

Supplemental Digital Content is Available in the Text. Protein arginine methyltransferase 7 modulates NaV1.9 currents and neuronal excitability by interacting with and methylating NaV1.9, whereas protein arginine methyltransferase 7 inhibitor (DS-437) inhibits the action potential frequency of dorsal root ganglion neurons in Scn11a A796G/A796G mice.
Keywords: NaV1.9, PRMT7, Dorsal root ganglia, Neuronal excitability, DS-437
Abstract
Human NaV1.9 (hNaV1.9), encoded by SCN11A, is preferentially expressed in nociceptors, and its mutations have been linked to pain disorders. NaV1.9 could be a promising drug target for pain relief. However, the modulation of NaV1.9 activity has remained elusive. Here, we identified a new candidate NaV1.9-interacting partner, protein arginine methyltransferase 7 (PRMT7). Whole-cell voltage-clamp recordings showed that coelectroporation of human SCN11A and PRMT7 in dorsal root ganglion (DRG) neurons of Scn11a −/− mice increased the hNaV1.9 current density. By contrast, a PRMT7 inhibitor (DS-437) reduced mNaV1.9 currents in Scn11a +/+ mice. Using the reporter molecule CD4, we observed an increased distribution of hLoop1 on the cell surface of PRMT7-overexpressing HKE293T cells. Furthermore, we found that PRMT7 mainly binds to residues 563 to 566 within the first intracellular loop of hNaV1.9 (hLoop1) and methylates hLoop1 at arginine residue 519. Moreover, overexpression of PRMT7 increased the number of action potential fired in DRG neurons of Scn11a +/+ mice but not Scn11a −/− mice. However, DS-437 significantly inhibited the action potential frequency of DRG neurons and relieved pain hypersensitivity in Scn11a A796G/A796G mice. In summary, our observations revealed that PRMT7 modulates neuronal excitability by regulating NaV1.9 currents, which may provide a potential method for pain treatment.
1. Introduction
Voltage-gated sodium channel subtype 1.9 (VGSC, NaV1.9) is predominantly expressed in nociceptive neurons, trigeminal neurons, and myenteric neurons. 9 NaV1.9 channels produce a persistent TTX-R sodium current and act as a threshold channel that contributes to the resting potential and prolongs the depolarization response to subthreshold stimuli, 13 lowering the threshold for single action potentials and increasing the frequency of action potential firing. 5,30 Previous evidence suggests that NaV1.9 acts as an effector of peripheral inflammatory pain in mice. In 2013, we first reported that SCN11A mutations were linked to familial episodic pain. 44 After that, many SCN11A mutations were found to be linked to human pain disorders. 7,21 Therefore, NaV1.9 could be a promising target for pain treatment because it is only associated with pain disorders and has not been reported to be involved in other pathological conditions.
To date, 10 α subunits of VGSCs (NaV1.1-1.9 and the atypical channel NaX) have been identified in mammals. NaV1.9 shows the lowest homology with other NaV family members at the amino acid sequence level, although it shares a common structural topology with other α subunit family members. These α subunits are composed of a long, single polypeptide chain that comprises 4 homologous repeats (domains I-IV). The 4 domains are linked by 3 intracellular loops, 9 which play an essential role in the regulation of VGSCs by binding with or modifying other proteins. However, the functions of intracellular domains in the NaV1.9 channel are only partially characterized, primarily because the expression of functional NaV1.9 in heterologous systems is difficult and studying this protein in isolated neurons is challenging. 10,22 To date, only a few studies have described the regulation of the kinetic properties and subcellular localization of NaV1.9, including posttranslational modifications (phosphorylation and glycosylation), 3,4,37 cytoplasmic domain–binding proteins (contactin and FHF1B), 23,24 and the G-protein pathway. 32,38 However, the detailed mechanism by which NaV1.9 mutations lead to pain disorders is unclear. Recently, we found that alcohol aggravated episodic pain in patients carrying both the NaV1.9 mutation and the ALDH2 polymorphism. We successfully constructed NaV1.9-knockin (KI) (Scn11a A796G/A796G) mice, and a COX2 inhibitor effectively relieved pain hypersensitivity in NaV1.9-KI mice. 42 We also found the first agonist of NaV1.9 (spider venom–derived peptide HpTx1) that induces hyperalgesia in NaV1.7-knockout mice. 46 However, selective NaV1.9 inhibitors have not yet been identified. Therefore, we aimed to find another strategy for the treatment of NaV1.9-related pain by inhibiting the interaction of NaV1.9 with other proteins.
In this study, we identified a novel interacting protein (protein arginine methyltransferase 7 [PRMT7]) with NaV1.9 by a yeast two-hybrid (Y2H) screening system. We found that NaV1.9 is a new nonhistone substrate of PRMT7 that binds loop1 of human NaV1.9 (hNaV1.9) and methylates arginine residue 519. Moreover, we found that PRMT7 regulates the current density of NaV1.9 and action potential firing of dorsal root ganglion (DRG) neurons. Importantly, the inhibition of PRMT7 significantly reduced the number of action potentials fired in DRG neurons and inhibited formalin-induced hyperalgesia in Scn11a A796G/A796G mice. Therefore, PRMT7 could serve as a potential target for new analgesic drug development by modulating NaV1.9.
2. Materials and methods
2.1. Mice
Scn11a −/− mice were kindly provided by Patrick Delmas. 27 Scn11A A796G/A796G mice were described earlier, 42 and Scn11a +/+ mice (C57BL/6J) were purchased from Hubei Provincial Center for Disease Control and Prevention (Wuhan, China). All animals were housed under 12-hour light/dark cycles with free access to food and water. All animal experiments were approved by the Ethics Committee on Animal Research of Huazhong University of Science and Technology (Wuhan, China) and the Animal Care and Use Committee of Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences (Shanghai, China).
2.2. Plasmid construction
The cDNA sequence encoding the first intracellular loop (residues 402-570) of mouse NaV1.9 (NM_011887.3) was subcloned into the pGBKT7 vector for use as “bait” in the Y2H screen. The first intracellular loop of human NaV1.9 (hLoop1) was fused to the pGEX-6P-1 vector for glutathione S-transferase (GST) fusion protein expression. The tagged eukaryotic proteins were correspondingly subcloned into the p3×FLAG-CMV-7.1 and pEGFP-C1 vectors. The pSIH1-H1-CopGFP-shRNA vector was used in the RNA interference experiment (Table S1, available at http://links.lww.com/PAIN/B444). The CD4-hLoop1 chimaeric construct was modified and generated as described. 12,45 The human CD4 (NM_000616.5) intracellular loop (residues 397-458) was replaced with loop 1 of hNaV1.9 (residues 404-572) using overlapping polymerase chain reaction (PCR). Then, a hemagglutinin (HA) tag was inserted after the signal peptide (residues 1-25).
For use in voltage-clamp recordings, the full-length cDNA of PRMT7 was subcloned into a pIRES2-EGFP expression vector such that the enhanced green fluorescent protein gene was downstream of the PRMT7 ATG and connected with an internal ribosome entry site (IRES) linker; the PRMT7 protein was produced from this vector as independent proteins from the same messenger RNA. The full-length cDNA of mouse PRMT7 (NM_145404.1) was subcloned into the pcDNA3.1 expression vector for use in current-clamp recordings.
2.3. Yeast two-hybrid assay
Mouse DRG cDNA library construction and Y2H screening were performed with Matchmaker Library Construction & Screening Kits according to the user manual (Clontech Laboratories, Inc, PT3955-1, Mountain View, CA). Candidate clones were identified by growth on quadruple dropout (QDO) medium lacking tryptophan, leucine, histidine, and adenine or on QDO-containing X-α-Gal medium. In yeast, α-galactosidase (MEL1) is expressed in response to direct GAL4-based Y2H interactions. This secreted enzyme hydrolyzes 5-bromo-4-chloro-3-indolyl-α-D-glucopyranoside (X-α-Gal), causing the yeast colonies to turn blue on QDO/X-α-Gal medium. The hybrid N/C terminal core of PRMT7 (NM_019023.3) and truncated loop1 of hNaV1.9 (NM_014139.2) were cloned into the 2-hybrid expression vectors pGBKT7 and pGADT7, respectively.
2.4. Cell culture and transfection
The human embryonic kidney cell line HEK293T was maintained under standard conditions in Dulbecco modified Eagle medium (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco, NSW, Australia) at 37°C in 5% CO2. For transient transfection, cells were transfected using LipoJet Transfection Reagent (SignaGen, Rockville, MD) according to the manufacturer's instructions.
2.5. Immunoprecipitation and immunoblotting
Transfected cells were lysed in IP lysis buffer for 30 minutes on ice. After lysis, the cells were centrifuged at 12,000 rpm for 10 minutes at 4°C. Total cell extracts were incubated with the appropriate antibodies on a rotator overnight at 4°C, after which protein A agarose beads (Merck Millipore, Billerica, MA) were added for a continued 4 hours incubation. Next, the immunoprecipitates were washed with IP lysis buffer and separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis. Proteins were detected using specific antibodies: mouse anti-GST antibody (AE001, ABclonal, Wuhan, China), mouse anti-HA antibody (AE008, ABclonal), mouse anti-GFP antibody (AE012, ABclonal), rabbit anti-PRMT7 (A12159, ABclonal), mouse anti-FLAG antibody (M185-3 L, MBL, Tokyo, Japan), rabbit anti-NaV1.9 antibody (ASC-017, Alomone Labs, Jerusalem, Israel), rabbit anti-methyl (mono) arginine antibody (ICP0801, ImmuneChem, Burnaby, Canada), control mouse immunoglobulin G (IgG), and rabbit IgG (B900620, 30000-0-AP, ProteinTech, Wuhan, China). For the endogenous immunoprecipitation experiment, proteins were extracted from Scn11a +/+ and Scn11a −/− mouse DRG tissues using radio immunoprecipitation assay (RIPA) cell lysis buffer (P0013B, Beyotime, Shanghai, China). The supernatant was then collected through centrifugation at 12,000 ×g for 10 minutes, followed by preclearance for 2 hours at 4°C with 20 μL of protein A. The treated supernatant was incubated overnight with rabbit anti-PRMT7 antibody, and we performed the subsequent steps described above.
2.6. Glutathione S-transferase pull-down assay
Glutathione S-transferase–tagged hLoop1 fusion vectors were transformed into Escherichia coli Rosetta cells, and expression was induced by the addition of 0.5 mM isopropyl-β-D-thiogalactoside at 30°C for 5 hours. GST fusion proteins were lysed using ultrasound, purified with glutathione–sepharose beads (Thermo Scientific, Waltham, MA) and subsequently incubated with GFP-PRMT7–overexpressing HEK293T cell lysates overnight at 4°C. After incubation, the beads were washed with IP lysis buffer (P0013, Beyotime, Shanghai, China), and the bound proteins that were pulled down were detected using immunoblotting.
2.7. Cell fractionation
HEK293T cells were cotransfected with FLAG-hLoop1 and GFP-PRMT7 or the empty GFP vector, harvested after 24 hours, and lysed with low-frequency ultrasound after which the membrane proteins were extracted using the Membrane and Cytosol Protein Extraction Kit (P0033, Beyotime, Shanghai, China). Western blotting was performed as described above.
2.8. Immunocytochemistry and immunohistochemistry
HEK293T cells transfected with the HA-CD4-hLoop1 fusion protein were fixed with 4% paraformaldehyde for 20 minutes and blocked with 5% albumin bovine V for 30 minutes at room temperature. Then, the cells were incubated with anti-HA antibody overnight at 4°C. Dissected DRG tissues were fixed with 4% paraformaldehyde overnight at 4°C and then immersed in a 20% to 30% sucrose gradient for 24 to 48 hours at 4°C for cryoprotection. Dorsal root ganglion sections (12 μm) were cut with a cryostat (Leica CM1950, Leica, Wetzlar, Germany) and mounted onto slides for immunofluorescence staining. The slides were exposed to an antigen retrieval solution before blocking with 5% albumin bovine V and 0.3% Triton X-100 for 2 hours at room temperature. The slides were incubated with mouse anti-PRMT7 (sc-376077, Santa Cruz Biotechnology, Dallas, TX) and rabbit anti-NaV1.9 primary antibodies overnight at 4°C. After 3 5-minute rinses, the HEK293T cells and slides were incubated with Alexa Fluor 594-labelled goat anti-mouse IgG (A11020, Life Technologies, Carlsbad, CA) or Alexa Fluor 488-labelled goat anti-rabbit IgG (A11070, Life Technologies) secondary antibodies for 2 hours at room temperature. Cell nuclei were stained with 4,6-diamidino-2-phenylindole. Images were captured using an Olympus FV1000 confocal microscope system.
2.9. Drug administration
The PRMT7 inhibitor DS-437 (GLPBIO, GC45927, Montclair, CA) was dissolved at 100 mM in DMSO and stored. The concentration of drug administered was 10 μM and 100 μM, 19,39 as obtained by diluting the stock solution (DMSO ≤ 0.1%). DS-437 was incubated with the neurons for 1 hour before current-clamp recordings. Whole-cell patch-clamp recordings were performed for up to 1 hour.
2.10. Formalin test, hot-plate test, and von Frey test
Seven to 8-week-old mice (n = 6 per group, the ratio of male to female mice was approximately 1-1) were used in the behaviour test. Before testing, the mice were placed in plastic testing chambers and habituated to the testing environment for 30 min. 33 The experiments were performed by investigators who were blinded to the drug administration conditions.
For the formalin test, DS-437 was diluted in saline and injected intraperitoneally. After 1 hour, formalin solution (5%, 20 μL) was injected into the surface of the hind paw. The duration of licking and biting behaviour was immediately recorded at 5-minute intervals for 45 minutes. The duration was analysed in 2 phases: the first phase (0-5 min) and the second phase (10-45 minutes).
For the hot-plate test, a hot or cold plate (Ugo Basile 35100, Gemonio, Italy) set to 52°C was used to test sensitivity to heat. 40 The mice were placed in the testing chamber, observed for nociceptive behaviour (licking, lifting paw, and flinching) which indicates sensitivity to heat, and quickly removed. The latencies were recorded 3 times. The mice were removed after a cutoff time of 60 seconds.
For the von Frey test, von Frey filaments (Ugo Basile 37277, Gemonio, Italy) were applied to test mechanical hyperalgesia using the “up–down” method as previously described. 8 The hind paws were struck by filaments of increasing stiffness. Filaments were carefully pressed upward at the sural nerve territory to induce a slight bend, and pressure was applied for more than 5 seconds. When an animal withdrew its hind paw from the fibre, the behaviour was scored as a response. All paws were tested 3 times, and the withdrawal latency was recorded as the average value.
2.11. Electrophysiology
Dorsal root ganglion neurons were dissected and subjected to conventional whole-cell patch-clamp recordings by using an Axopatch 200b amplifier as described in our previous studies. 42,44 Equal numbers of male and female mice were used. Protein arginine methyltransferase 7 or the control vector was mixed with hNaV1.9 at a ratio of 10:2 and then electroporated into Scn11a −/− mouse DRG neurons by using the Neon transfection system (Life Technologies). The bath solution for voltage-clamp recordings contained 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 10 mM HEPES, 0.1 mM CdCl2, 20 mM TEA-Cl, 0.001 mM TTX, and 10 to 30 mM glucose (pH 7.4, adjusted with NaOH) and the pipette contained 135 mM CsF, 10 mM NaCl, 2.5 mM MgCl2, 10 mM HEPES, 1 mM EGTA, 5 mM TEA-Cl, and 4 mM Mg-ATP (pH 7.4, adjusted with CsOH).
Fire-polished electrodes were fabricated with a resistance below 3 to 5 MΩ when filled with the pipette solution by PC-10 (Narishige). The pipette potential was zeroed before seal formation. Eighty percent to 90% series resistance compensation was applied to reduce voltage errors. Voltage-dependent currents were acquired at 5 minutes after establishing the whole-cell configuration.
Currents were elicited by 100-ms test pulses with the potential ranging from −120 to +10 mV at 5-mV increments. The activation curves were fitted to a Boltzmann function as follows: G/Gmax = 1/(1 + exp[Vm − V1/2)/k], where V1/2 is the midpoint of activation and k is the slope factor. Steady-state fast inactivation was evaluated with a series of 500-ms prepulses (−120 to −20 mV in 10-mV increments), followed by a 50-ms step depolarization to −50 mV, and steady-state inactivation curve were fitted with the function I/Imax = 1/(1 +1+ exp[(V1/2 − Vm)/k]).
Protein arginine methyltransferase 7 and the empty vector were individually electroporated into Scn11a +/+ mouse DRG neurons for current-clamp recordings. The pipette solution for current recordings contained 140 mM KCl, 0.5 mM EGTA, 5 mM HEPES, and 2 mM Mg-ATP (pH 7.3) with KOH (adjusted to 315 mOsm with dextrose), and the extracellular solution contained 140 mM NaCl, 3 mM KCl, 2 mM MgCl2, 2 mM CaCl2, and 10 mM HEPES (pH 7.3, adjusted with NaOH). The voltage threshold was calculated from the first peak on dV/dt vs V plot.
2.12. Statistical analysis
The data are presented as mean ± SEM. The data for the comparison of 2 groups were analysed using the independent samples t test, and one-way analysis of variance (ANOVA) and two-way ANOVA were used to compare data for 3 or more groups. Statistical analyses and graphic representations were performed using Origin software (OriginLab Corp, Northampton, MA).
3. Results
3.1. Identification of protein arginine methyltransferase 7 as a novel binding partner of mNaV1.9 using the yeast two-hybrid system
To identify proteins involved in regulating NaV1.9 channels through binding with the first loop (loop1), we screened the mouse DRG cDNA library with the Y2H system using the first intracellular loop of mouse NaV1.9 (mNaV1.9, residues 402-570) as bait (Fig. 1A). Through high-stringency selection on QDO agar plates, we acquired 258 positive yeast clones. Using the MEL1 reporter gene on QDO-containing X-α-Gal medium and bioinformatic analysis of the library plasmid inserts, one of the repetitive clones containing portions of the C-terminal region of PRMT7 was identified (Fig. 1B). This interaction manifested in yeast was reconfirmed by directed cotransformation of the purified library and bait plasmids (Fig. 1C). Pairwise BLAST analysis using the Clustal Omega program revealed strong homology between the mouse and human PRMT7 sequences (85.1% identity and 96.4% similarity overall) (Fig. 1B; and Fig. S1, available at http://links.lww.com/PAIN/B444) and loop1 of NaV1.9 proteins (76.3% identity and 93.5% similarity overall) (Fig. 1D). Therefore, we speculated that a functional relationship exists between human PRMT7 and NaV1.9-loop1.
Figure 1.

Yeast two-hybrid screen to identify proteins that interacted with NaV1.9. (A) Schematic structure of the NaV1.9 channel. Residues 402 to 570 of mNaV1.9 were used as bait for the yeast two-hybrid (Y2H) screen. (B) Location of the repeat-independent clones of PRMT7 identified by Y2H (blue lines, residues 594-692). The 2 domains (pink/green) and degrees of homology between full-length mouse and human PRMT7 are shown. (C) Residues 594 to 692 of mouse PRMT7 were purified from yeast clones for direct Y2H to confirm the interaction on QDO medium. (D) Amino acid sequence comparison of hLoop1 and mLoop1 using ClustalW (EMBL–European Bioinformatics Institute). Potential PRMT7 methylation motif, RXR, is boxed. PRMT7, protein arginine methyltransferase 7; QDO, quadruple dropout.
3.2. Protein arginine methyltransferase 7 binds NaV1.9 in vitro and in vivo
Next, we examined and validated the interaction between the loop1 fragment of hNav1.9 (hLoop1) and full-length PRMT7 by GST pull-down and reciprocal coimmunoprecipitation assays. The purified GST-hLoop1 fusion protein, but not GST alone, pulled down the GFP-PRMT7 fusion protein, indicating a direct association between hLoop1 and PRMT7 in vitro (Fig. 2A). Reciprocal coimmunoprecipitation with anti-FLAG or anti-GFP antibodies, but not the IgG control, immunoprecipitated the other tag-fused protein, indicating that full-length PRMT7 interacted with hLoop1 (Fig. 2B). Meanwhile, we tested the binding of PRMT7 to other cytoplasmic domains of hNaV1.9 by performing an immunoprecipitation assay in HEK293T cells (NT, residues 1-142; hLoop2, residues 815-1051; and CT, residues 1605-1791). We only detected binding between GFP-tagged PRMT7 and hLoop1 (Fig. 2C). In vivo, PRMT7 coimmunoprecipitated with its partner protein mNaV1.9 from Scn11a +/+ but not control IgG or Scn11a −/− mouse DRG tissues (Fig. 2D). Furthermore, we observed that the PRMT7 protein colocalized with mNaV1.9 in mouse DRG sections, and NaV1.9 expression in DRG neurons of WT mice showed an ∼35.9% overlap with the expression of PRMT7 (Figs. 2E–F). Our results indicated that this interaction also occurs endogenously in mouse DRG tissues.
Figure 2.

Interaction and coexpression of NaV1.9 with PRMT7. Interaction between hLoop1 and full-length hPRMT7 was analysed by GST pull-down assays (A) and reciprocal coimmunoprecipitation assays (B). (C) Interaction between the intracellular domains of hNaV1.9 and hPRMT7 was examined by coimmunoprecipitation assays. (D) Interaction of mPRMT7 with mNaV1.9 in mouse DRG tissues was assessed. Scn11a +/+ or Scn11a −/− mouse DRG tissue lysates were immunoprecipitated using anti-mPRMT7 antibodies and control IgG. The precipitates were immunoblotted with the indicated antibodies. (E) Immunohistochemical analysis of mPRMT7/mNaV1.9 expression in mouse DRG sections. The sections were stained for mPRMT7 (red) and mNaV1.9 (green) and DAPI (blue). mPRMT7 showed considerable colocalization with mNaV1.9 in mouse DRG neurons. Scale bars, 50 μm. (F) Percentages of mPRMT7-positive and mNaV1.9-positive neurons are shown. DAPI, 4,6-diamidino-2-phenylindole; DRG, dorsal root ganglion; GST, glutathione S-transferase; IgG, immunoglobulin G; hNaV1.9, human hNaV1.9; PRMT7, protein arginine methyltransferase 7.
3.3. Protein arginine methyltransferase 7 increases NaV1.9 currents by promoting its cell surface expression
We investigated the functional role of PRMT7 in the activity of the hNaV1.9 channel in mouse DRG neurons. PRMT7 plasmid or empty vector (mock) was cotransfected with SCN11A plasmid into Scn11a −/− mouse DRG neurons by electroporation. Voltage-clamp analysis revealed a significant increase in the hNaV1.9 peak current density in neurons overexpressing PRMT7 (−115 ± 16.8 pA/pF, n = 23) compared with the control empty vector (−66 ± 10.4 pA/pF, n = 25; P < 0.01) (Figs. 3A and B). To further confirm that PRMT7 is involved in hNaV1.9 current regulation, we then analysed the changes in mNaV1.9 channels in wild-type mouse DRG neurons incubated with or without DS-437 (PRMT7 inhibitor). The results showed that the inhibition of PRMT7 significantly reduced the current density of mNaV1.9 in DRG neurons (control: −199.30 ± 24.53 pA/pF, n = 16; 10 μM DS-437: −131.48 ± 20.55 pA/pF, n = 13, P = 0.078; 100 μM DS-437: −116.64 ± 21.96, n = 19, P<0.05) (Figs. 3C and D). However, neither condition affected the voltage-dependent activation curve or steady-state fast inactivation curve of NaV1.9 (Fig. S2 and Tables S2 and S3, available at http://links.lww.com/PAIN/B444).
Figure 3.

Protein arginine methyltransferase 7 affects the current density of NaV1.9 by promoting its accumulation on the cell membrane. (A) Representative whole-cell sodium currents evoked by voltage from Scn11a −/− mouse DRG neurons electroporated with SCN11A or SCN11A and PRMT7. (B) Current–voltage relationships in the indicated experimental groups. The peak current density (normalized by membrane capacitance) was statistically analysed (mock, n = 15; hNaV1.9, n = 25; and hNaV1.9 + hPRMT7, n = 23), and significant differences were tested by two-way ANOVA, followed by a post hoc Bonferroni test; infection × voltage: F(32, 948) = 5.846, P < 0.0001; infection: F(2, 948) = 66.28, P < 0.0001; voltage: F(15, 948) = 19.39, P<0.0001; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the mock group. (C) Representative whole-cell mNaV1.9 currents evoked by voltage from Scn11a +/+ mouse small DRG neurons treated without (control) or with DS-437. (D) Quantification of the peak mNaV1.9 current density in the experimental and control groups (control, n = 16; 10 μM DS-437, n = 13; 100 μM DS-437, n = 19). Significant differences were tested by two-way ANOVA, followed by a post hoc Bonferroni test; DS-437 × voltage: F(30, 693) = 3.237, P < 0.0001; DS-437: F(2, 693) = 27.49, P < 0.0001; voltage: F(15, 693) = 79.98, P < 0.0001; ***P < 0.001 compared with the control. (E) Schematic representation of the chimeric proteins. (F) HEK293T cells transiently expressing HA-CD4-hLoop1 and GFP-hPRMT7 or mock-transfected GFP cells were harvested and lysed, the whole-cell lysates (WCLs) were cleared of debris, and the postnuclear supernatant was fractionated into the membrane (Mem) and cytosolic (Cyto) fractions with a kit. Aliquots from all stages of fractionation were analysed by immunoblotting with the indicated antibodies. Na+K+-ATPase was used as a cell-surface–protein control. Quantification of Western blotting data showed that hPRMT7 significantly increased the amount of chimeras in plasma membranes (G) but did not affect the total expression level of the chimeric proteins (H). Data were statistically analysed by the unpaired Student t test; *P < 0.05 compared with GFP. All studies were repeated at least 3 times. DRG, dorsal root ganglion; hNaV1.9, human NaV1.9; PRMT7, protein arginine methyltransferase 7.
Given the important role of PRMT7 in the modulation of NaV1.9 currents, previous studies have suggested that methyltransferase is involved in protein translocation processes, including cell surface trafficking. 6,43 Thus, we examined whether PRMT7 affects the subcellular distribution of hLoop1. For this experiment, we constructed a chimeric protein as a reporter molecule that was present on the surface of HEK293T cells (Fig. 3E). Consequently, we examined the effects of PRMT7 on the membrane distribution of chimeric proteins. We performed subcellular fractionation of HEK293T cells coexpressing chimeric proteins and GFP-PRMT7 or the empty GFP vectors to examine the effects of PRMT7 on the membrane distribution of the chimeras. Compared with the empty vector, overexpression of PRMT7 significantly increased the amount of chimeric proteins in the membrane fractions (Fig. 3F-H). By nonpermeabilized immunofluorescence labelling of HEK293T cells, we found that PRMT7 increased the relative fluorescence intensity of chimeric proteins compared with the control (Fig. S3A and B, available at http://links.lww.com/PAIN/B444). These data suggest that PRMT7 facilitates the cell surface expression of hNaV1.9 to increase its currents.
3.4. Protein arginine methyltransferase 7 modulates NaV1.9 activity by binding and methylating hLoop1
Protein arginine methyltransferase 7 contains C-PRMT and N-PRMT core domains in tandem. The Y2H assay was repeated to verify that the interaction occurred mainly between hLoop1 and the C-terminal core domain (residues 364-692) of PRMT7 (Fig. S4A, available at http://links.lww.com/PAIN/B444). To define the PRMT7 binding site in hLoop1, we performed a series of truncated hLoop1 hybrid constructs with the C-terminal part of PRMT7 for Y2H assays. The results revealed that the deletion of proximal end residues of hLoop1 retained the interaction with PRMT7, whereas mutants in which distal end residues were deleted did not bind the PRMT7 C-terminal core. In particular, the deletion of residues 563 to 572 completely eliminated the binding of PRMT7 (Fig. S4B, available at http://links.lww.com/PAIN/B444). We then constructed a series of 4-residue alanine substitution mutations of hLoop1 among the 10 residues identified to be critical for binding with PRMT7 (Fig. S4C and D, available at http://links.lww.com/PAIN/B444). One of the 4 single alanine substitution clones (563-566A) showed a considerable growth defect on selective medium (Fig. 4A and Fig. S4C-D, available at http://links.lww.com/PAIN/B444). Further confirmation of the interactions between these mutants and the PRMT7-C domain was obtained by performing β-galactosidase assays, which allow direct visualization of protein interactions (Fig. S4D, available at http://links.lww.com/PAIN/B444). These Y2H experiments showed that the binding between PRMT7 C-terminal core and cytoplasmic loop1 of hNaV1.9 mainly requires residues 563 to 566 (Trp, Leu, Cys, and Val).
Figure 4.

Protein arginine methyltransferase 7 modulates NaV1.9 currents by binding and methylating hLoop1. (A) Schematic representation of one deletion mutant and 4 single-alanine–scanning constructs spanning the region between residues 563 and 572 used in the Y2H assay. The percentage of yeast transformant growth on QDO medium was analysed correspondingly by one-way ANOVA (n = 3). ***P < 0.001, n.s., no significance. (B) Immunoblotting analysis showing the enhanced monomethylation of hLoop1 in hPRMT7-overexpressing cells with the indicated antibodies. (C) Immunoblotting with the indicated antibodies showed decreased MMA levels of hLoop1 in hPRMT7-knockdown cells and increased MMA levels of hLoop1 in hPRMT7-overexpressing cells. (D) Immunoprecipitation of FLAG-tagged wild-type (WT) R519A, R521A, or R519A/R521A hLoop1 proteins expressed in HEK293T cells using anti-FLAG antibodies. The methylation signals and target bands were immunoblotted with the indicated antibodies. (E) Representative whole-cell hNaV1.9 currents evoked by voltage from Scn11a −/− mouse small DRG neurons electroporated with the indicated groups. (F) Current–voltage relationships of the indicated experimental groups. The peak current density (normalized by membrane capacitance) was statistically analysed (hNaV1.9-4A, n = 14; hNaV1.9-4A+hPRMT7, n = 12; hNaV1.9-R519A, n = 10; hNaV1.9-R519A+hPRMT7, n = 9), and significant differences were tested by two-way ANOVA. ANOVA, analysis of variance; DRG, dorsal root ganglion; hNaV1.9, human NaV1.9; MMA, monomethylarginine; PRMT7, protein arginine methyltransferase 7; QDO, quadruple dropout; WCLs, whole-cell lysates; Y2H, yeast two-hybrid.
Protein arginine methyltransferase 7, a class III methyltransferase, generates monomethylarginine residues of histone or nonhistone substrates (Fig. S5, available at http://links.lww.com/PAIN/B444). We observed that inhibition of PRMT7 altered the activity of NaV1.9 channels. Therefore, we tested whether PRMT7 methylates hLoop1 in vitro. HEK293T cells expressing FLAG-hLoop1 were transfected with control, GFP-PRMT7 or PRMT7 shRNA vectors, followed by immunoprecipitation with anti-FLAG antibodies and immunoblotting with antibodies against monomethylarginine (MMA). Protein arginine methyltransferase 7 overexpression enhanced hLoop1 methylation (Fig. 4B). Conversely, PRMT7 knockdown specifically decreased MMA-positive hLoop1 levels (Fig. 4C; and S6, available at http://links.lww.com/PAIN/B444), suggesting that hLoop1 methylation is sensitive to PRMT7 levels.
The RXR motif is a preferred motif for PRMT7 arginine methylation. 17,28,39 Loop1 of NaV1.9 contains a potential R519XR521 motif (Fig. 1D). Thus, the effects of arginine-to-alanine mutations at arginine residues 519 (R519A), 521 (R521A), and 519/R521 (R519A/R521A) in hLoop1 were examined. In HEK293T cells, only the R519A mutant showed reduced MMA immunoreactivity. None of the mutations affected the interaction with PRMT7 (Fig. 4D). These data indicated that PRMT7 binds residues 563 to 566 (Trp, Leu, Cys, and Val) of cytoplasmic loop1 and methylates hLoop1 at arginine 519 (R519me).
Moreover, the regulatory effect of PRMT7 on the current density of hNaV1.9 was weakened when we mutated the binding and methylation sites in hNaV1.9, although both mutants showed markedly lower current densities than wild-type hNaV1.9 (Fig. 4E and F). These results suggest that PRMT7 modulates NaV1.9 activity by binding and methylating NaV1.9-loop1.
3.5. Overexpression of protein arginine methyltransferase 7 contributed to hyperexcitability of Scn11a +/+ mouse dorsal root ganglion neurons
NaV1.9 channels mainly respond to subthreshold stimuli and modulate action potential firing. 13 We analysed the impact of mPRMT7 on neuronal excitability at basal mNaV1.9 protein levels in Scn11a +/+ mouse DRG neurons. Current-clamp recordings were performed on Scn11a +/+ mouse DRG neurons transfected with the empty vector or PRMT7 construct to investigate the effects on action potential firing. We injected currents ranging from 0 to 225 pA in 25-pA increments for 200 ms to transfected DRG neurons and measured the representative current-clamp responses to serial current pulse injection (Figs. 5A–D). Six parameters related to action potential firing were analysed: the resting membrane potential (RMP), rheobase, voltage threshold, amplitude, half-width, and after hyperpolarization (AHP). Protein arginine methyltransferase 7 overexpression significantly decreased rheobase (mock: 65.6 ± 8.5 pA, n = 16; mPRMT7: 36.1 ± 6.1 pA, n = 9) to evoke an action potential (Fig. 5E). However, no significant differences in RMP (mock: −44.8 ± 1.0 mV, n = 27; mPRMT7: −44.3 ± 0.6 mV, n = 27), voltage threshold (mock: −20.1 ± 0.7 mV, n = 27; mPRMT7: −21.1 ± 1.0 mV, n = 27), amplitude (mock: 108.5 ± 3.5 mV, n = 26; mPRMT7: 105.1 ± 3.5 mV, n = 27), half-width (mock: 2.14 ± 0.15 ms, n = 26; mPRMT7: 2.15 ± 0.15 ms, n = 27), or AHP (mock: −15.9 ± 2.3 mV, n = 26; mPRMT7: −15.8 ± 1.6 mV, n = 27) were observed in the DRG neurons (Fig. 5F-G; and Fig. S7A-C, available at http://links.lww.com/PAIN/B444). Importantly, the frequency of action potentials in Scn11a +/+ mouse DRG neurons transfected with the PRMT7 plasmid was significantly higher than that in mock-transfected cells in response to a similar stimulus from 50 pA to 225 pA, but not in Scn11a −/− mouse DRG neurons (Fig. 5H). These results indicate that mPRMT7 increases neuronal hyperexcitability by decreasing rheobase.
Figure 5.

mPRMT7 promoted the hyperexcitability of mouse DRG neurons in a SCN11A-dependent manner. Current-clamp responses to 200-ms depolarizing current steps of 50, 100, 150, and 225 pA in representative Scn11a −/− mouse DRG neurons (A-B) and Scn11a +/+ mouse DRG neurons (C-D) expressing the empty vector pcDNA3.1/GFP (mock) and pcDNA3.1-PRMT7/GFP (PRMT7). (E) Rheobase, (F) resting membrane potential (RMP), and (G) Vthreshold (the threshold at which AP takeoff occurs) were not significantly altered in mock-transfected DRG neurons. Data were statistically analysed by the unpaired Student t test; *P < 0.05 compared with the mock group. (H) Comparison of the average spike number of repetitive action potentials (APs) fired in response to the 200-ms current injection ranging from 0 to 225 pA in DRG neurons overexpressing the empty vector or PRMT7. (mNaV1.9+/+ + mock, n = 28; mNaV1.9+/+ + mPRMT7, n = 27; mNaV1.9−/− + mock, n = 41; mNaV1.9−/− + mPRMT7, n = 37). Significant differences were tested by two-way ANOVA, followed by a post hoc Bonferroni test; infection × current: F(27, 1027) = 1.815, P = 0.0069; infection: F(3, 1027) = 42.19, P < 0.0001; current: F(9, 1027) = 114.5, P < 0.0001; *P < 0.05 and **P < 0.01 compared with the mock group. ANOVA, analysis of variance; DRG, dorsal root ganglion; PRMT7, protein arginine methyltransferase 7.
3.6. A protein arginine methyltransferase 7 inhibitor reduced the excitability of Scn11a A796G/A796G mouse dorsal root ganglion neurons
To further test whether the inhibition of PRMT7 can reduce neuronal activity by decreasing NaV1.9 currents, we used Scn11a A796G/A796G mouse DRG neurons, which exhibited neuronal hyperexcitability through gain-of-function mNaV1.9 channels that lead to hyperalgesia. 42 We analysed the effects of the PRMT7 inhibitor DS-437 on neuronal excitability in DRG neurons from Scn11a A796G/A796G and Scn11a +/+ mice by performing current-clamp recordings and determining the representative current-clamp responses to serial current pulse injections (Fig. 6A). DS-437 (100 μM) significantly increased rheobase (control: 42.31 ± 4.37 pA, n = 31; 10 μM: 50.00 ± 4.35 pA, n = 22; 100 μM: 63.46 ± 6.71 pA, n = 21) in Scn11a A796G/A796G mouse DRG neurons (Fig. 6B). However, the other parameters, including the RMP (control: −48.10 ± 0.79 mV, n = 31; 10 μM: −49.68 ± 0.80 mV, n = 22; 100 μM: −49.67 ± 0.75 mV, n = 21), voltage threshold (control: −15.06 ± 0.74 mV, n = 31; 10 μM: −14.37 ± 0.47 mV, n = 22; 100 μM: −14.83 ± 1.12 mV, n = 21), amplitude (control: −131.07 ± 2.00 mV, n = 31; 10 μM: 127.62 ± 1.81 mV, n = 22; 100 μM: 131.27 ± 2.01 mV, n = 21), half-width (control: 1.04 ± 0.04 ms, n = 31; 10 μM: 0.98 ± 0.03 ms, n = 22; 100 μM: 0.98 ± 0.05 ms, n = 21), and AHP (control: −26.40 ± 0.69 mV, n = 31; 10 μM: −24.66 ± 1.20 mV, n = 22; 100 μM: −25.39 ± 1.27 mV, n = 21), were not significantly altered (Figs. 6C–D; and Fig. S7D-F, available at http://links.lww.com/PAIN/B444). In addition, DS-437 prominently attenuated the action potential firing in Scn11a A796G/A796G mouse DRG neurons (Fig. 6E). However, DS-437 did not affect the frequency of action potentials fired, rheobase (control: 56.67 ± 5.70 pA, n = 21; 100 μM: 65.79 ± 8.16 pA, n = 23), RMP (control: −49.00 ± 0.94 mV, n = 21; 100 μM: −49.61 ± 1.20, n = 23), voltage threshold (control: −14.83 ± 1.24 mV, n = 21; 100 μM: −15.17 ± 1.40, n = 23), amplitude (control: 126.40 ± 2.49 mV, n = 21; 100 μM: 126.29 ± 3.08 mV, n = 23), half-width (control: 1.48 ± 0.14 ms, n = 21; 100 μM: 1.46 ± 0.14 ms, n = 23), or AHP (control: −24.47 ± 1.03 mV, n = 21; 100 μM: −24.09 ± 1.69, n = 23) in Scn11a +/+ mouse DRG neurons (Figs. 6B–E; and Fig. S7D-F, available at http://links.lww.com/PAIN/B444).
Figure 6.

DS-437 reduced DRG neuronal excitability and relieved pain hypersensitivity in Scn11a A796G/A796G mice. (A) Current-clamp responses to a 200-ms depolarizing current of 200 pA in representative Scn11a +/+ mouse DRG neurons and Scn11a A796G/A796G mouse DRG neurons with DS-437 or control. (B) Rheobase, (C) resting membrane potential (RMP), and (D) Vthreshold (the threshold at which AP takeoff occurs) showed no significant changes in DRG neurons treated with or without DS-437. Data were statistically analysed by one-way ANOVA; *P < 0.05 compared with the control. (E) Comparison of the average spike numbers of repetitive action potentials (APs) fired in response to the 200-ms current injection ranging from 0 to 225 pA in DRG neurons (NaV1.9-WT + control, n = 21; NaV1.9-WT + 100 μM DS-437, n = 22; NaV1.9-KI + control, n = 30; NaV1.9-KI + 10 μM DS-437, n = 22; NaV1.9-KI + 100 μM DS-437, n = 24). Significant differences were tested by two-way ANOVA, followed by a post hoc Bonferroni test; DS-437 × current: F(36, 1041) = 2.049, P = 0.0003; DS-437: F(3, 1041) = 50.86, P < 0.0001; current: F(9, 1041) = 74.58, P < 0.0001; **P < 0.01 and ***P < 0.001 compared with the control. (F) The duration of licking and lifting behaviours in NaV1.9-KI mice and NaV1.9-WT in the 45 minutes after intraplantar administration of formalin to the hind paws binned at 5-min intervals. (G) Data from 2 phases of the formalin test are summarized. Phase I: 0 to 10 minutes. Phase II: 10 to 45 minutes (saline, n = 6; DS-437, n = 6). Significant differences were tested by one-way ANOVA; *P < 0.05 and **P < 0.01 compared with saline. (H) Heat threshold was assessed using the hot-plate test. (I) Mechanical withdrawal threshold was tested by applying von Frey filaments (saline, n = 6; DS-437, n = 6). Significant differences were tested by two-way ANOVA; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with saline. ANOVA, analysis of variance; DRG, dorsal root ganglion.
In preliminary in vivo experiments, the intraperitoneal injection of DS-437 (1.6 mg/kg) reduced the duration of licking and lifting of the hind paws in Scn11a +/+ and Scn11a A796G/A796G mice after intraplantar administration of 5% formalin in the first and second phases, and 0.8 mg/kg DS-437 also decreased paw licking and lifting time in the second phase in Scn11a A796G/A796G mice compared with the controls (Figs. 6F–G). In addition, 1.6 mg/kg DS-437 increased the hot palate latency and reduced mechanical sensitivity at 1 and 2 hours after intraperitoneal administration, and 0.8 mg/kg DS-437 reduced mechanical sensitivity in Scn11a A796G/A796G mice but not Scn11a +/+ mice (Figs. 6H–I).
Altogether, these results indicate that inhibiting the activity of PRMT7 may relieve pain hypersensitivity by decreasing DRG neuron excitability in Scn11a A796G/A796G mice.
4. Discussion
NaV1.9, as a threshold channel for action potential electrogenesis, is particularly important in inflammatory, 2 neuropathic, 31 and visceral 14 pain in rodent models, and variants of SCN11A in humans lead to congenital insensitivity to pain and painful syndromes. 10 However, the regulatory mechanisms of NaV1.9 channel activity are still largely uncharacterized, especially posttranslational modifications. In this study, we identified PRMT7 as a new binding partner of NaV1.9. Protein arginine methyltransferase 7 contains the C-terminal and N-terminal domains. We found that the C-terminal part of PRMT7 exhibits strong binding to hLoop1 (Fig. S4A, available at http://links.lww.com/PAIN/B444). As a member of the protein arginine methyltransferase family, PRMT7 binds loop1 of hNav1.9 through residues 563 to 566 (WLCV motif) (Figs. S4C and S4D, available at http://links.lww.com/PAIN/B444) and methylates arginine residue 519 (Fig. 4D). These data further confirmed that the C-terminal core domain of PRMT7 participates in protein methylation and revealed that the PRMT7 substrate-binding region is distinct from the arginine methylation site.
However, the detailed mechanism by which PRMT7 regulates the NaV1.9 current remains unknown. There may be 2 possible mechanisms: intracellular protein trafficking and protein stability. The mammalian PRMT family contains 9 highly related members (PRMT1-PRMT9) that share common structure and signature methyltransferase motifs, which have been shown to modulate the nucleus–cytoplasm translocation of RNA-binding proteins 25,35 and molecule cell surface trafficking. 6 PRMT3 or PRMT5 could increase the NaV1.5 current density by enhancing NaV1.5 cell surface expression. 6 In our study, by using reporter molecules, we found that PRMT7 increased the expression of the first loop of hNaV1.9 in the membrane compartment (Fig. 3F). On the other hand, arginine methylation by protein arginine methyltransferases could increase protein stability and reduce ubiquitination. 34 It would be interesting to determine whether PRMT7-methylated NaV1.9 channels exhibit increased stability with extended duration of localization at the cell surface because PRMT7 did not affect the electrical properties of the hNaV1.9 channel. In addition, a study reported that the RRR motif in the first intracellular loop of NaV1.8 is responsible for restricting its surface expression. 45 Although we cannot exclude the possibility that increased single-channel conductance may regulate current density, we propose the following hypothesis regarding the mechanism of NaV1.9 membrane targeting: Methylation of R519 of hLoop1 by PRMT7 is required for NaV1.9 trafficking and its subsequent accumulation on the cell surface (Fig. 7).
Figure 7.
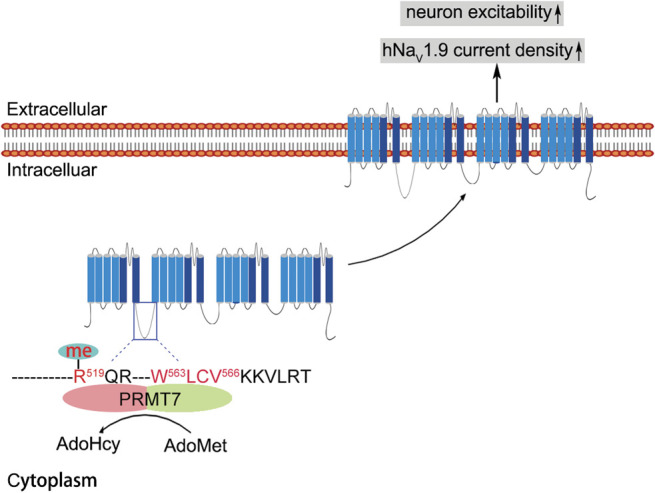
Proposed working model of PRMT7-mediated NaV1.9 trafficking and cellular excitability. The PRMT7 C-terminal domain interacts with residues 563 to 566 of hLoop1 and methylates arginine 519 (R519 me) in this loop using S-adenosyl-L-methionine (AdoMet) as a methyl donor to produce S-adenosylhomocysteine (AdoHcy). Human NaV1.9; R519 me is involved in the regulation of NaV1.9 trafficking to the cell surface through an undefined mechanism. Consequently, altered cell surface expression of NaV1.9 increases sodium current density, leading to hyperexcitability of DRG neurons. DRG, dorsal root ganglion; PRMT7, protein arginine methyltransferase 7.
NaV1.7, NaV1.8, and NaV1.9 are preferentially expressed in the peripheral nervous system and thereby regulate neuronal excitability and pain signals. 7 Multiple specific blockers that act on NaV1.7 and NaV1.8 have undergone clinical trials and have been validated as targets for pain treatment. 1 To date, a NaV1.9-specific inhibitor has not been developed because NaV1.9 exhibits very poor heterologous expression. However, NaV1.9-related familial episodic pain was remarkedly relieved by treatment with NSAIDs in patients. 20,29,44 According to a recent study, parecoxib can reduce NaV1.9 currents and inhibit neuronal excitability. 42 However, the detailed mechanism remains unclear. In this study, overexpression of PRMT7 increased hyperexcitability by decreasing rheobase in Scn11a +/+ mouse DRG neurons but not Scn11a −/− mouse DRG neurons. The increased rheobase in the presence of the PRMT7 inhibitor induced a decrease in neuronal excitability in Scn11a A796G/A796G mouse DRG neurons but not in Scn11a +/+ mouse DRG neurons (Figs. 6A–E). In a previous study, rheobase was somewhat greater in Scn11a −/− mouse DRG neurons, but action potential firing was not affected. 31 On the other hand, PRMT7 regulates the activation of p38 mitogen-activated protein kinase by interacting with p38 and stabilizing the active form, 16 but p-p38 was detected in a few DRG neurons without the application of tumor necrosis factor α (TNFα) 18 and shown to be associated with inflammatory and neuropathic pain. 26 In addition, PRMT7 regulates the number of hyperpolarization-activated cyclic nuleotide-gated (HCN) channels by controlling HCN1 protein expression. 19 However, HCN1 is expressed in medium to large DRG neurons, 11,36 and HCN expression was shown to increase with hind limb inflammation induced by complete Freund's adjuvant (CFA) in some of the small DRG neurons. 41 Therefore, we believe that PRMT7 regulates neuronal excitability mainly by modulating NaV1.9 activity in DRG neurons.
NaV1.9 mutations that cause large hyperpolarizing shifts in voltage dependence of activation of NaV1.9 cause large depolarizing shifts in RMP and silencing of nociceptors, whereas mutations that cause modest hyperpolarizing shifts in voltage dependence of activation of the channel cause smaller depolarizing shifts in RMP and induce hyperexcitability of nociceptors. 7,15 However, the inhibition of NaV1.9 attenuated hyperexcitability in Scn11a A796G/A796G mouse DRG neurons (Fig. 6E). NaV1.9 could be a promising strategy for treating peripheral inflammation and NaV1.9-related hyperalgesia. In this study, a PRMT7 inhibitor relieved formalin-induced pain hypersensitivity in WT and NaV1.9-KI mice. In addition, inhibiting PRMT7 activation attenuated mechanical and thermal sensitivity in NaV1.9-KI mice but not WT mice (Figs. 6F–I). Our results support the hypothesis that NaV1.9 contributes to pain signal processing under pathological conditions (inflammatory and neuropathic pain, as well as gain-of-function mutation). 2,20,31,44
In summary, we have identified a new signalling link between PRMT7 and NaV1.9 involved in the control of neuronal excitability and provided a mechanism underlying the prevention of neuronal hyperexcitability through sodium conductance. This study may also offer a new research area, namely, the posttranslational modification of NaV1.9, which contributes to the development of NaV1.9-related analgesic treatments.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B444.
Acknowledgments
The authors thank Dr. Patrick Delmas (Université de la Méditerranée, Marseille Cedex, France) for providing the Scn11a-knockout mice. This work was supported by the National Natural Science Foundation of China grants (31871262 and 31671301) to J.Y. Liu and (81741051) to X. Shi and Shanghai Municipal Science and Technology Major Project (grant No. 2018SHZDZX05) to J.Y. Liu The authors have declared that no conflict of interest exists.
Author contributions: J.Y. Liu and X. Shi conceived the study and designed the experiments. T. Ma, L. Li, R. Chen, and L. Yang analysed the data and wrote the manuscript. T. Ma and L. Yang conducted the patch-clamp recordings and animal behavioural experiments. L. Li conducted the protein interaction screen and molecular experiments. R. Chen, Z. Cao, H. Sun, S. Du., and X. Xu also conducted the molecular experiments. X. Zhang and L. Zhang participated in discussions and editing of the manuscript.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
T. Ma, L. Li, R. Chen, and L. Yang contributed equally to this work.
Contributor Information
Tingbin Ma, Email: m201571580@hust.edu.cn.
Lulu Li, Email: honeelulu@hust.edu.cn.
Rui Chen, Email: D201677466@hust.edu.cn.
Luyao Yang, Email: yangluyao@hust.edu.cn.
Hao Sun, Email: D201577463@hust.edu.cn.
Shiyue Du, Email: D201677475@hust.edu.cn.
Xuan Xu, Email: xxu@ion.ac.cn.
Zhijian Cao, Email: D201880563@hust.edu.cn.
Xianwei Zhang, Email: ourpain@163.com.
Luoying Zhang, Email: zhangluoying@hust.edu.cn.
Xiaoliu Shi, Email: shixl6@csu.edu.cn.
References
- [1]. Alsaloum M, Higerd GP, Effraim PR, Waxman SG. Status of peripheral sodium channel blockers for non-addictive pain treatment. Nat Rev Neurol 2020;16:689–705. [DOI] [PubMed] [Google Scholar]
- [2]. Amaya F, Wang H, Costigan M, Allchorne AJ, Hatcher JP, Egerton J, Stean T, Morisset V, Grose D, Gunthorpe MJ, Chessell IP, Tate S, Green PJ, Woolf CJ. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci 2006;26:12852–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]. Bai Q, Shao J, Cao J, Ren X, Cai W, Su S, George S, Tan Z, Zang W, Dong T. Protein kinase C-alpha upregulates sodium channel Nav1.9 in nociceptive dorsal root ganglion neurons in an inflammatory arthritis pain model of rat. J Cell Biochem 2020;121:768–78. [DOI] [PubMed] [Google Scholar]
- [4]. Baker MD. Protein kinase C mediates up-regulation of tetrodotoxin-resistant, persistent Na+ current in rat and mouse sensory neurones. J Physiol 2005;567:851–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Baker MD, Chandra SY, Ding Y, Waxman SG, Wood JN. GTP-induced tetrodotoxin-resistant Na+ current regulates excitability in mouse and rat small diameter sensory neurones. J Physiol 2003;548:373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]. Beltran-Alvarez P, Espejo A, Schmauder R, Beltran C, Mrowka R, Linke T, Batlle M, Perez-Villa F, Perez GJ, Scornik FS, Benndorf K, Pagans S, Zimmer T, Brugada R. Protein arginine methyl transferases-3 and -5 increase cell surface expression of cardiac sodium channel. FEBS Lett 2013;587:3159–65. [DOI] [PubMed] [Google Scholar]
- [7]. Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The role of voltage-gated sodium channels in pain signaling. Physiol Rev 2019;99:1079–151. [DOI] [PubMed] [Google Scholar]
- [8]. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53:55–63. [DOI] [PubMed] [Google Scholar]
- [9]. Dib-Hajj S, Black JA, Cummins TR, Waxman SG. NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci 2002;25:253–9. [DOI] [PubMed] [Google Scholar]
- [10]. Dib-Hajj SD, Black JA, Waxman SG. NaV1.9: a sodium channel linked to human pain. Nat Rev Neurosci 2015;16:511–19. [DOI] [PubMed] [Google Scholar]
- [11]. Doan TN, Stephans K, Ramirez AN, Glazebrook PA, Andresen MC, Kunze DL. Differential distribution and function of hyperpolarization-activated channels in sensory neurons and mechanosensitive fibers. J Neurosci 2004;24:3335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12]. Garrido JJ, Fernandes F, Giraud P, Mouret I, Pasqualini E, Fache MP, Jullien F, Dargent B. Identification of an axonal determinant in the C-terminus of the sodium channel Na(v)1.2. EMBO J 2001;20:5950–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Herzog RI, Cummins TR, Waxman SG. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J Neurophysiol 2001;86:1351–64. [DOI] [PubMed] [Google Scholar]
- [14]. Hockley JR, Winchester WJ, Bulmer DC. The voltage-gated sodium channel NaV 1.9 in visceral pain. Neurogastroenterol Motil 2016;28:316–26. [DOI] [PubMed] [Google Scholar]
- [15]. Huang J, Vanoye CG, Cutts A, Goldberg YP, Dib-Hajj SD, Cohen CJ, Waxman SG, George AL, Jr. Sodium channel NaV1.9 mutations associated with insensitivity to pain dampen neuronal excitability. J Clin Invest 2017;127:2805–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Jeong HJ, Lee HJ, Vuong TA, Choi KS, Choi D, Koo SH, Cho SC, Cho H, Kang JS. Prmt7 deficiency causes reduced skeletal muscle oxidative metabolism and age-related obesity. Diabetes 2016;65:1868–82. [DOI] [PubMed] [Google Scholar]
- [17]. Jeong HJ, Lee SJ, Lee HJ, Kim HB, Anh Vuong T, Cho H, Bae GU, Kang JS. Prmt7 promotes myoblast differentiation via methylation of p38MAPK on arginine residue 70. Cell Death Differ 2019;27:573–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Jin X, Gereau RW, IV. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci 2006;26:246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. Lee SY, Vuong TA, So HK, Kim HJ, Kim YB, Kang JS, Kwon I, Cho H. PRMT7 deficiency causes dysregulation of the HCN channels in the CA1 pyramidal cells and impairment of social behaviors. Exp Mol Med 2020;52:604–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]. Leipold E, Hanson-Kahn A, Frick M, Gong P, Bernstein JA, Voigt M, Katona I, Oliver Goral R, Altmuller J, Nurnberg P, Weis J, Hubner CA, Heinemann SH, Kurth I. Cold-aggravated pain in humans caused by a hyperactive NaV1.9 channel mutant. Nat Commun 2015;6:10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Leipold E, Liebmann L, Korenke GC, Heinrich T, Giesselmann S, Baets J, Ebbinghaus M, Goral RO, Stodberg T, Hennings JC, Bergmann M, Altmuller J, Thiele H, Wetzel A, Nurnberg P, Timmerman V, De Jonghe P, Blum R, Schaible HG, Weis J, Heinemann SH, Hubner CA, Kurth I. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat Genet 2013;45:1399–404. [DOI] [PubMed] [Google Scholar]
- [22]. Lin Z, Santos S, Padilla K, Printzenhoff D, Castle NA. Biophysical and pharmacological characterization of Nav1.9 voltage dependent sodium channels stably expressed in HEK-293 cells. PLoS One 2016;11:e0161450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23]. Liu C, Dib-Hajj SD, Waxman SG. Fibroblast growth factor homologous factor 1B binds to the C terminus of the tetrodotoxin-resistant sodium channel rNav1.9a (NaN). J Biol Chem 2001;276:18925–33. [DOI] [PubMed] [Google Scholar]
- [24]. Liu CJ, Dib-Hajj SD, Black JA, Greenwood J, Lian Z, Waxman SG. Direct interaction with contactin targets voltage-gated sodium channel Na(v)1.9/NaN to the cell membrane. J Biol Chem 2001;276:46553–61. [DOI] [PubMed] [Google Scholar]
- [25]. Lukong KE, Richard S. Arginine methylation signals mRNA export. Nat Struct Mol Biol 2004;11:914–15. [DOI] [PubMed] [Google Scholar]
- [26]. Mai L, Zhu X, Huang F, He H, Fan W. p38 mitogen-activated protein kinase and pain. Life Sci 2020;256:117885. [DOI] [PubMed] [Google Scholar]
- [27]. Maingret F, Coste B, Padilla F, Clerc N, Crest M, Korogod SM, Delmas P. Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J Gen Physiol 2008;131:211–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]. Miranda TB, Miranda M, Frankel A, Clarke S. PRMT7 is a member of the protein arginine methyltransferase family with a distinct substrate specificity. J Biol Chem 2004;279:22902–7. [DOI] [PubMed] [Google Scholar]
- [29]. Okuda H, Noguchi A, Kobayashi H, Kondo D, Harada KH, Youssefian S, Shioi H, Kabata R, Domon Y, Kubota K, Kitano Y, Takayama Y, Hitomi T, Ohno K, Saito Y, Asano T, Tominaga M, Takahashi T, Koizumi A. Infantile pain episodes associated with novel Nav1.9 mutations in familial episodic pain syndrome in Japanese families. PLoS One 2016;11:e0154827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. Ostman JA, Nassar MA, Wood JN, Baker MD. GTP up-regulated persistent Na+ current and enhanced nociceptor excitability require NaV1.9. J Physiol 2008;586:1077–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31]. Priest BT, Murphy BA, Lindia JA, Diaz C, Abbadie C, Ritter AM, Liberator P, Iyer LM, Kash SF, Kohler MG, Kaczorowski GJ, MacIntyre DE, Martin WJ. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci U S A 2005;102:9382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Rush AM, Waxman SG. PGE2 increases the tetrodotoxin-resistant Nav1.9 sodium current in mouse DRG neurons via G-proteins. Brain Res 2004;1023:264–71. [DOI] [PubMed] [Google Scholar]
- [33]. Shi Y, Chen Y, Wang Y. Kir2.1 channel regulation of glycinergic transmission selectively contributes to dynamic mechanical allodynia in a mouse model of spared nerve injury. Neurosci Bull 2019;35:301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Sivakumaran H, van der Horst A, Fulcher AJ, Apolloni A, Lin MH, Jans DA, Harrich D. Arginine methylation increases the stability of human immunodeficiency virus type 1 Tat. J Virol 2009;83:11694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35]. Souki SK, Gershon PD, Sandri-Goldin RM. Arginine methylation of the ICP27 RGG box regulates ICP27 export and is required for efficient herpes simplex virus 1 replication. J Virol 2009;83:5309–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Tu H, Deng L, Sun Q, Yao L, Han JS, Wan Y. Hyperpolarization-activated, cyclic nucleotide-gated cation channels: roles in the differential electrophysiological properties of rat primary afferent neurons. J Neurosci Res 2004;76:713–22. [DOI] [PubMed] [Google Scholar]
- [37]. Tyrrell L, Renganathan M, Dib-Hajj SD, Waxman SG. Glycosylation alters steady-state inactivation of sodium channel Nav1.9/NaN in dorsal root ganglion neurons and is developmentally regulated. J Neurosci 2001;21:9629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38]. Vanoye CG, Kunic JD, Ehring GR, George AL, Jr. Mechanism of sodium channel NaV1.9 potentiation by G-protein signaling. J Gen Physiol 2013;141:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39]. Vuong TA, Jeong HJ, Lee HJ, Kim BG, Leem YE, Cho H, Kang JS. PRMT7 methylates and suppresses GLI2 binding to SUFU thereby promoting its activation. Cell Death Differ 2019;27:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40]. Wang D, Stoveken HM, Zucca S, Dao M, Orlandi C, Song C, Masuho I, Johnston C, Opperman KJ, Giles AC, Gill MS, Lundquist EA, Grill B, Martemyanov KA. Genetic behavioral screen identifies an orphan anti-opioid system. Science 2019;365:1267–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41]. Weng X, Smith T, Sathish J, Djouhri L. Chronic inflammatory pain is associated with increased excitability and hyperpolarization-activated current (Ih) in C- but not Adelta-nociceptors. PAIN 2012;153:900–14. [DOI] [PubMed] [Google Scholar]
- [42]. Yang L, Li L, Tang H, Ma T, Li Y, Zhang X, Shi X, Liu JY. Alcohol-aggravated episodic pain in humans with SCN11A mutation and ALDH2 polymorphism. PAIN 2020;161:1470–82. [DOI] [PubMed] [Google Scholar]
- [43]. Zakrzewicz D, Didiasova M, Kruger M, Giaimo BD, Borggrefe T, Mieth M, Hocke AC, Zakrzewicz A, Schaefer L, Preissner KT, Wygrecka M. Protein arginine methyltransferase 5 mediates enolase-1 cell surface trafficking in human lung adenocarcinoma cells. Biochim Biophys Acta 2018;1864:1816–27. [DOI] [PubMed] [Google Scholar]
- [44]. Zhang XY, Wen J, Yang W, Wang C, Gao L, Zheng LH, Wang T, Ran K, Li Y, Li X, Xu M, Luo J, Feng S, Ma X, Ma H, Chai Z, Zhou Z, Yao J, Zhang X, Liu JY. Gain-of-function mutations in SCN11A cause familial episodic pain. Am J Hum Genet 2013;93:957–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45]. Zhang ZN, Li Q, Liu C, Wang HB, Wang Q, Bao L. The voltage-gated Na+ channel Nav1.8 contains an ER-retention/retrieval signal antagonized by the beta3 subunit. J Cell Sci 2008;121:3243–52. [DOI] [PubMed] [Google Scholar]
- [46]. Zhou X, Ma T, Yang L, Peng S, Li L, Wang Z, Xiao Z, Zhang Q, Wang L, Huang Y, Chen M, Liang S, Zhang X, Liu JY, Liu Z. Spider venom-derived peptide induces hyperalgesia in Nav1.7 knockout mice by activating Nav1.9 channels. Nat Commun 2020;11:2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B444.