

Abstract

A metal-free scalable synthesis of functionalized ketenimines from alkyl α-(aryl/heteroaryl)-α-diazoacetates and alkyl isocyanides induced by blue light irradiation has been developed. The reaction proceeds at room temperature without any photocatalyst and provides ketenimines in moderate to good yields. Density functional theory (DFT) calculations and the experimental study showed that aryl(alkoxycarbonyl)carbenes in both singlet and triplet states can react with isocyanides but only the reaction of the former leads to the smooth formation of ketenimines. The obtained ketenimines were used for the synthesis of functionalized amidines under mild metal-free conditions.
Introduction
Ketenimines are reactive compounds that are widely used in organic synthesis.1 They are often generated in the reaction mixture in situ and react further with an appropriate substrate to afford a target molecule. There are a number of reliable methods for the synthesis of ketenimines,2 which include the Wittig and aza-Wittig reactions, alkylation of nitrile anions, dehydrochlorination of imidoyl chlorides, dehydration of amides, etc. Obviously, to expand the versatility of ketenimine chemistry, functionalized ketenimines are in high demand. One of the attractive and perspective ways to access functionalized ketenimines is an addition of isocyanides to carbenes derived from diazocarbonyl compounds. Back in 1970, Ciganek reported the formation of a ketenimine under harsh thermolysis of methyl phenyldiazoacetate in tert-butyl isocyanide (Scheme 1a).3 In recent years, the coupling of isocyanides with diazo compounds has undergone a revival due to the application of transition metal catalysis (Scheme 1b). In particular, palladium catalysis has been the most actively studied.4 Several examples of the application of rhodium5 and cobalt6 catalysis have also been published. Despite the impressive progress in the metal-catalyzed synthesis of ketenimines, the development of a mild metal-free synthesis continues to be important, since removing trace amounts of toxic transition metals from products remains a long-standing problem in the pharmaceutical industry.7,8
Scheme 1. Synthesis of Ketenimines from Diazocarbonyl Compounds and Isocyanides.

As part of our continuing research interest in diazo chemistry,9 we addressed photochemical activation of diazo compounds in the reaction with isocyanides to develop an inexpensive, mild, and ecologically friendly method for the preparation of functionalized ketenimines. To the best of our knowledge, a photochemical synthesis of ketenimines from diazocarbonyl compounds and isocyanides has not been reported. It should be noted that several examples of the photochemical formation of ketenimines from diphenyldiazomethane and isocyanides using a mercury lamp with a Pyrex filter or 254 nm UV-light have been published (Scheme 1c).10 Currently, visible light activation of diazo compounds is actively studied.11 The advantages of this approach are simple light-emitting diode (LED) equipment, high selectivity of reactions, and easy workup of the reaction mixtures containing no metals.
In this work, we report a blue light-promoted metal-free synthesis of functionalized ketenimines from diazo esters and isocyanides (Scheme 1d).12 The mechanism of the reaction was studied by density functional theory (DFT) calculations and control experiments. The obtained ketenimines were converted to functionalized amidines under mild metal-free conditions.
Results and Discussion
Synthesis of Ketenimines
Optimization experiments were performed using readily available methyl phenyldiazoacetate (1a) and t-butyl isocyanide (2a), all reactions were conducted until full consumption of a diazo compound. In previous studies,11 it was shown that blue light can effectively promote the elimination of nitrogen from aryldiazoacetates in dichloromethane (DCM). The reaction of the diazo compound 1a with 2 equiv of isocyanide 2a under 450 nm LED light irradiation in DCM at room temperature led to the formation of ketenimine 3a, which was isolated by silica gel column chromatography in 43% yield (Table 1, entry 1). The reaction of the diazo compound 1a with 0.5 equiv of isocyanide 2a afforded ketenimine 3a in 45% yield (entry 2). Increasing the amount of isocyanide to 3 equiv led to a slight increase in the yield of ketenimine 3a (entry 3). The product yield is almost independent of the concentration of the reagents (entries 4 and 5). The addition of catalytic amounts of a transition metal catalyst such as zinc chloride, palladium acetate, or photocatalysts13 (fac-Ir(ppy)3 or [Ru(bpy)3]Cl2) (entries 6–9), as well as the use of LEDs with other wavelengths (390, 420, 475, 520 nm) (entries 10–13) did not lead to an increase in the ketenimine yield. We also tested several halogenated solvents [CHCl3, CCl4, 1,2-dichloroethane (DCE), and perfluorotoluene]. The yields of ketenimine 3a in these solvents were lower than those in DCM (entries 14–17). The use of a lower power LED (entry 18), solvent-free conditions (entry 19), or an argon atmosphere, as well as the slow addition of the diazo compound 1a to isocyanide 2a solution, did not improve the yield of ketenimine 3a. The optimal conditions were found to be irradiation of the diazo compound 1a (0.14 mol/L) with 450 nm LED light with 3 equiv of isocyanide 2a in DCM.
Table 1. Optimization of Photochemical Ketenimine Synthesisa.

| entry | equiv of 2a | additive (equiv) | solvent | concentration of 1a, M | LED, nm | yield of 3a,b % |
|---|---|---|---|---|---|---|
| 1 | 2 | DCM | 0.14 | 450 | 43 | |
| 2 | 0.5 | DCM | 0.28 | 450 | 45c | |
| 3 | 3 | DCM | 0.14 | 450 | 50 | |
| 4 | 3 | DCM | 0.21 | 450 | 44 | |
| 5 | 3 | DCM | 0.07 | 450 | 47 | |
| 6 | 3 | ZnCl2 (0.13) | DCM | 0.14 | 450 | 46 |
| 7 | 3 | Pd(OAc)2 (0.07) | DCM | 0.14 | 450 | 46 |
| 8 | 3 | fac-Ir(ppy)3 (0.05) | DCM | 0.14 | 450 | 47d |
| 9 | 3 | [Ru(bpy)3]Cl2 (0.05) | DCM | 0.14 | 450 | 45 |
| 10 | 3 | DCM | 0.14 | 390e | 29 | |
| 11 | 3 | DCM | 0.14 | 420 | 42 | |
| 12 | 3 | DCM | 0.14 | 475e | 50d | |
| 13 | 3 | DCM | 0.14 | 520f | 43d | |
| 14 | 3 | DCE | 0.14 | 450 | 36d | |
| 15 | 3 | CHCl3 | 0.14 | 450 | 35d | |
| 16 | 3 | CCl4 | 0.14 | 450 | 42d | |
| 17 | 3 | C6F5CF3 | 0.14 | 450 | 38d | |
| 18 | 3 | DCM | 0.14 | 450g | 46 | |
| 19 | 3 | 450h | 46 |
Reaction conditions: 1a (0.28 mmol), 2a (0.85 mmol), solvent (1–4 mL), 10W LED, rt, and 2 h.
Isolated yields.
Yield calculated on 2a.
Yield was determined by 1H NMR spectroscopy using CH2Br2 as the internal standard.
3 h.
15 h.
25 h, 0.5W LED.
6 h.
The moderate yield of ketenimine 3a may be associated with the low activity of the isocyanide toward the carbene species and, consequently, the low selectivity of the reaction (carbene tends to react with the diazo compound instead of the isocyanide). Indeed, a number of byproducts derived from the diazo compound 1a were identified in the reaction mixtures (see the Supporting Information). One more factor reducing the yield of the ketenimine is the degradation of the triplet ketenimine (see details below).
With the optimized conditions in hand, we examined the scope of the reaction (Scheme 2). The yields of ketenimines 3 in the reactions of tert-butyl isocyanide 2a with various aryldiazoacetates 1a–q were in the range of 27–64%. In general, the yields of ketenimines bearing ortho substituents in the phenyl ring were slightly higher, probably due to inhibition of side reactions of the ketenimine formed. Thus, the yields of ketenimines from ortho-chlorophenyl (1e), ortho-tolyl (1k), and 1-naphthyl (1m) diazoacetates were 52, 48, and 64%, respectively. No clear influence of the electronic effects of substituents on the course of the reaction was observed. The yield of ketenimine 3g turned out to be higher when the reaction was conducted under an argon atmosphere. A ketenimine was also obtained from a 3-pyridyl-substituted diazo compound 1l: the reaction was completed in 4.5 h to give ketenimine 3l in 48% yield. The reactions with ethyl 2-(2-pyridyl)- (using a 390 nm LED) and 2-(4-pyridyl)-2-diazoacetates resulted in a complex mixture of products. The ketenimine formation was not observed in the reaction of isocyanide 2a with 3-diazoindolin-2-one. The synthesis of ketenimines can be scaled up and that was demonstrated by a 2.5 mmol scale reaction of methyl phenyldiazoacetate (1a), which produced ketenimine 3a in 58% yield.
Scheme 2. Scope of Diazo Compounds and Isocyanides,

Reaction conditions: 1 (0.28 mmol), 2 (0.85 mmol), DCM (2 mL), 450 nm 10W LED, rt, and 2 h.
Isolated yields.
2.5 mmol scale.
Reaction was carried out under argon for 3 h.
4.5 h.
The yields of ketenimines 3r,s obtained from less sterically crowded isocyanides 2b,c turned out to be lower than from tert-butyl isocyanide 2a. It was found that the reaction was not effective for isocyanides with an active methylene group: ethyl isocyanoacetate (2d) gave ketenimine 3t in low yield whereas TosMIC did not afford the corresponding ketenimine at all. The reactions of phenyldiazoacetate 1a with aryl isocyanides (4-(dimethylamino)phenyl and 2-iodo-4-nitrophenyl isocyanides) led to the formation of complex mixtures of products.
Mechanism of the Reaction and DFT Calculations
As follows from the literature data,11 aryldiazoacetates can absorb light in the blue region that leads to photoexcitation of the diazo compound to the singlet state. The subsequent loss of nitrogen from the excited state of aryldiazoacetate generates a highly reactive singlet carbene. It can convert to a triplet carbene via intersystem crossing (ISC), which is generally a fast and reversible process.14a,14b It should be mentioned that the spin nature of several photochemically generated aryl(methoxycarbonyl)carbenes has been studied in previous studies by different methods.14 From these studies, two important points can be concluded: (a) a ground spin state of the carbenes changes from the triplet state in nonpolar solvents to the singlet state in polar solvents; (b) a spin state of the carbenes strongly depends upon the nature and position of substituents at the phenyl ring. Electron-donating substituents in ortho- and para-positions make the singlet state to be the ground state. In opposite, electron-withdrawing substituents stabilize the triplet state. Since in our work, ketenimines were obtained from diazo esters bearing both electron-donating and electron-withdrawing substituents at the phenyl ring, the mechanism of the reaction needs to be discussed.
Singlet carbenes can exhibit electrophilic properties due to the presence of a vacant p-type orbital (LUMO). On the other hand, they possess nucleophilic properties due to the presence of an occupied orbital (perpendicular to the empty p-orbital), which contains the lone pair of electrons at the divalent carbon atom (HOMO). The nature of the substituents has a decisive influence on the degree of electrophilicity and nucleophilicity of the carbene. The triplet carbene having two p-orbitals each occupied by an electron, behaves as a diradical species. In one of the previous studies, it was proposed that the ketenimine formation proceeds via “an electrophilic attack by isocyano-carbon upon the electron-rich carbon of diarylcarbene”.10 At the same time, due to the presence of an electron-withdrawing ester group, aryl(methoxycarbonyl)carbenes possess an electron-deficient nature. Thus, it is not clear which orbitals of aryl(methoxycarbonyl)carbenes and isocyanides interact in the reaction.
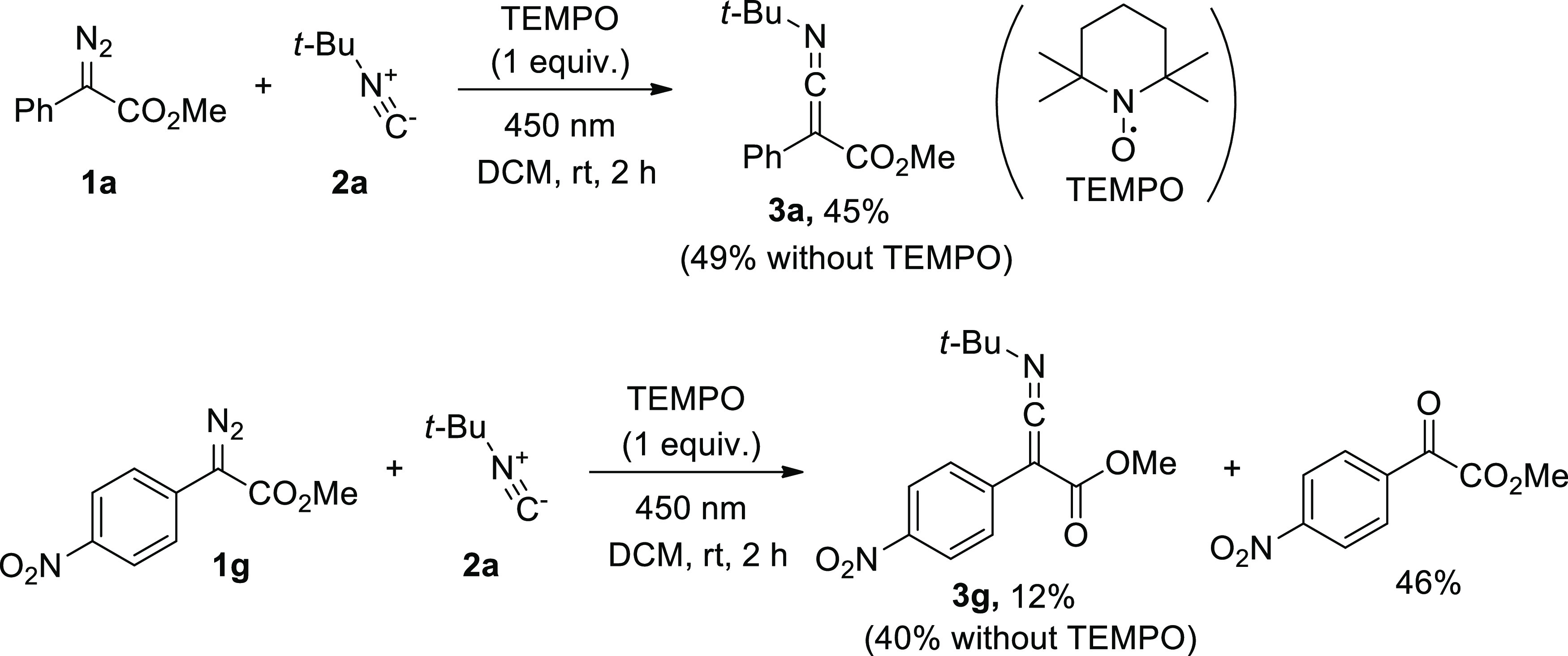
To get an insight into the mechanism of the ketenimine formation, we conducted control experiments and quantum-chemical calculations (DFT). It should be mentioned that a spin state of the carbenes involved in the blue light-induced reactions of diazo compounds has been discussed in a few studies.11a,11b,15 According to the literature,15 an influence of the radical scavenger (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) on the reactions of carbenes can be an indicator of the spin nature of a carbene. An addition of 1 equiv of TEMPO to the reaction of the diazo compound 1a with isocyanide 2a almost did not influence the yield of 3a, which was 45% (Scheme 3). This fact can provide evidence in favor of a singlet carbene being involved in the formation of ketenimine 3a. On the other hand, the analogous reaction of methyl (4-nitrophenyl)diazoacetate (1g) led to the formation of methyl 2-(4-nitrophenyl)-2-oxoacetate as a major product and to a decrease in the yield of ketenimine 3g from 40 to 12% (Scheme 3). These facts support the triplet carbene to be involved in this reaction. The same conclusion follows from the sensitivity of the yield of 3g to the presence of oxygen in the reaction mixture (see Scheme 2).
Scheme 3. Control Experiments with TEMPO.
According to the DFT calculations (B3LYP-D3/def2TZVP, PCM for dichloromethane, 298.15 K), phenyl(methoxycarbonyl)carbene 4 has a small singlet–triplet energy gap (Figure 1). The energy of singlet carbene 14 having a twisted geometry (CO2Me group is orthogonal to the PhC moiety) is lower than that for plane triplet carbene syn-34 (Ph and C=O groups are in the syn arrangement) by only 0.2 kcal/mol. The second conformer of triplet carbene, anti-34 (Ph and C=O groups are in the anti arrangement), is less stable than 14 by 1.7 kcal/mol. These results are in accordance with those previously published for phenyl(methoxycarbonyl)carbene in the gas phase (B3LYP/6-31G*).14a
Figure 1.

Energy diagrams for the addition of isocyanide 2a to carbenes 14, syn-34, and anti-34.
The calculated energy profiles for the reactions of tert-butyl isocyanide (2a) with carbenes 14, syn-34, and anti-34 are depicted in Figure 1. Isocyanide 2a can attack singlet carbene 14 in two ways: from the side of the methoxy group (TS1) or carbonyl group (TS2). Both reactions result in the formation of singlet ketenimine 3a but in different conformational states, 3a′ and 3a″, respectively. The energy of TS1 is slightly lower than that of TS2, both the energy barriers being quite low. As can be seen from the structures of TS1 and TS2, the isocyanide attacks carbene 14 almost perpendicularly to the plane of the phenyl ring (dihedral angles Cortho-Cipso-Ccarbene-Cisocyanide are 75.8 and 75.7° for TS1 and TS2, respectively). Such an approach corresponds to an interaction of p-type LUMO of the carbene with σ-type HOMO of the isocyanide. Two conformers of the triplet carbene, syn-34 and anti-34, react with 2a to afford two conformers of triplet ketenimine: twisted 33a and flat 33a′, respectively. The reactions of both triplet carbenes, syn-34 and anti-34, practically have the same energy barrier (TS3, ΔΔG‡ = 10.9 kcal/mol and TS4, ΔΔG‡ = 11.1 kcal/mol), which is very similar to that for the singlet carbene (TS1, ΔΔG‡ = 11.4 kcal/mol). The isocyanide attacks triplet carbenes syn-34 and anti-34 from the plane of the phenyl ring, an angle Ccarbene···C=N in transition states TS3 and TS4 being 116.5 and 119.8°, respectively. Such a geometry of these transition states implies the interaction of the p-type SOMO of the carbene with the π*-type LUMO of the isocyanide. The less stable triplet ketenimine 33a, derived from triplet carbene syn-34, very quickly transforms to the more stable triplet conformer 33a′ (TS5, ΔΔG‡ = 1.9 kcal/mol). The latter is extremely unstable due to its rapid homolytic fragmentation to radical 5 and the tert-butyl radical (TS6, ΔΔG‡ = 6.3 kcal/mol), which is typical of imidoyl radicals.16 This rapid reaction, in principle, can compete with the transformation of the triplet ketenimines to singlet ketenimine 3a via intersystem crossing.
Thus, it follows from the calculations that: (a) carbene 4 in both singlet and triplet states can be involved in the reaction; (b) the singlet and triplet carbenes 4 are comparable in their reactivity toward isocyanides; and (c) the triplet ketenimine 33 formed from the triplet carbene, in addition to conversion to the singlet ketenimine 13 through the intersystem crossing, can undergo the low-barrier homolytic fragmentation across the single C–N bond, leading to byproducts. We believe that the degradation of the triplet ketenimine formed is one of the reasons for moderate ketenimine yields in this reaction.
Synthesis of Amidines from Ketenimines
An amidine structural motif is of high relevance for medicinal chemistry.17 A notable example related to this work is napactadine (N,N′-dimethyl-2-naphthaleneethanimidamide hydrochloride), an antidepressant agent.18 Amidines, bearing an ester substituent at the α-position, are a practically unknown class of compounds and could be of great interest for drug development. A few examples of these compounds were just recently synthesized using transition metal catalysis.4a,6a To close this gap, we carried out a series of metal-free reactions of the ketenimines, obtained in this work, with primary and secondary amines in tetrahydrofuran (THF) (Scheme 4). It was found that the reactions of ketenimine 3a with primary aliphatic amines proceed under mild heating to afford amidines 6a,b in good yields. Compounds 6a,b exist in CDCl3 solution as an equilibrium mixture of two tautomers. Ketenimine 3a reacts with aniline in the presence of a strong base (DBU) at a higher temperature to give amidine 6c. As reported previously,4a ketenimines like 3 can react with morpholine under palladium catalysis to give an enamine tautomer of amidine. We found that the reaction of ketenimine 3a with morpholine can proceed effectively upon mild heating without any catalyst to afford the corresponding enamine 6d. The synthesis of enamines can be carried out in one-pot starting from the diazo compound and isocyanide that was demonstrated by the synthesis of enamines 6e,f.
Scheme 4. Synthesis of Functionalized Amidines.

Conclusions
We have developed a metal-free scalable synthesis of functionalized ketenimines from α-(aryl/heteroaryl)-α-diazoacetates and alkyl isocyanides induced by blue light. The reaction proceeds at room temperature without any photocatalyst and provides ketenimines in moderate to good yields. The mechanism of the reaction was studied by DFT calculations and control experiments, which demonstrated that both singlet and triplet forms of carbene can react with isocyanide. The obtained ketenimines were used for the synthesis of functionalized amidines under mild metal-free conditions.
Experimental Part
General Information
All solvents were distilled and dried prior to use. 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded on a Bruker AVANCE 400 spectrometer in the solvent indicated below. Chemical shifts (δ) are reported in ppm. Electrospray ionization (ESI) mass spectra were measured on a Shimadzu Nexera X2 LCMS-9030 mass spectrometer (on a Bruker micrOTOF for 3g). Thin-layer chromatography (TLC) was conducted on aluminum sheets precoated with SiO2 ALUGRAM SIL G/UV254. Irradiation was carried out with a 10W LED (spectral distribution of 420–490 nm with a maximum intensity at 447 nm, full width at half-maximum 17 nm) with 900 mA, 9–11 V power supply, and water-cooling system. Photochemical reactions were performed in a standard 4 mL glass vial. The distance from the diode to the vial was 25 mm. The vials were cooled with a 40 mm fan.
Diazo compounds 1a,b,c,d,e,f,191g,201h,o,211i,j,221k,231l,241m,251n,261p,9d and 1q(11a) are known compounds, which were prepared by the reported procedures.
General Procedure for the Synthesis of Ketenimines 3
A 4 mL vial was charged with a diazo compound (0.28 mmol, 1 equiv), an isocyanide (0.85 mmol, 3 equiv), and dichloromethane (2 mL). The vial was closed but not tightly to allow nitrogen to escape and irradiated with blue light (one 10W 450 nm LED) at room temperature under magnetic stirring until the diazo compound consumption (about 2 h, controlled by TLC). The reaction mixture was concentrated under reduced pressure, and the crude material was purified by column chromatography (eluent petroleum ether–EtOAc, from 5:1 to 2:1) to afford the corresponding ketenimine.
Methyl 3-(tert-butylimino)-2-phenylacrylate (3a)4b
Colorless oil (32 mg, 49%). Irradiation of a mixture of phenyldiazoacetate 1a (2.5 mmol), tert-butyl isocyanide 2a (7.5 mmol), and DCM (18 mL) with eight 10W 450 nm LEDs for 6 h gave ketenimine 3a in 58% yield (335 mg). 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 7.6 Hz, 2H), 7.42 (t, J = 7.7 Hz, 2H), 7.25 (t, J = 7.3 Hz, 1H), 3.88 (s, 3H), 1.59 (s, 9H); HRMS-ESI [M + H]+ calcd for C14H18NO2+ 232.1332, found 232.1332.
Methyl 3-(tert-butylimino)-2-(4-methoxyphenyl)acrylate (3b)
Colorless oil (30 mg, 41%). 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 8.9 Hz, 2H), 6.90 (d, J = 8.9 Hz, 2H), 3.82 (s, 3H), 3.79 (s, 3H), 1.49 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.9, 169.5, 157.8, 128.1, 123.0, 114.0, 68.2, 62.3, 55.2, 51.3, 30.4; HRMS-ESI [M + Na]+ calcd for C15H19NNaO3+ 284.1257, found 284.1258.
Methyl 3-(tert-butylimino)-2-(4-fluorophenyl)acrylate (3c)
Colorless oil (32 mg, 46%). 1H NMR (400 MHz, CDCl3) δ 7.51–7.46 (m, 2H), 7.05–7.01 (m, 2H), 3.79 (s, 3H), 1.50 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 30.4, 51.4, 62.6, 67.7, 115.3 (d, J = 21.5 Hz), 127.0 (d, J = 3.5 Hz), 128.2 (d, J = 8.1 Hz), 160.9 (d, J = 245.1 Hz), 169.2, 170.1; HRMS-ESI [M + H]+ calcd for C14H17FNO2+ 250.1238, found 250.1243.
Methyl 3-(tert-butylimino)-2-(4-chlorophenyl)acrylate (3d)
Colorless oil (30 mg, 40%). 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 8.6 Hz, 2H), 7.29 (d, J = 8.6 Hz, 2H), 3.79 (s, 3H), 1.51 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 168.9 (2C), 130.9, 130.0, 128.5, 127.6, 67.6, 62.9, 51.4, 30.5; HRMS-ESI [M + H]+ calcd for C14H1735ClNO2+ 266.0942, found 266.0945.
Methyl 3-(tert-butylimino)-2-(2-chlorophenyl)acrylate (3e)4a
Colorless oil (36 mg, 48%). 1H NMR (400 MHz, CDCl3) δ 7.46 (dd, J = 7.6 and 1.9 Hz, 1H), 7.42 (dd, J = 7.7 and 1.6 Hz, 1H), 7.29–7.20 (m, 2H), 3.79 (s, 3H), 1.48 (s, 9H); HRMS-ESI [M + H]+ calcd for C14H1735ClNO2+ 266.0942, found 266.0945.
Methyl 2-(4-bromophenyl)-3-(tert-butylimino)acrylate (3f)
Colorless oil (30 mg, 35%). 1H NMR (400 MHz, CDCl3) δ 7.44–7.43 (m, 4H), 3.79 (s, 3H), 1.51 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 168.8, 168.6, 131.5, 130.6, 127.9, 118.9, 67.6, 62.9, 51.4, 30.5; HRMS-ESI [M + H]+ calcd for C14H1779BrNO2+ 310.0437, found 310.0444.
Methyl 3-(tert-butylimino)-2-(4-nitrophenyl)acrylate (3g)
Orange solid (31 mg, 40%). This compound was obtained in 52% yield (40 mg) when the reaction was carried out under argon for 3 h. Mp: 85–86 °C (hexane). 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 9.1 Hz, 2H), 7.74 (d, J = 9.1 Hz, 2H), 3.82 (s, 3H), 1.57 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 168.0, 162.4, 144.6, 140.4, 125.3, 123.9, 66.9, 63.8, 51.5, 30.6; HRMS-ESI [M + Na]+ calcd for C14H16N2NaO4+ 299.1002, found 299.1008.
Methyl 3-(tert-butylimino)-2-(4-(trifluoromethyl)phenyl)acrylate (3h)
Colorless oil (37 mg, 44%). 1H NMR (400 MHz, CDCl3) δ 7.70–7.67 (m, 2H), 7.58–7.56 (m, 2H), 3.81 (s, 3H), 1.54 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 30.5, 51.4, 63.2, 67.4, 124.3 (q, J = 271.5 Hz), 125.3 (q, J = 3.7 Hz), 125.9, 127.1 (q, J = 32.4 Hz), 136.0, 166.6, 168.6; 19F NMR (376 MHz, CDCl3) δ −62.35; HRMS-ESI [M + H]+ calcd for C15H17F3NO2+ 300.1206, found 300.1212.
Ethyl 3-(tert-butylimino)-2-(p-tolyl)acrylate (3i)
Colorless oil (20 mg, 27%). 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 8.0 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 4.26 (q, J = 7.1 Hz, 2H), 2.35 (s, 3H), 1.50 (s, 9H), 1.32 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.7, 168.9, 135.2, 129.1, 128.1, 126.6, 68.8, 62.4, 59.9, 30.5, 21.0, 14.4; HRMS-ESI [M + H]+ calcd for C16H22NO2+ 260.1645, found 260.1650.
Ethyl 3-(tert-butylimino)-2-(m-tolyl)acrylate (3j)
Colorless oil (20 mg, 28%). 1H NMR (400 MHz, CDCl3) δ 7.42 (br.s, 1H), 7.33–7.31 (m, 1H), 7.23 (t, J = 7.7 Hz, 1H), 7.00–6.98 (m, 1H), 4.27 (q, J = 7.1 Hz, 2H), 2.37 (s, 3H), 1.51 (s, 9H), 1.33 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.0, 168.8, 138.0, 131.2, 128.3, 127.3, 126.3, 123.6, 68.9, 62.5, 59.9, 30.5, 21.5, 14.4; HRMS-ESI [M + H]+ calcd for C16H22NO2+ 260.1645, found 260.1650.
Ethyl 3-(tert-butylimino)-2-(o-tolyl)acrylate (3k)4a
Colorless oil (38 mg, 52%). 1H NMR (400 MHz, CDCl3) δ 7.31–7.28 (m, 1H), 7.24–7.20 (m, 3H), 4.25 (q, J = 7.1 Hz, 2H), 2.34 (s, 3H), 1.45 (s, 9H), 1.31 (t, J = 7.1 Hz, 3H); HRMS-ESI [M + H]+ calcd for C16H22NO2+ 260.1645, found 260.1647.
Ethyl 3-(tert-butylimino)-2-(pyridin-3-yl)acrylate (3l)
Colorless oil (33 mg, 48%). The reaction was irradiated for 4.5 h. 1H NMR (400 MHz, CDCl3) δ 8.64 (d, J = 1.8 Hz, 1H), 8.37 (dd, J = 4.8 and 1.8 Hz, 1H), 8.01 (dt, J = 8.1 and 2.0 Hz, 1H), 7.26–7.23 (m, 1H), 4.26 (q, J = 7.1 Hz, 2H), 1.52 (s, 9H), 1.32 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 14.3, 30.5, 60.2, 63.0, 65.3, 123.2, 128.4, 133.6, 146.3, 147.3, 167.1, 168.3; HRMS-ESI [M + H]+ calcd for C14H19N2O2+ 247.1441, found 247.1448.
Methyl 3-(tert-butylimino)-2-(naphthalen-1-yl)acrylate (3m)
Colorless oil (50 mg, 64%). 1H NMR (400 MHz, CDCl3) δ 8.06–8.04 (m, 1H), 7.91–7.86 (m, 2H), 7.58–7.49 (m, 4H), 3.82 (s, 3H), 1.47 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 169.8, 169.3, 133.9, 132.6, 129.1, 128.4, 128.3, 127.7, 126.0, 125.7, 125.4 (2C), 65.4, 61.7, 51.6, 30.3; HRMS-ESI [M + H]+ calcd for C18H20NO2+ 282.1489, found 282.1494.
Methyl 3-(tert-butylimino)-2-(naphthalen-2-yl)acrylate (3n)
Colorless oil (30 mg, 38%). 1H NMR (400 MHz, CDCl3) δ 8.19 (br.s, 1H), 7.84–7.79 (m, 3H), 7.55 (dd, J = 8.6 and 1.9 Hz, 1H); 7.50–7.41 (m, 2H), 3.85 (s, 3H), 1.55 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 170.1, 169.2, 133.8, 131.6, 128.6, 127.9, 127.8, 127.5, 126.0, 125.2, 124.9, 124.7, 68.8, 62.8, 51.4, 30.5; HRMS-ESI [M + H]+ calcd for C18H20NO2+ 282.1489, found 282.1489.
Methyl 3-(tert-butylimino)-2-(3,4-dimethoxyphenyl)acrylate (3o)
Colorless solid (26 mg, 32%). Mp: 70–71 °C (hexane). 1H NMR (400 MHz, CDCl3) δ 7.19 (d, J = 2.1 Hz, 1H), 7.01 (dd, J = 8.4 and 2.1 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 3.90 (s, 6H), 3.88 (s, 3H), 3.79 (s, 3H), 1.49 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.6, 169.4, 148.9, 147.1, 123.6, 118.9, 111.4, 110.5, 68.4, 62.4, 55.9, 55.8, 51.3, 30.5; HRMS-ESI [M + Na]+ calcd for C16H21NNaO4+ 314.1363, found 314.1373.
Methyl 3-(tert-butylimino)-2-(4-methoxy-3-methylphenyl)acrylate (3p)
Colorless oil (31 mg, 40%). 1H NMR (400 MHz, CDCl3) δ 7.33–7.27 (m, 2H), 6.82 (d, J = 8.5 Hz, 1H), 3.84 (s, 3H), 3.79 (s, 3H), 2.24 (s, 3H), 1.49 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 172.3, 169.6, 156.1, 129.5, 126.7, 125.5, 122.4, 110.1, 68.3, 62.2, 55.3, 51.3, 30.4, 16.3; HRMS-ESI [M + H]+ calcd for C16H22NO3+ 276.1594, found 276.1596.
Methyl 3-(tert-butylimino)-2-(3,4-dichlorophenyl)acrylate (3q)
Colorless oil (49 mg, 59%). 1H NMR (400 MHz, CDCl3) δ 7.71 (br.s, 1H), 7.37–7.36 (m, 2H), 3.79 (s, 3H), 1.52 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 168.5, 166.9, 132.5, 132.0, 130.2, 128.8, 127.7, 125.3, 66.7, 63.3, 51.5, 30.5; HRMS-ESI [M + H]+ calcd for C14H1635Cl2NO2+ 300.0553, found 300.0553.
Methyl 3-(benzylimino)-2-(3,4-dichlorophenyl)acrylate (3r)
Colorless oil (27 mg, 29%). 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 2.2 Hz, 1H), 7.43–7.38 (m, 5H), 7.32 (d, J = 8.5 Hz, 1H), 7.20 (dd, J = 8.5 and 2.2 Hz, 1H), 4.96 (s, 2H), 3.79 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 172.4, 168.1, 135.0, 132.4, 131.2, 130.1, 129.3, 129.0, 128.7, 128.3, 127.9, 125.9, 67.0, 54.9, 51.6; HRMS-ESI [M + Na]+ calcd for C17H1335Cl2NNaO2+ 356.0216, found 356.0224.
Methyl 2-(3,4-dichlorophenyl)-3-((3-methoxypropyl)imino)acrylate (3s)
Colorless oil (13 mg, 15%). 1H NMR (400 MHz, CDCl3) δ 7.66 (br.s, 1H), 7.38–7.37 (m, 2H), 3.97 (t, J = 6.6 Hz, 2H), 3.80 (s, 3H), 3.53 (t, J = 5.8 Hz, 2H), 3.36 (s, 3H), 2.10–2.04 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 168.4, 167.5, 132.4, 131.8, 130.2, 128.9, 128.0, 125.7, 71.6, 68.8, 58.8, 51.5, 47.8, 30.2; HRMS-ESI [M + Na]+ calcd for C14H1535Cl2NNaO3+ 338.0321, found 338.0325.
Methyl 2-(3,4-dichlorophenyl)-3-((2-ethoxy-2-oxoethyl)imino)acrylate (3t)
Colorless oil (11 mg, 12%). 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 2.1 Hz, 1H), 7.47 (dd, J = 8.5 and 2.1 Hz, 1H), 7.40 (d, J = 8.5 Hz, 1H), 4.55 (s, 2H), 4.31 (q, J = 7.1 Hz, 2H), 3.81 (s, 3H), 1.32 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 176.4, 168.0, 166.4, 132.4, 130.9, 130.2, 129.5, 128.7, 126.4, 66.7, 62.6, 51.8, 50.7, 14.1; HRMS-ESI [M + Na]+ calcd for C14H1335Cl2NNaO4+ 352.0114, found 352.0123.
General Procedure for the Synthesis of Amidines 6a,b,d
Ketenimine 3 (0.33 mmol), amine (0.33 mmol), and THF (3.0 mL) were placed successively into a screw cap glass tube and heated at 50 °C until full consumption of ketenimine was detected (about 12 h, controlled by TLC). The solvent was removed in vacuo, and the residue was purified by column chromatography on silica gel (eluent petroleum ether–EtOAc, from 5:1 to 2:1) to give compound 6.
Methyl 3-(benzylamino)-3-(tert-butylamino)-2-phenylacrylate (6a)
Colorless oil (69 mg, 62%). 1H NMR (400 MHz, CDCl3) δ 7.41–7.40 (m, 6H), 7.36–7.31 (m, 9H), 7.24–7.20 (m, 6H), 6.60 (br.s, 1H), 5.17 (br.s, 1H), 5.11 (s, 1.1H), 4.58 and 4.45 (AB-q, J = 16.3 Hz, 2H), 4.08 (d, J = 5.7 Hz, 2.2H), 3.77 (s, 3H), 3.62 (s, 3.3H), 1.49 (s, 9H), 1.33 (s, 10H); 13C{1H} NMR (100 MHz, CDCl3) δ 172.1, 170.7, 163.6, 150.5, 142.5, 138.7, 137.9, 135.0, 132.5, 128.9, 128.6, 128.1 (2C), 127.9, 127.7 (2C), 127.4, 127.1, 125.9, 125.8, 89.2, 53.7, 52.5, 52.0, 51.0, 50.8 (2C), 50.5, 30.6, 28.7; HRMS-ESI [M + H]+ calcd for C21H27N2O2+ 339.2069, found 339.2073.
Methyl 3-(tert-butylamino)-3-((4-methylbenzyl)amino)-2-phenylacrylate (6b)
Colorless oil (58 mg, 50%). 1H NMR (400 MHz, CDCl3) δ 7.36–7.05 (m, 22H), 6.70 (br.s, 1H), 5.41 (br.s, 1H), 5.09–5.07 (m, 1.9H), 4.50 and 4.37 (AB-q, J = 16.2 Hz, 2H), 3.98–3.97 (m, 2.9H), 3.76 (s, 3H), 3.59 (s, 4.3H), 2.35 (s, 7.3H), 1.44 (s, 9H), 1.30 (s, 13H); 13C{1H} NMR (100 MHz, CDCl3) δ 172.2, 170.7, 163.7, 150.4, 139.5, 138.1, 137.0, 135.7, 135.2, 135.1, 132.6, 129.3, 128.9, 128.7, 128.1 (2C), 127.7 (2C), 127.0, 125.9, 88.8, 53.6, 52.6, 51.8, 51.0, 50.8, 50.6, 50.5, 30.7, 28.7, 21.1, 21.0; HRMS-ESI [M + H]+ calcd for C22H29N2O2+ 353.2224, found 353.2227.
Methyl 3-(tert-butylamino)-3-morpholino-2-phenylacrylate (6d)
Colorless oil (95 mg, 91%). 1H NMR (400 MHz, CDCl3) δ 7.33–7.30 (m, 2H), 7.22–7.18 (m, 1H), 7.14–7.12 (m, 2H), 6.66 (br.s, 1H), 3.61 (s, 3H), 3.48–3.46 (m, 4H), 2.77 (br.s, 4H), 1.40 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 170.9, 165.8, 138.9, 132.1, 127.8, 125.6, 96.2, 66.1, 54.6, 51.0, 50.7, 30.5; HRMS-ESI [M + H]+ calcd for C18H27N2O3+ 319.2016, found 319.2021.
Synthesis of Methyl 3-(tert-butylamino)-2-phenyl-3-(phenylimino)propanoate (6c)
Ketenimine 3a (76 mg, 0.33 mmol), aniline (31 mg, 0.33 mmol), DBU (50 mg, 0.33 mmol), and THF (3.0 mL) were placed successively into a screw cap glass tube and heated at 70 °C until full consumption of ketenimine was detected (about 6 days, controlled by TLC). The solvent was removed in vacuo, and the residue was purified by column chromatography on silica gel (eluent petroleum ether–EtOAc, from 5:1 to 2:1) to give compound 6c. Colorless oil (34 mg, 32%). 1H NMR (400 MHz, CDCl3) δ 7.36–7.30 (m, 3H), 7.25–7.21 (m, 4H), 6.97 (t, J = 7.4 Hz, 1H), 6.68 (d, J = 7.3 Hz, 2H), 5.32 (s, 1H), 4.94 (s, 1H), 3.74 (s, 3H), 1.47 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 172.0, 150.4, 149.3, 135.6, 128.8, 128.7, 127.9, 127.6, 122.0, 121.6, 52.6, 51.4 (2C), 28.4; HRMS-ESI [M + H]+ calcd for C20H25N2O2+ 325.1911, found 325.1917.
General Procedure for the Synthesis of Enamines 6e,f
A 4 mL vial was charged with methyl phenyldiazoacetate (1a) (50 mg, 0.28 mmol, 1 equiv), isocyanide (0.85 mmol, 3 equiv), and dichloromethane (2 mL). The vial was closed but not tightly to allow nitrogen to escape and irradiated with blue light (one 10W 450 nm LED) at room temperature under magnetic stirring until the diazo compound consumption (about 2 h, controlled by TLC). The reaction mixture was concentrated under reduced pressure. The obtained crude ketenimine, morpholine (22 mg, 0.25 mmol), and THF (2.0 mL) were placed successively into a screw cap glass tube and heated at 50 °C until full consumption of ketenimine was detected (about 12 h, controlled by TLC). The solvent was removed in vacuo, and the residue was purified by column chromatography on silica gel (eluent petroleum ether–EtOAc, from 5:1 to 2:1) to give the enamine.
Methyl 3-(cyclohexylamino)-3-morpholino-2-phenylacrylate (6e)
Colorless oil (38 mg, 39%). 1H NMR (400 MHz, CDCl3) δ 7.70 (br.s, 1H), 7.31–7.28 (m, 2H), 7.16–7.13 (m, 3H), 3.62 (s, 3H), 3.45–3.43 (m, 4H), 3.24–3.16 (m, 1H), 2.78 (br.s, 4H), 1.99–1.96 (m, 2H), 1.86–1.83 (m, 2H), 1.65–1.63 (m, 1H), 1.45–1.25 (m, 4H), 0.93–0.82 (m, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.1, 165.6, 139.1, 132.1, 127.8, 125.1, 89.6, 66.3, 54.6, 50.6, 50.1, 33.8, 25.5, 25.3; HRMS-ESI [M + H]+ calcd for C20H29N2O3+ 345.2173, found 345.2179.
Methyl 3-(isopropylamino)-3-morpholino-2-phenylacrylate (6f)
Colorless oil (22 mg, 26%). 1H NMR (400 MHz, CDCl3) δ 7.57 (br.s, 1H), 7.32–7.28 (m, 2H), 7.18–7.14 (m, 3H), 3.62–3.53 (m, 4H), 3.46–3.44 (m, 4H), 2.79 (br.s, 4H), 1.28 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.1, 165.6, 139.0, 132.1, 127.8, 125.1, 89.8, 66.3, 50.6, 50.1, 47.2, 23.3; HRMS-ESI [M + H]+ calcd for C17H25N2O3+ 305.1860, found 305.1867.
Acknowledgments
We gratefully acknowledge the financial support of the Russian Science Foundation (grant No 19-73-10090). This research used resources of the Magnetic Resonance Research Centre, Chemical Analysis and Materials Research Centre, Computing Centre, and Chemistry Educational Centre of the Research Park of St. Petersburg State University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00367.
Calculation details and NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- For reviews, see; a Lu P.; Wang Y. The thriving chemistry of ketenimines. Chem. Soc. Rev. 2012, 41, 5687–5705. 10.1039/c2cs35159e. [DOI] [PubMed] [Google Scholar]; b Alajarin M.; Marin-Luna M.; Vidal A. Recent Highlights in Ketenimine Chemistry. Eur. J. Org. Chem. 2012, 2012, 5637–5653. 10.1002/ejoc.201200383. [DOI] [Google Scholar]; For some recent works, see; c Zhang R.; Zhang Z.; Wang K.; Wang J. Difluoroketenimine: Generation from Difluorocarbene and Isocyanide and Its [3 + 2] Cycloadditions with Alkenes or Alkynes. J. Org. Chem. 2020, 85, 9791–9800. 10.1021/acs.joc.0c01120. [DOI] [PubMed] [Google Scholar]; d Chen X.; Qiu G.; Liu R.; Chen D.; Chen Z. Divergent oriented synthesis (DOS) of aza-heterocyclic amides through palladium-catalyzed ketenimination of 2-iodo-N-(propa-1,2-dien-1-yl)anilines. Org. Chem. Front. 2020, 7, 890–895. 10.1039/C9QO01451A. [DOI] [Google Scholar]; e Huang J.; Li F.; Cui L.; Su S.; Jia X.; Li J. Palladium-catalyzed cascade reactions of enynones and isocyanides: access towards functionalized ketenimine and its application. Chem. Commun. 2020, 56, 4555–4558. 10.1039/C9CC09363J. [DOI] [PubMed] [Google Scholar]; f Jia X.; Zhang Z.; Gevorgyan V. Three-Component Visible-Light-Induced Palladium-Catalyzed 1,2-Alkyl Carbamoylation/Cyanation of Alkenes. ACS Catal. 2021, 11, 13217–13222. 10.1021/acscatal.1c04183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krow G. R. Synthesis and Reactions of Ketenimines. Angew. Chem., Int. Ed. 1971, 10, 435–528. 10.1002/anie.197104351. [DOI] [Google Scholar]
- Ciganek E. A ketenimine from the addition of a carbene to an isocyanide. J. Org. Chem. 1970, 35, 862 10.1021/jo00828a085. [DOI] [Google Scholar]
- a Liu Z.; Cao S.; Wu J.; Zanoni G.; Sivaguru P.; Bi X. Palladium(II)-Catalyzed Cross-Coupling of Diazo Compounds and Isocyanides to Access Ketenimines. ACS Catal. 2020, 10, 12881–12887. 10.1021/acscatal.0c02867. [DOI] [Google Scholar]; b Luo J.; Chen G.-S.; Chen S.-J.; Li Z.-D.; Zhao Y.-L.; Liu Y.-L. One-Pot Tandem Protocol for the Synthesis of 1,3-Bis(β-aminoacrylate)-Substituted 2-Mercaptoimidazole Scaffolds. Adv. Synth. Catal. 2020, 362, 3635–3643. 10.1002/adsc.202000789. [DOI] [Google Scholar]; c Chen G.-S.; Chen S.-J.; Luo J.; Mao X.-Y.; Chan A. S.-C.; Sun R. W.-Y.; Liu Y.-L. Tandem Cross-Coupling/Spirocyclization/Mannich-Type Reactions of 3-(2-Isocyanoethyl)indoles with Diazo Compounds toward Polycyclic Spiroindolines. Angew. Chem., Int. Ed. 2020, 59, 614–621. 10.1002/anie.201911614. [DOI] [PubMed] [Google Scholar]; d Chu H.; Dai Q.; Jiang Y.; Cheng J. Synthesis of 2-Amino-3-hydroxy-3H-indoles via Palladium-Catalyzed One-Pot Reaction of Isonitriles, Oxygen, and N-Tosylhydrazones Derived from 2-Acylanilines. J. Org. Chem. 2017, 82, 8267–8272. 10.1021/acs.joc.7b01195. [DOI] [PubMed] [Google Scholar]; e Dai Q.; Jiang Y.; Yu J.-T.; Cheng J. Palladium-Catalyzed Three-Component Reaction of N-Tosyl Hydrazones, Isonitriles and Amines Leading to Amidines. Chem. Commun. 2015, 51, 16645–16647. 10.1039/C5CC06771E. [DOI] [PubMed] [Google Scholar]; f Yan X.; Liao J.; Lu Y.; Liu J.; Zeng Y.; Cai Q. Pd-Catalyzed One-Pot Synthesis of Polysubstituted Acrylamidines from Isocyanides, Diazo Compounds, and Imines. Org. Lett. 2013, 15, 2478–2481. 10.1021/ol4009552. [DOI] [PubMed] [Google Scholar]; g Zhou F.; Ding K.; Cai Q. Palladium-Catalyzed Amidation of N-Tosylhydrazones with Isocyanides. Chem. - Eur. J. 2011, 17, 12268–12271. 10.1002/chem.201102459. [DOI] [PubMed] [Google Scholar]
- a Bu X.-B.; Wang Z.; Wang X.-D.; Meng X.-H.; Zhao Y.-L. Rhodium-Catalyzed Tandem Reaction of Isocyanides with Trifluorodiazoethane and Nucleophiles: Divergent Synthesis of Trifluoroethyl-Substituted Isoquinolines, Imidates, and Amidines. Adv. Synth. Catal. 2018, 360, 2945–2951. 10.1002/adsc.201800445. [DOI] [Google Scholar]; b Yu Y.; Zhang Y.; Wang Z.; Liang Y.-X.; Zhao Y.-L. A rhodium-catalyzed three-component reaction of arylisocyanides, trifluorodiazoethane, and activated methylene isocyanides or azomethine ylides: an efficient synthesis of trifluoroethyl-substituted imidazoles. Org. Chem. Front. 2019, 6, 3657–3662. 10.1039/C9QO00856J. [DOI] [Google Scholar]
- a Gu Z.-Y.; Han H.; Li Z.-Y.; Ji S.-J.; Xia J.-B. Catalytic synthesis of functionalized amidines via cobalt-carbene radical coupling with isocyanides and amines. Org. Chem. Front. 2021, 8, 1544–1550. 10.1039/D1QO00063B. [DOI] [Google Scholar]; b Grass A.; Dewey N. S.; Lord R. L.; Groysman S. Ketenimine Formation Catalyzed by a High-Valent Cobalt Carbene in Bulky Alkoxide Ligand Environment. Organometallics 2019, 38, 962–972. 10.1021/acs.organomet.8b00911. [DOI] [Google Scholar]
- Abernethy D. R.; DeStefano A. J.; Cecil T. L.; Zaidi K.; Williams R. L. USP Metal Impurities Advisory. Panel Metal Impurities in Food and Drugs. Pharm. Res. 2010, 27, 750–755. 10.1007/s11095-010-0080-3. [DOI] [PubMed] [Google Scholar]
- a Sun C.-L.; Shi Z.-J. Transition-Metal-Free Coupling Reactions. Chem. Rev. 2014, 114, 9219–9280. 10.1021/cr400274j. [DOI] [PubMed] [Google Scholar]; b Kloss F.; Neuwirth T.; Haensch V. G.; Hertweck C. Metal-Free Synthesis of Pharmaceutically Important Biaryls by Photosplicing. Angew. Chem., Int. Ed. 2018, 57, 14476–14481. 10.1002/anie.201805961. [DOI] [PubMed] [Google Scholar]
- a Strelnikova J. O.; Rostovskii N. V.; Khoroshilova O. V.; Khlebnikov A. F.; Novikov M. S. An Efficient Synthesis of Functionalized 2H-1,3,5-Oxadiazines via Metal-Carbenoid-Induced 1,2,4-Oxadiazole Ring Cleavage. Synthesis 2021, 53, 348–358. 10.1055/s-0040-1707278. [DOI] [Google Scholar]; b Koronatov A. N.; Rostovskii N. V.; Khlebnikov A. F.; Novikov M. S. Synthesis of 3-Alkoxy-4-Pyrrolin-2-ones via Rhodium(II)-Catalyzed Denitrogenative Transannulation of 1H-1,2,3-Triazoles with Diazo Esters. Org. Lett. 2020, 22, 7958–7963. 10.1021/acs.orglett.0c02893. [DOI] [PubMed] [Google Scholar]; c Koronatov A. N.; Rostovskii N. V.; Khlebnikov A. F.; Novikov M. S. Rh(II)-Catalyzed Ring Expansion of Pyrazoles with Diazocarbonyl Compounds as a Method for the Preparation of 1,2-Dihydropyrimidines. J. Org. Chem. 2018, 83, 9210–9219. 10.1021/acs.joc.8b01228. [DOI] [PubMed] [Google Scholar]; d Agafonova A. V.; Smetanin I. A.; Rostovskii N. V.; Khlebnikov A. F.; Novikov M. S. Expedient synthesis of 3-hydroxypyrroles via Bu3SnH-triggered ionic 5-exo-trig-cyclization of 5-chloro-3-azamuconoate derivatives. Org. Chem. Front. 2018, 5, 3396–3401. 10.1039/C8QO00982A. [DOI] [Google Scholar]
- a Boyer J. H.; Beverung W. N-cyclohexyldiphenylketenimine from cyclohexyl isocyanide and diphenyldiazomethane. J. Chem. Soc. D 1969, 1377–1378. 10.1039/c2969001377b. [DOI] [Google Scholar]; b Green J. A.; Singer L. A. Ketenimines via the photolysis of diphenyldiazomethane in the presence of isonitriles.. Tetrahedron Lett. 1969, 10, 5093–5095. 10.1016/S0040-4039(01)88892-4. [DOI] [Google Scholar]
- a Jurberg I. D.; Davies H. M. L. Blue light-promoted photolysis of aryldiazoacetates. Chem. Sci. 2018, 9, 5112–5118. 10.1039/C8SC01165F. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hommelsheim R.; Guo Y.; Yang Z.; Empel C.; Koenigs R. M. Blue-Light-Induced Carbene-Transfer Reactions of Diazoalkanes. Angew. Chem., Int. Ed. 2019, 58, 1203–1207. 10.1002/anie.201811991. [DOI] [PubMed] [Google Scholar]; c Yang Z.; Stivanin M. L.; Jurberg I. D.; Koenigs R. M. Visible light-promoted reactions with diazo compounds: a mild and practical strategy towards free carbene intermediates. Chem. Soc. Rev. 2020, 49, 6833–6847. 10.1039/D0CS00224K. [DOI] [PubMed] [Google Scholar]; d Durka J.; Turkowska J.; Gryko D. Lightening Diazo Compounds?. ACS Sustainable Chem. Eng. 2021, 9, 8895–8918. 10.1021/acssuschemeng.1c01976. [DOI] [Google Scholar]; e Xiao T.; Mei M.; He Y.; Zhou L. Blue light-promoted cross-coupling of aryldiazoacetates and diazocarbonyl compounds. Chem. Commun. 2018, 54, 8865–8868. 10.1039/C8CC04609C. [DOI] [PubMed] [Google Scholar]
- Related work on formation of ketenimines from isocyanides and bromides under visible-light photocatalysis has been recently published; Cannalire R.; Amato J.; Summa V.; Novellino E.; Tron G. C.; Giustiniano M. Visible-Light Photocatalytic Functionalization of Isocyanides for the Synthesis of Secondary Amides and Ketene Aminals. J. Org. Chem. 2020, 85, 14077–14086. 10.1021/acs.joc.0c01946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M. H.; Twilton J.; MacMillan D. W. C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Geise C. M.; Wang Y.; Mykhaylova O.; Frink B. T.; Toscano J. P.; Hadad C. M. Computational and Experimental Studies of the Effect of Substituents on the Singlet-Triplet Energy Gap in Phenyl(carbomethoxy)carbene. J. Org. Chem. 2002, 67, 3079–3088. 10.1021/jo0255330. [DOI] [PubMed] [Google Scholar]; b Wang J.; Burdzinski G.; Kubicki J.; Platz M. S. Ultrafast UV-Vis and IR Studies of p-Biphenylyl Acetyl and Carbomethoxy Carbenes. J. Am. Chem. Soc. 2008, 130, 11195–11209. 10.1021/ja803096p. [DOI] [PubMed] [Google Scholar]; c Wang Y.; Hadad C. M.; Toscano J. P. Solvent Dependence of the 2-Naphthyl(carbomethoxy)carbene Singlet-Triplet Energy Gap. J. Am. Chem. Soc. 2002, 124, 1761–1767. 10.1021/ja012139v. [DOI] [PubMed] [Google Scholar]; d Zhu Z.; Bally T.; Stracener L. L.; McMahon R. J. Reversible Interconversion between Singlet and Triplet 2-Naphthyl(carbomethoxy)carbene. J. Am. Chem. Soc. 1999, 121, 2863–2874. 10.1021/ja983277w. [DOI] [Google Scholar]; e Wang Y.; Yuzawa T.; Hamaguchi H.; Toscano J. P. Time-Resolved IR Studies of 2-Naphthyl(carbomethoxy)carbene: Reactivity and Direct Experimental Estimate of the Singlet/Triplet Energy Gap. J. Am. Chem. Soc. 1999, 121, 2875–2882. 10.1021/ja983278o. [DOI] [Google Scholar]
- Jana S.; Pei C.; Empel C.; Koenigs R. M. Photochemical Carbene Transfer Reactions of Aryl/Aryl Diazoalkanes–Experiment and Theory. Angew. Chem., Int. Ed. 2021, 60, 13271–13279. 10.1002/anie.202100299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanni D.Isonitriles: a Useful Trap in Radical Chemistry in Radicals in Organic Synthesis; Renaud P.; Sibi M. P., Eds.; Wiley, 2001. [Google Scholar]
- Greenhill J. V.; Lue P. Amidines and Guanidines in Medicinal Chemistry. Prog. Med. Chem. 1993, 30, 203–326. 10.1016/S0079-6468(08)70378-3. [DOI] [PubMed] [Google Scholar]
- McCarthy J. R.; Wright D. L.; Schuster A. J.; Abdallah A. H.; Shea P. J.; Eysters R. New Bicyclic Antidepressant Agent. Synthesis and Activity of Napactadine and Related Compounds. J. Med. Chem. 1985, 28, 1721–1727. 10.1021/jm00149a031. [DOI] [PubMed] [Google Scholar]
- Keipour H.; Ollevier T. Iron-Catalyzed Carbene Insertion Reactions of α-Diazoesters into Si–H Bonds. Org. Lett. 2017, 19, 5736–5739. 10.1021/acs.orglett.7b02488. [DOI] [PubMed] [Google Scholar]
- Arredondo V.; Hiew S. C.; Gutman E. S.; Premachandra I. D. U. A.; Van Vranken D. L. Enantioselective Palladiun-Catalyzed Carbene Insertion into the N–H Bonds of Aromatic Heterocycles. Angew. Chem., Int. Ed. 2017, 56, 4156–4159. 10.1002/anie.201611845. [DOI] [PubMed] [Google Scholar]
- Silva A. F.; Afonso M. A. S.; Cormanich R. A.; Jurberg I. D. Room Temperature Coupling of Aryldiazoacetates with Boronic Acids Enhanced by Blue Light Irradiation. Chem. - Eur. J. 2020, 26, 5648–5653. 10.1002/chem.201905812. [DOI] [PubMed] [Google Scholar]
- Ye F.; Qu S.; Zhou L.; Peng C.; Wang C.; Cheng J.; Hossain M. L.; Liu Y.; Zhang Y.; Wang Z.-X.; Wang J. Palladium-catalyzed C–H Functionalization of Acyldiazomethane and Tandem Cross-Coupling Reaction. J. Am. Chem. Soc. 2015, 137, 4435–4444. 10.1021/ja513275c. [DOI] [PubMed] [Google Scholar]
- Ye F.; Wang C.; Zhang Y.; Wang J. Synthesis of Aryldiazoacetates through Palladium(0)-Catalyzed Deacylative Cross-Coupling of Aryl Iodides with Acyldiazoacetates. Angew. Chem., Int. Ed. 2014, 53, 11625–11628. 10.1002/anie.201407653. [DOI] [PubMed] [Google Scholar]
- Santos F. M. F.; Rosa J. N.; André V.; Duarte M. T.; Veiros L. F.; Gois P. M. P. N-Heterocyclic Carbene Catalyzed Addition of Aldehydes to Diazo Compounds: Stereoselective Synthesis of N-Acylhydrazones. Org. Lett. 2013, 15, 1760–1763. 10.1021/ol400563w. [DOI] [PubMed] [Google Scholar]
- Santi M.; Ould D. M. C.; Wenz J.; Soltani Y.; Melen R. L.; Wirth T. Metal-Free Tandem Rearrangement/Lactonization: Access to 3,3-Disubstituted Benzofuran-2-(3H)-ones. Angew. Chem., Int. Ed. 2019, 58, 7861–7865. 10.1002/anie.201902985. [DOI] [PubMed] [Google Scholar]
- Hao J.; Xu Y.; Xu Z.; Zhang Z.; Yang W. Pd-Catalyzed Three-Component Domino Reaction of Vynil Benzoxazinanones for Regioselective and Stereoselective Synthesis of Allylic Sulfone-Containing Amino Acid Derivatives. Org. Lett. 2018, 20, 7888–7892. 10.1021/acs.orglett.8b03440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.