

Abstract
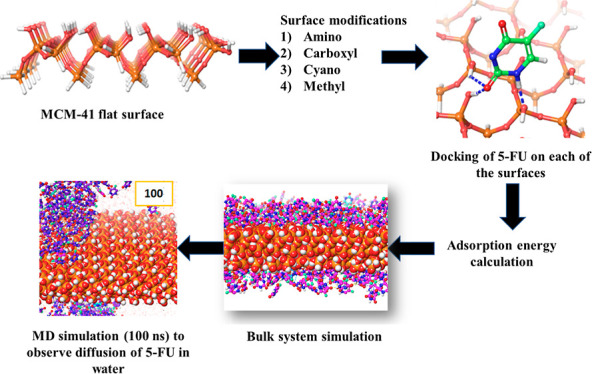
MCM-41, a type of mesoporous silica nanoparticle, has garnered widespread interests as a useful carrier for drug delivery wherein the drug gets adsorbed into the pores of the carrier. To understand the adsorption mechanism and release of the drug at the molecular level, in the current study, it was attempted to generate a computational model for the loading of 5-fluorouracil (5-FU), a chemotherapeutic agent into surface-modified MCM-41. The molecular surface models of the mesoporous silica (MCM-41) nanoparticle with different surface substitutions were created. In the first stage, molecular mechanics (MM) simulations were carried out to obtain the optimized surface structures. Subsequently, a 5-FU drug molecule in its different forms was docked on top of different MCM-41 surfaces to understand the adsorption orientation and energetics. To further validate the results, more accurate quantum mechanical (QM) calculations were also carried out, and the energetics between the QM and MM calculations are found to be similar. All the substitutions (−NH2, −CN, −COOH) except the methyl substitution exhibited favorable interactions compared to the unsubstituted MCM-41 surface which was in accordance with the experimental observations. The release rate of 5-FU from MCM-41 and aminopropyl-substituted MCM-41 (MCM-NH2) was studied using molecular dynamics simulations which revealed that the release rate of 5-FU from the MCM-NH2 surface was slower compared to that of plain MCM-41. The detailed surface characteristics and the adsorption energies from the molecular simulations correlating the loading capacity and release are reported in here.
1. Introduction
Nanoparticles have become the focus of the field of therapeutics owing to their more effective therapeutic intervention compared to the conventional therapy. Considerable efforts have been made, and significant progress has been achieved over the last few years in the field of nanotherapeutics. Nanocarriers such as liposomes, polymeric nanoparticles, and inorganic nanoparticles have been explored for their potential in delivering drugs.1 Over the past decade, mesoporous silica nanoparticles (MSNs) especially MCM-41 (Mobil Crystalline Material no. 41) are gaining wide interest among scientists as drug delivery systems. They are unique in terms of their uniform mesopores, feature of functionalization on external and internal surfaces, tunable pore size, and large pore volume compared to other carriers.2 These attributes can be widely exploited in modulating their loading efficiency and release characteristics. Researchers have explored the use of these nanocarriers for a loading variety of drugs. Experimentally, optimizing the carrier to achieve high drug loading is a tedious task, time-consuming, and expensive. Hence, simplification of the formulation process is more and more important in the pharmaceutical research. Drug loading depends on the various physicochemical and topological characteristics of the drug and carriers such as intermolecular interactions. These interactions can be manipulated by rational modifications to the system to enhance the drug–carrier interactions. Recent progress in the field of computational technology has led to the use of in silico modeling in predicting various properties of drugs, carriers, and drug-carrier systems which can be very well explored in formulation design. They can be well simulated using computational tools wherein an understanding of these interactions can be studied at the molecular level and can be effectively used as an approach in predicting the drug loading capacity.3,4 Computational tools such as molecular dynamics (MD), molecular docking along with the use of molecular mechanics (MM), and quantum mechanics (QM) theory can help predict the interaction between drug-carrier systems. MD simulation is an important tool which provides an idea of the behavior and conformational changes in molecules and complexes over a period of time. The forces acting on each atom are calculated by solving the classical Newton’s laws of motion. MD simulations have been widely used to study the behavior of biological molecules. However, it is seldom used in formulation design.5,6 Molecular docking is another virtual screening tool wherein the interaction between the drug and its binding site can be studied at the atomistic level. This tool aids in the screening of lead molecules during the drug discovery process and can also be used to study the non-covalent interactions between the drug and the drug–carrier complex.6,7
Many studies report the use of computational tools in studying the intermolecular interaction between a drug and various carriers like dendrimers,8,9 chitosan,10 and certain polymers.11,12 Modeling the interactions between mesoporous silica and ibuprofen, aspirin as the drug was studied in detail along with the effect of acidic and basic functionalization on the type of interactions.13,14 Surface functionalization is one of the methods to enhance the interaction between drugs and excipients thereby tailoring the loading and targeting efficiency. However, no attempts have been made so far to the best of our knowledge to study the effect of surface functionalization of MSNs on its loading capacity by computational modeling.
In the present study, a similar computational approach was used to analyze the effect of surface functionalization on the loading capacity of MSNs with 5-fluorouracil (5-FU) as the model drug. We studied the interactions on a flat surface of an MCM-41 model built using computational tools to approximate the interior of the pores of MCM-41. In addition, the release of 5-FU in water was also studied via simulations. Various tools, such as MD and docking, were used to study the drug–silica system. Research has shown that loading capacity of mesoporous silica can be enhanced by functionalization of the surface silanol groups with various modifications.15,16 In the current study, the available literature data was used to find out a correlation between the reported loading of 5-FU onto the surface-modified MCM-41 and the binding energy.16 This approach will reduce the experimentation efforts and time taken by scientists and formulators in designing a formulation. The theoretical approach will give a better understanding of the properties affecting the loading capacity of mesoporous silica.
2. Results and Discussion
This study involves an effort to generate the computational models to correlate and predict the effect of functionalization on the loading of 5-FU, a model drug, into the MSNs, MCM-41. Furthermore, attempts were also made to study the impact of molecular interactions on the release of 5-FU from the surface. The computational tools aid in understanding the adsorption and the release behavior at the molecular level which is difficult to study via an experimental approach which provides minimal information. Hence, these tools could be explored for the selection of a suitable solvent and surface functionalization during the formulation design of MCM-41 to optimize the drug loading and its release.
2.1. Ionic Forms of 5-FU
5-FU is a molecule which possesses two potential sites of deprotonation. The Epik tool of Schrödinger predicted two possible anionic forms of 5-FU viz., one where N1 is deprotonated (pKa to be 7.82) designated as 5-FU1 and N3, the deprotonated state (5-FU2) with a pKa of 8.19 (Figure 1). The 5-FU1 with a lower pKa was found to be closer to the reported pKa of 5-FU, that is, 8.02.17 The more acidic proton will be lost first in an aqueous solution, and hence, N1-deprotonated 5-FU has a higher percentage of existing at pH 7.0 which is in accordance with the paper by Markova and co-workers.18 However, in the present study, both the forms were taken into consideration due to the very close theoretical pKa of both the N–H groups, and at the given pH of 7.0, there are high chances that the 5-FU can coexist as 5-FU, 5-FU1, and 5-FU2. Figure 2 depicts the electrostatic potential (ESP) of 5-FU anionic species 5-FU1 and 5-FU2.
Figure 1.

Possible ionic forms of 5-FU at pH 7.0.
Figure 2.
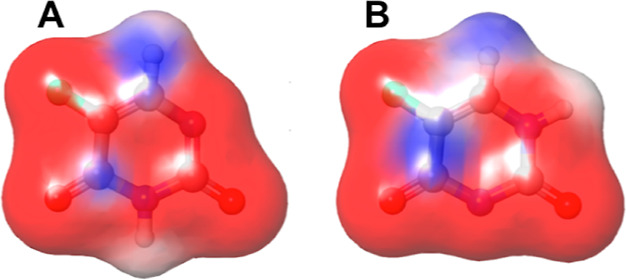
ESP of 5-FU anionic forms (A) 5-FU1 and (B) 5-FU2. The red color represents the electronegative region; blue indicates the electropositive region, and the gray color represents hydrophobic regions.
2.2. Electrostatic Surfaces
We have modeled the interaction of 5-FU with the silica surface (inner pore wall) through a periodic slab approach. We used a flat surface to model the interactions on MCM-41 in the present study which has an average pore diameter ranging from 2 to 5 to 4.5 nm. As 5-FU is a small molecule of ∼5.3 Å19 and its size is smaller compared to the diameter of the pore, a flat surface would be an approximation of what 5-FU sees inside the pores. The representative mesoporous silica structure was generated using an alpha-silica crystal unit cell wherein the oxygen-terminated slab of silica was generated, and hydrogen atoms were added to neutralize the surface. The generated model contained 44 silicon atoms, 88 oxygen atoms, and 84 terminal hydrogen atoms on either side of the surface and is labeled as MCM-41 in this paper (Figure 3). Six different surface terminations viz., 3-aminopropyl (MCM-NH2), 3-cyanopropyl (MCM-CN), 3-carboxypropyl (MCM-COOH), methyl (MCM-CH3), protonated 3-aminopropyl (MCM-NH3+), and deprotonated 3-carboxypropyl (MCM-COO–) were carried out to analyze and understand the impact of the surface modifications. Figure 4 depicts the representative structures of the modified surface. Three individual surfaces per substitution were prepared by randomly replacing the terminal hydrogens on the surface with the above-mentioned functional groups. The generated structures were energy minimized to obtain stable MCM structures as explained in Section 4.3. Figure 5 shows the ESP surface of all the seven MCM-41 surfaces considered. The positively charged areas are shown in blue color, and the negatively charged areas are marked in red color. The hydrophobic regions are gray in color. As expected, the plain MCM-41 has a systematic charge distribution on the surface and was flat in nature (Figure 5A). In all other modified surfaces, due to the substitutions, the surface has different charge densities (Figure 5B–G) leading to random corrugations on the surface at the substituted sites. The methyl-substituted surface exhibited predominant hydrophobic regions on the surface, and the methyl groups formed an average angle of 53.9° with a 3.7 Å ridge with the surface. The cyano-substituted MCM-41 had a negatively charged surface due to the CN group in combination with the hydrophobic areas; on the other hand, the deprotonated carboxyl-substituted surface was dominated by the strong electronegative regions. The carboxyl-substituted MCM-41 surface was also dominated by electronegative regions but exhibited electropositive and hydrophobic patches on the surface. The cyano- and the carboxyl-substituted surfaces demonstrated a bumpy appearance which was substantiated from the higher angle and corrugations of the substituents with the surface silanol groups. The cyano groups formed an angle of 63.3° with a groove of 5.05 Å, and the COOH group formed 70.9°, a groove of 4.16 Å, and COO– demonstrated a ridge of 5.3 Å with an angle of 51.4° with the surface. The neutral amine-substituted MCM-41 model had a combination of the electropositive and electronegative regions with corrugated hydrophobic regions, whereas the protonated amine-substituted model had predominant electropositive areas. Both the amino-modified surfaces formed an average angle of 66.6° with a corrugation of ∼5.8 Å with the surface silanol groups leading to the formation of deep pockets which also aided in better adsorption of the 5-FU molecule.
Figure 3.
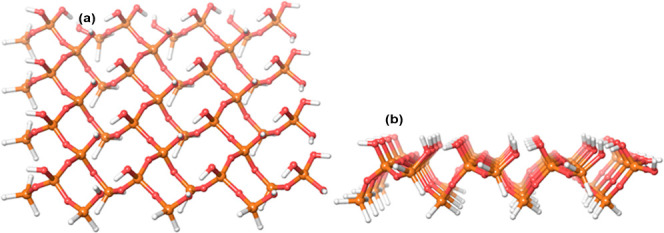
Top (A) and front (B) view of the model mesoporous surface constructed (MCM-41). The silicon atoms are shown in brown, the oxygen atoms are shown in red, and the hydrogen atoms are shown in white color.
Figure 4.

Representative structures of surface-modified MCMs.
Figure 5.

ESP surface of MCM-41 and its surface modifications with the adsorbed 5-FU molecule.
The positively charged area is shown in blue, the negatively charged area is shown in red, and hydrophobic regions are shown in gray color. The unionized 5-FU is depicted in yellow color; the ionized forms 5-FU1 and 5-FU2 are depicted in purple and green color, respectively. (A) represents the ESP surface with the adsorbed drug on the plain, (B) is 3-cyanopropyl-, (C) is methyl-, (D) is 3-aminopropyl-, (E) is protonated 3-aminopropyl-, (F) is 3-carboxypropyl-, and (G) is deprotonated 3-carboxypropyl-substituted MCM ESP surface model.
The partial charge densities were calculated for the unmodified MCM-41 surface and compared with the surface-modified MCM-41 to examine the changes in the partial charge density after modification. The silicon atom, oxygen atom, and hydrogen atom of the unmodified MCM-41 surface exhibited a partial charge density of 1.020, −0.659, and 0.404 electron unit (eu), respectively. In the case of different surface-modified MCM-41, the Si–O group attached showed on an average −0.441 eu partial charge density, while carbon attached next to the Si–O group exhibited 0.066 eu partial charge density in all modified groups. The hydrogen atoms attached to the carbon showed on an average partial charges density 0.060 eu. The nitrogen atom in 3-cyanopropyl-, 3-aminopropyl-, and protonated 3-aminopropyl-modified groups exhibited the 0.330, −0.897, and −0.331 eu partial charge density, respectively, while the carbon atom in methyl-, 3-carboxypropyl-, and deprotonated 3-carboxypropyl-modified groups exhibited 0.066, −0.331, and −0.331 eu partial charge density, respectively.
2.3. Adsorption of 5-FU on the Generated Surfaces
5-FU was docked on the surface with an aim to maximize the interactions between the silanol groups/substituted functional groups and the various functional groups of 5-FU using the “Glide” docking tool. Based on the results of the docking studies, the interactions observed are discussed below.
2.3.1. Non-Bonded Interactions
2.3.1.1. Hydrogen Bonds
Hydrogen bonding is the most dominant noncovalent interaction observed at the silica surfaces. Silanols can act as both a hydrogen bond donor and acceptor. In the present work, hydrogen bonds (donor and acceptor) were found to form between the surface and the 5-FU molecule. In the case of the neutral form (5-FU), all the surfaces were found to have at least one hydrogen bond and electrostatic interaction. The hydrogen atoms attached to the nitrogen at the 1st and 3rd position of the 5-FU molecule were found to form a donor-type interaction, whereas the carbonyl groups in the 2nd and 4th position formed an acceptor-type hydrogen bond interaction. In the anionic species, the ionized nitrogen was found to have an acceptor type of hydrogen bond because of its negative charge. The hydrogen bond interactions of 5-FU and its anionic forms with the generated MCM-41 surfaces have been shown in Figure 6.
Figure 6.
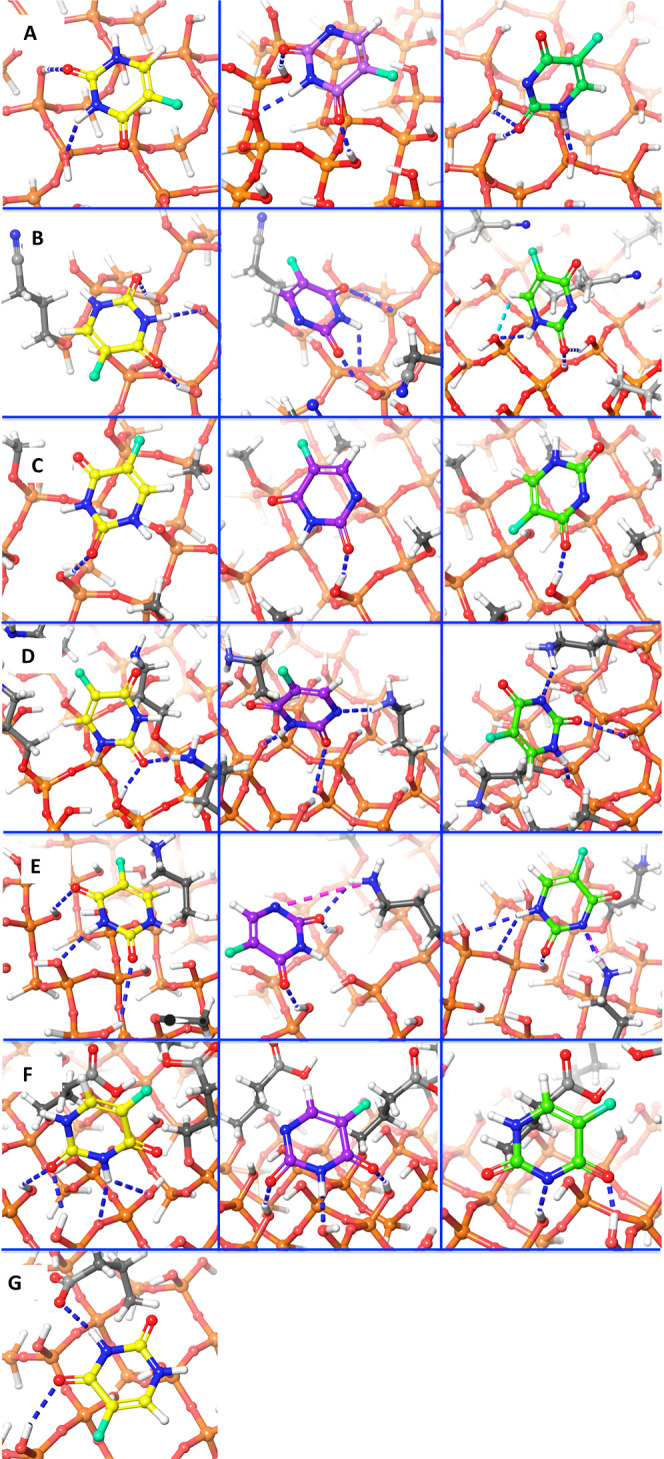
Hydrogen bond interactions of the adsorbed drug with MCM-41 and its surface modifications.
The hydrogen bonds are shown in blue-color dotted lines, and the ionic interactions are shown in pink-color dotted lines. The unionized 5-FU is depicted in yellow color (first column), the ionized 5-FU1 in purple (second column), and 5-FU2 are depicted in green color (third column). The column (A) represents plain MCM-41; (B) 3-cyanopropyl-; (C) methyl-; (D) 3-aminopropyl-; (E) protonated 3-aminopropyl-; (F) 3-carboxypropyl-; and (G) deprotonated 3-carboxypropyl-substituted MCM-41 surfaces.
The electrostatic surface for plain MCM-41, MCM-CN, and MCM-CH3 exhibited areas with a gray tinge indicating more of hydrophobic nature. MCM-NH2 and MCM-NH3+ surfaces showed the formation of small deep well-like pockets within which the 5-FU was adsorbed (Figure 5D,E). The MCM-NH3+ surface showed deep blue color tinges which indicated the strong electropositive nature of the surface. Ionic interactions were observed for the protonated surface with the ionic groups of the 5-FU. The electrostatic surface for MCM-COOH revealed light red and gray color patches corresponding to the silanol and neutral carboxypropyl groups on the surface. These carboxyl groups form better hydrogen bonds with polar compounds. Moreover, the carboxyl substituents mainly exist as O=C and O–H (trigonal planar) which renders a change in the electrostatic surface when compared to the plain MCM-41 resulting in deep corrugations, and the 5-FU molecule was found to get adsorbed in the pockets with greater interactions.
Table 1 presents the number of hydrogen bonds formed between 5-FU and the MCM-41 surfaces under consideration. It is evident from the values that the CH3-substituted surface had the lowest number of hydrogen bonding interactions with any of the forms of 5-FU. As expected, the negatively charged COO– surface had interactions only with the neutral 5-FU. The MCM-COO– surface undergoes charge repulsion interaction with the charged ionic forms of 5-FU, and hence, no adsorption was observed in these cases. With the positively charged NH3+ surface, the attractive electrostatic interactions led to a higher number of hydrogen bonds in all the three forms of 5-FU. The other neutral surfaces showed the same number of hydrogen bonds (nine) except the plain MCM-41 surface (which showed a total of seven hydrogen bonds).
Table 1. Types of hydrogen bonds (H-bonds) formed on different surfaces by 5-FU, 5-FU1, and 5-FU2.
| Bond
distance (Å) |
Total number of H-bonds | ||||||
|---|---|---|---|---|---|---|---|
| 5-FU |
5-FU1 |
5-FU2 |
|||||
| MCM-41 surfaces | Donor | Acceptor | Donor | Acceptor | Donor | Acceptor | |
| Plain | 2.25 | 2.30 | 1.87 | 2.49,1.77 | 1.65 | 1.61 | 7 |
| NH2 | 2.80 | 1.89, 2.22 | 1.91 | 1.52, 2.39 | 1.78 | 1.50, 2.43 | 9 |
| CN | 2.73 | 2.02, 2.20 | 2.65 | 1.73, 1.60 | 2.33 | 1.63, 3.20 | 9 |
| COOH | 2.57, 2.58 | 2.32, 2.25 | 1.75 | 1.55, 1.52 | 1.80 | 1.60 | 9 |
| CH3 | – | 2.07 | – | 1.54 | – | 1.54 | 3 |
| NH3+ | 2.20 | 2.30, 3.10 | 1.89 | 1.54, 2.37, 1.75 | 2.90, 2.89 | 1.54, 1.92 | 11 |
| COO– | 2.28 | 2.32 | – | – | – | – | 2 |
The values of the hydrogen bond distance suggested that the charged species of 5-FU formed stronger interactions leading to short hydrogen bonds (Table 1). The neutral form was found to have an average hydrogen bond distance of 1.94 Å. In all the cases, the results revealed that the drug adsorption on the MCM-41 surface was mainly driven by hydrogen bond interactions. Additionally, weak hydrophobic interactions were also observed between the ring carbons and the surface silica and the bridged oxygen atoms. In the case of the MCM-NH3+ surface, there was a favorable salt-bridge type of ionic interaction between the charged NH3+ group and the anionic forms of 5-FU. On the other hand, the MCM-COO– surface had negative interactions due to strong charge repulsion with the anionic forms.
2.3.1.2. Hydrophobic Interactions
Although the adsorption between the 5-FU molecule and the various silica surfaces was majorly driven by the hydrogen bonding, we also observed other non-bonded interactions which plausibly contributed to the adsorption of 5-FU.
The non-bonded interactions along with their adsorption energies are presented in Table 2.
Table 2. Non-Bonded Interactions Observed between 5-FU Molecule and Silica Surfacesa.
| Groups
on the surfaces showing interaction |
||||||||
|---|---|---|---|---|---|---|---|---|
| Groups on 5-FU showing interaction | MCM-41 | MCM-CN | MCM-CH3 | MCM-NH2 | MCM-NH3+ | MCM-COOH | MCM-COO- | |
| H-Bond Interactions | ||||||||
| 5-FU | ||||||||
| Acceptor | 2(C=O) | H–O–Si | H–O–Si | H–O–Si | H–O–Si, NH2–(C3H6)–Si | H–O–Si | with two H–O–Si groups | – |
| 4(C=O) | – | H–O–Si | – | – | H–O–Si | – | H–O–Si | |
| Donor | 3(N–H) | Si–O–H | Si–O–H | – | – | Si–O–H | Si–O–H, Si–O–Si | –OOC–(C3H6)–Si |
| 5-FU1 | ||||||||
| Acceptor | 2(C=O) | H–O–Si | H–O–Si | H–O–Si | H–O–Si | H–O–Si, NH3+–(C3H6)–Si | H–O–Si | – |
| 4(C=O) | H–O–Si | H–O–Si | – | – | H–O–Si | H–O–Si | – | |
| 1(N–) | – | – | – | NH2–(C3H6)–Si | – | – | – | |
| Donor | 3(N–H) | Si–O–H | Si–O–H | – | Si–O–H | Si–O–H | Si–O–H | – |
| 5-FU2 | ||||||||
| Acceptor | 2(C=O) | H–O–Si | H–O–Si | H–O–Si | H–O–Si | H–O–Si | – | – |
| 4(C=O) | – | – | – | – | – | H–O–Si | – | |
| 3(N–) | – | – | – | NH2-(C3H6)–Si | – | H–O–Si | – | |
| Donor | 1(N–H) | Si–O–H | – | – | Si–O–H | Si–O–H, Si–O–Si | – | – |
| Hydrophobic Interactions | ||||||||
| 5-FU, | ring carbons | Si, Si–O–Si | CN–(C3H6)–Si, | CH3–(C3H6)–, | NH2–(C3H6)–Si | NH3+–(C3H6)–Si, | HOOC–(C3H6)–Si, | –OOC–(C3H6)–Si |
| 5-FU1, | Si–O–Si, Si | Si–O–Si | Si–O–Si, Si | No interactions with ionized species of 5-FU | ||||
| 5-FU2 | ||||||||
| Adsorption Energy (kcal/mol) | ||||||||
| Molecular Mechanics (MM) by OPLS3 force field | ||||||||
| 5-FU | –14.318 | –20.227 | –12.736 | –15.320 | –12.839 | –16.057 | –20.286 | |
| 5-FU1 | –26.629 | –39.083 | –22.507 | –33.885 | –226.971 | –32.143 | 110.357 | |
| 5-FU2 | –19.483 | –28.497 | –19.329 | –23.796 | –234.593 | –32.557 | 99.735 | |
| Quantum Mechanics (QM) by Jaguar | ||||||||
| 5-FU | –17.026 | –28.160 | –13.986 | –18.331 | –23.810 | –19.755 | –12.544 | |
| 5-FU1 | –64.856 | –68.944 | –41.854 | –66.856 | –249.845 | –61.227 | 64.081 | |
| 5-FU2 | –31.844 | –48.319 | –55.653 | –42.433 | –253.184 | –44.096 | –41.804 | |
| Actual % loading from literature report16 | 18.34 | 22.54 | 12.73 | 28.89 | – | 20.73 | – | |
The numbers in bold outside brackets represent the position of the atom in the 5-FU molecule, the underline below the groups represents the groups involved in interaction.
2.4. Adsorption of 5-FU and Its Orientations
The adsorption of 5-FU on the MCM surfaces was predicted by docking. The Glide docking module uses the OPLS3 force field and molecular mechanic (MM) principles for potential energy calculations.21,−23 The adsorption models generated initially by Glide docking were further optimized and analyzed by the QM methods using the Jaguar module in the Schrödinger MS-Suite.24 The QM calculation offers a higher level of accuracy in the potential energy calculation than that by MM. Interestingly, in our study, we observed that in all the cases, the QM optimization led to only a minor change in the orientation. However, electrostatic and hydrophobic interactions remained unaltered.
Based on the docking results, the most stable pose of the 5-FU molecule was considered to study its interaction and orientation on the MCM surfaces. A similar trend was also observed on application of the “Boltzmann Population” function to the various 5-FU poses generated. The unionized 5-FU had a parallel orientation with the MCM-41 surface. In this orientation, 5-FU formed two hydrogen bonds and a weak hydrophobic interaction with the silica atoms and the bridging oxygen of MCM-41. Figure 7 shows the relaxation of the molecule during the QM optimization from the initial docked position. There was about an 11° angle tilt in the binding orientation away from the direction of the surface for 5-FU after QM optimization. A similar pattern was observed for other two ionic forms where the tilt was within a 5° angle.
Figure 7.

Adsorption pose orientation of 5-FU after QM optimization; the right-side column shows the side view and the left-side column the top view. (A) represents the plain MCM-41 surface, (B) is 3-aminopropyl-, and (C) is 3-carboxypropyl-modified surfaces.
The 5-FU molecule and its anionic forms were adsorbed preferentially onto the unsubstituted site favoring the formation of a hydrogen bond with the surface. The amino, cyano, and carboxyl surface modifications resulted in large corrugations or hills and valleys on the surfaces. The drug molecules preferred to get adsorbed on the pockets created by these corrugations. The hydrogen bonds were formed between the unmodified silanol groups and the different anionic forms of the drug molecule. In the case of carboxyl substitution, the highest difference between the MM and QM adsorbed poses was observed. The QM optimization led to an angle tilt of 16–26° with respect to the originally predicted adsorption pose in MM simulations. The protonated form of the amino-modified surface (NH3+) showed strong electrostatic interactions with the charged forms of the drug molecule (5-FU1 and 5-FU2). 5-FU and its ionic forms occupied a perpendicular position to the adsorption surface in the case of amino modifications.
2.5. Adsorption Energy
The interaction energy between 5-FU and the surface was estimated as the differential energy (ΔE) using the formula
| 1 |
where, E(MF)—energy of the MCM-41 and 5-FU complex, E(M)—energy of a fully relaxed MCM-41 model, and E(F)—energy of the fully optimized 5-FU molecule.
The values from MM and QM simulations are given in Table 2. Figure 8 shows the plot of adsorption energy calculated by the OPLS3 force field for different forms of 5-FU on MCM-41 and its different surface modifications. It can be seen from the graph that both MM energy calculations using the OPLS3 force field and the QM energy calculations for all the 5-FU species on various MCM-41-modified surfaces exhibited a similar trend.
Figure 8.

Comparative plot of adsorption energy calculated by MM using the OPLS3 force field and QM for different forms of 5-FU on MCM-41 and its different surface modifications.
Both the ionized 5-FU1 and 5-FU2 in general exhibited higher adsorption energy in both MM and QM energy calculations compared to 5-FU. This is due to a large contribution to adsorption energy from the electrostatic interaction of the ionized forms of 5-FU with the substituted surfaces. When the MCM-NH2 and MCM-NH3+ surfaces were studied for adsorption, we observed strong electrostatic interactions in the form of ionic interactions between the positively charged surface and the negatively charged drug molecules (5-FU1 and 5-FU2). The unionized form of the carboxyl surface model (COOH) exhibited stable binding adsorption energy for all the forms of 5-FU. The deprotonated form of the carboxyl surface model (COO–) showed stable adsorption energy for the unionized 5-FU, whereas the 5-FU1 and 5-FU2 exhibited unstable adsorption energies (reflected as the positive adsorption energy values).
In anionic forms of 5-FU, a negative charge is present on one of the nitrogen atoms present in the molecule. This leads to huge energy shifts during the adsorption process. In this case, once again the energies are comparable between the neutral surfaces. On the other hand, on the charged surfaces (protonated 3-aminopropyl and the deprotonated 3-carboxylpropyl), the situations change drastically. The positively charged surface (protonated 3-aminopropyl surface) attracts the molecule toward the surface, and strong non-covalent interactions are present between the positively charged ammonium ion and the negatively charged 5-FU. In the case of the deprotonated 3-carboxylpropyl surface, the reverse situation was observed. Here, the negatively charged carboxyl ion expels the negatively charged 5-FU molecule out of the surface which is reflected in the adsorption energies which were positive (Table 2). Moreover, the fluorine atom present in the 5-FU molecule is also highly electronegative because of which the interaction between the deprotonated carboxyl-modified silica surface and 5-FU forms may not be favorable.
The neutral 5-FU exhibited the following order of adsorption energy: CN > NH3+ > COOH > NH2 > Plain > CH3 > COO–. On the other hand, the trend in the case of both the charged forms is as follows: NH3+ > CN > NH2 > COOH > Plain > CH3 > COO–. The change in the trend can be addressed by the increased electrostatic interactions between the modified surface and the negatively charged forms. However, experimentally, the adsorption trend was found to be NH2 > CN > COOH > Plain > CH3.16 The difference between the experiment and the simulation can be explained by the different fractions of 5-FU that are possible under the experimental pH conditions. It is evident from the pKa calculations that all the three forms of 5-FU can co-exist at the given experimental pH conditions, that is, 7.0. The probability of formation of NH3+ is much higher than that of the formation of COO– ions on the respective modified MCM-41 surfaces. The formation of NH3+ is an exothermic process in the water phase, and the COO– formation at pH 7.0 is an endothermic reaction. Because the formation of the COO– ion is not feasible under experimental conditions, the contribution from the COO– can be safely ignored (any adsorption was not observed in this case for charged species in the studies). The COOH-terminated surface is better than plain and CH3-substituted surfaces. The highly modified CH3 surface renders it passive to any favorable interactions on the substituted sites thus making it a least preferred surface for the adsorption. The unmodified surface ousts the CH3 surface by allowing the drug molecule to form hydrogen bonding interactions with the silanol and bridged oxygen groups. Now, taking these energetic contributions, the adsorption trend can be rewritten as NH2 > CN > COOH > Plain > CH3 which correlated well with the published literature by She and co-workers wherein the experimental loading trend was found to be NH2 (28.89%) > CN (22.54%) > COOH (20.73%) > plain (18.34%) > CH3 (12.73%).16
From the single-molecule adsorption calculations, we could conclude that the MM calculations (force field) with OPLS3 yielded the same trend even though the results did not exactly match those from QM calculations. The QM calculations will be accurate but cannot be applied on the larger systems like the bulk simulations. The MM calculation trend revealed that the OPLS3 force field can be efficiently used for the bulk simulations in a reasonable time rather than using the more computationally expensive QM calculations.
2.6. Bulk System Simulation Comparison between Plain MCM-41 and MCM-NH2
In order to study the bulk system which exists in practice, several molecules of 5-FU were adsorbed on the plain MCM-41 and MCM-NH2. Among the modified surfaces studied, the MCM-NH2 surface was chosen for bulk simulations as it showed maximum adsorption of 5-FU on it, and the same was compared with plain MCM-41. The bulk simulation using MD was performed to understand the adsorption and the release phenomenon of 5-FU molecules from plain MCM-41 and MCM-NH2.
2.6.1. Adsorption of 5-FU on MCM-41 and MCM-NH2
At a formulation pH of 7.0, the majority of the 5-FU exists in the unionized form, and about 0.059% of the molecules exist in the ionized form (calculated based on the pH and pKa of 5-FU). Considering the MCM-41 as the substrate (in the “Disorder System Builder”), on each side of the surface of MCM, 500 molecules of the 5-FU were added. A total of thirty 5-FU molecules in the ionized form were added to each side of the surface. Because it is difficult to find how many would be in the 5-FU1 and 5-FU2 form, we considered an equal proportion (15 molecules) of both forms. The initially generated disorder system was equilibrated by following the protocol mentioned in Section 4.7. At formulation pH 7.0 as the majority of the amine groups exist in the protonated form (−NH3+), all the amine groups modified on the MCM-NH2 surface were considered in its protonated form except for one, which was in the neutral form. The density profile of the pure 5-FU was calculated from the last 20% segment of the trajectory from the MD simulation which was found to be 1.54 g/cm3.
The final frame saved from the MD trajectory was considered for the analysis of the adsorption pattern of 5-FU on both MCM-41 and MCM-NH2. Figure 9A depicts the density profile for 5-FU molecules adsorbed on the MCM-41 and MCM-NH2 surface in the simulation boxes. The two large curves represent the bilayer of 5-FU adsorbed on the generated silica surface (i.e., on either side of the generated silica bed). The dip observed in between these two peaks (around 55–65 Å) represents the thickness of the silica bed itself where there are no 5-FU molecules. The large curves comprise of small curves which depict the different adsorption layers of 5-FU molecules. The density profile for 5-FU on MCM-41 was found to be 1.50 g/cm3, whereas that on MCM-NH2 was found to be 1.52 g/cm3. From the density profile graph in Figure 9A, at least two to three distinct adsorption layers can be seen for 5-FU in both MCM-41 and MCM-NH2. The thickness of the first layer of adsorption for 5-FU on MCM-41 was about 4 Å, whereas it was found to be 6 Å on MCM-NH2. Figure 9B,C shows the number of H-bond counts between 5-FU in the first and second adsorption layer and between the 5-FU and surface of MCM which were counted in the structures saved in the trajectory from MD simulations. The higher number of H-bond interactions in the case with MCM-NH2 indicated a stronger binding of 5-FU with the surface. The plausible reason for the higher number of hydrogen bonds in the second layer of 5-FU adsorption may be due to the higher number of interactions between the 5-FU molecules itself. The number of molecular interactions observed for 5-FU molecules adsorbed on the MCM-NH2 was higher compared to the 5-FU molecules adsorbed on the MCM-41. This indicated that the molecule would prefer to be with the MCM-NH2 surface because of the better stabilization rather than going into the bulk.
Figure 9.

(A) depicts the density profile for 5-FU adsorbed on the MCM-41 and MCM-NH2 surface where the green and red curve represents 5-FU adsorbed on MCM-NH2 and MCM-41, respectively. The X-axis represents distance in Å units from the top side of the simulation box moving through the silica bed and the opposite side of the surface. The Y-axis represents the density of the 5-FU molecules on the generated silica bed. (B,C) are the number of H-bond counts between 5-FU in the first and second adsorption layer and between the 5-FU and surface of MCM-41 and MCM-NH2, respectively, counted in the structures saved in the trajectory from MD simulations (5-FU- 5-FU, bed—MCM surface, FAL-first adsorption layer, H-bond- hydrogen bond, and π–π -pi-pi stacking).
Figure 10 depicts the 5-FU adsorbed on the surface of the MCM-41 and MCM-NH2, the H-bond interactions between the surface and 5-FU, and the interactions and orientation of 5-FU on the surface of MCM and the first adsorption layer.
Figure 10.

Adsorption of 5-FU on the MCM-41 surface (left column A–G) and MCM-NH2 [right column (B–H)] where (A,B) are the lateral view of 5-FU adsorbed, (C,D) are the top view with the ESP surface, and (E–H) are zoomed-in top-view images showing different interactions between 5-FU and MCM surfaces; hydrogen bonding is represented in blue dotted lines, salt bridge interactions are represented by yellow dotted lines, and π–π interactions are represented by cyan dotted lines.
2.6.1.1. Interactions of 5-FU with Silica Surfaces
The presence of aminopropyl substitutions on the surface of MCM-NH2 led to the formation of corrugations which increased the surface area compared to the MCM-41. The total surface area calculated considering both the sides of the adsorption surface for the MCM-41 was found to be 8679.2 cubic Å, whereas that for MCM-NH2 was found to be 10,169.3 cubic Å. This greater surface area for MCM-NH2 might have also contributed to a better adsorption of 5-FU. The 5-FU molecules on the MCM-41 oriented themselves perpendicular to the bed with a partially tilted orientation. The fluorine atom of 5-FU was found to be oriented away from the surface. The carbonyl group (C=O) and the amine (−NH) group occupied positions in such a way that they could form the donor and acceptor type of H-bond interaction with the silanol hydroxyl groups. With this orientation, the amine group of one 5-FU could form intermolecular H-bond interaction with the carbonyl group of the nearby 5-FU molecule. A weak type of aromatic −CH intermolecular interaction with the carbonyl group of the neighboring 5-FU molecules was also observed. The 5-FU molecules on the MCM-NH2 occupied the corrugated surface, and they were oriented almost perpendicular to the surface of the bed. The carbonyl group (C=O) of 5-FU formed the H-bond interaction with the amine group from the surface. The fluorine favored the formation of the hydrophobic contact on the corrugated surface portion. The amine group and the carbonyl group formed an intermolecular interaction between the 5-FU molecules. Due to the perpendicular orientation of 5-FU molecules to the surface, a higher number of π–π stacking interactions were observed between the 5-FU molecules. The intermolecular aromatic −CH interactions with the neighboring 5-FU molecules were also higher in MCM-NH2 than those with MCM-41. Overall, the 5-FU molecules on MCM-NH2 exhibited a higher number of interactions which further substantiates the results obtained from single-molecule adsorption.
2.6.1.2. Single Molecule versus Bulk System Adsorption
The single molecule of 5-FU adsorption in the case of both plain MCM-41 and MCM-NH2 showed both parallel and perpendicular orientations. In the case of the protonated MCM-NH2 surface, a higher number of interactions led to the perpendicular orientation of the 5-FU molecules especially the ionic forms. When the bulk system simulation was carried out for plain MCM-41 and MCM-NH2, a similar orientation was observed in both the cases. In the case of plain MCM-41, 5-FU molecules showed a mix of parallel, perpendicular, and tilted orientations. The interactions mainly included the hydrogen bonding, ionic interactions between the 5-FU molecules and the silanol groups, and π–π interactions between the 5-FU molecules oriented parallel to one another. Similarly, in the case of 5-FU molecules adsorbed on MCM-NH2, the orientation was mostly perpendicular to the surface with a higher number of hydrogen bonds between the surface and 5-FU and between the 5-FU molecules. A greater number of π–π interactions were also observed between the 5-FU molecules oriented parallel to each other. Moreover, being a rigid molecule, 5-FU can plausibly have only a parallel or a perpendicular orientation on the silica surface. The same was also visualized in the trajectory of the MD simulation. In the case of bulk simulations, in the first adsorption layer, 5-FU can align itself in a parallel, perpendicular, or slightly tilted position, whereas from the next layer onward, they may lose their orientation and remain more spread-out forming intermolecular interactions due to weaker interaction with the surface.
2.6.2. Release of 5-FU from the MCM Surfaces
To study the release of 5-FU adsorbed from both the MCM surfaces, that is, MCM-41 and MCM-NH2, the surface-washing simulation studies were performed. To mimic the release, water was considered as the solvent. Generally during the loading, as only the first adsorbed layer of the drug gets retained because of its strong interaction with the surface, in the present study, we considered only the first adsorption layer of 5-FU on MCM-41 and MCM-NH2 for the washing simulations.
To check the release pattern, the first adsorption layer of 5-FU on MCM-41 and MCM-NH2 was subjected to the MD simulation. The system was built by adding 3700 molecules of the TIP3P water above the first adsorption layer of 5-FU molecules on either side of the MCM surface. The system was equilibrated as per the protocol mentioned in Section 4.7, and the production simulation was run for 100 ns time. During the simulation, 2000 frames were saved in the trajectory. The structures in the MS simulation trajectory were analyzed for various properties like intermolecular interactions and diffusion coefficients of 5-FU.
2.6.2.1. Hydrogen Bond Formation during the Simulation
Figure 11 presents the number of H-bonds observed between the MCM surfaces and 5-FU and the H-bond count between the MCM surfaces with the water molecules throughout the duration of MD simulations. In the beginning of the simulation, there were about 436 and 581 H-bonds observed between the 5-FU molecules with the MCM-41 surface and MCM-NH2 surface, respectively. The results suggest that 5-FU formed a lesser number of H-bonds with the MCM-41 surface when compared with MCM-NH2, and the number of H-bonds gradually reduced with the time of simulation. The reduction in the H-bond count was steep till 25 ns in the case of MCM-41, whereas in the case of MCM-NH2, a steady fall in the H-bond count was observed till 60 ns of the simulation (Figure 11A). Moreover, an increase in the H-bond count between the MCM-41 surfaces and the water molecules was observed till 25 ns, and it remained constant till the end of the 100 ns simulation. The water molecules stabilized both the MCM surfaces to a greater extent when compared to the 5-FU molecules which was evident from the increase in the number of H-bonds observed between the water and the MCM surfaces toward the end of the MD simulation when compared to the initial number of H-bonds between MCM surfaces and 5-FU molecules (Figure 11B). The water molecules formed about 700 H-bonds with the MCM-41 surface, whereas about 720 H-bonds counted at the last frame of MD simulations were formed with the MCM-NH2 surface. From the H-bond plots, it was observed that the water molecules replaced the 5-FU molecule faster from the surface of MCM-41 than compared with MCM-NH2 which further confirms the stronger adsorption capacity of the MCM-NH2 surface.
Figure 11.

H-bond counts during the water wash simulation. (A) H-bond between the bed and 5-FU molecules and (B) H-bond between the bed and water molecules.
2.6.2.3. Release Profile of 5-FU Molecules
To measure the rate of release of 5-FU molecules from the MCM surface, the number of the molecules diffused from the surface was determined from the trajectories of the MD simulation. 17 structures were saved from the MD trajectory at regular intervals, and the number of 5-FU molecules present in the first adsorption layer on the surface of MCM-41 and MCM-NH2 was calculated. The percentage cumulative of 5-FU molecules released into the bulk was plotted against the time. Figure 12A represents the cumulative number of 5-FU molecules which have been released from the surface into the bulk as observed from the MD simulation trajectory. The graph depicts the trend of the release of the molecules from the surface to the bulk which gives us an idea of the comparative affinity of 5-FU molecules to both the surfaces. About 57.9% of 5-FU molecules were released from the surface of MCM-NH2 at the end of the 100 ns simulation. Shani and co-workers reported the experimental release rate calculated for the 5-FU molecule in their work.25 They used magnetic MSNs with aminopropyl surface modification and plain MCM-41 without any surface modifications. They reported that the maximum percentage of 5-FU released at the end of 24 h was found to be 64% from the MCM-41 surface and 22% from MCM-NH2. In the computational simulations, 50% of the 5-FU molecules from the first adsorption layer were released from the MCM-41 surface in about 25 ns, whereas it took about 67 ns for the 5-FU molecules to be released from the MCM-NH2 surface. The release rate scenario calculated by simulations matched well with the experimental calculations. 72.55% of 5-FU molecules were released from the MCM-41 surface into water at 52.5 ns. It was difficult to calculate the rate of release of 5-FU from the MCM-41 surface after 72.55% of release as the molecule started forming the lumps.
Figure 12.

(A) Release rate of 5-FU from the MCM bed in presence of water where the green and red curve represents the % release of 5-FU molecules from MCM-NH2 and MCM-41, respectively; the density profile of 5-FU on (B) MCM-41 and (C) MCM-NH2 where the X-axis represents the depth of the bed in Å, and the Y-axis represents the density of 5-FU molecules. The different colored lines shown in the legend represent the density of the 5-FU molecules at different time intervals of MD simulations viz.,, 1, 2.5, 5, 15, 20, 25, 67.5, 82.5, and 100 ns. (D,E) Diffusion coefficient of the 5-FU molecules from the MCM-41 and MCM-NH2 surface over the simulation time period of 20 ns.
2.6.2.2. Density Profile and Diffusion Coefficient of 5-FU from the Silica Surfaces
The density of 5-FU in the first adsorption layer was 1.5 g/cm3 which was close to the density of 5-FU in the amorphous form measured by running the MD simulation. Two curves were observed in the density profile for 5-FU, corresponding to the 5-FU molecules adsorbed on the either side of the MCM surface. The valley between the two peaks corresponds to the thickness of the MCM bed. During the simulation, the 5-FU molecules diffused away from the surface of MCM. It could be seen that the density of 5-FU diminished at a faster rate from the surface of MCM-41, and the molecules diffused away from the surface to the bulk of the system when compared to the surface of MCM-NH2. During the simulation, it was observed that the water molecules displaced the 5-FU molecules. In MCM-NH2, the release of 5-FU was slow, and the density profile of 5-FU was concentrated near the bed as the amount of 5-FU release was less compared to that from the surface of MCM-41. The 5-FU molecules were concentrated initially near the surface of the MCM-41 until the 5-FU release from the surface was small. Once around 75% of the molecules diffused out into the bulk, the remaining 5-FU molecules clustered around which was indicated from the spread-out 5-FU density profile (Figure 12B,C). The density of 5-FU molecules on the surface reduces with time on both MCM-41 and MCM-NH2 surfaces which may be attributed to the release of the 5-FU molecules from the surface to the bulk of the system. In the case of the MCM-NH2 system, a relatively slower reduction in the density of the 5-FU molecules was observed when compared to the MCM-41 system. To further substantiate the results, we calculated the diffusion coefficient of the 5-FU molecules; we observed a slower diffusion coefficient of the 5-FU molecules from the MCM-NH2 when compared to the plain MCM-41 further suggesting higher affinity of the 5-FU molecules to the amino-modified MCM surface. Figure 12D,E depicts the mean square displacement of the 5-FU molecules from the MCM-41 and MCM-NH2 surfaces, respectively, over the simulation time period of 20 ns. The diffusion coefficient for 5-FU from MCM-41 was found to be 2.20 × 10–10 m2/s and that from MCM-NH2 was found to be 1.71 × 10–10 m2/s. This can be further correlated with the density profile of 5-FU on both the surfaces over the time period of MD simulations.
The higher affinity of the 5-FU molecules toward the MCM-NH2 system may be further correlated with the higher binding energy observed in single-molecule simulations which led to a slower diffusion or release of the 5-FU molecules into the bulk. Figures 13 and 14 give a pictorial visualization of a few frames from the MD simulation of 5-FU release in the presence of water from the MCM surface. It could be seen that in the beginning of the simulation, the surface of the plain and MCM-NH2 was fully covered by the 5-FU molecules. During the simulation, water molecules displaced the 5-FU molecules and reached out to the surface. The stronger interactions of 5-FU with the MCM-NH2 reduced the release of 5-FU molecules from the surface. In the presence of water, the 5-FU molecules reoriented and exhibited a higher number of H-bond counts with the bed compared to the amorphous 5-FU molecules. The presence of strong electrostatic interaction like a salt bridge and H-bond interaction between the amine groups of the bed which was missing in MCM-41 held the 5-FU molecules on the surface for a longer duration of time during the MD simulation in the case of MCM-NH2. We observed a higher number of π–π stacking interactions between the 5-FU molecules on MCM-NH2 compared to MCM-41 which may also further stabilize the 5-FU molecules leading to its slower diffusion into the bulk. Because of the weaker and lesser intermolecular interactions, the 5-FU molecules were released faster from MCM-41. The simulation box had a limited number of water molecules (7400), and since there were no enough water molecules to completely dissolve 5-FU, the environment around it was not favorable and resulted in its accumulation which was evident from the clusters of 5-FU molecules in the MD simulation. If a system with a larger number of water molecules was considered during the MD simulation, probably the 5-FU molecules would have solubilized, and the clusters would not have been formed. The accumulation of 5-FU molecules in water was not observed in the MD simulation with MCM-NH2 surface modification as the release rate of 5-FU from the surface was slower, and the cumulative amount of 5-FU released did not cross 60%. The accumulation of 5-FU was observed in the simulation once the cumulative amount of 5-FU molecules released was above 70%. The clusters formed were found to be near the MCM-41 surface, due to which in the density profile, scattering of the peaks near the surface was observed. This also rendered it impossible to correctly measure the rate of release of 5-FU molecules from the surface during the simulation, as the released molecule clusters moved close to the MCM surface.
Figure 13.

Trajectory time frames representing the release of 5-FU from the MCM-41 surface at different time intervals [upper pictures (A): top view; lower pictures (B): lateral view].
Figure 14.

Trajectory time frames representing the release of 5-FU from the MCM-NH2 surface shown at different time intervals [upper pictures (A): top view; lower pictures (B): lateral view].
3. Conclusions
The computational studies of MSNs established that the computational simulation can be used to study the adsorption and release behavior of 5-FU. The results revealed that the molecular simulations can be used to understand the experimental behavior of drug loading in MSNs (MCM-41). The computationally economical MM calculations with the OPLS3 force field were found to show similar behavior as that using the expensive QM calculations. It is evident from the simulations that the surface corrugations and the electrostatic charge distributions play a key role in molecular adsorptions. As expected, simulations showed the same trend as in experiments, but one should keep in mind that the charged fractions of both the surface and drug are a crucial factor in the adsorption process. The MD simulations were run to check the release rate of 5-FU from the MCM-41 and MCM-NH2 surface in the presence of water. The diffusion of the 5-FU molecules in the presence of water was found to be faster from the MCM-41 surface when compared to MCM-NH2 which further substantiates the stronger affinity of the 5-FU molecules to the MCM-NH2 surface. This trend matched with that of the experimental calculations reported earlier.25 The simulations could successfully predict the diffusion rate of drug molecules across different surface modifications considered. The present study will allow the formulation scientists to better understand the interactions at the molecular level and hence will help them in designing a rational carrier for drug delivery.
4. Computational Details
4.1. Formulation Data Collection for Effect of Functionalization on Loading Capacity
The published literature of 5-FU-loaded MSNs was taken as a model in the study.16 The loading capacity of 5-FU onto MCM-41 data were taken to generate and validate the computational model in the present study. She and co-workers synthesized hollow MSNs to form a mesoporous silica shell and investigated the effect of various surface functional groups on the loading capacity of the MSNs with respect to 5-FU. The nanoparticles were surface functionalized by post-synthesis grafting to introduce amino, methyl, cyano, and carboxyl chemical groups.16
4.2. Simulation Details
All the calculations have been performed using the Materials Science Suite (MS-Suite) 2019–04 of Schrödinger (Schrödinger, LLC, New York).
4.3. Construction and Optimization of Computational Models of MCM-41
The representative mesoporous silica structure was generated using an alpha-silica crystal unit cell. The unit cell was extended in all three directions (4 × 4 × 4) to build a bulk system. By using the slab and interface builder within the framework of the Schrödinger- Materials Science Suite, the oxygen-terminated slab of silica was generated, and hydrogen atoms were added to neutralize the surface. The generated model contained 44 silicon atoms, 88 oxygen atoms, and 84 terminal hydrogen atoms on either side of the surface.
The generated structure was subjected to Limited-memory Broyden-Fletcher-Goldfarb-Shanno (L-BFGS) minimization using the Optimized Potentials for Liquid Simulations 3 (OPLS3) force field to arrive at its lowest energy state.22,26 The system was then solvated using the orthorhombic box (dimensions of 5 Å) of the TIP4P water model. Furthermore, the MCM-41 structure was subjected to molecular dynamic (MD) simulations for 10 ns (i.e., 10,000 ps) at 300 K and 1.013 bar pressure under NPT conditions. A Nose–Hoover thermostat with a coupling constant of 2.0 ps was used for temperature control along with the Martyna–Tuckerman–Tobias–Klein (MTTK) barostat with a coupling constant of 5.0 ps for pressure control. The time step of the simulation was set to be 2.0 fs. The trajectory was recorded at every 40 ps interval. The trajectories generated from the simulations were analyzed for their energy, and structural changes in the form of root mean square deviations (rmsd) to the original structure were studied. From the equilibrated trajectory, the frame with the lowest energy was selected. This structure was used for all the further studies and will be henceforth referred to as plain MCM-41 in this paper.
Six different surface terminations viz., 3-aminopropyl (MCM-NH2), 3-cyanopropyl (MCM-CN), 3-carboxypropyl (MCM-COOH), methyl (MCM-CH3), protonated 3-aminopropyl (MCM-NH3+), and deprotonated 3-carboxypropyl (MCM-COO–) were carried out to analyze and understand the impact of the surface modifications. Three individual surfaces per substitution were prepared by randomly replacing the terminal hydrogens on the surface. The substitution of 12% w/w was directly taken from the published literature,16 and the corresponding molecular substitutions for each modification were calculated. Six substitutions were made for 3-aminopropyl, 3-cyanopropyl, and protonated-3-aminopropyl. In the case of 3-carboxypropyl and deprotonated 3-carboxypropyl, 4 groups were included on the corresponding molecular surfaces, and finally, in the case of the methyl surface, 22 substitutions were made. All the six modified surfaces were then minimized using the Macromodel routine using an OPLS3 force field. Subsequently, all the substituted surfaces were prepared using the same procedure as that used to equilibrate the plain MCM-41 surface. Figure 15 shows the rmsd plot of the frames enumerated from the MD trajectory. The rmsd within 0.2 Å indicated the formation of stable surface-modified MCM models.
Figure 15.
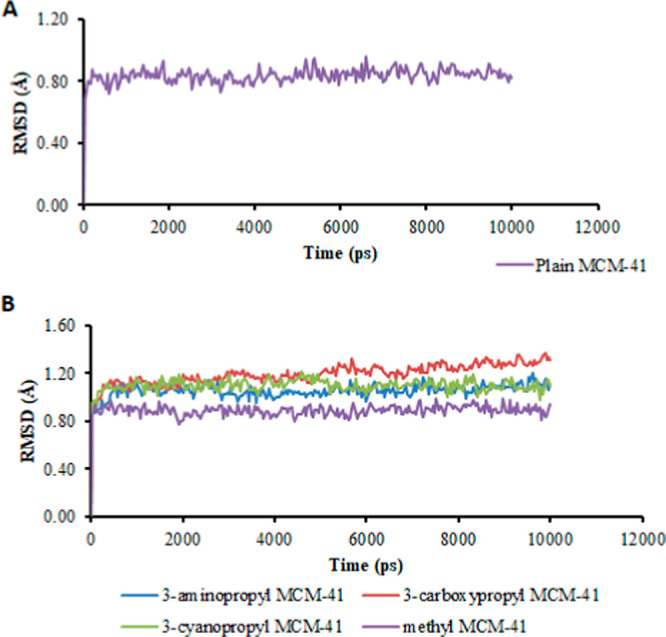
Graphs showing rmsd of (A) plain MCM-41 and (B) various surface-modified MCM-41.
4.4. Preparation of Chemical Structure of 5-FU
The ionized forms of 5-FU were analyzed at pH 7.0 using the Epik tool to understand the state of the drug which is an important factor influencing the adsorption pattern.27,28 All the structures generated were optimized with density functional theory using the Becke, three parameters, Lee–Yang–Parr (B3LYP) functional and split valence double-zeta basis sets plus polarization functions (6-31G**++).24
4.5. Generation of Initial States of Adsorption of 5-FU on MCM-41 Surfaces
To generate better initial adsorbed structures, molecular docking of the 5-FU molecule and its ionic forms was carried out using the Glide molecular docking tool.23 The receptor grid was generated by excluding the edges and covering 80% of the MM-optimized surface. Subsequently, the docking of 5-FU and its anionic forms was carried out on the plain and substituted MCM-41 surfaces. Thirty docked poses were saved and clustered based on the visual inspection. The clustering pattern for the plain MCM-41 surface is shown in Figure 16. From each cluster, the pose that is having the highest docking score was chosen for further calculations.
Figure 16.
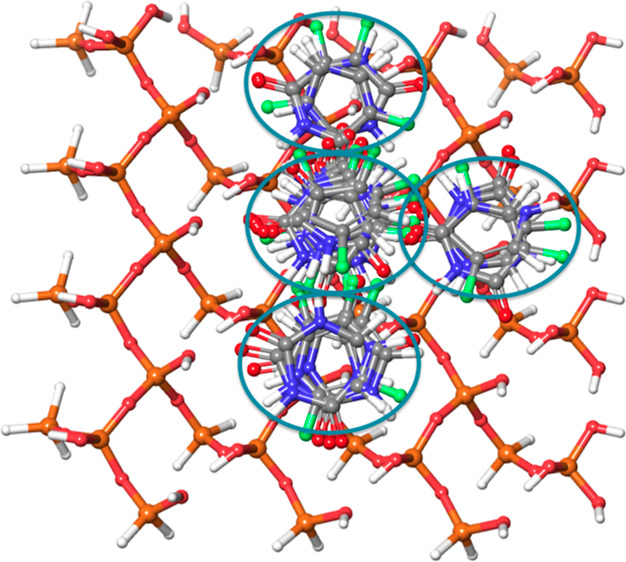
Clustering of 5-FU docking poses on the plain MCM-41 surface, based on the visual inspection. The different clusters are marked with blue color circles.
4.6. Adsorption Energy Calculations
4.6.1. Molecular Mechanics
The current potential energy of isolated 5-FU molecules and MCM-41 surfaces and selected adsorbed complexes was calculated using the Macromodel routine.
4.6.2. Quantum Mechanics
To calculate the accurate adsorption energies, QM optimization calculations were also carried out on the same MCM-41 surfaces, 5-FU and its ionic structures, and complexes that were used for MM calculations. To speed up the QM optimization, the MCM-41 surface atoms were kept rigid during the optimization process. The 6-311G** basis set with the B3LYP hybrid functional was used for all our calculations. The minimum energy cut-off for the convergence was set to 5 × 10–5 Hartree.
4.7. Bulk System Simulation
To further mimic the bulk properties like surface adsorption and the release of 5-FU from the plain MCM-41 and MCM-NH2 surface, the MD simulation was performed. As mentioned earlier, using the alpha quartz bed, a larger surface with the dimensions 54.07 × 19.66 × 59.45 cu Å was generated for both MCM-41 and MCM-NH2. To account for the 12% w/w aminopropyl substitution in MCM-NH2 as per the literature by She and group,16 about 22 groups were attached on each side of the MCM-41 surface to give the MCM-NH2 surface. The system was first prepared using the “Disorder System Builder” tool in MS suite. Considering the MCM as the substrate (in the “Disorder System Builder”), on each side of the surface of MCM, 500 molecules of 5-FU were added. A total of thirty 5-FU molecules (15 each of 5-FU1 and 5-FU2) in the ionized form were added to each side of the surface. The MD simulation was performed using the “Desmond” module of Schrödinger (Desmond, Schrödinger, LLC, New York), where one can specify the number of molecules to be included in the disordered system. It also allows one to generate the disordered system on the planar surface (either side) of a specified substrate. The system in the present study comprised of a total of 1000 molecules of 5-FU, with 60 molecules in the ionized form (thirty molecules each of 5-FU1 and 5-FU2). The generated system was then subjected to the MD simulation using the “Multistage Simulation Workflow” in MS suite, which included the multiple equilibration steps and the final production step. The protocol followed for the equilibration during the MD simulation is as mentioned below:
-
a)
The Molecular Mechanic (MM) minimization using the steepest descent method with 2000 maximum iterations and a 1.0 Kcal/mol/Å convergence threshold
-
b)
Brownian Minimization for 200 ps
-
c)
MD simulation with the NPT ensemble for 1.2 ns at 100 bar pressures at 300 K
-
d)
MD simulation with the NPT ensemble for 5.0 ns at 1.013 bar pressure at 300 K
-
e)
The production MD simulation was run for either 5/100/200 ns with below-mentioned specifications
-
f)
MD simulation with the NPT ensemble for 5/15/100 ns at 1.0 bar pressure at 300 K
During the simulation, the temperature was maintained with a Nose–Hoover Chain thermostat and the pressure using Martyna–Tobias–Klein Barostat methods with 100 ps relaxation time. A 2 fs time step was maintained during the simulation. All the MD simulations were run with the OPLS3e29 force field keeping the parameters as per the default options. 1000 structures were saved to the trajectory file. The “Simulation Event Analysis” tool was used for the analysis of the generated MD trajectory. The density profile and diffusion coefficient were calculated using the tools in MS suite.
Acknowledgments
This work was supported by the Science and Engineering Research Board, Dept. of Science and Technology, New Delhi, India (ref. no. EMR/2016/007006). Authors are thankful to Dr Pritesh Bhat and Dr Sudharsan Pandiyan, Schrödinger, Bengaluru, India, for their guidance in conducting this research work. Authors are thankful to Manipal Academy of Higher Education (MAHE), Manipal, India, for providing the Dr TMA Pai Fellowship to R.N.
The authors declare no competing financial interest.
References
- Rizzo L. Y.; Theek B.; Storm G.; Kiessling F.; Lammers T. Recent progress in nanomedicine: therapeutic, diagnostic and theranostic applications. Curr. Opin. Biotechnol. 2013, 24, 1159–1166. 10.1016/j.copbio.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.-H.; Hung Y.; Mou C.-Y. Mesoporous silica nanoparticles as nanocarriers. Chem. Commun. 2011, 47, 9972. 10.1039/c1cc11760b. [DOI] [PubMed] [Google Scholar]
- Li Y.; Yang L. Driving forces for drug loading in drug carriers. J. Microencapsulation 2015, 32, 255–272. 10.3109/02652048.2015.1010459. [DOI] [PubMed] [Google Scholar]
- Ouyang D.; Smith S. C.. Computational Pharmaceutics: Application of Molecular Modeling in Drug Delivery; John Wiley & Sons, 2015, ISBN 9781118573990. [Google Scholar]
- Allen M. P.Introduction to Molecular Dynamics Simulation; John von Neumann Institute for Computing, 2004; Vol. 23, pp 1–28. [Google Scholar]
- Yadav P.; Bandyopadhyay A.; Chakraborty A.; Sarkar K. Enhancement of anticancer activity and drug delivery of chitosan-curcumin nanoparticle via molecular docking and simulation analysis. Carbohydr. Polym. 2018, 182, 188–198. 10.1016/j.carbpol.2017.10.102. [DOI] [PubMed] [Google Scholar]
- Meng X.-Y.; Zhang H.-X.; Mezei M.; Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila-Salas F.; Sandoval C.; Caballero J.; Guiñez-Molinos S.; Santos L. S.; Cachau R. E.; González-Nilo F. D. Study of Interaction Energies between the PAMAM Dendrimer and Nonsteroidal Anti-Inflammatory Drug Using a Distributed Computational Strategy and Experimental Analysis by ESI-MS/MS. J. Phys. Chem. B 2012, 116, 2031–2039. 10.1021/jp2069122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara-Jaque A.; Comer J.; Monsalve L.; González-Nilo F. D.; Sandoval C. Computationally Efficient Methodology for Atomic-Level Characterization of Dendrimer-Drug Complexes: A Comparison of Amine- and Acetyl-Terminated PAMAM. J. Phys. Chem. B 2013, 117, 6801–6813. 10.1021/jp4000363. [DOI] [PubMed] [Google Scholar]
- Geetha P.; Sivaram A. J.; Jayakumar R.; Gopi Mohan C. Integration of in silico modeling, prediction by binding energy and experimental approach to study the amorphous chitin nanocarriers for cancer drug delivery. Carbohydr. Polym. 2016, 142, 240–249. 10.1016/j.carbpol.2016.01.059. [DOI] [PubMed] [Google Scholar]
- Metwally A. A.; Hathout R. M. Computer-Assisted Drug Formulation Design: Novel Approach in Drug Delivery. Mol. Pharm. 2015, 12, 2800–2810. 10.1021/mp500740d. [DOI] [PubMed] [Google Scholar]
- Patel S. K.; Lavasanifar A.; Choi P. Molecular dynamics study of the encapsulation capability of a PCL-PEO based block copolymer for hydrophobic drugs with different spatial distributions of hydrogen bond donors and acceptors. Biomaterials 2010, 31, 1780–1786. 10.1016/j.biomaterials.2009.11.060. [DOI] [PubMed] [Google Scholar]
- Delle Piane M.; Corno M.; Ugliengo P. Does Dispersion Dominate over H-Bonds in Drug-Surface Interactions? The Case of Silica-Based Materials As Excipients and Drug-Delivery Agents. J. Chem. Theory Comput. 2013, 9, 2404–2415. 10.1021/ct400073s. [DOI] [PubMed] [Google Scholar]
- Corno M.; Delle Piane M.; Monti S.; Moreno-Couranjou M.; Choquet P.; Ugliengo P. Computational Study of Acidic and Basic Functionalized Crystalline Silica Surfaces as a Model for Biomaterial Interfaces. Langmuir 2015, 31, 6321–6331. 10.1021/acs.langmuir.5b01828. [DOI] [PubMed] [Google Scholar]
- Sevimli F.; Yılmaz A. Surface functionalization of SBA-15 particles for amoxicillin delivery. Microporous Mesoporous Mater. 2012, 158, 281–291. 10.1016/j.micromeso.2012.02.037. [DOI] [Google Scholar]
- She X.; Chen L.; Li C.; He C.; He L.; Kong L. Functionalization of Hollow Mesoporous Silica Nanoparticles for Improved 5-FU Loading. J. Nanomater. 2015, 2015, 872035. 10.1155/2015/872035. [DOI] [Google Scholar]
- Fluorouracil; DrugBank; https://go.drugbank.com/drugs/DB00544. [Google Scholar]
- Markova N.; Enchev V.; Ivanova G. Tautomeric Equilibria of 5-Fluorouracil Anionic Species in Water. J. Phys. Chem. A 2010, 114, 13154–13162. 10.1021/jp1063879. [DOI] [PubMed] [Google Scholar]
- Al-Thawabeia R. A.; Hodali H. A. Use of Zeolite ZSM-5 for Loading and Release of 5-Fluorouracil. J. Chem. 2015, 2015, 403597. 10.1155/2015/403597. [DOI] [Google Scholar]
- Reddy G. D.; Kumar K. N. V. P.; Duganath N.; Divya R.; Amitha K. ADMET Docking studies & binding energy calculations of some Novel ACE - inhibitors for the treatment of Diabetic Nephropathy. Int. J. Drug Dev. Res. 2012, 4, 268. [Google Scholar]
- Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Bochevarov A. D.; Harder E.; Hughes T. F.; Greenwood J. R.; Braden D. A.; Philipp D. M.; Rinaldo D.; Halls M. D.; Zhang J.; Friesner R. A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. 10.1002/qua.24481. [DOI] [Google Scholar]
- Egodawatte S.; Dominguez S.; Larsen S. C. Solvent effects in the development of a drug delivery system for 5-fluorouracil using magnetic mesoporous silica nanoparticles. Microporous Mesoporous Mater. 2017, 237, 108–116. 10.1016/j.micromeso.2016.09.024. [DOI] [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- Shelley J. C.; Cholleti A.; Frye L. L.; Greenwood J. R.; Timlin M. R.; Uchimaya M. Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- Greenwood J. R.; Calkins D.; Sullivan A. P.; Shelley J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput.-Aided Mol. Des. 2010, 24, 591–604. 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- Roos K.; Wu C.; Damm W.; Reboul M.; Stevenson J. M.; Lu C.; Dahlgren M. K.; Mondal S.; Chen W.; Wang L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. 10.1021/acs.jctc.8b01026. [DOI] [PubMed] [Google Scholar]