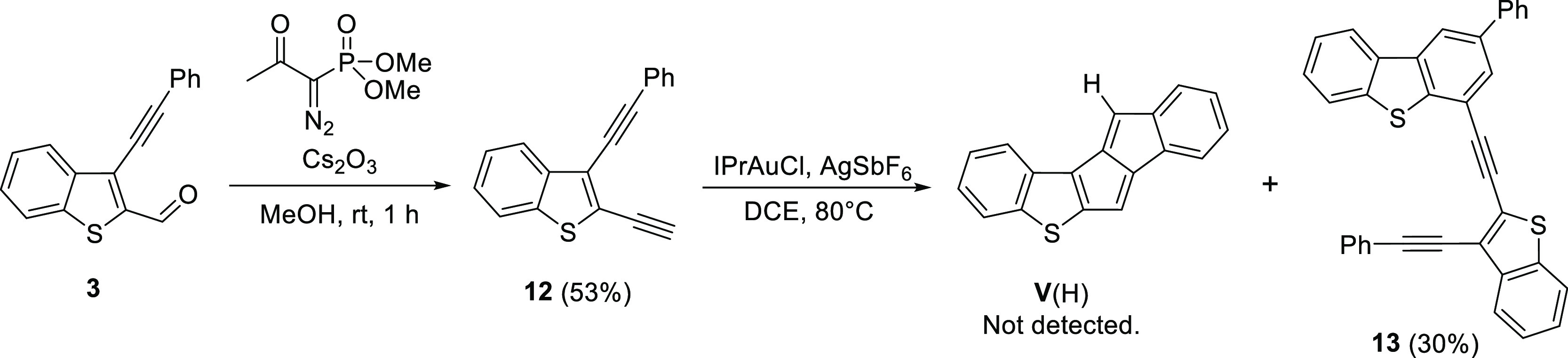
To eliminate the side reaction, we synthesized a benzothiophene derivative with two phenylacetylene moieties. For this, benzothiophene (14) was dibrominated with Br2, then product 15 was coupled with two equivalents of phenylacetylene. The gold-catalyzed pentalene formation from diacetylene 16 worked in this case; however, an inseparable and impure mixture of isomers of V(Ph) was obtained (Scheme 6).
Scheme 6. Attempted Synthesis of V(Ph).
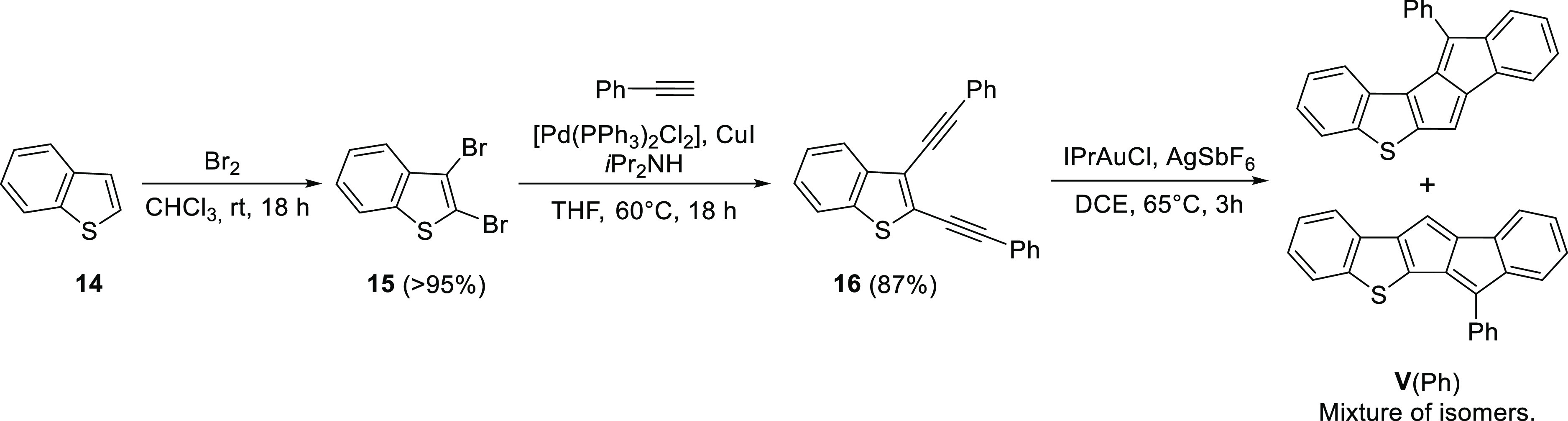
To control the selectivity of the reaction, one of the phenylacetylenes was replaced with 4-ethynylanisole that was expected to promote the regioselective annulation and facilitate the purification (Scheme 7).47 Indeed, after performing the gold-catalyzed annulation on compound 19, a single isomer of V(PMP) could be isolated as a green solid.
Scheme 7. Synthesis of V(PMP).
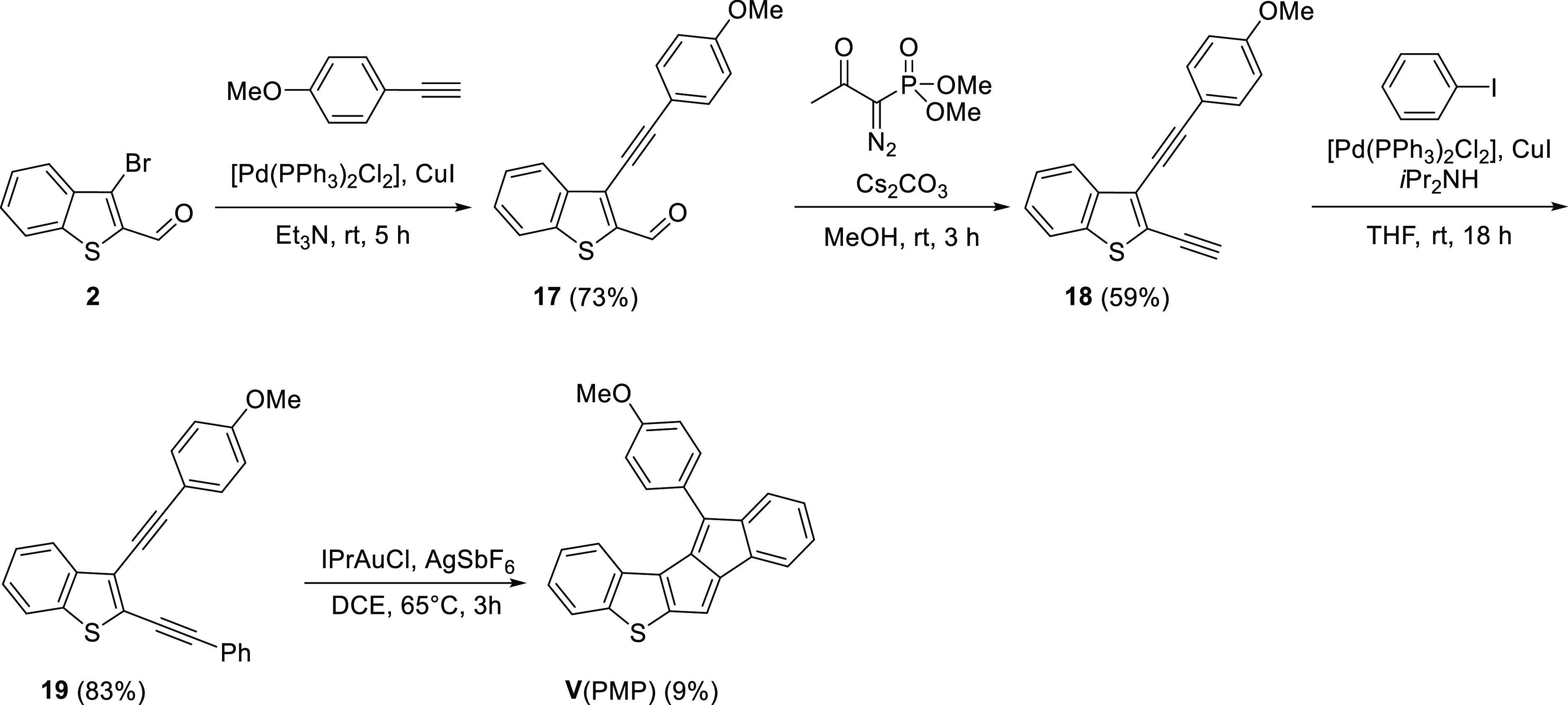
We also investigated the synthesis of thienopentalene VI (Scheme 8), in which the contribution of pentalene antiaromaticity is expected to be the least of all structures considered in this study due to the strong electronic stabilization. The comparably higher yielding annulation of diacetylene 20, giving VI as an orange solid, seemed to support this speculation.
Scheme 8. Synthesis of Compound VI.

Although not all the target molecules could be isolated, the unsuccessful syntheses provide important information. The compounds that could not be obtained represent limitations of the applied synthetic methodology on one hand and, likely, the stability of thienopentalene derivatives on the other hand.
1H NMR Spectroscopy
It is instructive to look at the chemical shifts of the protons connected to the pentalene moiety in the isolated molecules (Figure 2). As the substituent patterns around the free pentalene Hs are similar, it allows for the comparison of chemical shifts within the synthesized series of molecules and with related structures that have been reported earlier. In general, paratropic and diatropic ring current effects are reflected in upfield and downfield 1H NMR shifts, respectively.48
Figure 2.

1H NMR chemical shifts (CD2Cl2, 500 MHz, rt) of the protons attached to the pentalene unit within III, V(PMP), and VI. Values for BP and DBP are added for comparison.
The proton chemical shifts of the pentalene Hs within the synthesized molecules are decreasing in the order of VI (6.63 ppm) > III (6.53 ppm) > V(PMP) (6.09 ppm), which indicate an increasing olefinic double-bond character experienced by the protons of interest; although in III, the phenyl substituent could have some influence on the measured chemical shift through additional shielding.
Nevertheless, these values are in line with what would be expected intuitively, based on previously studied structures.34,49,50 In both III and VI, thiophene is fused through its 3,4 bond, which has a low double-bond character that lowers the antiaromatic character of the pentalene subunit. VI is further stabilized by the fusion of an additional phenyl ring that leads to an even more alleviated antiaromaticity. Although V(PMP) has a stabilizing benzene fusion as well, the other fused aryl system is a benzothiophene, in which the double-bond character of the thiophene ring contributes to the increased antiaromaticity; hence, we call this fusion destabilizing. The chemical shift of the pentalene proton in III (6.53 ppm) compared to that of in monobenzopentalene BP (6.35 ppm) suggests that thiophene fusion through its 3,4 bond stabilizes the antiaromatic system somewhat stronger than a single phenyl ring. This also holds when VI and dibenzopentalene DBP are compared. The contribution of benzothiophene fusion in V(PMP) to increased antiaromaticity is further confirmed by its analogy with DBP.
X-ray Crystallography
Among the synthesized molecules, only VI could be crystallized for X-ray single-crystal analysis (Figure 3 and Section S1, Supporting Information). In the asymmetric unit, a whole VI molecule was found. Stacks of thienopentalene conjugated cores are built up with π–π interactions within the crystal lattice. The disordered phenyl substituent is found between these stacks in the crystal layer. A general structural feature of antiaromatic molecules is bond length alternation within their conjugated core. This should be observed within the pentalene unit of VI, if its antiaromaticity is well preserved. However, by looking at the bond lengths within the molecule, it is clearly not the case. Within the pentalene unit, bond lengths l4 (1.356 Å) and l6 (1.354 Å) are considerably shorter than the other bonds; however, no pronounced bond length alternation can be observed that extends to the whole pentalene unit. Based on this, the π-system within the two fused five-membered rings has rather a butadiene-type character (l4–l3–l6), or, if the relatively short l8 is considered, it shows a fulvene-type conjugation pattern (l3–l6–l7–l8–l9–l4). In any case, based on the crystal structure, there is a relatively low pentalene-type 8π-character present in the molecule. This observation is in line with what was found for symmetric dithieno[a,e]pentalene derivatives having similarly fused thiophenes.32,34
Figure 3.

X-ray structure of VI (CCDC 2090075) and the corresponding bond lengths (Å) (ORTEP style representation is drawn at the 50% probability level. A disordered phenyl substituent was found in the asymmetric unit. Here, only the dominant conformation is presented, and disorder was omitted for clarity).
As a comparison, the replacement of a single benzene ring in DBP by a thiophene ring in VI did not drastically alter the solid-state packing properties of the two compounds. A very similar network of non-covalent interactions within the crystal lattices was found in both cases (Section S1.1, Supporting Information).
Opto-Electronic Properties
UV–Vis Spectroscopy
The optical properties of the synthesized pentalene derivatives were studied by UV–vis spectroscopy (Figure 4) in CHCl3. There is no pronounced difference between the compounds in terms of the position of the main absorption bands below 500 nm. The ε for V(PMP) in the whole spectrum is comparable to that of BP, suggesting that the benzopentalene subunit dominates the optical properties of the molecule regardless of the presence of five fused unsaturated rings. The spectra of VI and DBP are characterized by two distinct maxima in the 350–450 nm region, which are absent in the spectra of the other molecules. As a signature of the presence of the 8π subunit,16−19,24,26,30,33−35,43,44,49 broad, low-intensity absorptions were observed above 500 nm for all compounds measured, although these bands are rather shoulders of the stronger absorption bands below 500 nm in the case of VI and DBP (note that the spectra obtained for BP and DBP are added for comparison in Figure 4).
Figure 4.
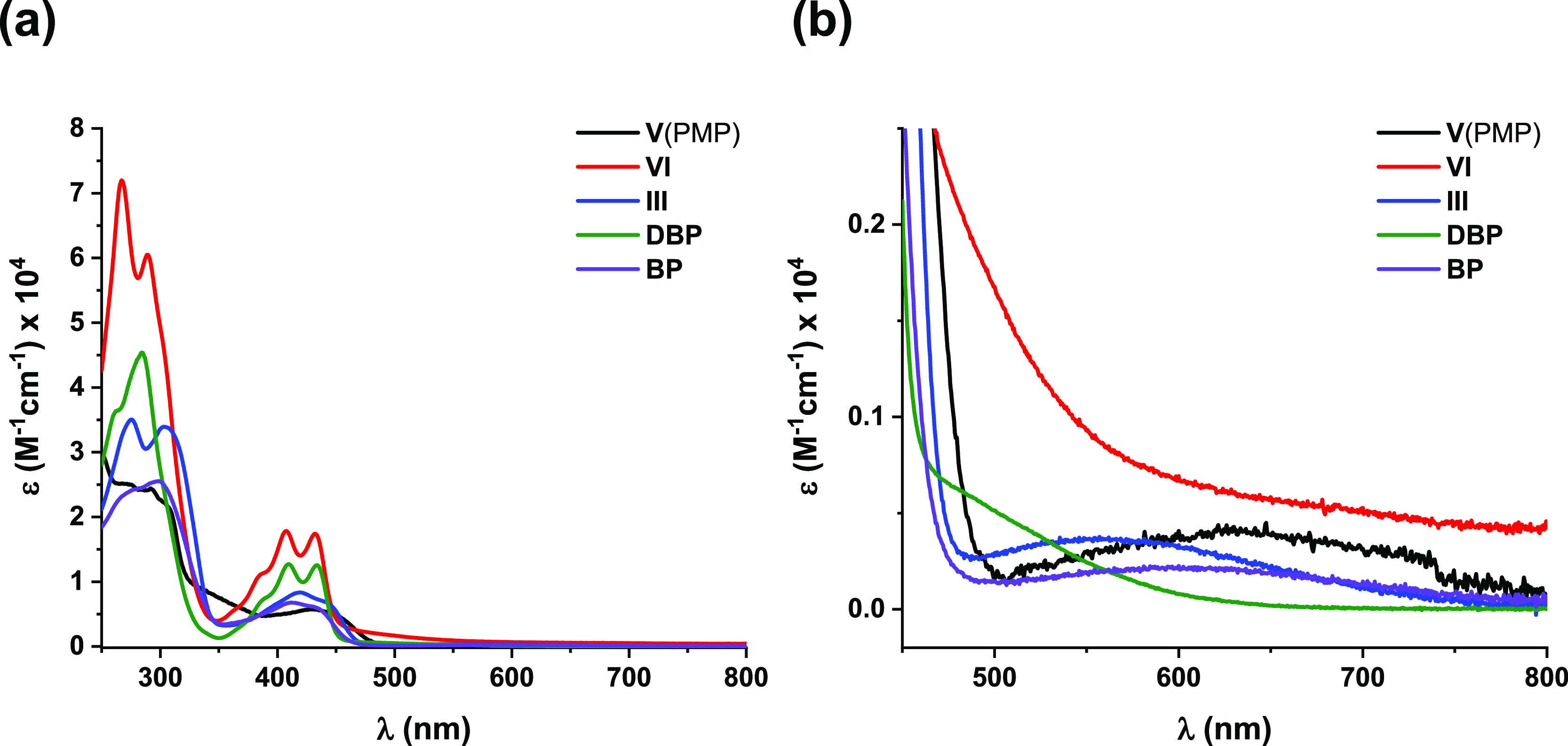
(a) UV–vis spectra of the synthesized new compounds and of DBP and BP for comparison; (b) partial UV–vis spectra showing the low-intensity, long wavelength absorptions. All spectra were recorded in CHCl3.
These long wavelength low-energy bands that are characteristic to pentalenes could be assigned as the symmetry-forbidden HOMO → LUMO transitions,16−19,24,26,30,33−35,43,44,49 whereas those in the 350–450 nm region could be assigned as the symmetry-allowed HOMO – 1 → LUMO transitions (Tables S7 and S8, Supporting Information). Clear maxima that could be obtained for III (549 nm) and V(PMP) (610 nm) correspond to an optical HOMO–LUMO gap of 2.26 and 2.05 eV, respectively (Table 1) (for higher-intensity spectra in this region, see Section S2, Supporting Information). For comparison, BP has a maximum approximately at 600 nm and a gap of 2.06 eV. As the HOMO–LUMO gap for VI could not be determined based on its UV–Vis spectrum, time-dependent density functional theory calculations [at B3LYP/6-311+G(d,p) level of theory] were performed. In agreement with the UV–vis spectra of VI that lacked the clear long-wavelength maximum, theory provided a larger HOMO–LUMO gap of 2.81 eV (441 nm, f = 0.015). It is noted that a fulvene-like conjugation pattern has a strong contribution to the HOMO of VI (Figure 5), which is in agreement with crystallographic bond lengths within the molecule. Such fulvene-like local structures have been identified in π-extended pentalenes, associated with strongly alleviated antiaromaticity.44,49 Furthermore, the pendant phenyl rings have considerable HOMO contribution in the case of III and VI, while the methoxyphenyl ring has negligible in V(PMP). Compared to VI, the HOMO of III is more pentalene-like, whereas the pentalene contribution is highest in V(PMP), which already suggests the extent of antiaromaticity within the series. In all three cases, the pentalene character can be clearly recognized in the LUMO of the molecules.
Table 1. Summary of Electrochemical, Optical, and Computational Data for III, V(PMP), and VI.
| entry | compound | Epa (E1/2,ox) [V]a | Epc (E1/2,red) [V]a | ΔEredox [eV]b | ΔEopt [eV] | ΔEcalc [eV]e |
|---|---|---|---|---|---|---|
| 1 | III | 0.65 (0.60)c | –1.68d | 1.95 | 2.26 | 2.25 |
| 2 | V(PMP) | 0.54 (0.51)c | –1.61 (−1.58)c | 1.96 | 2.05 | 1.99 |
| 3 | VI | 0.93d | –1.93d | 2.46 | n.d. | 2.81 |
Electrochemical measurements were carried out in 0.1 M Bu4NPF6 in dichloromethane at a scan rate of 0.1 V s–1 on a platinum wire working electrode. All potentials are given versus the Fc/Fc+ couple used as the internal standard (half-wave potentials in parenthesis).
Estimated from the differences between onset potentials.
Reversible first reduction or oxidation wave.
Irreversible first reduction or oxidation wave.
Calculations were performed on the B3LYP/6-311+G(d,p) level of theory.
Figure 5.
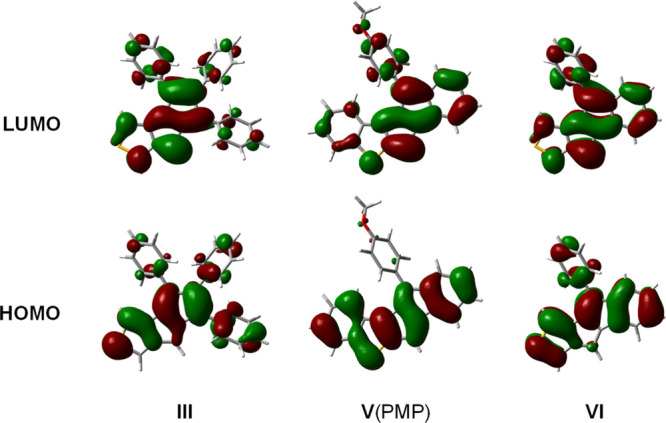
Calculated HOMOs and LUMOs of III, V(PMP), and VI (isosurface of 0.02 a.u. is used).
Electrochemistry
The electrochemical behavior of the synthesized thienopentalene derivatives was examined by cyclic voltammetry (CV) (for further details, see Section S3, Supporting Information). Notably, while for V(PMP) both the first oxidation and reduction processes were reversible, for III, only the oxidation step was reversible, and both redox processes were irreversible in the case of VI. The electrochemical energy gaps change in the order of V(PMP) (1.96 V) ≈ III (1.95 V) < VI (2.46 V) (Table 1). The similar ΔEredox values obtained for V(PMP) and III could partially come from the approximative determination of the onset potentials that are used for the calculations. Among the structures, VI can be compared with dibenzo[a,e]pentalenes and previously reported symmetric dithienopentalenes32,34 as it contains the same number of fused rings. The electrochemically determined HOMO–LUMO gap of VI (2.46 eV) is almost equal to what has been measured for TMS-substituted dibenzo[a,e]pentalene (2.48 eV),34 while significantly larger than that of pentaleno[1,2-b:4,5-b′]dithiophenes (1.87–1.97 eV).34 The electrochemical gap for pentaleno[1,2-c:4,5-c′]dithiophenes where both thiophene rings are fused through their 3,4(c) bond to the pentalene unit approaches 3 eV,32 which is somewhat larger compared to that of VI. Similar to pentaleno[1,2-c:4,5-c′]dithiophene derivatives,32VI polymerized during the measurements (Figure S16, Supporting Information).
Aromaticity Analysis
All geometry optimizations were made with the Gaussian 0951 package using the B3LYP52 hybrid functional and the 6-311+G(d,p)53 basis set. The (anti)aromatic character of the studied thienopentalene derivatives was evaluated by means of magnetic (NICS-XY scans54 and ACID plots55,56) and electronic indices (MCI57 and FLU58) in their ground (S0) state, computed at the B3LYP/6-311+G(d,p) level (for further details, see Section S4, Supporting Information). Aromatic rings are characterized by negative NICS values and clockwise ring currents revealed through ACID plots, whereas positive NICS values and anticlockwise ring currents are indicative of antiaromaticity.48 The higher the MCI and the lower the FLU, the more aromatic are the rings. For further details, see Section S4 in Supporting Information.
Magnetic-based aromaticity descriptors support the experimental findings. Among the synthesized compounds, based on the experimental data, the pentalene unit in VI is characterized by the lowest antiaromaticity, somewhat higher in III, while in V(PMP), it exhibits the highest. NICS-XY scans of the molecules support this trend (Figure 6a–c). Both in III and VI, the thiophene ring is essentially non-aromatic, although this non-aromaticity resides slightly on the antiaromatic side of the scale in III while on the aromatic side in VI. In the case of III, the small maximum in the scan over the thiophene ring could indicate a weak global paratropic current in the molecule.59 The antiaromaticity of the pentalene units is lowest in VI, which is stabilized by the 3,4-fusion of the thiophene ring on one hand and by the fused benzene ring on the other hand. In contrast to the non-aromatic thiophene unit, the benzene ring preserved a considerable level of aromaticity. The antiaromaticity of the pentalene unit was found the strongest in V(PMP) based on the NICS-XY scans. The fusion of a benzothiophene unit contributes to a preserved antiaromaticity due to the strong alkene character of the fused bond in the thiophene ring. This destabilizing interaction is compensated with the preserved aromaticity of the benzene ring within the benzothiophene unit. Likely due to the destabilizing fusion of the benzothiophene, the aromaticity of the benzene ring that is fused on the other side of the pentalene unit is strongly alleviated. This is in contrast to what was observed for VI, yet in that case, the thiophene fusion provided strong stabilization for the pentalene unit. Overall, the electronic nature of the fused system on one side of the antiaromatic unit influences the aromaticity of the fused ring on the other side of the same unit (Figure 6g–i). Destabilizing fusion on one side contributes to preserved antiaromaticity of the pentalene unit, which leads to alleviated aromaticity on the other side, such as in V(PMP) (Figure 6i). However, stabilizing fusion on one side contributes to alleviated antiaromaticity of the pentalene unit, which leads to preserved aromaticity on the other side, such as in VI (Figure 6h). Similar conclusions were found computationally for thiophene-fused benzocyclobutadienes recently.60 The possibility of tuning multiple electronic interactions in diarenopentalenes to alleviate antiaromaticity or to preserve aromaticity of the subunits is clearly an advantage that can be exploited in the molecular design. Due to their monoannelated structure, these possibilities are limited for monoarenopentalenes, which contributes to their strongly preserved antiaromaticity on one hand and could limit their synthetic accessibility on the other. The NICS-XY scans of the synthetically unfeasible molecules I, II, and IV support this explanation (see Section S4.2, Supporting Information). Complementary to NICS-XY scans, ACID plots (Figure 6d–f) reflect the extent of tropicities in each molecule, which is in agreement with the conclusions based on NICS calculations. For the ACID plots of structures I, II, and IV, see Section S4.2 in Supporting Information. The calculated transition energies (Table S7, Supporting Information) support the antiaromaticity trend [VI < III < V(PMP)] revealed by the NICS-XY scans, ACID, and MCI (λVI(HOMO → LUMO) = 440.86 nm, λIII(HOMO → LUMO) = 550.36 nm, and λV(PMP) (HOMO → LUMO) = 622.40 nm).
Figure 6.
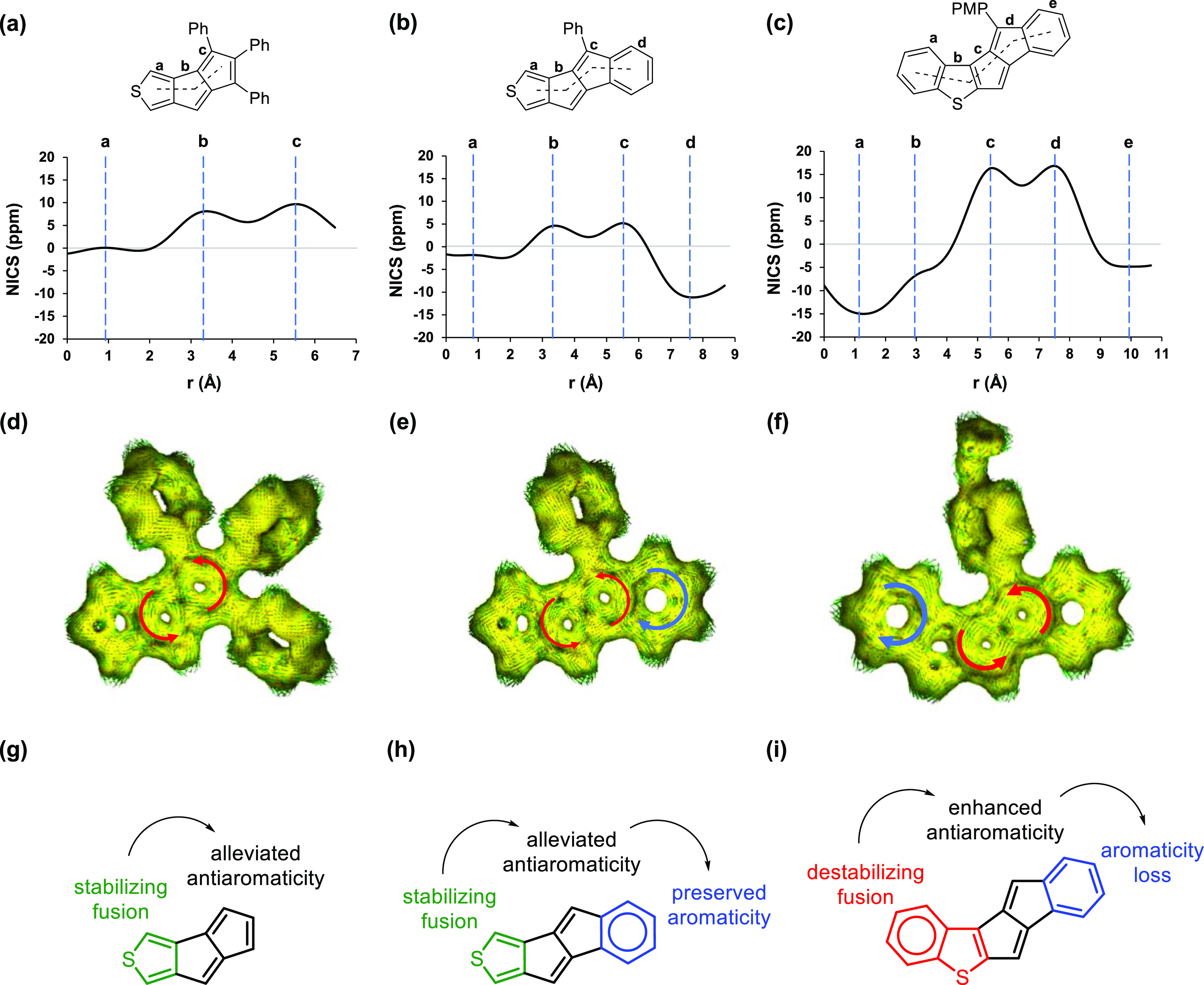
NICS-XY scans (a–c) and ACID plots (d–f) of compounds III, VI, and V(PMP) in their ground states. Antiaromaticity changes within the subunits of the molecules (substituents are removed for clarity) are also shown (g–i).
To complement the results of magnetic aromaticity calculations, we also analyzed the molecules by means of electronic indices. The antiaromaticity trend revealed by MCI data is in line with the results obtained from NICS-XY scans. The antiaromaticity of the pentalene unit is similar in III and VI (MCI = 0.0008 e and 0.0007 e, respectively), whereas it is enhanced in V(PMP) (MCI = 0.0004 e). As a comparison, pentalene itself has an MCI value of −0.005 e (for comparative FLU and DI analyses of the synthesized and proposed molecules, see Section S4.2 in Supporting Information).
Summary
In summary, we explored the synthesis and opto-electronic properties of unsymmetrical thienopentalenes. From a synthetic point, these molecules provided a ground for testing and exploiting recently developed methodologies in the preparation of novel pentalene derivatives. We showed that both the cascade carbopalladation reaction and the gold-catalyzed cyclization of diynes is a reliable methodology for the synthesis of unsymmetrical thienopentalenes. Notably, the stability of monoareno pentalenes reached its limit upon the destabilizing (2,3-) fusion of (benzo)thiophene (structures I and II). Stabilizing (3,4-) fusion of a single thiophene ring to pentalene (compound III) led to considerable alleviation of its antiaromaticity. In the case of the unsymmetrical diareno derivatives, we found that the nature of the (benzo)thiophene fusion on one edge of the pentalene unit affects the aromaticity of the fused benzene ring on the other edge. Destabilizing benzothiophene fusion resulted in aromaticity loss in the fused benzene ring (compound V) while stabilizing fusion of thiophene resulted in preserved aromaticity in the benzene unit (compound IV). In this latter case, due to the extensive stabilization of the pentalene unit, its antiaromaticity is essentially diminished and exhibited a diene/fulvene character.
Experimental Section
General Information
Commercial reagents, solvents, and catalysts (Aldrich, Fluorochem, VWR) were purchased as reagent grade and used without further purification. Solvents for extraction or column chromatography were of technical quality. Organic solutions were concentrated by rotary evaporation at 25–40 °C. Thin-layer chromatography (TLC) was carried out on SiO2-layered aluminum plates (60778-25EA, Fluka). Column chromatography was performed using SiO2-60 (230–400-mesh ASTM, 0.040–0.063 mm from Merck) at 25 °C or using a Teledyne Isco CombiFlash Rf+ automated flash chromatographer with silica gel (25–40 μm, Zeochem). Room temperature refers to 25(±1) °C. NMR spectra were acquired on a Varian 500 NMR spectrometer, running at 500 and 126 MHz for 1H and 13C, respectively. The residual solvent peaks were used as the internal reference. Chemical shifts (δ) are reported in ppm. The following abbreviations are used to indicate the multiplicity in 1H NMR spectra: s, singlet; d, doublet; t, triplet; q, quartet; h, heptet; and m, multiplet. 13C NMR spectra were acquired on a broad-band decoupled mode. Gas chromatography–mass spectrometry (GC–MS) analysis was performed on a Shimadzu GCMS-QP2010 UltraSystem operated in the electron impact ionization mode. High-resolution measurements were performed on a Sciex TripleTOF 5600+ high-resolution tandem mass spectrometer equipped with a DuoSpray ion source. Atmospheric-pressure chemical ionization was applied in the positive ion detection mode. Samples were dissolved in acetonitrile and flow injected into the acetonitrile/water 1:1 flow. The flow rate was 0.2 mL/min. The resolution of the mass spectrometer was 35000. UV–vis spectra were acquired on a Jasco V-750 spectrophotometer. Melting point was determined visually with a Boetius PHMK-05 hot stage microscope (VEB Kombinat Nagema, Germany).
General Procedures
General Procedure for Gem-dibromoolefination (GP1)
The aldehyde (1 equiv) and CBr4 (1.5 equiv) were stirred at 0 °C in CH2Cl2 (10 mL/mmol) under an inert atmosphere (N2). A solution of P(OiPr)3 (3 equiv) in CH2Cl2 was added dropwise to the mixture, and the reaction was stirred at 0 °C for 1–6 h. After the reaction was completed, the mixture was diluted with CH2Cl2 and washed with water twice, then the organic phase was dried on MgSO4. The solvent was evaporated under reduced pressure, and the product was purified with column chromatography (SiO2; n-hexane/EtOAc 12:1).
General Procedure for the Carbopalladation Cascade Reaction (GP2)
The dibromoolefin (0.22 mmol, 1 equiv), diphenylacetylene (200 mg, 1.10 mmol, 5 equiv), K2CO3 (61 mg, 0.44 mmol, 2 equiv), Pd(PPh3)2Cl2 (16 mg, 23 μmol 0.1 equiv), Zn powder (20 mg, 30 μmol, 1.4 equiv), and toluene (4 mL) were added to a sealed vial and stirred under an inert atmosphere for 1 h at 110 °C in an aluminum heating block. The reaction was cooled to rt, hydroquinone (30 mg, 0.27 mmol, 1.2 equiv) was added to the reaction mixture, and it was stirred at 110 °C for additional 16 h. After the reaction was completed, the mixture was diluted with EtOAc and washed with water twice and brine once. The organic phase was dried over MgSO4. The solvent was evaporated under reduced pressure, and the products were purified by column chromatography (SiO2; n-hexane/EtOAc, 12:1).
General Procedure for Seyferth–Gilbert Homologation (GP3)
The aldehyde (1 equiv) and Cs2CO3 (4 equiv) were stirred in methanol (10 mL/mmol) under a N2 atmosphere at rt. The Bestmann–Ohira reagent (1.5 equiv) was added dropwise to the reaction, which was stirred for 1–5 h at rt. The reaction was monitored by TLC. After the reaction was completed, the solvent was evaporated, and the product was purified with column chromatography (SiO2; n-hexane/EtOAc 12:1).
Synthesis of 3-Bromobenzo[b]thiophene-2-carbaldehyde (2)61,62
Bromine (1.33 g, 428 μL, 8.32 mmol) in CHCl3 (2 mL) was added dropwise to a stirred, ice cold solution of benzo[b]thiophene-2-carbaldehyde (1) (1.35 g, 8.32 mmol) in CHCl3 (10 mL). After the addition, the solution was allowed to warm to rt and was stirred for 10 h. The mixture was diluted with CHCl3, washed once with water, twice with 1 M NaOH, and once with brine. The organic phase was dried over MgSO4. The solvent was evaporated under reduced pressure. The crude product was recrystallized from acetic acid (4 mL). The product was obtained as pale yellow crystals. Yield: 1.25 g, 5.18 mmol, 63%. 1H NMR (500 MHz, CDCl3): δ 10.29 (s, 1H), 8.02 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.55 (dt, J = 14.9, 7.2 Hz, 2H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 184.8, 140.6, 138.2, 136.7, 129.4, 126.1, 125.2, 123.6, 118.9 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C9H6OSBr+, 240.9317; found, 240.9314.
Synthesis of 3-(Phenylethynyl)benzo[b]thiophene-2-carbaldehyde (3)63
3-Bromobenzo[b]thiophene-2-carbaldehyde (2) (300 mg, 1.24 mmol), phenylacetylene (152 mg, 164 μL, 1.5 mmol), Pd(PPh3)2Cl2 (16.4 mg, 23.4 μmol), and CuI (7 mg, 37 μmol) were stirred under an inert atmosphere in Et3N (10 mL) at rt for 3 h. After the reaction was completed, the mixture was diluted with EtOAc and washed once with water, twice with 10% HCl, and once with brine. The organic phase was dried over MgSO4, and the solvent was evaporated in vacuo. The crude product was purified by column chromatography (SiO2; n-hexane/EtOAc 12:1). The product was obtained as a yellow solid. Yield: 307 mg, 1.17 mmol, 94%. 1H NMR (500 MHz, CDCl3): δ 10.48 (s, 1H), 8.16 (d, J = 7.6 Hz, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.70–7.61 (m, 2H), 7.59–7.49 (m, 2H), 7.48–7.39 (m, 3H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 184.6, 143.6, 141.2, 139.6, 132.1 (2), 129.7, 129.0 (2), 128.8, 127.9, 125.7, 125.2, 123.5, 122.1, 99.2, 80.7 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C17H11OS+, 263.0525; found, 263.0522.
Synthesis of 2-(2,2-Dibromovinyl)-3-(phenylethynyl)benzo[b]thiophene (4)
This compound was synthesized from compound 3 according to GP1. The reaction was stirred for 0.5 h. The crude product was purified by column chromatography (SiO2; n-hexane), yielding a pale-yellow solid. Yield: 119 mg, 0.29 mmol, 78%. 1H NMR (500 MHz, CDCl3): δ 8.23 (s, 1H), 8.01 (dd, J = 5.2, 3.8 Hz, 1H), 7.80 (dd, J = 5.2, 3.7 Hz, 1H), 7.64 (dd, J = 6.5, 3.0 Hz, 2H), 7.48–7.38 (m, 5H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 140.4, 138.6, 138.2, 131.8 (2), 131.2, 129.0, 128.7 (2), 126.5, 125.4, 123.5, 122.9, 122.3, 120.5, 98.3, 91.2, 82.5 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C18H11SBr2+, 416.8942; found, 416.8957.
Synthesis of 1,2,3-Triphenylbenzo[b]pentaleno[1,2-d]thiophene (I)
The synthesis of this compound was attempted from compound 4 according to GP2. The desired product was not detected.
Synthesis of 3-(Phenylethynyl)thiophene-2-carbaldehyde (6)46
3-Bromothiophene-2-carbaldehyde (5) (400 mg, 2.09 mmol), phenylacetylene (256.6 mg, 276 μL, 2.51 mmol), Pd(PPh3)2Cl2 (44 mg, 62.8 μmol), and CuI (11.9 mg, 62.8 μmol) were stirred in THF (15 mL) at rt under an inert atmosphere (N2). After a few minutes, diisopropylamine (5 mL) was added to the reaction mixture. Following the addition, the reaction was stirred for 18 h at 60 °C in an aluminum heating block. The reaction was monitored by TLC (n-hexane/EtOAc 12:1). Upon completion, the mixture was diluted with EtOAc and washed twice with water and once with brine. The organic phase was dried over MgSO4. The crude product was purified by column chromatography [SiO2, n-hexane → n-hexane/EtOAc (12:1)]. The product was obtained as a yellow oil. Yield: 309 mg, 1.46 mmol, 69%. 1H NMR (500 MHz, CDCl3): δ 10.24 (d, J = 1.3 Hz, 1H), 7.69 (dd, J = 5.0, 1.3 Hz, 1H), 7.59–7.51 (m, 2H), 7.43–7.33 (m, 3H), 7.25 (d, J = 5.0 Hz, 1H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 183.1, 143.7, 134.0, 131.9 (2), 131.7, 131.1, 129.4, 128.7 (2), 122.1, 96.2, 81.7 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C13H9OS+, 213.0368; found, 213.0360.
Synthesis of 2-(2,2-Dibromovinyl)-3-(phenylethynyl)thiophene (7)
This product was prepared from compound 6 according to GP1. The reaction was stirred for 3 h. The product was obtained as a colorless oil. Yield: 469 mg, 0.839 mmol, 90%. 1H NMR (500 MHz, CDCl3): δ 8.06 (s, 1H), 7.57–7.52 (m, 2H), 7.40–7.33 (m, 4H), 7.13 (d, J = 5.2 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3): δ 140.1, 131.7 (2), 130.5, 129.3, 128.8, 128.6 (2), 126.4, 124.4, 122.9, 95.2, 88.8, 83.6 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C14H9SBr2+, 366.8786; found, 366.8799.
Synthesis of 4,5,6-Triphenylpentaleno[2,1-b]thiophene (II)
The synthesis of this compound was attempted from compound 7 according to GP2. The desired product was not detected.
Synthesis of 4-Bromothiophene-3-carbaldehyde (9)64
3,4-Dibromothiophene (8) (3.00 g, 1.37 mL, 12.4 mmol) was stirred in diethyl ether (15 mL) under an inert atmosphere (N2) at −78 °C (acetone/dry ice) for 5 min, then n-BuLi (5.21 mL, 2.5 M in hexane) was added dropwise to the reaction. The mixture was stirred for 40 min at −78 °C, then DMF (1.36 g, 1.45 mL, 18.6 mmol) was added, and the reaction was stirred for another 2 h. The reaction was quenched with saturated NH4Cl solution and allowed to warm to rt. The mixture was extracted twice with diethyl ether, and the combined organic phase was dried over MgSO4. The solvent was evaporated under reduced pressure. The crude product was purified by column chromatography [SiO2, n-hexane → n-hexane/EtOAc (9:1)]. The product was obtained as a yellow oil. Yield: 1.94 g, 10.15 mmol, 82%. 1H NMR (500 MHz, CDCl3): δ 9.95 (s, 1H), 8.16 (d, J = 3.4 Hz, 1H), 7.36 (d, J = 3.4 Hz, 1H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 184.8, 137.7, 134.7, 125.2, 111.5 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C5H4OSBr+, 190.9160; found, 190.9154.
Synthesis of 4-(Phenylethynyl)thiophene-3-carbaldehyde (10)
4-Bromothiophene-3-carbaldehyde (9) (500 mg, 2.62 mmol), phenylacetylene (321 mg, 345 μL, 3.14 mmol), Pd(PPh3)2Cl2 (55 mg, 78.5 μmol), and CuI (15 mg, 78.5 μmol) were stirred in THF (12 mL) at rt under an inert atmosphere (N2). After a few minutes, diisopropylamine (3 mL) was added to the mixture. After the addition, the reaction was stirred overnight at 60 °C in an aluminum heating block. The reaction was monitored by TLC [n-hexane/EtOAc (12:1)]. Upon the completion of the reaction, the mixture was diluted with EtOAc and washed twice with water and once with brine. The organic phase was dried over MgSO4. After evaporation of the solvent under reduced pressure, the crude product was purified by column chromatography [SiO2, n-hexane → n-hexane/EtOAc (12:1)]. The product was obtained as a brown solid. Yield: 502 mg, 2.36 mmol, 90%. 1H NMR (500 MHz, CDCl3): δ 10.15 (s, 1H), 8.14 (d, J = 3.2 Hz, 1H), 7.58–7.53 (m, 3H), 7.39–7.34 (m, 3H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 185.0, 140.7, 133.1, 131.8 (2), 130.3, 129.0, 128.6 (2), 123.4, 122.6, 93.1, 81.6 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C13H9OS+, 213.0368; found, 213.0359.
Synthesis of 3-(2,2-Dibromovinyl)-4-(phenylethynyl)thiophene (11)
The product was prepared from compound 10 according to GP1. The reaction was stirred for 3 h. The product was obtained as a pale yellow solid. Yield: 796 mg, 2.16 mmol, 92%. 1H NMR (500 MHz, CDCl3): δ 8.06 (d, J = 3.0 Hz, 1H), 7.71 (s, 1H), 7.57–7.51 (m, 2H), 7.50 (d, J = 3.1 Hz, 1H), 7.40–7.34 (m, 3H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 136.5, 131.8 (2), 130.3, 128.8, 128.6 (2), 128.1, 124.4, 123.6, 122.9, 92.9, 90.4, 82.9 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C14H9SBr2+, 366.8786; found, 366.8799.
Synthesis of 4,5,6-Triphenylpentaleno[1,2-c]thiophene (III)
This product was prepared from compound 11 according to GP2. The product was obtained as a green solid. Yield: 9 mg, 23.3 μmol, 11%. 1H NMR (500 MHz, CD2Cl2): δ 7.25–7.17 (m, 9H), 7.12–7.08 (m, 4H), 7.01–6.97 (m, 2H), 6.69 (s, 2H), 6.53 (s, 1H) ppm; 13C{1H} NMR (126 MHz, CD2Cl2): δ 160.8, 153.8, 148.7, 139.1, 139.1, 136.0, 135.2, 135.2, 134.5, 132.0, 130.2 (2), 129.2 (2), 129.2 (2), 129.0, 128.7 (2), 128.5 (2), 128.5 (2), 128.3, 128.0, 127.3, 118.8, 115.3 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C28H19S+, 387.1201; found, 387.1204.
Synthesis of 2-Ethynyl-3-(phenylethynyl)benzo[b]thiophene (12)46
This compound was synthesized from 3 according to GP3. The reaction was stirred for 1 h. The product was purified by column chromatography (SiO2; n-hexane) and obtained as a brownish yellow solid. Yield: 100 mg, 387.1 μmol, 53%. 1H NMR (500 MHz, CDCl3): δ 8.03–7.98 (m, 1H), 7.79–7.75 (m, 1H), 7.70–7.65 (m, 2H), 7.52–7.44 (m, 2H), 7.44–7.38 (m, 3H), 3.79 (s, 1H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 138.6, 138.5, 132.0 (2), 128.8, 128.5 (2), 126.7, 125.4, 124.8, 124.2, 123.8, 123.0, 122.3, 96.6, 87.4, 82.3, 76.7 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C18H11S+, 259.0575; found, 259.0568.
Synthesis of Benzo[b]benzo[4,5]pentaleno[1,2-d]thiophene [V(H)]
In a 20 mL vial, chloro[1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene]gold(I) (29 mg, 46.6 μmol) and AgSbF6 (20 mg, 58.2 μmol) were stirred in 1,2-dichloroethane (1 mL) for 5 min under an inert atmosphere (N2) at rt. 2-Ethynyl-3-(phenylethynyl)benzo[b]thiophene (12) (277.0 mg, 1.07 μmol) in 1,2-dichloroethane (7 mL) was added to the stirred solution, and the mixture was stirred for 16 h under an inert atmosphere (N2) at 80 °C in an aluminum heating block. The reaction was monitored with TLC (n-hexane). After the completion of the reaction, the mixture was diluted with CH2Cl2 and filtered through a pad of Celite. The solvent was evaporated in vacuo. The crude product was purified by column chromatography (SiO2, n-hexane). The title compound was not obtained; instead, a yellow solid product was isolated, which is suggested to be structure 13.43 Yield: 15 mg, 29.0 μmol, 30%. 1H NMR (500 MHz, CDCl3): δ 8.34 (d, J = 1.7 Hz, 1H), 8.23–8.19 (m, 1H), 8.06–8.03 (m, 1H), 7.96 (d, J = 1.7 Hz, 1H), 7.82–7.76 (m, 3H), 7.75–7.70 (m, 2H), 7.70–7.66 (m, 1H), 7.54–7.44 (m, 6H), 7.44–7.39 (m, 4H) ppm.
Synthesis of 2,3-Dibromobenzo[b]thiophene (15)65
Bromine (7.50 g, 2.35 mL, 46.93 mmol) in CHCl3 (10 mL) was added dropwise to a stirred solution of benzo[b]thiophene (14) (3.00 g, 22.36 mmol) in CHCl3 (50 mL) under an inert atmosphere (N2) at 0 °C. After the addition, the mixture was allowed to warm to rt, and it was stirred for an additional 18 h. The mixture was diluted with CHCl3 and washed once with water, twice with saturated NaHCO3, and once with brine. The organic phase was dried over MgSO4, and the solvent was evaporated in vacuo. The product was sufficiently pure to be used without any further purification. The product was obtained as a white solid. Yield: 6.48 g, 22.2 mmol, >95%. 1H NMR (500 MHz, CDCl3): δ 7.75 (d, J = 7.4 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.43 (td, J = 7.6, 1.2 Hz, 1H), 7.38 (td, J = 7.7, 1.3 Hz, 1H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 139.1, 137.7, 125.8, 125.7, 123.5, 122.0, 114.4, 111.9; HRMS (APCI) m/z: [M]+ calcd for C8H4SBr2+, 289.8400; found, 289.8403.
Synthesis of 2,3-Bis(phenylethynyl)benzo[b]thiophene (16)66
2,3-Dibromobenzo[b]thiophene (15) (600 mg, 2.05 mmol), phenylacetylene (504 mg, 542 μL, 4.93 mmol), Pd(PPh3)2Cl2 (72 mg, 103 μmol), and CuI (19.6 mg, 103 μmol) were stirred in THF (10 mL) at rt under a N2 atmosphere. After a few minutes, diisopropylamine (2 mL) was added to the reaction mixture. After the addition, the reaction was stirred for 18 h at 60 °C in an aluminum heating block. The reaction was monitored by TLC (n-hexane). The reaction mixture was diluted with EtOAc and washed once with 1 M HCl and once with brine. The organic phase was dried over MgSO4. The solvent was evaporated under reduced pressure. The crude product was purified by column chromatography (SiO2, n-hexane) and obtained as a yellow solid. Yield: 599 mg, 1.79 mmol, 87%. 1H NMR (500 MHz, CDCl3): δ 8.03–7.98 (m, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.70–7.65 (m, 2H), 7.65–7.60 (m, 2H), 7.51–7.37 (m, 8H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 138.8, 138.8, 131.9 (2), 131.8 (2), 129.1, 128.7, 128.6 (2), 128.6 (2), 126.4, 126.4, 125.3, 123.6, 123.3, 122.9, 122.8, 122.3, 99.8, 96.6, 83.0, 82.8 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C24H15S+, 335.0888; found, 335.0894.
Synthesis of 11-Phenylbenzo[b]benzo[4,5]pentaleno[1,2-d]thiophene and 6-phenylbenzo[b]benzo[4,5] pentaleno[2,1-d]thiophene [V(Ph)]
In a 40 mL vial, chloro[1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene]gold(I) (18.3 mg, 53.2 μmol) and AgSbF6 (33.1 mg, 53.2 μmol) were stirred in degassed dry 1,2-dichloroethane (2 mL) for 5 min under an inert atmosphere (N2). 2,3-Bis(phenylethynyl)benzo[b]thiophene (16) (356 mg, 1.06 mmol) in degassed dry 1,2-dichloroethane (8 mL) was added to the stirred solution, and the mixture was stirred for 6 h under an inert atmosphere (N2) at 65 °C in an aluminum heating block. The reaction was monitored with TLC (n-hexane). After the completion of the reaction, the mixture was diluted with CH2Cl2 and filtered through a pad of Celite. The solvent was evaporated in vacuo. The crude product was purified by column chromatography (SiO2, n-hexane). The products were obtained as an inseparable mixture of isomers and other impurities. HRMS (APCI) m/z: [M + H]+ calcd for C24H15S+, 335,0888; found, 335,0888.
Synthesis of 3-((4-Methoxyphenyl)ethynyl)benzo[b]thiophene-2-carbaldehyde (17)67
3-Bromobenzo[b]thiophene-2-carbaldehyde (2) (300 mg, 1.24 mmol), 1-ethynyl-4-methoxybenzene (197 mg, 1.5 mmol), Pd(PPh3)2Cl2 (26.4 mg, 37.6 μmol), and CuI (7 mg, 37.6 μmol) were stirred under an inert atmosphere (N2) in triethylamine (10 mL) at rt for 5 h. After the reaction was completed, the mixture was diluted with EtOAc and filtered through a pad of Celite. The solvent was evaporated in vacuo. The crude product was purified by column chromatography [SiO2, n-hexane → n-hexane/EtOAc (9:1)]. The product was obtained as a yellow solid. Yield: 265 mg, 0.88 mmol, 73%. 1H NMR (500 MHz, CDCl3): δ 10.44 (s, 1H), 8.16–8.10 (m, 1H), 7.87–7.82 (m, 1H), 7.58 (d, J = 8.8 Hz, 2H), 7.55–7.47 (m, 2H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 184.6, 160.8, 142.9, 141.2, 139.5, 133.7 (2), 128.9, 128.4, 125.6, 125.12, 123.4, 114.5 (2), 114.0, 99.6, 79.8, 55.5 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C18H13O2S+, 293.0630; found, 293.0632.
Synthesis of 2-Ethynyl-3-((4-methoxyphenyl)ethynyl)benzo[b]thiophene (18)46
This compound was synthesized from 17 according to GP3. The reaction was stirred for 3 h. The product was purified by column chromatography [SiO2, n-hexane → n-hexane/EtOAc (12:1)] and obtained as a brownish yellow solid. Yield: 154 mg, 0,534 mmol, 59%. 1H NMR (500 MHz, CDCl3): δ 8.00–7.95 (m, 1H), 7.77–7.72 (m, 1H), 7.59 (d, J = 8.8 Hz, 2H), 7.49–7.42 (m, 2H), 6.92 (d, J = 8.8 Hz, 2H), 3.85 (s, 3H), 3.76 (s, 1H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 160.2, 138.6, 138.6, 133.5 (2), 126.6, 125.3, 124.6, 124.0, 123.8, 122.3, 115.1, 114.2 (2), 96.8, 87.2, 81.2, 76.9, 55.5 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C19H13OS+, 289.0681; found, 289.0677.
Synthesis of 3-((4-Methoxyphenyl)ethynyl)-2-(phenylethynyl)benzo[b]thiophene (19)
2-Ethynyl-3-((4-methoxyphenyl)ethynyl)benzo[b]thiophene (18) (140 mg, 486 μmol), iodobenzene (149 mg, 81 μL, 728 μmol), Pd(PPh3)2Cl2 (10.2 mg, 14.6 μmol), and CuI (2.8 mg, 14.6 μmol) were stirred in THF (7 mL) at rt under an inert atmosphere (N2). After a few minutes, diisopropylamine (0.5 mL) was added to the reaction mixture. After the addition, the reaction was stirred overnight (16 h) at rt. The reaction was monitored by TLC (n-hexane/EtOAc 12:1). The reaction mixture was diluted with EtOAc and washed twice with water and once with brine. The organic phase was dried over MgSO4. The solvent was evaporated in vacuo. The crude product was purified by column chromatography [SiO2, n-hexane → n-hexane/EtOAc (12:1)]. The product was obtained as a yellow oil. Yield: 147 mg, 0.403 mmol, 83%. 1H NMR (500 MHz, CDCl3): δ 8.02–7.97 (m, 1H), 7.79–7.75 (m, 1H), 7.65–7.58 (m, 4H), 7.50–7.41 (m, 2H), 7.41–7.36 (m, 3H), 6.94 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 160.1, 138.9, 138.8, 133.4 (2), 131.8 (2), 129.0, 128.6 (2), 126.3, 125.6, 125.2, 123.6, 123.3, 122.8, 122.3, 115.4, 114.3 (2), 99.5, 96.8, 83.0, 81.7, 55.5 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C25H17OS+, 365.0994; found, 365.0996.
Synthesis of 11-(4-Methoxyphenyl)benzo[b]benzo[4,5]pentaleno[1,2-d]thiophene [V(PMP)]
In a 4 mL vial, chloro[1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene]gold(I) (6.7 mg, 10.8 μmol) and AgSbF6 (3.7 mg, 10.8 μmol) were stirred in degassed dry 1,2-dichloroethane (1 mL) under an inert atmosphere (N2) at rt. After 5 min, 3-((4-methoxyphenyl)ethynyl)-2-(phenylethynyl)benzo[b]thiophene (19) (78.7 mg, 216 μmol) in degassed dry 1,2-dichloroethane (1 mL) was added to the stirred solution, and the mixture was stirred for 3 h under an inert atmosphere (N2) at 65 °C in an aluminum heating block. The reaction was monitored with TLC (n-hexane). After the completion of the reaction, the mixture was diluted with CH2Cl2 and filtered through a pad of Celite. The solvent was evaporated in vacuo. The crude product was purified by column chromatography [SiO2, n-hexane → n-hexane/EtOAc (12:1)]. The product is a deep green solid. Yield: 7.2 mg, 19.8 μmol, 9%. 1H NMR (500 MHz, CD2Cl2): δ 7.60–7.52 (m, 3H), 7.09–7.05 (m, 2H), 7.05–7.00 (m, 3H), 6.81–6.73 (m, 2H), 6.72–6.65 (m, 2H), 6.10 (s, 1H), 3.91 (s, 3H) ppm; 13C{1H} NMR (126 MHz, CD2Cl2): δ 161.6, 152.8, 151.2, 150.7, 144.7, 143.9, 141.7, 137.7, 133.7, 132.8, 130.8 (2), 129.1, 128.3, 126.7, 125.5, 124.3, 123.8, 123.4, 122.7, 121.6, 119.5, 114.7 (2), 56.0 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C25H17OS+, 365.0994; found, 365.1001.
Synthesis of 3,4-Bis(phenylethynyl)thiophene (20)
3,4-Dibromothiophene (8) (750 mg, 343 μL 3.10 mmol), phenylacetylene (950 mg, 1.02 mL, 9.30 mmol), Pd(PPh3)2Cl2 (65 mg, 93 μmol), and CuI (18 mg, 93 μmol) were stirred in THF (12 mL) at rt under an inert atmosphere (N2). After a few minutes, diisopropylamine (3 mL) was added to the reaction mixture. After the addition, the reaction was stirred overnight at 60 °C in an aluminum heating block. The reaction was monitored by TLC (n-hexane). Upon completion, the mixture was diluted with EtOAc and washed twice with water and once with brine. The organic phase was dried over MgSO4. The crude product was purified by column chromatography (SiO2, n-hexane). The product was obtained as a yellow solid. Yield: 378 mg, 1.33 mmol, 43%. 1H NMR (500 MHz, CDCl3): δ 7.59–7.52 (m, 4H), 7.50 (s, 2H), 7.36–7.32 (m, 6H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 131.8 (4), 129.2 (2), 128.5 (4), 128.1 (2), 125.2 (2), 123.3 (2), 91.9 (2), 83.5 (2) ppm; HRMS (APCI) m/z: [M + H]+ calcd for C20H13S+, 285.0732; found, 285.0726.
Synthesis of 4-Phenylbenzo[4,5]pentaleno[1,2-c]thiophene (VI)
In a 4 mL vial, chloro[1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene]gold(I) (6.2 mg, 10.0 μmol) and AgSbF6 (3.4 mg, 10.0 μmol) were stirred in degassed dry 1,2-dichloroethane (1 mL) for 5 min under an inert atmosphere (N2). After that, 3,4-bis(phenylethynyl)thiophene (20) (57.0 mg, 200 μmol) in degassed dry 1,2-dichloroethane (1 mL) was added to the stirred solution, and the mixture was stirred for 3 h under an inert atmosphere (N2) at 65 °C in an aluminum heating block. The reaction was monitored by TLC (SiO2, hexane). After the completion of the reaction, the mixture was diluted with CH2Cl2 and filtered through a pad of Celite. The solvent was evaporated in vacuo. The crude product was purified by column chromatography (SiO2, n-hexane). The product was obtained as an orange solid. Yield: 25 mg, 87.9 μmol, 44%. mp 134.4–136.2 °C; 1H NMR (500 MHz, CD2Cl2): δ 7.73 (d, J = 7.2 Hz, 2H), 7.52 (t, J = 7.6 Hz, 2H), 7.43 (t, J = 7.4 Hz, 1H), 7.36–7.27 (m, 2H), 7.11–7.02 (m, 2H), 6.96 (d, J = 1.8 Hz, 1H), 6.72 (d, J = 1.8 Hz, 1H), 6.62 (s, 1H) ppm; 13C{1H} NMR (126 MHz, CD2Cl2): δ 155.3, 154.5, 148.6, 138.4, 137.5, 136.5, 135.0, 134.8, 129.4 (2), 129.2, 128.8, 128.5 (2), 127.3, 122.9, 122.7, 121.6, 117.0, 116.2 ppm; HRMS (APCI) m/z: [M + H]+ calcd for C20H13S+, 285.0732; found, 285.0726.
Acknowledgments
Financial support by the Lendület Program of the Hungarian Academy of Sciences (G.L.) and the National Research, Development and Innovation Office, Hungary [NKFIH grant no. and PD 128504 and K 124544 (T.H.)] is gratefully acknowledged. P.P.K. acknowledges support by the ÚNKP-20-2 New National Excellence Program of the Ministry for Innovation and Technology from the Source of the National Research, Development and Innovation Fund. M.P. acknowledges the professional support of the Doctoral Student Scholarship Program of the Co-operative Doctoral Program of the Ministry of Innovation and Technology financed from the National Research, Development and Innovation Fund. O.E.B. thanks the “Programa postdoctoral Beatriu de Pinós de la Secretaria d’Universitats i Recerca del Departament d’Empresa i Coneixement de la Generalitat de Catalunya” (2020-BP-00155). KIFÜ is acknowledged for computational resources. We are grateful to Krisztina Németh (MS Metabolomics Research Group, Instrumentation Center, Research Centre for Natural Sciences) for HRMS measurements. Dr. Tamás Pajkossy (Functional Interfaces Research Group, Research Centre for Natural Sciences) is acknowledged for his assistance with CV measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c05618.
The authors declare no competing financial interest.
Supplementary Material
References
- Saito M. Synthesis and Reactions of Dibenzo[a,e]pentalenes. Symmetry 2010, 2, 950–969. 10.3390/sym2020950. [DOI] [Google Scholar]
- Hopf H. Pentalenes – From Highly Reactive Antiaromatics to Substrates for Material Science. Angew. Chem., Int. Ed. 2013, 52, 12224–12226. 10.1002/anie.201307162. [DOI] [PubMed] [Google Scholar]
- Kawase T.; Nishida J.-i. π-Extended Pentalenes: The Revival of the Old Compound from New Standpoints. Chem. Rec. 2015, 15, 1045–1059. 10.1002/tcr.201402093. [DOI] [PubMed] [Google Scholar]
- Tobe Y. Non-Alternant Non-Benzenoid Aromatic Compounds: Past, Present, and Future. Chem. Rec. 2015, 15, 86–96. 10.1002/tcr.201402077. [DOI] [PubMed] [Google Scholar]
- Frederickson C. K.; Rose B. D.; Haley M. M. Explorations of the Indenofluorenes and Expanded Quinoidal Analogues. Acc. Chem. Res. 2017, 50, 977–987. 10.1021/acs.accounts.7b00004. [DOI] [PubMed] [Google Scholar]
- Dressler J. J.; Haley M. M. Learning how to Fine-Tune Diradical Properties by Structure Refinement. J. Phys. Org. Chem. 2020, 33, e4114 10.1002/poc.4114. [DOI] [Google Scholar]
- Pan M.-L.; Wu Y.-T. Synthesis of Biphenylenes and Their π-Extended Derivatives. Synlett 2020, 31, 97–101. 10.1055/s-0039-1690750. [DOI] [Google Scholar]
- Konishi A.; Yasuda M. Breathing New Life into Nonalternant Hydrocarbon Chemistry: Syntheses and Properties of Polycyclic Hydrocarbons Containing Azulene, Pentalene, and Heptalene Frameworks. Chem. Lett. 2021, 50, 195–212. 10.1246/cl.200650. [DOI] [Google Scholar]
- Breslow R. Antiaromaticity. Acc. Chem. Res. 1973, 6, 393–398. 10.1021/ar50072a001. [DOI] [Google Scholar]
- Hafner K. Structure and Aromatic Character of Non-benzenoid Cyclically Conjugated Systems. Angew. Chem., Int. Ed. Engl. 1964, 3, 165–173. 10.1002/anie.196401651. [DOI] [Google Scholar]
- Bally T.; Masamune S. Cyclobutadiene. Tetrahedron 1980, 36, 343–370. 10.1016/0040-4020(80)87003-7. [DOI] [Google Scholar]
- Bally T.; Chai S.; Neuenschwander M.; Zhu Z. Pentalene: Formation, Electronic, and Vibrational Structure. J. Am. Chem. Soc. 1997, 119, 1869–1875. 10.1021/ja963439t. [DOI] [Google Scholar]
- Chen W.; Yu F.; Xu Q.; Zhou G.; Zhang Q. Recent Progress in High Linearly Fused Polycyclic Conjugated Hydrocarbons (PCHs, n > 6) with Well-Defined Structures. Adv. Sci. 2020, 7, 1903766. 10.1002/advs.201903766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Guo Y.; Liu Y. Design and Synthesis of High Performance π-Conjugated Materials Through Antiaromaticity and Quinoid Strategy for Organic Field-effect Transistors. Mater. Sci. Eng. R 2019, 136, 13–26. 10.1016/j.mser.2018.10.003. [DOI] [Google Scholar]
- Liu C.; Xu S.; Zhu W.; Zhu X.; Hu W.; Li Z.; Wang Z. Diaceno[a,e]pentalenes: An Excellent Molecular Platform for High-Performance Organic Semiconductors. Chem.—Eur. J. 2015, 21, 17016–17022. 10.1002/chem.201502184. [DOI] [PubMed] [Google Scholar]
- Kawase T.; Fujiwara T.; Kitamura C.; Konishi A.; Hirao Y.; Matsumoto K.; Kurata H.; Kubo T.; Shinamura S.; Mori H.; Miyazaki E.; Takimiya K. Dinaphthopentalenes: Pentalene Derivatives for Organic Thin-Film Transistors. Angew. Chem., Int. Ed. 2010, 49, 7728–7732. 10.1002/anie.201003609. [DOI] [PubMed] [Google Scholar]
- Takahashi K.; Ito S.; Shintani R.; Nozaki K. Selective Synthesis of Unsymmetric Dibenzo[a,e]pentalenes by a Rhodium-Catalysed Stitching Reaction. Chem. Sci. 2017, 8, 101–107. 10.1039/c6sc04560j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner A. J.; Enright K. M.; Cole J. M.; Yuan K.; McGough J. S.; Ingleson M. J. Borylative Cyclisation of Diynes Using BCl3 and borocations. Org. Biomol. Chem. 2019, 17, 5520–5525. 10.1039/c9ob00991d. [DOI] [PubMed] [Google Scholar]
- Chen C.; Harhausen M.; Liedtke R.; Bussmann K.; Fukazawa A.; Yamaguchi S.; Petersen J. L.; Daniliuc C. G.; Fröhlich R.; Kehr G.; Erker G. Dibenzopentalenes from B(C6F5)3-Induced Cyclization Reactions of 1,2-Bis(phenylethynyl)benzenes. Angew. Chem., Int. Ed. 2013, 52, 5992–5996. 10.1002/anie.201300871. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Oniwa K.; Asao N.; Yamamoto Y.; Jin T. Pd-Catalyzed Cascade Crossover Annulation of O-Alkynylarylhalides and Diarylacetylenes Leading to Dibenzo[a,e]pentalenes. J. Am. Chem. Soc. 2013, 135, 10222–10225. 10.1021/ja403382d. [DOI] [PubMed] [Google Scholar]
- Hashmi A. S. K.; Wieteck M.; Braun I.; Nösel P.; Jongbloed L.; Rudolph M.; Rominger F. Gold-Catalyzed Synthesis of Dibenzopentalenes – Evidence For Gold Vinylidenes. Adv. Synth. Catal. 2012, 354, 555–562. 10.1002/adsc.201200086. [DOI] [Google Scholar]
- Wurm T.; Bucher J.; Duckworth S. B.; Rudolph M.; Rominger F.; Hashmi A. S. K. On the Gold-Catalyzed Generation of Vinyl Cations from 1,5-Diynes. Angew. Chem., Int. Ed. 2017, 56, 3364–3368. 10.1002/anie.201700057. [DOI] [PubMed] [Google Scholar]
- Sekine K.; Stuck F.; Schulmeister J.; Wurm T.; Zetschok D.; Rominger F.; Rudolph M.; Hashmi A. S. K. N-Heterocycle-Fused Pentalenes by a Gold-Catalyzed Annulation of Diethynyl-Quinoxalines and -Phenazines. Chem.—Eur. J. 2018, 24, 12515–12518. 10.1002/chem.201803096. [DOI] [PubMed] [Google Scholar]
- Wurm T.; Rüdiger E. C.; Schulmeister J.; Koser S.; Rudolph M.; Rominger F.; Bunz U. H. F.; Hashmi A. S. K. A Golden Access to Acenopentalenes. Chem.—Eur. J. 2018, 24, 2735–2740. 10.1002/chem.201705456. [DOI] [PubMed] [Google Scholar]
- Levi Z. U.; Tilley T. D. Versatile Synthesis of Pentalene Derivatives via the Pd-Catalyzed Homocoupling of Haloenynes. J. Am. Chem. Soc. 2009, 131, 2796–2797. 10.1021/ja809930f. [DOI] [PubMed] [Google Scholar]
- Kato S.-i.; Kuwako S.; Takahashi N.; Kijima T.; Nakamura Y. Benzo- and Naphthopentalenes: Syntheses, Structures, and Properties. J. Org. Chem. 2016, 81, 7700–7710. 10.1021/acs.joc.6b01409. [DOI] [PubMed] [Google Scholar]
- Dai G.; Chang J.; Zhang W.; Bai S.; Huang K.-W.; Xu J.; Chi C. Dianthraceno[a,e]pentalenes: Synthesis, Crystallographic Structures and Applications in Organic Field-Effect Transistors. Chem. Commun. 2015, 51, 503–506. 10.1039/c4cc07630c. [DOI] [PubMed] [Google Scholar]
- Dai G.; Chang J.; Jing L.; Chi C. Diacenopentalene Dicarboximides as New n-Type Organic Semiconductors for Field-Effect Transistors. J. Mater. Chem. C 2016, 4, 8758–8764. 10.1039/c6tc02601j. [DOI] [Google Scholar]
- Dai G.; Chang J.; Shi X.; Zhang W.; Zheng B.; Huang K.-W.; Chi C. Thienoacene-Fused Pentalenes: Syntheses, Structures, Physical Properties and Applications for Organic Field-Effect Transistors. Chem.—Eur. J. 2015, 21, 2019–2028. 10.1002/chem.201405652. [DOI] [PubMed] [Google Scholar]
- Wilbuer J.; Grenz D. C.; Schnakenburg G.; Esser B. Donor and Acceptor-Functionalized Dibenzo[a,e]pentalenes: Modulation of the Electronic Band Gap. Org. Chem. Front. 2017, 4, 658–663. 10.1039/c6qo00487c. [DOI] [Google Scholar]
- Grenz D. C.; Schmidt M.; Kratzert D.; Esser B. Dibenzo-[a,e]pentalenes with Low-Lying LUMO Energy Levels as Potential n-Type Materials. J. Org. Chem. 2018, 83, 656–663. 10.1021/acs.joc.7b02250. [DOI] [PubMed] [Google Scholar]
- Konishi A.; Fujiwara T.; Ogawa N.; Hirao Y.; Matsumoto K.; Kurata H.; Kubo T.; Kitamura C.; Kawase T. Pentaleno[1,2-c:4,5-c′]dithiophene Derivatives: First Synthesis, Properties, and a Molecular Structure. Chem. Lett. 2010, 39, 300–301. 10.1246/cl.2010.300. [DOI] [Google Scholar]
- Oshima H.; Fukazawa A.; Yamaguchi S. Facile Synthesis of Polycyclic Pentalenes with Enhanced Hückel Antiaromaticity. Angew. Chem., Int. Ed. 2017, 56, 3270–3274. 10.1002/anie.201611344. [DOI] [PubMed] [Google Scholar]
- Usuba J.; Hayakawa M.; Yamaguchi S.; Fukazawa A. Dithieno[a,e]pentalenes: Highly Antiaromatic Yet Stable π-Electron Systems without Bulky Substituents. Chem.—Eur. J. 2021, 27, 1638–1647. 10.1002/chem.202004244. [DOI] [PubMed] [Google Scholar]
- Cao J.; London G.; Dumele O.; von Wantoch Rekowski M.; Trapp N.; Ruhlmann L.; Boudon C.; Stanger A.; Diederich F. The Impact of Antiaromatic Subunits in [4n+2] π-Systems: Bispentalenes with [4n+2] π-Electron Perimeters and Antiaromatic Character. J. Am. Chem. Soc. 2015, 137, 7178–7188. 10.1021/jacs.5b03074. [DOI] [PubMed] [Google Scholar]
- Sekine K.; Schulmeister J.; Paulus F.; Goetz K. P.; Rominger F.; Rudolph M.; Zaumseil J.; Hashmi A. S. K. Gold-Catalyzed Facile Synthesis and Crystal Structures of Benzene-/Naphthalene-Based Bispentalenes as Organic Semiconductors. Chem.—Eur. J. 2019, 25, 216–220. 10.1002/chem.201805637. [DOI] [PubMed] [Google Scholar]
- Warren G. I.; Barker J. E.; Zakharov L. N.; Haley M. M. Enhancing the Antiaromaticity of s-Indacene through Naphthothiophene Fusion. Org. Lett. 2021, 23, 5012–5017. 10.1021/acs.orglett.1c01514. [DOI] [PubMed] [Google Scholar]
- Du C.; Guo Y.; Liu Y.; Qiu W.; Zhang H.; Gao X.; Liu Y.; Qi T.; Lu K.; Yu G. Anthra[2,3-b]benzo[d]thiophene: An Air-Stable Asymmetric Organic Semiconductor with High Mobility at Room Temperature. Chem. Mater. 2008, 20, 4188–4190. 10.1021/cm801305f. [DOI] [Google Scholar]
- Ogawa Y.; Yamamoto K.; Miura C.; Tamura S.; Saito M.; Mamada M.; Kumaki D.; Tokito S.; Katagiri H. Asymmetric Alkylthienyl Thienoacenes Derived from Anthra[2,3-b]thieno[2,3-d]thiophene for Solution-Processable Organic Semiconductors. ACS Appl. Mater. Interfaces 2017, 9, 9902–9909. 10.1021/acsami.6b15793. [DOI] [PubMed] [Google Scholar]
- Tang M. L.; Okamoto T.; Bao Z. High-Performance Organic Semiconductors: Asymmetric Linear Acenes Containing Sulphur. J. Am. Chem. Soc. 2006, 128, 16002–16003. 10.1021/ja066824j. [DOI] [PubMed] [Google Scholar]
- Tang M. L.; Reichardt A. D.; Miyaki N.; Stoltenberg R. M.; Bao Z. Ambipolar, High Performance, Acene-Based Organic Thin Film Transistors. J. Am. Chem. Soc. 2008, 130, 6064–6065. 10.1021/ja8005918. [DOI] [PubMed] [Google Scholar]
- Valiyev F.; Hu W.-S.; Chen H.-Y.; Kuo M.-Y.; Chao I.; Tao Y.-T. Synthesis and Characterization of Anthra[2,3-b]thiophene and Tetraceno[2,3-b]thiophenes for Organic Field-Effect Transistor Applications. Chem. Mater. 2007, 19, 3018–3026. 10.1021/cm070472d. [DOI] [Google Scholar]
- Rivera-Fuentes P.; Rekowski M. v. W.; Schweizer W. B.; Gisselbrecht J.-P.; Boudon C.; Diederich F. Cascade Carbopalladation Reaction Between Alkynes and Gem-Dibromoolefins: Facile Access to Monoannelated Pentalenes. Org. Lett. 2012, 14, 4066–4069. 10.1021/ol301670d. [DOI] [PubMed] [Google Scholar]
- London G.; von Wantoch Rekowski M.; Dumele O.; Schweizer W. B.; Gisselbrecht J.-P.; Boudon C.; Diederich F. Pentalenes with Novel Topologies: Exploiting the Cascade Carbopalladation Reaction Between Alkynes And gem-Dibromoolefins. Chem. Sci. 2014, 5, 965–972. 10.1039/c3sc52623b. [DOI] [Google Scholar]
- Lautens M.; Fang Y.-Q.; Lifchits O. Horner-Wadsworth-Emmons Modification for Ramirez gem-Dibromoolefination of Aldehydes and Ketones Using P(Oi-Pr)3. Synlett 2008, 413–417. 10.1055/s-2008-1032045. [DOI] [Google Scholar]
- Tšupova S.; Hansmann M. M.; Rudolph M.; Rominger F.; Hashmi A. S. K. Gold-Catalyzed Formal Cyclisation/Dimerization of Thiophene-Tethered Diynes. Chem.—Eur. J. 2017, 23, 5716–5721. 10.1002/chem.201700061. [DOI] [PubMed] [Google Scholar]
- Tavakkolifard S.; Sekine K.; Reichert L.; Ebrahimi M.; Museridz K.; Michel E.; Rominger F.; Babaahmadi R.; Ariafard A.; Yates B. F.; Rudolph M.; Hashmi A. S. K. Gold-Catalyzed Regiospecific Annulation of Unsymmetrically Substituted 1,5-Diynes for the Precise Synthesis of Bispentalenes. Chem.—Eur. J. 2019, 25, 12180–12186. 10.1002/chem.201902381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershoni-Poranne R.; Stanger A. Magnetic Criteria of Aromaticity. Chem. Soc. Rev. 2015, 44, 6597–6615. 10.1039/c5cs00114e. [DOI] [PubMed] [Google Scholar]
- Mayer P. J.; El Bakouri O.; Holczbauer T.; Samu G. F.; Janáky C.; Ottosson H.; London G. Structure–Property Relationships in Unsymmetric Bis(antiaromatics): Who Wins the Battle between Pentalene and Benzocyclobutadiene?. J. Org. Chem. 2020, 85, 5158–5172. 10.1021/acs.joc.9b03119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson C. K.; Zakharov L. N.; Haley M. M. Modulating Paratropicity Strength in Diareno-Fused Antiaromatics. J. Am. Chem. Soc. 2016, 138, 16827–16838. 10.1021/jacs.6b11397. [DOI] [PubMed] [Google Scholar]
- Gaussian 09, Revision E.01 (for full reference, see Supporting Information).
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. A 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Krishnan R.; Binkley J. S.; Seeger R.; Pople J. A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- Gershoni-Poranne R.; Stanger A. The NICS-XY-Scan: Identification of Local and Global Ring Currents in Multi-Ring Systems. Chem.—Eur. J. 2014, 20, 5673–5688. 10.1002/chem.201304307. [DOI] [PubMed] [Google Scholar]
- Herges R.; Geuenich D. Delocalization of Electrons in Molecules. J. Phys. Chem. A 2001, 105, 3214–3220. 10.1021/jp0034426. [DOI] [Google Scholar]
- Geuenich D.; Hess K.; Köhler F.; Herges R. Anisotropy of the Induced Current Density (ACID), a General Method to Quantify and Visualize Electronic Delocalization. Chem. Rev. 2005, 105, 3758– 3772. 10.1021/cr0300901. [DOI] [PubMed] [Google Scholar]
- Bultinck P.; Rafat M.; Ponec R.; Van Gheluwe B.; Carbó-Dorca R.; Popelier P. Electron Delocalization and Aromaticity in Linear Polyacenes: Atoms in Molecules Multicenter Delocalization Index. J. Phys. Chem. A 2006, 110, 7642–7648. 10.1021/jp0609176. [DOI] [PubMed] [Google Scholar]
- Matito E.; Duran M.; Solà M. The aromatic fluctuation index (FLU): A new aromaticity index based on electron delocalization. J. Chem. Phys. 2005, 122, 014109. 10.1063/1.1824895. [DOI] [PubMed] [Google Scholar]
- Stanger A.; Monaco G.; Zanasi R. NICS-XY-Scan Predictions of Local, Semi-Global, and Global Ring Currents in Annulated Pentalene and s-Indacene Cores Compared to First-Principles Current Density Maps. ChemPhysChem 2020, 21, 65–82. 10.1002/cphc.201900952. [DOI] [PubMed] [Google Scholar]
- Hashimoto S.; Tahara K. Theoretical Study on the Geometry, Aromaticity, and Electronic Properties of Benzo[3,4]cyclobutathiophenes and Their Homologues. J. Org. Chem. 2019, 84, 9850–9858. 10.1021/acs.joc.9b00661. [DOI] [PubMed] [Google Scholar]
- Ried W.; Bender H. Notiz über einige neue Abkömmlinge des Thionaphthens. Chem. Ber. 1955, 88, 34–38. 10.1002/cber.19550880107. [DOI] [Google Scholar]
- Shigeno M.; Fujii Y.; Kajima A.; Nozawa-Kumada K.; Kondo Y. Catalytic Deprotonative α-Formylation of Heteroarenes by an Amide Base Generated in Situ from Tetramethylammonium Fluoride and Tris(trimethylsilyl)amine. Org. Process Res. Dev. 2019, 23, 443–451. 10.1021/acs.oprd.8b00247. [DOI] [Google Scholar]
- Verma A. K.; Kotla S. K. R.; Choudhary D.; Patel M.; Tiwari R. K. Silver-Catalyzed Tandem Synthesis of Naphthyridines and Thienopyridines via Three-Component Reaction. J. Org. Chem. 2013, 78, 4386–4401. 10.1021/jo400400c. [DOI] [PubMed] [Google Scholar]
- Meyer C.; Neue B.; Schepmann D.; Yanagisawa S.; Yamaguchi J.; Würthwein E.-U.; Itami K.; Wünsch B. Exploitation of an Additional Hydrophobic Pocket of σ1 Receptors: Late-Stage Diverse Modifications of Spirocyclic tThiophenes by C–H Bond Functionalization. Org. Biomol. Chem. 2011, 9, 8016–8029. 10.1039/c1ob06149f. [DOI] [PubMed] [Google Scholar]
- Mitsudo K.; Tanaka S.; Isobuchi R.; Inada T.; Mandai H.; Korenaga T.; Wakamiya A.; Murata Y.; Suga S. Rh-Catalyzed Dehydrogenative Cyclization Leading to Benzosilolothiophene Derivatives via Si–H/C–H Bond Cleavage. Org. Lett. 2017, 19, 2564–2567. 10.1021/acs.orglett.7b00878. [DOI] [PubMed] [Google Scholar]
- Ried W.; Suarez-Rivero E.; Äthinierungsreaktionen X. X. I. Notiz zur Äthinierung von o-Chinonen. Chem. Ber. 1963, 96, 1475–1477. 10.1002/cber.19630960544. [DOI] [Google Scholar]
- Rustagi V.; Tiwari R.; Verma A. K. AgI-Catalyzed Cascade Strategy: Regioselective Access to Diversely Substituted Fused Benzimidazo[2,1-a]isoquinolines, Naphthyridines, Thienopyridines, and Quinoxalines in Water. Eur. J. Org. Chem. 2012, 4590–4602. 10.1002/ejoc.201200546. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.