

ABSTRACT
Although the therapeutic efficacy and commercial success of monoclonal antibodies (mAbs) are tremendous, the design and discovery of new candidates remain a time and cost-intensive endeavor. In this regard, progress in the generation of data describing antigen binding and developability, computational methodology, and artificial intelligence may pave the way for a new era of in silico on-demand immunotherapeutics design and discovery. Here, we argue that the main necessary machine learning (ML) components for an in silico mAb sequence generator are: understanding of the rules of mAb-antigen binding, capacity to modularly combine mAb design parameters, and algorithms for unconstrained parameter-driven in silico mAb sequence synthesis. We review the current progress toward the realization of these necessary components and discuss the challenges that must be overcome to allow the on-demand ML-based discovery and design of fit-for-purpose mAb therapeutic candidates.
KEYWORDS: Machine learning, artificial intelligence, antibody, antigen, developability, drug design
1. Introduction
1.1. mAb discovery remains anchored to legacy technologies
Monoclonal antibody (mAb) based therapeutics continue to top the chart for best-selling drugs worldwide. In 2021, the sales figures for the top 10 antibody therapeutics are forecasted to reach more than $110 billion and almost double in 2024.1,2 Despite the major commercial success, antibody discovery has remained anchored to time- and cost-intensive legacy technologies, namely display libraries, animal immunization,3,4 and comparatively low-throughput antibody modeling.5–10 Indeed, although effective, monoclonal antibody therapies cost up to 100,000 USD per year.11 As such, there is a critical need for developing novel in silico, and specifically ML-based, antibody discovery tools, to achieve fast, inexpensive, and on-demand generation of fit-for-purpose antibodies.
1.2. Three technological pillars for ML-based on-demand generation of mAbs: learnability, modularity, and unconstrained generation of novel sequences
In recent years, ML has taken the center stage in various fields due to its ability to recognize latent patterns in data, allowing a constructive extrapolation of such patterns to unseen new data.5,7,12–16 A particularly potent type of ML is deep learning where layers of interconnected computing units (neurons) work in tandem to detect signals in the data, enabling the model to discriminate between groups (classification) or to synthesize new data points that share particular traits with the original data (generation).17–19
Based on current literature, we identify and review recent progress and challenges in the three pillars that are instrumental to a successful realization of the long-sought immunobiotechnological vision of on-demand antigen-specific antibody generation (Figure 1): 1) the presence of rules underlying antibody–antigen interactions and developability (learnability),20,21 2) the capacity for the modular and non-linear optimization of interdependent antibody design parameters, e.g., plasma half-life, is affected by multiple regions of the antibody interdependently (modularity),22–24 and 3) the capability to synthesize a virtually limitless quantity of new antibodies that are distinct from the training data yet possess affinity and developability parameters (feature-controlled) that match, exceed, or extend those of the training dataset (unconstrained generation).25–27
Figure 1.

Overview of progress and challenges within the three technological pillars for ML-based on-demand generation of mAb therapeutic candidates, namely learnability, modularity, and unconstrained generation. We highlight three key optimizable design parameters for in silico on-demand mAb design: (i) the AA residues at the surface of the antigen (epitope) that engage the antibody residues (paratope) at the interaction interface, (ii) the strength of an antibody–antigen interaction (affinity), and (iii) the extent to which the mAb successfully progresses from the discovery to the development phase (developability). We discuss these design parameters from the perspective of three technological pillars: (i) learnability indicates the presence of rules underlying antibody–antigen interactions as well as antibody developability, (ii) modularity signifies that antibody design parameters could be impacted by multiple regions on the antibody and the extent to which they can be recombined interdependently, and (iii) unconstrained generation signifies the capacity of high-throughput in silico synthesis of fit-for-purpose mAb candidates.
1.3. Learning from nature: considering biological complexity in computational antibody design
Although antibody-antigen binding is a subset of protein–protein interactions, several important differences exist that render the prediction and design of antibody-antigen binding even more challenging than the prediction of typical protein–protein interaction. These differences are: 1) the immense diversity of antibody sequences, 2) many-to-many binding due to the high extent of non-linear sequence dependencies, and 3) inter-dependence of affinity and pharmacokinetic parameters. These specific characteristics need to be considered in antibody design and inform computational design constraints.
Diversity: V(D)J recombination and somatic hypermutation jointly create a potential immunoglobulin (Ig) diversity of >1014,28,29 as compared to a non-immune protein diversity of 105–106.30 Although there exists antigen-driven (or functional) repertoire convergence (formation of similar antibodies in different individuals undergoing an identical antigen challenge),28,31,32 the extent of antigen-specific convergence is antigen-dependent33 and overall comparatively low (on average, <10% pairwise overlap of antigen-specific Ig sequences).34 Thus, the vast observed diversity of (antigen-specific) antibodies implies that the discovery space for target-specific mAbs is rather large and amenable to constraint-based sequence design approaches.
Additional mechanisms to diversify antibody repertoires include insertion and deletion of amino acid (AA) sequences into the V region, the use of non-protein cofactor molecules, and post-translational modifications.35 On the one hand, the insertion, and deletion of AAs and the use of non-protein cofactors (e.g., metal ion or haem) are suggested to be strategies toward diversifying specificity against pathogens.35–37 On the other hand, post-translational modifications, such as O- and N-glycosylation, phosphorylation, and oxidation in the antibody structure may affect the pharmacokinetics, solubility, stability, modulation of effector functions as well as receptor-binding properties.38–42 These modes of antibody diversification are not fully understood and require further investigations to enable favorable in vivo binding and transport properties as well as optimal manufacturability and storage formulations.
Many-to-many binding: Antibody-antigen binding is mediated by the interaction of AAs at the paratope–epitope interface of the complex. Antibody binding to the epitope is mainly formed by the three hypervariable regions termed complementarity-determining regions (CDRs) situated in each of the antibody heavy and light chains.43 The CDR3 on the heavy chain (CDR3H) is obligate for epitope binding and is on average 15-17 AAs long.44,45 Given that the diversity of antigens is even larger, the recognition of the majority of antigens encountered is ensured by antibody cross-reactivity, which means they may bind multiple epitopes on different proteins with high affinity.46 Epitope binding is therefore encoded in higher-order complex dependencies (correlations between spatially distant AAs in the CDRH3 enabling the binding of conformational epitopes, allowing a higher combination of binding motifs) in the low dimensionality of the antibody sequence space. These strong dependencies reflect 3D binding, where residues that are distant along the sequence can be close in the folded 3D structure. Indeed, the majority of antibody epitopes are thought to be conformational47 – although 85% of epitopes contain one or several contiguous (linear) epitope stretches.45,48 Therefore, to learn the rules of antibody-antigen binding, approaches need to be developed that untangle the non-linear sequence dependencies that govern the antibody, antigen, and antibody-antigen structures in both bound49,50 and unbound51 states.
Interdependence of antibody design parameters: Antibody design parameters can be broadly categorized into binding parameters (paratope, epitope, affinity) as well as developability parameters (e.g., plasma half-life, thermal stability, solubility, aggregation propensity, and immunogenicity). Traditionally, it was thought that antibody design categories may be optimized independently. However, recent reports suggest that, for example, the plasma half-life is not only a function of the antibody isotype and constant fragment crystallized (Fc) region, but also sequence variations in the CDRs.22–24 Therefore, antibody design parameters are interdependent and thus require modular optimization and bioengineering techniques (Figure 1).
Taken together, nature succeeded in devising an extraordinary antibody repertoire that combines diversity, specificity, and modularity. Hence, leveraging data sources that combine these properties and developing computational models that can take advantage of multi-property optimization would be the key to define the fundamental principles that can guide tailored antibody design.
1.4. Augmenting scarce experimental data with simulated data that account for the biological complexity of antibody–antigen interaction
For the design and discovery of mAbs, available experimental datasets are particularly scarce in comparison to the biological complexity of antibody-antigen binding. To date, one of the largest developability studies on mAbs remains very restricted at 137 samples (Figure 2).52 Similarly, 3D structures, which are useful in defining residues of the antibody (paratope) that engage the residues of the antigen (epitope) at the interaction interface, are limited to 1200 non-redundant antibody–(protein)antigen complexes.55 Sequence data, however, can be produced at larger scales, higher efficiencies, and at markedly reduced costs, making it the leading choice to study antibody-antigen binding albeit at a reduced resolution where paratope-epitope information often is not available (Figure 2). At present, the Observed Antibody Space (OAS)57 database contains over one billion antibody sequences curated from 79 studies, while the iReceptor database contains almost four billion sequences and 6013 repertoires from three remote repositories, 49 research labs, and 60 studies.53 Such large sequence datasets have been used, for example, to generate latent representations of phenotypically similar antibodies,58,59 prior to training ML models on small-scale structural datasets.
Figure 2.

Overview of public datasets on antibody developability, experimental, or synthetic sequence and structural antibody(-antigen) data. The available sequence and structural datasets were queried from Europe pubmed central (europepmc.org) using keywords “antibody” and “database” and filtered for publications that contain these keywords in the title in addition to manual literature curation (codes and data are available as mentioned in the Code availability section of this manuscript). The datasets are visualized with respect to the sequence or structure, and the availability of binding affinity, antigen annotation, developability parameters, or paratope and epitope information. Sequence (red), structure (blue), synthetic structural data (purple) and developability (gray) are color-coded. Each circle corresponds to a specific type of data. The outer circles correspond to the global data (sequences, structures, synthetic structures, and developability), and the inner ones – to the subdata (antibody-antigen complexes, Ig repertoire, mAbs, and paratope and epitope). A separate outer circle for developability is used as its data types differ from the others. Since there is not a single database containing quantitative information about the available developability parameters, we used the data from52 as an example for visualizing the scarcity of available experimental developability information. The outer red ring represents the number of antibody sequences in the iReceptor database (the largest publicly available sequence data,53 the outer purple ring the number of synthetic antibody–antigen binding structures from Absolut! (the largest publicly available synthetic antibody-antigen structural dataset),54 the outer blue ring displays the number of structures from AbDb (curated antibody–antigen structural data55 obtained from the protein data bank),56 and the outer grey ring represents developability information.52 inner rings illustrate information about antibody-antigen complexes, ig repertoire, therapeutic antibodies, and paratope and epitope data. For a curated overview of available databases, see Focus Box 1.
Recently, we presented our efforts to increase the amount of 3D-structure data by six orders of magnitude larger than the 1200 structures available experimentally (Figure 2)55 via simulating virtual coarse-grained docking of billions of antibody-antigen pairs with several layers of biological complexity.54 We complemented this data with in silico predicted developability parameters to create datasets that encompass the three aforementioned key design parameters: paratope-epitope binding, affinity, and developability.27 Such efforts have begun to increase the number of datasets to a level where the benchmarking of data-intensive methods, such as deep learning to study antibody-antigen binding at the paratope-epitope level as well as deep learning-based antibody sequence generation, started to become feasible.27,54 More generally, large-scale 3D-atomistic resolution data generation may represent the next major step where abundantly available antibody sequence data will be leveraged to obtain large quantities of antibody-antigen complexes via recent advances in computational structural biology methods such as antibody modeling,59–63 molecular docking,64–67 and molecular dynamics.68,69
Focus Box 1 | Databases that curate antibody sequences or structures (see Figure 2).
- AB‐Bind70 (https://github.com/sarahsirin/AB-Bind-Database) is a dataset containing experimental results for wild-type and mutant antibodies and antigens, including the change in Gibbs free energy of binding (ΔΔG), linked to crystal structures of the parent complexes. Year of publication: 2016.
- ABCD71 (https://web.expasy.org/abcd/) database is a manually curated depository of sequenced antibodies. Year of publication: 2019.
- AbDb55 (http://www.abybank.org/abdb/) is a compilation of antibodies (including nanobodies) extracted from the PDB56 with standard numbering schemes applied and redundancy information. Year of publication: 2018.
- abYsis72 (http://www.abysis.org/abysis/) is a web-based antibody research system that includes an integrated database of antibody sequence and structure data. Year of publication: 2017.
- AgAbDb73 (http://196.1.114.46:8080/agabdb2/home.jsp) is a derived knowledge base archive of molecular interactions of protein and peptide antigens characterized by co-crystal structures. Year of publication: 2014.
- bNAber74 (http://bnaber.org/) is a database of HIV broadly neutralizing antibodies providing neutralization profiles, sequences and three-dimensional structures. Year of publication: 2013.
- cAb-Rep75 (https://cab-rep.c2b2.columbia.edu/) is a database of curated human B cell immunoglobulin sequence repertoires. Year of publication: 2019.
- CoV-AbDab76 (http://opig.stats.ox.ac.uk/webapps/covabdab/) is a database of published or patented binding antibodies and nanobodies to coronaviruses, including SARS-CoV2, SARS-CoV1, and MERS-CoV. Year of publication: 2021.
- IEDB77 (https://www.iedb.org/) is a resource of experimental data on humans, non-human primates, and other animal species antibody and T cell epitopes. Year of publication: 2018.
- IMGT78 (http://www.imgt.org/) is a sequence, genome and structure knowledge resource specialized in the immunoglobulins, T cell receptors, MHC, and in the immunoglobulin and MH superfamilies, and related proteins of the immune system of vertebrates and invertebrates. Year of publication: 2018.
- OAS57 (http://opig.stats.ox.ac.uk/webapps/oas/) is a database of annotated immune repertoires. Year of publication: 2018.
- PROXiMATE79 (https://www.iitm.ac.in/bioinfo/PROXiMATE/) is a database of interaction kinetics and thermodynamics data (including wild-type vs mutant KD and ΔΔG) for mutations in protein–protein complexes including antibody-antigen complexes, collected from literature and previously published databases. Year of publication: 2017.
- SAbDab80 (http://opig.stats.ox.ac.uk/webapps/sabdab) is a database containing annotated antibody and nanobody structures available in the PDB.56 SAbDab also contains affinity data for antibody-antigen complexes, taken from the PDBbind database.81 Year of publication: 2013.
- sdAb-DB82 (http://www.sdab-db.ca/) is a dedicated single-domain antibody repository and database. Year of publication: 2018.
- SKEMPI 2.083 (https://life.bsc.es/pid/skempi2/) is a database contains kinetics and energetics data upon mutation, for protein–protein interactions including antibody-antigen complexes of which structure are available in the PDBe.84 Year of publication: 2019.
- Thera-SAbDab85 (http://opig.stats.ox.ac.uk/webapps/newsabdab/therasabdab/search/) is a database curating WHO recognized antibody-related therapeutics. Year of publication: 2020.
2. Learnability of antibody–antigen binding
The hurdles of antibody–antigen binding prediction may be subdivided into five ML challenges. Figure 3 illustrates how these challenges are intertwined with each other in a typical ML workflow. We group these challenges as the ‘learnability’ problem, i.e., the capacity of an ML method for a certain type of dataset and biological question to achieve generalization and provide surrogate rules responsible for its predictions from only a limited set of examples (the training dataset). We discuss herein aspects of learnability pertaining to antibody-antigen binding (affinity), while the following sections review aspects of learnability from the perspective of modularity, developability, and unconstrained sequence generation.
Figure 3.

Major ML components that could enable the identification of the rules that govern antibody design parameters (binding, paratope-epitope, and developability). These components relate to the five ML challenges namely (1) predictability, (2) generalization, (3) interpretability, (4) model uncertainty, and (5) data completeness. Multiplexing (integration and augmentation) of data with varying degrees of information may improve the completeness of the training data which would consequently produce an informed representation (learned or otherwise) and allows for data-driven mAb design. As synthetic data tend to be superior (crisp icons) in comparison to experimental data (fuzzy icons) with respect to quantity and the extent of completeness (the parameters and rules underlying the data are known), the augmentation of sparse experimental data with synthetic data may yield a dataset that contains a fuller degree of completeness than either subset thereof. The training of advanced deep learning architectures on informed representation (containing sequence, developability, affinity, linguistic [Focus Box 2], and paratope-epitope feature) either via online (continuous) or batch (one-off bulk data) learning would result in high accuracy models that may well be capable of generalization. Importantly, the mapping of features that are critical for the predictive performance of the model (interpretability) must be undertaken to allow for rule inference, and consequently, to allow rule-driven design.
ML challenge 1: Predictability. The capacity to predict properties of antibody-antigen binding with high accuracy is the sine qua non prerequisite for computationally aided mAb discovery. The predictability of antibody-antigen binding is often obscured by the biological complexity and the limited information content of the considered datasets.
ML challenge 2: Generalization. Specifically, ML-based mAb discovery relies on the generalizability of the models, i.e., information learned from “dataset A” will be valid for predicting binding in “dataset B”, provided that the two datasets are “similar enough” (Figure 3). In general, antibody sequence similarity is not necessarily associated with phenotypic similarity since sequence-similar antibodies may bind different antigens.86 Therefore, the similarity of antibodies should be considered both in terms of sequence similarity and binding behavior (function). Of note, while transfer learning aims at performing a new task from subsets of a pre-trained model that is further trained for a certain task, generalization refers to performing the same task with the same model (without additional training) on different datasets.
ML challenge 3: Interpretability. When it comes to prospective antibody engineering as well as clinical practice, the most beneficial setting would be an interpretable ML model that proposes rules to explain the reasons underlying its generalization.4,59,87,88 This would both decrease the risk of predicting dataset-dependent properties, and provide guidelines to generate new possible antibody sequences based on those rules. So far, rule inference via, for instance, attribution methods, remains a challenge and is poorly standardized.54,89
ML challenge 4. Model (also called epistemic) uncertainty.90 This descirbes the situation where multiple models predict a dataset with equally high accuracy while relying on different sets of rules that might or might not be equivalent. For ML, as the training data is often noisy and sparse, exhaustive learning of the “rules” is likely intractable. Instead, we argue that a successful learning model for antibody-antigen binding could converge to an approximate (surrogate) set of rules, such that its predictions are sufficiently accurate across multiple datasets, indicating that these rules have only retained minimal dataset bias.
ML challenge 5: Dataset completeness. Intuitively, the presence of instances in the dataset illustrating a certain rule is required to infer this rule. We refer to the “completeness” of a dataset as the amount of information it contains, in comparison to the information needed to infer the rules that we believe are underlying the properties of the dataset. Currently, it remains unclear how to determine if a dataset is complete to infer (surrogate) rules (see ML challenge 3). If the rules are explicit (for instance by learning a scoring function or using interpretable ML architectures), it can be easier to test the completeness of a dataset.91 When the rules are not directly interpretable, it can become difficult to assess dataset completeness, except by practically testing the extent of the data coverage of the rules.92 Therefore, interpretable ML methods are preferable for assessing dataset completeness.
These ML challenges stem from the theoretical foundations of computational learning theory, which has been applied to natural language (linguistics). Focus Box 2 provides further discussion of the theoretical background and the parallels with linguistics.
Altogether, learnability and availability of suitable data ensure high prediction accuracy on new tasks.
Focus Box 2 | A formal language perspective of learnability applied to antibody discovery problems.
Multiple articles relate linguistics to the study of proteins by applying neural network (NN) language models to protein sequences.58,93–100 Another fruitful way to relate linguistics and biological research is to translate formal perspectives from linguistic research into biological research questions, such as computational learning theory. It provides a precise way to define learnability,101 and it has been used for computationally defining natural language learning.102 Below, we adapt learning theory to antibody discovery problems: we formalize antibody discovery questions as formal language learning questions and discuss aspects of learnability as they pertain to antibody discovery.
A language is a formal system that consists of a potentially infinite set of structures, built from a finite set of elements (the alphabet) with a finite set of rules (a grammar). 103 In the context of antibody discovery, the language to be learned depends on the research question. If the goal is to predict if input antibodies bind to a given antigen (a binary classification problem),54 then the language is the set of encoded representations of antibody structures that bind to the antigen, and the learner’s task is to discriminate between the structures that are part of the language and ones that are not. If the goal is to predict the set of antigens that bind to input antibodies (a multiclass classification problem),54 the language consists of antigen–antibody pairs that exhibit high affinity. A learner in this case has to be able to recognize these pairs. For both types of research questions, the target grammar to be learned are the physicochemical rules that govern antibody structures and antibody–antigen interactions.
When learning a language, the learner maps from an observable subset of all possible data (examples) to a grammar that describes the language.102 The learner can be either a black box learner that can answer whether an input belongs in a language (or give a probability for it) without showing the grammar it operates on, or an interpretable one that returns the full grammar it learned. It is possible, though often difficult, to extract the grammar from black-box learners. We argue that for successful antibody discovery, it is crucial to have access to the rules, and have an interpretable model. A fully interpretable model enables more informed antibody discovery because it clarifies which properties might be entirely dataset dependent versus which properties might be generalizable across datasets.
Another question raised by learning theory is the definition of successful learning. The criteria for successful learning can range from exact convergence to some defined value of approximate convergence. Exact convergence requires the inferred grammar to be identical to the target grammar of antibody discovery problems, while approximate convergence means that the inferred grammar is not completely identical, but “close enough” to the target grammar.102 Approximate convergence is easier to achieve and is thus a more practical criterion for successful learning. This is especially appropriate for antibody discovery problems, where the target grammar is a set of highly complex and largely unknown physicochemical rules, and currently, available data is too limited and noisy for exact convergence to be feasible. Moreover, it is difficult to assess how approximate any inferred ruleset is to the target. It can therefore be useful to use synthetic data to study the performance of a given ML algorithm first, as synthetic data are generated with explicit rules.54,104,105
Lastly, there is a possibility that the data presented to a learner is not complete. A complete dataset contains every kind of structure from the target language that would be sufficient for the learner to converge successfully to the target grammar, while incomplete datasets might lead to alternative grammars that only account for a subset of the examples but not all.102 If the dataset is not complete, the learner might reach high prediction accuracy for dataset-specific properties rather than converging to the more fundamental grammar that describes general binding specificity. The completeness of the dataset is only loosely related to its size: a very large dataset can still be incomplete if it lacks crucial data points for inferring the target grammar, and a complete dataset can be relatively small as long as it has everything necessary for successful learning. It is therefore important to aim for completeness rather than merely size in the dataset in order to achieve successful learning.
In conclusion, formalizing antibody-antigen binding questions as formal language learning questions helps clarify various aspects of learnability. It particularly draws attention to the nature of the learner, defining the standard for successful learning, and the completeness of the dataset.
2.1. Formalization of antibody–antigen binding problems
Three main types of prediction problems have been investigated using ML in antibody-antigen binding: 1) prediction of the antibody-antigen binding interface (paratope, epitope or paratope-epitope prediction), 2) prediction of binding affinity (in particular following AA substitution), and 3) prediction of binding partners (binary and/or many-to-many).54 In view of the above ML challenges, we delineate to which extent these studies have achieved the first proof of principle steps of antibody-antigen learnability. We focus on the type and size of datasets they use and how antibody-antigen sequence or structural data are embedded into data representations.
2.2. Epitope prediction
Epitope prediction may be divided into two different application areas. Antibody-agnostic epitope prediction seeks to identify the most probable epitopes without prior knowledge of the corresponding antibody(ies), and antibody-aware epitope prediction seeks to identify the epitope to which a known antibody will bind.
2.2.1. Antibody-agnostic epitope prediction
Early epitope prediction methods infer contiguous epitope residues based on a few hundred linear epitopes via propensity scales (e.g., PREDITOP,106 BEPITOPE,107 BcePred,108 see,109,110 for more details). ABCPred used a Jordan network (a version of RNN) to perform binary classification on segments of the antigen via sliding windows.111 Another ensemble method, iBCE-EL, used physicochemical properties, AA composition, and combined extremely randomized tree (ERT) and gradient boosting (GB) to predict linear epitope with higher accuracies.112 However, the vast majority of described epitopes are conformational,113 hence, linear epitopes may not account for non-flanking residues as they may only represent contiguous subsequences of the full epitopes.
Other prediction tools used support-vector machines (SVM) to classify each antigen residue as epitope or non-epitope residue (Söllner and Mayer114 BCPred,115 BEST,116 EPSVR,117 Chen et al.118). These tools combined physicochemical properties with sequence conservation, similarity to other known epitopes, predicted 2D structural features, or even structural properties of similar known sequences. SePre119 first predicts individual immunogenic residues then clusters them as an epitope in a second step. These methods reported high prediction accuracy, indicating that sequence information only allowed the prediction of non-contiguous epitopes. The inclusion of sequence conservation makes it challenging to understand which information in the (explicit) training dataset versus (implicit) alignment was important for high prediction accuracy.
In parallel, structure-based discontinuous epitope prediction methods have been trained on antibody-antigen structures and then tested on the antigen structures alone (i.e., antibody agnostic) to predict the epitopes, using AA propensity scales as above but adding geometric predictors such as the number of neighbors according to different distance thresholds, triangle-based propensity measures, or ellipsoids (SEPPA,120 Discotope,121 PEPITO/BEpro,122 ElliPro123) and reported ~0.75 AUC on 75 antibody-antigen structures. Moreover, Lu et al.124 combined a graph convolution network to leverage local spatial neighborhood information with an attention-based long short-term memory-recurrent neural network (LSTM-RNN). They examined whether spatially distant information on the antigen sequence can improve prediction accuracy and reported an AUC of ~0.8.
Nevertheless, we argue that antibody-agnostic epitope prediction is an ill-defined problem 125,126 because only in the context of an antibody (a paratope) does an epitope become functional and vice versa. Indeed, it is now a general consensus that nearly any surface accessible region of an antigen may be recognized by an antibody.127 In addition, epitopic and other surface residues were found to be mostly indistinguishable in their amino-acid composition.128
2.2.2. Antibody-aware epitope prediction
Bepar129 utilizes correlations of AAs usage on sliding windows between the antigen and the CDR loops of the antibody in antibody-antigen complexes to predict epitope residues from the antibody and antigen sequences only.
Several structure-based studies attempted to improve the quality of antibody-antigen docking by including geometrical features on both antibody and antigen to re-rank the list of predicted possible poses. For instance, EpiPred130 measures the conformational matching of an input pair of antibody and antigen structures. DLAB-Re131 models the antibody structure from its sequence,132 generates docking to the antigen structure and uses a convolutional neural network (CNN) to predict the paratope-epitope complementarity of a pose as a re-ranking score, therefore predicting both epitope and paratope.
PEASE133,134 takes the antibody and antigen structures, calculates a solvent accessibility score per residue, predicts the pairs of interacting epitope-paratope residues using random forest, followed by patch reconstruction in order to reconstruct the epitope. Another study20 has defined antibody and antigen surface patches using a Monte Carlo method that includes or excludes neighboring residues with a probability defined from features initially learned from antibody-antigen complexes. From the observation that matching paratope and epitope patches share correlated features in shape or AA composition, a deep feed-forward network was built to predict whether a paratope patch would bind an epitope patch. PECAN135 used CNNs with an attention layer directly from the antibody and antigen structures to predict the binding interfaces of antibody and antigen structures. We discuss DLAB131 and PECAN135 in detail in the paratope prediction section below.
Altogether, these studies have shown that, at present, in silico epitope prediction tools yield moderately accurate predictions and that structural information of the antibody or the paratope is critical to improving epitope prediction performance.
2.3. Paratope prediction
Although paratope prediction may look like the symmetric reverse problem of epitope prediction, paratope residues are both sequentially and spatially close to each other as they are most often contained within the CDR loops,45 in contrast to epitope residues that can be spatially close but sequentially distant over the span of the antigen length.45 Further, the AA usage of paratopes is distinct to those of epitopes45 as each CDR has its own preferential AA usage, and the subset of epitope residues bound by a CDR also have a preferential AA usage specific to which CDR it was bound to.128
2.3.1. Sequence-based antigen-agnostic paratope prediction
Parapred136 uses either an LSTM-RNN-based or a deep NN-based architecture on top of a CNN to predict the 1D paratope, starting from the antibody sequence alone. In this process, only the antibody CDRs are considered and one-hot encoding is combined with biochemical encoding for each residue. proABC137 outperformed Parapred using a random forest model with additional features on the full antibody sequence to predict 1D epitope. Briefly, along with the one-hot encoding of the full variable heavy (VH) and variable light (VL) chains, proABC includes information on the species of origin, the inferred germline VH and VL families, the predicted canonical structure associated with each CDR sequence, and predicts the binding status of each of the residues. In a refined version proABC-2,138 a CNN architecture has been implemented as a replacement to the random forest, following the same data processing and problem formulation as pro-ABC. The authors showed that the output of proABC-2 (for instance, including the predicted types of interactions) can be used as additional constraints when later performing docking of the antibody to the antigen.
Paratope prediction tools have also been leveraged to identify novel binders that originate from different clonotypes. In immune repertoire mining, for example, known binders are typically used to identify new binders via clonotyping 34,139 (i.e., finding sequences with close genetic history). By design, this approach limits the diversity of the identified binders. In contrast, an approach called paratyping aimed at identifying convergent binders from different clonotypes by using the predicted paratope to cluster antigen-specific antibodies that originate from diverse clonotypes.140 Re-epitoping, on the other hand, used ML to predict AA substitutions that would improve the complementarity of the resulting paratope to the epitope of interest.141 Another application is the mapping of sequence features, or combinations of subsets thereof, to discern phenotypic traits such as inhibitors or non-inhibitors.142
2.3.2. Structure-based antigen-agnostic paratope prediction
Paratome web server143 uses structural alignment to identify consensus antigen-binding regions on a given antibody sequence or structure. The server uses the structural consensus regions from multiple structure alignment of a reference set of antibody-antigen complexes to identify binding regions of antibodies.
AntibodyInterfacePrediction combined 3D Zernike Descriptors (3DZDs) and SVM to predict antibody–antigen interface.144 It firstly obtains geometrical representation, physicochemical and biological characteristics of the residues on the antibody surface starting from an input of the antibody 3D structure. A rotationally invariant local descriptor is calculated for each uniform spherical patch sampled from the antibody surface. On the 3DZDs, randomized logistic regression was used to decrease the overall number of features. SVMs were employed as a classifier to distinguish the paratope interface LSPs from the non-interface ones. As a result, AntibodyInterfacePrediction outperformed Parapred,136 Paratome,143 and Antibody i-Patch.145 However, Parapred remains competitive against AntibodyInterfacePrediction as it does not require structural data.
As discussed in antibody-agnostic epitope prediction earlier, we reaffirm our assessment that paratope prediction without the context from the epitope may not be very insightful.
2.3.3. Sequence-based antigen-aware paratope prediction
The subsequent update of Parapred, called AG-Fast-Parapred21 makes use of the six CDR sequences and the sequence of the cognate bound antigen, with each AA encoded separately with their AA and seven chemical features as descriptors, and returns a binary vector of the binding status of each position in the CDR3 (linear paratope prediction) to train an architecture combining an “à trous” CNN with an attention layer. They compared antibody-only prediction (i.e., antigen-agnostic) against prediction including antigen information using cross-modal attention. The new architecture moderately improved the accuracy (AUC = 0.90) compared to Parapred (AUC = 0.88).21
Lu et al.124 proposed a sequence-based paratope prediction tool from the antigen sequence by separately predicting the probability of each antibody residue to be a paratope residue (binary classifications). The antigen and antibody sequences were transformed into 80 predicted structural features, including evolutionary information, secondary structure prediction,146 solvent accessibility, and backbone dihedral angles with NetSurfP2.0.147 Antibody and antigen information is then processed by two parallel attention-LSTM-RNN architecture, while a CNN leverages local information on the antibody side, and fully connected layers transform the CNN and LSTM-RNN outputs into binary prediction per antibody residue. Their method showed moderate improvement in accuracy by including the partner antigen sequence, as observed in AG-Fast-Parapred.
2.3.4. Structure-based antigen-aware paratope prediction
Antibody i-Patch, relies on the structures of antibody and antigen as input to predict the paratope.145 Antibody i-Patch, annotates each residue with a binding likelihood score rather than providing an entire binding region as Paratome, and outperformed Paratome in precision. In addition, the usage of Antibody i-Patch prediction with the fast docking algorithm, ZDOCK,148 increased the number of near-native poses.
Furthermore, Paratope and Epitope prediction with graph Convolution Attention Network (PECAN) is a deep learning framework that predicts the binding interfaces of antibody-antigen-antibody complexes.135 The local spatial connections of the interfaces were captured using graph convolutions while an attention layer connects distant information, and transfer learning was performed using a base network trained on generic protein–protein interactions. PECAN outperformed EpiPred130 and DiscoTope121 in epitope prediction and AntibodyInterfacePrediction144 in paratope prediction. The attention layer showed only a little improvement in paratope prediction performance over convolution, probably because paratopes are mostly located around CDRs, while it improved epitope prediction significantly. From the observation that PECAN sometimes predicts spatially too distant epitope residues, a new strategy termed Contiguous Epitope – Sub-sampled Convolution Attention Network (CE-SCAN149) was proposed. CE-SCAN succeeded in predicting localized epitopes, while leveraging long-distance information from multiple patches and sequentially distant residues, and provided a small increase in prediction accuracy compared to PECAN.
Schneider et al.131 modeled 3D antibody structures from their sequence using ABodyBuilder60 and performed docking using ZDOCK150 on their known cognate antigen structure. Interestingly, docking the modeled structure was a harder task in comparison to using the known bound antibody structure, and the authors developed a CNN-based strategy (DLAB-Re) to re-rank the docking poses proposed from ZDOCK to prioritize those with the correct epitope. DLAB-Re takes as input a proposed antibody-antigen docking pose, transforms the binding interface in voxels, and learns a ‘compatibility score’ based on the 3D distribution of the AAs along the voxels.
Vecchio et al.151 used epitope-paratope message passing (EPMP) for paratope-epitope prediction. Considering that epitope residues are distant and antigen-dependent, the architecture combines a paratope model (Para-EPMP), sequentially processing antibody input features and followed by a graph structure, and an epitope model (Epi-EPMP), where only structural features are used with GNN layers and substantially merged with contextual cues from the cognate antibody.
More recently, geometric deep learning (GDL) has emerged as one of the most promising advances to generate a molecular representation for the prediction of interacting interfaces (e.g., antibody–antigen interface).152,153 The method extends neural networks to allow for the incorporation of geometric priors (structure and symmetry) of the input in order to improve the quality of the signal captured by the model. GDL has been used for instance in developing molecular surface interaction fingerprints (MaSIF).154 MaSIF was mostly trained on non-immune protein–protein interaction including antibody-antigen data. The authors note that geometric models, such as MaSIF, are able to capture geometric matching across protein–protein interfaces that may extrapolate to paratope–epitope interfaces pending further validation. Hence, it would be of interest to benchmark MaSIF against antibody-antigen binding prediction tools as the model is increasingly being used to study antibody–antigen interface.155 Briefly, MaSIF starts with a mesh representation of a protein surface where each point on the surface is annotated with both geometric and chemical features that capture degrees of curvature, concavity, electrostatic potential, hydrophobicity, and hydrogen potential. Subsequently, a set of geodesic filters generate a one-dimensional embedding of the protein surface. MaSIF was used to classify interacting versus non-interacting residues with satisfactory performance. A GDL model with a simpler surface representation for large-scale learning has also recently been made available.156 However, GDL, or any other deep learning-based tool, has yet to be configured to account for the dynamics of the interaction at the interfaces. In particular, antibodies sample multiple conformations even at the unbound stage,157 and bound antibody structures differs from their corresponding unbound structures.49,158 Incorporating the dynamics and conformational changes upon binding at the interfaces between two molecules remains one of the major challenges in protein design in general, as well as in the design of antigen-specific antibodies.159
2.4. From single paratope-epitope pair to many-to-many binding partner prediction
In addition to paratope or epitope prediction of already known binding pairs, learning the rules for paratope-epitope matching, and generating all possible binding partners of an antibody or antigen represents a difficult challenge. Leveraging antibody-antigen complexes in the database AbDb, graph theory, and deep learning, we45 discovered a set of antibody-antigen structural interaction motifs that demonstrates the potential predictability of antibody–antigen interaction in general, and the prediction of paratope-epitope pairs more specifically. Indeed, these interaction motifs were shared across unrelated antibody-antigen complexes (but were largely distinct from non-immune protein–protein interaction motifs), suggesting the existence of a general interaction vocabulary of antibody–antigen interfaces that may help, in the future, learn antibody–antigen interaction rules. However, the lack of large structure and affinity datasets for antibody-antigen hinders the exhaustive benchmarking of deep learning-based many-to-many binding and affinity prediction.
By generating large-scale synthetic antibody-antigen structural datasets,54 we investigated the relative influence of structural and sequence-based features on the accuracy of paratope-epitope prediction (i.e., predicting a compatible epitope of an epitope). Both an encoder-decoder with attention, and the transformer architectures yielded accuracies of ≈90% at generating the cognate epitopes using at least 2000 to 10,000 unique encoded paratope-epitope training pairs. In contrast, sequence information alone led to unsatisfactory accuracy even with 200,000 distinct paratope-epitope pairs in the training dataset. Interestingly, the binding degree of paratope and epitope residues (number of binding residues on the other protein) was the structural feature that contributed most to increase prediction accuracy.
Ab-Ligity158 uses the ABodyBuilder tool60 to reconstruct the paratope structure of the full antigen-binding fragment (Fab) region (based on CDR sequences and homology modeling) and to cluster the antibody sequences that would bind the same epitope. This is done by hashed encoding of the physicochemical property of the binding (paratope-epitope) pairs of residues and their distance binned per groups of 1.0 Å. A binding similarity site is then calculated from the encoded paratope–epitope interaction code. As such, it was possible to identify dissimilar antibody sequences that would bind the same epitope, a task that is usually very challenging. With a conceptually similar goal, Ripoll et al.160 computationally constructed 3D-models of epitope-specific antibody sequences to train image-based deep neural networks for antibody-epitope classification showing a potential route towards applying image recognition techniques to sequence-based datasets for antigen specificity discovery.
DLAB-VS (Virtual Screening) transforms paratope or epitope prediction into binding prediction131 by virtual screening of a docked antibody-antigen pair and using a CNN to predict their compatibility. The CNN was trained on the best poses of known cognate antibody-antigen pairs as the positive class, while two types of negative pairs were selected: docking of non-cognate pairs, and the lowest range (FNAT < 0.1) of docked poses for a binding pair.
Xu et al.161 used a structure-based clustering of CDRH3 sequences to cluster supposedly phenotypically similar sequences and based on this created a group of sequences that bind the same epitope on human immunodeficiency virus (HIV) or influenza virus. They predicted whether sequences bind the same epitope with SVM.
Altogether, the different strategies for paratope and epitope prediction have shown that antibody-antigen binding is predictable and ML may be able to learn complex rules that govern antibody–antigen interaction. In light of the main learnability challenges, we would argue that prediction accuracy is not the only goal of concern. A major open question is the robustness of the prediction accuracies against information that was either absent from the data (for instance cross-reactive antibodies or antigens), or that has leaked between test and training datasets (i.e., the separation of sequence-similar or homologous sequences between both datasets).54,162
2.5. Learnability of sequence-induced affinity change
The prediction of the binding affinity of antibody sequences toward antigens (binding prediction) is among the major applications of deep learning in antibody research. Affinity is the strength of the interactions between an antibody and an antigen. It is typically governed by proximity, contact surface area, and the distribution of charged, polar, and hydrophobic groups. When an associated antibody-antigen complex is favored, the antibody is categorized as high affinity.163 Thus, in the sequence to affinity setting, a deep learning model maps the sequence space to the affinity space.
Experimentally, the affinity of an antibody toward a target antigen is measured as the change in free energy of binding (ΔG) and can be determined by techniques such as surface plasmon resonance (SPR), amplified luminescence homogeneous assay (AlphaScreen), enzyme-linked immunosorbent assay (ELISA), phage display ELISA (phage ELISA), yeast surface display flow cytometry, isothermal titration calorimetry (ITC), biolayer interferometry (BLI) and enzymatic assays.70,86,164 Alternatively, when the structure of an antibody in complex with an antigen is resolved, the free energy of binding can be inferred with knowledge-based scoring functions (statistical potential) or molecular mechanics force fields,165,166 which are frequently used in molecular docking and molecular dynamics studies.167
Guest et al.49 built a docking benchmark dataset comprising antibody-antigen complex structures (single domain or multiple domain antibodies) for which the unbound antibody and antigen structures were also known (N = 67 antibody-antigen pairs). The dataset allows the testing of the performance of docking strategies to predict the docking pose or binding affinity to the target antigen knowing only their unbound conformation. They showed a high discrepancy in docking methods’ capacity to predict the correct docking pose and a wide variety of correlations between 20 affinity prediction tools and the experimental ΔG of antibody-antigen binding. These observations underline the challenge in the prediction of the affinity, the binding pose, and consequently the interacting interface (paratope-epitope) of an antibody binding to its cognate antigen.
Lippow et al.168 proposed a computational design alternative to directed evolution for affinity maturation by studying the effect of CDR single AA substitutions on electrostatic-binding contributions. Briefly, using a classical physics-based energy function combined with a hierarchical search indexing, single AA substitutions were performed to replace each of the CDR positions with the 20 common side chains, excluding proline and cysteine. Combining multiple AA substitutions led to a 10-fold affinity improvement to an anti-epidermal growth factor receptor IgG1 antibody (cetuximab), and similarly, a 140-fold improvement in affinity was observed for an anti-lysozyme IgG1 antibody.168
If antibody–antigen interaction is predictable, it must follow that affinity could also be predicted by leveraging the combination of sequence and structural data. Indeed, Kurumida et al.164 used single AA substitutions from SiPMAB dataset169 to train an ensemble of ML-based predictors for affinity prediction and reported notable improvements over molecular mechanics-based affinity scoring function. Pires and Ascher170 built the mCSM-AB webserver to predict the effect of AA substitutions, trained on structural signature and pharmacophore count differences between wild-type and mutant residue, together with experimental affinity difference from the AB-Bind dataset.70 mCSM-AB2171 was trained on an expanded dataset including mutant variants with binding affinities obtained from the AB-Bind, PROXiMATE,79 and SKEMPI 2.083 databases. mCSM-AB2 uses graph-based signatures (pharmacophore and distance pattern) for the wild-type residue, structural-based signatures (distance changes, interatomic interactions, solvent-accessible area), evolutionary score and potential energy difference calculated using FoldX.172 mCSM-AB2 achieved a higher Pearson’s correlation coefficient than the previous version, between predicted and experimental ΔΔG. mmCSM-AB173 analyzes the effect of multi-point mutations on antigen-binding affinity, using graph-, sequence- and structure-based signatures. Topology-based network tree (TopNetTree) was developed to predict changes in protein–protein interaction (PPI) affinity upon engineering,174 and was built by combining the CNN with gradient-boosting trees (GBT). The TopNetTree model outperformed TopGBT (topology-based GBT), TopCNN (topology-based CNN) models, and previously published methods on the AB-Bind dataset and SKEMPI database. A similar method, GeoPPI175 consists of two components, a graph neural network trained on topology features from protein structure via self-supervised learning and a gradient-boosting tree (GBT) trained on learned features of both wild-type residue and its mutant to predict ΔΔG upon AA replacement.
2.6. High-throughput experimental methods to generate data for the prediction of antibody-antigen binding using ML
Sequence data, as opposed to 3D structures, can be produced at larger scales, higher efficiencies, and at markedly reduced costs making it the leading choice to study antibody-antigen binding. Typically, the utility of sequence data for studying antibody–antigen binding is restricted to the prediction of binders and non-binders, as it does not afford a sufficient resolution to recover paratope-epitope information. Deep mutational scanning, for instance, can be paired with screening tools, such as ELISA or SPR-based platforms, to obtain large collections of binding and non-binding sequences for an antigen of interest. For example, Mason et al.86 combined deep mutational scanning, ELISA, and CNN to discover new antibody candidates. Specifically, they used CRISPR-Cas9-mediated homology-directed repair mutagenesis to create 104 antibody variants, which were subsequently screened for binding against human epidermal growth factor receptor 2 (HER-2). The resulting binders and non-binders were used to train a deep learning model, which was subsequently used to screen a much larger (108) in silico library, inaccessible to experimental exploration, of antibody variants for HER-2 binders.
Sequencing technologies that study, at high-throughput, many antibodies against many antigens (a logical step forward to the many antibodies against a single antigen as described in Mason et al.86) have begun to emerge as well. Setliff et al.176 developed Linking B-cell receptor to antigen specificity through sequencing (LIBRA-seq) and demonstrated the utility of LIBRA-seq in high throughput screening of antibodies (103) against nine antigens (five HIV envelope proteins and four influenza hemagglutinins) and its efficacy to discover broadly neutralizing antibodies. Briefly, the methods use DNA-barcoded antigens to tag B cells which are then subsequently single-cell sequenced to recover the B-cell receptor (BCR) transcripts and the antigen barcodes, and thus providing a direct readout of BCR-antigen binding. Importantly, the LIBRA-seq score was shown to correlate well with the observations from ELISA making it a useful metric to partition the resulting data as binder and non-binders for subsequent ML training and exploration. LIBRA-seq has also been used to delineate cross-reactive antibodies against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with distinct epitopes and Fc effector functions177 as well as rapid profiling of SARS-CoV-2 specific memory B cells.178
The selection of many antibodies (or variants thereof) against many antigens (or variants thereof) in parallel has been coupled to a display-based library on library (L-o-L) screening platforms. While L-o-L screening represents a key technology for the large-scale symmetric antibody-antigen binding generation, progress in that direction has been slow. Briefly, Hu et al.179 screened a phage-based human antibody library against an active mutant library of Mac-1 inserted domain displayed on the yeast surface. The library enrichment process was bridged with a yeast two-hybrid system for the final quantitative selection of antibody-antigen pairs. A similar L-o-L screening approach has been used to screen an antigen library of the HIV-1 gp160 protein against an antibody library generated from an HIV-1 infected individual.180 Further, Younger et al.181 developed a yeast synthetic agglutination-based improved single pot L-o-L screening platform, which enables high throughput methods for screening protein–protein interactions by reprogramming yeast mating where they quantitatively characterized 7000 distinct protein–protein interactions. Recently, a high-throughput yeast-based synthetic agglutination assay (AlphaSeq) was used to characterize the binding profiles of tens of antibodies against thousands of SARS-CoV-2 receptor-binding domain (RBD) variants. Specifically, 178,760 protein–protein interactions were measured between 33 antibodies and single AA substitutions corresponding to 165 binding sites within the panel of RBD variants.182
In summary, emerging high-throughput experimental assays that are capable of generating large (developability-adjusted) antibody-antigen binding data in the order of 104–105 have begun to unlock the potential of ML for the prediction of antibody-antigen binding.3,176,183,184 However, for the prediction or generation of paratope-epitope pairs on the sequence level without any structure-aided encoding, much larger data at a higher resolution may still be necessary, as previously suggested by us.54
2.7. Leveraging ground-truth synthetic data to establish lower bounds on learnability
The generation of synthetic data via simulations is a crucial, but yet under-explored tool in computational antibody design.185 Estimating (ML prediction) error requires us to know the ground truth about the training data. We define ground truth as a system in which any parameter (and the value thereof) that contributed to training data generation is known and controlled – this is the case for synthetic data but usually not the case for experimental data. Only if we know how the training data has been generated, can we benchmark ML methods not only with respect to accuracy but also with respect to feature discovery and interpretability. To objectively benchmark ML approaches, special care should be put on the distribution of elements and property of elements in the datasets as to faithfully represent experimental datasets because method benchmarking on simulated data is only useful if conclusions gained on simulated data are transferable to experimental data. By distributions we mean, in the case of simulation of antibody-antigen binding data, for example, parameters such as positional amino acid frequency, antibody and antigen topology, sequence dependencies.185 Specifically, simulations that allow to precisely define different antibody-antigen binding problems, which requires explicit datasets with all required levels of annotation allowing any kind of encoding, are not yet available in experimental data (Figure 1, challenges in Learnability). Commonly used data encodings can be divided into sequence-based and structure-based ones, while hybrid formalizations leverage both types of datasets.54 To summarize, define, and compare ML approaches on the same basis, it is critical that simulated data represent 3D features of antibody-antigen binding (especially for defining paratopes and epitopes), allow the generation of large-scale datasets, and the integration with other data types, such as sequence-based datasets. Integration of structure and sequence-based datasets is especially important given the large imbalance in the availability of sequence and structural experimental data (Figure 1, challenges in learnability).
We define synthetic datasets as computer-generated datasets that mimic a set of observed properties of experimental datasets that are the most important in determining the biological outcome to predict. Synthetic datasets can be generated by data augmentation, for instance, by starting from experimental antibody-antigen structures, and generating other possible docking poses that are added to the dataset,131 or structures that are calculated based on physical-based simplified models.54,186 Alternatively, structure-independently, antibody sequences may be simulated according to the principles of V(D)J recombination and, partially, somatic hypermutation.105,187–191
Sequence-based Ig simulation tools, such as IGoR187,192 and immuneSIM,105 enable the generation of large numbers of Ig sequences with moderate computational needs. They have the advantage of generating sequence data that is native-like, which means that data generation is performed, to a large extent, in agreement with the rules of V(D)J recombination, resulting in the generation of data that are largely indistinguishable from experimental data. Importantly, immuneSIM also allows the insertion of sequence motifs (“immune signals”) into the generated sequences, which may be used to model motifs implicated in antigen binding. Therefore, such simulated data can be used for exploring antibody-specificity prediction tasks where in a binary or multi-class/label fashion, sequences are to be classified for their antigen-binding behavior (see Use Cases 1 and 2 in ref.54). Of note, simulations with implanted motifs have also been used for repertoire-based ML with applications to immunodiagnostics.104,193 Independently of established simulation frameworks, experimental-based simulations for training sequence classifiers may also be performed to augment Ig sequence data by reflecting experimentally determined AA position bias.86
A current drawback in these simulation frameworks is the lack of nuance pertaining to VH-VL pairing. Since the rules of VH-VL pairing remain underexplored, chain pairing is either not simulated at all or implemented by simple random pairing of VH and VL chains.105,187,189 Although it has been shown that the CDRH3 is the most important site for antigen binding,44,45 considering chain pairing is crucial to fully reflect the biological complexity of antigen binding.69,194 Pioneering work on jointly modeling TCRalpha/beta chain pairing may potentially be ported to VH-VL modeling.195,196
To summarize the advantages of synthetic data for the development of computational and machine learning applications for antibody engineering: we agree that the discovery of novel biology can only be performed using experimental data (unless synthetic data perfectly reflects biology complexity). Rather, the advantage of synthetic data over experimental data, if carefully designed, is that due to its arbitrary size and specification, it enables the exploration of the capacity and limits of computational methods as well as the ranking of methods for a given task.185 In other words, synthetic data allows the development and refinement of computational methods in the absence of suitably large and complete experimental data.197
Large-scale synthetic structural antibody-antigen datasets mimicking key aspects of natural antibody-antigen (i.e., paratope–epitope interaction) are needed to develop and benchmark antibody-adapted ML approaches. Therefore, we have recently established the computational framework “Absolut!” for simulating in silico antibody–epitope interaction datasets.54 This framework enables automatic conversion of antibody-antigen structure into a (3D)-lattice representation followed by modeling of 2D/3D antibody-antigen binding of each antibody sequence around the discretized antigens using structural lattice affinity computational method based on experimentally derived coarse-grained amino–acid interaction potentials.186,198 The Absolut! framework was mainly developed to address the issue of antibody-antigen binding data availability for ML method development, formalization, and benchmarking. The simulated binding structures incorporate a range of physiological properties of antibody-antigen binding (a large number of possible binding structures, AA composition and surface topologies, complex positional AA dependencies in binding antibody sequences, existence of immunogenic binding hotspots, and complexity of the paratope-epitope binding network) and allow for the exploration of various types of negative control datasets that are largely infeasible to create experimentally. Using Absolut!, we have generated close to one billion antibody-antigen structures. To further close in on the physiological reflection of Absolut!-generated structures (or any other framework that aims to simulate antibody-antigen binding), further work is needed to establish 1) full VH-VL chain binding (so far, we can only model CDRH3-antigen binding), 2) smaller angle grid in the lattice as our framework was limited to integer positions in a 3D grid and 3) adding constraints at the CDR3 ends in order to reproduce the anchoring of the CDR chains to the framework region (FR) of the antibody. In the even more long-term future, atomistic and molecular dynamics resolution are needed to add further biological complexity to the Absolut! antibody–antigen binding simulation framework.51,54,131 Of note, given that Absolut! simulations are based on physics-based (“equation-based”) principles, Absolut!-generated datasets can also be used to develop novel deep learning approaches such as end-to-end differentiable ML architectures that combine mathematical equations specific to a particular domain (in this case, for example, antibody-antigen affinity) with general-purpose, machine-learnable components.197
Although much progress has been made in the learnability of antibody-antigen binding and developability (see next section), key challenges such as interpretability as well as data completeness have only begun to be addressed.
On interpretability. Interpretability encompasses the effort to infer the rules underlying the data. However, there is not yet a way to mechanistically and comprehensively map the rules that govern antibody-antigen interaction due to the combination of large search space and scarcity of data (see the Section entitled “Learning from nature: considering biological complexity in computational antibody design”). As the immunology field begins to accumulate more data (experimental as well as synthetic), we will become increasingly reliant on large ML models to infer these rules. Drawing parallels from the natural language processing (NLP) field where large transformer-based models (Figure 3) continue to advance the state-of-the-art results in many different problems and benchmark studies at the expense of building larger and larger models.199 It begs the question of whether continuing along the lines of building large and more sophisticated architecture will perpetuate the improvements we have seen thus far at the cost of interpretability. For instance, the Bidirectional Encoder Representations from Transformers (BERT),200 a prominent language representation model, has grown from 110 million parameters to 17 billion parameters in Turing-NLG and 175 billion parameters in GPT-3.201 The massive complexity of these models gave birth to a subspecialty that focuses exclusively to study BERT models – BERTology.202 Emphasis has been particularly given on the overparameterization of these large models as they do not seem to use the parameters to their fullest potential. Accumulating evidence reveals that many BERT models can be pruned without impacting their predictive prowess, i.e., most heads in the same layers converge to a similar attention pattern, and thus many layers can be consolidated into a single head.203,204 In biology, attention layers of transformer-based models, including BERT, have been shown to capture long-range interaction in protein and antibody folding by folding AAs that are distant in 1D sequence but spatially adjacent in the 3D structure, to identify active sites and to capture the hierarchy of complex biophysical properties with increasing layer depths100,205 – properties that are also critical for antibody-antigen binding. Nevertheless, as in NLP, these models remain susceptible to overparameterization and lack of interpretability. Future deep learning methods would benefit greatly from architectures that accommodate the mapping of rules underlying the data instead of merely focusing on prediction accuracy.
On data completeness. Our immune system is, at least partially, a reactive system where germline gene base diversity can be expanded via stochastic recombinations, insertion, deletion, and mutation. By that definition, our collective antibody repertoires expand or converge to the prevailing landscape of pathogens. Consequently, generating experimental datasets that contain exhaustive multiparameter information (a complete dataset) for the purpose of training and benchmarking ML models remains challenging (Figure 3). In addition to having access to key design parameters (Figure 4) on the same dataset (something that has not been achieved thus far), even more “exotic” data on biological parameters would be crucial, such as structural information on bound and unbound state,49 chain flexibility,206 molecular dynamics simulations.69 Indeed, only large and exhaustive data will allow us to perform subsampling studies27,54 for determining the minimal dataset size necessary to achieve satisfactory prediction accuracy on a given prediction task. In order to reach data completeness faster, it may be interesting to explore experimentally to what degree some parameters may be set constant, such as for example only working on the CDRH386 or with single-chain antibodies207 or only with linear epitopes (or antigen immunizations with simple peptides).208,209
Figure 4.

Mapping of developability parameters to the antibody regions. The high-level developability parameters are shown in bold font and placed within black boxes with respective mapped antibody regions listed in brackets below each box and referred to with dashed black arrows. The widely used low-level physicochemical developability parameters are also shown in grey text and connected to respective high-level developability parameters with solid grey arrows (detailed further in Table 1). Antibody regions are color-coded as follows; Fc: grey, VH: red, VL: purple, CDRs: blue. High-level developability parameters. Viscosity, solubility, and aggregation propensity of mAbs are mainly linked to the surface-exposed regions of mAb molecules. Antigen specificity and binding affinity, on the other hand, are mainly associated with the CDRs as well as thermal stability. All regions of the antibody can impact half-life and immunogenicity. Low-level developability parameters. Viscosity has been reported to be influenced by charge, hydrophobicity, atomic/diffusion interaction, and the isoelectric point (pI) of the mAb molecule. Solvent exposure area and AA composition are frequently reported to impact the solubility of the antibody. Charge and hydrophobicity were also found to affect antibody preparation aggregation likelihood together with stability and spatial aggregation propensity (SAP) measures. The binding affinity of the Fc region to FcRn significantly impacts mAb PK, in addition to the reported role of poly-specificity, charge, and pI on mAbs half-life. The likelihood of a mAb to elicit an immune response (immunogenicity) is linked to the non-human AA sequence content of the mAb, in addition to the way it is processed (digested) into smaller peptides by APCs, bound to the human leukocyte antigen II (HLA II) and presented to T-helper cells. The hydrophobicity and AA composition of mAb CDRs were often reported to affect its thermal stability.
Furthermore, we and others have shown that learned representation from one problem (e.g., one antigen) can be leveraged to improve the predictive capability of a deep learning model that was built for a different problem (a different antigen) by way of transfer learning.27,97,210,211 Large complete knowledge datasets have been conceived by simulating large antibody-antigen pairs,54 although possible at reduced resolution, the computational bottleneck to efficiently simulate these pairs at full atomistic resolution presents yet another challenge. Future ML models may benefit from integrating continuous streams of data (either experimental or simulated) in an online fashion where the data comes in a sequential manner, and the model is updated constantly to allow it to evolve along with the prevailing scenario, in contrast to the typical batch learning where the model is trained once (often with incomplete data) and is expected to generalize well (Figure 3).212
3. Capacity to modularly learn antibody design parameters
3.1. Modularity of antibodies and developability parameters
mAb therapeutic candidates need to pass several developability hurdles for feasible commercial-scale manufacturing and clinical application.213,214 The developability of an antibody encompasses the likelihood of the antibody to successfully progress to the clinical phase, which is assessed based on several biophysical properties including a tendency to aggregate, stability, immunogenicity, and plasma half-life (Figure 4).215 The conventional approach for antibody design is focused on segregating different biophysical properties to different components of antibody.8,43 However, in our view, a modular approach for antibody design would need to consider the interdependence and non-linear optimization of the antibody design parameters (both developability and antigen-binding related), which will lead to the desired function and functionality (Figure 4). In the following section of this review, we extensively review critical knowledge of the developability and pharmacokinetics of mAbs. An experienced reader in antibody developability and mAbs therapeutics might find it more convenient to directly resume reading from the section entitled “Designing antibodies with desirable efficacy and developability remains challenging”.
3.2. Background: therapeutic mAbs
Among the five isotypes of human Igs – IgA, IgD IgE, IgG, and IgM, the gamma class (IgGs) comprises all clinically approved mAb therapeutics.2,216–218 This is due to the combined features of distinct effector functions with advantageous pharmacokinetic properties of the IgG subclasses. In addition, the high abundance of endogenous IgGs in humans (10–12 mg/ml in blood, accounting for up to 80% of the native antibody repertoire219 and 60% of serum Igs217 intravenous or subcutaneous injection and robust manufacturing processes well established at an industrial scale220–222 makes them suitable for therapeutic applications.
Four subclasses of IgG exist in humans, named in decreasing serum abundance, IgG1, IgG2, IgG3, and IgG4.223 Although they share high similarity in their structural architecture and AA composition, they have distinct differences that dictate unique effector molecule binding and pharmacokinetic properties.219,224 Specifically, while IgG1 and IgG3 trigger potent immune responses upon engagement of antigen, IgG2 and IgG4 induce more subtle responses.216,225 Thus, for the development of an antigen-specific therapeutic mAb candidate, it is a prerequisite to select the most preferable subclass.224–227
Early developability screening for fit-to-manufacture properties is crucial to minimize the cost and time used for the selection of lead mAb candidates.215,228 For this purpose, major efforts have been invested to develop in silico tools and ML algorithms that could ultimately improve antibody design parameters by implementing modular learning strategies (Table 1).4,215,228 Here, we discuss each of the developability parameters with the main focus on computational developability prediction tools.
Table 1.
| Overview of in silico methods for antibody developability parameter computation. We summarize the most prominent computational tools to predict the value of high-level developability parameters (thermal stability, solubility, aggregation, viscosity, immunogenicity and half-life shown in Figure 4). We detail the methodology used in each tool, the corresponding lower-level developability parameters, and method availability
| Method name | Methodology/approach | Main low-level parameter(s) | Availability |
|---|---|---|---|
| Thermal stability | |||
| Spatial aggregation propensity (SAP)229 | Custom code, MD simulation | Surface hydrophobicity | Mathematical equation |
| Bekker et al.230 | MD simulation | Fraction of native contacts (Q-value) | NA |
| ANN model231 | ML model | AA composition | NA |
| Aggregation | |||
| Developability Index (DI)232 | Custom code | Charge, spatial aggregation propensity (SAP) |
Mathematical equation |
| AbsoluRATE233 | ML model for aggregation kinetics prediction | Environmental conditions, disorderness, aggregation related properties etc. |
https://web.iitm.ac.in/bioinfo2/absolurate-pred/ |
| Therapeutic Antibody Profiler (TAP)213 | Developability rules as per authors | CDR length, surface hydrophobicity, charge |
http://opig.stats.ox.ac.uk/webapps/newsabdab/sabpred/tap |
| VLAmY-Pred234 | ML model | Charge, hydrophobicity, Disorderness, β-propensity |
https://web.iitm.ac.in/bioinfo2/vlamy-pred/ |
| Solubis235 | Custom code | Aggregation propensity, Stability |
https://solubis.switchlab.org/ |
| Solubility | |||
| SOLpro236 | ML model | 23 sequence-based features | http://scratch.proteomics.ics.uci.edu/ |
| Protein-Sol237 | Regression model | 35 sequence-based features | https://protein-sol.manchester.ac.uk/ |
| CamSol238 | Custom code | solvent exposure, intrinsic solubility profile | http://www-vendruscolo.ch.cam.ac.uk/camsolmethod.html |
| SoDoPE239 | Custom code Solubility-Weighted Index (SWI) | AA composition | https://tisigner.com/sodope |
| DeepSol240 | Convolutional neural network, Deep learning | 57 sequence-structure features | https://zenodo.org/record/1162886#.YQvaxJNKhaQ |
| Viscosity | |||
| Fv region-based qualitative screening profile241 | Developability rules, MD simulation | Charge, ξ-potential, isoelectric point (pI) | Mathematical equation |
| Sharma et al. 242 | Developability rules, MD simulation, principal component regression | Hydrophobicity, dipole distribution, charge | Mathematical equation |
| Tomar et al.243 | Regression model | Surface hydrophobicity, charge, hinge regions | Mathematical equation |
| Nicholas et al.244 | Mutational analysis | Negatively charged surface patches | NA |
| High viscosity index (HVI)245 | ML model | Charge, AA composition | Mathematical equation |
| spatial charge map (SCM)246 | Custom code | Partial charge of the atom | Mathematical equation |
| Lai et al.247 | MD simulation, ML model, concentration dependent | Charge, Hamaker constant (short-range interaction parameter) | Mathematical equation |
| Tilegenova et al.248 | MD simulation, Mutational study | Aromatic interaction (cation-π and/or π-π) | NA |
| Schwenger et al.249 | Ross-Minton model | Temperature and concentration dependent | Mathematical equation |
| Immunogenicity & tolerability | |||
| netMHCIIpan(T-cell epitope prediction method)250 | ML model | Antigen (mAb) processing, HLA II peptide binding and presentation | https://services.healthtech.dtu.dk/service.php?NetMHCIIpan-4.0 |
| Hu-mAb(mAb humanization method)251 | ML model | Non-human sequence content | http://opig.stats.ox.ac.uk/webapps/newsabdab/sabpred/humab |
| BioPhi(mAb humanness evaluation, humanisation and design)252 | ML model | Non-human sequence content | https://biophi.dichlab.org/ |
| Humanness (T20) score253 | Quantitative distance measure | Non-human sequence content | https://dm.lakepharma.com/bioinformatics/ |
| Half-life | |||
| Combinatorial LASSO approach254 | ML model | FcRn affinity at pH 7, thermal stability | Multiple regression & mathematical equation (methods) |
| Random forest approach255 | ML model | pI (in silico) and poly-specificity (in vitro) | Clearance (PK) classification thresholds |
| Multiple high-level parameter prediction/calculation methods | |||
| Aggrescan3D (A3D) For aggregation and solubility256 |
Custom code | Residue wise aggregation propensity scale, relative surface accessibility |
http://biocomp.chem.uw.edu.pl/A3D2/ |
3.3. Tailoring the plasma half-life of therapeutic mAbs
Although the IgG plasma half-life is threea weeks on average in humans, the half-life of therapeutic IgG mAbs is actually in the range of 6–32 days.254,257 Importantly, large differences are not necessarily a direct effect of target-mediated clearance as half-life variation is also measured for IgG1 mAbs against pathogens.258 An illustrating example is briakinumab and ustekinumab, both IgG1s targeting the same antigen (interleukin-12/23), which in humans have half-lives of 9 and 23 days, respectively.259,260 Understanding the molecular basis for these striking differences is the key to predict in vivo pharmacokinetic properties. However, the parameters that determine the pharmacokinetics of mAbs are multifactorial, including target-mediated clearance, nonspecific off-target binding as well as specific off-target binding via liver receptors and charge characteristics.24,261 While IgG mAbs have a size above the renal clearance threshold, which excludes renal filtration, FcRn operates as a global half-life regulator as discussed above. Thus, Fc-engineering for improved pH-dependent FcRn binding has resulted in the generation of Fc technologies that can extend the half-life beyond that of natural IgG.224
While the Fc technologies can extend the half-life of IgG, each mAb will have a unique pharmacokinetic profile, as a result of its variable domain sequence composition, which is determined by the targeting-binding properties, but also cellular handling (in an FcRn-independent but also likely in an FcRn-dependent manner). As such, the distinct sequences generally have a unique pharmacokinetics profile. In this regard, physicochemical properties of the variable region, such as hydrophobicity, isoelectric point (pI), and charge patches may have a major influence on mAb pharmacokinetics (Figure 4).24,216,261 For instance, positively charged antibodies more readily interact with the negatively charged plasma membranes, and therefore, be more susceptible to cellular uptake.262,263 In accordance with this, generating mAb with increased charge in the variable domains has been shown to result in increased nonspecific binding and consequently faster clearance.261,263 On the contrary, reducing the pI or balancing the charge distribution of the CDRs has been shown to extend the plasma half-life by reducing non-specific binding.22,264
While the principal binding site for FcRn is at the Fc elbow region, recent findings support that charge features of the variable domains may modulate engagement of the receptor, and as such affect cellular transport and half-life.24,42,265,266 This layer of complexity is far from understood, and thus is under intense investigation.
Although non-human primates generally offer reliable pharmacokinetic parameters that can be translated to a human setting, they are not suitable surrogates for screening of panels of mAbs due to impractical handling and ethical considerations. As such, mice are easier to use, but differences exist in how mouse FcRn binds human IgG compared with the human receptor, which must carefully be taken into consideration.267,268 Thus, mouse strains expressing human FcRn have become the state-of-the-art preclinical standard for investigating the pharmacokinetic properties of IgG mAbs.269,270 Furthermore, Grevys et al.271 have developed a human endothelial cell-based recycling assay (HERA) that can be used to screen IgG mAbs for their ability to be taken up and sorted in an FcRn-dependent manner, which has been demonstrated to correlate with half-life values obtained in human-FcRn transgenic mice.
In silico screening of antibody candidates for favorable PK properties prior to pre-clinical investigations can increase the convenience and cost-effectiveness of mAbs development. However, identifying the most impactful parameters that affect mAbs PK is challenging due to its multifactorial nature (Figure 4). In this context, Goulet et al.254 used a LASSO (least-absolute shrinkage and selection operator) ML strategy to identify the combination of parameters that best correlate with mAbs clinical clearance data. They reported that FcRn affinity together with mAb thermal stability is the most powerful parameters for mAb half-life prediction. Most recently, Grinshpun et al.255 investigated the effects of 40 physicochemical parameters (12 measured in vitro, 28 calculated in silico) on 48 IgG1 mAbs clearance profiles. They implemented a random forest ML algorithm and identified in silico computed sequence-based features “pI” and in vitro measured feature “binding poly-specificity reagent (PSR)” as the top two ranked parameters based on 10,000 repeated runs of the random forest model.255 These findings align with the previously reported impact of pI and PSR measures on mAbs PK.22,264,272
In summary, the half-life of IgG mAbs can be tailored via the optimization of numerous physicochemical parameters, including FcRn affinity, thermal stability, pI, and PSR. ML and the increasing availability of mAb PK data have been aiding the identification of the parameters and their threshold values to facilitate in silico estimation of mAb PK.
3.4. Improving the stability of mAbs
The stability term of antibodies comprises thermodynamic or conformational stability (thermal stability) and colloidal stability (solubility, viscosity, and aggregation), which are physically related and sometimes used interchangeably. These parameters are in particular important to consider during development and manufacturing processes as part of risk assessment, to reduce the need for cold-chain storage, extend shelf life, and expand the range of applications for practical use. The applicability of experimental assays is limited for antibody development due to the high mAb concentration requirement for some of these steps (i.e., >50 mg/mL) and the lack of high throughput methods.273,274 These drawbacks may limit the complete screening of all parameters during early-stage development. However, there has been significant progress in the development of high-throughput computational methods to compensate for the time-consuming lab-based biophysical experiments (Table 1).
The generally recommended temperature for storing biopharmaceuticals is in the range of 2°C – 8°C. However, protein denaturation can occur during freezing and freeze-thawing cycles, affecting both conformational and colloidal stability of proteins.275,276 A study on eight human IgG mAbs (6 IgG1 and 2 IgG4) suggests that focusing on strategies that increase the thermal unfolding temperature of the Fab arms is an attractive approach to improve storage stability.277 The development of generic antibody fragments to improve the stability of antibodies against a range of denaturing conditions (e.g., temperature, denaturants, polar and non-polar solvents, surfactants, and proteases) while maintaining antibody-specificity, is widely studied experimentally.278–280
Among the computational approaches, molecular dynamics (MD) approaches are widely used to assess the stability of antibodies in the context of different solvent conditions,281 spatial aggregation propensity (SAP),229 fraction of native contacts (Q-value).230 In comparison to simulation studies, ML-based models are still at a nascent state to predict the stability parameters of antibodies. However, an ANN-based model has been developed using the AA composition as a feature for studying melting temperature (Tm), aggregation onset temperature (Tagg), and diffusion interaction parameters (kD) as a function of pH and salt concentration.231 Jia et al.282 used a sequence-consensus approach combined with structural residue pair covariance methods to predict the thermostability of antibodies. In another study, authors used the Rosetta platform for protein design to achieve thermal stabilization of anti-HA33 (Clostridium botulinum hemagglutinin protein) antibody, through clusters of mutation in the FR region, and verified the results through experiments.283 In summary, antibody-specific MD simulations are widely accepted as a more robust way to study thermal stability, in comparison to generalized ML/mathematical model development (Table 1).
Aggregation describes the accumulation of denatured antibodies into large clusters due to high concentration or environmental factors (such as temperature, pH, salt concentration, additives, etc.). In general, aggregation-prone region (APR) prediction methods are widely used to predict the aggregation capability of proteins, including antibodies. Several studies on antibodies have identified potential aggregation-prone regions in the relatively exposed CDR of the VH domains.234,284–286 However, a recent study observed that almost all the latest APR prediction algorithms perform poorly on identifying aggregation.287 This can be attributed to (1) limited overall variations in the antibody sequence (except for CDRs) leading to higher sequence conservation and (2) low sensitivity of these algorithms toward similar protein sequences. Importantly, these algorithms do not account for aggregation-related environmental factors and protein concentration. While aggregation kinetics prediction methods account for these external factors, the lack of significant dataset size for training limits their robustness.233,288,289
In antibody-specific studies, the developability index (DI) allows the prediction of aggregation propensity and long-term stability based on the antibody structure and AA sequence charge information.232 In an attempt to define success limits for developability parameters, Jain et al.52 assessed 12 assay-based biophysical properties of 137 mAbs that had reached at least phase II clinical trials and observed that decreased protein stability, an increased disposition for protein aggregation, and polyreactivity are linked to poor developability. This study established thresholds for desirable drug-like developability measures, suggesting practical rules for mAb candidates. Subsequently, in an effort to recreate a ‘Lipinski’s rule of five’ for antibodies, Raybould et al.213 examined five developability properties of 242 post-phase-I clinical-stage IgG1 antibodies and implemented them into Therapeutic Antibody Profiler (TAP) online tool. Specifically, TAP scores take into account developability factors, such as the length of the CDRs, hydrophobicity, and the presence of charge patches, which are linked to polyspecificity, aggregation, and viscosity of mAb preparations. Rawat et al.234 developed an ML-based light chain aggregation prediction method and highlighted that lambda light chains are inherently more aggregation-prone. Notably, most approved IgG mAbs harbor the kappa light chain, and the pool of human IgG in serum has about 2-fold more of kappa light chain than the lambda. Van der Kant et al.290 combined the aggregation propensity and thermodynamic stability prediction methods to rationally improve the developability parameters of mAbs, which was implemented as the Solubis server.235 Another example is the aggrescan3D (A3D) 2.0 web server, primarily developed to predict aggregation propensity of proteins, which recently was implemented for simultaneous prediction of change in solubility and stability for improved antibody developability.256 In conclusion, in silico studies on antibody-specific aggregation have emerged in the past few years, whereas generalized protein aggregation predictions have matured for over decades (Table 1).
Solubility is another aspect of colloidal stability, which is inversely proportional to aggregation. SOLpro is one of the early solubility prediction methods that used 23 groups of features computed from the primary sequence to design a two-stage support vector machine (SVM) architecture.236 Another method, Protein-Sol, combines 35 sequence-based properties including AA composition and other conventional solubility/aggregation related properties (i.e., hydropathy, charge, disorder, β-strand propensity, etc.) in a linear model to predict the solubility of proteins.237 Further, CamSol is a structure-based method to generate the intrinsic solubility profile of proteins. Similar to aggregation prediction algorithms, CamSol also identifies low solubility patches in protein structures that may elicit the self-assembly process.238,291 A recent paper discusses ‘Solubility-Weighted Index’ (SWI), which is derived from a simple sequence composition scoring method, to predict the solubility of proteins.239 The availability of a large dataset(s) for solubility led to the implementation of advanced deep learning models as well. For example, Khurana et al.240 used a convolutional neural network that exploits k-mer structure and additional sequence and structural features extracted from the protein sequence to develop the DeepSol model. Solubility prediction methods in general are relatively more robust compared to other developability parameters due to the availability of large-scale datasets. However, antibody-specific solubility data is still scarce.
Concentration-dependent viscosity is the part of colloidal stability that may depend on pairwise and higher-order intermolecular interactions, non-native aggregation, and concentration-dependent fluctuations of distinct structural regions of antibodies.292 An increase in viscosity has been a challenge for concentrated antibody formulation, which can reduce the volume of antibody dose, increase dose interval by improved pharmacokinetic profile, reduce the healthcare cost, and improve the bioprocessing of drugs during downstream ultrafiltration and diafiltration steps.293 Most of the in silico studies on limited datasets correlated viscosity with sequence-structural properties, such as net charge, spatial charge map (SCM), pI, zeta-potential, hydrophobic parameters, AA composition/aggregation propensity of the fragment variable (Fv) region.241–246 Although MD simulation-based parameters, such as short-range interactions, van der Waals attractions and electrostatic repulsions are also used to develop models to predict viscosity of antibodies under a wide range of concentration and ionic strength.247,294 A mutagenesis study using MD simulations and experiments showed that replacing surface-exposed aromatic AA residues reduces the viscosity of antibodies.248 Schwenger et al.249 measured the viscosity as a function of concentration using Ross-Minton model and temperature using the Arrhenius equation and tested it on four mAbs in the range of potential clinical formulation. An interesting study on a relatively large dataset of 59 mAbs showed that diffusion interaction parameter (kD), a dilute-solution measure of colloidal self-interaction, can predict solution viscosity with high accuracy.295 Viscosity of antibodies is dependent on intermolecular interaction, and therefore, MD simulation-based studies are heavily exploited. The low-level computational parameters related to viscosity are still at a preliminary stage of development and should be explored further on large datasets.
3.5. Reducing the immunogenicity of therapeutic antibodies
All protein-based therapeutics, including mAbs, may potentially be immunogenic and elicit immune responses when administered to humans, resulting in the generation of anti-drug antibodies (ADAs). ADAs may affect the therapeutic efficacy of mAbs by neutralizing their activity and accelerating their circulatory clearance.2,253,296 For instance, ADA formation occurs in up to 35% of inflammatory bowel disease patients treated with the anti-tumor necrosis factor-α (anti-TNF-α) adalimumab (Humira), subsequently resulting in a loss of clinical response within 12 months of treatment initiation.297,298 ADAs could also result in adverse effects ranging from topical rashes to systematic fatal inflammatory reactions.253,299 Thus, immunogenicity is a key concern for mAb development.300,301
Similarly to exogenous proteins, mAbs may be internalized by antigen-presenting cells (APCs), processed (digested) into shorter peptides, and subsequently bound to the major histocompatibility molecule II (MHC II) and presented for T-helper cells on the surface of APCs.302 Anti-drug immunogenic responses only occur when these complexes (termed as T-cell epitopes) are recognized by T-helper cell receptors, thereafter activating the adaptive immune cascade and leading to the production of ADAs against the mAbs.303,304
While the first approved therapeutic mAb was a mouse IgG2 antibody,305 mAbs have evolved to include an increasing proportion of human sequences to avoid the generation of ADAs.299,300 Thus, murine mAbs were followed by the engineering of (1) chimeric versions where the constant regions of the mAb are of human origin, (2) humanized antibodies where only the CDRs are of murine origin, and finally (3) fully human mAbs where murine sequences are completely absent from the mAb sequence as it is obtained from human cell libraries.299,306 mAb humanization has been widely implemented due its advantageous in vivo tolerability.307 In fact, almost 50% of approved or investigational therapeutic mAbs in the EU or US were humanized antibodies as of 10th July 2021 making them the leading class of mAbs in development.308
Humanization of a mAb refers to the partial replacement of murine sequences in an antibody sequence precursor that has been initially identified using animal (often, mouse) models with human sequences, to improve their tolerability while maintaining their specificity, affinity, and stability (Figure 4).306 Antibody humanization is usually achieved by selecting and grafting the murine CDRs from the antibody precursor into human FR regions.309 In this process, human FRs are selected from the human germline Ig genes that produce the most homologous FRs to the original murine. While humanization seems straightforward in principle, further AA substitutions are often required to retrieve the desirable properties of the antibody that were lost in the grafting process.306,309 AA substitutions are usually performed on a trial-and-error basis until an antibody sequence with desirable immunogenicity and binding properties is identified, which can be both time- and resource-intensive.309 A recent study envisaged that human antibody repertoires can be a useful predictive tool for mAb development.310 They analyzed the AA substitutions in mAbs using position-specific scoring matrices (PSSMs) and observed that positions with high frequency of AA alteration may potentially reduce immunogenicity and improve other developability parameters.
To measure the extent of “humanness”, Gao et al.253 introduced the “humanness score” as a quantitative measure to reflect the distance between the mAb sequence and the human consensus sequence. Also, several in silico tools have been developed that could potentially accelerate mAb humanization (Table 1).251 For example, Hu-mAb is a computational tool built on an extensively trained ML model on native human and mouse repertoires to compare an input sequence to the closest human germline Ig gene, and suggests as few AA substitutions as possible on the FRs in order to achieve maximal sequence humanness score while reducing the likelihood of impacting the efficacy of the mAb.251 Hu-mAb humanness predictions have shown to be interpretable relative to clinical immunogenicity data when tested on a set of 217 mAbs. In their study, Marks and colleagues illustrated that high Hu-mAb humanness scores were linked with a low proportion of patients with observed ADAs titers.251 Most recently, Prihoda et al.57,252 devised an in silico platform BioPhi that offers three complementary tools: 1) OASis, short for Observed Antibody Space (OAS) identity search, is an interpretable humanness scoring system based on an exact 9-mer peptide search within the OAS database, capable of accurately distinguishing human and non-human sequences with clinical immunogenicity correlation; 2) Sapiens, is an ML-based humanization method trained on the OAS human database using language modeling to recognize and substitute non-human sequences with human native equivalents in FR regions to improve sequence humanness (the OASis score); and 3) an interactive interface, to incorporate AA substitutions in the sequence and visualization.57,252 In their study, Prihoda et al.57,252 compared the humanization performance of Sapiens on 152 precursor sequences of humanized mAbs against Hu-mAb (computational) and mutation-based humanization (experimental). They reported that Sapiens achieved higher humanness improvement than Hu-mAb and comparable results to experimental methods, suggesting AA substitutions that were experimentally validated to be advantageous for sequence humanization, while maintaining mAb specificity and binding affinity. In summary, BioPhi is an open platform based on deep learning from the human native antibody repertoire providing in silico tools for antibody design, humanization, and humanness evaluation with a graphical interface aiming for user-friendliness.
In contrast to expectations, mAbs completely derived from human sequences (human mAbs) can still be immunogenic which invites further immunogenicity investigations.300,311,312 Another approach to estimate mAb immunogenicity is inspired by the adaptive immune system activation mechanism. It has been suggested that analyzing the peptidic pool presented by the MHC II molecules to T-helper cells, termed as immunopeptidome, could provide valuable insights for immunogenicity estimation.313 However, the complexity of the human MHC II immunopeptidome is amplified by the large genetic pool coding for structurally distinct MHC II molecules, termed as human leukocyte antigen (HLA) molecules.314 Over 8000 human allelic forms HLA class II have been identified (EBI IMGT/HLA: accessed 20th July 2021315), and each person typically expresses up to eight different HLA II allelic forms.316
Experimental data from EL assays have been empowering the construction of immunopeptidome public databases (IEDB, accessed 20th July 2021, last updated 11 July 2021317 and others reviewed by Doneva et al.301). These databases have been implemented for the development of in silico tools that can predict protein immunogenicity based on the content of immunogenic peptides. These tools, as reviewed by Doneva et al.301 could use protein sequence or structural data to predict its potency to stimulate a T-cell response. Due to the limited availability and high cost of generating structural data, in silico immunogenicity prediction methods that rely on sequence input for motif search and ML-based approaches are heavily investigated.301 Among alternative methods reviewed by Doneva et al.,301 netMHCIIpan provides an ANN-mediated holistic approach to predict peptide processing, presentation, and binding to any human MHC II molecule (Table 1).250 Importantly, netMHCIIpan benefits from the advanced abilities of the NNAlign_MA ML algorithm to handle peptide ligands with multiple potential HLA allele annotation to produce pan-specific T-cell epitope predictions.250,318 The most recent version of netMHCIIpan (4.0) has been trained with extensive multi-allele EL datasets and showed a significant improvement when benchmarked against state-of-the-art T-cell epitope prediction methods.250 This tool allows the prediction of binding affinity of AIRR-seq and stretches of mAb-derived AA sequences to selected HLA II alleles.27,86 netMHCIIpan can be implemented for the prediction of global immunogenicity by specifying the HLA II supertypes found in the majority of the human population in the command arguments.86 Of note, HLA II supertypes refer to just over 25 HLA II alleles that were found to be responsible for T-cell epitope presentation in over 98% of the universal human population.319
3.6. Designing antibodies with desirable efficacy and developability remains challenging
In silico prediction of mAb developability parameters have been evolving in efficiency and accuracy, however, several challenges remain. Specifically, computational immunogenicity predictions cannot yet fully replace the in vitro animal testing due to the safety element associated with this particular parameter, as discussed above.301 Also, considering the fact that even fully human antibodies may be immunogenic, it might be difficult to solely rely on in silico tools for immunogenicity assessment until the biological rules for immunogenicity are better understood.320 Indeed, we believe it will be of interest to explore whether mimicking antibody design parameter combinations found in natural antibody repertoires would improve in silico antibody development.213,321 Furthermore, some tools do not allow including essential variables that might change the developability prediction outcome. For example, most of the solubility and aggregation prediction tools that have been developed for proteins, in general, do not take the concentration of the molecule into consideration which is a critical factor for mAbs (Table 1).288,322 Additionally, the current state-of-the-art in silico tools (discussed in this review) are mostly mono-parameter tools, implying the need for setting up a multi-parameter pipeline to compute all the necessary parameters. Moreover, some computational tools developed by pharmaceutical businesses are not made publicly available, which can hinder their implementation and further assessment.243,323 More generally, the lack of a comprehensive atlas mapping different developability parameters to different regions hinders computational antibody design (Figure 4). Finally, finding an equilibrium for all developability parameters in one antibody, and thus achieving the longstanding goal of modular antibody design, has proven to be a challenging task as working on improving one developability measure might result in compromising another (Figure 4). For example, Kuroda and Tsumoto argued that improved antibody stability may be accompanied by increased aggregation.320 One experimental solution that may contribute to improving the overall percentage of candidates with a fitting developability profile prior to computation and optimization is the development of developability-optimized screening libraries.183 An intriguing computational solution to optimizing the number of mAb candidates with respect to multiple design parameters is the combination of ML models trained on data from different experimental campaigns.324
4. Unconstrained parameter-driven in silico antibody sequence synthesis
4.1. Antibody (sequence) generation with deep generative models
The term “deep generative model” describes a set of deep learning-based methods that enable the learning of data distributions and subsequent sampling of new unseen points (Figure 5). Currently, there exist, to our knowledge, eight reports that take advantage of deep generative models for antibody sequence generation with diverse objectives: optimizing binding affinity toward a specific antigen, generating new antibodies that replicate developability parameters of the original distribution, realistic backbone structure sampling, unsupervised identification of antibodies in latent space.25–27,94,325–328 These studies each used one of the three most popular architectures namely Variational Autoencoders (VAE),329 Generative Adversarial Networks (GAN),330 and Autoregressive models (AR) namely long short-term memory recurrent neural network (LSTM-RNN) and transformer (Table 2).
Figure 5.
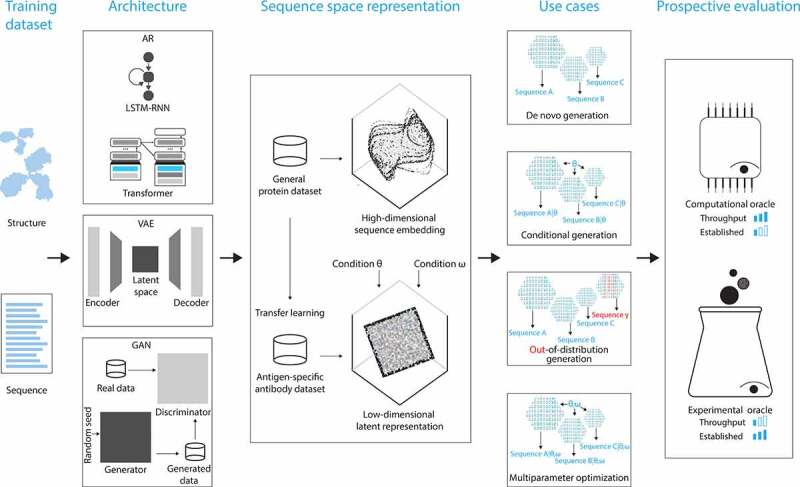
Generative models can be trained on generic or custom-designed datasets to obtain sequence space representation and to generate new sequences for a variety of use cases in antibody design. AR models enable the generation of highly diverse proteins and can be used to obtain meaningful sequence embeddings, circumventing the need for hand-crafted features. VAEs and GANs have been employed in protein generation in a similar manner to generate functionally relevant leads, obtain biologically meaningful latent representations, and condition them on additional features (e.g., solubility). As such, these models can be employed in de novo generation of sequences, conditional, or out-of-distribution generation, as well as optimization of multiple parameters. Evaluating the specificity (or any other design parameter of interest) of the in silico designed antibody sequences requires either computational or experimental oracles. As deep generative models output a large number of sequences, experimental prospective evaluation methods may not possess the time- and cost-efficiency to evaluate these sequences at scale, thus creating considerable demand for in silico oracles (Figure 5). Transfer learning may be leveraged to infer higher-order, functionally specific interactions from a small number of available sequences (low N). Integrating computational and experimental oracles or directly conditioning the generative models on additional features would enable high-yield multiparameter optimization of machine-learning engineered antibody sequences.
Table 2.
| (Dis)Advantages of the three most common generative methods (GAN, VAE, AR) with respect to five properties. Polyspecific training objective indicates if the training objective assumes only one possible target. Supervision indicates whether the model can be trained in a completely unsupervised manner or if the definition of an identity error function is required. Categorical indicates whether the model can deal with categorical data (most common datatype in protein sequence generation problems). The difficulty of training indicates the tendency of the model to experience numerical instabilities during training
| pecific training objective | Approximate training objective | Supervision | Categorical | Difficulty of training | |
|---|---|---|---|---|---|
| GAN | Yes | Direct | Unsupervised | Not out-of-the-box | 3/3 |
| VAE | No | Lower Bound | Unsupervised (supervised Identity function) |
Yes | 2/3 |
| AR | No | Direct | Unsupervised (supervised Identity function) |
Yes | 1/3 |
4.2. GAN-based approaches for antibody design
GANs aim to learn the potential distribution of the actual data by setting up a generator and a discriminator in a zero-sum game. Equilibrium is achieved when the discriminator can no longer discriminate the generator’s outputs from the actual distribution.331 For biological sequences, GANs are usually used to generate sequences with a particular phenotype of interest. GANs have also been used to generate novel samples (out-of-distribution sampling) from existing distributions (Table 2 and Figure 5).332
4.2.1. Demonstrating the capability of GANs to generate feature-controlled antibody sequences
Amimeur et al.25 demonstrated that GANs are a viable option for novel antibody design. They trained a GAN on the sequences of the light and heavy chain variable regions from the Observed Antibody Space (OAS) database.57 Subsequently, they sampled new sequences from the model and showed that the model can generate large and diverse libraries of novel antibodies that mimic the features (CDRH3 length, isoelectric point, representative maximum patch size, and predicted affinity to MHCII) of the antibodies from the OAS. They also demonstrated that they can bias the model to generate feature-controlled antibody sequences with a lower binding affinity toward MHCII or a higher isoelectric point by conditioning the training dataset on the respective variable. The authors experimentally validated their method by expressing newly generated antibody sequences in vivo.
4.2.2. GAN-based approaches for general protein design
Repecka et al.333 trained a GAN architecture with temporal convolutional networks and a self-attention layer, named ProteinGAN, to generate novel malate dehydrogenase (MDH) sequences. Their 20,000 generated sequences had a median sequence identity of 64.6% when compared to the best matching training sample, which was similar to the median of the training data against the validation set (64.9%), indicating that GANs can generate realistic sequences. The resulting sequences were also four times more diverse than the training subset at 75% sequence identity. A sequence with 106 substituted AAs was identified among the functional subset – an example of the exhaustive exploration of the functional sequence space that GANs are capable of.
Additionally, GANs can also be conditioned on a secondary input, which is often a set of categorical labels.334 Combining conditioned models with multiple computational oracles (classifiers for key design parameters) may enable fast multi-parameter/multi-objective optimization (Figure 5). Integrating conditional labels remains a challenging undertaking.335,336 Despite their realistic sequence or image generation, GANs can often suffer from mode collapse, where the generator is stuck in a local minimum of a few valid samples. There are, however, methods to circumvent mode collapse such as implementing a different loss function.337
4.3. VAE-based approaches for antibody design
VAEs provide a unique avenue to interpretable protein design via their latent representation where functionally similar sequences group together due to their conserved residues, and novel proteins can be obtained by decoding points nearby these clusters.338 Thus, VAEs can be used for lowering the dimensionality of a dataset, obtaining biologically meaningful representations and clusters, sampling to generate de novo sequences, and interpolating in the latent space to obtain proteins with the desired function (navigating from one sequence to another in the latent space), all in the same model (Table 2 and Figure 5).
4.3.1. Generative modeling of antibody backbone structure
Eguchi et al.326 used VAEs to generate new backbone structures of antibodies that are filtered for desirable properties by evaluating them with simulators. This resulted in new randomly sampled structures that were different from the training data and constrained optimization with Rosetta339 does not lead to large changes to the overall structure. They also verified generated structures by linearly interpolating the latent representation of existing antibody structures. The authors sampled 5000 antibody structures from the IG-VAE and identified candidates with binding affinity for the SARS-CoV-2 RBD with PatchDock.340
4.3.2. Clustering antibody sequences by applying Gaussian Mixture Models to the latent space of VAEs
Friedensohn et al.26 used a VAE that incorporated Gaussian mixture models in the latent space to identify clusters of potentially antigen-specific antibody sequences, which were obtained from antigen-immunized mice. Then, they sampled from the VAE new antibodies from those putatively antigen-associated clusters. Twelve antibodies from one cluster were recombinantly expressed and all 12 were confirmed to be antigen specific.
4.4. VAE-based approaches for general protein design
An initial study in VAE-generated protein sequences was conducted by Costello and Martin.341 Their architecture (BioSeqVAE) employed residual networks, dilated convolutions, and an autoregressive layer on top of the decoder, resulting in a latent space that captures residue interactions along the entire sequence. Furthermore, they used the latent space to generate proteins with the desired phenotype by training a classifier on the latent representation of a protein dataset with a desirable function, then sampling points from the latent space until they were validated by the classifier. They also showed that multiple phenotypes could be integrated by training other classifiers, further extending the idea of multi-parameter optimization as a key step in highly specific protein generation. Additionally, Gane et al.342 provide a benchmark for protein design on synthetic data that investigates VAEs among other models.
4.5. AR-based approaches for antibody design
Autoregressive models are inherently sequential, thus, they are ideally suited for modeling biological sequences as they decompose into a fixed ordering (one AA after the other). Although they can be powerful estimators for the distribution of interest, the training and generation can be exceedingly slow as they are done in a sequential manner (Table 2).343 A comprehensive benchmark over three different AR-based approaches to mAb generation, conditioned on protein structure can be found in the work of Melnyk et al. where “causal convolutions”, GNNs, and transformer-based generation methods are compared. The authors also provide guidelines as to which of the three AR-based approaches are best suited to specific antibody design tasks (e.g., CDR3 grafting, broad or narrow sequence diversity generation).328
4.5.1. Optimizing the binding affinity of antibody sequences with LSTM-RNNs
Saka etal.325 used LSTM-RNNs to examine the capacity of deep generative models to improve the affinity of kynurenine-binding antibodies. The authors generated a dataset of kynurenine-binding antibodies through two rounds of phage-display panning. An LSTM-RNN was trained on the sequence data of the Fv region of the heavy chain. They found that the predicted likelihood values of the generated sequences correlate well with binding affinity (R2 = 0.52) and the best LSTM-RNN generated sequence yielded over 1800-fold lower dissociation constant over the original kynurenine-binding antibody.
4.5.2. Demonstrating the capability of LSTM-RNNs to learn distributions of CDRH3 sequences with a wide variety of antibody design parameters
We have recently demonstrated the feasibility of in silico antibody design with deep generative methods.27 Briefly, we trained LSTM-RNNs on ground truth synthetic data antibody (CDRH3) sequences where for each CDRH3 sequence, the design parameters (affinity, epitope, developability) were known54 and showed that LSTM-RNN can generate new CDRH3 sequences that match and/or exceed and extend the antibody design parameters of the training dataset, but greatly differ sequence-wise from those sequences contained in the training dataset. Additionally, we showed that pre-training models for transfer learning can improve prediction results. Finally, we validated the antibody-design conclusions reached from ML training on simulated antibody-antigen binding data by training on experimental antibody sequence data and evaluating the generated sequences using an experimentally validated computational oracle published by Mason etal.27,86
4.5.3. AR models for nanobody design
Shin et al.94 demonstrated the utility of causal CNN models to generate new nanobody sequences. This was done by training a causal CNN model to learn a conditional distribution over CDRH3 sequences, given the CDRH1 and CDRH2 sequences, to avoid CDRH3 sequences that are chemically incompatible with the other CDRHs. They also showed that the generated CDRH3s have a similar distribution to the developability parameters hydrophobicity and isoelectric point of the reference CDRH3s. In addition, they evaluated the method by training on a llama dataset344 which contains 1.2 million nanobody sequences from seven different immune repertoires, and evaluated the hydrophobicity and isoelectric point by computational methods.345,346
4.5.4. Improved AR-based models through simultaneous estimation of structure
Jin et al. developed an AR-based model that generates new AAs in a sequence while iteratively refining the sequence’s predicted global structure. Simultaneously, the inferred structure guides subsequent residue choices. The structure is modeled with a graph representation that models the position of the AAs and the angles that define the backbone structure. Edges within the graph are defined through proximity.327
4.6. AR based approaches for general protein design
4.6.1. On the impracticality of simple AR-based models
The capacity of Ar-based models to generate highly diverse sequences was illustrated in an antimicrobial peptide design task via an LSTM-RNN model: out of 2000 generated peptides, 1747 were unique.347 These were further assessed for their antimicrobial activity via a random forest classifier (computational oracle) against both the training dataset and against randomly sampled sequences using the original AA distribution: de novo peptides had a higher probability of being antimicrobial compared to the randomly sampled group and as high as the training ones.
However, AR models are often impractical on multidimensional (vast sequence space), multi-feature data: incorporating other physicochemical, affinity, or structural properties to direct the sequence generation would require pre-trained vector embeddings of protein sequences,210,211 or carefully engineered features (by hand). These embeddings often require a much larger sequence database for training. For example, Alley et al.97 used 24 million UniRef50 sequences to construct their embeddings, which was later used in an in silico optimization framework to improve the fluorescence of a GFP.348
4.6.2. Advanced AR-based approaches for general protein design – sequence embeddings (self-supervised learning)
The above-mentioned problems (lack of a functionally meaningful latent space, which could represent structural/physicochemical information and high-dimensionality of the sequence space) could be addressed with LSTM-RNNs97 or Transformer models.58 These models were trained on large unlabeled sequence databases and can capture the secondary and tertiary structures, AA biochemical properties, homology, and function of a protein in a learned, sequence embedding (self-supervised learning). Moreover, Villegas-Morcillo et al.210 discovered that such embeddings solely obtained from sequence data outperform even well-engineered features on classification tasks, as well as other structure-based protein embeddings via distance maps (as designed by ref. 349). Surprisingly, when combining their sequence embedding with contact maps, their classifier performed similarly (AUC-ROC = 0.76) to when such information was omitted (AUC-ROC = 0.77), indicating that the sequence embedding encompassed enough of the structural information. Sequence embeddings provide, therefore, a biologically interpretable reduction of the vast protein sequence space.
It is of interest to investigate how such sequence embeddings can be used in protein generation. A first insight was recently gained by ref. 348 into how this might be possible for in silico directed protein evolution: first, an LSTM-RNN network was trained on 20 million general protein sequences to obtain a general embedding, which was later fine-tuned on sequences evolutionarily related to the protein to optimize, followed by the sampling of a small number of mutants (low-N engineering), quantifying their specific functional property (e.g., the fluorescence of a GFP) and building a linear regression model (inputs = embedding representation, outputs = quantified property of protein). The starting sequence was mutated, embedded, then fed into the linear regression model – sequences with large enough values can be functionally assessed in vitro.
4.6.3. Transformer networks for general protein design
To tackle the problem of low receptive fields (i.e., the size of the regions in the input that informs the output350) in RNNs, Transformer or general attention models have been increasingly used for various NLP tasks and for protein sequence modeling as well, achieving state-of-the-art results.351 For example, Wu et al.352 trained a 5-layer Transformer network to generate signal peptide sequences, a task where (self)-attention is advantageous to scrutinize the entire sequence of an instance. In total, 25,000 paired proteins without signal peptides were fed into the model along with their respective signal peptides, in an attempt to translate protein sequences to specific peptides. The generated signal peptides had a 73% sequence similarity to their corresponding BLAST result from SwissProt, therefore showing some diversity, yet had poor AUC-ROC values on a signal peptide deep learning classifier (AUC-ROC = 0.59, almost equal to baseline classification).
Recently, the ProGen model353 was able to generate label-conditioned sequences: by training a Transformer network on sequences with a conditioning tag prefix (e.g., organism, function, location, etc.), their model learned conditional probabilities on both the previous residue and the label of interest. Integrating such conditioning tags allowed for protein generation without any starting residue: using the tags Flavoprotein and FMN, they were able to sample a 400 AA protein which matched numerous other similar proteins (oxidoreductases). The work above showed that conditional transformers for text generation can be applied to protein engineering problems. The extent to which sequences generated from conditional labels differ from similar ones in the training data may be used to gauge the potential for out-of-distribution generation in these models. Another example on conditioning transformers for protein design is the work from Ingraham et al, who condition a transformer on folding information by utilizing a graph neural network as representation.354
4.7. Remaining challenges in generative antibody design
Limited application of generative ML approaches to antibody design: The current literature on generative modeling of antibodies already incorporates many approaches currently used in generative protein design, such as sampling of 3D backbone structure of antibodies for finding new antibodies with relevant properties,326 sampling of antibody sequences for optimization,325 interpolation of a latent space for antibody property design,326 and generation of novel and highly diverse antibodies that faithfully reproduce developability parameter distributions. However, there are still several approaches that have not been explored in the antibody generation domain, such as using learned amino-acid vector representations,210,211 combining adversarial training with modern autoregressive models (e.g., transformers), and conditioning models directly on developability parameters.353
High-throughput prospective evaluation: In a typical ML study, a dataset is split into training, validation, and test sets to allow for the model to be retrospectively evaluated with the validation and test sets upon the completion of the training. In such a setup the data comes in the format of input-output pair, thus during evaluation, the correct label for a sample either in the validation or the test set is known a priori. In generative learning, however, the label (i.e., binding affinity, developability, and plasma half-life or a subset thereof) is not known a priori as the model generates new sequences that may or may not overlap with the training data. Thus, generative learning necessitates external (computational or experimental) validators (oracles) to evaluate its output as the evaluation process is performed post-generation (prospective evaluation; Figure 5).
An experimental prospective evaluation workflow usually involves the expression and testing of 101–102 binders.26 A computational validation workflow might involve, for example, the sequence-based modeling of the antibody structure with tools such as ABodyBuilder,132 followed by molecular docking,66,355,356 or MD simulation (all-atom simulation; computationally expensive357) to validate whether the generated sequence overlaps with the desired antibody-antigen binding pose. However, given that de novo docking approaches remain at low accuracy, such computational validation workflows require further refinement. Furthermore, so far, there exist only a few ML-based experimentally validated computational oracles published.86 Recent studies27,54 led by us were among the first efforts to tackle the high-throughput prospective evaluation problem in antibody generative learning by leveraging a high-throughput oracle in the form of virtual (coarse-grained) docking albeit at a reduced lattice resolution.
Out-of-distribution generation (functional novelty): A common challenge in deep generative learning is that the model tends to reproduce the training data extremely faithfully, a phenomenon known as the copy problem.358 Such a model remains useful when the objective of the study is to generate new samples that are very similar to the training data. In antibody design, however, sequence similarity may not reflect binding behavior faithfully. It has been shown, for example in HER2 binding antibodies, that two very similar sequences (Levenshtein distance < 2) had opposing binding behavior.86 Secondly, it is often desirable to discover new modes of binding (novel target epitopes) when designing antibodies for a target of interest, these functionally novel antibodies represent out-of-distribution samples as the novel epitopes were never learned from the training data. A naïve strategy to obtain out-of-distribution samples is to couple a simple architecture with unconstrained generation, i.e., the simple architecture reduces the risk of overfitting the training data (reduces the risk of copy problem) and unconstrained generation allows the model to explore a larger sampling space. Indeed, we employed such a strategy to obtain novel epitopes and a diverse set of developability parameter combinations rather successfully.27 A more sophisticated strategy may include conditioning the model in such a way that the output is biased toward out-of-distribution samples.359
All-round optimization – conditioning simultaneously on multiple developability parameters in a single model: In the two pioneering studies from Amimeur et al.25,86 and Mason et al.,25,86 the developability optimization step is a separate entity. Next-generation antibody design tools must be developed with all-round optimization in mind where multiple developability parameters and binding affinity are simultaneously optimized. For instance, models such as conditional VAEs have been deployed to generate drug-like molecules where five target properties were simultaneously optimized during training.360 Another challenge is that different developability parameters localize in different regions of the antibody (Figure 4) whereas many studies such as ours and Mason et al.25,86 conveniently focus on the most important segment for antigen engagement, the CDRH3. In summary, a holistic all-round antibody generator represents a crucial component for the on-demand generation of fit-for-purpose mAbs.
5. Concluding remarks
In this review, we outlined strategies toward ML-based mAb design and the associated necessary computational and experimental steps required. We argue that a resolution to the in silico antibody design problem lies in the development of novel experimental and computational technologies for large-scale generation combined with a screening of antibody, antigen, and antibody-antigen parameters. Furthermore, self-supervised learning may provide a means to leverage large amounts of unlabeled data to boost in silico protein design efficiency.197 As a bridge between experimental and simulated data, more investment is needed in the development of data augmentation algorithms that can expand training dataset sizes. Correspondingly, powerful simulation frameworks to generate ground-truth synthetic data are mission critical for testing the accuracy and performance of novel in silico antibody specificity prediction and generation approaches. Furthermore, for maximum generalizability, it will be paramount to learn from and combine in vitro and in vivo data since these datasets underlie different generative distributions (e.g., in vitro antibody libraries may display broader diversity that have not undergone biology self-reactivity-driven selection).3,184 Finally, we believe the antibody design field requires closer collaboration with ML experts. As was witnessed in the case of protein structure prediction, the infusion of domain-specific ML knowledge can propel an entire field substantially forward.361,362
Funding Statement
We acknowledge generous support by The Leona M. and Harry B. Helmsley Charitable Trust (#2019PG-T1D011, to VG), UiO World-Leading Research Community (to VG), UiO:LifeScience Convergence Environment Immunolingo (to VG), EU Horizon 2020 iReceptorplus (#825821) (to VG), a Research Council of Norway FRIPRO project (#300740, to VG), a Research Council of Norway IKTPLUSS project (#311341, to VG), a Norwegian Cancer Society Grant (#215817, to VG), Research Council of Norway (#287927, to JTA and KFK) and a grant from the South-Eastern Norway Regional Health Authority (#2021069, to JTA and KFK).
Abbreviations
3DZD: 3D Zernike Descriptor; AA: Amino Acid; AB‐Bind: Antibody-Bind dataset; ABCD: AntiBodies Chemically Defined Database; ADA: Anti-Drug Antibody; ADCC: Antibody-Dependent Cell-mediated Cytotoxicity; ADCP: Antibody-Dependent Cellular Phagocytosis; AgAbDb: Antigen–Antibody Interaction Database; AlphaScreen: Amplified luminescence homogeneous assay; ANN: Artificial Neural Network; anti-TNF-α: anti-Tumor Necrosis Factor-α; APC: Antigen Presenting Cell; APR: Aggregation Prone Region; AR: AutoRegressive model; AUC: Area Under the Curve; AUC-ROC: Area Under the Receiver Operating Curve; BCR: B-cell Receptor; BERT: Bidirectional Encoder Representations from Transformers; BLI: BioLayer Interferometry; bNAber: broadly Neutralizing Antibodies electronic resource; cAb-Rep: Curated human B cell immunoglobulin sequence repertoires; CDC: Complement-Dependent Cytotoxicity; CDR: Complementarity-Determining Region; CDR3H: Complementarity-Determining Region 3 on the heavy chain; CNN: Convolutional Neural Network; CoV-AbDab: Coronavirus Antibody Database; DI: Developability Index; EL: Eluted Ligand; ELISA: Enzyme-Linked Immunosorbent assay; EPMP: Epitope-Paratope Message Passing; ERT: Extremely Randomized Tree; Fab: Fragment antigen-binding; Fc: Fragment crystallized; FcRn: Neonatal Fc receptor; FR: Framework Region; GAN: Generative Adversarial Network; GB: Gradient Boosting; GBT: Gradient-Boosting Trees; GDL: Geometric Deep Learning; HER-2: Human Epidermal growth factor Receptor 2; HERA: Human Endothelial cell-based Recycling Assay; HLA: Human Leukocyte Antigen; HVI: High Viscosity Index; IEDB: Immune Epitope Database; Ig: Immunoglobulin; IMGT: International ImMunoGeneTics information system; ITC: Isothermal Titration Calorimetry; kD: diffusion interaction parameter; L-o-L: library-on-Library; LASSO: Least-Absolute Shrinkage and Selection Operator; LIBRA-seq: Linking B-cell Receptor to Antigen specificity through sequencing; LSP: Local Surface Patches; LSTM-RNN: Long Short-Term Memory-Recurrent Neural Network; mAb: monoclonal Antibody; MaSIF: Molecular Surface Interaction Fingerprints; MD: Molecular Dynamics; MERS-CoV: Middle East Respiratory Syndrome Coronavirus; MDH: Malate Dehydrogenase; MHC II: Major Histocompatibility Molecule II; ML: Machine Learning; MS: Mass Spectrometry; NLP: Natural Language Processing; NN: Neural Network; OAS: Observed Antibody Space; PECAN: Paratope and Epitope prediction with graph Convolution Attention Network; pH: Potential hydrogen; pI: Isoelectric point; PK: PharmacokineticsPPI: Protein–Protein Interaction; PROXiMATE: PROtein–protein compleX MutAtion ThErmodynamics Database; PSR: Poly-Specificity Reagent; PSSM: Position-Specific Scoring Matrix; Q-value: Fraction of native contacts; RBD: Receptor-Binding Domain; RNN: Recurrent Neural Network; SAbDab: Structural Antibody Database; SAP: Spatial Aggregation Propensity; SARS-CoV-1: Severe Acute Respiratory Syndrome CoronaVirus 1; SARS-CoV-2: Severe Acute Respiratory Syndrome CoronaVirus 2; SCM: Spatial Charge Map; sdAb-DB: Single-Domain Antibody Database; SiPMAB: Single-Point Mutant Antibody Binding; SKEMPI: Structural Kinetic and Energetic database of Mutant Protein Interactions; SPR: Surface Plasmon Resonance; SVM: Support-Vector Machines; SWI: Solubility-Weighted Index; Tagg: Aggregation onset temperature; TAP: Therapeutic Antibody Profiler; Thera-SAbDab: Therapeutic Structural Antibody Database; Tm: Melting temperature; TopCNN: Topology-based Convolutional Neural Network; TopGBT: Topology-based Gradient-Boosting Trees; TopNetTree: Topology-based Network Tree; VAE: Variational Autoencoder; VH: Variable Heavy; VL: Variable Light; WHO: World Health Organization; ΔG: Gibbs free energy of binding; ΔΔG: The change in Gibbs free energy of binding
Disclosure statement
V.G. declares advisory board positions in aiNET GmbH and Enpicom B.V.G. is a consultant for Roche/Genentech.
Data and code availability
Datasets, codes, and figures are available at https://github.com/csi-greifflab/manuscript_progress_challenges_on-demand_antibody. Plots were generated in Matplotlib (version 3.3.4)363 and R (version 3.6.1).364
Author contributions
Conceptualization: V.G. and R.A.; Writing - Original Draft: R.A., P.A.R., E.S., H.B., P.R., M.H.V., J.T.A., K.F.K., B.B.M., T.Z., J.G.M., R.F., T.S.C., V.G.: Writing - Review & Editing: R.A., P.A.R., E.S., H.B., P.R., M.H.V., K.F.K., B.B.M., T.Z., J.G.M., R.F., T.S.C., F.L.J., J.T.A., V.G.; Visualization: V.G., E.S., R.A., H.B., T.S.C; Supervision: V.G., P.A.R., and R.A. The authorship order was determined alphabetically within each tier of contribution (H and L). Tier H: R.A., H.B., P.R., P.A.R., and E.S. Tier L: T.S.C., K.F.K., R.F., B.B.M., M.H.V., and T.Z. Within each tier authors are free to list their names first in their C.V. All authors contributed and approved the submitted version of the article.
References
- 1.Urquhart L. Top product forecasts for 2021 [Internet]. Nature Reviews Drug Discovery. 2021;20:10–35. doi: 10.1038/d41573-020-00219-5. [DOI] [PubMed] [Google Scholar]
- 2.Lu R-M, Hwang Y-C, Liu I-J, Lee -C-C, Tsai H-Z, Li H-J, Wu H-C.. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27(1):1–30. doi: 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laustsen AH, Greiff V, Karatt-Vellatt A, Muyldermans S, Jenkins TP. Animal Immunization, in Vitro Display Technologies, and Machine Learning for Antibody Discovery [Internet]. Trends Biotechnol. 2021; 39:1263–73. doi: 10.1016/j.tibtech.2021.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Narayanan H, Dingfelder F, Butté A, Lorenzen N, Sokolov M, Arosio P. Machine Learning for Biologics: Opportunities for Protein Engineering, Developability, and Formulation [Internet]. Trends Pharmacol Sci. 2021;42:151–65. doi: 10.1016/j.tips.2020.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Norman RA, Ambrosetti F, Bonvin AMJJ, Colwell LJ, Kelm S, Kumar S, Krawczyk K. Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief Bioinform. 2020;21:1549–67. doi: 10.1093/bib/bbz095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leman JK, Weitzner BD, Lewis SM, Adolf-Bryfogle J, Alam N, Alford RF, Aprahamian M, Baker D, Barlow KA, Barth P, et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat Methods. 2020;17:665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown AJ, Snapkov I, Akbar R, Pavlović M, Miho E, Sandve GK, Greiff V. Augmenting adaptive immunity: progress and challenges in the quantitative engineering and analysis of adaptive immune receptor repertoires. Mol Syst Des Eng. 2019;4:701–36. [Google Scholar]
- 8.Sormanni P, Aprile FA, Vendruscolo M. Third generation antibody discovery methods in silico rational design. Chem Soc Rev. 2018;47(24):9137–57. doi: 10.1039/C8CS00523K. [DOI] [PubMed] [Google Scholar]
- 9.Riahi S, Lee JH, Wei S, Cost R, Masiero A, Prades C, Olfati-Saber R, Wendt M, Park A, Qiu Y, et al. Application of an integrated computational antibody engineering platform to design SARS-CoV-2 neutralizers [Internet]. bioRxiv2021 [cited 2021 Aug 12]; 2021.03.23.436613. https://www.biorxiv.org/content/ 10.1101/2021.03.23.436613v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang T, Chen H, Yuan J, Jiang C, Hao Y, Wang Y, Feng Z, Xie X-Q. IsAb: a computational protocol for antibody design. Brief Bioinform [Internet]. 2021;22. doi: 10.1093/bib/bbab143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernandez I, Bott SW, Patel AS, Wolf CG, Hospodar AR, Sampathkumar S, Shrank WH. Pricing of monoclonal antibody therapies: higher if used for cancer? Am J Manag Care. 2018;24:109–12. [PubMed] [Google Scholar]
- 12.Graves J, Byerly J, Priego E, Makkapati N, Parish SV, Medellin B, Berrondo M. A review of deep learning methods for antibodies. Antibodies (Basel) [Internet]. 2020;9(2). 10.3390/antib9020012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bengio Y. Learning Deep Architectures for AI. Now Publishers Inc; 2009. [Google Scholar]
- 14.Glorot X, Bengio Y. Understanding the difficulty of training deep feedforward neural networks. In: Teh YW, Titterington M, editors. Proceedings of the Thirteenth International Conference on Artificial Intelligence and Statistics. Chia Laguna Resort, Sardinia, Italy: PMLR; 2010. 249–56. [Google Scholar]
- 15.Greiff V, Yaari G, Cowell LG. Mining adaptive immune receptor repertoires for biological and clinical information using machine learning. Current Opinion in Systems Biology. 2020;24:109–19. doi: 10.1016/j.coisb.2020.10.010. [DOI] [Google Scholar]
- 16.Pertseva M, Gao B, Neumeier D, Yermanos A, Reddy ST. Applications of Machine and Deep Learning in Adaptive Immunity. [cited 2021 Apr 26]; https://www.annualreviews.org/doi/abs/ 10.1146/annurev-chembioeng-101420-125021 [DOI] [PubMed]
- 17.Jebara T. Machine Learning: Discriminative and Generative. Springer Science & Business Media; 2012. [Google Scholar]
- 18.Ching T, Himmelstein DS, Beaulieu-Jones BK, Kalinin AA, Do BT, Way GP, Ferrero E, Agapow P-M, Zietz M, Hoffman MM, et al. Opportunities and obstacles for deep learning in biology and medicine. J R Soc Interface [Internet]. 2018; 15. 10.1098/rsif.2017.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ovchinnikov S, Huang P-S. Structure-based protein design with deep learning. Curr Opin Chem Biol. 2021;65:136–44. doi: 10.1016/j.cbpa.2021.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jespersen MC, Mahajan S, Peters B, Nielsen M, Marcatili P. Antibody Specific B-Cell Epitope Predictions: Leveraging Information From Antibody-Antigen Protein Complexes. Front Immunol. 2019;10:298. doi: 10.3389/fimmu.2019.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deac A, Veličković P, Sormanni SP. Attentive Cross-Modal Paratope Prediction. J Comput Biol. 2019;26(6):536–45. doi: 10.1089/cmb.2018.0175. [DOI] [PubMed] [Google Scholar]
- 22.Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. MAbs. 2015;7:483–93. doi: 10.1080/19420862.2015.1016696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the “high-hanging fruit.”. Nat Rev Drug Discov. 2018;17:197–223. doi: 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- 24.Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci U S A. 2015;112:5997–6002. doi: 10.1073/pnas.1408766112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amimeur T, Shaver JM, Ketchem RR, Taylor JA. Designing feature-controlled humanoid antibody discovery libraries using generative adversarial networks. bioRxiv [Internet]. 2020; https://www.biorxiv.org/content/ 10.1101/2020.04.12.024844v2.abstract [DOI] [Google Scholar]
- 26.Friedensohn S, Neumeier D, Khan TA, Csepregi L. Convergent selection in antibody repertoires is revealed by deep learning. bioRxiv [Internet]. 2020; https://www.biorxiv.org/content/ 10.1101/2020.02.25.965673v1.abstract [DOI] [Google Scholar]
- 27.Akbar R, Robert PA, Weber CR, Widrich M, Frank R, Pavlović M, Scheffer L, Chernigovskaya M, Snapkov I, Slabodkin A, et al. In silico proof of principle of machine learning-based antibody design at unconstrained scale [Internet]. bioRxiv2021 [cited 2021 Jul 11]; 2021.07.08.451480. https://www.biorxiv.org/content/ 10.1101/2021.07.08.451480v1.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greiff V, Menzel U, Miho E, Weber C, Riedel R, Cook S, Valai A, Lopes T, Radbruch A, Winkler TH, et al. Systems Analysis Reveals High Genetic and Antigen-Driven Predetermination of Antibody Repertoires throughout B Cell Development. Cell Rep. 2017;19:1467–78. doi: 10.1016/j.celrep.2017.04.054. [DOI] [PubMed] [Google Scholar]
- 29.Elhanati Y, Sethna Z, Marcou Q, Jr CCG, Mora T, Walczak AM. Inferring processes underlying B-cell repertoire diversity. Philos Trans R Soc Lond B Biol Sci [Internet]. 2015;370. 10.1098/rstb.2014.0243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tress ML, Abascal F, Valencia A. Alternative Splicing May Not Be the Key to Proteome Complexity. Trends Biochem Sci. 2017;42:98–110. doi: 10.1016/j.tibs.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galson JD, Pollard AJ, Trück J, Kelly DF. Studying the antibody repertoire after vaccination: practical applications. Trends Immunol. 2014;35(7):319–31. doi: 10.1016/j.it.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 32.Raybould MIJ, Rees AR, Deane CM. Current strategies for detecting functional convergence across B-cell receptor repertoires. MAbs. 2021;13:1996732. doi: 10.1080/19420862.2021.1996732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang F, Nielsen SCA, Hoh RA, Röltgen K, Wirz OF, Haraguchi E, Jean GH, Lee J-Y, Pham TD, Jackson KJL, et al. Shared B cell memory to coronaviruses and other pathogens varies in human age groups and tissues. Science. 2021;372:738–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trück J, Ramasamy MN, Galson JD, Rance R, Parkhill J, Lunter G, Pollard AJ, Kelly DF. Identification of antigen-specific B cell receptor sequences using public repertoire analysis. J Immunol. 2015;194(1):252–61. doi: 10.4049/jimmunol.1401405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanyavuz A, Marey-Jarossay A, Lacroix-Desmazes S, Dimitrov JD. Breaking the law: unconventional strategies for antibody diversification. Nat Rev Immunol. 2019;19(6):355–68. doi: 10.1038/s41577-019-0126-7. [DOI] [PubMed] [Google Scholar]
- 36.Pieper K, Tan J, Piccoli L, Foglierini M, Barbieri S, Chen Y, Silacci-Fregni C, Wolf T, Jarrossay D, Anderle M, et al. Public antibodies to malaria antigens generated by two LAIR1 insertion modalities. Nature. 2017;548:597–601. doi: 10.1038/nature23670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan J, Pieper K, Piccoli L, Abdi A, Perez MF, Geiger R, Tully CM, Jarrossay D, Maina Ndungu F, Wambua J, et al. A LAIR1 insertion generates broadly reactive antibodies against malaria variant antigens. Nature. 2016;529:105–09. doi: 10.1038/nature16450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Werner RG, Kopp K, Schlueter M. Glycosylation of therapeutic proteins in different production systems. Acta Paediatr. 2007;96:17–22. doi: 10.1111/j.1651-2227.2007.00199.x. [DOI] [PubMed] [Google Scholar]
- 39.Jennewein MF, Alter G. The Immunoregulatory Roles of Antibody Glycosylation. Trends Immunol. 2017;38:358–72. doi: 10.1016/j.it.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 40.Choe H, Li W, Wright PL, Vasilieva N, Venturi M, Huang -C-C, Grundner C, Dorfman T, Zwick MB, Wang L, et al. Tyrosine sulfation of human antibodies contributes to recognition of the CCR5 binding region of HIV-1 gp120. Cell. 2003;114:161–70. doi: 10.1016/S0092-8674(03)00508-7. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, Doud E, Stone T, Xin L, Hong W, Li Y. Rapid global characterization of immunoglobulin G1 following oxidative stress. MAbs. 2019;11:1089–100. doi: 10.1080/19420862.2019.1625676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schlothauer T, Rueger P, Stracke JO, Hertenberger H, Fingas F, Kling L, Emrich T, Drabner G, Seeber S, Auer J, et al. Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. MAbs. 2013;5:576–86. doi: 10.4161/mabs.24981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sela-Culang I, Kunik V, Ofran Y. The structural basis of antibody-antigen recognition. Front Immunol. 2013;4:302. doi: 10.3389/fimmu.2013.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu JL, Davis MM. Diversity in the CDR3 Region of VH Is Sufficient for Most Antibody Specificities. Immunity. 2000;13:37–45. doi: 10.1016/S1074-7613(00)00006-6. [DOI] [PubMed] [Google Scholar]
- 45.Akbar R, Robert PA, Pavlović M, Jeliazkov JR, Snapkov I, Slabodkin A, Weber CR, Scheffer L, Miho E, Haff IH, et al. A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. Cell Rep. 2021;34:108856. doi: 10.1016/j.celrep.2021.108856. [DOI] [PubMed] [Google Scholar]
- 46.Wucherpfennig KW, Allen PM, Celada F, Cohen IR, Boer RD, Garcia KC, Goldstein B, Greenspan R, Hafler D, Hodgkin P, et al. Polyspecificity of T cell and B cell receptor recognition. Semin Immunol. 2007;19:216–24. doi: 10.1016/j.smim.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barlow DJ, Edwards MS, Thornton JM. Continuous and discontinuous protein antigenic determinants. Nature. 1986;322:747–48. doi: 10.1038/322747a0. [DOI] [PubMed] [Google Scholar]
- 48.Kringelum JV, Nielsen M, Padkjær SB, Lund O. Structural analysis of B-cell epitopes in antibody: proteincomplexes. Mol Immunol. 2013;53:24–34. doi: 10.1016/j.molimm.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guest JD, Vreven T, Zhou J, Moal I, Jeliazkov JR, Gray JJ, Weng Z, Pierce BG. An expanded benchmark for antibody-antigen docking and affinity prediction reveals insights into antibody recognition determinants. Structure [Internet]. 2021;doi: 10.1016/j.str.2021.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boughter CT, Borowska MT, Guthmiller JJ, Bendelac A, Wilson PC, Roux B, Adams EJ. Biochemical patterns of antibody polyreactivity revealed through a bioinformatics-based analysis of CDR loops. Elife [Internet]. 2020; 9. Available from. ;: 10.7554/eLife.61393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernández-Quintero ML, Loeffler JR, Kraml J, Kahler U, Kamenik AS, Liedl KR. Characterizing the Diversity of the CDR-H3 Loop Conformational Ensembles in Relationship to Antibody Binding Properties. Front Immunol. 2018;9:3065. doi: 10.3389/fimmu.2018.03065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114:944–49. doi: 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corrie BD, Marthandan N, Zimonja B, Jaglale J, Zhou Y, Barr E, Knoetze N, Breden FMW, Christley S, Scott JK, et al. iReceptor: A platform for querying and analyzing antibody/B-cell and T-cell receptor repertoire data across federated repositories. Immunol Rev. 2018;284:24–41. doi: 10.1111/imr.12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robert PA, Akbar R, Frank R, Pavlović M, Widrich M, Snapkov I, Chernigovskaya M, Scheffer L, Slabodkin A, Mehta BB, et al. One billion synthetic 3D-antibody-antigen complexes enable unconstrained machine-learning formalized investigation of antibody specificity prediction [Internet]. bioRxiv2021 [cited 2021 Jul 22]; 2021.07.06.451258. https://www.biorxiv.org/content/ 10.1101/2021.07.06.451258v2 [DOI] [Google Scholar]
- 55.Ferdous S, Martin ACR. AbDb: antibody structure database—a database of PDB-derived antibody structures. Database [Internet]. [cited 2021 Apr 20]; 2018. https://academic.oup.com/database/article-abstract/doi/ 10.1093/database/bay040/4989324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berman HM, Westbrook J, Feng Z, Westbrook J, Feng Z, Feng Z. The protein data bank. Nucleic acids [Internet]. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kovaltsuk A, Leem J, Kelm S, Snowden J, Deane CM, Krawczyk K. Observed antibody space: a resource for data mining next-generation sequencing of antibody repertoires. J Immunol. 2018;201:2502–09. doi: 10.4049/jimmunol.1800708. [DOI] [PubMed] [Google Scholar]
- 58.Rives A, Meier J, Sercu T, Goyal S, Lin Z, Liu J, Guo D, Ott M, Lawrence Zitnick C, Ma J, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci U S A [Internet]. 2021. [cited 2021 Jul 20];118. https://www.pnas.org/content/118/15/e2016239118.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruffolo JA, Sulam J, Gray JJ. Antibody structure prediction using interpretable deep learning [Internet]. bioRxiv2021 [cited 2021 Jul 20]; 2021.05.27.445982. https://www.biorxiv.org/content/ 10.1101/2021.05.27.445982v3.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leem J, Dunbar J, Georges G, Shi J, Deane CM. ABodyBuilder: automated antibody structure prediction with data-driven accuracy estimation. MAbs. 2016;8:1259–68. doi: 10.1080/19420862.2016.1205773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schritt D, Li S, Rozewicki J, Katoh K, Yamashita K, Volkmuth W, Cavet G, Standley DM. Repertoire Builder: high-throughput structural modeling of B and T cell receptors. Molecular Systems Design & Engineering. 2019;4:761–68. doi: 10.1039/C9ME00020H. [DOI] [Google Scholar]
- 62.Lapidoth G, Parker J, Prilusky J, Fleishman SJ, Valencia A. AbPredict 2: a server for accurate and unstrained structure prediction of antibody variable domains. Bioinformatics. 2019;35:1591–93. doi: 10.1093/bioinformatics/bty822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kovaltsuk A, Raybould MIJ, Wong WK, Marks C, Kelm S, Snowden J, Trück J, Deane CM, Ofran Y. Structural diversity of B-cell receptor repertoires along the B-cell differentiation axis in humans and mice. PLoS Comput Biol. 2020;16:e1007636. doi: 10.1371/journal.pcbi.1007636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Glanville J, Ives S, J-p B, Pettus C, Peñate KR, Hovde R, Bayless N, Shanghavi D, Glanville K, Calgua E, et al. A general solution to broad-spectrum vaccine design for rapidly mutating viruses. 2020; https://www.researchsquare.com/article/rs-100459/latest.pdf
- 65.Ambrosetti F, Jiménez-García B, Roel-Touris J, Bonvin AMJJ. Modeling antibody-antigen complexes by information-driven docking. Structure. 2020;28(1):119–29.e2. doi: 10.1016/j.str.2019.10.011. [DOI] [PubMed] [Google Scholar]
- 66.Jeliazkov JR, Frick R, Zhou J, Gray JJ. Robustification of rosettaantibody and rosetta snugdock. PLoS One. 2021;16(3):e0234282. doi: 10.1371/journal.pone.0234282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hua CK, Gacerez AT, Sentman CL, Ackerman ME, Choi Y, Bailey-Kellogg C. Computationally-driven identification of antibody epitopes. Elife [Internet]. 2017; 6. 10.7554/eLife.29023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Waibl F, Fernández-Quintero ML, Kamenik AS, Kraml J, Hofer F, Kettenberger H, Georges G, Liedl KR. Conformational ensembles of antibodies determine their hydrophobicity. Biophys J. 2021;120(1):143–57. doi: 10.1016/j.bpj.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fernández-Quintero M, Kroell K, Bacher L, Loeffler J, Quoika PK, Georges G, Bujotzek A, Kettenberger H, Liedl KR. Germline-dependent antibody paratope states and pairing specific VH-VL interface dynamics. Front Immunol. 2021;12:2741. doi: 10.3389/fimmu.2021.675655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sirin S, Apgar JR, Bennett EM, Keating AE. AB-Bind: Antibody binding mutational database for computational affinity predictions: antibody-antigen affinity database and computational benchmarks. Protein Sci. 2016;25:393–409. doi: 10.1002/pro.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lima WC, Gasteiger E, Marcatili P, Duek P, Bairoch A, The CP. ABCD database: a repository for chemically defined antibodies. Nucleic Acids Res. 2020;48:D261–4. doi: 10.1093/nar/gkz714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swindells MB, Porter CT, Couch M, Hurst J, Abhinandan KR, Nielsen JH, Macindoe G, Hetherington J, Martin ACR. abYsis: Integrated Antibody Sequence and Structure-Management, Analysis, and Prediction. J Mol Biol. 2017;429:356–64. doi: 10.1016/j.jmb.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 73.Kulkarni-Kale U, Raskar-Renuse S, Natekar-Kalantre G, Saxena SA. Antigen-antibody interaction database (AgAbDb): a compendium of antigen-antibody interactions. Methods Mol Biol. 2014;1184:149–64. [DOI] [PubMed] [Google Scholar]
- 74.Eroshkin AM, LeBlanc A, Weekes D, Post K, Li Z, Rajput A, Butera ST, Burton DR, Godzik A. bNAber: database of broadly neutralizing HIV antibodies. Nucleic Acids Res. 2014;42:D1133–9. doi: 10.1093/nar/gkt1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guo Y, Chen K, Kwong PD, Shapiro L, Sheng Z. cAb-Rep: A Database of Curated Antibody Repertoires for Exploring Antibody Diversity and Predicting Antibody Prevalence. Front Immunol. 2019;10:2365. doi: 10.3389/fimmu.2019.02365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raybould MIJ, Kovaltsuk A, Marks C, Deane CM, Wren J. CoV-AbDab: the coronavirus antibody database. Bioinformatics. 2021;37:734–35. doi: 10.1093/bioinformatics/btaa739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mahajan S, Vita R, Shackelford D, Lane J, Schulten V, Zarebski L, Jespersen MC, Marcatili P, Nielsen M, Sette A, et al. Epitope Specific Antibodies and T Cell Receptors in the Immune Epitope Database. Front Immunol. 2018;9:2688. doi: 10.3389/fimmu.2018.02688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lefranc M-P, Ehrenmann F, Kossida S, Giudicelli V, Duroux P. Use of IMGT® Databases and Tools for Antibody Engineering and Humanization. Methods Mol Biol. 2018;1827:35–69. [DOI] [PubMed] [Google Scholar]
- 79.Jemimah S, Yugandhar K, Michael Gromiha M, Valencia A. PROXiMATE: a database of mutant protein–protein complex thermodynamics and kinetics. Bioinformatics. 2017;33(17):2787–88. doi: 10.1093/bioinformatics/btx312. [DOI] [PubMed] [Google Scholar]
- 80.Dunbar J, Krawczyk K, Leem J, Baker T, Fuchs A, Georges G, Shi J, Deane CM. SAbDab: the structural antibody database. Nucleic Acids Res. 2014;42(D1):D1140–6. doi: 10.1093/nar/gkt1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang R, Fang X, Lu Y, Yang C-Y C-Y, Wang S. The PDBbind Database: methodologies and Updates. J Med Chem. 2005;48(12):4111–19. doi: 10.1021/jm048957q. [DOI] [PubMed] [Google Scholar]
- 82.Wilton EE, Opyr MP, Kailasam S, Kothe RF, Wieden H-J. sdAb-DB: The Single Domain Antibody Database. ACS Synthetic Biology. 2018;7(11):2480–84. doi: 10.1021/acssynbio.8b00407. [DOI] [PubMed] [Google Scholar]
- 83.Jankauskaite J, Jiménez-García B, Dapkunas J, Fernández-Recio J, Moal IH, Xenarios I. SKEMPI 2.0: an updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics. 2019;35(3):462–69. doi: 10.1093/bioinformatics/bty635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mir S, Alhroub Y, Anyango S, Armstrong DR, Berrisford JM, Clark AR, Conroy MJ, Dana JM, Deshpande M, Gupta D, et al. PDBe: towards reusable data delivery infrastructure at protein data bank in Europe. Nucleic Acids Res. 2018;46(D1):D486–92. doi: 10.1093/nar/gkx1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raybould MIJ, Marks C, Lewis AP, Shi J, Bujotzek A, Taddese B, Deane CM. Thera-SAbDab: the therapeutic structural antibody database. Nucleic Acids Res. 2020;48(D1):D383–8. doi: 10.1093/nar/gkz827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mason DM, Friedensohn S, Weber CR, Jordi C, Wagner B, Meng SM, Ehling RA, Bonati L, Dahinden J, Gainza P, et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. [Internet]. Nature Biomedical Engineering2021; 10.1038/s41551-021-00699-9. [DOI] [PubMed] [Google Scholar]
- 87.Karimi M, Wu D, Wang Z, Shen Y, Valencia A. DeepAffinity: interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks. Bioinformatics. 2019;35(18):3329–38. doi: 10.1093/bioinformatics/btz111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sundararajan M, Taly A, Yan Q. Axiomatic attribution for deep networks [Internet]. arXiv [cs.LG]2017. http://arxiv.org/abs/1703.01365 [Google Scholar]
- 89.Adebayo J, Gilmer J, Muelly M, Goodfellow I, Hardt MKim B. Sanity Checks for Saliency Map [Internet]. arXiv [cs.CV]2018. http://arxiv.org/abs/1810.03292 [Google Scholar]
- 90.Abdar M, Pourpanah F, Hussain S, Rezazadegan D, Liu L, Ghavamzadeh M, Fieguth P, Cao X, Khosravi A, Rajendra Acharya U, et al. A Review of Uncertainty Quantification in Deep Learning: Techniques, Applications and Challenges. arxiv [Internet]. 2021;https://arxiv.org/abs/2011.06225 [Google Scholar]
- 91.Josse J, Prost N, Scornet E, Varoquaux G. On the consistency of supervised learning with missing values. 2019. [cited 2021 Aug 6]; Available from:
- 92.O’Leary DS. Medicine’s metaphysical morass: how confusion about dualism threatens public health. Synthese. 2020;1–32. doi: 10.1007/s11229-020-02869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Madani A, Krause B, Greene ER, Subramanian S, Mohr BP, Holton JM, Olmos JL, Xiong C, Sun ZZ, Socher R, et al. Deep neural language modeling enables functional protein generation across families. bioRxiv. 2021 07 18.452833. [Google Scholar]
- 94.Shin J-E, Riesselman AJ, Kollasch AW, McMahon C, Simon E, Sander C, Manglik A, Kruse AC, Marks DS. Protein design and variant prediction using autoregressive generative models. Nat Commun. 2021;12(1):2403. doi: 10.1038/s41467-021-22732-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brandes N, Ofer D, Peleg Y, Rappoport N, ProteinBERT: LM. A universal deep-learning model of protein sequence and function. bioRxiv. 2021 05 24.445464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Y, You Z-H, Yang S, Li X, Jiang T-H ZX, High Efficient A. Biological Language Model for Predicting Protein–Protein Interactions. Cells. 2019/2(8):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alley EC, Khimulya G, Biswas S, AlQuraishi M, Church GM. Unified rational protein engineering with sequence-based deep representation learning. Nat Methods. 2019;16(12):1315–22. doi: 10.1038/s41592-019-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ostrovsky-Berman M, Frankel B, Polak P, Yaari G. Immune2vec: Embedding B/T cell receptor sequences in N using natural language processing. Front Immunol [Internet]. 2021; 12. https://www.frontiersin.org/articles/ 10.3389/fimmu.2021.680687/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ofer D, Brandes N, Linial M. The language of proteins: NLP, machine learning & protein sequences. Comput Struct Biotechnol J. 2021;19:1750–58. doi: 10.1016/j.csbj.2021.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Leem J, Mitchell LS, Farmery JHR, Barton J, Galson JD. Deciphering the language of antibodies using self-supervised learning[Internet]. bioRxiv2021 [cited 2021 Nov 16]; 2021.11.10.468064. https://www.biorxiv.org/content/ 10.1101/2021.11.10.468064v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Angluin D. Computational learning theory: survey and selected bibliography. In: Proceedings of the twenty-fourth annual ACM symposium on Theory of Computing. New York, NY, USA: Association for Computing Machinery; 1992. page; 351–69. [Google Scholar]
- 102.Heinz J. Computational theories of learning and developmental psycholinguistics. In: Jl L, Snyder W, Pater J editors. The Oxford Handbook of Developmental Linguistics. Oxford University Press; p. 633–63. 2016. [Google Scholar]
- 103.Moll RN, Arbib MA, Kfoury AJ. An Introduction to Formal Language Theory. Springer Science & Business Media; 2012. [Google Scholar]
- 104.Kanduri C, Pavlović M, Scheffer L, Motwani K. Profiling the baseline performance and limits of machine learning models for adaptive immune receptor repertoire classification. bioRxiv [Internet]. 2021; https://www.biorxiv.org/content/ 10.1101/2021.05.23.445346v1.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weber CR, Akbar R, Yermanos A, Pavlović M, Snapkov I, Sandve GK, Reddy ST, Greiff V, Schwartz R. immuneSIM: tunable multi-feature simulation of B- and T-cell receptor repertoires for immunoinformatics benchmarking. Bioinformatics [Internet]. 2020;36(11):3594–96. doi: 10.1093/bioinformatics/btaa158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pellequer JL, Westhof E. PREDITOP: a program for antigenicity prediction. J Mol Graph [Internet]. 1993. [cited 2021 Aug 12]; 11. https://pubmed.ncbi.nlm.nih.gov/7509182/. [DOI] [PubMed] [Google Scholar]
- 107.Odorico M, Pellequer J-L. BEPITOPE: predicting the location of continuous epitopes and patterns in proteins. J Mol Recognit [Internet]. 2003. [cited 2021 Aug 12];16(1):20–22. doi: 10.1002/jmr.602. [DOI] [PubMed] [Google Scholar]
- 108.Saha S, Raghava GPS. BcePred: Prediction of Continuous B-Cell Epitopes in Antigenic Sequences Using Physico-chemical Properties. In: Artificial Immune Systems. Springer, Berlin, Heidelberg; 2004. p. 197–204. [Google Scholar]
- 109.Soria-Guerra RE, Nieto-Gomez R, Govea-Alonso DO, Rosales-Mendoza S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. J Biomed Inform. 2015;53:405–14. doi: 10.1016/j.jbi.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 110.El-Manzalawy Y, Honavar V. Recent advances in B-cell epitope prediction methods. Immunome Res. 2010;6(Suppl 2):S2. doi: 10.1186/1745-7580-6-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Saha S, Raghava GP. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins [Internet]. 2006. [cited 2021 Aug 12];65. https://pubmed.ncbi.nlm.nih.gov/16894596/ [DOI] [PubMed] [Google Scholar]
- 112.Manavalan B, Govindaraj RG, Shin TH, Kim MO, Lee G. iBCE-EL: A new ensemble learning framework for improved linear b-cell epitope prediction. Front Immunol [Internet]. 2018. [cited 2021 Aug 16]; 10.3389/fimmu.2018.01695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Huang J, Honda W. CED: a conformational epitope database. BMC Immunol. 2006;7:1–8. doi: 10.1186/1471-2172-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Söllner J, Mayer B. Machine Learning Approaches for Prediction of Linear B-cell Epitopes on Proteins. J Mol Recognit. 2006;19:200–08. doi: 10.1002/jmr.771. [DOI] [PubMed] [Google Scholar]
- 115.El-Manzalawy Y, Dobbs D, Honavar V. Predicting linear B-cell epitopes using string kernels. J Mol Recognit [Internet]. 2008. [cited 2021 Aug 13];21:243–55. doi: 10.1002/jmr.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gao J, Faraggi E, Zhou Y, Ruan J, Kurgan KL. BEST: Improved Prediction of B-Cell Epitopes from Antigen Sequences. PLoS One. 2012;7(6):e40104. doi: 10.1371/journal.pone.0040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liang S, Zheng D, Standley DM, Yao B, Zacharias M, Zhang C. EPSVR and EPMeta: prediction of antigenic epitopes using support vector regression and multiple server results. BMC Bioinformatics [Internet]. 2010. [cited 2021 Aug 6]; 11(1). 10.1186/1471-2105-11-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen J, Liu H, Yang J, Chou K-C. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids [Internet]. 2007. [cited 2021 Aug 13];33. (3):423–28. doi: 10.1007/s00726-006-0485-9. [DOI] [PubMed] [Google Scholar]
- 119.Ren J, Song J, Ellis J, Li J. Staged heterogeneity learning to identify conformational B-cell epitopes from antigen sequences. BMC Genomics. 2017;18(S2):1–13. doi: 10.1186/s12864-017-3493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sun J, Wu D, Xu T, Wang X, Xu X, Tao L, Li YX, Cao ZW. SEPPA: a computational server for spatial epitope prediction of protein antigens. Nucleic Acids Res. 2009;37:W612–6. doi: 10.1093/nar/gkp417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Andersen PH, Nielsen M, Lund O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006;15:2558. doi: 10.1110/ps.062405906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sweredoski MJ, Baldi P. PEPITO: improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics [Internet] 2008. [cited 2021 Aug 6];24:1459–60. doi: 10.1093/bioinformatics/btn199. [DOI] [PubMed] [Google Scholar]
- 123.Ponomarenko J, Bui -H-H, Li W, Fusseder N, Bourne PE, Sette A, Peters B. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics [Internet]. 2008. [cited 2021 Aug 6];9(1). 10.1186/1471-2105-9-514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lu S, Li Y, Nan XZhang S. A Structure-based B-cell Epitope Prediction Model Through Combing Local and Global Features [Internet]. bioRxiv2021 [cited 2021 Aug 13]; 2021.07.13.452188. https://www.biorxiv.org/content/ 10.1101/2021.07.13.452188v1.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Greenbaum JA, Andersen PH, Blythe M, Bui -H-H, Cachau RE, Crowe J, Davies M, Kolaskar AS, Lund O, Morrison S, et al. Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J Mol Recognit. 2007;20:75–82. doi: 10.1002/jmr.815. [DOI] [PubMed] [Google Scholar]
- 126.Blythe MJ, Flower DR. Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci. 2005–1(14):246–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sela-Culang I, Ofran Y, Peters B. Antibody specific epitope prediction—emergence of a new paradigm. Curr Opin Virol. 2015;11:98–102. doi: 10.1016/j.coviro.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kunik V, Ofran Y. The indistinguishability of epitopes from protein surface is explained by the distinct binding preferences of each of the six antigen-binding loops. Protein Eng Des Sel [Internet]. 2013. [cited 2021 Aug 13];26:599–609. doi: 10.1093/protein/gzt027. [DOI] [PubMed] [Google Scholar]
- 129.Zhao L, Li J. Mining for the antibody-antigen interacting associations that predict the B cell epitopes. BMC Struct Biol [Internet]. 2010. [cited 2021 Aug 13];101:S6. doi: 10.1186/1472-6807-10-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Krawczyk K, Liu X, Baker T, Shi J, Deane CM. Improving B-cell epitope prediction and its application to global antibody-antigen docking. Bioinformatics [Internet]. 2014. [cited 2021 Aug 6];30(16):2288–94. doi: 10.1093/bioinformatics/btu190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schneider C, Buchanan A, Taddese B, Deane CM, Valencia A. DLAB: deep learning methods for structure-based virtual screening of antibodies. Bioinformatics [Internet]. 2021; https://www.biorxiv.org/content/ 10.1101/2021.02.12.430941v1.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Leem J, Deane CM. High-Throughput Antibody Structure Modeling and Design Using ABodyBuilder [Internet]. Methods in Molecular Biology2019 367–80. 10.1007/978-1-4939-8736-8_21 [DOI] [PubMed] [Google Scholar]
- 133.Sela-Culang I, Ashkenazi S, Peters B, Ofran Y. PEASE: predicting B-cell epitopes utilizing antibody sequence. Bioinformatics. 2015;31(8):1313–15. doi: 10.1093/bioinformatics/btu790. [DOI] [PubMed] [Google Scholar]
- 134.Sela-Culang I, Benhnia MEI, Matho MH, Kaever T, Maybeno M, Schlossman A, Nimrod G, Li S, Xiang Y, Zajonc D, et al. Using a combined computational-experimental approach to predict antibody-specific B cell epitopes. Structure [Internet]. 2014. [cited 2021 Aug 13];22(4):646–57. doi: 10.1016/j.str.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 135.Pittala S, Bailey-Kellogg C, Elofsson A. Learning context-aware structural representations to predict antigen and antibody binding interfaces. Bioinformatics. 2020;36(13):3996–4003. doi: 10.1093/bioinformatics/btaa263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liberis E, Velickovic P, Sormanni P, Vendruscolo M, Liò P, Hancock J. Parapred: antibody paratope prediction using convolutional and recurrent neural networks. Bioinformatics. 2018;34(17):2944–50. doi: 10.1093/bioinformatics/bty305. [DOI] [PubMed] [Google Scholar]
- 137.Olimpieri PP, Chailyan A, Tramontano A, Marcatili P. Prediction of site-specific interactions in antibody-antigen complexes: the proABC method and server. Bioinformatics. 2013;29(18):2285–91. doi: 10.1093/bioinformatics/btt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ambrosetti F, Olsen TH, Olimpieri PP, Jiménez-García B, Milanetti E, Marcatilli P, Bonvin AMJJ, Ponty Y. proABC-2: PRediction of AntiBody contacts v2 and its application to information-driven docking. Bioinformatics. 2020;36(20):5107–08. doi: 10.1093/bioinformatics/btaa644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Galson JD, Trück J, Fowler A, Clutterbuck EA, Münz M, Cerundolo V, Reinhard C, van der Most R, Pollard AJ, Lunter G, et al. Analysis of B Cell Repertoire Dynamics Following Hepatitis B Vaccination in Humans, and Enrichment of Vaccine-specific Antibody Sequences. EBioMedicine. 2015;2(12):2070–79. doi: 10.1016/j.ebiom.2015.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Richardson E, Galson JD, Kellam P, Kelly DF, Smith SE, Palser A, Watson S, Deane CM. A computational method for immune repertoire mining that identifies novel binders from different clonotypes, demonstrated by identifying anti-pertussis toxoid antibodies. MAbs. 2021;13(1):1869406. doi: 10.1080/19420862.2020.1869406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nimrod G, Fischman S, Austin M, Herman A, Keyes F, Leiderman O, Hargreaves D, Strajbl M, Breed J, Klompus S, et al. Computational Design of Epitope-Specific Functional Antibodies. Cell Rep. 2018;25(8):2121–31.e5. doi: 10.1016/j.celrep.2018.10.081. [DOI] [PubMed] [Google Scholar]
- 142.Li X, Van Deventer JA, Hassoun HS, Ghersi D. ASAP-SML: An antibody sequence analysis pipeline using statistical testing and machine learning. PLoS Comput Biol. 2020;16(4):e1007779. doi: 10.1371/journal.pcbi.1007779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kunik V, Ashkenazi S, Ofran Y. Paratome: an online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure. Nucleic Acids Res. 2012;40(W1):W521–4. doi: 10.1093/nar/gks480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Daberdaku S, Ferrari C, Valencia A. Antibody interface prediction with 3D Zernike descriptors and SVM. Bioinformatics. 2019;35(11):1870–76. doi: 10.1093/bioinformatics/bty918. [DOI] [PubMed] [Google Scholar]
- 145.Krawczyk K, Baker T, Shi J, Deane CM. Antibody i-Patch prediction of the antibody binding site improves rigid local antibody-antigen docking. Protein Eng Des Sel. 2013;26(10):621–29. doi: 10.1093/protein/gzt043. [DOI] [PubMed] [Google Scholar]
- 146.Meiler J, Zeidler A, Schmschke F, Mller M. Generation and evaluation of dimension-reduced amino acid parameter representations by artificial neural networks. Journal of Molecular Modeling. 2001;7(9):360–69. doi: 10.1007/s008940100038. [DOI] [Google Scholar]
- 147.Klausen MS, Jespersen MC, Nielsen H, Jensen KK, Jurtz VI, Sønderby CK, Sommer MOA, Winther O, Nielsen M, Petersen B, et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins: Structure, Function, and Bioinformatics [Internet]. 2019. [cited 2021 Aug 16]; 87. (6):520–27. doi: 10.1002/prot.25674. [DOI] [PubMed] [Google Scholar]
- 148.Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins: Structure, Function, and Genetics. 2003;52(1):80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 149.Oh L, Dai B, Bailey-Kellogg C. A multi-resolution graph convolution network for contiguous epitope prediction. BCB 21: Proceedings of the 12th ACM Conference on Bioinformatics, Computational Biology, and Health. 2021; 1–10. [Google Scholar]
- 150.Pierce BG, Hourai Y, Weng Z, Keskin O. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS One. 2011;6(9):e24657. doi: 10.1371/journal.pone.0024657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Del Vecchio A, Deac A, Liò P, Veličković P. Neural message passing for joint paratope-epitope prediction [Internet]. arXiv [q-bio.QM]2021. http://arxiv.org/abs/2106.00757 [Google Scholar]
- 152.Bronstein MM, Bruna J, Cohen T, Geometric Deep VP. Learning: Grids, Groups, Graphs, Geodesics, and Gauges [Internet]. arXiv [cs.LG]2021. http://arxiv.org/abs/2104.13478 [Google Scholar]
- 153.Atz K, Grisoni F, Schneider G. Geometric Deep Learning on Molecular Representations [Internet]. arXiv [physics.chem-ph]2021. http://arxiv.org/abs/2107.12375 [Google Scholar]
- 154.Gainza P, Sverrisson F, Monti F, Rodolà E, Boscaini D, Bronstein MM, Correia BE. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat Methods. 2020;17(2):184–92. doi: 10.1038/s41592-019-0666-6. [DOI] [PubMed] [Google Scholar]
- 155.Nunez-Castilla J, Stebliankin V, Baral P, Balbin CA, Sobhan M, Cickovski T, Mondal AM, Narasimhan G, Chapagain P, Mathee K, et al. Molecular mimicry between Spike and human thrombopoietin may induce thrombocytopenia in COVID-19 [Internet]. bioRxiv2021 [cited 2021 Aug 18]; 2021.08.10.455737. https://www.biorxiv.org/content/ 10.1101/2021.08.10.455737v1 [DOI] [Google Scholar]
- 156.Sverrisson F, Feydy J, Correia BE, Bronstein MM. Fast end-to-end learning on protein surfaces [Internet]. bioRxiv2020 [cited 2021 Aug 3]; 2020.Dec.28.424589. https://www.biorxiv.org/content/ 10.1101/2020.12.28.424589v1 [DOI] [Google Scholar]
- 157.Fernández-Quintero ML, Heiss MC, Pomarici ND, Math BA, Liedl KR. Antibody CDR loops as ensembles in solution vs. canonical clusters from X-ray structures. MAbs [Internet]. 2020. [cited 2021 Aug 13]; 12. (1):1744328. doi: 10.1080/19420862.2020.1744328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wong WK, Robinson SA, Bujotzek A, Georges G, Lewis AP, Shi J, Snowden J, Taddese B, Deane CM. Ab-Ligity: identifying sequence-dissimilar antibodies that bind to the same epitope. MAbs. 2021;13(1):1873478. doi: 10.1080/19420862.2021.1873478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Woolfson DN. A brief history of de novo protein design: minimal, rational, and computational. J Mol Biol. 2021;433(20):167160. doi: 10.1016/j.jmb.2021.167160. [DOI] [PubMed] [Google Scholar]
- 160.Ripoll DR, Chaudhury S, Wallqvist A, Deane CM. Using the antibody-antigen binding interface to train image-based deep neural networks for antibody-epitope classification. PLoS Comput Biol. 2021;17:e1008864. doi: 10.1371/journal.pcbi.1008864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Xu Z, Li S, Rozewicki J, Yamashita K, Teraguchi S, Inoue T, Shinnakasu R, Leach S, Kurosaki T, Standley DM. Functional clustering of B cell receptors using sequence and structural features. Mol Syst Des Eng. 2019;4:769–78. doi: 10.1039/C9ME00021F. [DOI] [Google Scholar]
- 162.Petti S, Eddy SR. Constructing benchmark test sets for biological sequence analysis using independent set algorithms [Internet]. bioRxiv2021 [cited 2021 Oct 3]; 2021.09.29.462285. https://www.biorxiv.org/content/ 10.1101/2021.09.29.462285v1.abstract [DOI] [Google Scholar]
- 163.Aikawa E. 3.316 - Immunohistochemistry. In: Ducheyne P, editor. Comprehensive Biomaterials. Oxford: Elsevier; 2011. p. 277–90. [Google Scholar]
- 164.Kurumida Y, Saito Y, Kameda T. Predicting antibody affinity changes upon mutations by combining multiple predictors. Sci Rep. 2020;10:19533. doi: 10.1038/s41598-020-76369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol. 2000;295:337–56. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- 166.Murcko MMA. Computational methods to predict binding free energy in ligand-receptor complexes. J Med Chem. 1995;38(26):4953–67. doi: 10.1021/jm00026a001. [DOI] [PubMed] [Google Scholar]
- 167.Weitzner BD, Jeliazkov JR, Lyskov S, Marze N, Kuroda D, Frick R, Adolf-Bryfogle J, Biswas N, Dunbrack DRL, Gray JJ. Modeling and docking of antibody structures with Rosetta. Nat Protoc. 2017;12(2):401–16. doi: 10.1038/nprot.2016.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25(10):243–55. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sulea T, Vivcharuk V, Corbeil CR, Deprez C, Purisima EO. Assessment of Solvated Interaction Energy Function for Ranking Antibody–Antigen Binding Affinities. J Chem Inf Model. 2016;56(7):1292–303. doi: 10.1021/acs.jcim.6b00043. [DOI] [PubMed] [Google Scholar]
- 170.Pires DEV, Ascher DB. mCSM-AB: a web server for predicting antibody–antigen affinity changes upon mutation with graph-based signatures. Nucleic Acids Res. 2016;44:W469–73. doi: 10.1093/nar/gkw458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Myung Y, Rodrigues CHMAscher DB, Dev P. mCSM-AB2: guiding rational antibody design using graph-based signatures [Internet]. Bioinformatics2019. 10.1093/bioinformatics/btz779 [DOI] [PubMed] [Google Scholar]
- 172.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–8. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Myung Y, Pires DEV, Ascher DB. mmCSM-AB: guiding rational antibody engineering through multiple point mutations. Nucleic Acids Res. 2020;48:W125–31. doi: 10.1093/nar/gkaa389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang M, Cang Z, Wei G-W. A topology-based network tree for the prediction of protein-protein binding affinity changes following mutation. Nat Mach Intell. 2020;2:116–23. doi: 10.1038/s42256-020-0149-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu X, Luo Y, Li P, Song S, Peng J, Dunbrack RL. Deep geometric representations for modeling effects of mutations on protein-protein binding affinity. PLoS Comput Biol. 2021;17:e1009284. doi: 10.1371/journal.pcbi.1009284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Setliff I, Shiakolas AR, Pilewski KA, Murji AA, Mapengo RE, Janowska K, Richardson S, Oosthuysen C, Raju N, Ronsard L, et al. High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell. 2019;179:1636–46.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Shiakolas AR, Kramer KJ, Wrapp D, Richardson SI, Schäfer A, Wall S, Wang N, Janowska K, Pilewski KA, Venkat R, et al. Cross-reactive coronavirus antibodies with diverse epitope specificities and Fc effector functions. Cell Rep. 2021;2:100313. doi: 10.1016/j.xcrm.2021.100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.He B, Liu S, Wang Y, Xu M, Cai W, Liu J, Bai W, Ye S, Ma Y, Hu H, et al. Rapid isolation and immune profiling of SARS-CoV-2 specific memory B cell in convalescent COVID-19 patients via LIBRA-seq. Signal Transduct Target Ther. 2021;6:195. doi: 10.1038/s41392-021-00610-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hu X, Kang S, Lefort C, Kim M, Jin MM. Combinatorial libraries against libraries for selecting neoepitope activation-specific antibodies. Proc Natl Acad Sci U S A. 2010;107:6252–57. doi: 10.1073/pnas.0914358107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bowley DR, Jones TM, Burton DR, Lerner RA. Libraries against libraries for combinatorial selection of replicating antigen-antibody pairs. Proc Natl Acad Sci U S A. 2009;106:1380–85. doi: 10.1073/pnas.0812291106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Younger D, Berger S, Baker D. High-throughput characterization of protein–protein interactions by reprogramming yeast mating. Proceedings of the [Internet]. 2017; https://www.pnas.org/content/114/46/12166.short [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Engelhart E, Lopez R, Emerson R, Lin C, Shikany C, Guion D, Kelley M, Younger D. Massively Multiplexed Affinity Characterization of Therapeutic Antibodies Against SARS-CoV-2 Variants [Internet]. bioRxiv2021 [cited 2021 Aug 1]; 2021.04.27.440939. https://www.biorxiv.org/content/ 10.1101/2021.04.27.440939v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Erasmus MF, D’Angelo S, Ferrara F, Naranjo L, Teixeira AA, Buonpane R, Stewart SM, Nastri HG, Bradbury ARM. A single donor is sufficient to produce a highly functional in vitro antibody library. Commun Biol. 2021;4:350. doi: 10.1038/s42003-021-01881-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bradbury ARM, Dübel S, Knappik A, Plückthun A. Animal- versus in vitro-derived antibodies: avoiding the extremes. MAbs. 2021;13:1950265. doi: 10.1080/19420862.2021.1950265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Robert PA, Akbar R, Frank R, Pavlović M, Widrich M, Snapkov I, Chernigovskaya M, Scheffer L, Slabodkin A, Mehta BB, et al. One billion synthetic 3D-antibody-antigen complexes enable unconstrained machine-learning formalized investigation of antibody specificity prediction [Internet]. 10.1101/2021.07.06.451258. [DOI] [Google Scholar]
- 186.Robert PA, Arulraj T, Ymir: M-HM. A 3D structural affinity model for multi-epitope vaccine simulations. iScience [Internet]. 2021. [cited 2021 Aug 19];https://www.cell.com/iscience/fulltext/S2589-0042(21)00947-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Marcou Q, Mora T, Walczak AM. High-throughput immune repertoire analysis with IGoR. Nat Commun. 2018;9:561. doi: 10.1038/s41467-018-02832-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yang X, Zhu Y, Zeng H, Chen S, Guan J, Wang Q, Lan C, Sun D, Yu X, Zhang Z. Knowledge-based antibody repertoire simulation, a novel allele detection tool evaluation and application[Internet] . bioRxiv2021 [cited 2021 Jul 27]; 2021.07.01.450681. https://www.biorxiv.org/content/ 10.1101/2021.07.01.450681v1 [DOI] [Google Scholar]
- 189.Han J, Kuhn R, Papadopoulou C, Agrafiotis A, Kreiner V, Shlesinger D, Dizerens R, Hong K-L, Weber C, Greiff V, et al. Echidna: integrated simulations of single-cell immune receptor repertoires and transcriptomes [Internet]. bioRxiv2021 [cited 2021 Jul 27]; 2021.07.17.452792. https://www.biorxiv.org/content/ 10.1101/2021.07.17.452792v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yermanos A, Greiff V, Krautler NJ, Menzel U, Dounas A, Miho E, Oxenius A, Stadler T, Reddy ST, Kelso J. Comparison of methods for phylogenetic B-cell lineage inference using time-resolved antibody repertoire simulations (AbSim). Bioinformatics [Internet]. 2017. [cited 2017 Sep 1]; 33:3938–46. doi: 10.1093/bioinformatics/btx533. [DOI] [PubMed] [Google Scholar]
- 191.Hasan MM, Kahveci T. Indexing a protein-protein interaction network expedites network alignment. Bioinformatics. 2015;16:326. doi: 10.1186/s12859-015-0756-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sethna Z, Elhanati Y, Jr CCG, Am W, Mora T, Berger B. OLGA: fast computation of generation probabilities of B- and T-cell receptor amino acid sequences and motifs. Bioinformatics [Internet]. 2019;35:2974–81. doi: 10.1093/bioinformatics/btz035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Pavlovic M, Scheffer L, Motwani K, Kanduri C. immuneML: an ecosystem for machine learning analysis of adaptive immune receptor repertoires. bioRxiv [Internet]. 2021; https://www.biorxiv.org/content/ 10.1101/2021.03.08.433891v2.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.D’Angelo S, Ferrara F, Naranjo L, Erasmus MF, Hraber P, Bradbury ARM. Many Routes to an Antibody Heavy-Chain CDR3: Necessary, Yet Insufficient, for Specific Binding. Front Immunol. 2018;9:395. doi: 10.3389/fimmu.2018.00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dupic T, Marcou Q, Walczak AM, Mora T, Chain B. Genesis of the αβ T-cell receptor. PLoS Comput Biol. 2019;15:e1006874. doi: 10.1371/journal.pcbi.1006874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Shcherbinin DS, Belousov VA, Shugay M, Matsen FA. Comprehensive analysis of structural and sequencing data reveals almost unconstrained chain pairing in TCRαβ complex. PLoS Comput Biol. 2020;16:e1007714. doi: 10.1371/journal.pcbi.1007714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.AlQuraishi M, Sorger PK. Differentiable biology: using deep learning for biophysics-based and data-driven modeling of molecular mechanisms. Nat Methods. 2021;18:1169–80. doi: 10.1038/s41592-021-01283-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Miyazawa S, Jernigan RL. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J Mol Biol. 1996;256:623–44. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 199.Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, Ł K, Polosukhin I. Attention is all you need. Advances in neural information processing systems. 2017. page 5998–6008. [Google Scholar]
- 200.Devlin J, Chang M-W, Lee K, Bert: TK. Pre-training of Deep Bidirectional Transformers for Language Understanding. arXiv [cs CL] [Internet]. 2018; http://arxiv.org/abs/1810.04805 [Google Scholar]
- 201.Brown TB, Mann B, Ryder N, Subbiah M, Kaplan J, Dhariwal P, Neelakantan A, Shyam P, Sastry G, Askell A, et al. Language Models are Few-Shot Learners. arXiv [cs CL] [Internet]. 2020; http://arxiv.org/abs/2005.14165 [Google Scholar]
- 202.Rogers A, Kovaleva O, Rumshisky A. A Primer in BERTology: What we know about how BERT works. arXiv [cs CL] [Internet]. 2020; http://arxiv.org/abs/2002.12327 [Google Scholar]
- 203.Clark K, Khandelwal U, Levy O, Manning CD. What Does BERT Look At? An Analysis of BERT’s Attention. arXiv [cs CL] Internet]. 2019; http://arxiv.org/abs/1906.04341 [Google Scholar]
- 204.Michel P, Levy O, Neubig G. Are Sixteen Heads Really Better than One? ArXiv [cs CL] [Internet]. 2019; http://arxiv.org/abs/1905.10650 [Google Scholar]
- 205.Vig J, Madani A, Varshney LR, Xiong C, Socher R, Rajani NF. BERTology Meets Biology: Interpreting Attention in Protein Language Models. arXiv [cs CL] [Internet]. 2020;http://arxiv.org/abs/2006.15222 [Google Scholar]
- 206.Babor M, Kortemme T. Multi-constraint computational design suggests that native sequences of germline antibody H3 loops are nearly optimal for conformational flexibility. Proteins: Structure, Function, and Bioinformatics. 2009;75(4):846–58. doi: 10.1002/prot.22293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Deszynski P, Mlokosiewicz J, Volanakis A, Jaszczyszyn I, Castellana N, Bonissone S, Ganesan R, Indi KK. Integrated Nanobody Database for Immunoinformatics [Internet]. bioRxiv2021. http://medrxiv.org/lookup/doi/ 10.1101/2021.08.04.21261581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Halperin RF, Stafford P, Johnston SA. Exploring Antibody Recognition of Sequence Space through Random-Sequence Peptide Microarrays. Mol Cell Proteomics [Internet] März. 2011; 01, [cited 2011 Oct 31];10(3):M110.000786. 10.1074/mcp.M110.000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Greiff V, Redestig H, Lück J, Bruni N, Valai A, Hartmann S, Rausch S, Schuchhardt J, Or-Guil M. A minimal model of peptide binding predicts ensemble properties of serum antibodies. BMC Genomics. 2012;13(1):79. doi: 10.1186/1471-2164-13-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Villegas-Morcillo A, Makrodimitris S, Rchj VH, Gomez AM, Sanchez V, Reinders MJT, Elofsson A. Unsupervised protein embeddings outperform hand-crafted sequence and structure features at predicting molecular function. Bioinformatics. 2021;37:162–70. doi: 10.1093/bioinformatics/btaa701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yang KK, Wu Z, Bedbrook CN, Arnold FH. Learned protein embeddings for machine learning. Bioinformatics. 2018;34:4138. doi: 10.1093/bioinformatics/bty455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ó F-R, Guijarro-Berdiñas B, Martinez-Rego D, Pérez-Sánchez B, Online Machine P-BD. Learning. Efficiency and Scalability Methods for Computational Intellect. IGI Global. 2013. page 27–54. [Google Scholar]
- 213.Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, Deane CM. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci U S A. 2019;116:4025–30. doi: 10.1073/pnas.1810576116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Xu Y, Wang D, Mason B, Rossomando T, Li N, Liu D, Cheung JK, Xu W, Raghava S, Katiyar A, et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11(2):239–64. doi: 10.1080/19420862.2018.1553476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting Antibody Developability Profiles Through Early Stage Discovery Screening. MAbs. 2020;12(1):1743053. doi: 10.1080/19420862.2020.1743053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT: Pharmacometrics and Systems Pharmacology. 2017;6:576–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kang TH, Jung ST. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp Mol Med. [Internet] 2019; 51. (11):1–9. 10.1038/s12276-019-0345-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Snapkov I, Chernigovskaya M, Sinitcyn P, Lê Quý K, Nyman TA, Greiff V. Progress and challenges in mass spectrometry-based analysis of antibody repertoires. Trends Biotechnol [Internet]. 2021; doi: 10.1016/j.tibtech.2021.08.006. [DOI] [PubMed] [Google Scholar]
- 219.Foltz IN, Karow M, Wasserman SM. Evolution and emergence of therapeutic monoclonal antibodies what cardiologists need to know. Circulation. 2013;127:2222–30. doi: 10.1161/CIRCULATIONAHA.113.002033. [DOI] [PubMed] [Google Scholar]
- 220.Farid SS, Baron M, Stamatis C, Nie W, Coffman J. Benchmarking biopharmaceutical process development and manufacturing cost contributions to R&D. MAbs. 2020;12:1754999. doi: 10.1080/19420862.2020.1754999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Khanal O, Lenhoff AM. Developments and opportunities in continuous biopharmaceutical manufacturing. MAbs. 2021;13:1903664. doi: 10.1080/19420862.2021.1903664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Yang X, Xu W, Dukleska S, Benchaar S, Mengisen S, Antochshuk V, Cheung J, Mann L, Babadjanova Z, Rowand J, et al. Developability studies before initiation of process development: improving manufacturability of monoclonal antibodies. MAbs. 2013;5:787–94. doi: 10.4161/mabs.25269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: From structure to effector functions. Front Immunol. 2014;5:1–17. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol. 2019;10:1–20. doi: 10.3389/fimmu.2019.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Dumet C, Pottier J, Gouilleux-Gruart V, Watier H. Insights into the IgG heavy chain engineering patent landscape as applied to IgG4 antibody development. MAbs. 2019;11:1341–50. doi: 10.1080/19420862.2019.1664365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Brezski RJ, Georgiou G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr Opin Immunol. 2016;40:62–69. doi: 10.1016/j.coi.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 227.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29:457–66. doi: 10.1093/protein/gzw040. [DOI] [PubMed] [Google Scholar]
- 228.Chen X, Dougherty T, Hong C, Schibler R, Cong Y, Reza Z. Predicting Antibody Developability from Sequence using Machine Learning. biorxiv. 2020; 2–8. [Google Scholar]
- 229.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci U S A. 2009;106(29):11937–42. doi: 10.1073/pnas.0904191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Bekker G-J, Ma B, Kamiya N. Thermal stability of single-domain antibodies estimated by molecular dynamics simulations. Protein Sci. 2019;28:429–38. doi: 10.1002/pro.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Gentiluomo L, Roessner D, Augustijn D, Svilenov H, Kulakova A, Mahapatra S, Winter G, Streicher W, Å R, Peters GHJ, et al. Application of interpretable artificial neural networks to early monoclonal antibodies development. Eur J Pharm Biopharm. 2019;141:81–89. doi: 10.1016/j.ejpb.2019.05.017. [DOI] [PubMed] [Google Scholar]
- 232.Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability Index: A Rapid In Silico Tool for the Screening of Antibody Aggregation Propensity. J Pharm Sci. 2012;101(1):2271–80. doi: 10.1002/jps.22758. [DOI] [PubMed] [Google Scholar]
- 233.Rawat P, Prabakaran R, Kumar S, Gromiha MM. AbsoluRATE: An in-silico method to predict the aggregation kinetics of native proteins. Biochim Biophys Acta: Proteins Proteomics. 2021;1869:140682. doi: 10.1016/j.bbapap.2021.140682. [DOI] [PubMed] [Google Scholar]
- 234.Rawat P, Prabakaran R, Kumar S, Gromiha MM. Exploring the sequence features determining amyloidosis in human antibody light chains. Sci Rep. 2021;11:13785. doi: 10.1038/s41598-021-93019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Van Durme J, De Baets G, Van Der Kant R, Ramakers M, Ganesan A, Wilkinson H, Gallardo R, Rousseau F, Schymkowitz J. Solubis: a webserver to reduce protein aggregation through mutation. Protein Eng Des Sel. 2016;29(8):285–89. doi: 10.1093/protein/gzw019. [DOI] [PubMed] [Google Scholar]
- 236.Magnan CN, Randall A, Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics. 2009;25(17):2200–07. doi: 10.1093/bioinformatics/btp386. [DOI] [PubMed] [Google Scholar]
- 237.Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J. Protein–Sol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33:3098–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sormanni P, Vendruscolo M. Protein Solubility Predictions Using the CamSol Method in the Study of Protein Homeostasis. CSH Perspect Biol [Internet]. 2019;11. 10.1101/cshperspect.a033845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Bhandari BK, Gardner PP, Lim CS. Solubility-Weighted Index: fast and accurate prediction of protein solubility. Bioinformatics. 2020;36:4691–98. doi: 10.1093/bioinformatics/btaa578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Khurana S, Rawi R, Kunji K, Chuang G-Y, Bensmail H, Mall R. DeepSol: a deep learning framework for sequence-based protein solubility prediction. Bioinformatics. 2018;34:2605–13. doi: 10.1093/bioinformatics/bty166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Li L, Kumar S, Buck PM, Burns C, Lavoie J, Singh SK, Warne NW, Nichols P, Luksha N, Boardman D. Concentration dependent viscosity of monoclonal antibody solutions: explaining experimental behavior in terms of molecular properties. Pharm Res. 2014;31:3161–78. doi: 10.1007/s11095-014-1409-0. [DOI] [PubMed] [Google Scholar]
- 242.Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A. 2014;111:18601–06. doi: 10.1073/pnas.1421779112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Tomar DS, Li L, Broulidakis MP, Luksha NG, Burns CT, Singh SK, Kumar S. In-silico prediction of concentration-dependent viscosity curves for monoclonal antibody solutions. MAbs. 2017;9:476–89. doi: 10.1080/19420862.2017.1285479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S, Balthazor B, Conley TR, Sek D, Allen MJ. Rational design of viscosity reducing mutants of a monoclonal antibody: hydrophobic versus electrostatic inter-molecular interactions. MAbs. 2015;7:212–30. doi: 10.4161/19420862.2014.985504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Lai P-K, Fernando A, Cloutier TK, Gokarn Y, Zhang J, Schwenger W, Chari R, Calero-Rubio C, Trout BL. Machine Learning Applied to Determine the Molecular Descriptors Responsible for the Viscosity Behavior of Concentrated Therapeutic Antibodies. Mol Pharm. 2021;18:1167–75. doi: 10.1021/acs.molpharmaceut.0c01073. [DOI] [PubMed] [Google Scholar]
- 246.Agrawal NJ, Helk B, Kumar S, Mody N, Sathish HA, Samra HS, Buck PM, Li L, Trout BL. Computational tool for the early screening of monoclonal antibodies for their viscosities. MAbs. 2016;8:43–48. doi: 10.1080/19420862.2015.1099773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lai P-K, Swan JW, Trout BL. Calculation of therapeutic antibody viscosity with coarse-grained models, hydrodynamic calculations and machine learning-based parameters. MAbs. 2021;13:1907882. doi: 10.1080/19420862.2021.1907882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Tilegenova C, Izadi S, Yin J, Huang CS, Wu J, Ellerman D, Hymowitz SG, Walters B, Salisbury C, Carter PJ. Dissecting the molecular basis of high viscosity of monospecific and bispecific IgG antibodies. MAbs. 2020;12:1692764. doi: 10.1080/19420862.2019.1692764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Schwenger W, Pellet C, Attonaty D, Authelin J-R. An Empirical Quantitative Model Describing Simultaneously Temperature and Concentration Effects on Protein Solution Viscosity. J Pharm Sci. 2020;109:1281–87. doi: 10.1016/j.xphs.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 250.Reynisson B, Barra C, Kaabinejadian S, Hildebrand WH, Peters B, Nielsen M. Improved Prediction of MHC II Antigen Presentation through Integration and Motif Deconvolution of Mass Spectrometry MHC Eluted Ligand Data. J Proteome Res. 2020;19:2304–15. doi: 10.1021/acs.jproteome.9b00874. [DOI] [PubMed] [Google Scholar]
- 251.Marks C, Hummer AM, Chin M, Deane CM, Martelli PL. Humanization of antibodies using a machine learning approach on large-scale repertoire data. Bioinformatics [Internet]. 2021; doi: 10.1093/bioinformatics/btab434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Prihoda D, Maamary J, Waight A, Juan V, Fayadat-Dilman L, Svozil D, Bitton DA. BioPhi: A platform for antibody design, humanization and humanness evaluation based on natural antibody repertoires and deep learning [Internet]. bioRxiv2021 [cited 2021 Aug 11]; 2021.08.08.455394. https://www.biorxiv.org/content/ 10.1101/2021.08.08.455394v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Gao SH, Huang K, Tu H, Adler AS. Monoclonal antibody humanness score and its applications. BMC Biotechnol. 2013;13:55. doi: 10.1186/1472-6750-13-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Goulet DR, Watson MJ, Tam SH, Zwolak A, Chiu ML, Atkins WM, Nath A. Toward a Combinatorial Approach for the Prediction of IgG Half-Life and Clearance. Drug Metab Dispos. 2018;46:1900–07. doi: 10.1124/dmd.118.081893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Grinshpun B, Thorsteinson N, Pereira JN, Rippmann F, Nannemann D, Sood VD, Fomekong Nanfack Y. Identifying biophysical assays and in silico properties that enrich for slow clearance in clinical-stage therapeutic antibodies. MAbs. 2021;13:1932230. doi: 10.1080/19420862.2021.1932230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kuriata A, Iglesias V, Pujols J, Kurcinski M, Kmiecik S, Ventura S. Aggrescan3D (A3D) 2.0: prediction and engineering of protein solubility. Nucleic Acids Res. 2019;47:W300–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Suzuki T, Ishii-Watabe A, Tada M, Kobayashi T, Kanayasu-Toyoda T, Kawanishi T, Yamaguchi T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: a comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J Immunol. 2010;184:1968–76. doi: 10.4049/jimmunol.0903296. [DOI] [PubMed] [Google Scholar]
- 258.Shehata L, Maurer DP, Wec AZ, Lilov A, Champney E, Sun T, Archambault K, Burnina I, Lynaugh H, Zhi X, et al. Affinity Maturation Enhances Antibody Specificity but Compromises Conformational Stability. Cell Rep. 2019;28(4):3300–08. doi: 10.1016/j.celrep.2019.08.056. [DOI] [PubMed] [Google Scholar]
- 259.Zhou H, Hu C, Zhu Y, Lu M, Liao S, Yeilding N, Davis HM. Population-based exposure-efficacy modeling of ustekinumab in patients with moderate to severe plaque psoriasis. J Clin Pharmacol. 2010;50:257–67. doi: 10.1177/0091270009343695. [DOI] [PubMed] [Google Scholar]
- 260.Gandhi M, Alwawi E, Gordon KB. Anti-p40 antibodies ustekinumab and briakinumab: blockade of interleukin-12 and interleukin-23 in the treatment of psoriasis. Semin Cutan Med Surg. 2010;29:48–52. doi: 10.1016/j.sder.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 261.Ovacik M, Lin K. Tutorial on Monoclonal Antibody Pharmacokinetics and Its Considerations in Early Development. Clin Transl Sci. 2018;11:540–52. doi: 10.1111/cts.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Putnam WS, Prabhu S, Zheng Y, Subramanyam M, Wang Y-MC. Pharmacokinetic, pharmacodynamic and immunogenicity comparability assessment strategies for monoclonal antibodies. Trends Biotechnol. 2010;28:509–16. doi: 10.1016/j.tibtech.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 263.Bumbaca Yadav D, Sharma VK, Boswell CA, Hotzel I, Tesar D, Shang Y, Ying Y, Fischer SK, Grogan JL, Chiang EY, et al. Evaluating the use of antibody variable region (Fv) charge as a risk assessment tool for predicting typical Cynomolgus monkey pharmacokinetics. J Biol Chem. 2015;290:29732–41. doi: 10.1074/jbc.M115.692434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel. 2010;23:385–92. doi: 10.1093/protein/gzq009. [DOI] [PubMed] [Google Scholar]
- 265.Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos. 2011;39:1469–77. doi: 10.1124/dmd.111.039453. [DOI] [PubMed] [Google Scholar]
- 266.Piche-Nicholas NM, Avery LB, King AC, Kavosi M, Wang M, O’Hara DM, Tchistiakova L, Katragadda M. Changes in complementarity-determining regions significantly alter IgG binding to the neonatal Fc receptor (FcRn) and pharmacokinetics. MAbs. 2018;10:81–94. doi: 10.1080/19420862.2017.1389355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Sally Ward E. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc Natl Acad Sci U S A. 2006;103:18709–14. doi: 10.1073/pnas.0606304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Andersen JT, Daba MB, Berntzen G, Michaelsen TE, Sandlie I. Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J Biol Chem. 2010;285:4826–36. doi: 10.1074/jbc.M109.081828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Roopenian DC, Christianson GJ, Proetzel G, Sproule TJ. Human FcRn Transgenic Mice for Pharmacokinetic Evaluation of Therapeutic Antibodies. Methods Mol Biol. 2016;1438:103–14. [DOI] [PubMed] [Google Scholar]
- 270.Nilsen J, Sandlie I, Roopenian DC, Andersen JT. Animal models for evaluation of albumin-based therapeutics. Curr Opin Chem Eng. 2018;19:68–76. doi: 10.1016/j.coche.2017.11.007. [DOI] [Google Scholar]
- 271.Grevys A, Nilsen J, Kmk S, Mb D, Øynebråten I, Bern M, Mb M, Foss S, Schlothauer T, Te M, et al. A human endothelial cell-based recycling assay for screening of FcRn targeted molecules. Nat Commun. 2018;9:621. doi: 10.1038/s41467-018-03061-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kelly RL, Sun T, Jain T, Caffry I, Yu Y, Cao Y, Lynaugh H, Brown M, Vásquez M, Wittrup KD, et al. High throughput cross-interaction measures for human IgG1 antibodies correlate with clearance rates in mice. MAbs. 2015;7:770–77. doi: 10.1080/19420862.2015.1043503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Chai Q, Shih J, Weldon C, Phan S, Jones BE. Development of a high-throughput solubility screening assay for use in antibody discovery. MAbs. 2019;11:747–56. doi: 10.1080/19420862.2019.1589851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Lowe D, Dudgeon K, Rouet R, Schofield P, Jermutus L, Christ D. Aggregation, stability, and formulation of human antibody therapeutics. Adv Protein Chem Struct Biol. 2011;84:41–61. [DOI] [PubMed] [Google Scholar]
- 275.Liu L, Braun LJ, Wang W, Randolph TW, Carpenter JF. Freezing-induced perturbation of tertiary structure of a monoclonal antibody. J Pharm Sci. 2014;103:1979–86. doi: 10.1002/jps.24013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Le Basle Y, Chennell P, Tokhadze N, Astier A, Sautou V. Physicochemical Stability of Monoclonal Antibodies: A Review. J Pharm Sci. 2020;109(1):169–90. doi: 10.1016/j.xphs.2019.08.009. [DOI] [PubMed] [Google Scholar]
- 277.Brader ML, Estey T, Bai S, Alston RW, Lucas KK, Lantz S, Landsman P, Maloney KM. Examination of thermal unfolding and aggregation profiles of a series of developable therapeutic monoclonal antibodies. Mol Pharm. 2015;12:1005–17. doi: 10.1021/mp400666b. [DOI] [PubMed] [Google Scholar]
- 278.Dooley H, Grant SD, Harris WJ, Porter AJ. Stabilization of antibody fragments in adverse environments. Biotechnol Appl Biochem. 1998;28:77–83. [PubMed] [Google Scholar]
- 279.Ad M, Spasojevich V, Jl M, Ip K, Chen A, Jc S, Berkebile A, Ra H, Neben S, Dj K, et al. An integrated approach to extreme thermostabilization and affinity maturation of an antibody. Protein Eng Des Sel. 2013;26:151–64. doi: 10.1093/protein/gzs090. [DOI] [PubMed] [Google Scholar]
- 280.Ad M, Zhang X, Jl M, Chau B, Jc S, Rahmanian S, Hare E, Spasojevic V, Ra H, Dj K, et al. A general approach to antibody thermostabilization. MAbs. 2014;6:1274–82. doi: 10.4161/mabs.29680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Al Qaraghuli MM, Kubiak-Ossowska K, Mulheran PA. Thinking outside the Laboratory: Analyses of Antibody Structure and Dynamics within Different Solvent Environments in Molecular Dynamics (MD) Simulations. Antibodies (Basel) [Internet]. 2018; 7. doi: 10.3390/antib7030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Jia L, Jain M, Sun Y. Improving antibody thermostability based on statistical analysis of sequence and structural consensus data. bioRxiv [Internet]. 2021; https://www.biorxiv.org/content/ 10.1101/2021.01.28.428721v1.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Lee J, Der BS, Karamitros CS, Li W, Marshall NM, Lungu OI, Miklos AE, Xu J, Kang TH, Lee C-H, et al. Computer-based Engineering of Thermostabilized Antibody Fragments. AIChE J [Internet]. 2020; 66(3): 10.1002/aic.16864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wang X, Singh SK, Kumar S. Potential aggregation-prone regions in complementarity-determining regions of antibodies and their contribution towards antigen recognition: a computational analysis. Pharm Res. 2010;27:1512–29. doi: 10.1007/s11095-010-0143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Perchiacca JM, Bhattacharya M, Tessier PM. Mutational analysis of domain antibodies reveals aggregation hotspots within and near the complementarity determining regions. Proteins. 2011;79:2637–47. doi: 10.1002/prot.23085. [DOI] [PubMed] [Google Scholar]
- 286.Wang X, Das TK, Singh SK, Kumar S. Potential aggregation prone regions in biotherapeutics: A survey of commercial monoclonal antibodies. MAbs. 2009;1:254–67. doi: 10.4161/mabs.1.3.8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Prabakaran R, Rawat P, Kumar S, Gromiha MM. Evaluation of in silico tools for the prediction of protein and peptide aggregation on diverse datasets. Brief Bioinform [Internet]. 2021; doi: 10.1093/bib/bbab240. [DOI] [PubMed] [Google Scholar]
- 288.Prabakaran R, Rawat P, Thangakani AM, Kumar S, Gromiha MM. Protein aggregation: in silico algorithms and applications. Biophys Rev. 2021;13:71–89. doi: 10.1007/s12551-021-00778-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Rawat P, Prabakaran R, Sakthivel R, Mary Thangakani A, Kumar S, Gromiha MMCPAD. CPAD 2.0: a repository of curated experimental data on aggregating proteins and peptides. Amyloid. 2020;27(2):128–33. doi: 10.1080/13506129.2020.1715363. [DOI] [PubMed] [Google Scholar]
- 290.van der Kant R, Karow-Zwick AR, Van Durme J, Blech M, Gallardo R, Seeliger D. Aßfalg K, Baatsen P, Compernolle G, Gils A, et al. Prediction and Reduction of the Aggregation of Monoclonal Antibodies. J Mol Biol. 2017;429:1244–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Sormanni P, Amery L, Ekizoglou S, Vendruscolo M, Popovic B. Rapid and accurate in silico solubility screening of a monoclonal antibody library. Sci Rep. 2017;7:8200. doi: 10.1038/s41598-017-07800-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Tomar DS, Kumar S, Singh SK, Goswami S, Li L. Molecular basis of high viscosity in concentrated antibody solutions: Strategies for high concentration drug product development. MAbs. 2016;8:216–28. doi: 10.1080/19420862.2015.1128606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Goswami S, Wang W, Arakawa T, Developments OS. Challenges for mAb-Based Therapeutics. Antibodies. 2013;2:452–500. doi: 10.3390/antib2030452. [DOI] [Google Scholar]
- 294.Chowdhury A, Bollinger JA, Dear BJ, Cheung JK, Johnston KP, Truskett TM. Coarse-Grained Molecular Dynamics Simulations for Understanding the Impact of Short-Range Anisotropic Attractions on Structure and Viscosity of Concentrated Monoclonal Antibody Solutions. Mol Pharm. 2020;17:1748–56. doi: 10.1021/acs.molpharmaceut.9b00960. [DOI] [PubMed] [Google Scholar]
- 295.Kingsbury JS, Saini A, Auclair SM, Fu L, Lantz MM, Halloran KT, Calero-Rubio C, Schwenger W, Airiau CY, Zhang J, et al. A single molecular descriptor to predict solution behavior of therapeutic antibodies. Sci Adv. 2020;6:eabb0372. doi: 10.1126/sciadv.abb0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Zinsli LV, Stierlin N, Loessner MJ, Schmelcher M. Deimmunization of protein therapeutics - Recent advances in experimental and computational epitope prediction and deletion. Comput Struct Biotechnol J. 2021;19:315–29. doi: 10.1016/j.csbj.2020.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Roda G, Jharap B, Neeraj N, Colombel J-F. Loss of response to anti-TNFs: Definition, epidemiology, and management. Clin Transl Gastroenterol. 2016;7:e135. doi: 10.1038/ctg.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Reinhold I, Blümel S, Schreiner J, Boyman O, Bögeholz J, Cheetham M, Rogler G, Biedermann L, Scharl M. Clinical Relevance of Anti-TNF Antibody Trough Levels and Anti-Drug Antibodies in Treating Inflammatory Bowel Disease Patients. Inflamm Intest Dis. 2021;6:38–47. doi: 10.1159/000511296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Hwang WYK, Foote J. Immunogenicity of engineered antibodies. Methods. 2005. 5;36:3–10. doi: 10.1016/j.ymeth.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 300.Liang S, Zhang C, Gill AC. Prediction of immunogenicity for humanized and full human therapeutic antibodies. PLoS One. 2020;15:e0238150. doi: 10.1371/journal.pone.0238150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Doneva N, Doytchinova I, Dimitrov I. Predicting Immunogenicity Risk in Biopharmaceuticals. Symmetry. 2021;13:388. doi: 10.3390/sym13030388. [DOI] [Google Scholar]
- 302.Unanue ER, Turk V, Neefjes J. Variations in MHC Class II Antigen Processing and Presentation in Health and Disease. Annu Rev Immunol. 2016;34(1):265–97. doi: 10.1146/annurev-immunol-041015-055420. [DOI] [PubMed] [Google Scholar]
- 303.Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F, Freund C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front Immunol [Internet]. 2017. [cited 2021 Jul 19]; 8. doi: 10.3389/fimmu.2017.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Enrico D, Paci A, Chaput N, Karamouza E, Besse B. Antidrug Antibodies Against Immune Checkpoint Blockers: Impairment of Drug Efficacy or Indication of Immune Activation? Clin Cancer Res. 2020;26:787–92. doi: 10.1158/1078-0432.CCR-19-2337. [DOI] [PubMed] [Google Scholar]
- 305.Todd PA, Brogden RN. Muromonab CD3. Drugs. 1989;37:871–99. doi: 10.2165/00003495-198937060-00004. [DOI] [PubMed] [Google Scholar]
- 306.Clavero-Álvarez A, Di Mambro T, Perez-Gaviro S, Magnani M, Bruscolini P. Humanization of Antibodies using a Statistical Inference Approach. Sci Rep. 2018;8:1–11. doi: 10.1038/s41598-018-32986-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256–65. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.The Antibody Society . Therapeutic monoclonal antibodies approved or in review in the EU or US. [Internet]. The Antibody Society2021 [cited 2021 Jul 19]; https://www.antibodysociety.org/antibody-therapeutics-product-data/ [Google Scholar]
- 309.Williams DG, Matthews DJ, Jones T. Humanising Antibodies by CDR Grafting. In: Kontermann R, Dübel S, editors. Antibody Engineering. Berlin, Heidelberg: Springer Berlin Heidelberg; 2010. p. 319–39. [Google Scholar]
- 310.Petersen BM, Ulmer SA, Rhodes ER, Gonzalez MFG. Regulatory approved monoclonal antibodies contain framework mutations predicted from human antibody repertoires. bioRxiv [Internet]. 2021; https://www.biorxiv.org/content/ 10.1101/2021.06.22.449488v1.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Almagro JC, Daniels-Wells TR, Perez-Tapia SM, Penichet ML. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front Immunol. 2018;8:1751. doi: 10.3389/fimmu.2017.01751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Doevendans E, Schellekens H. Immunogenicity of Innovative and Biosimilar Monoclonal Antibodies. Antibodies (Basel) [Internet]. 2019;8(1). 10.3390/antib8010021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Nl D, Np C, Rb S, Sh R, Aw P. A Systems Approach to Understand Antigen Presentation and the Immune Response. In: Reinders J, editor. Proteomis in Systems Biology. New York, NY: Springer New York; 2016. p. 189–209. [Google Scholar]
- 314.Caron E, Kowalewski D, Chiek Koh C, Sturm T, Schuster H, Aebersold R. Analysis of Major Histocompatibility Complex (MHC) Immunopeptidomes Using Mass Spectrometry. Mol Cell Proteomics 12/2015. 14:3105–17. doi: 10.1074/mcp.O115.052431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE. IPD-IMGT/HLA Database. Nucleic Acids Res. 2020;48:D948–55. doi: 10.1093/nar/gkz950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Cole DK. The ultimate mix and match: making sense of HLA alleles and peptide repertoires. Immunol Cell Biol 07/2015. 93:515–16. doi: 10.1038/icb.2015.40. [DOI] [PubMed] [Google Scholar]
- 317.Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, Wheeler DK, Sette A, Peters B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019;47:D339–43. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Alvarez B, Reynisson B, Barra C, Buus S, Ternette N, Connelley T, Andreatta M, Nielsen M. NNAlign_MA; MHC Peptidome Deconvolution for Accurate MHC Binding Motif Characterization and Improved T-cell Epitope Predictions. Mol Cell Proteomics. 2019. 12;18(18):2459–77. doi: 10.1074/mcp.TIR119.001658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics 6/2011. 63:325–35. doi: 10.1007/s00251-011-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Kuroda D, Engineering Stability TK. Viscosity, and Immunogenicity of Antibodies by Computational Design. J Pharm Sci. 2020;109:1631–51. doi: 10.1016/j.xphs.2020.01.011. [DOI] [PubMed] [Google Scholar]
- 321.Krawczyk K, Buchanan A, Marcatili P. Data mining patented antibody sequences. MAbs. 2021;13:1892366. doi: 10.1080/19420862.2021.1892366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Habibi N, Mohd Hashim SZ, Norouzi A, Samian MR. A review of machine learning methods to predict the solubility of overexpressed recombinant proteins in Escherichia coli. BMC Bioinformatics. 2014;15:134. doi: 10.1186/1471-2105-15-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Delmar JA, Buehler E, Chetty AK, Das A, Quesada GM, Wang J, Chen X. Machine learning prediction of methionine and tryptophan photooxidation susceptibility. Mol Ther Methods Clin Dev. 2021;21:466–77. doi: 10.1016/j.omtm.2021.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Liu G, Zeng H, Mueller J, Carter B, Wang Z, Schilz J, Horny G, Birnbaum ME, Ewert S, Gifford DK. Antibody complementarity determining region design using high-capacity machine learning. Bioinformatics. 2020;36:2126–33. doi: 10.1093/bioinformatics/btz895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Saka K, Kakuzaki T, Metsugi S, Kashiwagi D, Yoshida K, Wada M, Tsunoda H, Teramoto R. Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Sci Rep. 2021;11:5852. doi: 10.1038/s41598-021-85274-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Eguchi RR, Anand N, Choe CA, Huang PS. Ig-VAE: generative modeling of immunoglobulin proteins by direct 3D coordinate generation. bioRxiv [Internet]. 2020; https://www.biorxiv.org/content/ 10.1101/2020.08.07.242347v1.abstract [DOI] [Google Scholar]
- 327.Jin W, Wohlwend J, Barzilay R, Jaakkola T. Iterative Refinement Graph Neural Network for Antibody Sequence-Structure Co-design. arXiv [q-bio.BM] [Internet]. 2021; http://arxiv.org/abs/2110.04624 [Google Scholar]
- 328.Melnyk I, Das P, Chenthamarakshan V, Lozano A. Benchmarking deep generative models for diverse antibody sequence design. arXiv [q-bio.BM] [Internet]. 2021; http://arxiv.org/abs/2111.06801 [Google Scholar]
- 329.Kingma DP, Welling M. Auto-Encoding Variational Bayes. arXiv [stat.ML] [Internet]. 2013; http://arxiv.org/abs/1312.6114v10 [Google Scholar]
- 330.Goodfellow I, Pouget-Abadie J, Mirza M, Xu B, Warde-Farley D, Ozair S, Courville A, Bengio Y. Generative Adversarial Nets. In: Ghahramani Z, Welling M, Cortes C, Lawrence ND, Weinberger KQ editors. Advances in Neural Information Processing Systems 27. Curran Associates, Inc; 2014. p. 2672–80. [Google Scholar]
- 331.Gonog L, Zhou Y. A Review: Generative Adversarial Networks. In: 2019 14th IEEE Conference on Industrial Electronics and Applications (ICIEA). 2019. 505–10. [Google Scholar]
- 332.Dan Y, Zhao Y, Li X, Li S, Hu M, Hu J. Generative adversarial networks (GAN) based efficient sampling of chemical composition space for inverse design of inorganic materials. npj Computational Materials. 2020;6(1):1–7. doi: 10.1038/s41524-020-00352-0. [DOI] [Google Scholar]
- 333.Repecka D, Jauniskis V, Karpus L, Rembeza E, Rokaitis I, Zrimec J, Poviloniene S, Laurynenas A, Viknander S, Abuajwa W, et al. Expanding functional protein sequence spaces using generative adversarial networks. Nat Mach Intell. 2021;3(4):324–33. doi: 10.1038/s42256-021-00310-5. [DOI] [Google Scholar]
- 334.Mirza M, Osindero S. Conditional Generative Adversarial Nets. arXiv [cs.LG] [Internet]. 2014; http://arxiv.org/abs/1411.1784 [Google Scholar]
- 335.Odena A, Olah C, Shlens J. Conditional image synthesis with auxiliary classifier gans. In: International conference on machine learning. PMLR; 2017. page 2642–51. [Google Scholar]
- 336.Miyato T, Koyama M. cGANs with Projection Discriminator. arXiv [cs.LG] [Internet]. 2018; http://arxiv.org/abs/1802.05637 [Google Scholar]
- 337.Arjovsky M, Chintala S, Bottou L. Wasserstein generative adversarial networks. In: International conference on machine learning. PMLR; 2017. page 214–23. [Google Scholar]
- 338.Sinai S, Kelsic E, Church GM, Nowak MA. Variational auto-encoding of protein sequences. arXiv [q-bio.QM] [Internet]. 2017; http://arxiv.org/abs/1712.03346 [Google Scholar]
- 339.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33(Web Server):W363–7. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Costello Z, Martin HG. How to Hallucinate Functional Proteins. arXiv [q-bio.QM] [Internet]. 2019; http://arxiv.org/abs/1903.00458 [Google Scholar]
- 342.Hamed M, Logan A, Gruszczyk AV, Beach TE, James AM, Dare AJ, Barlow A, Martin J, Georgakopoulos L, Gane AM. Mitochondria-targeted antioxidant MitoQ ameliorates ischaemia-reperfusion injury in kidney transplantation models[Internet]. [cited 2021 Sep 11]; The British journal of surgery. 108(9): 1072–81. doi: 10.1093/bjs/znab108 [DOI] [PubMed] [Google Scholar]
- 343.Bond-Taylor S, Leach A, Long Y, Willcocks CG. Deep Generative Modelling: A Comparative Review of VAEs, GANs, Normalizing Flows, Energy-Based and Autoregressive Models. arXiv [cs.LG] [Internet]. 2021; http://arxiv.org/abs/2103.04922 [DOI] [PubMed] [Google Scholar]
- 344.McCoy LE, Rutten L, Frampton D, Anderson I, Granger L, Bashford-Rogers R, Dekkers G, Strokappe NM, Seaman MS, Koh W, et al. Molecular evolution of broadly neutralizing Llama antibodies to the CD4-binding site of HIV-1. PLoS Pathog. 2014;10(12):e1004552. doi: 10.1371/journal.ppat.1004552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–32. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 346.Bjellqvist B, Hughes GJ, Pasquali C, Paquet N, Ravier F, Sanchez J-C, Frutiger S, Hochstrasser D. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis. 1993;14(1):1023–31. doi: 10.1002/elps.11501401163. [DOI] [PubMed] [Google Scholar]
- 347.Müller AT, Hiss JA, Schneider G. Recurrent Neural Network Model for Constructive Peptide Design. J Chem Inf Model. 2018;58(2):472–79. doi: 10.1021/acs.jcim.7b00414. [DOI] [PubMed] [Google Scholar]
- 348.Biswas S, Khimulya G, Alley EC, Esvelt KM, Church GM. Low-N protein engineering with data-efficient deep learning. Nat Methods. 2021;18(4):389–96. doi: 10.1038/s41592-021-01100-y. [DOI] [PubMed] [Google Scholar]
- 349.Liu Y, Ye Q, Wang L, Peng J. Learning structural motif representations for efficient protein structure search. Bioinformatics. 2018;34(17):i773–80. doi: 10.1093/bioinformatics/bty585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Araujo A, Norris W, Sim J. Computing receptive fields of convolutional neural networks. Distill [Internet]. 2019;4(11). https://distill.pub/2019/computing-receptive-fields [Google Scholar]
- 351.Shanehsazzadeh A, Belanger D, Dohan D. Is Transfer Learning Necessary for Protein Landscape Prediction? ArXiv [q-bio.BM] [Internet]. 2020;http://arxiv.org/abs/2011.03443 [Google Scholar]
- 352.Wu Z, Yang KK, Liszka MJ, Lee A, Batzilla A, Wernick D, Weiner DP, Arnold FH. Signal Peptides Generated by Attention-Based Neural Networks. ACS Synth Biol. 2020;9:2154–61. doi: 10.1021/acssynbio.0c00219. [DOI] [PubMed] [Google Scholar]
- 353.Madani A, McCann B, Naik N, Keskar NS, Anand N, Eguchi RR, P-s H, Socher R. ProGen: Language Modeling for Protein Generation [Internet]. 2020;DOI: 10.1101/2020.03.07.982272. [DOI] [Google Scholar]
- 354.Ingraham J, Garg VK, Barzilay R, Jaakkola T. Generative Models for Graph-Based Protein Design [Internet]. 2019. cited 2021 Sep 1. https://openreview.net/pdf?id=ByMEAHrgLB [Google Scholar]
- 355.Sircar A, Gray JJ, Kortemme T. SnugDock: paratope structural optimization during antibody-antigen docking compensates for errors in antibody homology models. PLoS Comput Biol. 2010;6:e1000644. doi: 10.1371/journal.pcbi.1000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Pierce BG, Wiehe K, Hwang H, Kim B-H, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771–73. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Bekker G-J, Fukuda I, Higo J, Kamiya N. Mutual population-shift driven antibody-peptide binding elucidated by molecular dynamics simulations. Sci Rep. 2020;10:1406. doi: 10.1038/s41598-020-58320-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Renz P, Van Rompaey D, Wegner JK, Hochreiter S, Klambauer G, Sung K-J, Jia H, Johnson JM, Saeed M, Mace CR. On Failure Modes of Molecule Generators and Optimizers. ChemRxiv [Internet]. [cited 2021 Jul 28]; doi: 10.26434/chemrxiv.14442785. [DOI] [Google Scholar]
- 359.Lotfollahi M, Naghipourfar M, Theis FJ, Wolf FA. Conditional out-of-distribution generation for unpaired data using transfer VAE. Bioinformatics. 2020;36:i610–7. doi: 10.1093/bioinformatics/btaa800. [DOI] [PubMed] [Google Scholar]
- 360.Lim J, Ryu S, Kim JW, Kim WY. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J Cheminform. 2018;10(1):31. doi: 10.1186/s13321-018-0286-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. Highly accurate protein structure prediction with AlphaFold. Nature [Internet]. 2021; doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–76. doi: 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Hunter JD. Matplotlib: A 2D Graphics Environment. Comput Sci Eng. 2007;9:90–95. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 364.R Core Team . R: A language and environment for statistical computing [Internet]. 2014; http://www.R-project.org/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Datasets, codes, and figures are available at https://github.com/csi-greifflab/manuscript_progress_challenges_on-demand_antibody. Plots were generated in Matplotlib (version 3.3.4)363 and R (version 3.6.1).364